
Drug Repositioning via Graph Neural Networks: Identifying Novel JAK2 Inhibitors from FDA-Approved Drugs through Molecular Docking and Biological Validation



 and
and
Abstract
1. Introduction
2. Results and Discussions
2.1. JAK2 Active and Decoy Datasets and Its Preprocessing Using RDKit
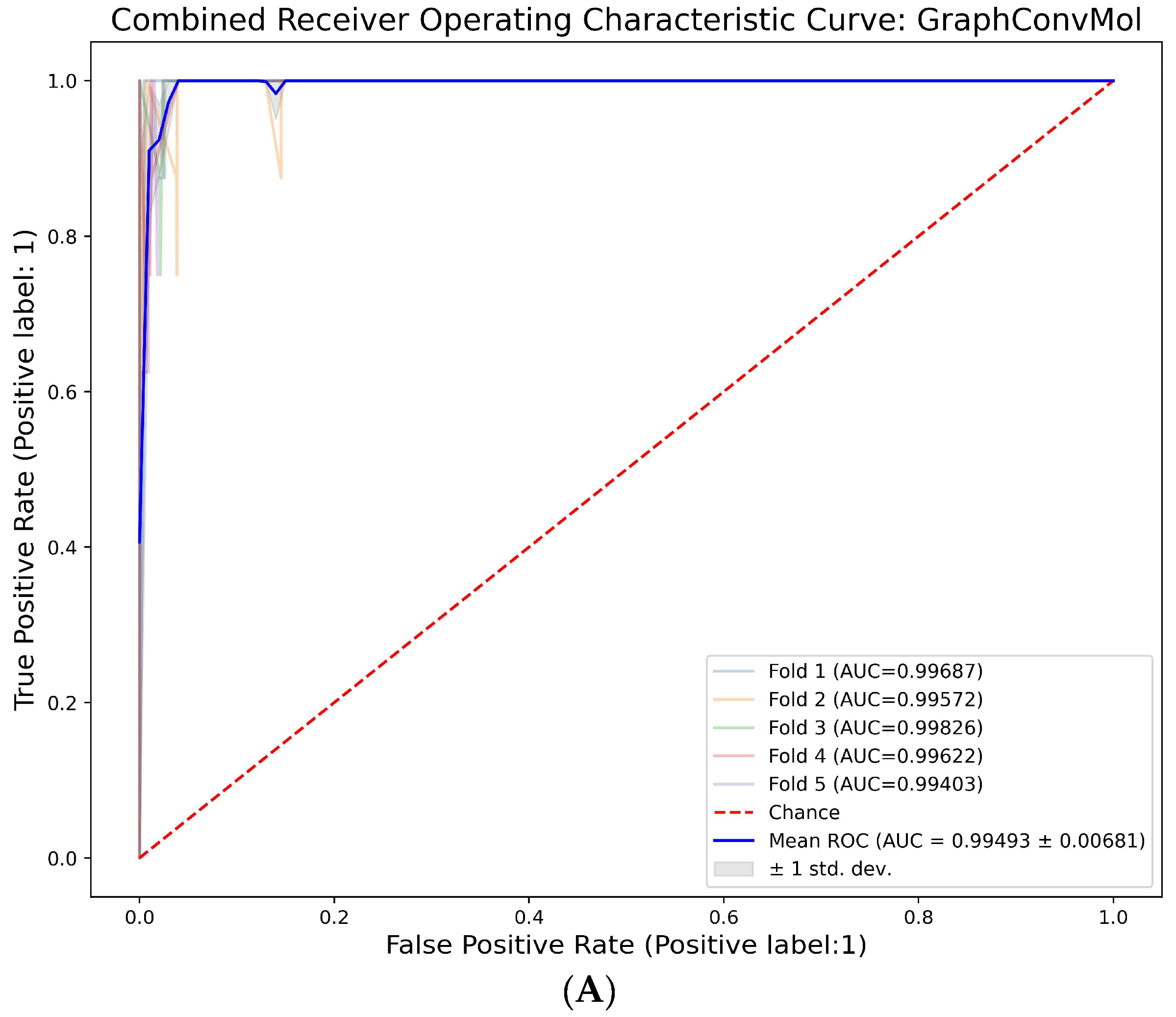
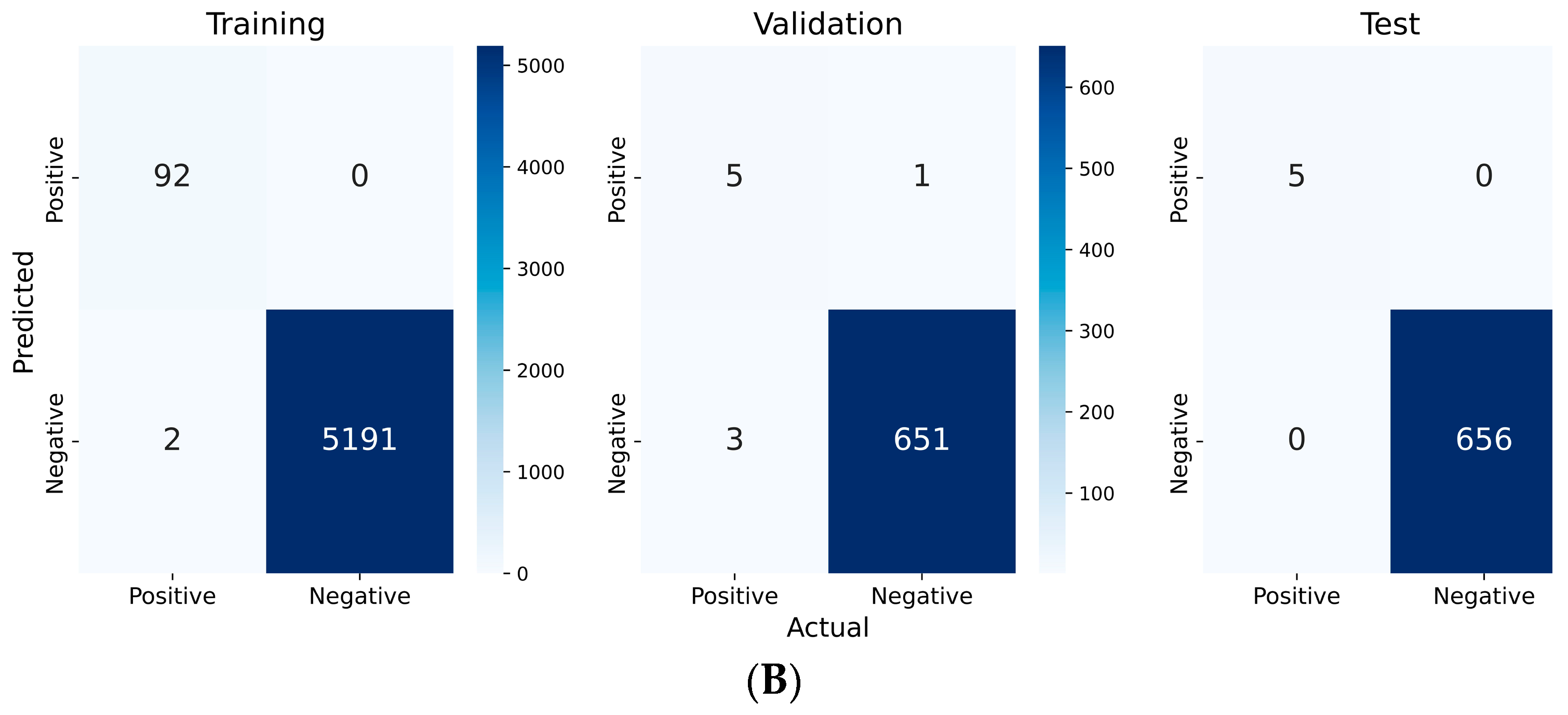
2.2. Deep-Learning Model Setup, Training, and Evaluation
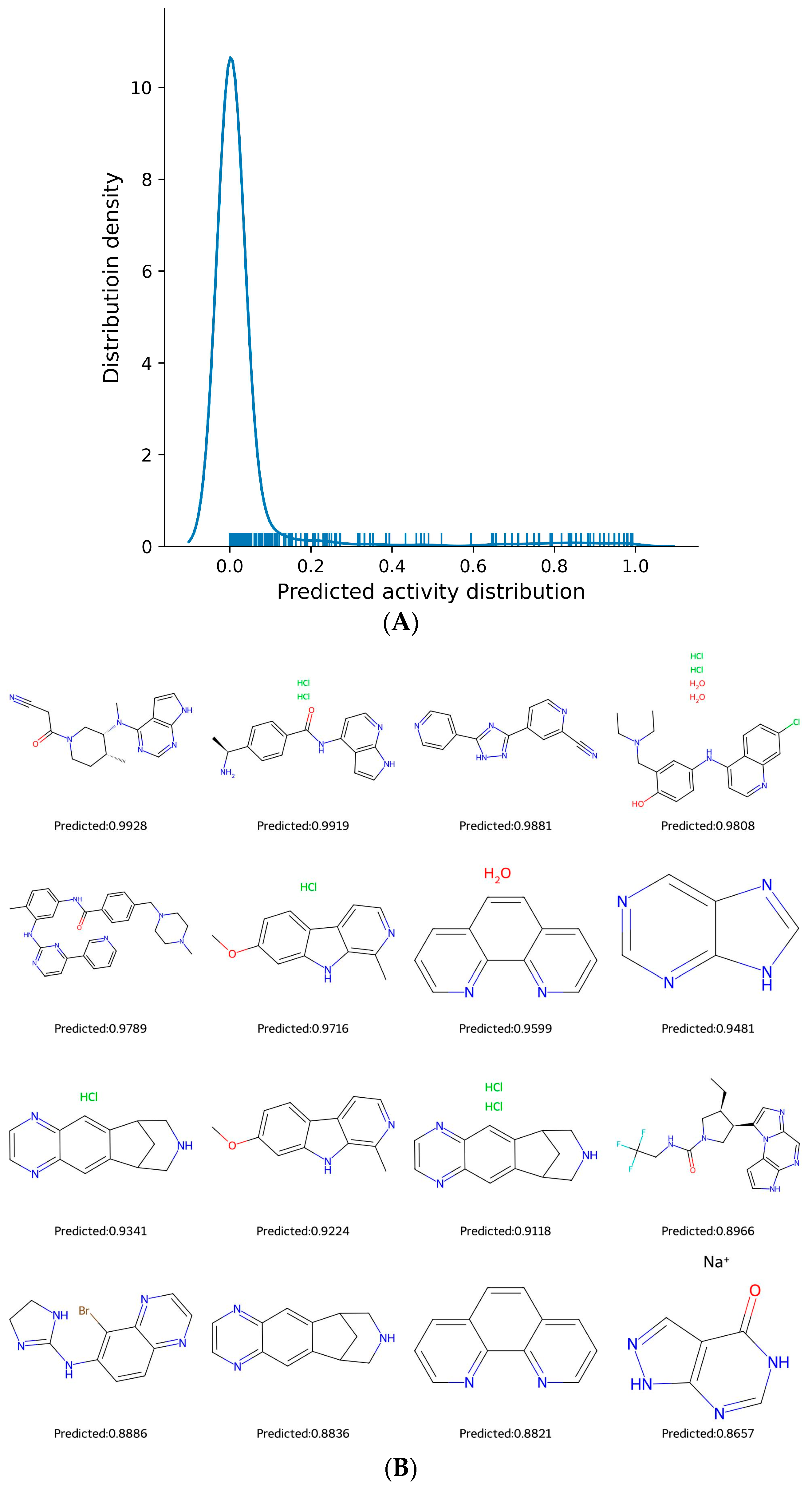
2.3. Prediction of JAK2 Inhibitory Potential from FDA-Approved Drugs
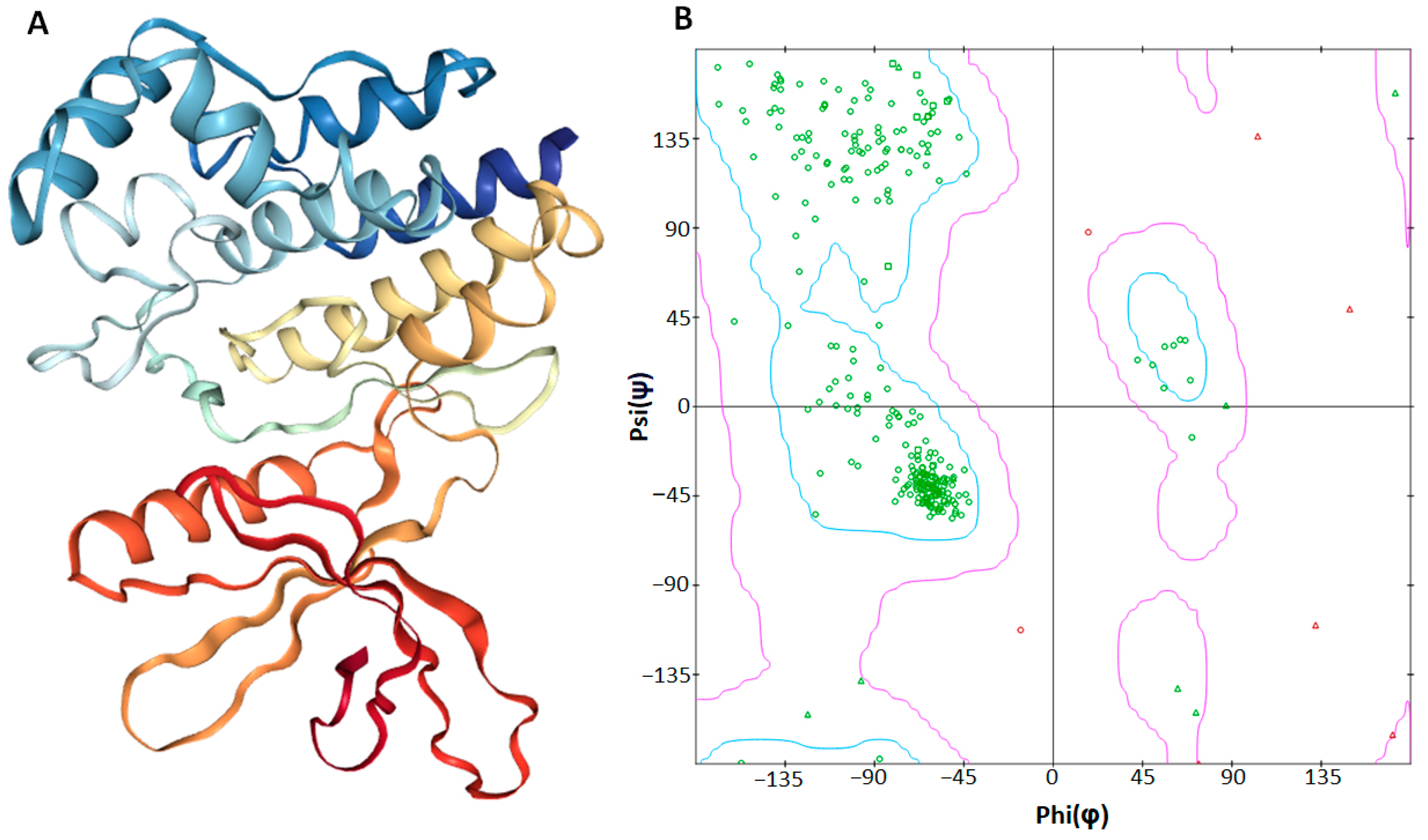
2.4. Structural Analysis of the JAK2 Protein
2.5. The Binding Pocket Analysis
2.6. Molecular Docking Analysis
2.7. Binding Interaction Analysis against JAK2
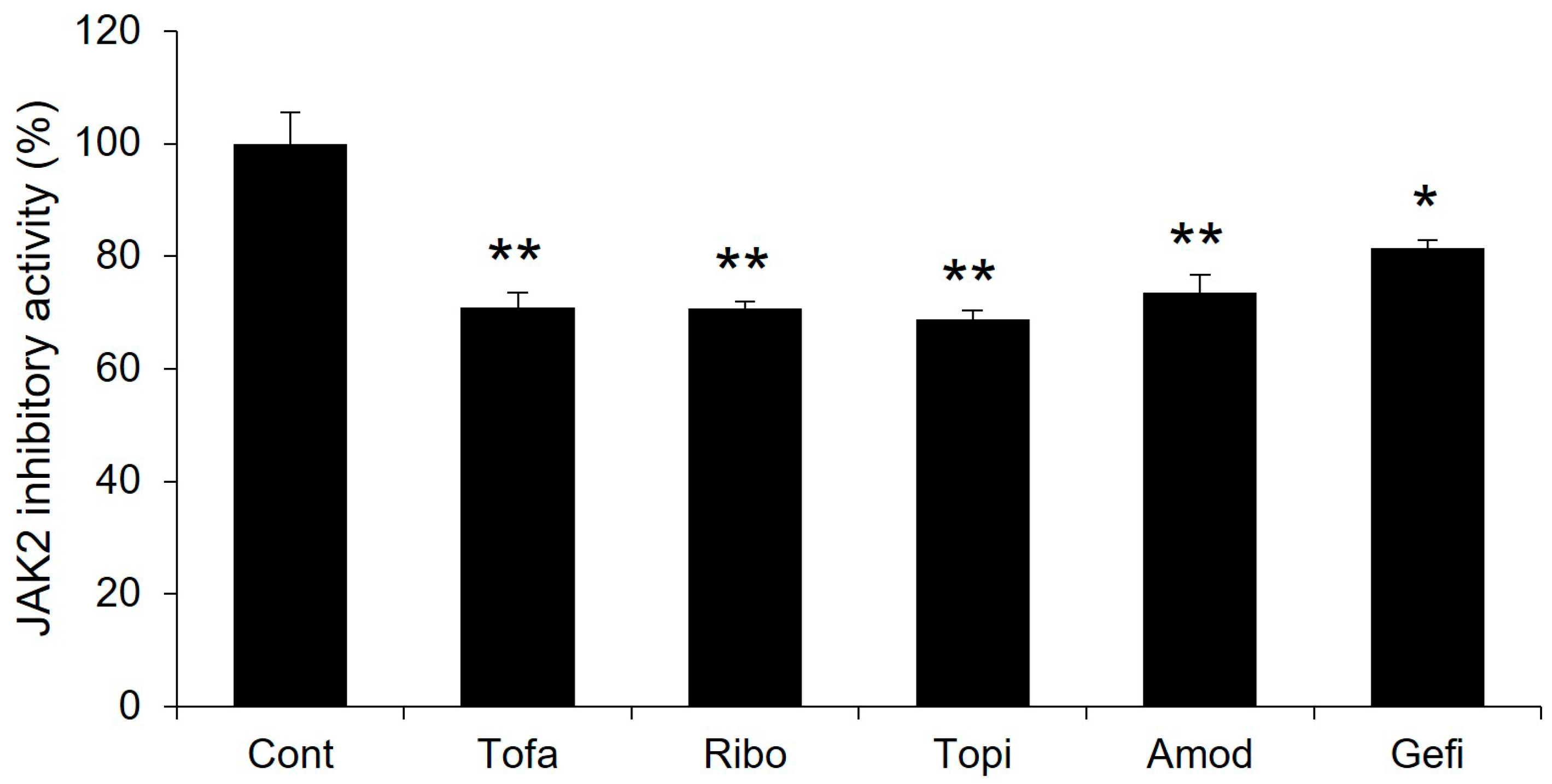
2.8. Experimental Validation
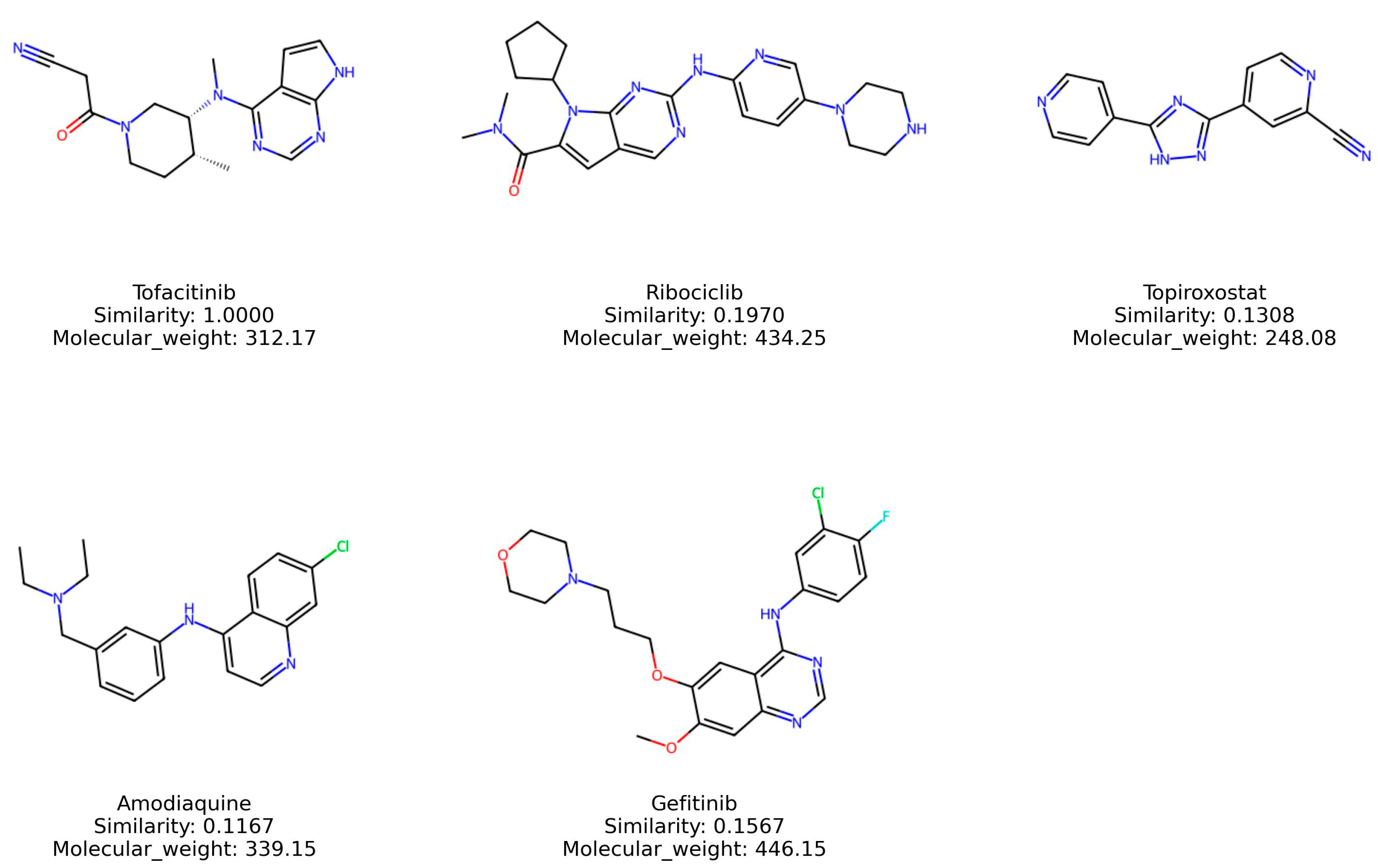
2.9. Structural Evaluation and Similarity Comparison
3. Methodology
3.1. JAK2 Datasets and FDA-Approved Drug Library
3.2. Molecular Descriptor Generation Using RDKit
3.3. Deep Learning Architecture
3.4. JAK2 Structure Retrieval
3.5. Prediction of Active Binding Site
3.6. Molecular Docking
3.7. Binding Interaction Analysis
3.8. JAK2 Kinase Inhibitory Activity Assay
3.9. Statistical Analysis
4. Conclusions
5. Limitations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jourdan, J.P.; Bureau, R.; Rochais, C.; Dallemagne, P. Drug repositioning: A brief overview. J. Pharm. Pharmacol. 2020, 72, 1145–1151. [Google Scholar] [CrossRef]
- Parvathaneni, V.; Kulkarni, N.S.; Muth, A.; Gupta, V. Drug repurposing: A promising tool to accelerate the drug discovery process. Drug Discov. Today 2019, 24, 2076–2085. [Google Scholar] [CrossRef]
- Zhao, L.; Ciallella, H.L.; Aleksunes, L.M.; Zhu, H. Advancing computer-aided drug discovery (CADD) by big data and data-driven machine learning modeling. Drug Discov. Today 2020, 25, 1624–1638. [Google Scholar] [CrossRef]
- Ramsay, R.R.; Popovic-Nikolicb, M.R.; Nikolic, K.; Uliassi, E.; Bolognesi, M.L. A perspective on multi-target drug discovery and design for complex diseases. Clin. Transl. Med. 2018, 7, 3. [Google Scholar] [CrossRef]
- Bechelane-Maia, E.H.; Assis, L.C.; Alves de Oliveira, T.; Marques da Silva, A.; Gutterres Taranto, A. Structure-based virtual screening: From classical to artificial intelligence. Front. Chem. 2020, 8, 343. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Zhang, K.; Bagheri, M.; Burken, J.G.; Gu, A.; Li, B.; Ma, X.; Marrone, B.L.; Ren, Z.J.; Schrier, J.; et al. Machine learning: New ideas and tools in environmental science and engineering. Environ. Sci. Technol. 2021, 55, 12741–12754. [Google Scholar] [CrossRef]
- Howard, J. Artificial intelligence: Implications for the future of work. Am. J. Ind. Med. 2019, 62, 917–926. [Google Scholar] [CrossRef]
- Dara, S.; Dhamercherla, S.; Jadav, S.S.; Babu, C.M.; Ahsan, M.J. Machine Learning in Drug Discovery: A Review. Artif. Intell. Rev. 2022, 55, 1947–1999. [Google Scholar] [CrossRef] [PubMed]
- Nag, S.; Baidya, A.T.K.; Mandal, A.; Mathew, A.T.; Das, B.; Devi, B.; Kumar, R. Deep learning tools for advancing drug discovery and development. 3 Biotech 2022, 12, 110. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.I.; Mitchell, T.M. Machine learning: Trends, perspectives, and prospects. Science 2015, 349, 255–260. [Google Scholar] [CrossRef]
- Wei, J.; Chu, X.; Sun, X.; Xu, K.; Deng, H.; Chen, J.; Wei, Z.; Lei, M. Machine learning in materials science. InfoMat 2019, 1, 338–358. [Google Scholar] [CrossRef]
- Stephenson, N.; Shane, E.; Chase, J.; Rowland, J.; Ries, D.; Justice, N.; Zhang, J.; Chan, L.; Cao, R. Survey of machine learning techniques in drug discovery. Curr. Drug Metab. 2019, 20, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, S.; Cheema, K.S.; Raychaudhuri, S.K.; Raychaudhuri, S.P. Janus kinase–signal transducers and activators of transcription cell signaling in Spondyloarthritis: Rationale and evidence for JAK inhibition. Curr. Opin. Rheumatol. 2021, 33, 348–355. [Google Scholar] [CrossRef]
- Sopjani, M.; Morina, R.; Uka, V.; Xuan, N.T.; Dërmaku-Sopjani, M. JAK2-mediated intracellular signaling. Curr. Mol. Med. 2021, 21, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Ojha, A.A.; Srivastava, A.; Votapka, L.W.; Amaro, R.E. Selectivity and ranking of tight-binding JAK-STAT inhibitors using Markovian milestoning with Voronoi tessellations. J. Chem. Inf. Model. 2022, 63, 2469–2482. [Google Scholar] [CrossRef]
- Spiewak, T.A.; Patel, A. User’s guide to JAK inhibitors in inflammatory bowel disease. Curr. Res. Pharmacol. Drug Discov. 2022, 3, 100096. [Google Scholar] [CrossRef]
- Desai, J.; Patel, B.; Gite, A.; Panchal, N.; Gite, S.; Argade, A.; Kumar, J.; Sachchidanand, S.; Bandyopadhyay, D.; Ghoshdastidar, K.; et al. Optimisation of momelotinib with improved potency and efficacy as pan-JAK inhibitor. Bioorganic Med. Chem. Lett. 2022, 66, 128728. [Google Scholar] [CrossRef]
- Lin, C.M.; Cooles, F.A.; Isaacs, J.D. Basic mechanisms of JAK inhibition. Mediterr. J. Rheumatol. 2020, 31 (Suppl. S1), 100. [Google Scholar] [CrossRef] [PubMed]
- Furumoto, Y.; Gadina, M. The arrival of JAK inhibitors: Advancing the treatment of immune and hematologic disorders. BioDrugs 2013, 27, 431–438. [Google Scholar] [CrossRef]
- Czech, J.; Cordua, S.; Weinbergerova, B.; Baumeister, J.; Crepcia, A.; Han, L.; Maié, T.; Costa, I.G.; Denecke, B.; Maurer, A.; et al. JAK2V617F but not CALR mutations confer increased molecular responses to interferon-α via JAK1/STAT1 activation. Leukemia 2019, 33, 995–1010. [Google Scholar] [CrossRef]
- Chen, C.; Lu, D.; Sun, T.; Zhang, T. JAK3 inhibitors for the treatment of inflammatory and autoimmune diseases: A patent review (2016–present). Expert Opin. Ther. Pat. 2022, 32, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Spivak, J.L. Narrative review: Thrombocytosis, polycythemia vera, and JAK2 mutations: The phenotypic mimicry of chronic myeloproliferation. Ann. Intern. Med. 2010, 152, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Geetha, J.P.; Arathi, C.A.; Shalini, M.; Srinivasa Murthy, A.G. JAK2 Negative Polycythemia Vera. J. Lab. Physicians 2010, 2, 114–116. [Google Scholar] [PubMed]
- Losdyck, E.; Hornakova, T.; Springuel, L.; Degryse, S.; Gielen, O.; Cools, J.; Constantinescu, S.N.; Flex, E.; Tartaglia, M.; Renauld, J.C.; et al. Distinct Acute Lymphoblastic Leukemia (ALL)-associated Janus Kinase 3 (JAK3) Mutants Exhibit Different Cytokine-Receptor Requirements and JAK Inhibitor Specificities. J. Biol. Chem. 2015, 290, 29022–29034. [Google Scholar] [CrossRef] [PubMed]
- McLornan, D.; Percy, M.; McMullin, M.F. JAK2 V617F: A single mutation in the myeloproliferative group of disorders. Ulst. Med. J. 2006, 75, 112–119. [Google Scholar]
- Hu, M.; Xu, C.; Yang, C.; Zuo, H.; Chen, C.; Zhang, D.; Shi, G.; Wang, W.; Shi, J.; Zhang, T. Discovery and evaluation of ZT55, a novel highly-selective tyrosine kinase inhibitor of JAK2V617F against myeloproliferative neoplasms. J. Exp. Clin. Cancer Res. 2019, 38, 49. [Google Scholar] [CrossRef]
- Perner, F.; Perner, C.; Ernst, T.; Heidel, F.H. Roles of JAK2 in aging, inflammation, hematopoiesis and malignant transformation. Cells 2019, 8, 854. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Zhang, Y.; Vass, M.; Shi, D.; Abualrous, E.; Chambers, J.M.; Chopra, N.; Higgs, C.; Kasavajhala, K.; Li, H.; Nandekar, P.; et al. Benchmarking refined and unrefined AlphaFold2 structures for hit discovery. J. Chem. Inf. Model. 2023, 63, 1656–1667. [Google Scholar] [CrossRef]
- Sabe, V.T.; Ntombela, T.; Jhamba, L.A.; Maguire, G.E.; Govender, T.; Naicker, T.; Kruger, H.G. Current trends in computer aided drug design and a highlight of drugs discovered via computational techniques: A review. Eur. J. Med. Chem. 2021, 224, 113705. [Google Scholar] [CrossRef]
- Minnich, A.J.; McLoughlin, K.; Tse, M.; Deng, J.; Weber, A.D.; Murad, N.; Madej, B.D.; Ramsundar, B.; Rush, T.; Calad-Thomson, S.; et al. AMPL: A data-driven modeling pipeline for drug discovery. J. Chem. Inf. Model. 2020, 60, 1955–1968. [Google Scholar] [CrossRef]
- Ramsundar, B.; Eastman, P.; Walters, P.; Pande, V. Deep Learning for the Life Sciences: Applying Deep Learning to Genomics, Microscopy, Drug Discovery, and More; O’Reilly Media, Inc.: Sebastopol, CA, USA, 2019. [Google Scholar]
- Grebner, C.; Matter, H.; Kofink, D.; Wenzel, J.; Schmidt, F.; Hessler, G. Application of deep neural network models in drug discovery programs. ChemMedChem 2021, 16, 3772–3786. [Google Scholar] [CrossRef] [PubMed]
- Grebner, C.; Matter, H.; Hessler, G. Artificial Intelligence in Drug Design; Methods in Molecular Biology; Humana: New York, NY, USA, 2022; pp. 349–382. [Google Scholar]
- Dielschneider, R.F.; Xiao, W.; Yoon, J.-Y.; Noh, E.; Banerji, V.; Li, H.; Marshall, A.J.; Johnston, J.B.; Gibson, S.B. Gefitinib targets ZAP-70-expressing chronic lymphocytic leukemia cells and inhibits B-cell receptor signaling. Cell Death Dis. 2014, 5, e1439. [Google Scholar] [CrossRef] [PubMed]
- Seifert, R.; Küper, A.; Tewes, M.; Heuschmid, M.; Welt, A.; Fendler, W.P.; Herrmann, K.; Decker, T. [18F]-Fluorodeoxyglucose positron emission tomography/CT to assess the early metabolic response in patients with hormone receptor-positive HER2-negative metastasized breast cancer treated with cyclin-dependent 4/6 kinase inhibitors. Oncol. Res. Treat. 2021, 44, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Doharey, P.K.; Singh, V.; Gedda, M.R.; Sahoo, A.K.; Varadwaj, P.K.; Sharma, B. In silico study indicates antimalarials as direct inhibitors of SARS-CoV-2-RNA dependent RNA polymerase. J. Biomol. Struct. Dyn. 2022, 40, 5588–5605. [Google Scholar] [CrossRef] [PubMed]
- Maghsoud, Y.; Dong, C.; Cisneros, G.A. Computational Characterization of the Inhibition Mechanism of Xanthine Oxidoreductase by Topiroxostat. ACS Catal. 2023, 13, 6023–6043. [Google Scholar] [CrossRef]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef] [PubMed]
- Yasir, M.; Park, J.; Han, E.-T.; Park, W.S.; Han, J.-H.; Kwon, Y.-S.; Lee, H.-J.; Hassan, M.; Kloczkowski, A.; Chun, W. Exploration of Flavonoids as Lead Compounds against Ewing Sarcoma through Molecular Docking, Pharmacogenomics Analysis, and Molecular Dynamics Simulations. Molecules 2023, 28, 414. [Google Scholar] [CrossRef]
- Wu, Y.; Brooks, C.L., III. Flexible CDOCKER: Hybrid Searching Algorithm and Scoring Function with Side Chain Conformational Entropy. J. Chem. Inf. Model. 2021, 61, 5535–5549. [Google Scholar] [CrossRef]
- Yasir, M.; Park, J.; Han, E.-T.; Park, W.S.; Han, J.-H.; Kwon, Y.-S.; Lee, H.-J.; Chun, W. Computational Exploration of Licorice for Lead Compounds against Plasmodium vivax Duffy Binding Protein Utilizing Molecular Docking and Molecular Dynamic Simulation. Molecules 2023, 28, 3358. [Google Scholar] [CrossRef]
- Yasir, M.; Park, J.; Han, E.T.; Park, W.S.; Han, J.H.; Kwon, Y.S.; Lee, H.J.; Chun, W. Machine Learning-Based Drug Repositioning of Novel Janus Kinase 2 Inhibitors Utilizing Molecular Docking and Molecular Dynamic Simulation. J. Chem. Inf. Model. 2023, 63, 6487–6500. [Google Scholar] [PubMed]
- Cavallo, G.; Metrangolo, P.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef] [PubMed]
- Sanachai, K.; Mahalapbutr, P.; Choowongkomon, K.; Poo-Arporn, R.P.; Wolschann, P.; Rungrotmongkol, T. Insights into the binding recognition and susceptibility of tofacitinib toward janus kinases. ACS Omega 2020, 5, 369–377. [Google Scholar] [CrossRef]
- Studio, D.J.A. Discovery studio. Accelrys 2008, 9. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Hassan, M.; Yasir, M.; Shahzadi, S.; Kloczkowski, A. Exploration of Potential Ewing Sarcoma Drugs from FDA-Approved Pharmaceuticals through Computational Drug Repositioning, Pharmacogenomics, Molecular Docking, and MD Simulation Studies. ACS Omega 2022, 7, 19243–19260. [Google Scholar] [CrossRef]
- Yasir, M.; Park, J.; Han, E.-T.; Park, W.S.; Han, J.-H.; Kwon, Y.-S.; Lee, H.-J.; Hassan, M.; Kloczkowski, A.; Chun, W. Investigation of Flavonoid Scaffolds as DAX1 Inhibitors against Ewing Sarcoma through Pharmacoinformatic and Dynamic Simulation Studies. Int. J. Mol. Sci. 2023, 24, 9332. [Google Scholar] [CrossRef] [PubMed]
- Yasir, M.; Park, J.; Lee, Y.; Han, E.T.; Park, W.S.; Han, J.H.; Kwon, Y.S.; Lee, H.J.; Chun, W. Discovery of GABA Aminotransferase Inhibitors via Molecular Docking, Molecular Dynamic Simulation, and Biological Evaluation. Int. J. Mol. Sci. 2023, 24, 16990. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Precision | Recall | F1 score | Accuracy | Specificity | |
|---|---|---|---|---|---|
| Training | 1.000 | 0.979 | 0.989 | 0.999 | 1.000 |
| Validation | 0.833 | 0.625 | 0.714 | 0.994 | 0.999 |
| Test | 1.000 | 1.000 | 1.000 | 1.000 | 1.000 |
| Smiles | Neg | Pos | Name | Target | Use |
|---|---|---|---|---|---|
| N#CC[C@H](C1CCCC1)n1ccc2ncnc3[nH]ccc23)cn1 | 0.0002 | 0.9998 | Ruxolitinib | JAK Inhibitor | Myelofibrosis, Anti cancer drug |
| COc1cc2ncnc(Nc3ccc(F)c(Cl)c3)c2cc1OCCCN1CCOCC1.Cl | 0.0004 | 0.9996 | Gefitinib | Tyrosine Kinase, EGFR inhibitor | Non-small cell lung carcinoma |
| Nc1ncnc2[nH]cnc12 | 0.0007 | 0.9993 | Adenine | Nucleobase | Nucleotide |
| c1ncc2nc[nH]c2n1 | 0.0007 | 0.9993 | Purine | Heterocyclic aromatic organic compound | DNA and RNA formation |
| CC[C@H](Nc1ncnc2[nH]cnc12)c1nc2cccc(F)c2c(=O)n1-c1ccccc1 | 0.0011 | 0.9989 | Idelalisib | Phosphoinositide 3-kinase inhibitor | Blood cancer |
| C#Cc1cccc(Nc2ncnc3cc4c(cc23)OCCOCCOCCO4)c1 | 0.0021 | 0.9979 | Icotinib | Epidermal growth factor receptor tyrosine kinase inhibitor (EGFR-TKI) | Non-small cell lung cancer |
| c1coc(CNc2ncnc3nc[nH]c23)c1 | 0.0031 | 0.9969 | Kinetin | Proapoptotic anti-proliferative plant growth regulator | Cell division |
| Cl.O=C(O)c1cn(-c2ccc(F)cc2)c2cc(N3CCNCC3)c(F)cc2c1=O | 0.0031 | 0.9969 | Sarafloxcin | Quinolone antibiotic drug | Antibiotic |
| CCS(=O)(=O)N1CC(CC#N)(n2cc(-c3ncnc4[nH]ccc34)cn2)C1 | 0.0033 | 0.9967 | Baricitinib | JAK2 inhibitor | Rheumatoid arthritis |
| C[C@@H]1CCN(C(=O)CC#N)C[C@@H]1N(C)c1ncnc2[nH]ccc12 | 0.0033 | 0.9967 | Tofacitinib | JAKs inhibitor | Rheumatoid arthritis |
| Nc1ncnc2c1ncn2[C@@H]1O[C@H](CO)[C@@H](O)[C@@H]1O.O | 0.0039 | 0.9961 | Vidarabine | Human herpesvirus 1 DNA polymerase | Antiviral |
| Nc1ncnc2c1ncn2[C@H]1C[C@H](O)[C@@H](CO)O1.O | 0.0039 | 0.9961 | 2′-Deoxyadenosine | Phosphodiesterase inhibitor | Energy source |
| CN(C)C(=O)c1cc2cnc(Nc3ccc(N4CCNCC4)cn3)nc2n1C1CCCC1 | 0.0045 | 0.9955 | Ribociclib | CDK4/CDK6 kinase inhibitor | Metastatic breast cancer |
| CCN(CC)CCCC(C)Nc1ccnc2cc(Cl)ccc12 | 0.0051 | 0.9949 | Chloroquine | Heme polymerase inhibitor | Malaria, Rheumatoid arthritis |
| COc1cc2nc(N3CCN(C(=O)C4CCCO4)CC3)nc(N)c2cc1OC.Cl.O.O | 0.0074 | 0.9926 | Terazosin | Alpha 1-adrenergic receptor inhibitor | Adrenaline blocker |
| CCN(CC)Cc1cc(Nc2ccnc3cc(Cl)ccc23)ccc1O.Cl.Cl.O.O | 0.0077 | 0.9923 | Amodiaquine | Heme polymerase inhibitor | Malaria |
| C[C@H](Nc1ncnc2[nH]cnc12)c1cc2cccc(Cl)c2c(=O)n1-c1ccccc1 | 0.0081 | 0.9919 | Duvelisib | PI3K inhibitor | Chronic lymphocytic leukemia |
| CC[C@@H]1CN(C(=O)NCC(F)(F)F)C[C@@H]1c1cnc2cnc3[nH]ccc3n12 | 0.0082 | 0.9918 | Upadacitinib | JAK inhibitor | Rheumatoid arthritis |
| c1cnc2c(c1)ccc1cccnc12 | 0.0086 | 0.9914 | 1,10-Phenanthroline | Fe(II) chelator | Metal chelator |
| C[C@H](Cn1cnc2c(N)ncnc21)OCP(=O)(O)O.O | 0.0100 | 0.9900 | Tenofovir | Nucleotide reverse transcriptase inhibitor | HIV |
| Cc1cc(/C=C/C#N)cc(C)c1Nc1ccnc(Nc2ccc(C#N)cc2)n1 | 0.0117 | 0.9883 | Rilpivirine | Transcriptase inhibitor | HIV |
| C#Cc1cccc(Nc2ncnc3cc(OCCOC)c(OCCOC)cc23)c1 | 0.0122 | 0.9878 | Erlotinib | Tyrosine kinase, EGFR inhibitor | Non-small cell lung cancer (NSCLC), pancreatic cancer |
| Cc1ccc(NC(=O)c2ccc(CN3CCN(C)CC3)cc2)cc1Nc1nccc(-c2cccnc2)n1 | 0.0172 | 0.9828 | Imatinib | Tyrosine kinase, Bcr-abl inhibitor | Chronic myeloid leukemia |
| N#Cc1cc(-c2n[nH]c(-c3ccncc3)n2)ccn1 | 0.0177 | 0.9823 | Topiroxostat | Xanthine oxidase inhibitor | Hyperuricemia (gout) |
| O=c1[nH]cnc2[nH]ncc12.[Na+] | 0.0196 | 0.9804 | Allopurinol | Xanthine oxidase inhibitor | Hyperuricemia (gout) |
| O.S=c1nc[nH]c2nc[nH]c12 | 0.0298 | 0.9702 | 6-Mercaptopurine | Purine nucleotide synthesis inhibitor | Antimetabolite, Antineoplastic |
| C=C[C@H]1CN2CC[C@H]1C[C@H]2[C@H](O)c1ccnc2ccc(OC)cc12.Cl.O.O | 0.0301 | 0.9699 | Quinine | Potassium channel blocker | Antimalarial, Analgesic |
| Cl.Cl.c1cnc2cc3c(cc2n1)C1CNCC3C1 | 0.0337 | 0.9663 | Varenicline | Nicotinic receptor blocker | Smoking cessation |
| O=c1[nH]cnc2nc[nH]c12 | 0.0365 | 0.9635 | Hypoxanthine | Nucleic acid synthesis | Malaria parasite cultures |
| CN[C@@H]1C[C@H]2O[C@@](C)([C@@H]1OC)n1c3ccccc3c3c4c(c5c6ccccc6n2c5c31)C(=O)NC4 | 0.0414 | 0.9586 | Staurosporine | PKC inhibitor | Cancer |
| Compounds | Cdocker Interaction Energy (kcal/mol) | CDocker Energy (kcal/mol) |
|---|---|---|
| Ribociclib | −58.0 | −5.3 |
| Imatinib | −52.6 | −24.3 |
| Staurosporine | −52.2 | 100.1 |
| Gefitinib | −50.6 | −15.5 |
| Adiphenine | −47.5 | −32.4 |
| Difloxacin | −47.2 | −26.1 |
| Amodiaquine | −44.4 | −19.8 |
| Naratriptan | −42.5 | −31.8 |
| Y-33075 | −42.0 | −31.1 |
| Rilpivirine | −41.3 | −31.8 |
| Tofacitinib | −40.0 | −29.3 |
| Dibucaine | −39.8 | −17.6 |
| Amsacrine | −39.6 | −8.2 |
| Chloroprocaine | −33.4 | −21.5 |
| Topiroxostat | −28.8 | −22.6 |
| Pinacidil | −28.6 | −20.6 |
| Varenicline | −25.6 | 19.9 |
| Phenanthroline | −23.0 | −4.8 |
| Pargyline | −22.5 | −18.9 |
| Allopurinol | −18.6 | −4.0 |
| Similarity | Tofacitinib | Ribociclib | Topiroxostat | Amodiaquine | Gefitinib |
|---|---|---|---|---|---|
| Tofacitinib | - | 0.196970 | 0.130841 | 0.114754 | 0.156716 |
| Ribociclib | 0.196970 | - | 0.146154 | 0.146853 | 0.180645 |
| Topiroxostat | 0.130841 | 0.146154 | - | 0.186916 | 0.131783 |
| Amodiaquine | 0.114754 | 0.146853 | 0.186916 | - | 0.257812 |
| Gefitinib | 0.156716 | 0.180645 | 0.131783 | 0.257812 | - |
| Name | LogP | Solubility | GI Absorption | BBB Permeation | CYP2D6 Inhibition | Lipinski Violation |
|---|---|---|---|---|---|---|
| Tofacitinib | 1.22 | −3.34 | High | No | No | 0 |
| Ribociclib | 2.12 | −5.51 | High | No | Yes | 0 |
| Topiroxostat | 1.38 | −5.24 | High | No | Yes | 0 |
| Amodiaquine | 4.6 | −8.18 | High | Yes | Yes | 0 |
| Gefitinib | 3.92 | −7.94 | High | Yes | Yes | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yasir, M.; Park, J.; Han, E.-T.; Park, W.S.; Han, J.-H.; Chun, W. Drug Repositioning via Graph Neural Networks: Identifying Novel JAK2 Inhibitors from FDA-Approved Drugs through Molecular Docking and Biological Validation. Molecules 2024, 29, 1363. https://doi.org/10.3390/molecules29061363
Yasir M, Park J, Han E-T, Park WS, Han J-H, Chun W. Drug Repositioning via Graph Neural Networks: Identifying Novel JAK2 Inhibitors from FDA-Approved Drugs through Molecular Docking and Biological Validation. Molecules. 2024; 29(6):1363. https://doi.org/10.3390/molecules29061363
Chicago/Turabian StyleYasir, Muhammad, Jinyoung Park, Eun-Taek Han, Won Sun Park, Jin-Hee Han, and Wanjoo Chun. 2024. "Drug Repositioning via Graph Neural Networks: Identifying Novel JAK2 Inhibitors from FDA-Approved Drugs through Molecular Docking and Biological Validation" Molecules 29, no. 6: 1363. https://doi.org/10.3390/molecules29061363
APA StyleYasir, M., Park, J., Han, E.-T., Park, W. S., Han, J.-H., & Chun, W. (2024). Drug Repositioning via Graph Neural Networks: Identifying Novel JAK2 Inhibitors from FDA-Approved Drugs through Molecular Docking and Biological Validation. Molecules, 29(6), 1363. https://doi.org/10.3390/molecules29061363