
A Thiourea Derivative of 2-[(1R)-1-Aminoethyl]phenol as a Chiral Sensor for the Determination of the Absolute Configuration of N-3,5-Dinitrobenzoyl Derivatives of Amino Acids
 , , , and
, , , and
Abstract
1. Introduction
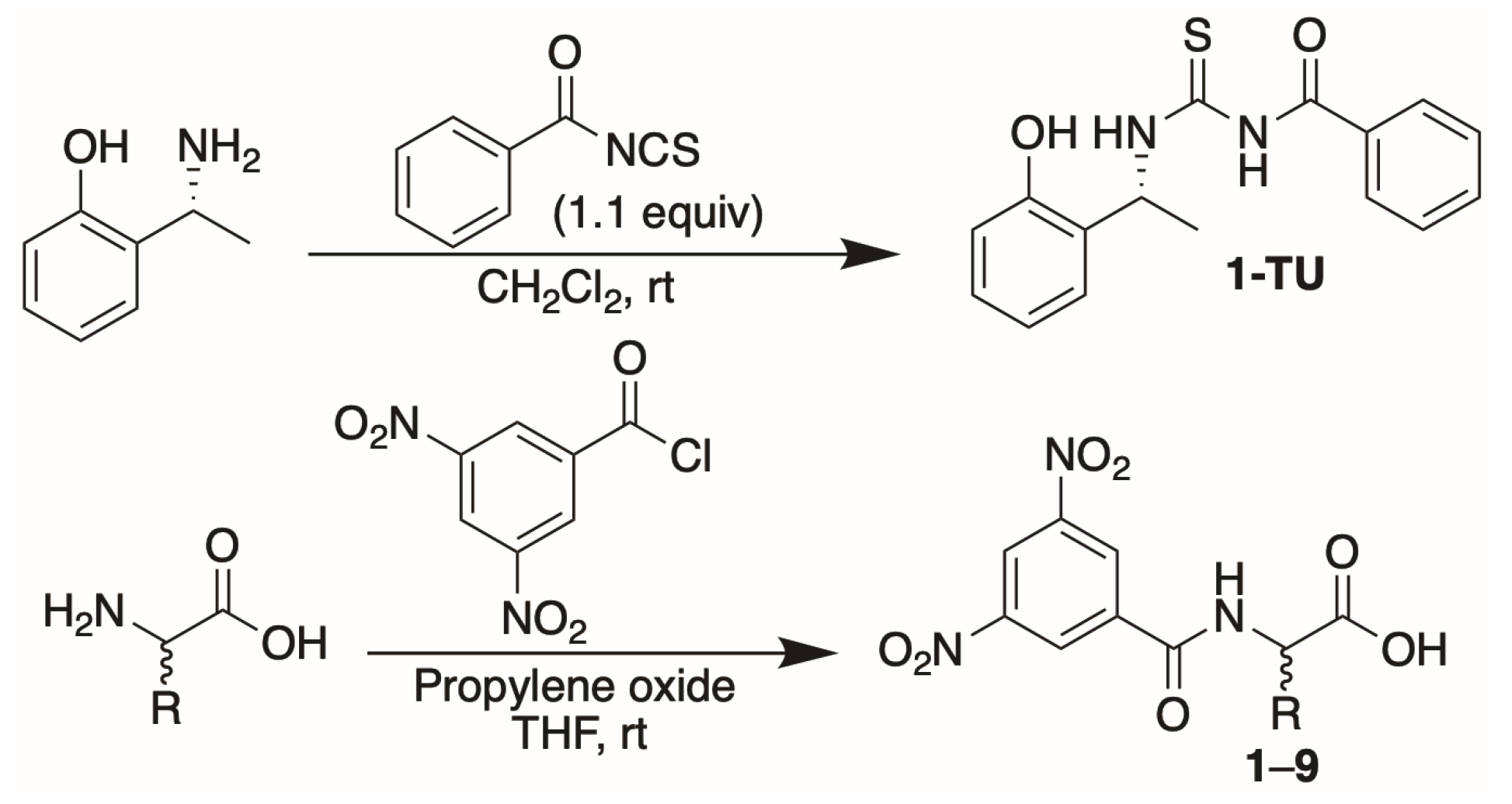
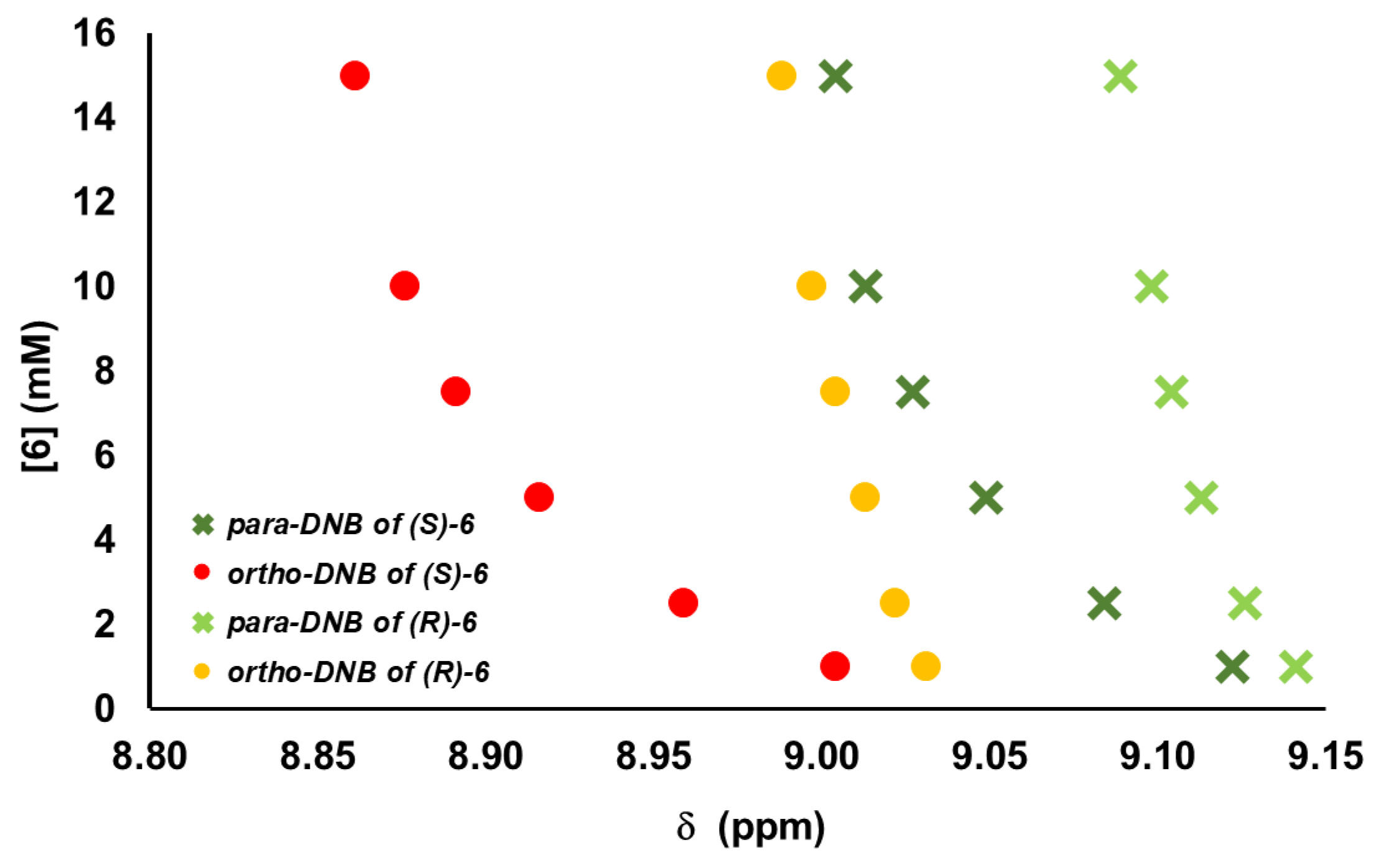
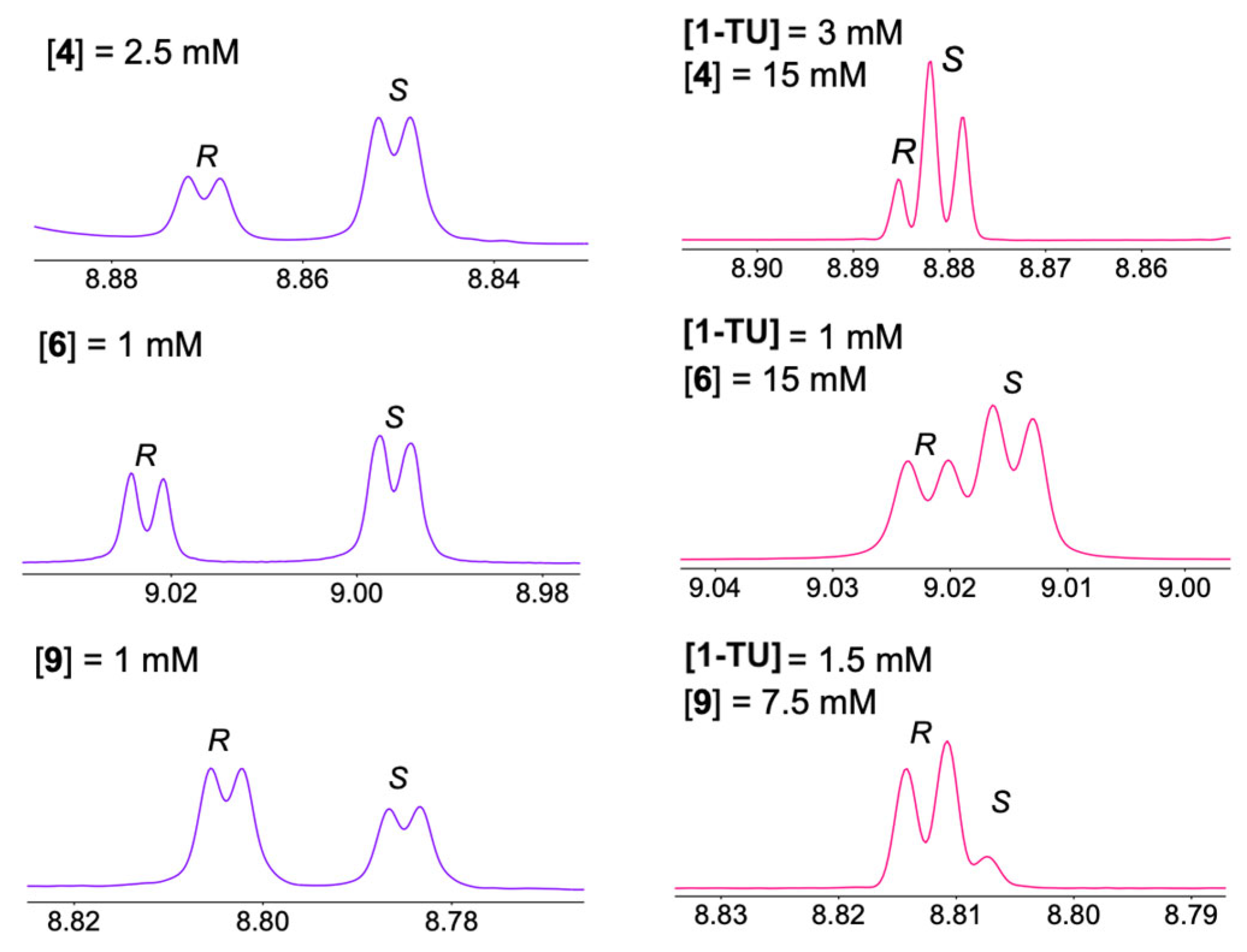
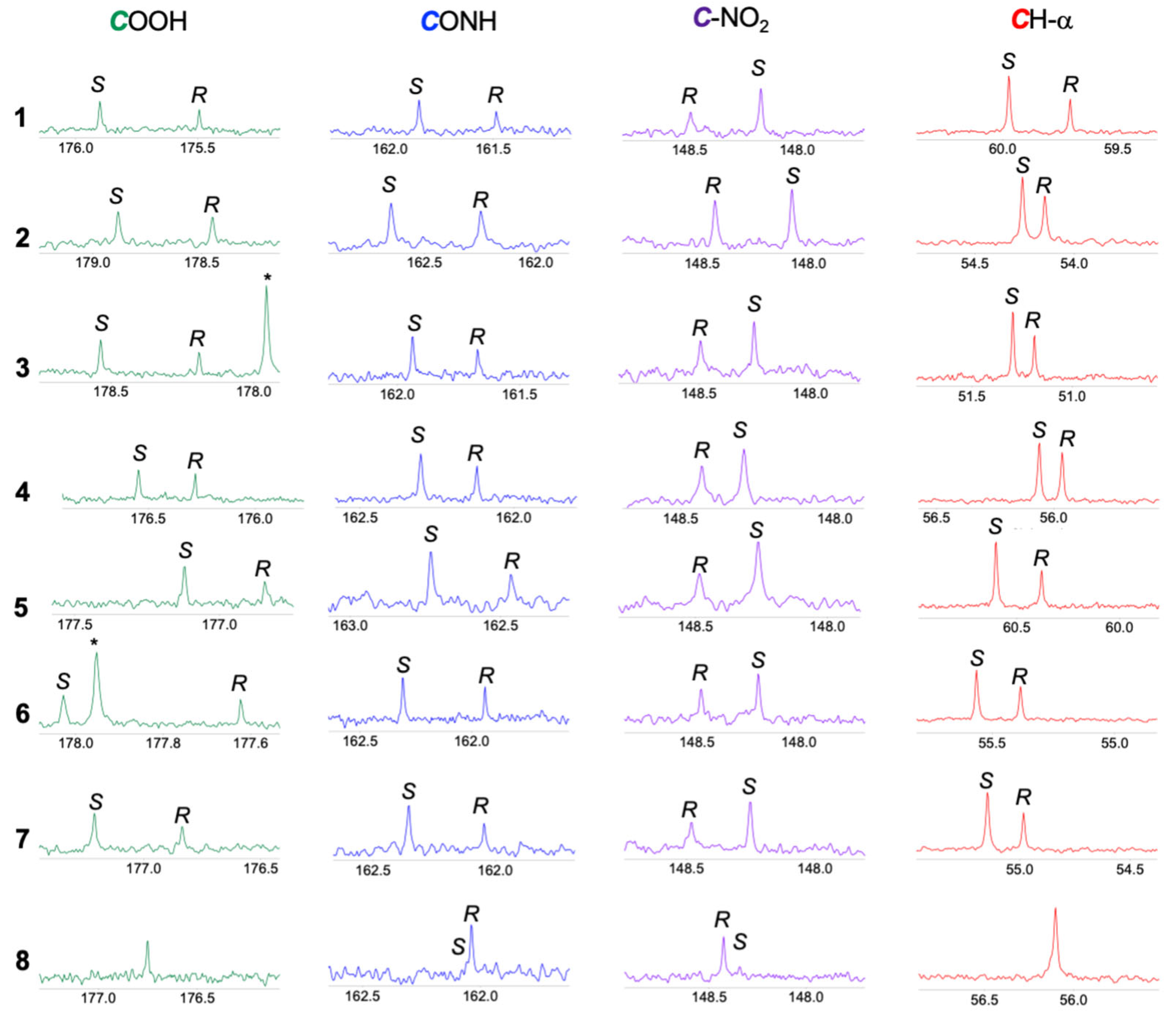
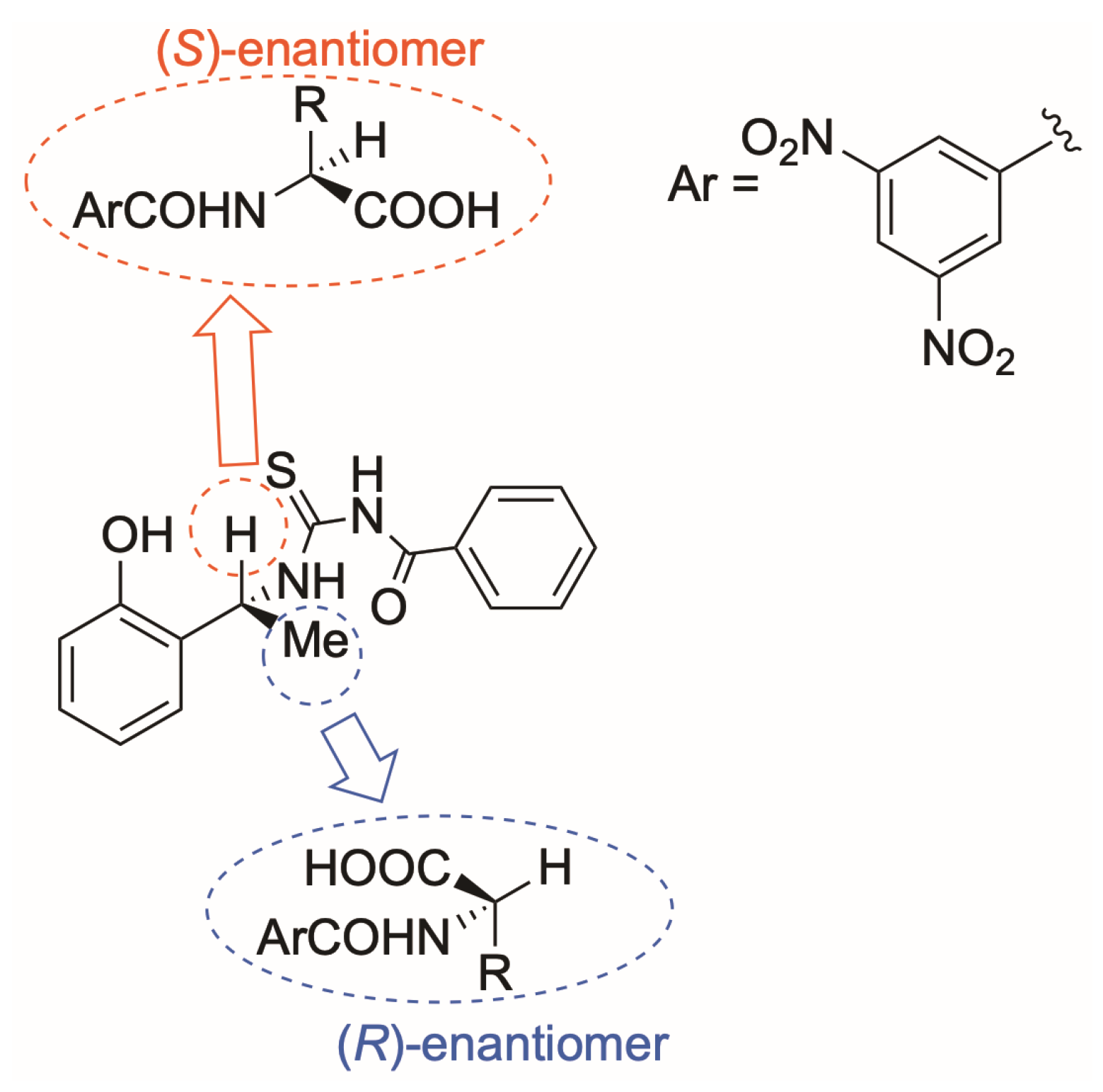
2. Results and Discussions
3. Materials and Methods
3.1. Materials
3.2. Methods
3.3. Synthesis of N-3,5-Dinitrobenzoyl Amino Acids 6–9
3.4. Preparation of Enantiomerically Enriched Mixtures for 1H and 13C NMR Experiments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dutot, L.; Wright, K.; Gaucher, A.; Wakselman, M.; Mazaleyrat, J.-P.; De Zotti, M.; Peggion, C.; Formaggio, F.; Toniolo, C. The Bip Method, Based on the Induced Circular Dichroism of a Flexible Biphenyl Probe in Terminally Protected-Bip-Xaa*-Dipeptides, for Assignment of the Absolute Configuration of β-Amino Acids. J. Am. Chem. Soc. 2008, 130, 5986–5992. [Google Scholar] [CrossRef] [PubMed]
- Bentley, K.W.; Nam, Y.G.; Murphy, J.M.; Wolf, C. Chirality Sensing of Amines, Diamines, Amino Acids, Amino Alcohols, and α-Hydroxy Acids with a Single Probe. J. Am. Chem. Soc. 2013, 135, 18052–18055. [Google Scholar] [CrossRef] [PubMed]
- Gholami, H.; Chakraborty, D.; Zhang, J.; Borhan, B. Absolute Stereochemical Determination of Organic Molecules through Induction of Helicity in Host–Guest Complexes. Acc. Chem. Res. 2021, 54, 654–667. [Google Scholar] [CrossRef] [PubMed]
- Harada, N. Chiral Molecular Science: How Were the Absolute Configurations of Chiral Molecules Determined? “Experimental Results and Theories”. Chirality 2017, 29, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Flack, H.D. Absolute-Structure Determination: Past, Present and Future. Chimia 2014, 68, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Jin, Z.; Wang, X.; Zeng, S.; Sun, C.; Pan, Y. Pair of Stereodynamic Chiral Benzylicaldehyde Probes for Determination of Absolute Configuration of Amino Acid Residues in Peptides by Mass Spectrometry. Anal. Chem. 2017, 89, 11902–11907. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, Z.; Li, S.; Rong, X.; Bu, J.; Liu, Q.; Ouyang, Z. Differentiating Enantiomers by Directional Rotation of Ions in a Mass Spectrometer. Science 2024, 383, 612–618. [Google Scholar] [CrossRef]
- Phyo, Y.Z.; Ribeiro, J.; Fernandes, C.; Kijjoa, A.; Pinto, M.M.M. Marine Natural Peptides: Determination of Absolute Configuration Using Liquid Chromatography Methods and Evaluation of Bioactivities. Molecules 2018, 23, 306. [Google Scholar] [CrossRef]
- Harada, N. HPLC Separation of Diastereomers: Chiral Molecular Tools Useful for the Preparation of Enantiopure Compounds and Simultaneous Determination of Their Absolute Configurations. Molecules 2016, 21, 1328. [Google Scholar] [CrossRef]
- Balzano, F.; Uccello-Barretta, G.; Aiello, F. Chiral Analysis by NMR Spectroscopy: Chiral Solvating Agents. In Chiral Analysis, 2nd ed.; Polavarapu, P.L., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; Chapter 9; pp. 367–427. [Google Scholar] [CrossRef]
- Wenzel, T.J. Differentiation of Chiral Compounds Using NMR Spectroscopy, 2nd ed.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2018; ISBN 978-1-119-32391-4. [Google Scholar]
- Uccello Barretta, G.; Wenzel, T.J.; Balzano, F. Spectroscopic Analysis: NMR and Shift Reagents. In Reference Collection in Chemistry, Molecular Sciences and Chemical Engineering; Elsevier: Amsterdam, The Netherlands, 2022. [Google Scholar] [CrossRef]
- Silva, M.S. Recent Advances in Multinuclear NMR Spectroscopy for Chiral Recognition of Organic Compounds. Molecules 2017, 22, 247. [Google Scholar] [CrossRef]
- Mishra, S.K.; Suryaprakash, N. Some New Protocols for the Assignment of Absolute Configuration by NMR Spectroscopy Using Chiral Solvating Agents and CDAs. Tetrahedr. Asymm. 2017, 28, 1220–1232. [Google Scholar] [CrossRef]
- Wenzel, T.J. Strategies for Using NMR Spectroscopy to Determine Absolute Configuration. Tetrahedr. Asymm. 2017, 28, 1212–1219. [Google Scholar] [CrossRef]
- Seco, J.M.; Riguera, R. NMR Methods for the Assignment of Absolute Stereochemistry of Bioactive Compounds. In eMagRes; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2015; pp. 1–30. [Google Scholar] [CrossRef]
- Dale, J.A.; Mosher, H.S. Nuclear Magnetic Resonance Nonequivalence of Diastereomeric Esters of Alpha-Substituted Phenylacetic Acids for the Determination of Stereochemical Purity. J. Am. Chem. Soc. 1968, 90, 3732–3738. [Google Scholar] [CrossRef]
- Uccello-Barretta, G.; Balzano, F.; Caporusso, A.M.; Iodice, A.; Salvadori, P. Permethylated Beta-Cyclodextrin as Chiral Solvating Agent for the NMR Assignment of the Absolute Configuration of Chiral Trisubstituted Allenes. J. Org. Chem. 1995, 60, 2227–2231. [Google Scholar] [CrossRef]
- Du, G.; Li, Y.; Ma, S.; Wang, R.; Li, B.; Guo, F.; Zhu, W.; Li, Y. Efficient Determination of the Enantiomeric Purity and Absolute Configuration of Flavanones by Using (S)-3,3′-Dibromo-1,1′-Bi-2-Naphthol as a Chiral Solvating Agent. J. Nat. Prod. 2015, 78, 2968–2974. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, S.R.; Suryaprakash, N. Ternary Ion-Pair Complexation: A Protocol for Chiral Discrimination and the Assignment of Absolute Configuration of Chiral Hydroxy Acids. New J. Chem. 2013, 37, 4025–4030. [Google Scholar] [CrossRef]
- Wenzel, T.J.; Bourne, C.E.; Clark, R.L. (18-Crown-6)-2,3,11,12-Tetracarboxylic Acid as a Chiral NMR Solvating Agent for Determining the Enantiomeric Purity and Absolute Configuration of β-Amino Acids. Tetrahedr. Asymm. 2009, 20, 2052–2060. [Google Scholar] [CrossRef]
- Lesot, P.; Aroulanda, C.; Zimmermann, H.; Luz, Z. Enantiotopic Discrimination in the NMR Spectrum of Prochiral Solutes in Chiral Liquid Crystals. Chem. Soc. Rev. 2015, 44, 2330–2375. [Google Scholar] [CrossRef]
- Aroulanda, C.; Lesot, P. Molecular Enantiodiscrimination by NMR Spectroscopy in Chiral Oriented Systems: Concept, Tools, and Applications. Chirality 2022, 34, 182–244. [Google Scholar] [CrossRef]
- Bian, G.; Fan, H.; Huang, H.; Yang, S.; Zong, H.; Song, L.; Yang, G. Highly Effective Configurational Assignment Using Bisthioureas as Chiral Solvating Agents in the Presence of DABCO. Org. Lett. 2015, 17, 1369–1372. [Google Scholar] [CrossRef]
- Chen, Z.; Fan, H.; Yang, S.; Bian, G.; Song, L. Chiral Sensors for Determining the Absolute Configurations of α-Amino Acid Derivatives. Org. Biomol. Chem. 2018, 16, 8311–8317. [Google Scholar] [CrossRef] [PubMed]
- Recchimurzo, A.; Micheletti, C.; Uccello-Barretta, G.; Balzano, F. Thiourea Derivative of 2-[(1R)-1-Aminoethyl]Phenol: A Flexible Pocket-like Chiral Solvating Agent (CSA) for the Enantiodifferentiation of Amino Acid Derivatives by NMR Spectroscopy. J. Org. Chem. 2020, 85, 5342–5350. [Google Scholar] [CrossRef] [PubMed]
- Castañar, L. Pure Shift 1H NMR: What Is Next? Magn. Reson. Chem. 2017, 55, 47–53. [Google Scholar] [CrossRef]
- Duengo, S.; Muhajir, M.I.; Hidayat, A.T.; Musa, W.J.A.; Maharani, R. Epimerisation in Peptide Synthesis. Molecules 2023, 28, 8017. [Google Scholar] [CrossRef] [PubMed]
- McCudden, C.R.; Kraus, V.B. Biochemistry of Amino Acid Racemization and Clinical Application to Musculoskeletal Disease. Clin. Biochem. 2006, 39, 1112–1130. [Google Scholar] [CrossRef]
- Benfodda, Z.; Bénimélis, D.; Jean, M.; Naubron, J.-V.; Rolland, V.; Meffre, P. Synthesis, Resolution, and Determination of Absolute Configuration of Protected α-Ethynylphenylalanine Enantiomers. Amino Acids 2015, 47, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Reason, A.J. Validation of Amino Acid Analysis Methods. In The Protein Protocols Handbook; Walker, J.M., Ed.; Springer Protocols Handbooks; Humana Press: Totowa, NJ, USA, 2009; pp. 1015–1028. [Google Scholar] [CrossRef]
- Vargas-Caporali, J.; Juaristi, E. Fundamental Developments of Chiral Phase Chromatography in Connection with Enantioselective Synthesis of β-Amino Acids. Isr. J. Chem. 2017, 57, 896–912. [Google Scholar] [CrossRef]
- Bhushan, R. Enantioselective and Chemoselective Optical Detection of Chiral Organic Compounds without Resorting to Chromatography. Chem. Asian J. 2023, 18, e202300825. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sub | ΔΔδ | ||
|---|---|---|---|
| ortho-DNB | para-DNB | CH-α | |
| 1 * | 0.156 | 0.096 | 0.039 |
| 2 * | 0.132 | 0.082 | 0.004 |
| 3 * | 0.134 | 0.095 | 0.037 |
| 4 * | 0.034 | 0.016 | 0.026 |
| 5 * | 0.084 | 0.055 | 0.008 |
| 6 | 0.129 | 0.085 | 0.017 |
| 7 | 0.102 | 0.070 | 0.019 |
| 8 | 0.055 | 0.019 | 0.018 |
| 9 | 0.054 | 0.041 | 0.026 |
| Sub | ΔδR | ΔδS | ||||
|---|---|---|---|---|---|---|
| ortho-DNB | para-DNB | CH-α | ortho-DNB | para-DNB | CH-α | |
| 1 | −0.002 | −0.016 | 0.057 | −0.158 | −0.112 | 0.096 |
| 2 | 0.041 | −0.011 | 0.076 | −0.091 | −0.093 | 0.080 |
| 3 | −0.003 | −0.033 | 0.019 | −0.137 | −0.128 | 0.056 |
| 4 | −0.051 | −0.044 | 0.007 | −0.085 | −0.060 | 0.033 |
| 5 | 0.013 | −0.045 | 0.062 | −0.071 | −0.100 | 0.070 |
| 6 | −0.012 | −0.036 | −0.015 | −0.141 | −0.121 | 0.002 |
| 7 | 0.021 | 0.005 | 0.011 | −0.081 | −0.065 | 0.030 |
| 8 | −0.008 | −0.037 | 0.056 | −0.063 | −0.056 | 0.074 |
| 9 | −0.032 | −0.046 | 0.041 | −0.086 | −0.087 | 0.067 |
| Sub | ΔΔδ | |||
|---|---|---|---|---|
| COOH | CONH | C-NO2 | CH-α | |
| 1 | 0.405 | 0.377 | 0.328 | 0.267 |
| 2 | 0.436 | 0.403 | 0.360 | 0.115 |
| 3 | 0.345 | 0.269 | 0.241 | 0.107 |
| 4 | 0.266 | 0.183 | 0.138 | 0.102 |
| 5 | 0.275 | 0.266 | 0.222 | 0.222 |
| 6 | 0.405 | 0.367 | 0.284 | 0.184 |
| 7 | 0.374 | 0.314 | 0.230 | 0.170 |
| 8 | 0 | 0.055 | 0.023 | 0 |
| Sub | ΔδR | ΔδS | ||||||
|---|---|---|---|---|---|---|---|---|
| COOH | CONH | C-NO2 | CH-α | COOH | CONH | C-NO2 | CH-α | |
| 1 | 0.275 | 0.406 | −0.004 | 0.509 | 0.680 | 0.783 | −0.332 | 0.776 |
| 2 | 0.404 | 0.542 | −0.068 | 0.512 | 0.840 | 0.945 | −0.428 | 0.627 |
| 3 | 0.251 | 0.304 | −0.012 | 0.305 | 0.596 | 0.573 | −0.253 | 0.412 |
| 4 | 0.289 | 0.449 | −0.085 | 0.189 | 0.555 | 0.632 | −0.223 | 0.291 |
| 5 | 0.444 | 0.366 | −0.082 | 0.460 | 0.719 | 0.632 | −0.304 | 0.682 |
| 6 | 0.511 | 0.046 | −0.108 | 0.856 | 0.916 | 0.413 | −0.392 | 1.040 |
| 7 | −0.069 | 0.399 | 0.015 | 0.257 | 0.305 | 0.713 | −0.215 | 0.427 |
| 8 | −0.458 | −0.152 | −0.109 | 0.046 | −0.458 | −0.207 | −0.132 | 0.046 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aiello, F.; Recchimurzo, A.; Balzano, F.; Uccello Barretta, G.; Cefalì, F. A Thiourea Derivative of 2-[(1R)-1-Aminoethyl]phenol as a Chiral Sensor for the Determination of the Absolute Configuration of N-3,5-Dinitrobenzoyl Derivatives of Amino Acids. Molecules 2024, 29, 1319. https://doi.org/10.3390/molecules29061319
Aiello F, Recchimurzo A, Balzano F, Uccello Barretta G, Cefalì F. A Thiourea Derivative of 2-[(1R)-1-Aminoethyl]phenol as a Chiral Sensor for the Determination of the Absolute Configuration of N-3,5-Dinitrobenzoyl Derivatives of Amino Acids. Molecules. 2024; 29(6):1319. https://doi.org/10.3390/molecules29061319
Chicago/Turabian StyleAiello, Federica, Alessandra Recchimurzo, Federica Balzano, Gloria Uccello Barretta, and Federica Cefalì. 2024. "A Thiourea Derivative of 2-[(1R)-1-Aminoethyl]phenol as a Chiral Sensor for the Determination of the Absolute Configuration of N-3,5-Dinitrobenzoyl Derivatives of Amino Acids" Molecules 29, no. 6: 1319. https://doi.org/10.3390/molecules29061319
APA StyleAiello, F., Recchimurzo, A., Balzano, F., Uccello Barretta, G., & Cefalì, F. (2024). A Thiourea Derivative of 2-[(1R)-1-Aminoethyl]phenol as a Chiral Sensor for the Determination of the Absolute Configuration of N-3,5-Dinitrobenzoyl Derivatives of Amino Acids. Molecules, 29(6), 1319. https://doi.org/10.3390/molecules29061319