Abstract
A practical metal-free and additive-free approach for the synthesis of 6/7/8-membered oxacyclic ketone-fused isoxazoles/isoxazolines tetracyclic or tricyclic structures is reported through Csp3–H bond radical nitrile oxidation and the intramolecular cycloaddition of alkenyl/alkynyl-substituted aryl methyl ketones. This convenient approach enables the simultaneous formation of isoxazole/isoxazoline and 6/7/8-membered oxacyclic ketones to form polycyclic architectures by using tert-butyl nitrite (TBN) as a non-metallic radical initiator and N–O fragment donor.
1. Introduction
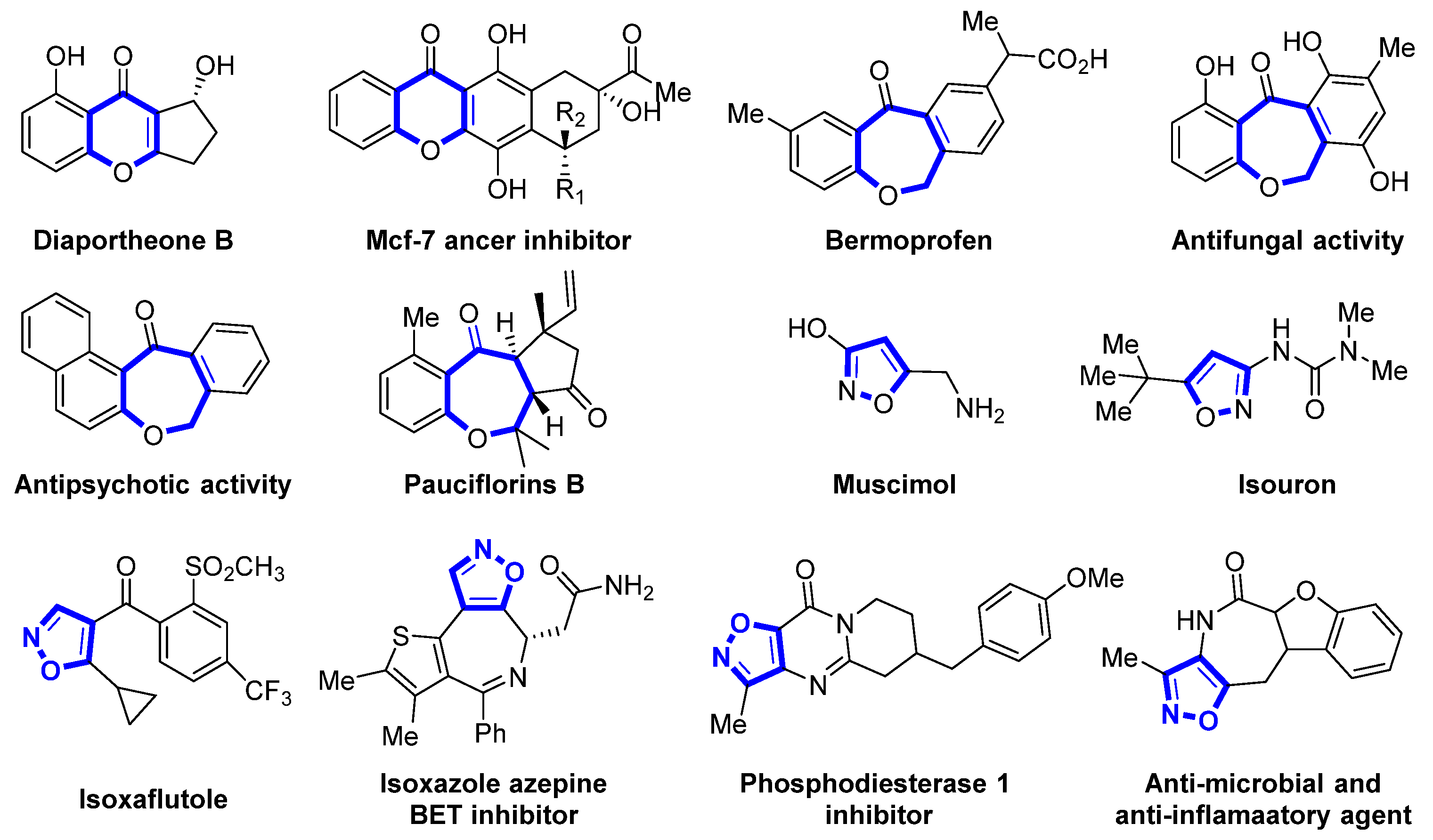
Polycyclic structures containing heteroatoms are regarded as important structural motifs in the realm of organic chemistry and pharmaceuticals. They are present in various natural products, agrochemicals, and physiologically active molecules and play a significant role in drug synthesis and discovery [1,2]. The benzo oxacyclic ketone skeleton is an important scaffold for multiring structures, such as benzochromones and their derivatives. These structures are found in numerous natural products and pharmaceuticals, playing a pivotal role in the formation of polycyclic systems (Figure 1) [3,4,5,6,7,8].
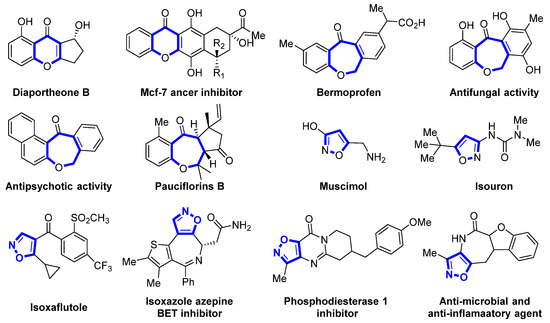
Figure 1.
Some drugs with pharmacological activity containing benzo[b]oxygenes or isoxazole frameworks.
Isoxazole/isoxazoline, a five-membered heterocyclic ring, is present in numerous biologically significant compounds known for their anti-inflammatory, antifungal, anticancer, and antimicrobial properties. Its ability to interact with the target protein through multiple non-covalent bonds makes it a pivotal drug component in various pharmaceutical formulations [9,10,11,12].
Due to the significant biological activities associated with the benzo oxacyclic ketone and isoxazole/isoxazoline skeletons, the development of efficient methods to merge these two entities is highly significant and desirable in the realms of medicinal and synthetic chemistry. Fusing two or more heterocycles to form a tricyclic or tetracyclic fused heterocycle is of interest to access polycyclic architectures. These polycyclic architectures demonstrate enhanced biological activity [13,14].
Numerous methods have been reported for synthesizing small ring (3–6 membered) and large ring (≥12 membered) compounds, including the Diels Alder reaction, Corey Nicolaou macrocycle esterification reaction, Keck macrocycle esterification reaction, and olefin metathesis reaction. Advancements in transition-metal-catalyzed closed-loop metathesis, olefin reactions, small ring cycloaddition, and hydrogenation acylation have led to progress in synthesizing medium-sized ring (7–11 membered) compounds. The intermolecular cycloaddition reaction is also effective for the formation of medium-sized rings [15]. However, predicting the reactivity of these compounds is challenging due to unfavorable cross-ring tension and entropy effects, making their synthesis both difficult and intriguing. Medium-sized rings, particularly seven- and eight-membered ones, pose significant challenges in synthesis [16,17,18,19].
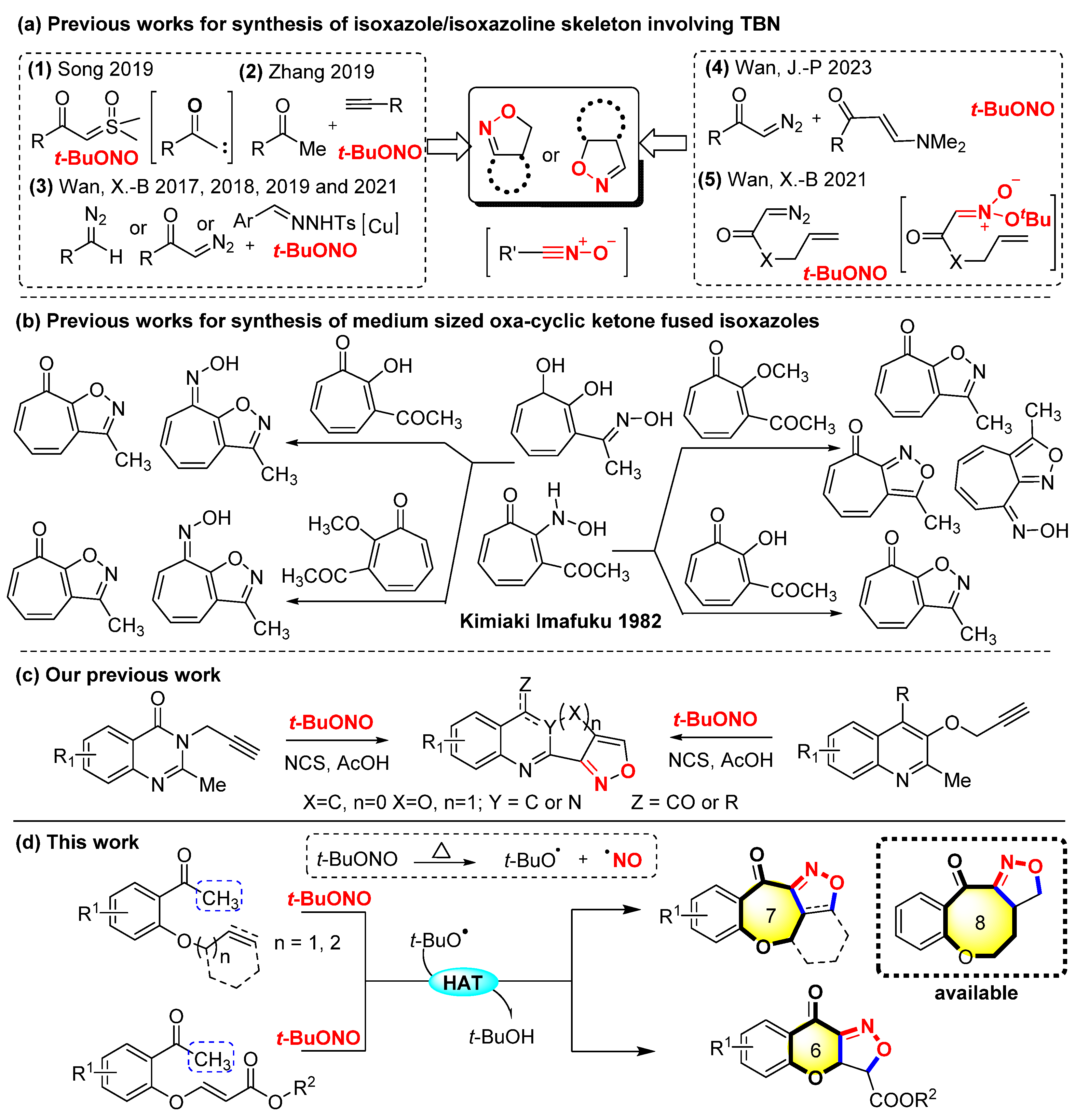
Reactions employing tert-butyl nitrite (TBN) as both a free radical initiator and N–O fragment donor have emerged as an important tool for isoxazole/isoxazoline synthesis over the past few years [20,21,22,23,24,25,26]. Song et al. developed a new [2 + 1 + 1 + 1] annulation reaction of sulfoxonium ylides with TBN for the first time to synthesize furoxans and isoxazoles [27]. Zhang et al. reported the graceful synthesis of isoxazoles from methyl ketones, terminal alkynes, and TBN under catalyst-free conditions [28]. Wan, X.-B. et al. reported the graceful cycloaddition reactions for the synthesis of isoxazoles from diazo compounds or N-tosylhydrazones with alkenes or β-keto esters activated by tert-butyl nitrite [29,30,31]. These approaches are robust and can deliver fully substituted isoxazoles. In a recent study, Wan, J.-P. et al. reported a refined metal-catalyzed strategy for the synthesis of isomeric isoxazoles through the reactions of enaminones, diazo compounds, and TBN under different Cu- and Ag-catalyzed conditions [32]. The synthesis of isoxazoline-fused bicyclic compounds poses challenges, particularly under transition-metal-free conditions. Instead, Wan, X.-B. et al. used intramolecular acyclic nitronate olefin cycloaddition reactions via the in situ generated acyclic nitronates combined with cascade [3 + 2] cycloaddition and tert-butyloxy group elimination to enable the formation of diverse γ-lactone-fused isoxazolines and even tricyclic isoxazolines (Scheme 1a) [33]. A metal-free method had already been used to synthesize 3-methyl-1,8-dihydrocycloheptapyrazol-8-one derivatives and isoxazole-fused seven-membered oxacyclic ketones by Imafuku in 1982 (Scheme 1b) [34].
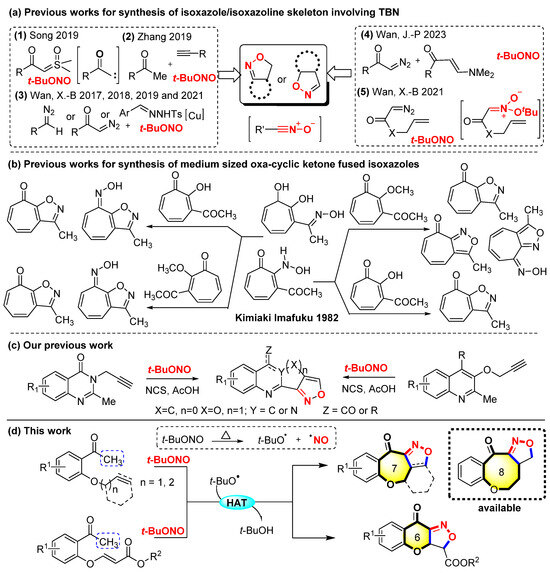
Scheme 1.
Strategy for the synthesis of skeleton-fused isoxazole/isoxazoline. (HAT: Hydrogen Atom Transfer) [27,28,29,30,31,32,33,34].
Recently, our group successfully demonstrated an efficient synthetic method to synthesize diverse isoxazole-fused tricyclic quinazoline alkaloids and their derivatives (Scheme 1c) [35]. We gained inspiration from the synthesis of the 3-acyl-isoxazoles and Δ2-isoxazolines series compounds reported by Zhang et al. [34] based on their previous research. Drawing inspiration from these investigations, a metal-free and additive-free method for Csp3–H bond radical nitrile oxidation and the intramolecular cycloaddition of alkenyl/alkynyl-substituted aryl methyl ketones to synthesize 6/7/8-membered oxacyclic ketone-fused isoxazoles/isoxazolines tetracyclic or tricyclic structures is reported. This convenient approach enables the simultaneous formation of the isoxazole/isoxazoline and 6/7/8-membered oxacyclic ketone, thereby leading to the formation of the polycyclic architectures using TBN as a non-metallic radical initiator and N−O fragment donor. (Scheme 1d).
2. Results and Discussion
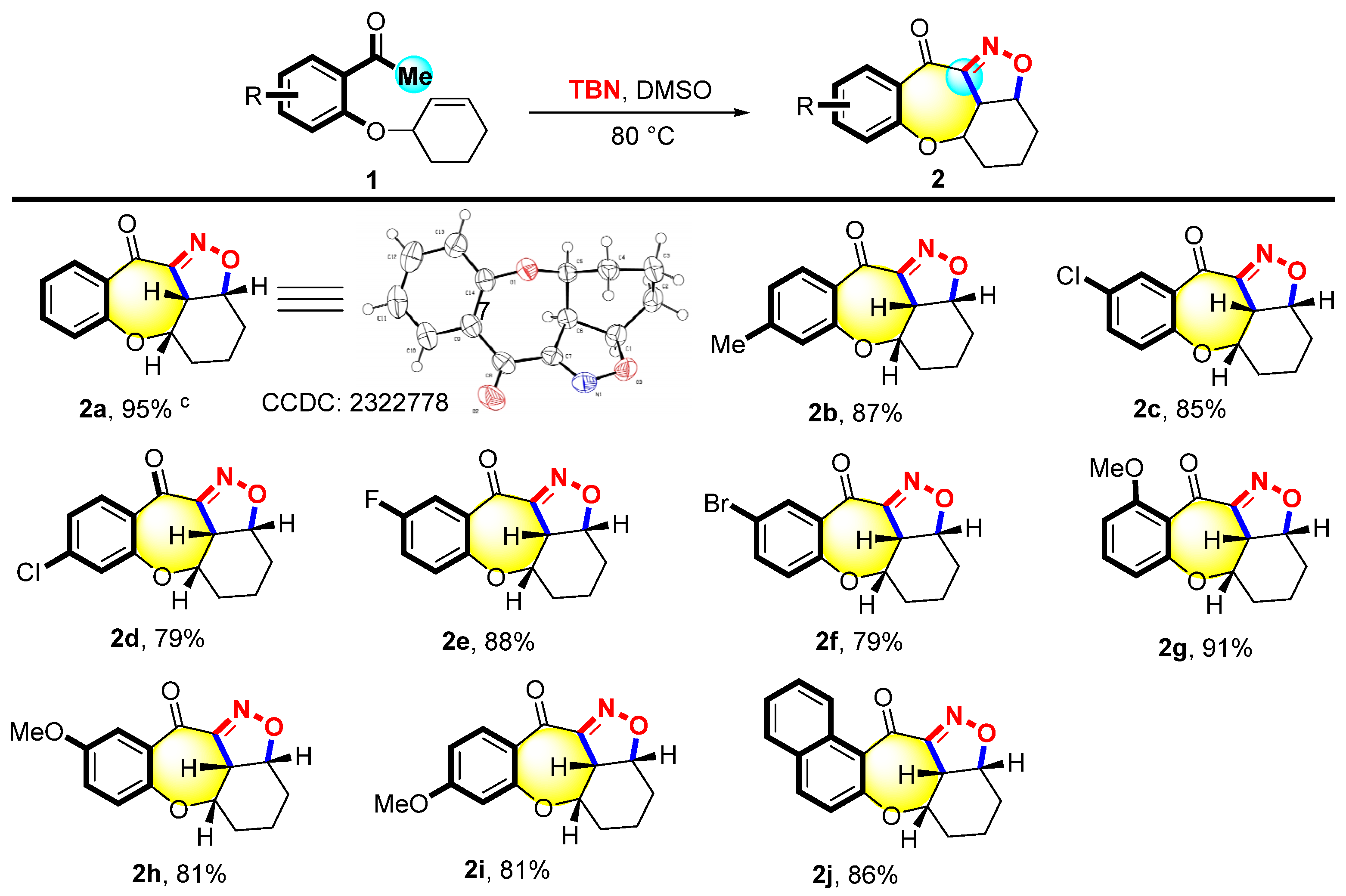
Firstly, the reaction conditions were optimized, and the results are summarized in Tables S2–S4 (supporting information). The substrate scope of 2 was investigated under optimized conditions. As shown in Figure 2, the method displayed excellent tolerance for structure 1, substituted with electron-donating groups, and can yield the desired products 2b, 2g–2i. A series of substrates with a methyl group at the C4 (1b) and the methoxyl group at the C4 (1i), C5 (1h), and C6 (1g) positions led to the corresponding products with yields ranging from 81% to 91%. On the other hand, structure 1 substituted with electron-withdrawing groups such as Cl, Br, and F at the C4 or C5 position performed the reaction smoothly to give the desired products 2c–2f in good yields (79–88%). The naphthalenyl-substituted substrate 1j was also suitable for this reaction to deliver the desired product 2j with an 86% yield. X-ray single crystal diffraction was employed to determine the crystal structure of product 2a.
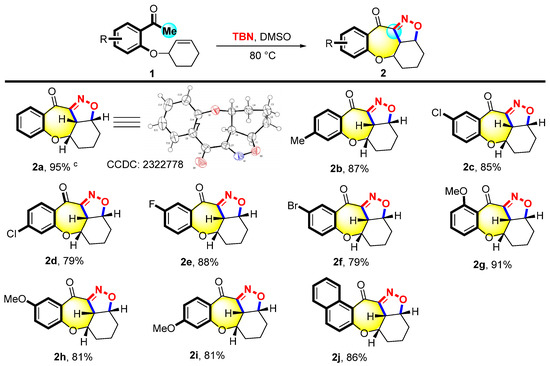
Figure 2.
Scope of 3-(2-acetophenoxy) cyclohexene a,b. a Reaction condition: 1 (0.1 mmol) and TBN (0.4 mmol) were heated in DMSO (2 mL) at 80 °C for 10 h. b Isolated yields. c The molecular structure of 2a with ellipsoids at the 50% probability level.
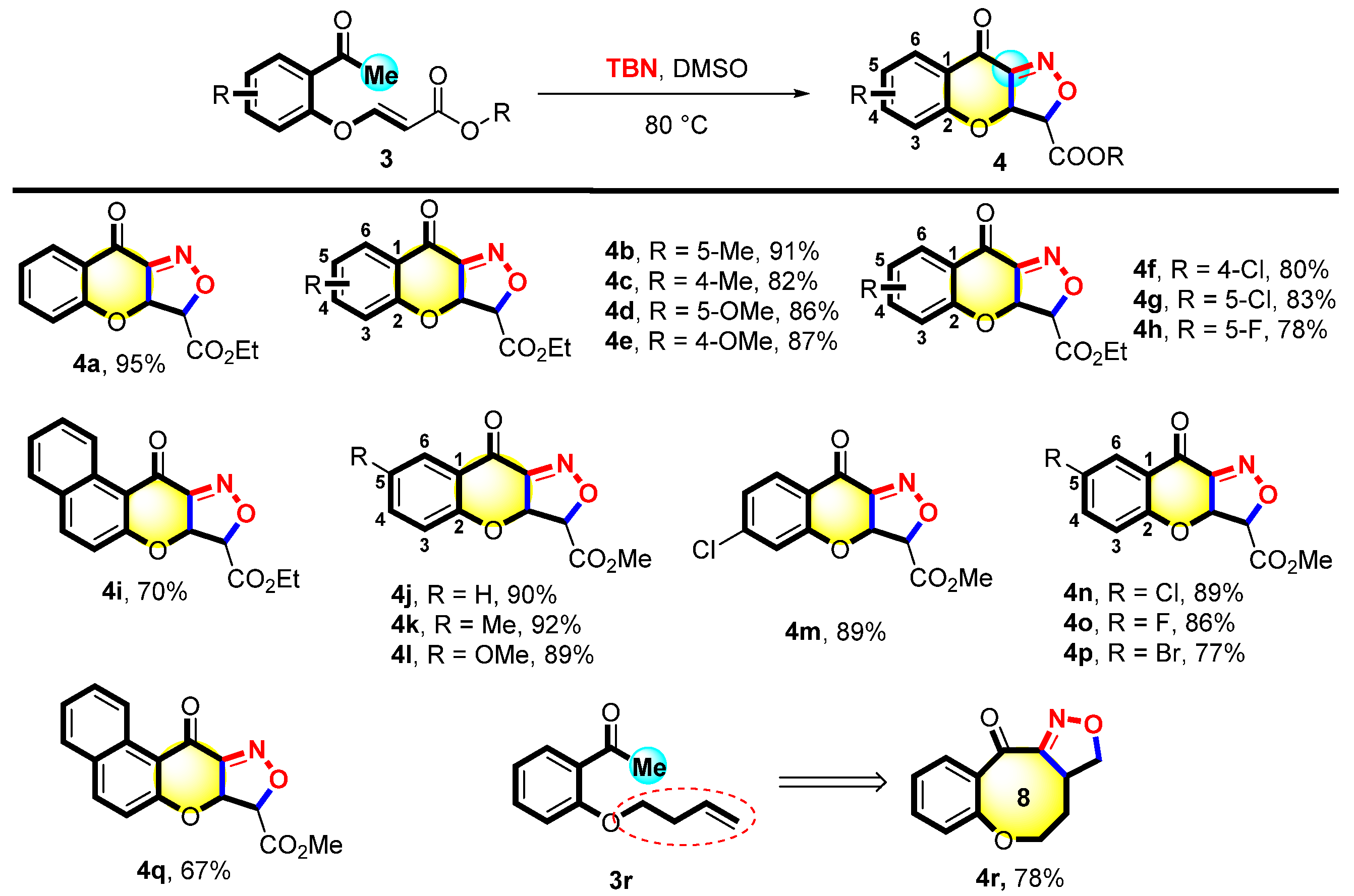
Moreover, the reaction between various acrylates 3 with TBN was explored. It is evident from Figure 3 that 3a was successfully converted into the expected product 4a with a 95% yield. Surprisingly, different acetophenones 3b–3e with electron-donating substituents (such as 4-Me, 5-Me, 4-OMe, and 5-OMe) reacted analogously, yielding the corresponding products 4b–4e with 82–91% yields. Halogen–halogen atom substrates formed the corresponding products (4f–4h) with 78–83% yields. Furthermore, the side chain ethyl ester was converted to methyl ester and proceeded under standard conditions, yielding the desired products (4j–4q) within 67–92% yields. When the benzene ring of the template substrate 3 became a naphthalene ring, 3i and 3q yielded the corresponding products 4i and 4q in 70% and 67% yields, respectively. We synthesized the raw material O-acetylphenoxybutene (3r). Subsequently, 3r performed the reaction under the optimal conditions to give a polycyclic compound containing an eight-membered ring (4r) with a 78% yield.
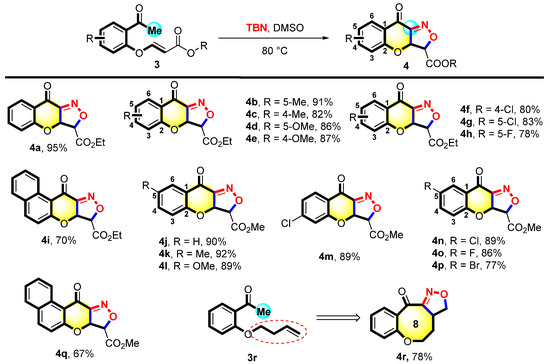
Figure 3.
Scope of 3-(2-acetylphenoxy)acrylates a,b. a Reaction condition: 3 (0.1 mmol) and TBN (0.7 mmol) were heated in DMSO (2 mL) at 80 °C for 10 h. b Isolated yields.
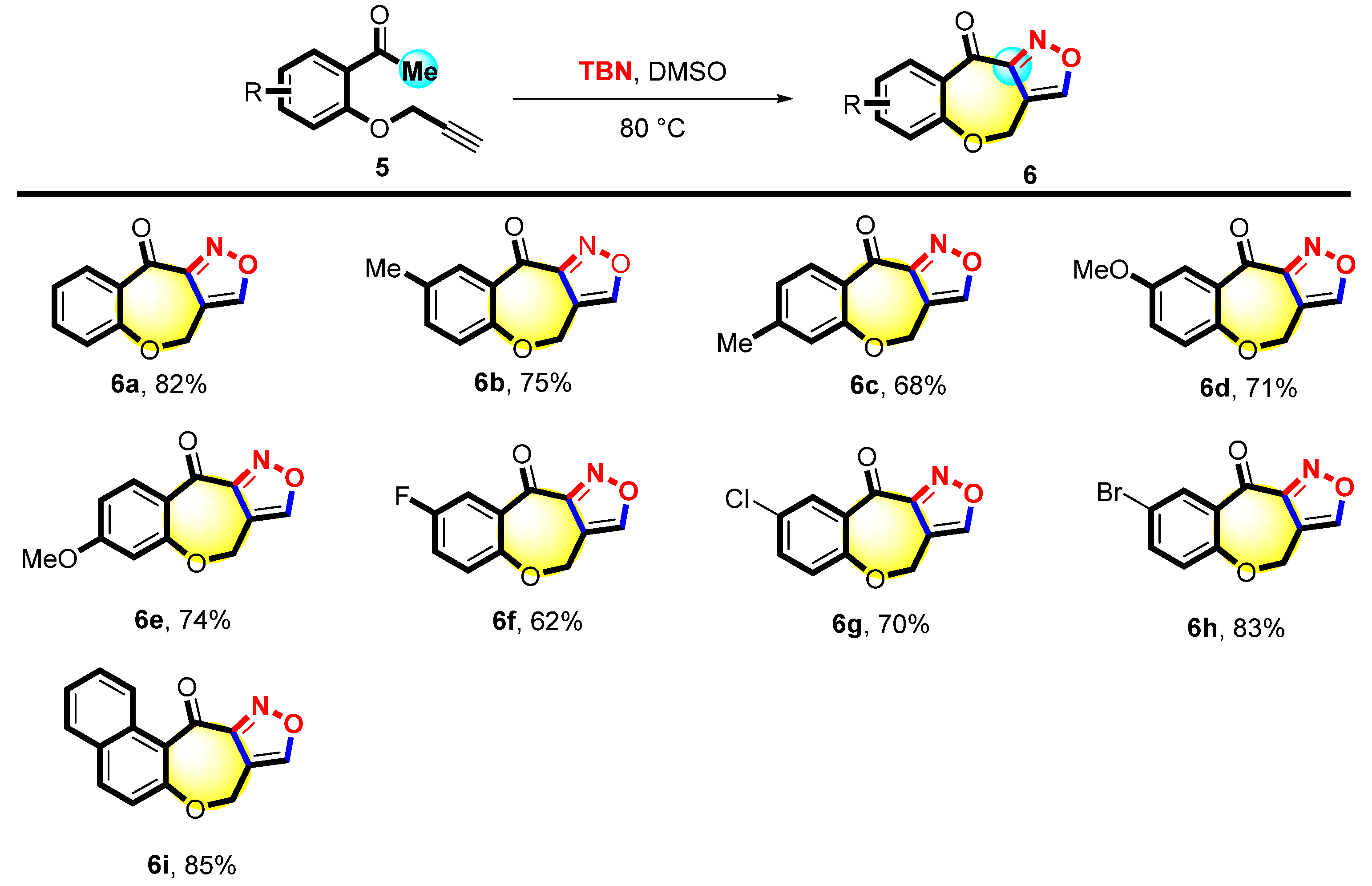
Next, substrate 5 was explored to obtain a series of derivatives with an isoxazole structure, and the reaction conditions were further optimized for the synthesis (Table S4). The scope of 6 was studied under the optimal conditions. As shown in Figure 4, substrate 5a was smoothly transformed into the corresponding product with a yield of 82%. A series of 5 with different substitutions (4-Me, 5-Me 4-OMe, and 4-OMe) was investigated. The desired products 6b–6e were obtained with 68%-75% yields. The 1-(2-(prop-2-yn-1-yloxy)phenyl)ethan-1-ones (5f–5h) attached with halogen atoms (e.g., 5-F, 5-Cl, and 5-Br) were also tolerated in the reaction, yielding the corresponding products 6f–6h with 62%-83% yields. Substrate 5i was also found to be suitable for this reaction, giving the desired product 6i with an 85% yield.
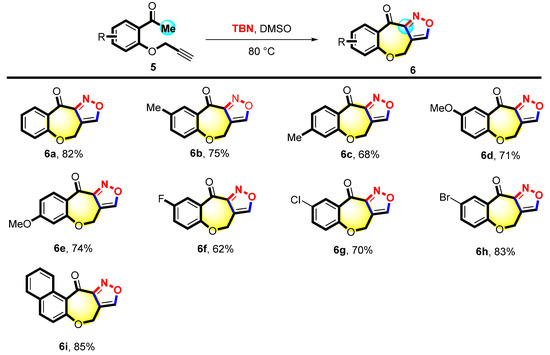
Figure 4.
Scope of 3-(2-acetophenoxy) propyne a,b. a Reaction condition: 5 (0.1 mmol) and TBN (0.5 mmol) were heated in DMSO (2 mL) at 80 °C for 10 h. b Isolated yields.
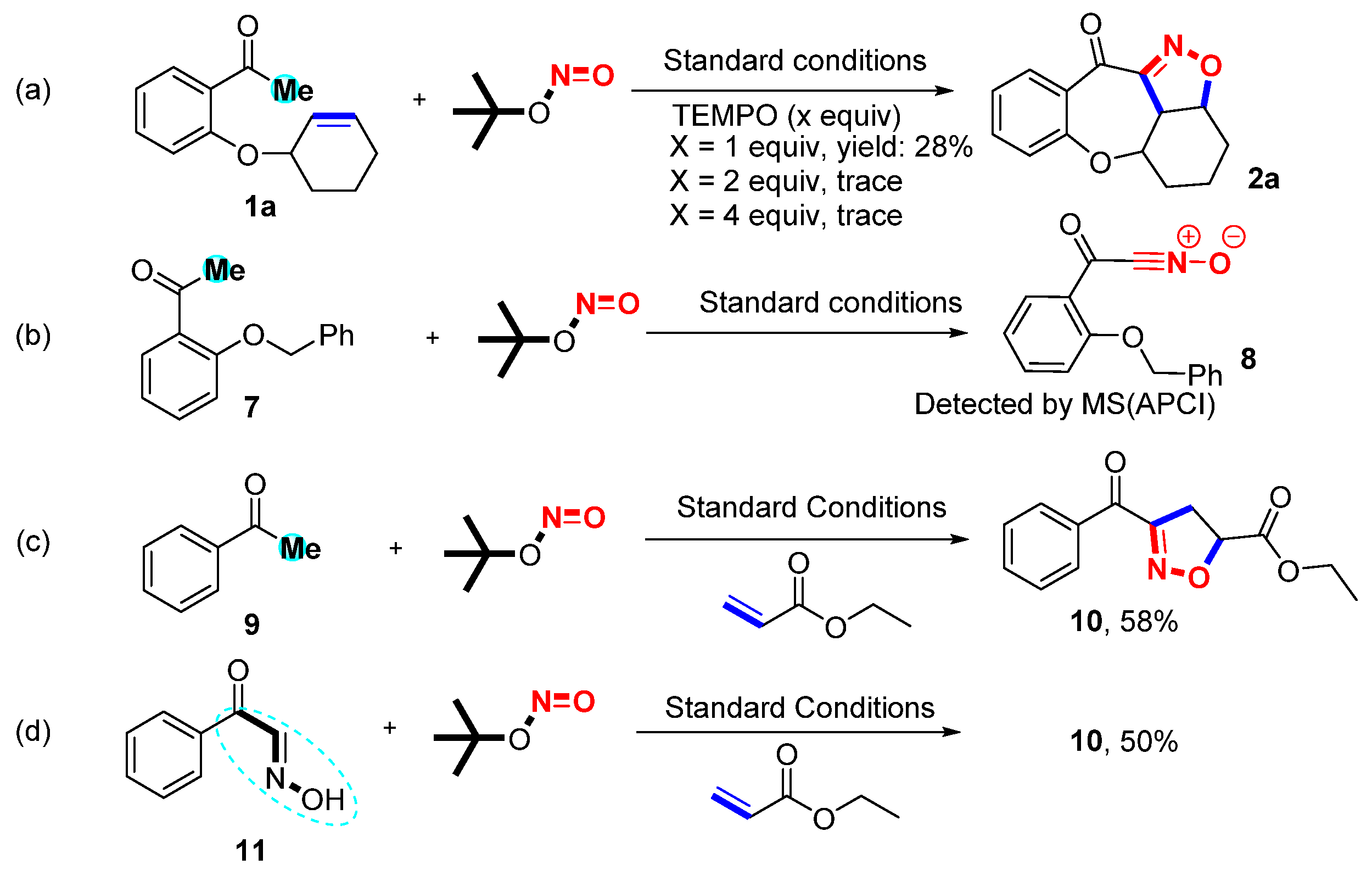
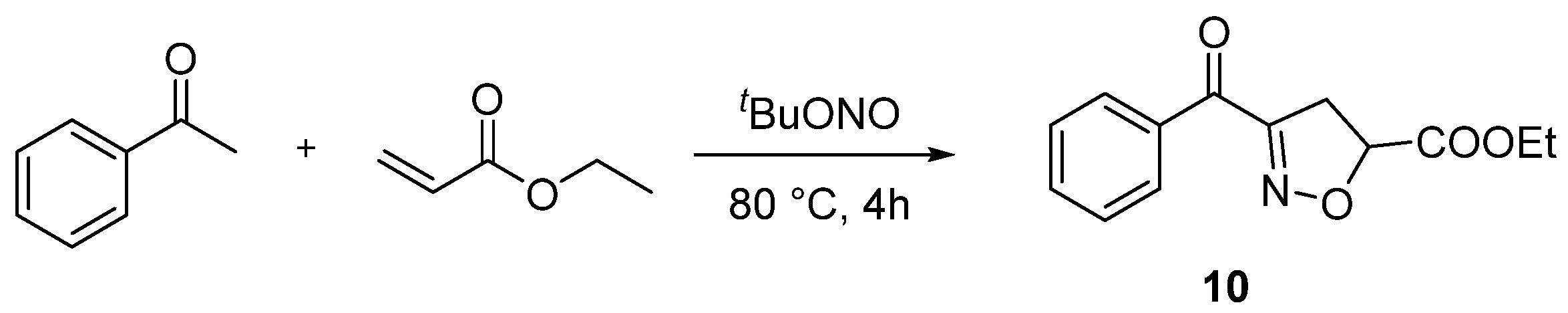
Several control experiments were carried out to investigate the reaction mechanism (Scheme 2) [36,37,38]. The reaction was restrained completely and trace amounts of 2a were observed when a 2.0-equivalent radical scavenger 2,2,6,6-tetramethyl-1-piperidinyl (TEMPO) was added to the standard reaction. This result revealed that the reaction proceeded through a radical pathway. Next, 1a and TBN were reacted under standard conditions for 20 min to identify the possible intermediates. However, only 2a was detected by MS (APCI) because the intermediate nitrile oxide E shares the same relative molecular mass as 2a. The result of MS is ambiguous because the masses 2a and E are the same. Alternative approaches were performed to confirm this by subjecting substrates 7 to standard conditions for 20 min to detect 8 nitrile oxides via MS (APCI). A group of intermolecular reactions was used to further explore the reaction mechanism by using 9, 11, and ethyl acrylate. Under the optimal conditions, the desired product 10 was produced with yields of 58% and 50% from 9 and 11. These results disclosed that nitrile oxide was the potential intermediate for this protocol.
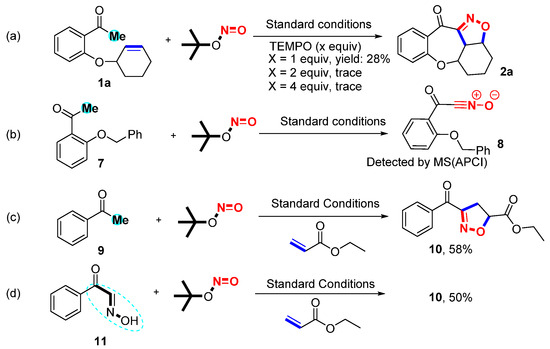
Scheme 2.
Control experiments. (a) Radical capture. (b) Formation of intermediate 8. (c) Formation of 4,5-dihydroisoxazole 8b via intermolecular cycloaddition under standard conditions. (d) Formation of 10 from intermediate 11 under standard conditions.
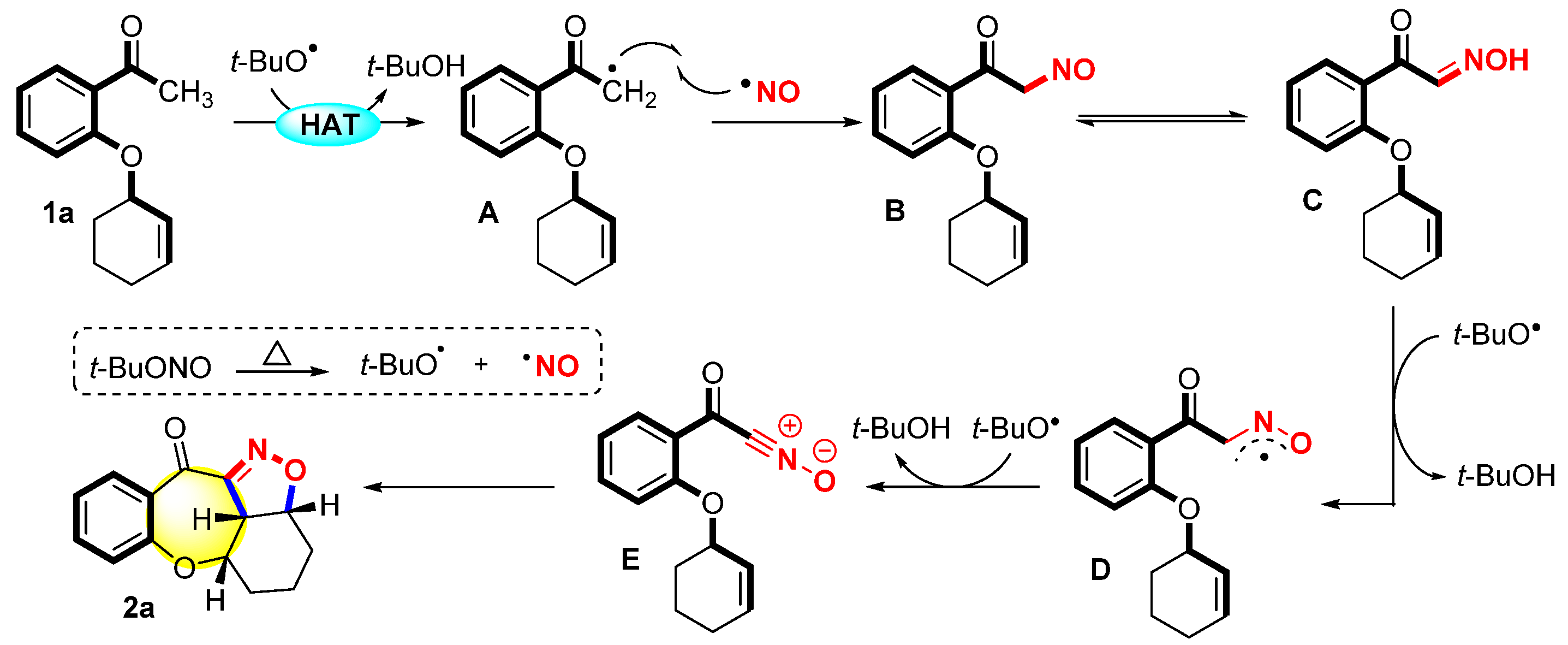
Based on the evidence presented above and the related literature [28,35,36,39,40,41], a plausible reaction pathway was proposed (Scheme 3). First, TBN was transformed into NO and tBuO radicals through thermal homolysis. The substrate 1a underwent hydrogen abstraction with the tBuO radical to afford intermediate A. Then, intermediate A and the NO radical performed radical cross-coupling to produce intermediate B. Intermediate B underwent tautomerization to generate oxime C, which was further conducted two times via hydrogen abstraction with the tBuO radical to generate the nitrile oxide intermediate E. Finally, the nitrile oxide intermediate E underwent 1,3-dipolar cycloaddition with an intramolecular alkene to produce the final product 2a.
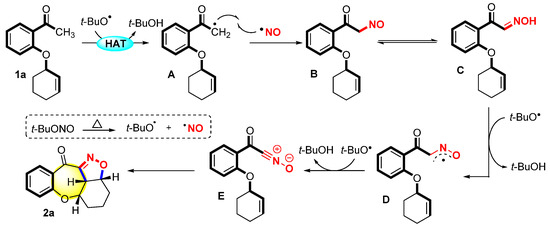
Scheme 3.
Proposed mechanism.
3. Materials and Methods
3.1. General Information
Analytical thin layer chromatography (TLC) was performed by using pre-coated silica gel HF254 glass plates. Column chromatography was performed by using silica gel (200–300 mesh). The 1H NMR and 13C NMR spectra were recorded on a Bruker Advance 500 MHz instrument at 500 MHz (1H NMR) and 126 MHz (13C NMR). We used the residual solvent peak in CDCl3 as an internal reference (δ = 7.26 for 1H and δ = 77.0 for 13C{1H}). Chemical shifts (δ) are reported in ppm relative to the internal standard of tetramethylsilane (TMS). The coupling constants (J) are quoted in Hz (hertz). Resonances are described as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), br (broad), or combinations thereof. High-resolution mass spectra (HRMS) were obtained on Thermo Scientific Q-Exactive (ESI mode, Q-Exactive Orbitrap MS system). The melting points were measured with the SGW X-4 apparatus. Data collection for the crystal structure was performed by using Mo Kα radiation on a Bruker Smart APEX CCD area-detector diffractometer.
3.2. Synthetic Procedures
Compounds 1a–1j were prepared according to the referenced literature [42]. To a solution of 1-(2-hydroxyphenyl)ethan-1-one) (1.0 equiv.) and Cs2CO3 (3.0 equiv.) in CH2Cl2 (0.1 M), a solution of 3-bromocyclohex-1-ene (2.0 equiv.) in CH2Cl2 (0.5 M) was added dropwise at room temperature and stirred for 10 h. After the reaction was completed, 50 mL of water was added to the mixture and then extracted with DCM 3 times (3 × 50 mL). The extract was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude residues were purified by column chromatography using an ethyl acetate/petroleum ether mixture to obtain the desired products (Scheme 4).
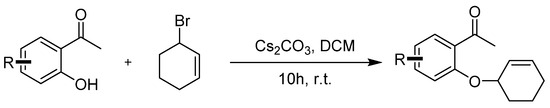
Scheme 4.
General procedure for synthesis of 1-(2-(cyclohex-2-en-1-yloxy)phenyl)ethan-1-one 1a–1j.
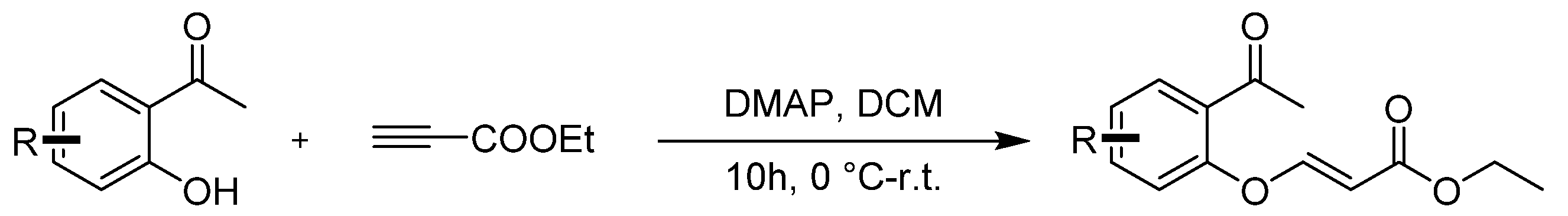
Compounds 3a–3q were prepared according to the referenced literature [39,40]. To a solution of 1-(2-hydroxyphenyl)ethan-1-one) (1.0 equiv.) and DMAP (0.1 equiv.) in CH2Cl2 (0.1 M), a solution of ethyl acetylenecarboxylate (2.0 equiv.) was added dropwise at room temperature and stirred for 10 h. After the reaction was completed, 50 mL of water was added to the mixture and then extracted with DCM 3 times (3 × 50 mL). The extract was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude residues were purified by column chromatography using an ethyl acetate/petroleum ether mixture to obtain the desired products (Scheme 5).
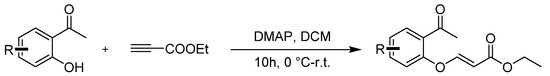
Scheme 5.
General procedure for synthesis of ethyl (E)-3-(2-acetylphenoxy)acrylate 3a–3q.
Compounds 5a–5i were prepared according to the referenced literature [43,44,45]. To a solution of 1-(2-hydroxyphenyl)ethan-1-one) (1.0 equiv.) and K2CO3 (3.0 equiv.) in CH2Cl2 (0.1 M), a solution of 3-bromoprop-1-yne (2.0 equiv.) in CH2Cl2 (0.5 M) was added dropwise at room temperature and stirred for 10 h. After the reaction was completed, 50 mL of water was added to the mixture and then extracted with DCM 3 times (3 × 50 mL). The extract was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude residues were purified by column chromatography using an ethyl acetate/petroleum ether mixture to obtain the desired products (Scheme 6).
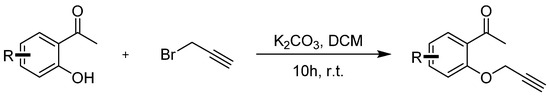
Scheme 6.
General procedure for synthesis of 1-(2-(prop-2-yn-1-yloxy)phenyl)ethan-1-one 5g–5i.
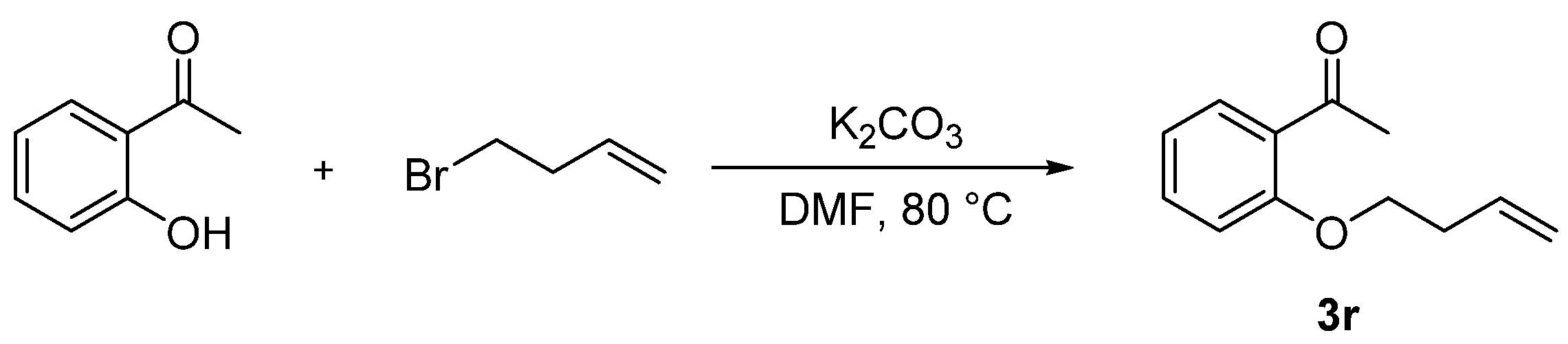
Compound 3r was prepared according to the referenced literature [46]. To a solution of 1-(2-(but-3-en-1-yloxy)phenyl)ethan-1-one (1.0 equiv.) and K2CO3 (1.0 equiv.), a solution of 4-bromo-1-butene (1.2 equiv.) in DMF (4 mL) was added dropwise at 80 °C and stirred for 24 h. After the reaction was completed, 50 mL of water was added to the mixture and then extracted with DCM 3 times (3 × 50 mL). The extract was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude residues were purified by column chromatography using an ethyl acetate/petroleum ether mixture to obtain the desired products (Scheme 7).
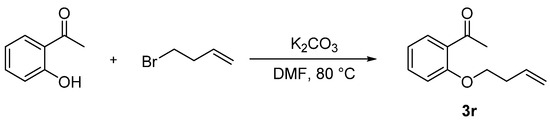
Scheme 7.
General procedure for synthesis of 1-(2-(but-3-en-1-yloxy)phenyl)ethan-1-one (3r).
Compound 10 was prepared according to the referenced literature [28]. A mixture of acetophenone (1 equiv.), ethyl acrylate (3 equiv.), and tBuONO (3 equiv.) was dissolved in DMSO (2.0 mL). Then, the mixture was reacted under 80 °C for 4 h. After the reaction was completed, 50 mL of water was added to the mixture and then extracted with EtOAc 3 times (3 × 50 mL). The extract was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude residues were purified by column chromatography by using an ethyl acetate/petroleum ether mixture to obtain the desired product (Scheme 8).
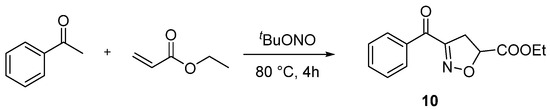
Scheme 8.
General procedure for synthesis of ethyl 3-benzoyl-4,5-dihydroisoxazole-5-carboxylate (10).
Compound 11 was prepared according to the referenced literature [36,41]. A mixture of acetophenone (1.0 equiv.) and I2 (1.6 equiv.) was reacted under 110 °C for 10 h. Phenyl glyoxal was afforded without further purification. Then, hydroxylamine hydrochloride (1.0 equiv.) was added to a solution of phenyl glyoxal in THF (40 mL), and the reaction mixture was reacted under 24 °C for 12 h. After the reaction was completed, 50 mL of water was added to the mixture and then extracted with EtOAc 3 times (3 × 50 mL). The extract was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude residues were purified by column chromatography using an ethyl acetate/petroleum ether mixture to obtain the desired product (Scheme 9).
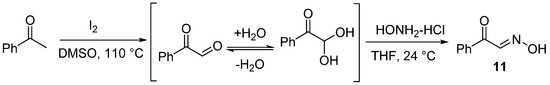
Scheme 9.
General procedure for synthesis of (E)-2-oxo-2-phenylacetaldehyde oxime (11).
3.3. Characterization of Products
- 2a,2a1,3,4,5,5a-Hexahydro-11H-2,6-dioxa-1-azadibenzo[cd,g]azulen-11-one (2a), 38 mg, 95%, white solid, m.p.: 118–119 °C. 1H NMR (500 MHz, CDCl3) δ (ppm) 7.99 (dd, J = 7.9, 1.8 Hz, 1H), 7.57 (td, J = 7.7, 1.8 Hz, 1H), 7.31–7.21 (m, 1H), 7.12 (dd, J = 8.1, 1.1 Hz, 1H), 4.87 (dt, J = 10.4, 4.4 Hz, 1H), 4.31 (td, J = 6.7, 4.1 Hz, 1H), 3.70 (dd, J = 10.4, 7.2 Hz, 1H), 2.26–2.00 (m, 2H), 1.96–1.80 (m, 2H), 1.79–1.54 (m, 2H). 13C NMR (126 MHz, CDCl3) δ (ppm) 184.9, 160.7, 158.4, 136.1, 129.9, 129.8, 124.9, 123.3, 81.3, 78.0, 49.2, 27.8, 23.7, 14.8. HRMS (ESI): m/z [M + H]+ calcd for C14H14NO3: 244.0968; found: 244.0966.
- 8-Methyl-2a,2a1,3,4,5,5a-hexahydro-11H-2,6-dioxa-1-azadibenzo[cd,g]azulen-11-one (2b), (silica gel: 200–300 mesh, solvent system: petroleum ether/ethyl acetate = 10:1–5:1), 31 mg, 87%, light yellow solid, m.p.: 135–136 °C. 1H NMR (500 MHz, CDCl3) δ (ppm) 7.90 (d, J = 8.0 Hz, 1H), 7.05 (dd, J = 8.0, 1.6 Hz, 1H), 6.92 (d, J = 1.6 Hz, 1H), 4.83 (dt, J = 10.3, 4.4 Hz, 1H), 4.27 (ddd, J = 7.1, 5.8, 4.0 Hz, 1H), 3.69 (dd, J = 10.4, 7.1 Hz, 1H), 2.38 (s, 3H), 2.14 (dddd, J = 37.4, 13.8, 10.9, 5.7 Hz, 2H), 1.95–1.75 (m, 3H), 1.74–1.53 (m, 2H). 13C NMR (126 MHz, CDCl3) δ (ppm) 184.5, 161.1, 158.5, 147.9, 129.8, 126.9, 125.9, 123.6, 80.9, 77.8, 49.6, 27.9, 23.9, 21.7, 14.7. HRMS (ESI): m/z [M + H]+ calcd for C15H16NO3: 258.1125; found: 258.1123.
- 9-Chloro-2a,2a1,3,4,5,5a-hexahydro-11H-2,6-dioxa-1-azadibenzo[cd,g]azulen-11-one (2c), (silica gel: 200–300 mesh, solvent system: petroleum ether/ethyl acetate = 10:1–5:1), 24 mg, 85%, light brown solid, m.p.: 135–136 °C. 1H NMR (500 MHz, CDCl3) δ (ppm) 7.89 (d, J = 2.7 Hz, 1H), 7.48 (dd, J = 8.6, 2.6 Hz, 1H), 7.06 (d, J = 8.6 Hz, 1H), 4.86 (dt, J = 10.3, 4.4 Hz, 1H), 4.29 (td, J = 6.7, 4.1 Hz, 1H), 3.70 (dd, J = 10.4, 7.2 Hz, 1H), 2.23–1.97 (m, 2H), 1.84 (ddq, J = 23.1, 9.2, 4.5, 3.9 Hz, 2H), 1.77–1.54 (m, 2H). 13C NMR (126 MHz, CDCl3) δ (ppm) 183.6, 158.9, 157.9, 135.7, 130.7, 130.5, 129.2, 125.0, 81.6, 78.4, 48.9, 27.6, 23.5, 14.7. HRMS (ESI): m/z [M + Na]+ calcd for C14H12ClNO3Na: 300.0398; found: 300.0398.
- 8-Chloro-2a,2a1,3,4,5,5a-hexahydro-11H-2,6-dioxa-1-azadibenzo[cd,g]azulen-11-one (2d), (silica gel: 200–300 mesh, solvent system: petroleum ether/ethyl acetate = 10:1–5:1), 24 mg, 79%, white solid, m.p.: 179–181 °C. 1H NMR (500 MHz, CDCl3) δ (ppm) 7.94 (d, J = 8.5 Hz, 1H), 7.24 (dd, J = 8.5, 2.0 Hz, 1H), 7.15 (d, J = 2.0 Hz, 1H), 4.87 (dt, J = 10.5, 4.4 Hz, 1H), 4.33 (ddd, J = 7.1, 6.1, 4.0 Hz, 1H), 3.71 (dd, J = 10.4, 7.1 Hz, 1H), 2.2 –2.04 (m, 2H), 1.93–1.79 (m, 2H), 1.78–1.60 (m, 2H). 13C NMR (101 MHz, CDCl3) δ (ppm) 183.6, 161.2, 158.0, 141.9, 131.0, 128.1, 125.5, 123.7, 81.3, 78.5, 49.3, 27.7, 23.7, 14.7. HRMS (ESI): m/z [M + H]+ calcd for C14H13ClNO3: 278.0578; found: 278.0580.
- 9-Fluoro-2a,2a1,3,4,5,5a-hexahydro-11H-2,6-dioxa-1-azadibenzo[cd,g]azulen-11-one (2e), (silica gel: 200–300 mesh, solvent system: petroleum ether/ethyl acetate = 10:1–5:1), 33 mg, 88%, white solid, m.p.: 132–133 °C. 1H NMR (500 MHz, CDCl3) δ (ppm) 7.62 (dd, J = 8.3, 3.2 Hz, 1H), 7.30–7.22 (m, 1H), 7.11 (dd, J = 8.9, 4.4 Hz, 1H), 4.88 (dt, J = 10.4, 4.3 Hz, 1H), 4.29 (td, J = 6.7, 4.0 Hz, 1H), 3.70 (dd, J = 10.4, 7.3 Hz, 1H), 2.23–2.00 (m, 2H), 1.93–1.78 (m, 3H), 1.81–1.54 (m, 3H). 13C NMR (126 MHz, CDCl3) δ (ppm) 183.9, 159.2 (d, Jc–f = 246.8 Hz), 158.0, 156.7 (d, Jc–f = 2.7 Hz), 130.8 (d, Jc–f = 7.2 Hz), 125.1 (d, Jc–f = 8.1 Hz), 123.0 (d, Jc–f = 23.3 Hz), 115.4 (d, Jc–f = 24.1 Hz), 81.5, 78.3, 48.8, 27.7, 23.6, 14.7. HRMS (ESI): m/z [M + H]+ calcd for C14H13FNO3: 262.0874; found: 262.0873.
- 9-Bromo-2a,2a1,3,4,5,5a-hexahydro-11H-2,6-dioxa-1-azadibenzo[cd,g]azulen-11-one (2f), (silica gel: 200–300 mesh, solvent system: petroleum ether/ethyl acetate = 10:1–5:1), 25 mg, 79%, white solid, m.p.: 134–135 °C. 1H NMR (500 MHz, CDCl3) δ (ppm) 8.07 (d, J = 2.6 Hz, 1H), 7.64 (dd, J = 8.6, 2.6 Hz, 1H), 7.01 (d, J = 8.6 Hz, 1H), 4.88 (dt, J = 10.4, 4.4 Hz, 1H), 4.30 (td, J = 6.8, 4.0 Hz, 1H), 3.70 (dd, J = 10.4, 7.2 Hz, 1H), 2.21–2.00 (m, 2H), 1.93–1.80 (m, 2H), 1.79–1.57 (m, 3H). 13C NMR (126 MHz, CDCl3) δ (ppm) 183.5, 159.4, 157.8, 138.7, 132.4, 131.1, 125.3, 118.0, 81.6, 78.3, 49.0, 27.6, 23.6, 14.8. HRMS (ESI): m/z [M + H]+ calcd for C14H13BrNO3: 322.0073; found: 322.0073.
- 10-Methoxy-2a,2a1,3,4,5,5a-hexahydro-11H-2,6-dioxa-1-azadibenzo[cd,g]azulen-11-one (2g), (silica gel: 200–300 mesh, solvent system: petroleum ether/ethyl acetate = 10:1–5:1), 38 mg, 91%, white solid, m.p.: 194–195 °C. 1H NMR (500 MHz, CDCl3) δ (ppm) 7.43 (t, J = 8.3 Hz, 1H), 6.80 (dd, J = 8.5, 0.9 Hz, 1H), 6.73 (dd, J = 8.1, 0.8 Hz, 1H), 4.92 (dt, J = 10.5, 4.4 Hz, 1H), 4.25 (ddd, J = 10.7, 7.6, 4.9 Hz, 1H), 3.86 (s, 3H), 3.64 (dd, J = 10.5, 7.6 Hz, 1H), 2.10–1.96 (m, 2H), 1.95–1.78 (m, 2H), 1.77–1.52 (m, 2H). 13C NMR (126 MHz, CDCl3) δ (ppm) 183.0, 159.2, 158.2, 157.4, 134.6, 122.0, 115.0, 108.7, 83.4, 78.0, 56.3, 47.4, 26.4, 22.5, 16.2. HRMS (ESI): m/z [M + H]+ calcd for C15H16NO4: 274.1074; found: 274.1074.
- 9-Methoxy-2a,2a1,3,4,5,5a-hexahydro-11H-2,6-dioxa-1-azadibenzo[cd,g]azulen-11-one (2h), (silica gel: 200–300 mesh, solvent system: petroleum ether/ethyl acetate = 10:1–5:1), 29 mg, 81%, white solid, m.p.: 137–139 °C. 1H NMR (500 MHz, CDCl3) δ (ppm) 7.41 (d, J = 3.2 Hz, 1H), 7.11 (dd, J = 8.8, 3.2 Hz, 1H), 7.04 (d, J = 8.8 Hz, 1H), 4.85 (dt, J = 10.3, 4.3 Hz, 1H), 4.23 (ddd, J = 7.3, 6.1, 4.1 Hz, 1H), 3.83 (s, 3H), 3.66 (dd, J = 10.4, 7.3 Hz, 1H), 2.23–2.01 (m, 2H), 1.93–1.78 (m, 2H), 1.75–1.55 (m, 2H). 13C NMR (101 MHz, CDCl3) δ (ppm) 184.9, 158.4, 156.5, 154.9, 130.0, 124.4, 123.8, 111.2, 81.3, 78.1, 55.8, 49.1, 27.9, 23.7, 14.8. HRMS (ESI): m/z [M + H]+ calcd for C15H16NO4: 274.1074; found: 274.1072.
- 8-Methoxy-2a,2a1,3,4,5,5a-hexahydro-11H-2,6-dioxa-1-azadibenzo[cd,g]azulen-11-one (2i), (silica gel: 200–300 mesh, solvent system: petroleum ether/ethyl acetate = 10:1–5:1), 26 mg, 81%, light yellow solid, m.p.: 98–100 °C. 1H NMR (500 MHz, CDCl3) δ (ppm) 7.99 (d, J = 8.8 Hz, 1H), 6.77 (dd, J = 8.9, 2.5 Hz, 1H), 6.56 (d, J = 2.4 Hz, 1H), 4.80 (dt, J = 10.4, 4.5 Hz, 1H), 4.28 (ddd, J = 7.0, 5.0, 3.9 Hz, 1H), 3.85 (s, 3H), 3.70 (dd, J = 10.4, 7.0 Hz, 1H), 2.16 (ddq, J = 18.8, 14.5, 4.9 Hz, 2H), 1.92–1.73 (m, 2H), 1.72–1.56 (m, 2H). 13C NMR (126 MHz, CDCl3) δ (ppm) 183.3, 166.2, 163.7, 158.6, 131.6, 122.3, 111.9, 107.1, 80.3, 77.9, 55.8, 50.0, 28.1, 24.1, 14.4. HRMS (ESI): m/z [M + H]+ calcd for C15H16NO4: 274.1074; found: 274.1074.
- 2a,2a1,3,4,5,5a-Hexahydro-13H-2,6-dioxa-1-azabenzo[cd]naphtho[2,1-g]azulen-13-one (2j), (silica gel: 200–300 mesh, solvent system: petroleum ether/ethyl acetate = 10:1–5:1), 29 mg, 86%, brown solid, m.p.: 147–150 °C. 1H NMR (500 MHz, CDCl3) δ (ppm) 8.65 (d, J = 8.7 Hz, 1H), 7.99 (d, J = 8.8 Hz, 1H), 7.82 (dd, J = 8.2, 1.3 Hz, 1H), 7.60 (ddd, J = 8.5, 6.7, 1.4 Hz, 1H), 7.49 (ddd, J = 8.0, 6.8, 1.2 Hz, 1H), 7.23 (d, J = 8.8 Hz, 1H), 4.91 (dt, J = 10.5, 4.2 Hz, 1H), 4.34 (ddd, J = 9.0, 7.6, 4.8 Hz, 1H), 3.63 (dd, J = 10.5, 7.6 Hz, 1H), 2.07 (dddd, J = 26.6, 13.3, 10.1, 5.4 Hz, 2H), 1.91 (dddd, J = 15.2, 13.3, 7.4, 3.8 Hz, 2H), 1.85–1.54 (m, 2H). 13C NMR (126 MHz, CDCl3) δ (ppm) 186.1, 158.3, 158.0, 136.0, 131.3, 131.0, 129.0, 128.3, 126.0, 126.0, 124.9, 122.0, 83.1, 78.0, 47.5, 26.9, 22.7, 15.9. HRMS (ESI): m/z [M + H]+ calcd for C18H16NO3: 294.1124; found: 294.1123.
- Ethyl 9-oxo-3,3a-dihydro-9H-chromeno[3,2-c]isoxazole-3-carboxylate (4a), (silica gel: 200–300 mesh, solvent system: petroleum ether/ethyl acetate = 5:1–2:1), 35 mg, 95%, yellow oil. 1H NMR (500 MHz, CDCl3) δ (ppm) 8.04 (dd, J = 7.9, 1.8 Hz, 1H), 7.61 (ddd, J = 8.7, 7.2, 1.8 Hz, 1H), 7.19 (ddd, J = 8.1, 7.2, 1.0 Hz, 1H), 7.09 (dd, J = 8.4, 1.0 Hz, 1H), 6.03 (d, J = 6.9 Hz, 1H), 5.35 (d, J = 6.9 Hz, 1H), 4.36 (q, J = 7.1 Hz, 2H), 1.37 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 174.4, 166.2, 159.2, 151.4, 137.6, 128.0, 123.6, 123.4, 118.7, 86.7, 85.3, 63.1, 14.0. HRMS (ESI): m/z [M + H]+ calcd for C13H12NO5: 274.0710; found: 274.0712.
- Ethyl 7-methyl-9-oxo-3,3a-dihydro-9H-chromeno[3,2-c]isoxazole-3-carboxylate (4b), (silica gel: 200–300 mesh, solvent system: petroleum ether/ethyl acetate = 5:1–2:1), 31 mg, 91%, yellow solid, m.p.: 129–130 °C. 1H NMR (500 MHz, CDCl3) δ (ppm) 7.83–7.77 (m, 1H), 7.41 (dd, J = 8.5, 2.4 Hz, 1H), 6.98 (d, J = 8.4 Hz, 1H), 5.98 (d, J = 7.0 Hz, 1H), 5.32 (d, J = 7.0 Hz, 1H), 4.35 (q, J = 7.1 Hz, 2H), 2.35 (s, 3H), 1.37 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 174.5, 166.3, 157.3, 151.6, 138.7, 133.4, 127.4, 123.0, 118.4, 86.7, 85.2, 63.0, 20.4, 14.0. HRMS (ESI): m/z [M + H]+ calcd for C14H14NO5: 276.0867; found: 276.0867.
- Ethyl 6-methyl-9-oxo-3,3a-dihydro-9H-chromeno[3,2-c]isoxazole-3-carboxylate (4c), (silica gel: 200–300 mesh, solvent system: petroleum ether/ethyl acetate = 5:1–2:1), 26 mg, 82%, white solid, m.p.: 95–97 °C. 1H NMR (500 MHz, CDCl3) δ (ppm) 7.88 (d, J = 8.1 Hz, 1H), 6.98 (dd, J = 8.2, 1.5 Hz, 1H), 6.86 (s, 1H), 5.97 (d, J = 7.0 Hz, 1H), 5.30 (d, J = 7.0 Hz, 1H), 4.34 (q, J = 7.1 Hz, 2H), 2.39 (s, 3H), 1.36 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, CDCl3)) δ (ppm) 174.0, 166.3, 159.2, 151.5, 149.8, 127.7, 124.9, 121.1, 118.6, 86.7, 85.1, 63.0, 22.0, 14.0. HRMS (ESI): m/z [M + H]+ calcd for C14H14NO5: 276.0867; found: 274.0867.
- Ethyl 7-methoxy-9-oxo-3,3a-dihydro-9H-chromeno[3,2-c]isoxazole-3-carboxylate (4d), 30 mg, 86%, yellow solid, m.p.: 167–168 °C. 1H NMR (500 MHz, CDCl3) δ (ppm) 7.40 (d, J = 3.1 Hz, 1H), 7.19 (dd, J = 9.1, 3.2 Hz, 1H), 7.01 (d, J = 9.1 Hz, 1H), 5.97 (d, J = 7.0 Hz, 1H), 5.31 (d, J = 7.1 Hz, 1H), 4.35 (q, J = 7.1 Hz, 2H), 3.83 (s, 3H), 1.37 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ (ppm) 174.3, 166.3, 155.6, 153.9, 151.7, 126.9, 123.5, 120.0, 107.8, 86.8, 85.2, 63.1, 55.9, 14.1. HRMS (ESI): m/z [M + H]+ calcd for C14H14NO6: 292.0816; found: 292.0816.
- Ethyl 6-methoxy-9-oxo-3,3a-dihydro-9H-chromeno[3,2-c]isoxazole-3-carboxylate (4e), 29 mg, 87%, white solid, m.p.: 120–121 °C. 1H NMR (500 MHz, CDCl3) δ (ppm) 7.93 (d, J = 8.9 Hz, 1H), 6.71 (dd, J = 8.9, 2.4 Hz, 1H), 6.49 (d, J = 2.3 Hz, 1H), 5.98 (d, J = 7.1 Hz, 1H), 5.28 (d, J = 7.2 Hz, 1H), 4.34 (q, J = 7.1 Hz, 2H), 3.86 (s, 3H), 1.36 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ (ppm) 172.9, 167.3, 166.3, 161.4, 151.4, 129.6, 117.1, 111.9, 101.7, 86.9, 84.9, 63.0, 55.9, 14.0. HRMS (ESI): m/z [M + H]+ calcd for C14H14NO6: 292.0816; found: 292.0817.
- Ethyl 7-bromo-9-oxo-3,3a-dihydro-9H-chromeno[3,2-c]isoxazole-3-carboxylate (4f), 24 mg, 80%, yellow solid, m.p.: 139–142 °C. 1H NMR (500 MHz, CDCl3) δ (ppm) 11.76 (s, 1H), 8.72 (d, J = 2.5 Hz, 1H), 7.66 (d, J = 11.4 Hz, 1H), 7.41 (s, 1H), 6.98 (d, J = 8.9 Hz, 1H), 4.49 (q, J = 7.1 Hz, 2H), 1.45 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ (ppm) 187.3, 163.0, 161.8, 161.5, 156.0, 140.7, 135.3, 120.5, 119.5, 111.4, 110.0, 62.9, 14.1. HRMS (ESI): m/z [M + H]+ calcd for C13H11BrNO5: 339.9815; found: 339.9817.
- Ethyl 7-chloro-9-oxo-3,3a-dihydro-9H-chromeno[3,2-c]isoxazole-3-carboxylate (4g), 27 mg, 83%, yellow solid, m.p.: 142–144 °C. 1H NMR (500 MHz, CDCl3) δ (ppm) 7.97 (d, J = 2.7 Hz, 1H), 7.54 (dd, J = 8.9, 2.7 Hz, 1H), 7.05 (d, J = 8.9 Hz, 1H), 6.03 (d, J = 6.9 Hz, 1H), 5.35 (d, J = 6.9 Hz, 1H), 4.36 (q, J = 7.2 Hz, 2H), 1.37 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ (ppm) 173.3, 166.0, 157.6, 150.8, 137.4, 129.3, 127.1, 124.1, 120.4, 86.7, 85.4, 63.2, 14.0. HRMS (ESI): m/z [M + H]+ calcd for C13H11ClNO5: 296.0320; found: 296.0320.
- Ethyl 7-fluoro-9-oxo-3,3a-dihydro-9H-chromeno[3,2-c]isoxazole-3-carboxylate (4h), 20 mg, 78%, white solid, m.p.: 149–151 °C. 1H NMR (500 MHz, CDCl3) δ (ppm) 7.66 (dd, J = 8.0, 3.1 Hz, 1H), 7.32 (ddd, J = 9.0, 7.4, 3.2 Hz, 1H), 7.08 (dd, J = 9.1, 4.1 Hz, 1H), 6.02 (d, J = 6.9 Hz, 1H), 5.34 (d, J = 6.9 Hz, 1H), 4.35 (q, J = 7.1 Hz, 2H), 1.37 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ (ppm) 173.6, 166.0, 159.2 (d, Jc–f = 246.1 Hz), 155.4, 151.0, 125.1 (d, Jc–f = 24.8 Hz), 124.0 (d, Jc–f = 7.3 Hz), 120.5 (d, Jc–f = 7.4 Hz), 113.1 (d, Jc–f = 24.5 Hz), 86.8, 85.3, 63.1, 14.0. HRMS (ESI): m/z [M + H]+ calcd for C13H11FNO5: 280.0616; found: 280.0616.
- Ethyl 11-oxo-7a,8-dihydro-11H-benzo[5,6]chromeno[3,2-c]isoxazole-8-carboxylate (4i), 14 mg, 70%, yellow solid, m.p.: 194–195 °C. 1H NMR (500 MHz, CDCl3) δ (ppm) 9.47 (d, J = 8.7 Hz, 1H), 8.04 (d, J = 9.0 Hz, 1H), 7.81 (d, J = 8.1 Hz, 1H), 7.76–7.70 (m, 1H), 7.53 (t, J = 7.5 Hz, 1H), 7.16 (d, J = 9.0 Hz, 1H), 6.09 (d, J = 6.9 Hz, 1H), 5.40 (d, J = 6.9 Hz, 1H), 4.38 (q, J = 7.1 Hz, 2H), 1.39 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ (ppm) 175.0, 166.3, 161.8, 152.1, 139.5, 131.4, 130.7, 129.9, 128.7, 126.3, 126.2, 118.5, 115.7, 86.4, 85.3, 63.1, 14.1. HRMS (ESI): m/z [M + H]+ calcd for C17H14NO5: 312.0866; found: 312.0866.
- Methyl 9-oxo-3,3a-dihydro-9H-chromeno[3,2-c]isoxazole-3-carboxylate (4j), 29 mg, 90%, white solid, m.p.: 138–139 °C. 1H NMR (400 MHz, CDCl3) δ (ppm) 8.04 (dd, J = 7.9, 1.8 Hz, 1H), 7.61 (ddd, J = 8.4, 7.2, 1.8 Hz, 1H), 7.19 (ddd, J = 8.1, 7.2, 1.1 Hz, 1H), 7.08 (dd, J = 8.4, 1.1 Hz, 1H), 6.03 (d, J = 6.9 Hz, 1H), 5.37 (d, J = 7.0 Hz, 1H), 3.91 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 174.3, 166.6, 159.1, 151.4, 137.6, 1287.0, 123.6, 123.4, 118.7, 86.7, 85.1, 53.6. HRMS (ESI): m/z [M + H]+ calcd for C12H10NO5: 248.0554; found: 248.0553.
- Methyl 7-methyl-9-oxo-3,3a-dihydro-9H-chromeno[3,2-c]isoxazole-3-carboxylate (4k), 32 mg, 92%, yellow solid, m.p.: 160–162 °C. 1H NMR (400 MHz, CDCl3) δ (ppm) 7.80 (d, J = 2.4 Hz, 1H), 7.40 (dd, J = 8.5, 2.4 Hz, 1H), 6.97 (d, J = 8.5 Hz, 1H), 5.98 (d, J = 7.2 Hz, 1H), 5.34 (d, J = 7.1 Hz, 1H), 3.90 (s, 3H), 2.35 (s, 3H). 13C NMR (101 MHz, CDCl3) δ (ppm) 174.4, 166.8, 157.3, 151.6, 138.7, 133.4, 127.4, 123.0, 118.4, 86.7, 85.0, 53.5, 20.4. HRMS (ESI): m/z [M + H]+ calcd for C13H12NO5: 262.0710; found: 262.0710.
- Methyl 7-methoxy-9-oxo-3,3a-dihydro-9H-chromeno[3,2-c]isoxazole-3-carboxylate (4l), 28 mg, 89%, yellow solid, m.p.: 193–194 °C. 1H NMR (500 MHz, CDCl3) δ (ppm) 7.41 (d, J = 3.2 Hz, 1H), 7.20 (dd, J = 9.1, 3.2 Hz, 1H), 7.01 (d, J = 9.0 Hz, 1H), 5.98 (d, J = 7.2 Hz, 1H), 5.34 (d, J = 7.2 Hz, 1H), 3.91 (s, 3H), 3.84 (s, 3H). 13C NMR (126 MHz, CDCl3) δ (ppm) 174.2, 166.8, 155.6, 153.9, 151.7, 126.9, 1235, 120.0, 107.8, 86.8, 85.0, 55.9, 53.6. HRMS (ESI): m/z [M + H]+ calcd for C13H12NO6: 278.0659; found: 278.0657.
- Methyl 6-chloro-9-oxo-3,3a-dihydro-9H-chromeno[3,2-c]isoxazole-3-carboxylate (4m), 29 mg, 89%, yellow solid, m.p.: 121–123 °C. 1H NMR (500 MHz, CDCl3) δ (ppm) 11.97 (d, J = 1.5 Hz, 1H), 8.55 (dd, J = 8.8, 1.5 Hz, 1H), 7.43 (d, J = 1.4 Hz, 1H), 7.08 (t, J = 1.8 Hz, 1H), 6.98 (dd, J = 8.8, 2.0 Hz, 1H), 4.03 (d, J = 1.5 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ (ppm) 187.2, 164.6, 162.1, 161.0, 156.5, 144.4, 134.4, 120.5, 118.5, 117.0, 110.3, 53.3. HRMS (ESI): m/z [M + H]+ calcd for C12H9ClNO5: 282.0164; found: 282.0164.
- Methyl 7-chloro-9-oxo-3,3a-dihydro-9H-chromeno[3,2-c]isoxazole-3-carboxylate (4n), 29 mg, 89%, yellow solid, m.p.: 176–178 °C. 1H NMR (500 MHz, CDCl3) δ (ppm) 7.98 (d, J = 2.7 Hz, 1H), 7.55 (dd, J = 8.9, 2.6 Hz, 1H), 7.05 (d, J = 8.8 Hz, 1H), 6.03 (d, J = 7.0 Hz, 1H), 5.38 (d, J = 6.9 Hz, 1H), 3.91 (s, 3H). 13C NMR (126 MHz, CDCl3) δ (ppm) 173.3, 166.5, 157.6, 150.9, 137.4, 129.4, 127.2, 124.1, 120.4, 86.8, 85.2, 53.7. HRMS (ESI): m/z [M + H]+ calcd for C12H9ClNO5: 282.0164; found: 282.0164.
- Methyl 11-oxo-7a,8-dihydro-11H-benzo[5,6]chromeno[3,2-c]isoxazole-8-carboxylate (4o), 30 mg, 86%, light yellow solid, m.p.: 145–146 °C. 1H NMR (500 MHz, CDCl3) δ (ppm) 7.68 (dd, J = 7.9, 3.1 Hz, 1H), 7.33 (ddd, J = 9.1, 7.4, 3.2 Hz, 1H), 7.09 (dd, J = 9.1, 4.1 Hz, 1H), 6.03 (d, J = 6.9 Hz, 1H), 5.38 (d, J = 6.9 Hz, 1H), 3.92 (s, 3H). 13C NMR (126 MHz, CDCl3) δ (ppm) 173.5, 166.5, 159.3, 155.4, 151.0, 125.3, 124.1, 120.56, 113.2, 86.8, 85.2, 53.7. HRMS (ESI): m/z [M + H]+ calcd for C12H9FNO5: 266.0459; found: 266.0459.
- Methyl 7-bromo-9-oxo-3,3a-dihydro-9H-chromeno[3,2-c]isoxazole-3-carboxylate (4p), 18 mg, 77%, yellow solid, m.p.: 173–174 °C. 1H NMR (500 MHz, CDCl3) δ (ppm) 8.13 (d, J = 2.6 Hz, 1H), 7.68 (dd, J = 8.8, 2.6 Hz, 1H), 6.99 (d, J = 8.8 Hz, 1H), 6.03 (d, J = 6.9 Hz, 1H), 5.38 (d, J = 6.9 Hz, 1H), 3.91 (s, 3H). 13C NMR (126 MHz, CDCl3) δ (ppm) 173.2, 166.4, 158.0, 150.8, 140.2, 130.3, 124.5, 120.6, 116.5, 86.7, 85.2, 53.7. HRMS (ESI): m/z [M + H]+ calcd for C12H9BrNO5: 325.9658; found: 325.9658.
- Methyl 11-oxo-7a,8-dihydro-11H-benzo[5,6]chromeno[3,2-c]isoxazole-8-carboxylate (4q), 20 mg, 67%, yellow solid, m.p.: 183–184 °C. 1H NMR (500 MHz, CDCl3) δ 9.48 (d, J = 8.7 Hz, 1H), 8.05 (d, J = 8.9 Hz, 1H), 7.82 (d, J = 8.1 Hz, 1H), 7.73 (ddd, J = 8.6, 6.9, 1.5 Hz, 1H), 7.54 (t, J = 7.5 Hz, 1H), 7.17 (d, J = 9.2 Hz, 1H), 6.10 (d, J = 6.9 Hz, 1H), 5.43 (d, J = 6.8 Hz, 1H), 3.93 (s, 3H). 13C NMR (126 MHz, Chloroform-d) δ 174.9, 166.8, 161.8, 152.1, 139.5, 131.4, 130.8, 129.9, 128.7, 126.3, 126.2, 118.4, 115.8, 86.42, 9.12, 53.6. HRMS (ESI): m/z [M + H]+ calcd for C16H12NO5: 298.07100; found:298.07100.
- 3,3a,4,5-Tetrahydro-11H-benzo[7,8]oxocino[5,4-c]isoxazol-11-one (4r), 22 mg, 78%, light brown solid, m.p.: 178–179 °C. 1H NMR (400 MHz, CDCl3) δ (ppm) 7.78 (dd, J = 7.8, 1.7 Hz, 1H), 7.55 (ddd, J = 8.1, 7.4, 1.8 Hz, 1H), 7.21 (td, J = 7.5, 1.0 Hz, 1H), 7.11 (dd, J = 8.2, 1.0 Hz, 1H), 5.08 (dd, J = 8.5, 4.7 Hz, 1H), 4.57 (dt, J = 9.4, 2.7 Hz, 1H), 3.83 (ddd, J = 12.0, 9.7, 2.3 Hz, 1H), 3.45 (ddd, J = 14.7, 8.5, 1.2 Hz, 1H), 3.32 (dd, J = 14.5, 0.9 Hz, 1H), 2.03–1.78 (m, 2H). 13C NMR (101 MHz, CDCl3) δ (ppm) 190.4, 161.2, 159.0, 135.5, 132.4, 128.7, 124.2, 121.4, 79.4, 70.3, 41.8, 34.5. HRMS (ESI): m/z [M + H]+ calcd for C12H12NO3: 218.0811; found: 218.0813.
- 4H,10H-Benzo[6,7]oxepino[4,3-c]isoxazol-10-one (6a), 22 mg, 82%, white solid, m.p.: 166–167 °C. 1H NMR (500 MHz, CDCl3) δ (ppm) 8.57 (s, 1H), 8.26 (dd, J = 8.1, 1.8 Hz, 1H), 7.58 (ddd, J = 8.5, 7.2, 1.8 Hz, 1H), 7.31–7.23 (m, 1H), 7.16 (dd, J = 8.1, 1.2 Hz, 1H), 5.15 (s, 2H). 13C NMR (126 MHz, CDCl3) δ (ppm) 180.2, 160.1, 155.6, 136.3, 132.1, 127.7, 124.5, 122.8, 117.5, 63.6. HRMS (ESI): m/z [M + H]+ calcd for C11H8NO3: 202.0498; found: 202.0498.
- Methyl-4H,10H-benzo[6,7]oxepino[4,3-c]isoxazol-10-one (6b), 15 mg, 75%, light yellow solid, m.p.: 193–196 °C. 1H NMR (500 MHz, CDCl3) δ (ppm) 8.56 (s, 1H), 8.02 (d, J = 2.5 Hz, 1H), 7.37 (dd, J = 8.3, 2.4 Hz, 1H), 7.05 (d, J = 8.2 Hz, 1H), 5.10 (s, 2H), 2.37 (s, 3H). 13C NMR (126 MHz, CDCl3) δ (ppm) 180.4, 160.0, 158.0, 155.6, 137.2, 134.2, 131.8, 127.4, 122.6, 117.7, 63.6, 20.5. HRMS (ESI): m/z [M + H]+ calcd for C12H10NO3: 216.0655; found: 216.0655.
- 7-Methyl-4H,10H-benzo[6,7]oxepino[4,3-c]isoxazol-10-one (6c), 10 mg, 68%, white solid, m.p.: 196–197 °C. 1H NMR (500 MHz, CDCl3) δ (ppm) 8.56 (s, 1H), 8.02 (d, J = 2.5 Hz, 1H), 7.37 (dd, J = 8.3, 2.4 Hz, 1H), 7.05 (d, J = 8.2 Hz, 1H), 5.10 (s, 2H), 2.37 (s, 3H). 13C NMR (126 MHz, CDCl3) δ (ppm) 179.7, 160.2, 160.1, 155.5, 148.2, 132.2, 125.6, 125.1, 122.9, 117.4, 63.4, 21.5. HRMS (ESI): m/z [M + H]+ calcd for C12H10NO3: 216.0655; found: 216.0654.
- 8-Methoxy-4H,10H-benzo[6,7]oxepino[4,3-c]isoxazol-10-one (6d), 32 mg, 71%, yellow solid, m.p.: 157–159 °C. 1H NMR (500 MHz, CDCl3) δ (ppm) 8.56 (s, 1H), 7.66 (d, J = 3.2 Hz, 1H), 7.14 (dd, J = 8.9, 3.1 Hz, 1H), 7.09 (d, J = 8.9 Hz, 1H), 5.15–5.04 (m, 2H), 3.86 (s, 3H). 13C NMR (126 MHz, CDCl3) δ (ppm) 180.1, 159.7, 156.1, 155.6 154.3, 128.3, 124.4, 124.1, 117.8, 113.1, 63.8, 55.9. HRMS (ESI): m/z [M + H]+ calcd for C12H10NO4: 232.0604; found: 232.0605.
- 7-Methoxy-4H,10H-benzo[6,7]oxepino[4,3-c]isoxazol-10-one (6e), 20 mg, 74%, white solid, m.p.: 206–207 °C. 1H NMR (500 MHz, CDCl3) δ (ppm) 8.56 (s, 1H), 8.27 (d, J = 9.0 Hz, 1H), 6.80 (dd, J = 9.1, 2.5 Hz, 1H), 6.59 (d, J = 2.5 Hz, 1H), 5.12 (d, J = 0.7 Hz, 2H), 3.88 (s, 3H). 13C NMR (126 MHz, CDCl3) δ (ppm) 178.5, 166.2, 162.3, 160.4, 155.5, 134.3, 120.7, 117.0, 111.8, 106.1, 63.3, 55.8. HRMS (ESI): m/z [M + H]+ calcd for C12H10NO4: 232.0604; found: 232.0604.
- Fluoro-4H,10H-benzo[6,7]oxepino[4,3-c]isoxazol-10-one (6f), 15 mg, 62%, white solid, m.p.: 187–189 °C. 1H NMR (500 MHz, CDCl3) δ (ppm) 8.59 (s, 1H), 7.91 (dd, J = 9.2, 3.3 Hz, 1H), 7.31–7.26 (m, 1H), 7.16 (dd, J = 8.9, 4.6 Hz, 1H), 5.13 (s, 2H). 13C NMR (126 MHz, CDCl3) δ (ppm) 179.1, 159.4, 159.4 (d, Jc–f = 245.3 Hz), 156.2 (d, Jc–f = 2.6 Hz), 155.8, 128.9 (d, Jc–f = 7.3 Hz), 124.7 (d, Jc–f = 7.4 Hz), 123.5 (d, Jc–f = 23.3 Hz), 117.5 (d, Jc–f = 24.9 Hz), 63.8. HRMS (ESI): m/z [M + H]+ calcd for C11H7FNO3: 220.0405; found: 220.0404.
- 8-Chloro-4H,10H-benzo[6,7]oxepino[4,3-c]isoxazol-10-one (6g), 20 mg, 70%, white solid, m.p.: 202–203 °C. 1H NMR (500 MHz, CDCl3) δ (ppm) 8.59 (s, 1H), 8.22 (d, J = 2.8 Hz, 1H), 7.51 (dd, J = 8.7, 2.8 Hz, 1H), 7.13 (d, J = 8.7 Hz, 1H), 5.14 (s, 2H). 13C NMR (126 MHz, CDCl3) δ (ppm) 178.9, 159.6, 158.6, 155.9, 136.1, 131.4, 130.2, 128.5, 124.6, 117.2, 63.7. HRMS (ESI): m/z [M + H]+ calcd for C11H7ClNO3: 236.0109; found: 236.0109.
- 8-Bromo-4H,10H-benzo[6,7]oxepino[4,3-c]isoxazol-10-one (6h), 31 mg, 83%, white solid, m.p.: 193–194 °C. 1H NMR (500 MHz, CDCl3) δ (ppm) 8.59 (s, 1H), 8.36 (d, J = 2.6 Hz, 1H), 7.65 (dd, J = 8.7, 2.6 Hz, 1H), 7.06 (d, J = 8.6 Hz, 1H), 5.14 (s, 2H). 13C NMR (126 MHz, CDCl3) δ (ppm) 178.8, 159.6, 159.1, 155.9, 139.9, 134.4, 128.8, 124.8, 117.5, 117.2, 63.6. HRMS (ESI): m/z [M + H]+ calcd for C11H7BrNO3: 279.9603; found: 279.9602.
- 8H,12H-Naphtho[1′,2′:6,7]oxepino[4,3-c]isoxazol-12-one (6i), 27 mg, 85%, light brown solid, m.p.: 156–158 °C. 1H NMR (500 MHz, CDCl3) δ (ppm) 8.53–8.42 (m, 2H), 7.98 (d, J = 8.8 Hz, 1H), 7.82 (dd, J = 8.1, 1.4 Hz, 1H), 7.59 (ddd, J = 8.7, 6.9, 1.5 Hz, 1H), 7.49 (ddd, J = 8.1, 6.9, 1.2 Hz, 1H), 7.28 (d, J = 8.8 Hz, 1H), 5.18 (d, J = 1.2 Hz, 2H). 13C NMR (126 MHz, CDCl3) δ (ppm) 184.2, 159.9, 158.1, 154.8, 135.9, 131.6, 131.5, 128.8, 128.3, 126.5, 126.1, 125.5, 121.6, 117.8, 65.2. HRMS (ESI): m/z [M + H]+ calcd for C15H10NO3: 252.0655; found: 252.0653.
- 1-(2-(But-3-en-1-yloxy)phenyl)ethan-1-one (3r), 24 mg, 67%, light yellow oil. 1H NMR (400 MHz, CDCl3) δ (ppm) 7.73 (dd, J = 7.7, 1.9 Hz, 1H), 7.43 (ddd, J = 8.3, 7.3, 1.9 Hz, 1H), 7.05–6.84 (m, 2H), 5.90 (ddt, J = 16.9, 10.2, 6.6 Hz, 1H), 5.30–5.02 (m, 2H), 4.12 (t, J = 6.4 Hz, 2H), 2.63–2.57 (m, 5H). 13C NMR (101 MHz, CDCl3) δ (ppm) 199.8, 158.2, 134.3, 133.6, 130.4, 128.2, 120.5, 117.4, 112.1, 67.6, 33.6, 32.1. HRMS (ESI): m/z [M + H]+ calcd for C12H15NO2: 191.1066; found: 191.1066.
- Ethyl 3-benzoyl-4,5-dihydroisoxazole-5-carboxylate (10), 27 mg, 58%, light yellow oil, m.p.: 156–158 °C. 1H NMR (400 MHz, CDCl3) δ (ppm) 8.25–8.16 (m, 2H), 7.60 (td, J = 7.3, 1.5 Hz, 1H), 7.47 (td, J = 7.9, 1.7 Hz, 2H), 5.22–5.14 (m, 1H), 4.32–4.22 (m, 2H), 3.73–3.56 (m, 2H), 1.32 (td, J = 7.1, 1.6 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ (ppm) 185.4, 169.1, 156.8, 135.4, 133.8, 130.3, 128.4, 78.9, 62.2, 38.4, 14.0. HRMS (ESI): m/z [M + H]+ calcd for C13H14NO4: 248.0917; found: 248.0918.
- (E)-2-Oxo-2-phenylacetaldehyde oxime (11), 24 mg, 67%, light yellow solid, m.p.: 150–151 °C. 1H NMR (400 MHz, CDCl3) δ (ppm) 8.68 (s, 1H), 8.07–8.04 (m, 2H), 8.03 (d, J = 1.4 Hz, 1H), 7.64–7.58 (m, 1H), 7.48 (dd, J = 8.4, 7.2 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ (ppm) 188.6, 148.7, 135.8, 133.6, 129.9, 128.5. HRMS (ESI): m/z [M + H]+ calcd for C8H8NO2: 150.0550; found: 150.0550.
4. Conclusions
In summary, an effective and metal-free method for the synthesis of 6/7/8-membered ketone-fused isoxazoles/isoxazolines tetra- or tricyclic compounds was developed while employing TBN as a radical initiator and NO source. In this protocol, TBN activated the Csp3–H bond of aryl methyl ketones to produce α-carbonyl nitrile oxide intermediates in situ through cascade Hydrogen Atom Transfer (HAT) and the radical coupling process, which then underwent [3 + 2] cycloaddition with alkenyl/alkynyl groups. The present approach overcomes the entropic effects and ring strain associated with the conventional synthesis of densely fused polycyclic compounds. The protocol has a wide substrate scope and diverse possible products, with the additional merits of being metal-catalyst-free and additive-free.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/molecules29061202/s1. X-ray crystal structure and crystallographic data; optimization of reaction conditions; and characterization data for product 2a–2j, 4a–4r, and 6a–6i, including 1H-NMR and 13C-NMR.
Author Contributions
Conceptualization, Y.-P.Z.; methodology, Y.-P.Z.; investigation, J.-K.C. and T.-Z.C.; data curation, Q.-W.Y., Y.M., C.-M.Y., H.-X.Z., Y.-C.L., Q.-K.D. and Y.-Y.S.; writing—original draft preparation, J.-K.C. and Y.-Y.S.; writing—review and editing, Y.-P.Z.; visualization, J.-K.C.; supervision, Y.-P.Z.; project administration, Y.-P.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by Yantai’s “Double Hundred Plan”, the Talent Induction Program for Youth Innovation Teams in Colleges and Universities of Shandong Province, and the Foundation of Anhui Laboratory of Molecule-Based Materials (fzj22022). The Graduate Innovation Foundation of Yantai University (KGIFYTU2312) is gratefully acknowledged (for J.-K. Cao).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in article and Supplementary Materials.
Acknowledgments
The authors thank the Talent Induction Program for Youth Innovation Teams in Colleges and Universities of Shandong Province.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Morita, T.; Fukuhara, S.; Fuse, S.; Nakamura, H. Gold(I)-Catalyzed Intramolecular SEAr Reaction: Efficienct Synthesis of Isoxazole-Containing Fused Heterocycles. Org. Lett. 2018, 20, 433–436. [Google Scholar] [CrossRef]
- Ibrahim, K.T.; Neetha, M.; Anilkumar, G. Advancements in the synthesis of oxazolines. Monatsh. Chem. 2022, 153, 837–871. [Google Scholar] [CrossRef]
- Jiang, B.; Dai, M. Concise Total Syntheses of the 6–7–5 Hamigeran Natural Products. J. Am. Chem. Soc. 2023, 145, 18731–18736. [Google Scholar] [CrossRef]
- Jiang, B.; Dai, M. Synthetic Studies toward the Hamigerans with a 6–7–5 Tricyclic Core. Org. Lett. 2020, 22, 4176–4179. [Google Scholar] [CrossRef]
- Guo, Y.; Xiang, Y.; Wei, L.; Wan, J.-P. Thermoinduced Free-Radical C–H Acyloxylation of Tertiary Enaminones: Catalyst-Free Synthesis of Acyloxyl Chromones and Enaminones. Org. Lett. 2018, 20, 3971–3974. [Google Scholar] [CrossRef]
- Lin, Y.; Wan, J.-P.; Liu, Y. Cascade in Situ Iodination, Chromone Annulation, and Cyanation for Site-Selective Synthesis of 2-Cyanochromones. J. Org. Chem. 2023, 88, 4017–4023. [Google Scholar] [CrossRef]
- Luo, T.; Wan, J.-P.; Liu, Y. Toward C2-nitrogenated chromones by copper-catalyzed β-C(sp2)–H N-heteroarylation of enaminones. Org. Chem. Front. 2020, 7, 1107–1112. [Google Scholar] [CrossRef]
- Yu, Q.; Liu, Y.; Wan, J.-P. Transition metal-free synthesis of 3-trifluoromethyl chromones via tandem C–H trifluoromethylation and chromone annulation of enaminones. Org. Chem. Front. 2020, 7, 2770–2775. [Google Scholar] [CrossRef]
- Haji, M.; Hosseinzadeh, M. Cyclohepta[b]pyran: An important scaffold in biologically active natural products. Med. Chem. Res. 2022, 31, 2059–2073. [Google Scholar] [CrossRef]
- Liu, Y.-W.; Wang, M.-M.; Zhang, Y.-Q.; Xu, H.; Dai, H.-X. Construction of Indole-Fused Seven- and Eight-Membered Azaheterocycles via a Tandem Pd/NBE-Catalyzed Decarbonylation and Dual C–H Activation Sequence. Org. Lett. 2023, 25, 5406–5410. [Google Scholar] [CrossRef] [PubMed]
- Osipyan, A.; Sapegin, A.; Novikov, A.S.; Krasavin, M. Rare Medium-Sized Rings Prepared via Hydrolytic Imidazoline Ring Expansion (HIRE). J. Org. Chem. 2018, 83, 9707–9717. [Google Scholar] [CrossRef]
- Gao, M.; Gan, Y.; Xu, B. From Alkenes to Isoxazolines via Copper-Mediated Alkene Cleavage and Dipolar Cycloaddition. Org. Lett. 2019, 21, 7435–7439. [Google Scholar] [CrossRef]
- Meng, L.; Liu, H.; Lin, Z.; Wang, J. Synthetic and Computational Study of the Enantioselective [3 + 2]-Cycloaddition of Chromones with MBH Carbonates. Org. Lett. 2022, 24, 5890–5895. [Google Scholar] [CrossRef]
- Krstić, G.; Saidu, M.B.; Bombicz, P.; De, S.; Ali, H.; Zupkó, I.; Berkecz, R.; Gallah, U.S.; Rédei, D.; Hohmann, J. Pauciflorins A–E, Unexpected Chromone–Monoterpene-Derived Meroterpenoids from Centrapalus pauciflorus. J. Nat. Prod. 2023, 86, 891–896. [Google Scholar] [CrossRef] [PubMed]
- Rao, B.; Tang, J.; Wei, Y.; Zeng, X. Ring Expansion via Cleavage of Benzylic C−C Bonds Enabling Direct Synthesis of Medium Ring-Fused Benzocarbocycles. Chem. Asian J. 2016, 11, 991–995. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Qi, T.; He, S.; Huang, W.; Peng, C.; Zhan, G.; Han, B. Catalyst-Controlled Switchable (5 + 4)/(3 + 4) Cycloadditions for the Divergent Synthesis of Pyrazole-Fused Seven- and Nine-Membered Heterocycles. ACS Catal. 2023, 13, 10694–10704. [Google Scholar] [CrossRef]
- Reyes, R.L.; Iwai, T.; Sawamura, M. Construction of Medium-Sized Rings by Gold Catalysis. Chem. Rev. 2021, 121, 8926–8947. [Google Scholar] [CrossRef] [PubMed]
- Pan, G.; Lu, L.; Zhuang, W.; Huang, Q. Synthesis of Indole-Fused Six-, Seven-, or Eight-Membered N,O-Heterocycles via Rhodium-Catalyzed NH-Indole-Directed C–H Acetoxylation/Hydrolysis/Annulation. J. Org. Chem. 2021, 86, 16753–16763. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.-Z.; Guan, Y.-L.; Huang, Q.-W.; Qi, T.; Xiang, P.; Zhang, X.; Leng, H.-J.; Li, J.-L. Temperature-Controlled Divergent Asymmetric Synthesis of Indole-Based Medium-Sized Heterocycles through Palladium Catalysis. ACS Catal. 2023, 13, 1164–1172. [Google Scholar] [CrossRef]
- Li, P.; Jia, X. tert-Butyl Nitrite (TBN) as a Versatile Reagent in Organic Synthesis. Synthesis 2018, 50, 711–722. [Google Scholar] [CrossRef]
- Khaligh, G.N. Recent Advances and Applications of tert-Butyl Nitrite (TBN) in Organic Synthesis. Mini-Rev. Org. Chem. 2020, 17, 3–25. [Google Scholar] [CrossRef]
- Dahiya, A.; Sahoo, A.K.; Alam, T.; Patel, B.K. tert-Butyl Nitrite (TBN), a Multitasking Reagent in Organic Synthesis. Chem. Asian J. 2019, 14, 4454–4492. [Google Scholar] [CrossRef]
- Guo, X.; Xu, G.; Zhou, L.; Yan, H.; Hao, X.-Q.; Wang, Q. Synthesis and application of α-carbonyl nitrile oxides. Org. Chem. Front. 2020, 7, 2467–2473. [Google Scholar] [CrossRef]
- Pan, J.; Li, X.; Lin, F.; Liu, J.; Jiao, N. Chemoselective Nitrosylation of Anilines and Alkynes via Fragmentary or Complete NO Incorporation. Chem 2018, 4, 1427–1442. [Google Scholar] [CrossRef]
- Wang, X.-D.; Zhu, L.-H.; Liu, P.; Wang, X.-Y.; Yuan, H.-Y.; Zhao, Y.-L. Copper-Catalyzed Cascade Cyclization Reactions of Diazo Compounds with tert-Butyl Nitrite and Alkynes: One-Pot Synthesis of Isoxazoles. J. Org. Chem. 2019, 84, 16214–16221. [Google Scholar] [CrossRef] [PubMed]
- Xiong, M.; Liang, X.; Gao, Z.; Lei, A.; Pan, Y. Synthesis of Isoxazolines and Oxazines by Electrochemical Intermolecular [2 + 1 + n] Annulation: Diazo Compounds Act as Radical Acceptors. Org. Lett. 2019, 21, 9300–9305. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Zhou, Y.; Song, Q. Synthesis of Furoxans and Isoxazoles via Divergent [2 + 1 + 1 + 1] Annulations of Sulfoxonium Ylides and tBuONO. Org. Lett. 2019, 21, 5273–5276. [Google Scholar] [CrossRef] [PubMed]
- Dai, P.; Tan, X.; Luo, Q.; Yu, X.; Zhang, S.; Liu, F.; Zhang, W.-H. Synthesis of 3-Acyl-isoxazoles and Δ2-Isoxazolines from Methyl Ketones, Alkynes or Alkenes, and tert-Butyl Nitrite via a Csp3–H Radical Functionalization/Cycloaddition Cascade. Org. Lett. 2019, 21, 5096–5100. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Jin, F.; Cheng, X.; Tao, S.; Jiang, G.; Li, X.; Yang, J.; Bao, X.; Wan, X. [2 + 2 + 1] Cycloaddition of N-tosylhydrazones, tert-butyl nitrite and alkenes: A general and practical access to isoxazolines. Chem. Sci. 2021, 12, 9823–9830. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zhao, Y.; Fang, S.; Long, W.; Sun, H.; Wan, X. Coupling Reaction of Cu-Based Carbene and Nitroso Radical: A Tandem Reaction to Construct Isoxazolines. Org. Lett. 2017, 19, 5896–5899. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Ogunlana, A.A.; Fang, S.; Long, W.; Sun, H.; Bao, X.; Wan, X. In situ generation of nitrile oxides from copper carbene and tert-butyl nitrite: Synthesis of fully substituted isoxazoles. Org. Biomol. Chem. 2018, 16, 4683–4687. [Google Scholar] [CrossRef]
- Song, W.; Liu, Y.; Yan, N.; Wan, J.-P. Tunable Key [3 + 2] and [2 + 1] Cycloaddition of Enaminones and α-Diazo Compounds for the Synthesis of Isomeric Isoxazoles: Metal-Controlled Selectivity. Org. Lett. 2023, 25, 2139–2144. [Google Scholar] [CrossRef]
- Ma, L.; Kou, L.; Jin, F.; Cheng, X.; Tao, S.; Jiang, G.; Bao, X.; Wan, X. Acyclic nitronate olefin cycloaddition (ANOC): Regio- and stereospecific synthesis of isoxazolines. Chem. Sci. 2021, 12, 774–779. [Google Scholar] [CrossRef]
- Sudoh, Y.; Jin, Z.-T.; Imafuku, K.; Matsumura, H. Reactions of 3-acetyltropolone and its methyl ethers with hydroxylamine. Formation of 8H-cyclohept[d]isoxazol-8-one and 8H-cyclohept[c]isoxazol-8-one. J. Heterocycl. Chem. 1982, 19, 525–528. [Google Scholar] [CrossRef]
- Wang, Z.; Zhao, Y.; Chen, J.; Chen, M.; Li, X.; Jiang, T.; Liu, F.; Yang, X.; Sun, Y.; Zhu, Y. One-Pot Synthesis of Isoxazole-Fused Tricyclic Quinazoline Alkaloid Derivatives via Intramolecular Cycloaddition of Propargyl-Substituted Methyl Azaarenes under Metal-Free Conditions. Molecules 2023, 28, 2787. [Google Scholar] [CrossRef]
- Zhao, P.; Wu, X.; Zhou, Y.; Geng, X.; Wang, C.; Wu, Y.-d.; Wu, A.-X. Direct Synthesis of 2,3-Diaroyl Quinolines and Pyridazino[4,5-b]quinolines via an I2-Promoted One-Pot Multicomponent Reaction. Org. Lett. 2019, 21, 2708–2711. [Google Scholar] [CrossRef]
- Huang, H.-M.; Bellotti, P.; Daniliuc, C.G.; Glorius, F. Radical Carbonyl Propargylation by Dual Catalysis. Angew. Chem. Int. Ed. 2021, 60, 2464–2471. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, A.R.; Manna, M.S.; Mukherjee, S. Nitro-enabled catalytic enantioselective formal umpolung alkenylation of β-ketoesters. Chem. Sci. 2017, 8, 6686–6690. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-J.; Wang, Z.; Zhang, H.; Gao, J.-J.; Yang, K.-R.; Fan, W.-Y.; Wu, R.-X.; Feng, M.-L.; Zhu, W.; Zhu, Y.-P. Iodine-Mediated Domino Cyclization for One-Pot Synthesis of Indolizine-Fused Chromones via Metal-Free sp3 C–H Functionalization. J. Org. Chem. 2022, 87, 835–845. [Google Scholar] [CrossRef] [PubMed]
- Shang, Z.-H.; Zhang, X.-J.; Li, Y.-M.; Wu, R.-X.; Zhang, H.-R.; Qin, L.-Y.; Ni, X.; Yan, Y.; Wu, A.-X.; Zhu, Y.-P. One-Pot Synthesis of Chromone-Fused Pyrrolo[2,1-a]isoquinolines and Indolizino[8,7-b]indoles: Iodine-Promoted Oxidative [2 + 2 + 1] Annulation of O-Acetylphenoxyacrylates with Tetrahydroisoquinolines and Noreleagnines. J. Org. Chem. 2021, 86, 15733–15742. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Dang, H.; Rose, J.A.; Rablen, P.; Herzon, S.B. Hydroheteroarylation of Unactivated Alkenes Using N-Methoxyheteroarenium Salts. J. Am. Chem. Soc. 2017, 139, 5998–6007. [Google Scholar] [CrossRef]
- Sutariya, T.R.; Labana, B.M.; Parmar, B.D.; Parmar, N.J.; Kant, R.; Gupta, V.K. A domino synthetic approach for new, angular pyrazol- and isoxazol-heterocycles using [DBU][Ac] as an effective reaction medium. RSC Adv. 2015, 5, 23519–23529. [Google Scholar] [CrossRef]
- Shaikh, M.H.; Subhedar, D.D.; Khedkar, V.M.; Jha, P.C.; Khan, F.A.K.; Sangshetti, J.N.; Shingate, B.B. 1,2,3-Triazole tethered acetophenones: Synthesis, bioevaluation and molecular docking study. Chin. Chem. Lett. 2016, 27, 1058–1063. [Google Scholar] [CrossRef]
- Purushothaman, S.; Prasanna, R.; Raghunathan, R. Regioselective synthesis of spiropyrrolidine/spiropyrrolizidine/spirothiazolidine-grafted macrocycles through 1,3-dipolar cycloaddition methodology. Tetrahedron 2013, 69, 9742–9750. [Google Scholar] [CrossRef]
- Dimirjian, C.A.; Castiñeira Reis, M.; Balmond, E.I.; Turman, N.C.; Rodriguez, E.P.; Di Maso, M.J.; Fettinger, J.C.; Tantillo, D.J.; Shaw, J.T. Synthesis of Spirobicyclic Pyrazoles by Intramolecular Dipolar Cycloadditions/[1s, 5s] Sigmatropic Rearrangements. Org. Lett. 2019, 21, 7209–7212. [Google Scholar] [CrossRef] [PubMed]
- James, M.J.; Schwarz, J.L.; Strieth-Kalthoff, F.; Wibbeling, B.; Glorius, F. Dearomative Cascade Photocatalysis: Divergent Synthesis through Catalyst Selective Energy Transfer. J. Am. Chem. Soc. 2018, 140, 8624–8628. [Google Scholar] [CrossRef] [PubMed]
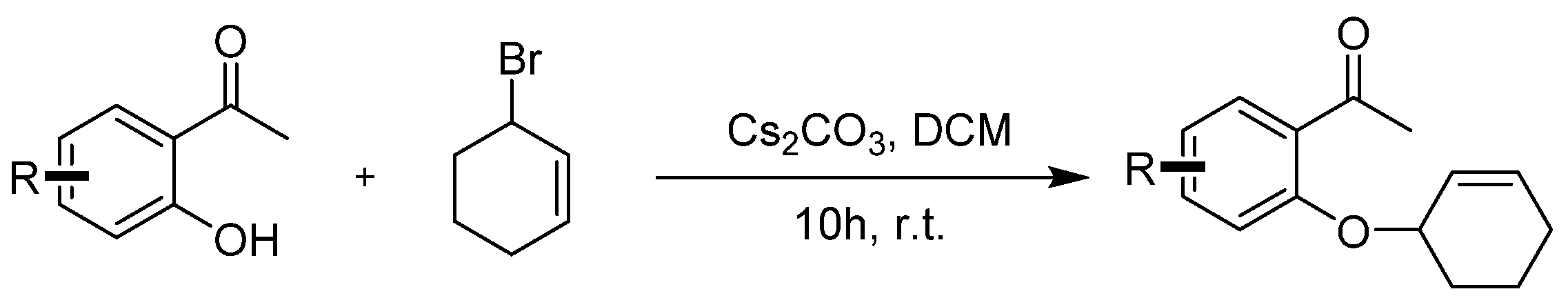
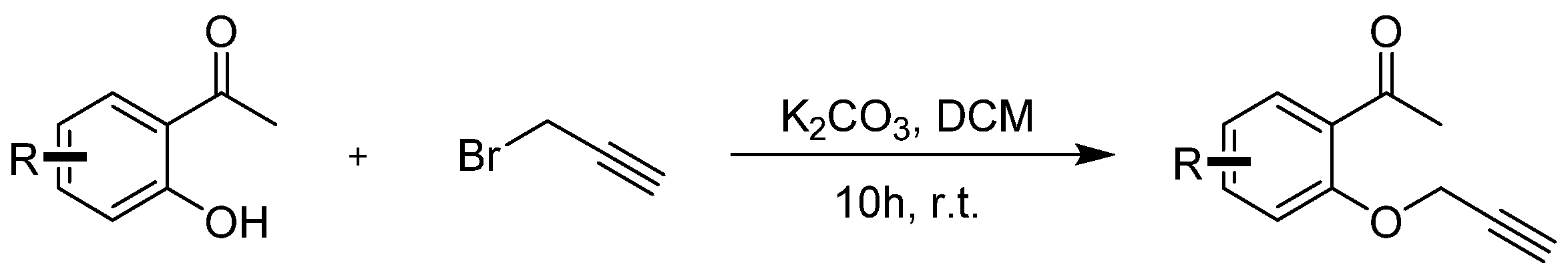
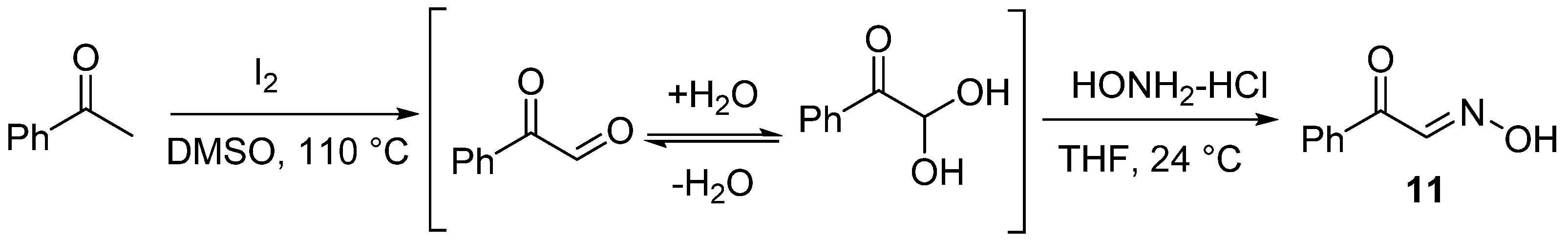
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).