

Enzymes in “Green” Synthetic Chemistry: Laccase and Lipase


Abstract
1. Introduction
1.1. Enzymes: History and Distinctiveness
1.1.1. Enzyme Catalytic Mechanism
1.1.2. Enzyme Kinetics
1.1.3. Influence of Additives on Enzyme Catalysis
1.1.4. Current Enzyme Usage in Synthetic Chemistry
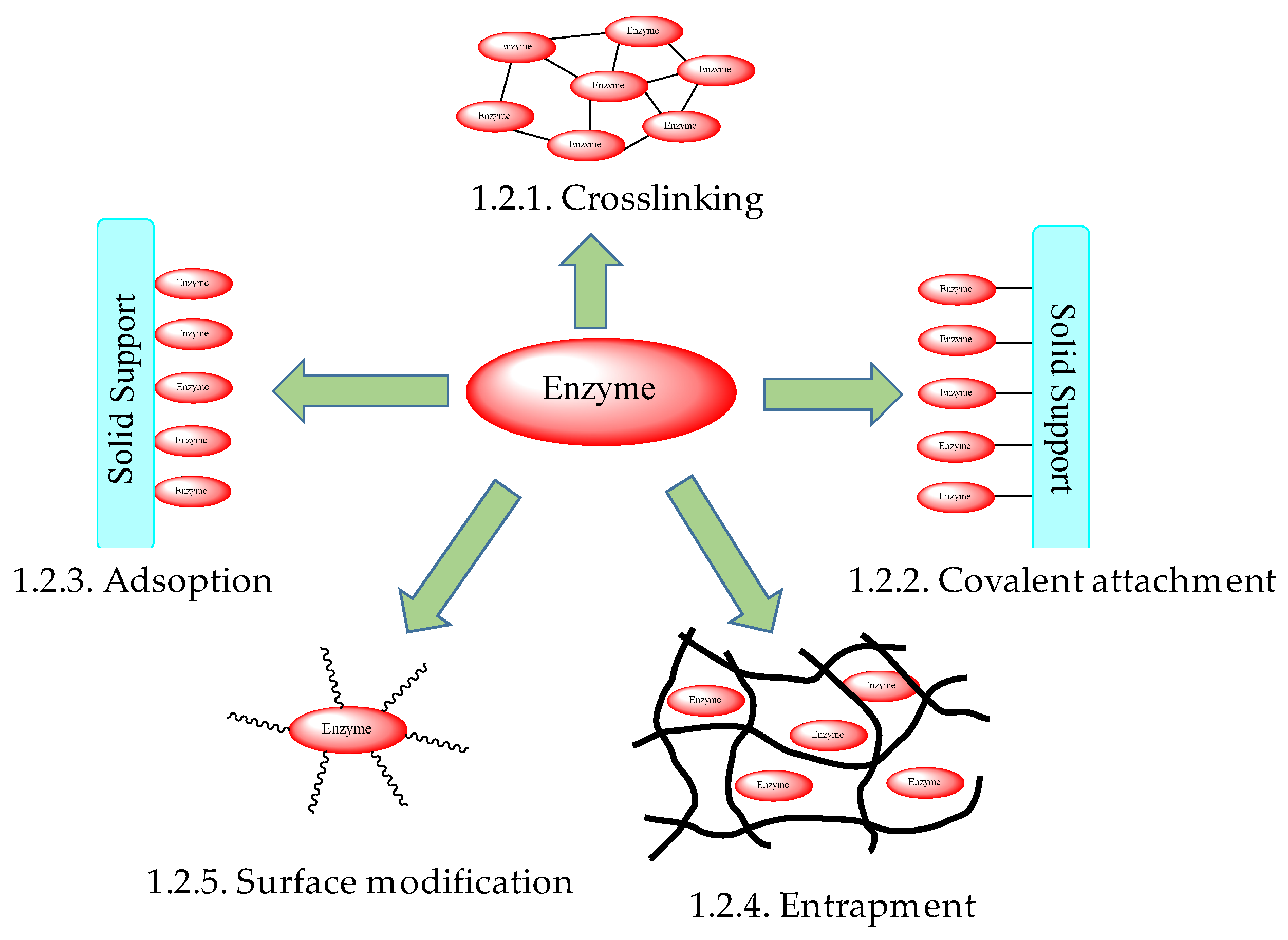
1.2. Enzyme Immobilizations
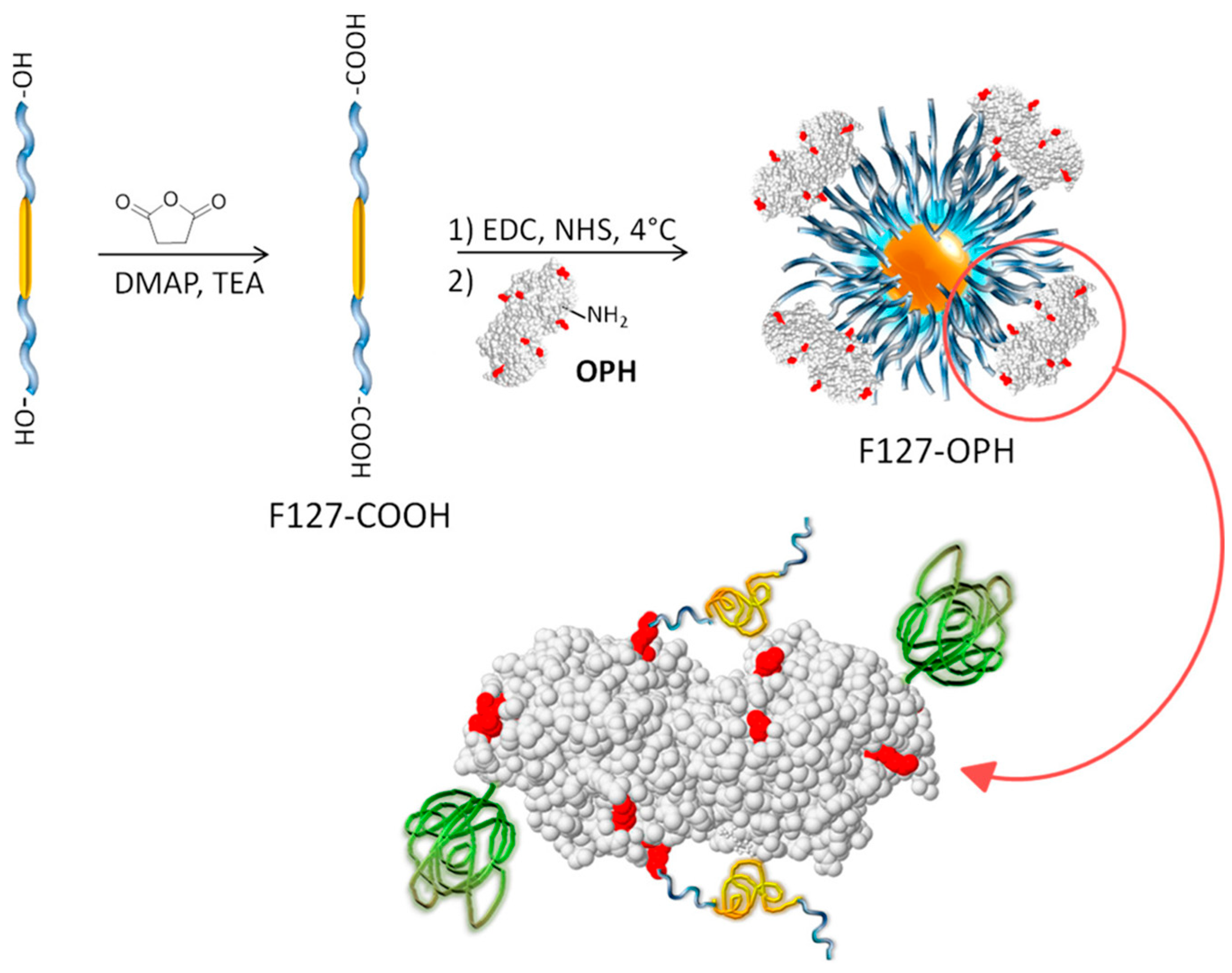
1.2.1. Crosslinking of Enzymes
1.2.2. Covalent Attachment of Enzymes to Solid Scaffolds
1.2.3. Adsorption of Enzymes onto Solid Supports
1.2.4. Enzyme Entrapment in Polymeric Supports
1.2.5. Enzyme Stabilization in Micelles and by Surface Polymeric Modification
1.2.6. Immobilized Enzymes as Polymerization Catalysts
2. Laccase
2.1. Laccase Catalysts in Nature

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
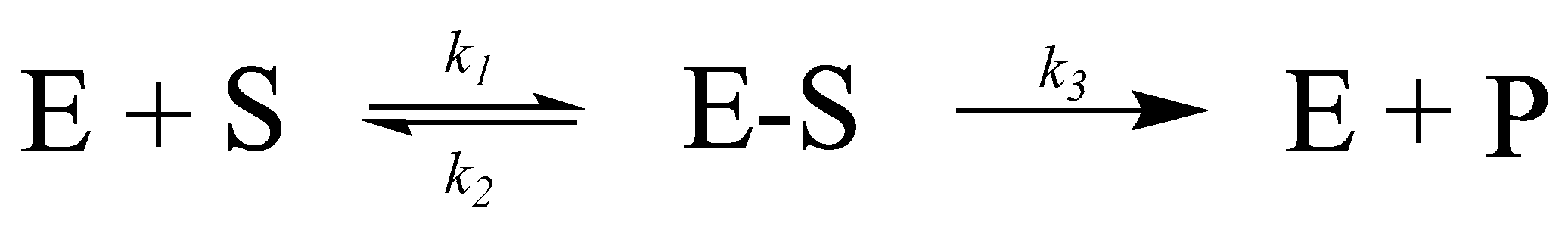{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Source | Molecular Mass (kDa) | Natural Function | Reference | |
|---|---|---|---|---|
| Fungal | Scytalidium thermophilum | 82 | Lignin degradation Pigment formation | [109] |
| Rhizoctonia solani | 70–85 | Lignin degradation Pigment formation | [109] | |
| Trametes versicolor | 58 | Lignin degradation Pigment formation | [110] | |
| Bacterial | Strepotomyces antibioticus | 67.5 | Phenoxazinone synthesis | [111] |
| Bacillus subtilis | 65 | Spores pigmentation UV and H2O2 resistance | [112] | |
| Pseudomonas syringae | 72 | Copper resistance | [113] | |
| Plant | Acer pseudoplatanus | 59.9 | Lignin Synthesis | [114] |
| Malus domestica | 74 | Lignin Synthesis | [115] | |
| Rhus vernicifera | 110 | Lignin Synthesis | [116] | |
| Insect | Manducta sexta | 220–280 | Cuticle Scleritization | [117] |
| Sarcophaga bullata | 90–100 | Cuticle Scleritization | [118] | |
| Periplaneta americana | 185 | Cuticle Scleritization | [119] | |
2.2. Laccase Structure and Catalytic Pathway
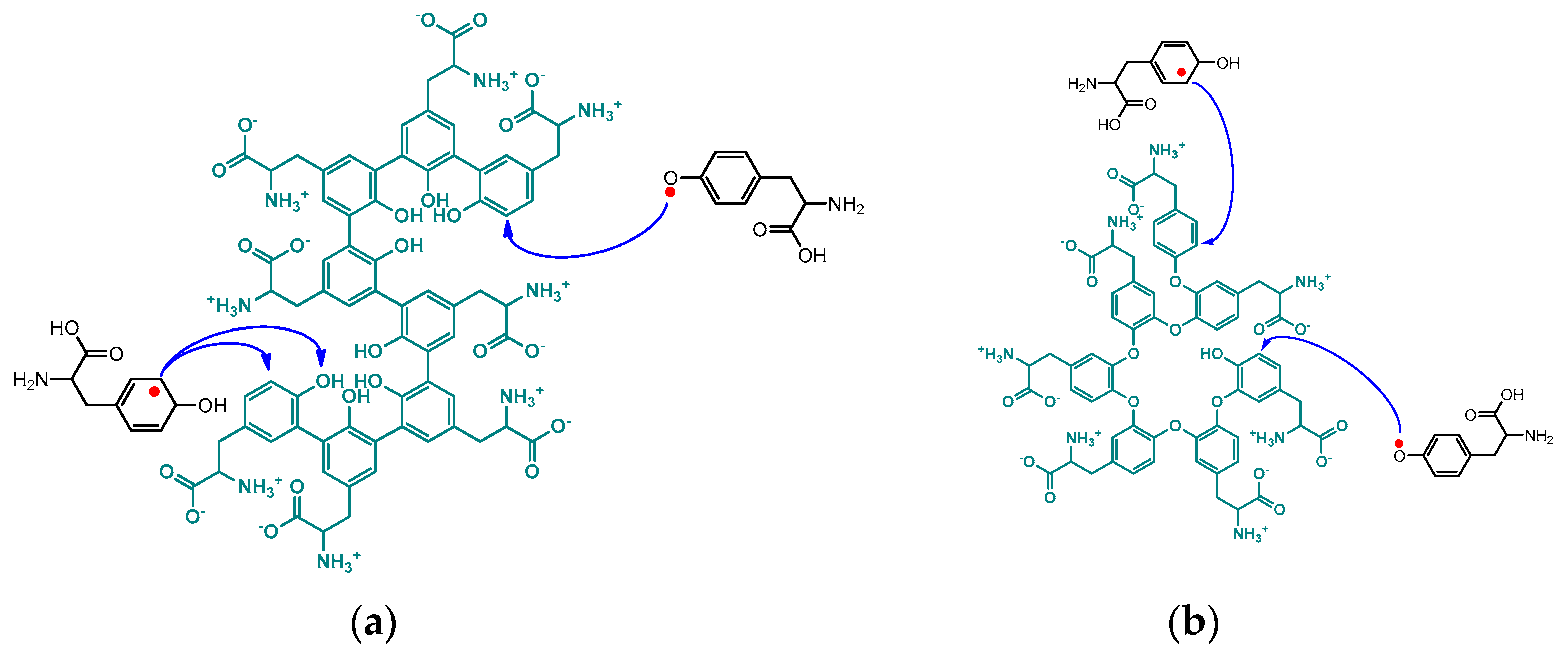
2.2.1. Radical Mediators in Laccase Catalysis
2.3. Laccase Catalysis: Commercial Applications
2.4. Laccase Use in Bioremediation Applications
2.5. Laccase-Catalyzed Polymerizations
3. Lipase
3.1. Lipase Catalysts in Nature
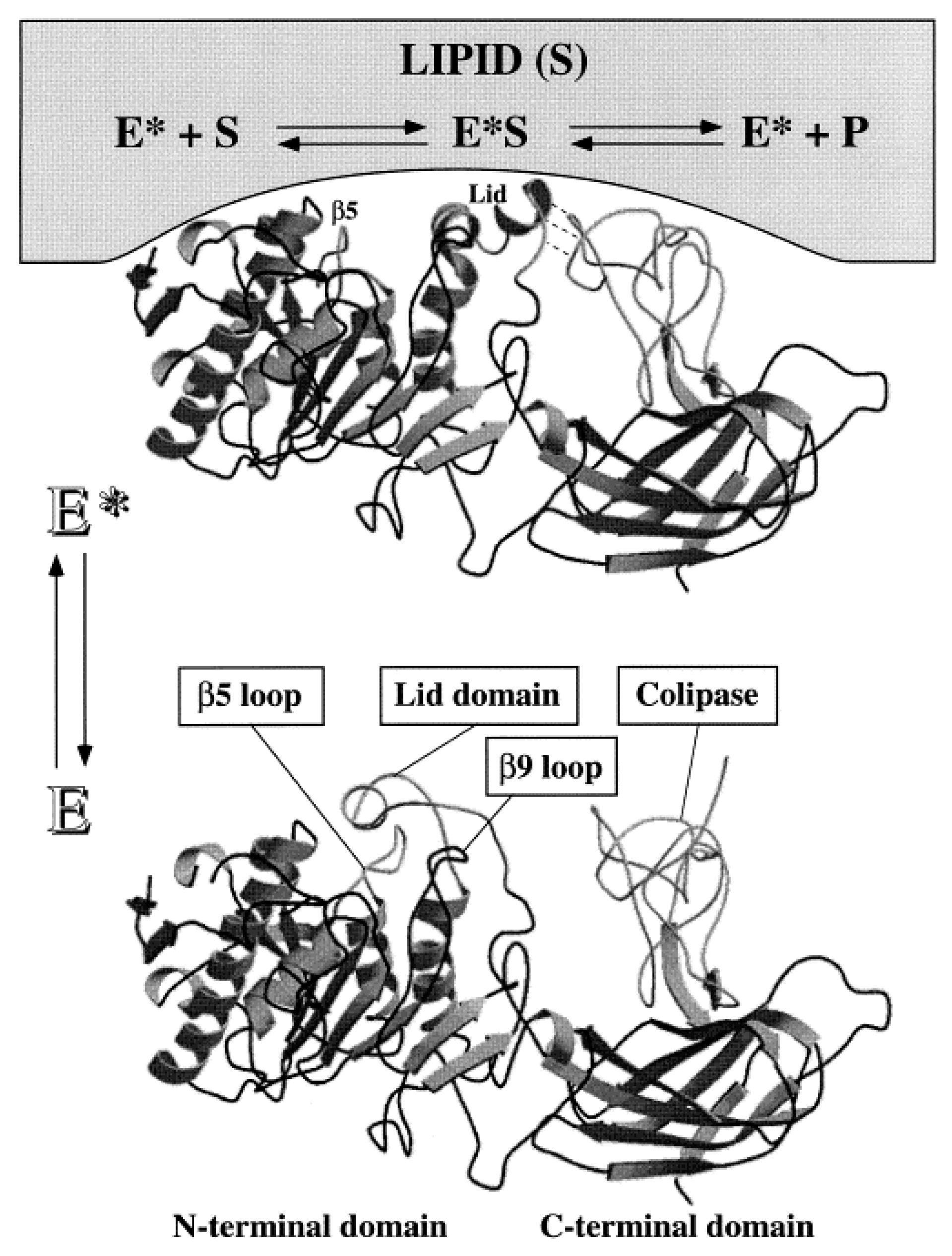
3.2. Lipase Structure and Catalytic Pathway
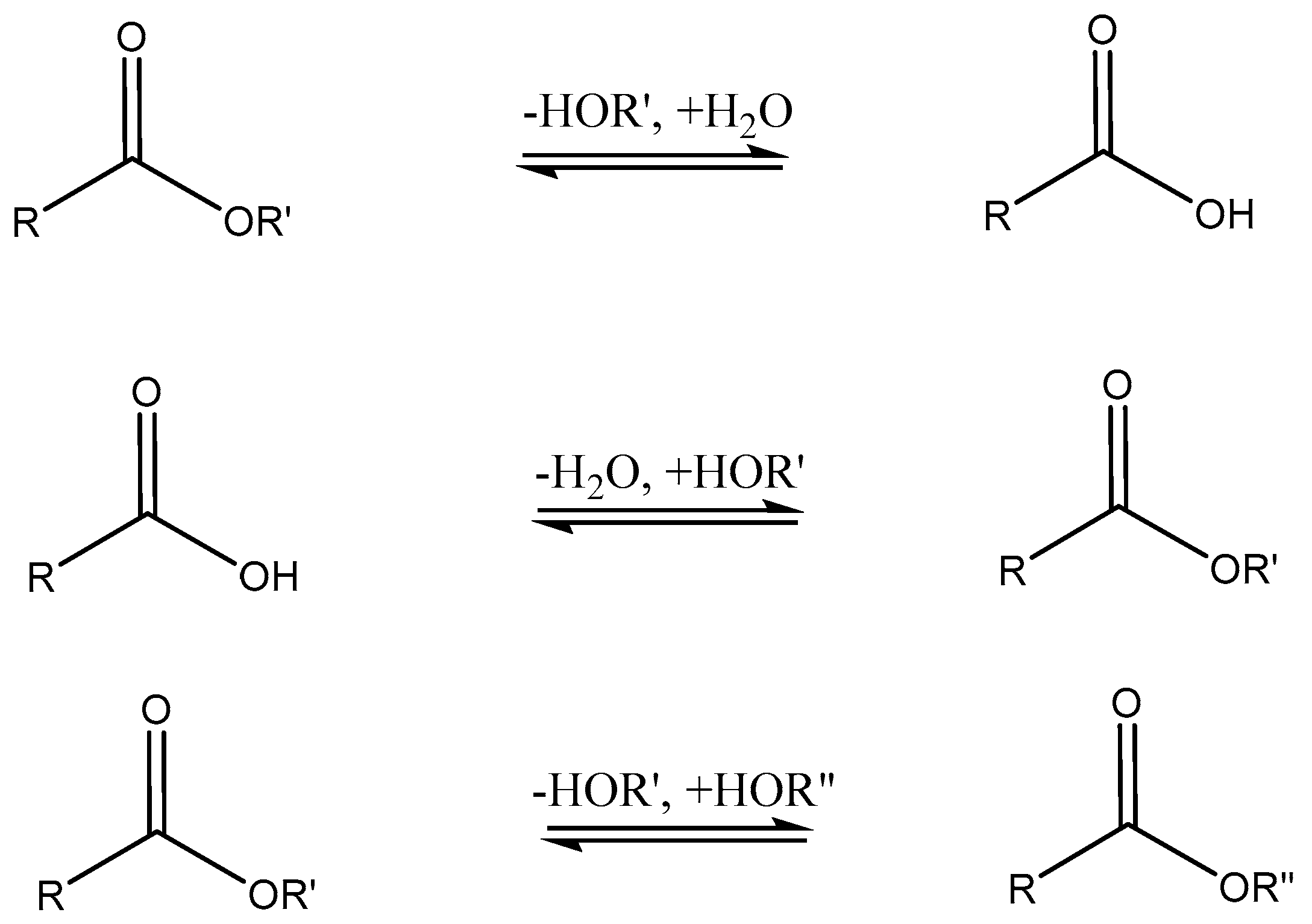
3.3. Lipase Applications in Organic Chemistry
3.4. Lipase-Catalyzed Polymerizations
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Buller, R.; Lutz, S.; Kazlauskas, R.J.; Snajdrova, R.; Moore, J.C.; Bornscheuer, U.T. From nature to industry: Harnessing enzymes for biocatalysis. Science 2023, 382, eadh8615. [Google Scholar] [CrossRef] [PubMed]
- Anwar, A.; Imran, M.; Iqbal, H.M.N. Smart chemistry and applied perceptions of enzyme-coupled nano-engineered assemblies to meet future biocatalytic challenges. Coord. Chem. Rev. 2023, 493, 215329. [Google Scholar] [CrossRef]
- Payen, P.A.; Persoz, J.F. Memoir on diastase, the principal products of its reactions, and their applications to the industrial arts. Ann. Chim. Phys. 1833, 53, 73–92. [Google Scholar]
- Kühne, W. Über das Verhalten verschiedener organisirter und sog. Ungeformter Fermente. Verh. Heidelb. Naturhist.-Med. Ver. Neue Folge 1877, 1, 190–193. [Google Scholar]
- Sumner, J.B. The Isolation and Crystallization of the Enzyme Urease. J. Biol. Chem. 1926, 69, 435–441. [Google Scholar] [CrossRef]
- Liu, J.; Ren, H.; Tang, T.; Wang, J.; Fang, J.; Huang, C.; Zheng, Z.; Qin, B. The Biocatalysis in Cancer Therapy. ACS Catal. 2023, 13, 7730–7755. [Google Scholar] [CrossRef]
- Gurung, N.; Ray, S.; Bose, S.; Rai, V. A Broader View: Microbial Enzymes and Their Relevance in Industries, Medicine, and Beyond. BioMed Res. Int. 2013, 2013, 329121. [Google Scholar] [CrossRef] [PubMed]
- Mustafa, A.; Faisal, S.; Ahmed, I.A.; Munir, M.; Cipolatti, E.P.; Manoel, E.A.; Pastore, C.; di Bitonto, L.; Hanelt, D.; Nitbani, F.O.; et al. Has the time finally come for green oleochemicals and biodiesel production using large-scale enzyme technologies? Current status and new developments. Biotechnol. Adv. 2023, 69, 108275. [Google Scholar] [CrossRef]
- Westley, J. Enzymic Catalysis; Harper & Row: New York, NY, USA, 1969; pp. 5–15. [Google Scholar]
- Koshland, D.E. Application of a Theory of Enzyme Specificity to Protein Synthesis. Proc. Natl. Acad. Sci. USA 1958, 44, 98–104. [Google Scholar] [CrossRef]
- Johnson, K.A.; Goody, R.S. The Original Michaelis Constant: Translation of the 1913 Michaelis–Menten Paper. Biochemistry 2011, 50, 8264–8269. [Google Scholar] [CrossRef]
- Reetz, M.T.; Sun, Z.; Qu, G. Enzyme Engineering: Selective Catalysts for Applications in Biotechnology, Organic Chemistry, and Life Science; Wiley-VCH GmbH: Weinheim, Germany, 2023. [Google Scholar]
- Reetz, M.T.; Qu, G.; Sun, Z. Engineered enzymes for the synthesis of pharmaceuticals and other high-value products. Nat. Synth. 2024, 3, 19–32. [Google Scholar] [CrossRef]
- Arnodo, D.; Maffeis, E.; Marra, F.; Nejrotti, S.; Prandi, C. Combination of enzymes and deep eutectic solvents as powerful toolbox for organic synthesis. Molecules 2023, 28, 516. [Google Scholar] [CrossRef]
- Kries, H.; Trottmann, F.; Hertweg, C. Novel Biocatalysts from Specialized Metabolism. Angew. Chem. Int. Ed. 2024, 63, e202309284. [Google Scholar] [CrossRef]
- Wirz, B.; Spurr, P.; Pfleger, C. Enantioselective synthesis of (1R,2S,4S)-7-oxabicyclo[2.2.1]heptan-2-exo-carboxylic acid. Tetrahedron Asym. 2010, 22, 159–161. [Google Scholar] [CrossRef]
- Zhu, Z.; Sun, F.; Zhang, X.; Zhang, Y.-H. Deep oxidation of glucose in enzymatic fuel cells through a synthetic enzymatic pathway containing a cascade of two thermostable dehydrogenases. Biosens. Bioelectron. 2012, 36, 110–115. [Google Scholar] [CrossRef]
- Takayama, S.; McGarvey, G.J.; Wong, C.-H. Microbial Aldolases and Transketolases: New Biocatalytic Approaches to Simple and Complex Sugars. Annu. Rev. Micro. 1997, 51, 285–310. [Google Scholar] [CrossRef]
- Taylor, N.G. Cellulose biosynthesis and deposition in higher plants. New Phytologist. 2008, 178, 239–252. [Google Scholar] [CrossRef]
- Bulawa, C.E. Genetics and molecular biology of chitin synthesis in fungi. Annu. Rev. Microbiol. 1993, 47, 505–534. [Google Scholar] [CrossRef] [PubMed]
- Bouskila, M.; Hunter, R.W.; Ibrahim, A.F.M.; Delattre, L.; Peggie, M.; Diepen, J.A.; Voshol, P.J.; Jensen, J.; Sakamoto, K. Allosteric Regulation of Glycogen Synthase Controls Glycogen Synthesis in Muscle. Cell Metabol. 2010, 12, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, A.J.F.; Miller, J.H.; Suzuki, D.T. An Introduction to Genetic Analysis, 7th ed.; W. H. Freeman: New York, NY, USA, 2000. [Google Scholar]
- Vanholme, R.; Demedts, B.; Morreel, K.; Ralph, J.; Boerjan, W. Lignin Biosynthesis and Structure. Plant Physiol. 2010, 153, 895–905. [Google Scholar] [CrossRef]
- Kobayashi, S.; Makino, A. Enzymatic Polymer Synthesis: An Oportunity for Green Polymer Chemistry. Chem. Rev. 2009, 109, 5288–5353. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Puskas, J.E. Green Polymer Chemistry: Enzyme Catalysis for Polymer Functionalization. Molecules 2015, 20, 9358–9379. [Google Scholar] [CrossRef] [PubMed]
- Castro-Rodríguez, J.A.; Rodríguez-Sotres, R.; Farrés, A. Determinants for an Efficient Enzymatic Catalysis in Poly (Ethylene Terephthalate) Degradation. Catalysts 2023, 13, 591. [Google Scholar] [CrossRef]
- Zdarta, J.; Meyer, A.S.; Jesionowski, T.; Pinelo, M. A General Overview of Support Materials for Enzyme Immobilization: Characteristics, Properties, Practical Utility. Catalysts 2018, 8, 92. [Google Scholar] [CrossRef]
- Bolivar, J.M.; Woodley, J.M.; Fernadez-Lafuente, R. Is enzyme immobilization a mature discipline? Some critical considerations to capitalize on the benefits of immobilization. Chem. Soc. Rev. 2022, 51, 6251–6290. [Google Scholar] [CrossRef] [PubMed]
- Almeida, F.L.C.; Prata, A.S.; Forte, M.B.S. Enzyme immobilization: What have we learned in the past five years? Biof. Bioprod. Bioref. 2022, 16, 587–608. [Google Scholar] [CrossRef]
- Maghraby, Y.R.; El-Shabasy, R.M.; Ibrahim, A.H.; El-Said Azzazy, H.M. Enzyme Immobilization Technologies and Industrial Applications. ACS Omega 2023, 8, 5184–5196. [Google Scholar] [CrossRef] [PubMed]
- Sicard, C. In Situ Enzyme Immobilization by Covalent Organic Frameworks. Angew. Chem. Int. Ed. 2023, 62, e202213405. [Google Scholar] [CrossRef]
- Johansson, A.C.; Mosbach, K. Acrylic copolymers as matrixes for the immobilization of 150 enzymes. II. Effect of a hydrophobic microenvironment on enzyme reactions studied with alcohol dehydrogenase immobilized to different acrylic copolymers. Biochim. Biophys. Acta 1974, 370, 348–353. [Google Scholar] [CrossRef]
- Miwa, N.; Ohtomo, K. Enzyme immobilization in the presence of substrates and inhibitors. Jpn. Kokai Tokkyo Koho 1975, 56, 591. [Google Scholar]
- Chen, N.; Chang, B.; Shi, N.; Yan, W.; Lu, F.; Liu, F. Cross-linked enzyme aggregates immobilization: Preparation, characterization, and applications. Crit. Rev. Biotechnol. 2023, 43, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Costa, I.O.; Morais, J.R.F.; Dantas, J.M.M.; Gonçalves, L.R.B.; Santos, E.S.; Rios, N.S. Enzyme immobilization technology as a tool to innovate in the production of biofuels: A special review of the Cross-Linked Enzyme Aggregates (CLEAs) strategy. Enzyme Microb Technol. 2023, 170, 110300. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, R.C.; Ortiz, C.; Berenguer-Murcia, Á.; Torres, R.; Fernández-Lafuente, R. Modifying enzyme activity and selectivity by immobilization. Chem. Soc. Rev. 2013, 42, 6290–6307. [Google Scholar] [CrossRef] [PubMed]
- Isanapong, J.; Lohawet, K.; Kumnorkaew, P. Optimization and Characterization of Immobilized Laccase on Titanium Dioxide Nanostructure and Its Application in Removal of Remazol Brilliant Blue R. Biocatal. Agric. Biotechnol. 2021, 37, 102186. [Google Scholar] [CrossRef]
- Heilmann, A.; Teuscher, N.; Kiesow, A.; Spohn, U. Nanoporous aluminum oxide as a novel support material for enzyme biosensors. J. Nanosci. Nanotechnol. 2003, 3, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Huckel, M.; Wirth, H.J.; Hearn, M.T. Porous zirconia: A new support material for enzyme immobilization. J. Biochem. Biophys. Meth. 1996, 31, 165–179. [Google Scholar] [CrossRef] [PubMed]
- Stranix, B.R.; Darling, G.D. Functional polymers from (vinyl)polystyrene. Enzyme immobilization through a cysteinyl-S-ethyl spacer. Biotechnol. Tech. 1995, 9, 75–80. [Google Scholar] [CrossRef]
- Shinde, P.; Musameh, M.; Gao, Y.; Robinson, A.J.; Kyratzis, I. Immobilization and stabilization of alcohol dehydrogenase on polyvinyl alcohol fibre. Biotechnol. Rep. 2018, 19, e00260. [Google Scholar] [CrossRef]
- Grotzky, A.; Altamura, E.; Adamcik, J.; Carrara, P.; Stano, P.; Mavelli, F.; Nauser, T.; Mezzenga, R.; Schlüter, A.D.; Walde, P. Structure and Enzymatic Properties of Molecular Dendronized Polymer-Enzyme Conjugates and Their Entrapment inside Giant Vesicles. Langmuir 2013, 29, 10831–10840. [Google Scholar] [CrossRef]
- Biro, E.; Nemeth, A.S.; Sisak, C.; Feczko, T.; Gyenis, J. Preparation of chitosan particles suitable for enzyme immobilization. J. Biochem. Biophys. Methods 2008, 70, 1240–1246. [Google Scholar] [CrossRef]
- Hanachi, P.; Jafary, F.; Jafary, F.; Motamedi, S. Immobilization of the Alkaline Phosphatase on Collagen Surface via Cross-Linking Method. Iran. J. Biotechnol. 2015, 13, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Erramuspe, I.; Fazeli, E.; Nareoja, T.; Trygg, J.; Hanninen, P.; Heinze, T.; Fardim, P. Advanced Cellulose Fibers for Efficient Immobilization of Enzymes. Biomacromolecules 2016, 17, 3188–3197. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.C.; Juo, Y.Y.; Liang, C.F.; Chein, W.T.; Wu, H.T.; Chang, T.C.; Jan, F.D.; Lin, C.C. Site-Specific Immobilization of Enzymes on Magnetic Nanoparticles and Their Use in Organic Synthesis. Bioconjug. Chem. 2012, 23, 714–724. [Google Scholar] [CrossRef] [PubMed]
- Zahidah, K.A.; Kakooei, S.; Ismail, M.C.; Raja, P.B. Halloysite nanotubes as nanocontainer for smart coating application: A review. Progr. Org. Coat. 2017, 111, 175–185. [Google Scholar] [CrossRef]
- Liu, X.; Gao, Z.; Wang, D.; Yu, F.; Du, B.; Gitsov, I. Improving the Protection Performance of Waterborne Coatings with a Corrosion Inhibitor Encapsulated in Polyaniline-Modified Halloysite Nanotubes. Coatings 2023, 13, 1677. [Google Scholar] [CrossRef]
- Tully, J.; Yendluri, R.; Lvov, Y. Halloysite Clay Nanotubes for Enzyme Immobilization. Biomacromolecules 2016, 17, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Kumar-Krishnan, S.; Hernandez-Rangel, A.; Pal, U.; Ceballos-Sanchez, O.; Flores-Ruiz, F.J.; Prokhorov, E.; Arias de Fuentes, O.; Esparza, R.; Meyyappan, M. Surface functionalized halloysite nanotubes decorated with silver nanoparticles for enzyme immobilization and biosensing. J. Mater. Chem. B 2016, 4, 2553–2560. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Ding, W.; Zhou, K.; Guo, S.; Zhang, Q.; Haddleton, D.M. Coating Titania Nanoparticles with Epoxy-Containing Catechol Polymers via Cu(0)-Living Radical Polymerization as Intelligent Enzyme Carriers. Biomacromolecules 2018, 19, 2979–2990. [Google Scholar] [CrossRef]
- Gómez, J.L.; Bódalo, A.; Gómez, E.; Bastida, J.; Hidalgo, A.M.; Gómez, M. Immobilization of peroxidases on glass beads: An improved alternative for phenol removal. Enzym. Microb. Technol. 2006, 39, 1016–1022. [Google Scholar] [CrossRef]
- Kim, M.I.; Ham, H.O.; Oh, S.-D.; Park, H.G.; Chang, H.N.; Choi, S.-H. Immobilization of Mucor javanicus lipase on effectively functionalized silica nanoparticles. J. Mol. Catal. B Enzym. 2006, 39, 62–68. [Google Scholar] [CrossRef]
- Gao, Y.; Kyratzis, I. Covalent Immobilization of Proteins on Carbon Nanotubes Using the Cross-Linker 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide—A Critical Assessment. Bioconjug. Chem. 2008, 19, 1945–1950. [Google Scholar] [CrossRef] [PubMed]
- Koneracka, M.; Kočansky, P.; Antalik, M.; Timko, M.; Ramchand, C.N.; Lobo, D.; Mehta, R.V.; Upadhyay, R.V. Immobilization of proteins and enzymes to fine magnetic particles. J. Magn. Magn. Mater. 1999, 201, 427–430. [Google Scholar] [CrossRef]
- Velasco-Lozano, S.; López-Gallego, F.; Vázquez-Duhalt, R.; Mateos-Díaz, J.C.; Guisán, J.M.; Favela-Torres, E. Carrier-free immobilization of lipase from candida rugosa with polyethyleneimines by carboxyl-activated cross-linking. Biomacromolecules 2014, 15, 1896–1903. [Google Scholar] [CrossRef]
- Mohamad, N.R.; Merzuki, N.H.C.; Buang, N.A.; Huyop, F.; Wahab, R.A. An overview of technologies for immobilization of enzymes and surface analysis techniques or immobilized enzymes. Biotechnol. Biotechnol. Equip. 2015, 29, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, J.L.; Nicolaus, T.; Neuert, G.; Blank, K. Thiol-based, site-specific and covalent immobilization of biomolecules for single-molecule experiments. Nat. Protoc. 2010, 5, 975–986. [Google Scholar] [CrossRef] [PubMed]
- Bailes, J.; Gazi, S.; Ivanova, R.; Soloviev, M. Effect of Gold Nanoparticle Conjugation on the Activity and Stability of Functional Proteins. In Nanoparticles in Biology and Medicine. Methods in Molecular Biology (Methods and Protocols); Soloviev, M., Ed.; Humana Press: Totowa, NJ, USA, 2012; p. 906. [Google Scholar]
- Heck, T.; Faccio, G.; Richter, M.; Thöny-Meyer, L. Enzyme-catalyzed protein crosslinking. Appl. Microbiol. Biotechnol. 2013, 97, 461. [Google Scholar] [CrossRef]
- Minamihata, A.; Goto, M.; Kamiya, N. Site-Specific Protein Cross-Linking by Peroxidase-Catalyzed Activation of a Tyrosine-Containing Peptide Tag. Bioconjug. Chem. 2011, 22, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Matijošytė, I.; Arends, I.; de Vries, S.; Sheldon, R.A. Preparation and use of cross-linked enzyme aggregates (CLEAs) of laccases. J. Mol. Catal. B Enzym. 2010, 62, 142–148. [Google Scholar] [CrossRef]
- Faccio, G.; Kämpf, M.M.; Piatti, C.; Thöny-Meyer, L.; Richter, M. Tyrosinase-catalyzed site-specific immobilization of engineered C-phycocyanin to surface. Sci. Rep. 2014, 4, 5370. [Google Scholar] [CrossRef]
- Nelson, J.M.; Griffin, E.G. Adsorption of invertase. J. Am. Chem. Soc. 1916, 38, 1109–1115. [Google Scholar] [CrossRef]
- Ahmad, R.; Sardar, M. Immobilization of cellulose on TiO2 nanoparticles by physical and covalent methods: A comparative study. Indian J. Biochem. Biophys. 2014, 51, 314–320. [Google Scholar] [PubMed]
- He, H.; Han, H.; Shi, H.; Tian, Y.; Sun, F.; Song, Y.; Li, Q.; Zhu, G. Construction of thermophilic lipase-embedded metal-organic frameworks via biomimetic mineralization: A biocatalyst for ester hydrolysis and kinetic resolution. ACS Appl. Mater. Interf. 2016, 8, 24517–24524. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Ni, Q.; Mu, J.; Fan, W.; Liu, L.; Wang, Z.; Ling, L.; Tang, W.; Liu, Y.; Cheng, Y.; et al. Solvent-assisted self-assembly of a metal-organic framework based biocatalyst for cascade reaction driven photodynamic therapy. J. Am. Chem. Soc. 2020, 142, 6822–6832. [Google Scholar] [CrossRef] [PubMed]
- Dencheva, N.; Oliveira, S.; Braz, J.; Getya, D.; Malfois, M.; Denchev, Z.; Gitsov, I. Magnetically responsive PA6 microparticles with immobilized laccase show high catalytic efficiency in the enzymatic treatment of catechol. Catalysts 2021, 11, 239. [Google Scholar] [CrossRef]
- Monteiro, R.R.C.; Lima, P.J.M.; Pinheiro, B.; Freire, T.M.; Dutra, L.M.U.; Fechine, L.; Gonçalves, L.R.B.; De Souza, M.C.M.; Dos Santos, J.C.S.; Fernández-Lafuente, R. Immobilization of lipase A from Candida antarctica onto chitosan-coated magnetic nanoparticles. Int. J. Mol. Sci. 2019, 20, 4018. [Google Scholar] [CrossRef] [PubMed]
- Bilal, M.; Asgher, M.; Parra, R.; Hu, H.; Wang, W.; Zhang, X.; Iqbal, H.M. Immobilized ligninolytic enzymes: An innovative and environmental responsive technology to tackle dye-based industrial pollutants—A review. Sci. Total Environ. 2016, 576, 646–659. [Google Scholar] [CrossRef] [PubMed]
- Manoel, E.A.; dos Santos, J.C.S.; Freire, D.M.G.; Rueda, N.; Fernández-Lafuente, R. Immobilization of lipases on hydrophobic supports involves the open form of the enzyme. Enzyme Microb. Technol. 2015, 71, 53–57. [Google Scholar] [CrossRef]
- Ortiz, C.; Ferreira, M.L.; Barbosa, O.; Dos Santos, J.C.S.; Rodriguez, R.C.; Berenguer-Murcia, Á.; Brand, L.E.; Fernández-Lafuente, R. Novozym 435: The “perfect” lipase immobilized biocatalyst? Catal. Sci. Technol. 2019, 9, 2380–24520. [Google Scholar] [CrossRef]
- Galgali, A.; Gawas, S.D.; Rathod, V.K. Ultrasound assisted synthesis of citronellol laurate by using Novozym 435. Catal. Today 2018, 309, 133–139. [Google Scholar] [CrossRef]
- Liu, C.; Jiang, Z.; Decatur, J.; Xie, W.; Gross, R.A. Chain growth and branch structure formation during lipase-catalyzed synthesis of aliphatic polycarbonate polyols. Macromolecules 2011, 44, 1471–1479. [Google Scholar] [CrossRef]
- Wu, W.-X.; Liu, Z. Novozym 435-Catalyzed Synthesis of Well-Defined Hyperbranched Aliphatic Poly(β-thioether ester). Molecules 2020, 25, 687. [Google Scholar] [CrossRef] [PubMed]
- Dencheva, N.; Braz, J.; Scheibel, D.; Malfois, M.; Denchev, Z.; Gitsov, I. Polymer-assisted biocatalysis: Polyamide 4 microparticles as promising carriers of enzymatic function. Catalysts 2020, 10, 767. [Google Scholar] [CrossRef]
- Scheibel, D.M.; Gitsov, I. Polymer-assisted biocatalysis: Effects of macromolecular architectures on the stability and catalytic activity of immobilized enzymes toward water-soluble and water-insoluble substrates. ACS Omega 2018, 3, 1700–1709. [Google Scholar] [CrossRef] [PubMed]
- Betigeri, S.S.; Neau, S.H. Immobilization of lipase using hydrophilic polymers in the form of hydrogel beads. Biomaterials 2002, 23, 3627–3636. [Google Scholar] [CrossRef] [PubMed]
- Cosnier, S.; Mousty, C.; Gondran, C.; Lepellec, A. Entrapment of enzyme within organic and inorganic materials for biosensor applications: Comparative study. Mater. Sci. Eng. C 2006, 26, 442–447. [Google Scholar] [CrossRef]
- Virgen-Ortíz, J.J.; Tacias-Pascacio, V.G.; Hirata, D.B.; Torrestiana-Sanchez, B.; Rosales-Quintero, A.; Fernández-Lafuente, R. Relevance of substrates and products on the desorption of lipases physically adsorbed on hydrophobic supports. Enzyme Microb. Technol. 2017, 96, 30–35. [Google Scholar] [CrossRef]
- Bose, A.L.; Bhattacharjee, D.; Goswami, D. Mixed Micelles and Bicontinuous Microemulsions: Promising Media for Enzymatic Reactions. Colloids Surf. B Biointerfaces 2022, 209, 112193. [Google Scholar] [CrossRef]
- Gitsov, I.; Hamzik, J.; Ryan, J.; Simonyan, A.; Nakas, J.P.; Omori, S.; Krastanov, A.; Cohen, T.; Tanenbaum, S.W. Enzymatic nanoreactors for environmentally benign biotransformations—1. Formation and catalytic activity of supramolecular complexes of laccase and linear-dendritic blockcopolymers. Biomacromolecules 2008, 9, 804–811. [Google Scholar] [CrossRef]
- Silva, C.; Martins, M.; Jing, S.; Fu, J.; Cavaco-Paulo, A. Practical insights on enzyme stabilization. Crit. Rev. Biotechnol. 2018, 38, 335–350. [Google Scholar] [CrossRef]
- Márquez-Villa, J.M.; Mateos-Díaz, J.C.; Rodríguez, J.A.; Camacho-Ruíz, R.M. Lipase B from Candida antarctica in Highly Saline AOT-Water-Isooctane Reverse Micelle Systems for Enhanced Esterification Reaction. Catalysts 2023, 13, 492. [Google Scholar] [CrossRef]
- Chen, H.; Liu, L.-H.; Wang, L.-S.; Ching, C.-B.; Yu, H.-W.; Yang, Y.-Y. Thermally Responsive Reversed Micelles for Immobilization of Enzymes. Adv. Funct. Mater. 2008, 18, 95–102. [Google Scholar] [CrossRef]
- DeSantis, G.; Jones, J.B. Chemical modification of enzymes for enhanced functionality. Curr. Opin. Biotechnol. 1999, 10, 324–330. [Google Scholar] [CrossRef]
- Kagliwal, L.D.; Singhal, R.S. Enzyme-polysaccharide interaction: A method for improved stability of horseradish peroxidase. Int. J. Biol. Macromol. 2014, 69, 329–335. [Google Scholar] [CrossRef]
- Gauthier, M.A.; Klok, H.-A. Polymer-protein conjugates: An enzymatic activity perspective. Polym. Chem. 2010, 1, 1352–1373. [Google Scholar] [CrossRef]
- Ito, Y.; Kotoura, M.; Chung, D.J.; Imanishi, Y. Trypsin modification by vinyl polymers with variable solubilities in response to external signals. Bioconjug. Chem. 1993, 4, 358–361. [Google Scholar] [CrossRef]
- Moreno-Perez, S.; Orrego, A.H.; Romero-Fernandez, M.; Trobo-Maseda, L.; Martins-DeOliveira, S.; Munilla, R.; Fernandez-Lorente, G.; Guisan, J.M. Intense PEGylation of Enzyme Surfaces: Relevant Stabilizing Effects. Methods Enzymol. 2016, 571, 55–72. [Google Scholar]
- Suthiwangcharoen, N.; Nagarajan, R. Enhancing enzyme stability by construction of polymer–enzyme conjugate micelles for decontamination of organophosphate agents. Biomacromolecules 2014, 15, 1142–1152. [Google Scholar] [CrossRef]
- Perrotta, J.A.; Tanenbaum, S.W.; Lai, Y.-Z.; Nakas, J.P. Modification of laccase for use in the forest industry. ESPRA Res. Rep. 1999, 111, 115–119. [Google Scholar]
- Schoffelen, S.; van Hest, J.C.M. Chemical approaches for the construction of multi-enzyme reaction systems. Curr. Opin. Struct. Biol. 2013, 23, 613–621. [Google Scholar] [CrossRef]
- Mackenzie, K.J.; Francis, M.B. Recyclable thermoresponsive polymer–cellulase bioconjugates for biomass depolymerization. J. Am. Chem. Soc. 2013, 135, 293–300. [Google Scholar] [CrossRef]
- Nakamura, I.; Horikawa, Y.; Makino, A.; Sugiyama, J.; Kimura, S. Enzymatic Polymerization Catalyzed by Immobilized Endoglucanase on Gold. Biomacromolecules 2011, 12, 785–790. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, I.; Makino, A.; Horikawa, Y.; Sugiyama, J.; Ohmae, M.; Kimura, S. Preparation of Fibrous Cellulose by Enzymatic Polymerization Using Cross-Linked Mutant Endoglucanase II. Chem. Commun. 2011, 47, 10127–10129. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nanayakkara, S.; Zhao, Z.; Patti, A.F.; He, L.; Saito, K. Immobilized Horseradish Peroxidase (I-HRP) as Biocatalyst for Oxidative Polymerization of 2,6-Dimethylphenol. ACS Sustain. Chem. Eng. 2014, 2, 1947–1950. [Google Scholar] [CrossRef]
- Xu, P.; Singh, A.; Kaplan, D.L. Enzymatic Catalysis in the Synthesis of Polyanilines and Derivatives of Polyanilines. In Enzyme-Catalyzed Synthesis of Polymers. Advances in Polymer Science; Kobayashi, S., Ritter, H., Kaplan, D., Eds.; Springer: Berlin/Heidelberg, Germany, 2005; Volume 194, pp. 69–94. [Google Scholar]
- Kausaite-Minkstimiene, A.; Mazeiko, V.; Ramanaviciene, A.; Ramanavicius, A. Enzymatically Synthesized Polyaniline Layer for Extension of Linear Detection Region of Amperometric Glucose Biosensor. Biosens. Bioelectron. 2010, 26, 790–797. [Google Scholar] [CrossRef]
- Junker, K.; Gitsov, I.; Quade, N.; Walde, P. Preparation of aqueous polyaniline-vesicle suspensions with class III peroxidases. Comparison between horseradish peroxidase isoenzyme C and soybean peroxidase. Chem. Pap. 2013, 67, 1028–1047. [Google Scholar] [CrossRef]
- Zhao, Q.; Sun, J.Z.; Ren, H.; Zhou, Q.Y.; Lin, Q.C. Horseradish Peroxidase Immobilized in Macroporous Hydrogel for Acrylamide Polymerization. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 2222–2232. [Google Scholar] [CrossRef]
- Janusz, G.; Pawlik, A.; Świderska-Burek, U.; Polak, J.; Sulej, J.; Jarosz-Wilkołazka, A.; Paszczyński, A. Laccase Properties, Physiological Functions, and Evolution. Int. J. Mol. Sci. 2020, 21, 966. [Google Scholar] [CrossRef]
- Yoshida, H. Chemistry of Lacquer (Urushi). J. Chem. Soc. Trans. 1883, 43, 472–486. [Google Scholar] [CrossRef]
- Kang, K.H.; Dec, J.; Park, H.; Bollag, J.M. Transformation of the fungicide cyprodinil by a laccase of Trametes villosa in the presence of phenolic mediators and humic acid. Water Res. 2002, 36, 4907–4915. [Google Scholar] [CrossRef]
- Nagai, M.; Kawata, M.; Watanabe, H.; Ogawa, M.; Saito, K.; Takesawa, T. Important role of fungal intracellular laccase for melanin synthesis: Purification and characterization of an intracellular laccase from Lentinula edodes fruit bodies. Microbiology 2003, 149, 2455–2462. [Google Scholar] [CrossRef]
- Claus, H. Laccases and their occurrences in prokaryotes. Arch. Microbiol. 2003, 179, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-Y.; Zhang, S.; Yu, Y.; Luo, Y.-C.; Liu, Q.; Ju, C.; Zhang, Y.-C.; Qu, L.-H.; Lucas, W.J.; Wang, X.; et al. MiR397b regulates both lignin content and seed number in Arabidopsis via modulating a laccase involved in lignin biosynthesis. Plant Biotchnol. J. 2014, 12, 1132–1142. [Google Scholar] [CrossRef] [PubMed]
- Soares, M.P.M.; Silva-Torres, F.A.; Elias-Neto, M.; Nunes, F.M.F.; Simoes, Z.L.P.; Bitondi, M.M.G. Ecdysteroid-Dependent Expression of the Tweedle and Peroxidase Genes during Adult Cuticle Formation in the Honey Bee, Apis mellifera. PLoS ONE 2011, 6, e20513. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Berka, R.M.; Wahleithner, J.A.; Nelson, B.A.; Shuster, J.R.; Brown, S.H.; Palmer, A.E.; Solomon, E.I. Site-directed mutations in fungal laccase: Effect on redox potential, activity, and pH profile. Biochem. J. 1998, 334, 63–70. [Google Scholar] [CrossRef]
- Xu, F.; Palmer, A.E.; Yaver, D.S.; Berka, R.M.; Gambetta, G.A.; Brown, S.H. Targeted mutations in a Trametes villosa laccase. Axial perturbations of the T1 copper. J. Biol. Chem. 1999, 274, 12372–12375. [Google Scholar] [CrossRef]
- Freeman, J.C.; Nayar, P.G.; Begley, T.P.; Villafranca, J.J. Stoichiometry and spectroscopic identity of copper centers in phenoxazonine synthase: A new addition for the blue copper oxidase family. Biochemistry 1993, 32, 4826–4830. [Google Scholar] [CrossRef]
- Hullo, M.F.; Moszer, I.; Danchin, A.; Martin-Verstraete, I. CotA of Bacillus substilis is a copper-dependent laccase. J. Bacteriol. 2001, 183, 5426–5430. [Google Scholar] [CrossRef]
- Cha, J.; Cooksey, D.A. Copper resistance in Pseudomonas syringae by periplasmic and outer membrane proteins. Proc. Natl. Acad. Sci. USA 1991, 88, 8915–8919. [Google Scholar] [CrossRef]
- LaFeyette, P.R.; Erikson, K.-E.L.; Dean, J. Nucleotide sequence of a cDNA cloned encoding and acidic laccase from sycamore maple (Acer pseudoplatanus L.). Plant Physiol. 1995, 107, 667–668. [Google Scholar] [CrossRef]
- Slomczynski, D. Biochemical Studies of Laccase from Botrytis cinerea. Masters’ Thesis, State University of New York—ESF, Syracuse, NY, USA, 1993. [Google Scholar]
- Musci, G.; Tosi, L.; Desideri, A.; Morpungo, L.; Garnier-Suillerot, A. Effects of laser radiation on Rhus vernicifera laccase, type 2 Cu-depleted laccase and stellacyanin. J. Inorg. Biochem. 1984, 20, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Thomas, B.R.; Yonekura, M.; Morgan, T.D.; Czapula, T.H.; Hopkins, T.L.; Kramer, K.J. A trypsin solubilized laccase from pharate pupal integument of the tobacco hornworm, Manduca sexta. Insect Biochem. 1989, 19, 611–622. [Google Scholar] [CrossRef]
- Sugumaran, M.; Giglio, L.; Kundzicz, H.; Saul, S.; Semensi, V. Studies on the enzymes involved in puparial cuticle sclerotization in Drosophila melanogaster. Arch. Insect Biochem. Physiol. 1992, 19, 271–283. [Google Scholar] [CrossRef] [PubMed]
- Sugumaran, M.; Nellaiappan, K. On the latency and nature of phenoloxidase present in the left collecterial gland of the cockroach Periplaneta americana. Arch. Insect Biochem. Physiol. 1990, 15, 165–181. [Google Scholar] [CrossRef]
- Piontek, K.; Antorini, M.; Choinowski, T. Crystal Structure of a Laccase from the Fungus Trametes versicolor at 1.90-A Resolution Containing a Full Complement of Coppers. J. Biol. Chem. 2002, 277, 37663–37669. [Google Scholar] [CrossRef]
- Slomczynski, D.; Nakas, J.P.; Tanenbaum, S.W. Production and Characterization of Laccase from Botrytis cinerea. Appl. Environ. Microbiol. 1995, 61, 907–912. [Google Scholar] [CrossRef] [PubMed]
- Kallio, J.P.; Auer, S.; Jänis, J.; Andberg, M.; Kruus, K.; Rouvinen, J.; Koivula, A.; Hakulinen, N. Structure–Function Studies of a Melanocarpus albomyces Laccase Suggest a Pathway for Oxidation of Phenolic Compounds. J. Mol. Biol. 2009, 392, 895–909. [Google Scholar] [CrossRef]
- Baldrian, P. Fungal laccases—Occurrence and properties. FEMS Microbiol. Rev. 2006, 30, 215–242. [Google Scholar] [CrossRef]
- Jones, S.; Solomon, E.I. Electron transfer and reaction mechanism of laccases. Cell. Mol. Life Sci. 2015, 72, 869–883. [Google Scholar] [CrossRef]
- Augustine, A.J.; Kjaergaard, C.; Qayyum, M.; Ziegler, L.; Kosman, D.J.; Hodgson, K.O.; Hedman, B.; Solomon, E.I. Systematic Perturbation of the Trinuclear Copper Cluster in the Multicopper Oxidases: The Role of Active Site Asymmetry in Its Reduction of O2 to H2O. J. Am. Chem. Soc. 2010, 132, 6057–6067. [Google Scholar] [CrossRef]
- Cañas, A.I.; Camarero, S. Laccases and their natural mediators: Biotechnological tools for sustainable eco-friendly processes. Biotechnol. Adv. 2010, 28, 694–705. [Google Scholar] [CrossRef]
- Gitsov, I.; Lambrych, K.; Lu, P.; Nakas, J.; Ryan, J.; Tanenbaum, S. Nondestructive Regioselective Modification of Laccase by Linear-Dendritic Copolymers. Enhanced Oxidation of Benzo-α-Pyrene in Water. In Polymer Biocatalysis and Biomaterials; Cheng, H.N., Gross, R.A., Eds.; American Chemical Society: Washington, DC, USA, 2005; ACS Symp. Ser.; Volume 900, pp. 80–94. [Google Scholar]
- Cannatelli, M.D.; Ragauskas, A.J. Two Decades of Laccases: Advancing Sustainability in the Chemical Industry. Chem. Rec. 2017, 17, 122–140. [Google Scholar] [CrossRef]
- Mate, D.M.; Alcalde, M. Laccase: A multipurpose biocatalysts at the forefront of biotechnology. Microb. Biotechnol. 2017, 10, 1457–1467. [Google Scholar] [CrossRef]
- Curran, L.M.C.L.K.; Pham, L.T.M.; Sale, K.L.; Simmons, B.A. Review of advances in the development of laccases for the valorization of lignin to enable the production of lignocellulosic biofuels and bioproducts. Biotechnol. Adv. 2022, 54, 107809. [Google Scholar]
- Minussi, R.C.; Pastore, G.M.; Durán, N. Potential applications of laccase in the food industry. Trends Food Sci. Technol. 2002, 13, 205–216. [Google Scholar] [CrossRef]
- Macheix, J.J.; Fleuriet, A.; Sapis, J.C.; Lee, C.Y. Phenolic compounds and polyphenoloxidase in relation to browning in grapes and wines. Crit. Rev. Food Sci. Nutr. 1991, 30, 441–486. [Google Scholar] [CrossRef]
- Li, H.; Guo, A.; Wang, H. Mechanisms of oxidative browning of wine. Food Chem. 2008, 108, 1–13. [Google Scholar] [CrossRef]
- Brenna, O.; Bianchi, E. Immobilised laccase for phenolic removal in must and wine. Biotechnol. Lett. 1994, 16, 35–40. [Google Scholar] [CrossRef]
- Minussi, R.C.; Rossi, M.; Bologna, L.; Rotilio, D.; Pastore, G.M.; Durán, N. Phenols removal in musts: Strategy for wine stabilization by laccase. J. Mol. Catal. B Enzym. 2007, 45, 102–107. [Google Scholar] [CrossRef]
- Zinnai, A.; Venturi, F.; Sanmartin, C.; Quartacci, M.; Andrich, G. Chemical and Laccase catalysed oxidation of gallic acid: Determination of kinetic parameters. Res. J. Biotechnol. 2013, 8, 62–65. [Google Scholar]
- Valaer, P. Methods of Analysis of Wine. J. Assoc. Off. Agric. Chem. 1947, 30, 327–331. [Google Scholar] [CrossRef]
- Gamella, M.; Campuzano, S.; Reviejo, A.J.; Pingarrón, J.M. Electrochemical estimation of the polyphenol index in wines using a laccase biosensor. J. Agric. Food Chem. 2006, 54, 7960–7967. [Google Scholar] [CrossRef]
- Montereali, M.R.; Seta, L.D.; Vastarella, W.; Pilloton, R. A disposable Laccase-Tyrosinase based biosensor for amperometric detection of phenolic compounds in must and wine. J. Mol. Catal. B Enzym. 2010, 64, 189–194. [Google Scholar] [CrossRef]
- Gil, D.M.A.; Rebelo, M.J.F. Evaluating the antioxidant capacity of wines: A laccase-based biosensor approach. Eur. Food Res. Technol. 2010, 231, 303–308. [Google Scholar] [CrossRef]
- Di Fusco, M.; Tortolini, C.; Deriu, D.; Mazzei, F. Laccase-based biosensor for the determination of polyphenol index in wine. Talanta 2010, 81, 235–240. [Google Scholar] [CrossRef]
- Chawla, S.; Rawal, R.; Kumar, D.; Pundir, C.S. Amperometric determination of total phenolic content in wine by laccase immobilized onto silver nanoparticles/zinc oxide nanoparticles modified gold electrode. Anal. Biochem. 2012, 430, 16–23. [Google Scholar] [CrossRef]
- Magyar, I. Botrytized Wines. Adv. Food Nutr. Res. 2011, 63, 147–206. [Google Scholar]
- Thudichum, J.L.W.; Dupré, A. A Treatise on the Origin, Nature, and Varieties of Wine: Being a Complete Manual of Viticulture and Œnology; Macmillan and Co.: London, UK; New York, NY, USA, 1872; p. 760. [Google Scholar]
- Jackson, R.S. Specific and Distinctive Wine Styles in Wine Science: Principles and Applications; Academic Press: London, UK, 2008; pp. 520–576. [Google Scholar]
- Modesti, M.; Alfieri, G.; Chieffo, C.; Mencarelli, F.; Vannini, A.; Catalani, A.; Chilosi, G.; Bellincontro, A. Destructive and non-destructive early detection of postharvest noble rot (Botrytis cinerea) in wine grapes aimed at producing high-quality wines. J. Sci. Food Agric. 2023; early view. [Google Scholar] [CrossRef]
- Magyar, I.; Soos, J. Botrytized wines—Current perspectives. Int. J. Wine Res. 2016, 8, 29–39. [Google Scholar] [CrossRef]
- Bailly, S.; Jerkovic, V.; Marchand-Brynaert, J.; Collin, S. Aroma extraction dilution analysis of sauternes wines. Key role of polyfunctional thiols. J. Agric. Food Chem. 2006, 54, 7227–7234. [Google Scholar] [CrossRef]
- Semenov, A.N.; Lomonosova, I.V.; Berezin, V.I.; Titov, M.I. Peroxidase and laccase as catalysts for removal of the phenylhydrazide protecting group under mild conditions. Biotechnol. Bioeng. 1993, 42, 1137–1141. [Google Scholar] [CrossRef]
- Liu, J.; Wu, S.; Li, Z. Recent advances in enzymatic oxidation of alcohols. Curr. Opin. Chem. Biol. 2018, 43, 77–86. [Google Scholar] [CrossRef]
- Xu, F. Catalysis of novel enzymatic iodide oxidation by fungal laccase. Appl. Biochem. Biotechnol. 1996, 59, 221–230. [Google Scholar] [CrossRef]
- Chauhan, P.S.; Kumarasamy, M.; Sosnik, A.; Danino, D. Enhanced Thermostability and Anticancer Activity in Breast Cancer Cells of Laccase Immobilized on Pluronic-Stabilized Nanoparticles. ACS Appl. Mater. Interfaces 2019, 11, 39436–39448. [Google Scholar] [CrossRef]
- Mikolasch, A.; Hildebrandt, O.; Schlüter, R.; Hammer, E.; Witt, S.; Lindequist, U. Targeted synthesis of novel β-lactam antibiotics by laccase-catalyzed reaction of aromatic substrates selected by pre-testing for their antimicrobial and cytotoxic activity. Appl. Microbiol. Biotechnol. 2016, 100, 4885–4899. [Google Scholar] [CrossRef]
- Uhnáková, B.; Petrícková, A.; Biedermann, D.; Homolka, L.; Vejvoda, V.; Bednár, P.; Papousková, B.; Sulc, M.; Martínková, L. Biodegradation of brominated aromatics by cultures and laccase of Trametes versicolor. Chemosphere 2009, 76, 826–832. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, X.; Xu, Z.; Chen, H.; Yang, Y. Degradation of chlorophenols catalyzed by laccase. Int. Biodeter. Biodegr. 2008, 61, 351–356. [Google Scholar] [CrossRef]
- Gitsov, I.; Lambrych, K.R.; Nakas, J.; Lu, P.; Ryan, J.; Omori, S.; Tanenbaum, S.W. Nondestructive Regioselective Modification of Laccase by Linear-Dendritic Copolymers. Enhanced Oxidation of Polyaromatic Hydrocarbons in Water. Polym. Prepr. 2003, 44, 143–144. [Google Scholar]
- Gitsov, I.; Simonyan, A.; Wang, L.; Krastanov, A.; Tanenbaum, S.W.; Kiemle, D. Polymer-assisted biocatalysis: Unprecedented enzymatic oxidation of fullerene in aqueous medium. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 119–126. [Google Scholar] [CrossRef]
- Chiang, L.Y.; Upasani, R.B.; Swirczewski, J.W.; Soled, S. Evidence of hemiketals incorporated in the structure of fullerols derived from aqueous acid chemistry. J. Am. Chem. Soc. 1993, 115, 5453–5457. [Google Scholar] [CrossRef]
- Lin, H.; Yu, Z.J.; Wang, Q.; Liu, Y.J.; Jiang, L.; Xu, C.; Xian, M. Application of Laccase Catalysis in Bond Formation and Breakage: A Review. Catalysts 2023, 13, 750. [Google Scholar] [CrossRef]
- Gitsov, I.; Simonyan, A. “Green” Synthesis of Bisphenol Polymers and Copolymers, Mediated by Supramolecular Complexes of Laccase and Linear-Dendritic Block Copolymers. In Green Polymer Chemistry: Biocatalysis and Materials II; Cheng, H.N., Gross, R.A., Eds.; American Chemical Society: Washington, DC, USA, 2013; ACS Symposium Series; Volume 1144, pp. 121–139. [Google Scholar]
- Novotný, Č.; Vyas, B.R.M.; Erbanová, P.; Kubátová, A.; Šašek, A. Removal of PCBs by various white rot fungi in liquid cultures. Folia Microbiol. 1997, 42, 136–142. [Google Scholar] [CrossRef]
- Mori, T.; Kondo, R. Oxidation of chlorinated dibenzo-p-dioxin and dibenzofuran by white-rot fungus, Phlebia lindtneri. FEMS Microbiol. Lett. 2002, 216, 223–227. [Google Scholar] [CrossRef]
- Gitsov, I.; Simonyan, A.; Krastanov, A.; Tanenbaum, S. Green oxidation of steroids in nano-reactors assembled from laccase and linear-dendritic copolymers. In Polymer Biocatalysis and Biomaterials II; Cheng, H.N., Gross, R.A., Eds.; American Chemical Society: Washington, DC, USA, 2008; ACS Symposium Series; Volume 999, pp. 110–128. [Google Scholar]
- Yang, J.; Li, W.; Ng, T.B.; Deng, X.; Lin, J.; Ye, X. Laccases: Production, Expression Regulation and Application in Pharmaceutical Biodegradation. Front. Microbiol. 2017, 8, 832. [Google Scholar] [CrossRef]
- Janusz, G.; Skwarek, E.; Pawlik, A. Potential of Laccase as a Tool for Biodegradation of Wastewater Micropollutants. Water 2023, 15, 3770. [Google Scholar] [CrossRef]
- Kanagaraj, J.; Senthilvelan, T.; Panda, R. Degradation of azo dyes by laccase: Biological method to reduce pollution load in dye wastewater. Clean Technol. Environ. Policy 2015, 17, 1443–1456. [Google Scholar] [CrossRef]
- Chhabra, M.; Mishra, S.; Sreekrishnan, T.R. Laccase/mediator assisted degradation of triarylmethane dyes in a continuous membrane reactor. J. Biotechnol. 2009, 143, 69–78. [Google Scholar] [CrossRef]
- Campos, R.; Kandelbauer, A.; Robra, K.H.; Cavaco-Paulo, A.; Gubitz, G.M. Indigo degradation with purified laccases from Trametes hirsuta and Sclerotium rolfsii. J. Biotechnol. 2001, 89, 131–139. [Google Scholar] [CrossRef]
- Soares, G.M.; Costa-Ferreira, M.; Pessoa, M.T. Decolorization of an anthraquinone-type dye using a laccase formulation. Bioresour. Technol. 2001, 79, 171–177. [Google Scholar] [CrossRef]
- Itoh, K.; Yatome, C. Decolorization and Degradation of Xanthene Dyes by a White Rot Fungus, Coriolus Versicolor. J. Environ. Sci. Health Part A Tox. Haz. Subst. Environ. Eng. 2011, 39, 2382–2389. [Google Scholar] [CrossRef]
- Gitsov, I.; Wang, L.; Vladimirov, N.; Simonyan, A.; Kiemle, D.J.; Schutz, A. “Green” Synthesis of Unnatural Poly (Amino Acid)s with Zwitterionic Character and pH-Responsive Solution Behavior, Mediated by Linear–Dendritic Laccase Complexes. Biomacromolecules 2014, 15, 4082–4095. [Google Scholar] [CrossRef]
- Liu, X.; Wang, L.; Gitsov, I. Novel Amphiphilic Dendronized Copolymers Formed by Enzyme-Mediated “Green” Polymerization. Biomacromolecules 2021, 22, 1706–1720. [Google Scholar] [CrossRef]
- Scheibel, D.M.; Guo, D.; Luo, J.; Gitsov, I. A single enzyme mediates the “quasi-living” formation of multiblock copolymers with a broad biomedical potential. Biomacromolecules 2020, 21, 2132–2146. [Google Scholar] [CrossRef]
- Aktas, N.; Tanyolac, A. Reaction conditions for laccase catalyzed polymerization of catechol. Bioresour. Technol. 2003, 87, 209–214. [Google Scholar] [CrossRef]
- Marjasvaara, A.; Torvinen, M.; Kinnunen, H.; Vainiotalo, P. Laccase-Catalyzed Polymerization of Two Phenolic Compounds Studied by Matrix-Assisted Laser Desorption/Ionization Time-of-Flight and Electrospray Ionization Fourier Transform Ion Cyclotron Resonance Mass Spectrometry with Collision-Induced Dissociation Experiments. Biomacromolecules 2006, 7, 1604–1609. [Google Scholar]
- Alvarez, S.; Manolache, S.; Denes, F. Synthesis of polyaniline using horseradish peroxidase immobilized on plasma-functionalized polyethylene surfaces as initiator. J. Appl. Polym. Sci. 2003, 88, 369–379. [Google Scholar] [CrossRef]
- Gospodinova, N.; Terlemezyan, L. Conducting polymers prepared by oxidative polymerization: Polyaniline. Progr. Polym. Sci. 1998, 23, 1443–1484. [Google Scholar] [CrossRef]
- Karamyshev, A.V.; Shleev, S.V.; Koroleva, O.V.; Yaropolov, A.I.; Sakharov, I.Y. Laccase-catalyzed synthesis of conducting polyaniline. Enzyme Microb. Technol. 2003, 33, 556–564. [Google Scholar] [CrossRef]
- Walde, P.; Kashima, K.; Ćirić-Marjanović, G. Synthesizing Polyaniline with Laccase/O2 as Catalyst. Front. Bioeng. Biotechnol. 2019, 77, 165. [Google Scholar] [CrossRef]
- Fodor, C.; Gajewska, B.; Rifaie-Graham, O.; Apedende, E.A.; Pollard, J.; Bruns, N. Laccase-catalyzed controlled radical polymerization of N-vinylimidazole. Polym. Chem. 2016, 7, 6617–6625. [Google Scholar] [CrossRef]
- Ikeda, R.; Taraka, H.; Uyama, H.; Kobayashi, S. Laccase-catalyzed polymerization of acrylamide. Macromol. Rapid Commun. 1998, 19, 423–425. [Google Scholar] [CrossRef]
- Tsujimoto, T.; Uyama, H.; Kobayashi, S. Polymerization of Vinyl Monomers Using Oxidase Catalysis. Macromol. Biosci. 2001, 1, 228–232. [Google Scholar] [CrossRef]
- Mai, C.; Schormann, W.; Huttermann, A. Chemo-enzymatically induced copolymerization of phenolics with acrylate compounds. Appl. Microbiol. Biotechnol. 2001, 55, 177–186. [Google Scholar] [CrossRef]
- Elegir, G.; Kindl, A.; Sadocco, P.; Orlandi, M. Development of antimicrobial cellulose packaging through laccase-mediated grafting of phenolic compounds. Enzym. Microb. Technol. 2008, 43, 84–92. [Google Scholar] [CrossRef]
- Bernard, C. Sur les usages du suc pancréatique. L’Institut 1848, 16, 137–138. [Google Scholar]
- Anthonsen, H.W.; Baptista, A.; Drabløs, F.; Martel, P.; Petersen, S.B.; Sebastião, M.; Vaz, L. Lipases and esterases: A review of their sequences, structure and evolution. Biotechnol. Ann. Rev. 1995, 1, 315–371. [Google Scholar]
- Reis, P.; Holmberg, K.; Watzke, H.; Leser, M.E.; Miller, R. Lipases at interfaces: A review. Adv. Colloid Interface Sci. 2009, 147, 237–250. [Google Scholar] [CrossRef]
- Khan, F.I.; Lan, D.; Durrani, R.; Huan, W.; Zhao, Z.; Wang, Y. The Lid Domain in Lipases: Structural and Functional Determinant of Enzymatic Properties. Front. Bioeng. Biotechnol. 2017, 5, 16. [Google Scholar] [CrossRef]
- Carrière, F.; Withers-Martinez, C.; Van Tilbeurgh, H.; Roussel, A.; Cambillau, C.; Verger, R. Structural basis for the substrate selectivity of pancreatic lipases and some related proteins. Biochim. Biophys. Acta Rev. Biomembr. 1998, 1376, 417–432. [Google Scholar] [CrossRef]
- Sarda, L.; Desnuelle, P. Action de la lipase pancréatique sur les esters en émulsion. Biochim. Biophys. Acta 1958, 30, 513–521. [Google Scholar] [CrossRef]
- Hamdan, S.H.; Maiangwa, J.; Ali, M.S.M.; Normi, Y.M.; Sabri, S.; Leow, T.C. Thermostable lipases and their dynamics of improved enzymatic properties. Appl. Microbiol. Biotechnol. 2021, 105, 7069–7094. [Google Scholar] [CrossRef]
- Karmee, S.K. Lipase catalyzed synthesis of ester-based surfactants from biomass derivatives. Biofuels Bioprod. Bioref. 2008, 2, 144–154. [Google Scholar] [CrossRef]
- Ghanem, A.; Aboul-Enein, H. Application of lipases in kinetic resolution of racemates. Chirality 2005, 17, 1–15. [Google Scholar] [CrossRef]
- Cortez, D.V.; Reis, C.; Perez, V.H.; De Castro, H.F. The realm of lipases in biodiesel production. In Sustainable Biotechnology-Enzymatic Resources of Renewable Energy; Singh, O.V., Chandel, A.K., Eds.; Springer International Publishing AG: New York, NY, USA, 2018; pp. 247–288. [Google Scholar]
- Uyama, H.; Kuwabara, M.; Tsujimoto, T.; Kobayashi, S. Enzymatic Synthesis and Curing of Biodegradable Epoxide-Containing Polyesters from Renewable Resources. Biomacromolecules 2003, 4, 211–215. [Google Scholar] [CrossRef]
- Kapoor, M.; Majumder, A.; Gupta, M. Promiscuous Lipase-Catalyzed C-C Bond Formation Reactions Between 4-Nitrobenzaldehyde and 2-Cyclohexen-1-one in Biphasic Medium: Aldol and Morita-Baylis-Hillman Adduct Formations. Catal. Lett. 2014, 145, 527–532. [Google Scholar] [CrossRef]
- Witayakran, S.; Ragauskas, A. Cocatalytic Enzyme System for the Michael Addition Reaction of in-situ-Generated ortho-Quinones. Eur. J. Org. Chem. 2009, 3, 358–363. [Google Scholar] [CrossRef]
- Ortega-Rojas, M.; Rivera-Ramirez, J.; Avila-Ortiz, C.; Juaristi, E.; Gonzalez-Munoz, F.; Castillo, E.; Escalante, J. One-Pot Lipase-Catalyzed Enantioselective Synthesis of (R)-(−)-N-Benzyl-3-(benzylamino)butanamide: The Effect of Solvent Polarity on Enantioselectivity. Molecules 2017, 22, 2189. [Google Scholar] [CrossRef]
- Albuqueque, T.; Silva, C.; Oliviera, A.; Santos, B.; Silva, B.; Katla, R.; Rocha, M.; Dominques, N. Lipase catalyzed 1,2-addition of thiols to imines under mild conditions. New J. Chem. 2018, 42, 1642–1645. [Google Scholar] [CrossRef]
- Brahmachari, G. Lipase-catalyzed organic transformations: A recent update. In Biotechnology of Microbial Enzymes, 2nd ed.; Brahmachari, G., Ed.; Academic Press: Cambridge, MA, USA, 2023; Chapter 13; pp. 297–321. [Google Scholar]
- Uyama, H.; Takeya, K.; Kobayashi, S. Synthesis of Polyesters by Enzymatic Ring-Opening Copolymerization Using Lipase Catalyst. Proc. Jpn. Acad. 1993, 69, 203–207. [Google Scholar] [CrossRef]
- Kobayashi, S.; Uyama, H.; Kimura, S. Enzymatic Polymerization. Chem. Rev. 2001, 101, 3793–3814. [Google Scholar] [CrossRef]
- Scandola, M.; Focarete, M.L.; Gross, R. Polymers from Biocatalysis: Materials with a Broad Spectrum of Physical Properties. In Green Polymer Chemistry: Biocatalysis and Biomaterials; Cheng, H., Gross, R., Eds.; American Chemical Society: Washington, DC, USA, 2010; Volume 1043, pp. 201–211. [Google Scholar]
- Kobayashi, S. Lipase-catalyzed polyester synthesis—A green polymer chemistry. Proc. Jpn. Acad. Ser. B 2010, 86, 338–365. [Google Scholar] [CrossRef]
- Spinella, S.; Ganesh, M.; Lo-Re, G.; Zhang, S.; Raquez, J.-M.; Dubois, P.; Gross, R.A. Enzymatic reactive extrusion: Moving towards continuous enzyme-catalyzed polyester polymerization and processing. Green Chem. 2015, 17, 4146–4150. [Google Scholar] [CrossRef]
- Svirkin, Y.Y.; Xu, J.; Gross, R.A.; Kaplan, D.L.; Swift, G. Enzyme-catalyzed stereoelective ring-opening polymerization of α-methyl-β-propiolactone. Macromolecules 1996, 29, 4591–4597. [Google Scholar] [CrossRef]
- Scheibel, D.M.; Gitsov, I. Unprecedented Enzymatic Synthesis of Perfectly Structured Alternating Copolymers via “Green” Reaction Cocatalyzed by Laccase and Lipase Compartmentalized within Supramolecular Complexes. Biomacromolecules 2019, 20, 927–936. [Google Scholar] [CrossRef]
- Wang, K.; Li, C.; Man, L.; Zhang, M.; Jia, Y.-G.; Zhu, X.X. Lipase-catalyzed ring-opening polymerization of natural compound-based cyclic monomers. Chem. Commun. 2023, 59, 9182–9194. [Google Scholar] [CrossRef]
- Schutz, C.; Dwars, T.; Kragl, U. Investigation on the Biocatalytic Oligomerization of Glycidol. Lett. Org. Chem. 2006, 3, 679–684. [Google Scholar] [CrossRef]
- Merlani, M.; Scheibel, D.M.; Barbakadze, V.; Gogilashvili, L.; Amiranashvili, L.; Amiranashvili, L.; Geronikaki, A.; Catania, V.; Schillaci, D.; Gallo, G.; et al. Enzymatic Synthesis and Antimicrobial Activity of Oligomer Analogues of Medicinal Biopolymers from Comfrey and Other Species of the Boraginaceae Family. Pharmaceutics 2022, 14, 115. [Google Scholar] [CrossRef]
- Şen, M.Y. Green Polymer Chemistry: Functionalization of Polymers Using Enzymatic Catalysis. Ph.D. Thesis, University of Akron, Akron, OH, USA, 2009. [Google Scholar]
- Uyama, H.; Kuwabara, M.; Tsujimoto, T.; Kobayashi, S. High Performance Immobilized Lipase catalyst for Polyester Synthesis. Polym. J. 2002, 34, 970–972. [Google Scholar] [CrossRef]
- Noda, S.; Kamiya, N.; Goto, M.; Nakashio, F. Enzymatic polymerization catalyzed by surfactant-coated lipases in organic media. Biotechnol. Lett. 1997, 19, 307–309. [Google Scholar] [CrossRef]
- Liu, X.; Kokare, C. Microbial enzymes of use in industry. In Biotechnology of Microbial Enzymes: Production, Biocatalysis, and Industrial Application, 2nd ed.; Brahmachari, G., Ed.; Academic Press: Cambridge, MA, USA, 2023; Chapter 17; pp. 405–444. [Google Scholar]
- Patil, P.D.; Kelkar, R.K.; Patil, N.P.; Pise, P.V.; Patil, S.P.; Patil, A.S.; Kulkarni, N.S.; Tiwari, M.S.; Phirke, A.N.; Nadar, S.S. Magnetic nanoflowers: A hybrid platform for enzyme immobilization. Crit. Rev. Biotechnol. 2023; early view. [Google Scholar] [CrossRef]
- Du, P.; Xu, S.; Xu, Z.-K.; Wang, Z.-G. Bioinspired Self-Assembling Materials for Modulating Enzyme Functions. Adv. Func. Mater. 2021, 31, 2104819. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Q. Enzyme-Laden Bioactive Hydrogel for Biocatalytic Monitoring and Regulation. Acc. Chem. Res. 2021, 54, 1274–1287. [Google Scholar] [CrossRef]





















| Enzyme Class | Catalytic Function | Selected Enzymes |
|---|---|---|
| Oxidoreductase, EC1 | Oxidation-reduction reactions involving intermolecular electron transfer | Laccase Horseradish Peroxidase Glucose Oxidase Cytochrome Oxidase |
| Transferase, EC2 | Transfer of functional groups from donor molecules to acceptor molecules | Butyrate Kinase DNA Methyltransferase Glucosyltransferase Thiaminase |
| Hydrolase, EC3 | Hydrolysis | Amylase Lipase Pepsin Urease |
| Lyase, EC4 | Non-hydrolytic bond cleavage | Adenylate Cyclase Carbonic Anhydrase Tryptophan Synthase |
| Isomerase, EC5 | Molecular isomerization | Photoisomerase Beta-carotene Isomerase Glucose isomerase |
| Ligase, EC6 | Large molecules coupling | Pyruvate Carboxylase DNA Ligase CTP Synthase |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scheibel, D.M.; Gitsov, I.P.I.; Gitsov, I. Enzymes in “Green” Synthetic Chemistry: Laccase and Lipase. Molecules 2024, 29, 989. https://doi.org/10.3390/molecules29050989
Scheibel DM, Gitsov IPI, Gitsov I. Enzymes in “Green” Synthetic Chemistry: Laccase and Lipase. Molecules. 2024; 29(5):989. https://doi.org/10.3390/molecules29050989
Chicago/Turabian StyleScheibel, Dieter M., Ioan Pavel Ivanov Gitsov, and Ivan Gitsov. 2024. "Enzymes in “Green” Synthetic Chemistry: Laccase and Lipase" Molecules 29, no. 5: 989. https://doi.org/10.3390/molecules29050989
APA StyleScheibel, D. M., Gitsov, I. P. I., & Gitsov, I. (2024). Enzymes in “Green” Synthetic Chemistry: Laccase and Lipase. Molecules, 29(5), 989. https://doi.org/10.3390/molecules29050989