
Synthesis and Morphology Characteristics of New Highly Branched Polycaprolactone PCL

 , , , , , and
, , , , , and
Abstract
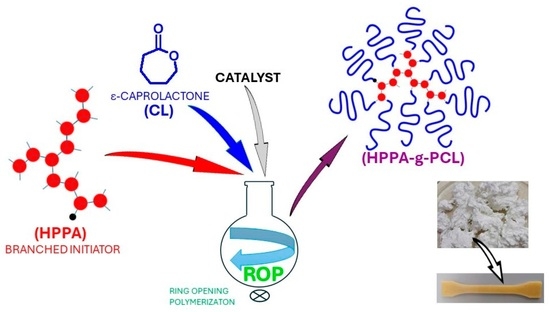
1. Introduction
2. Results
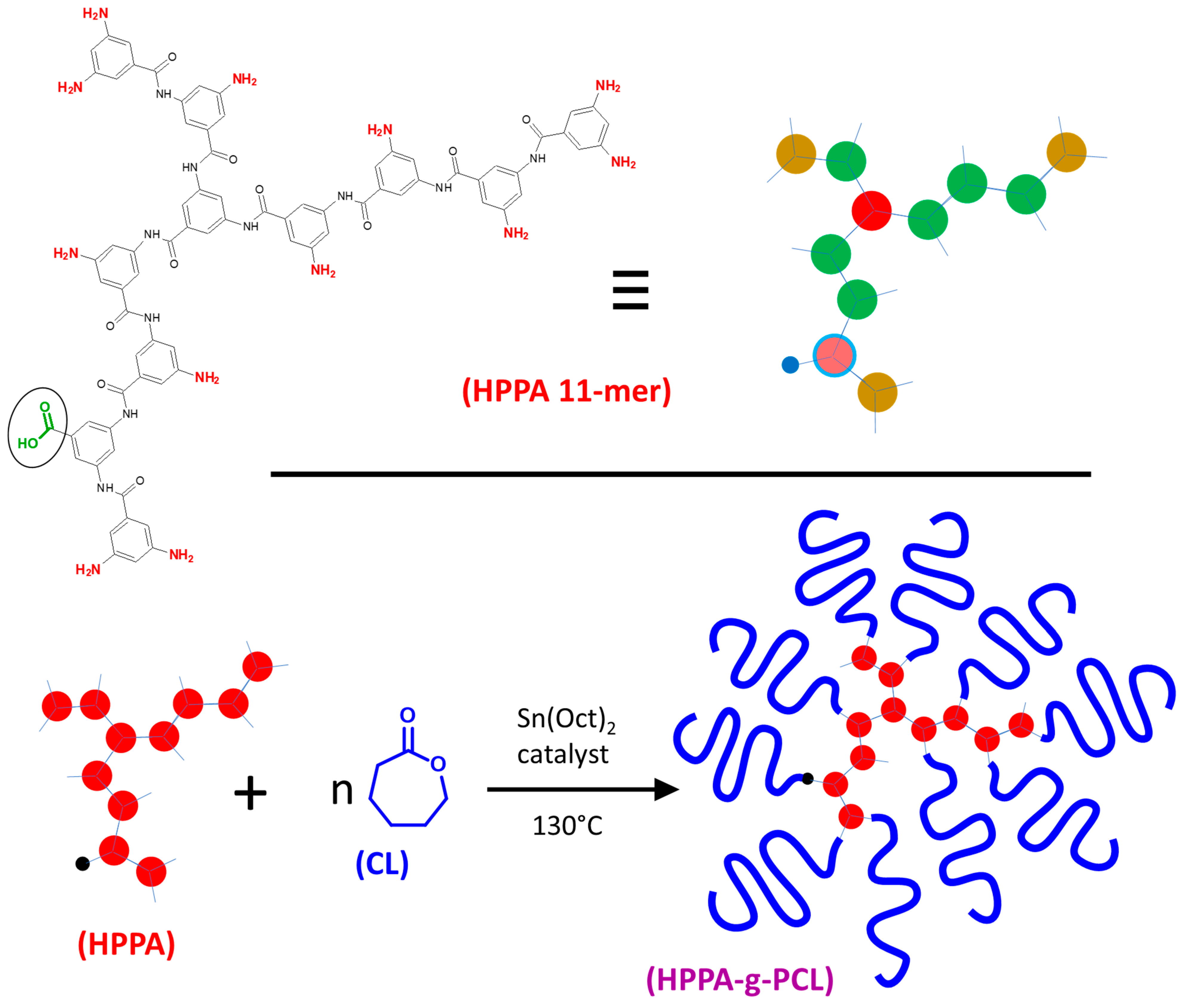
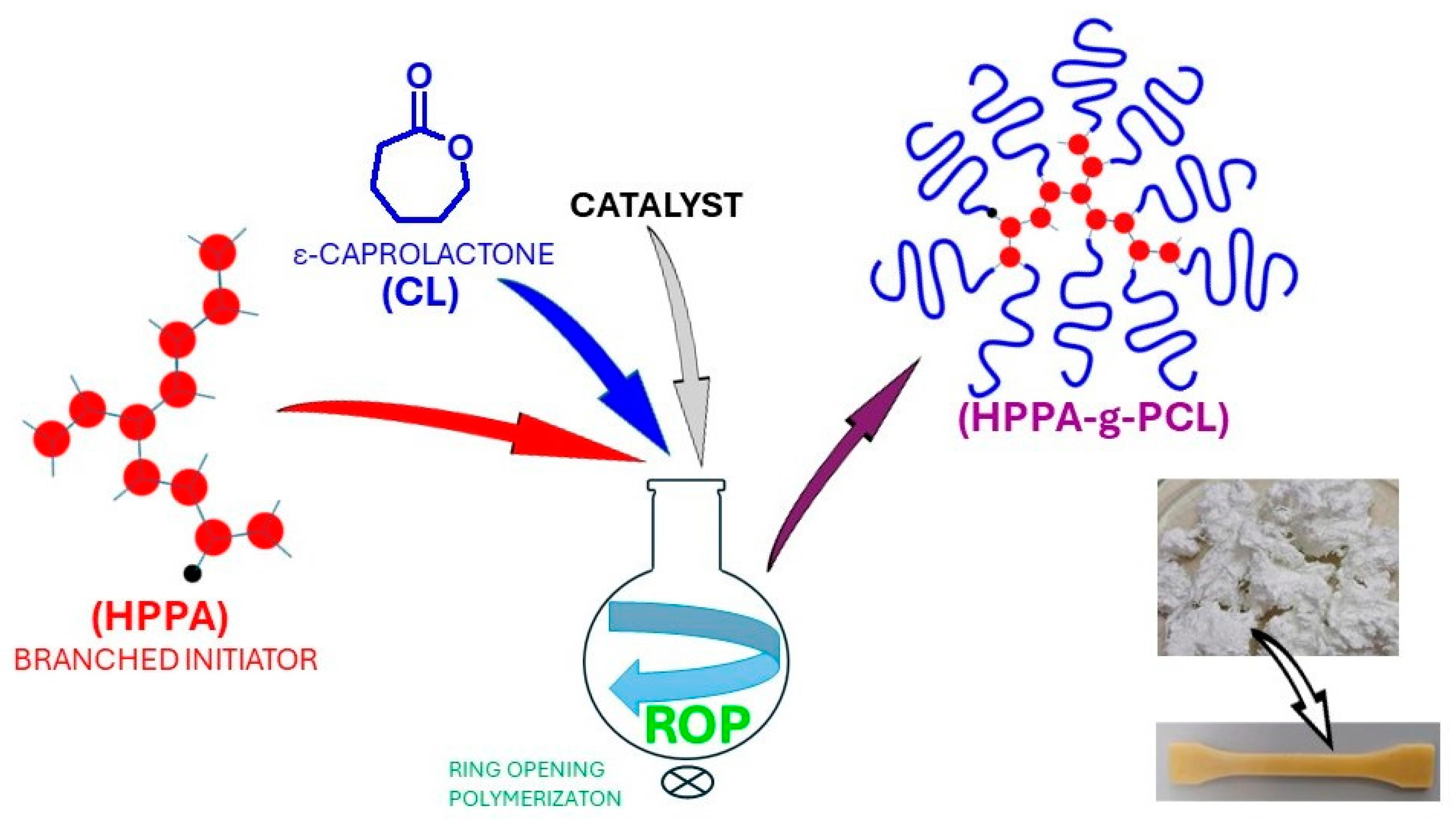
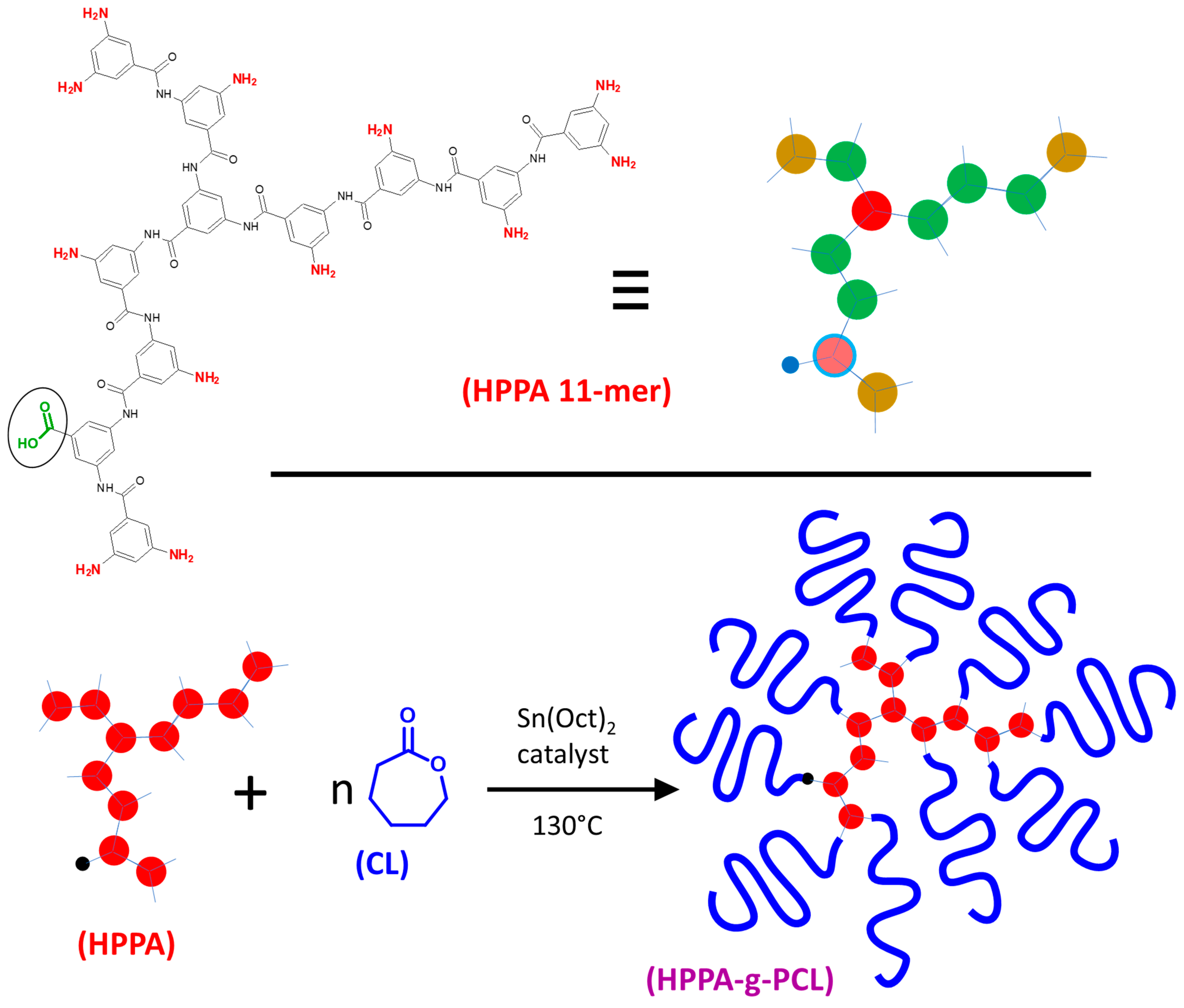
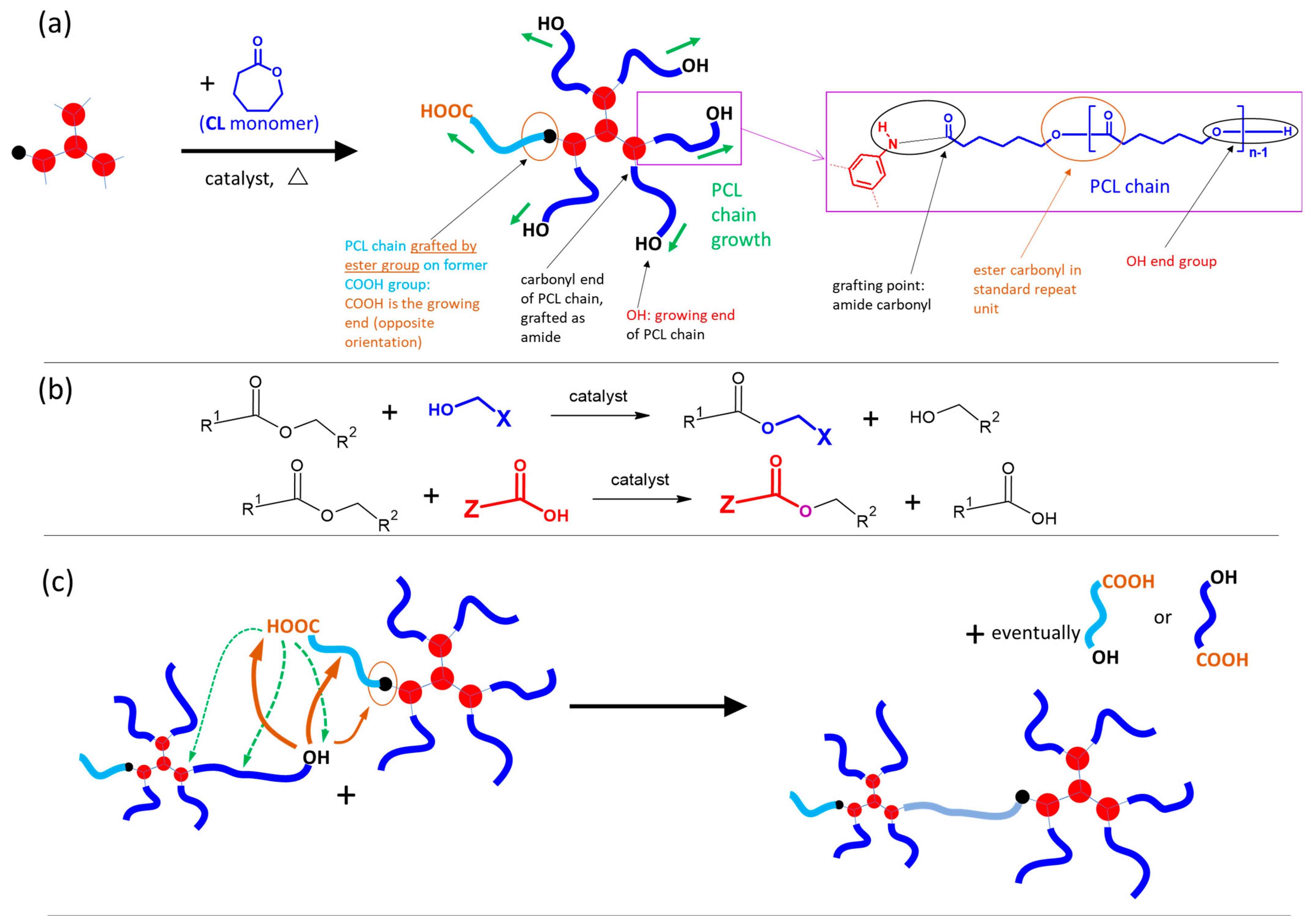
2.1. Synthesis and Basic Characterization of the Highly Branched PCL Polymers
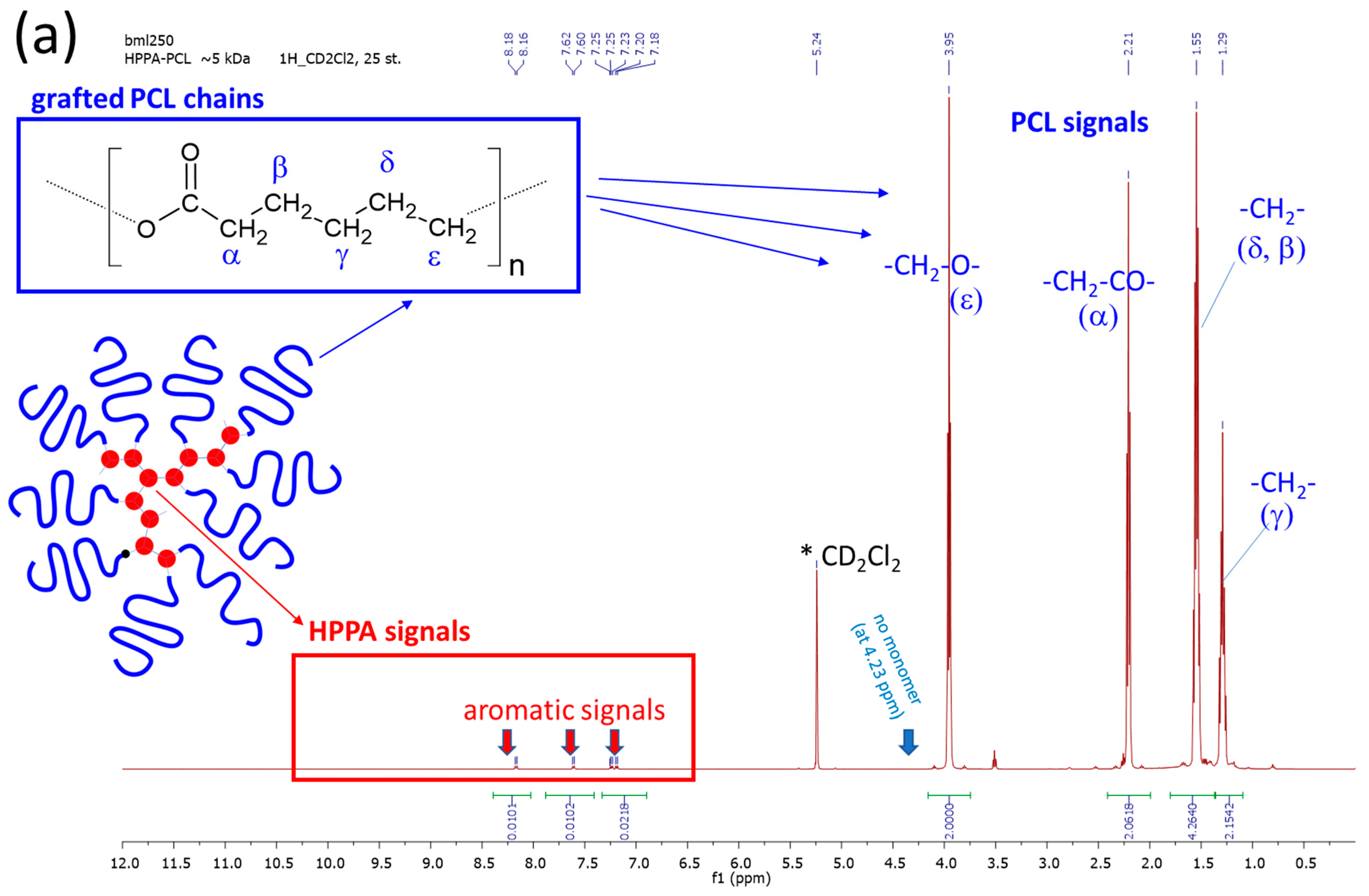
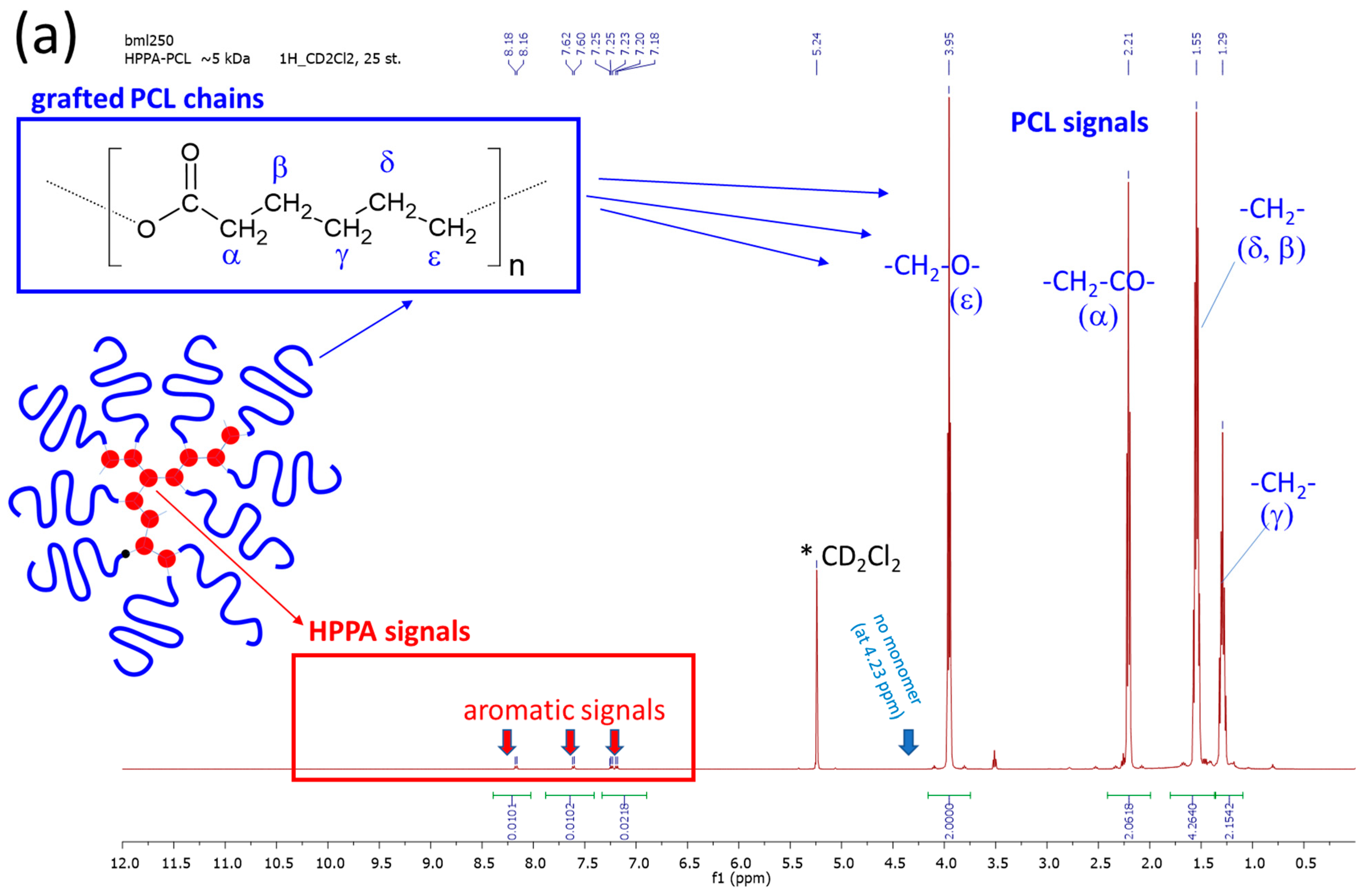
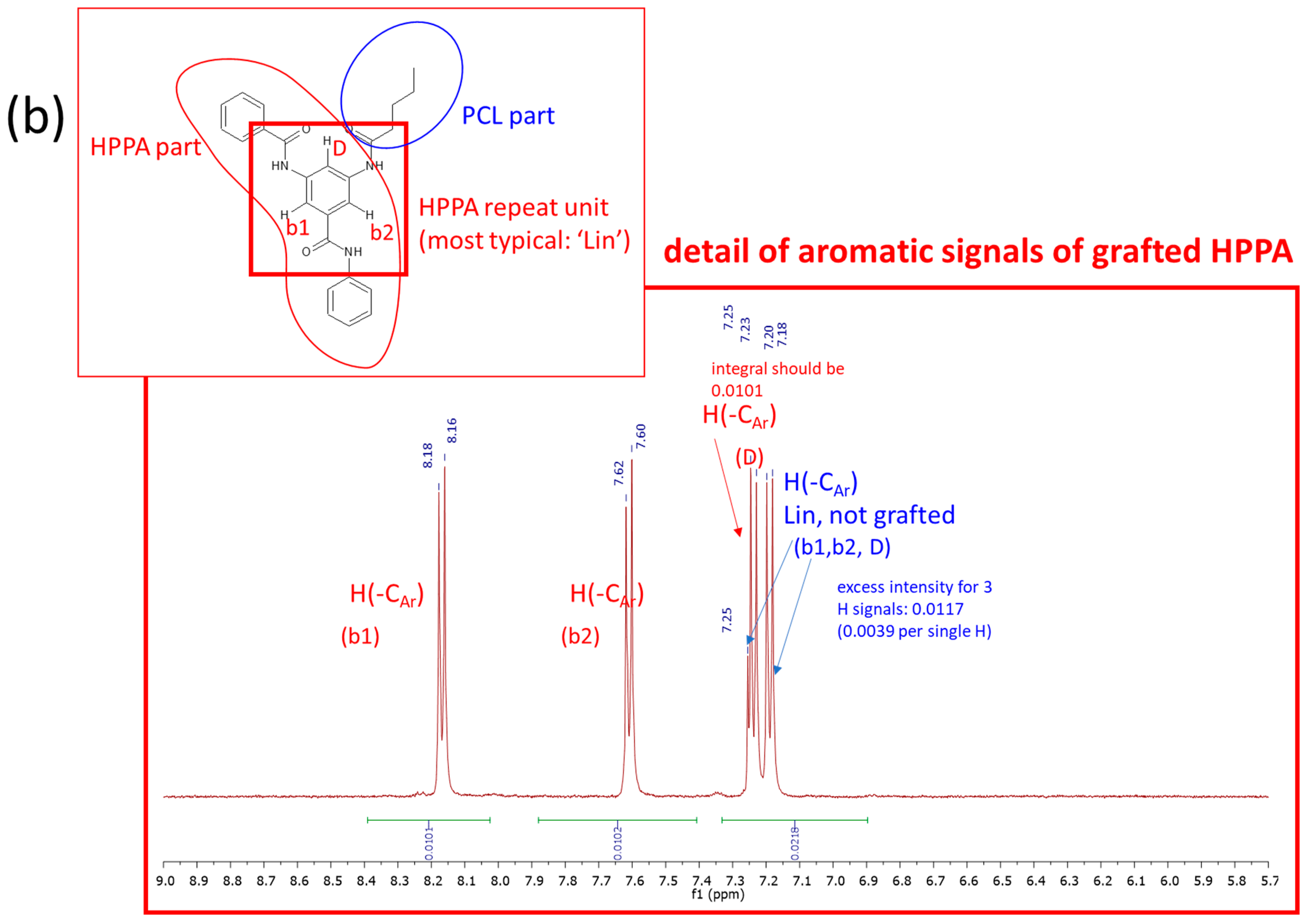
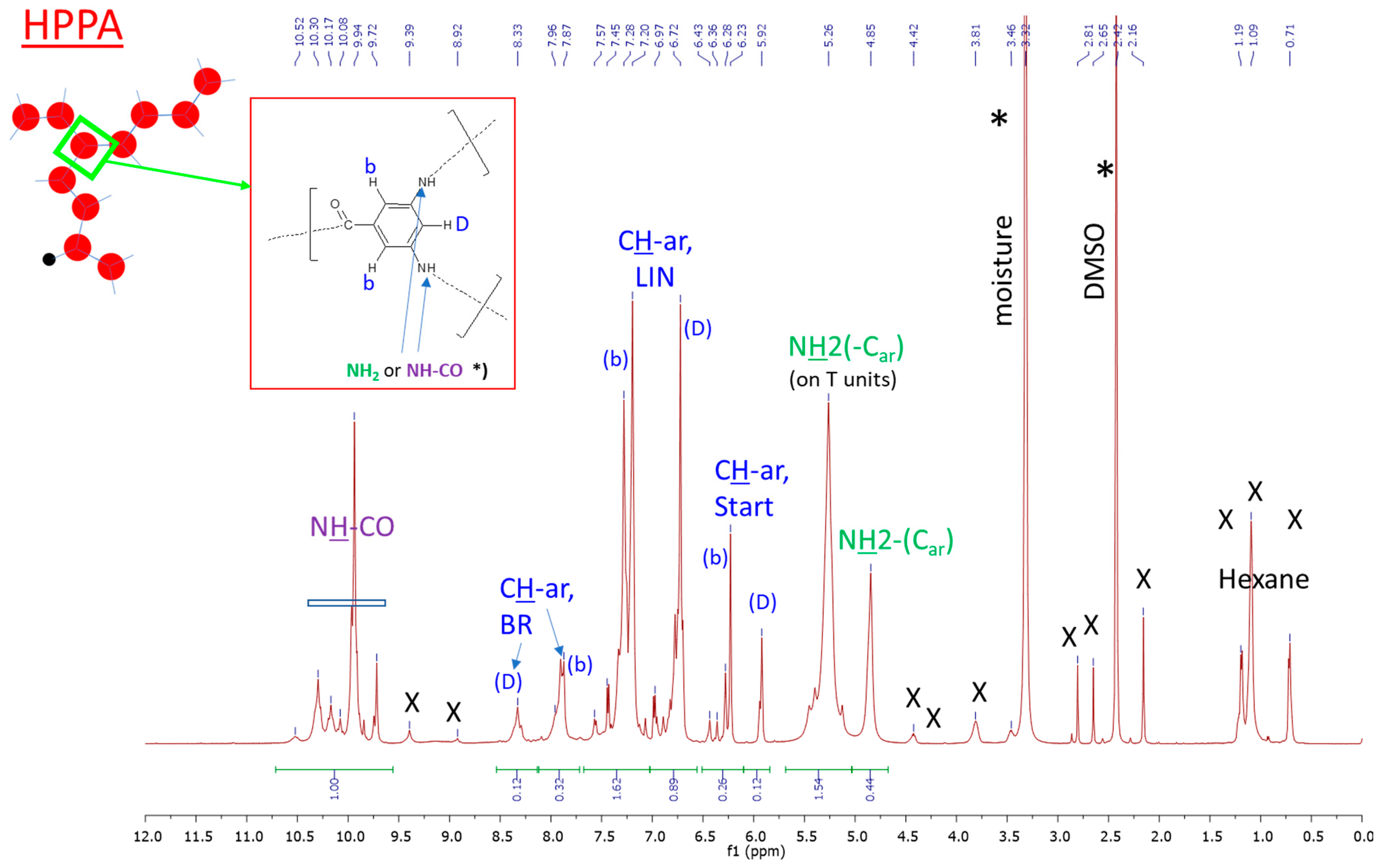
2.1.1. Primary Structure Analyzed by Means of 1H NMR and FTIR Spectroscopy
2.1.2. Nitrogen from HPPA Proven by Elemental Analysis
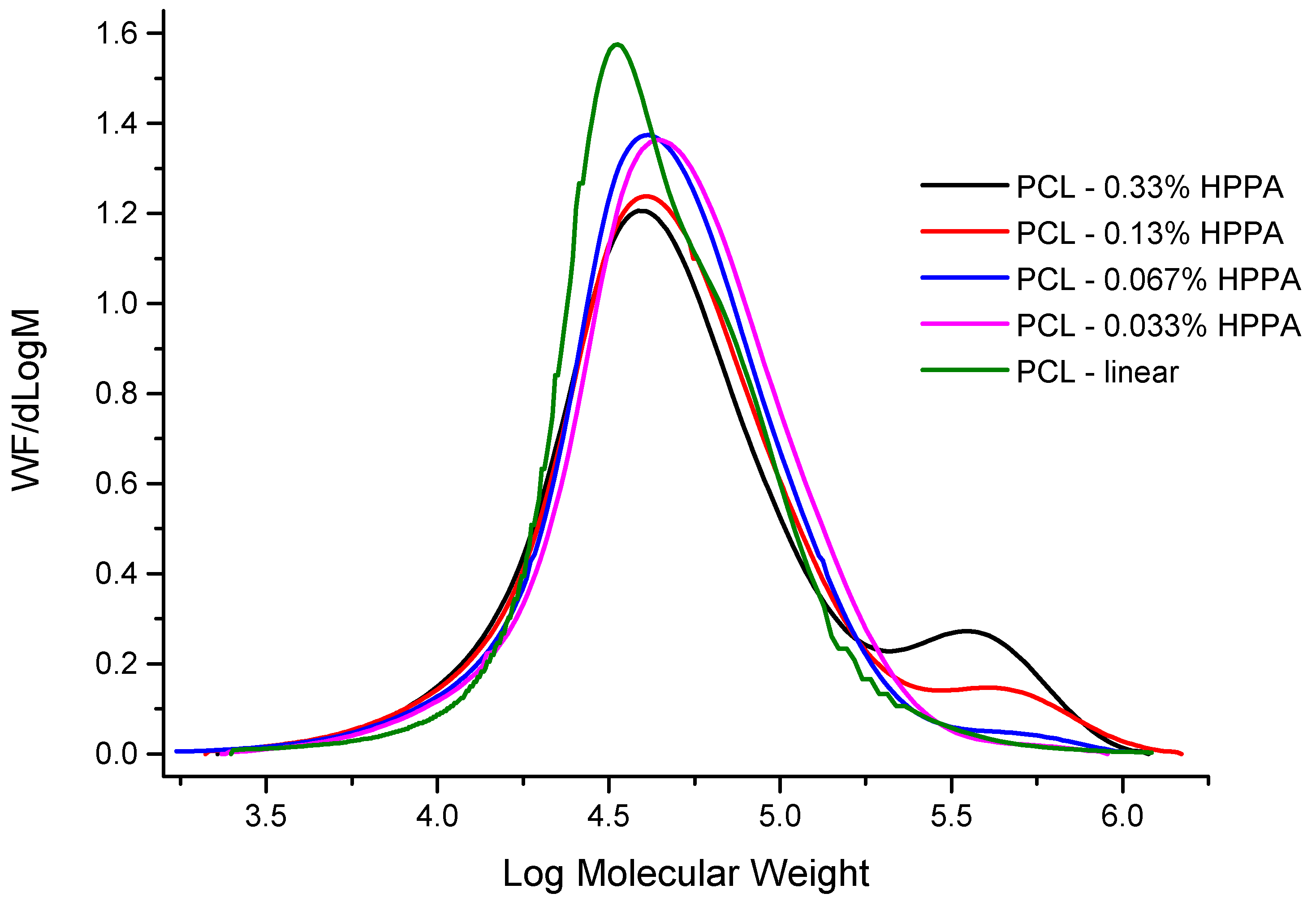
2.1.3. Molecular Masses and Their Distribution
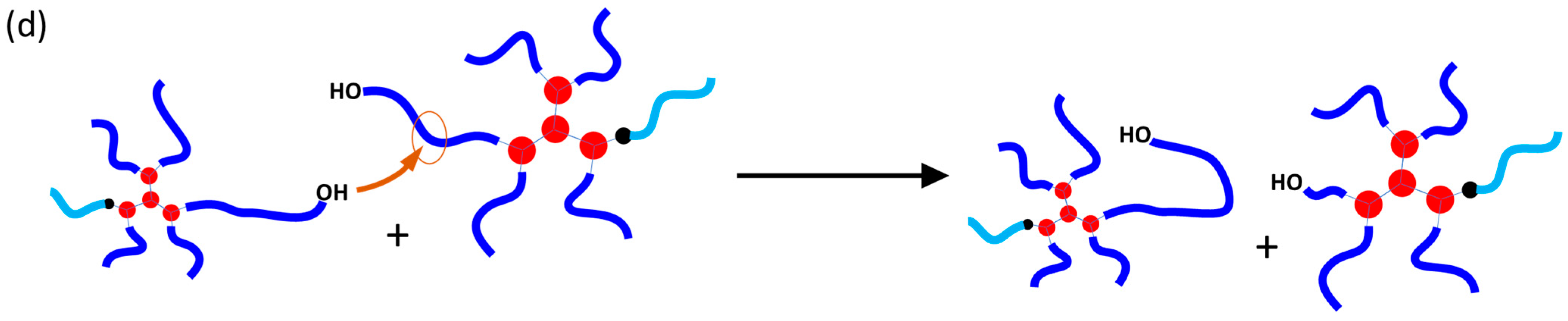
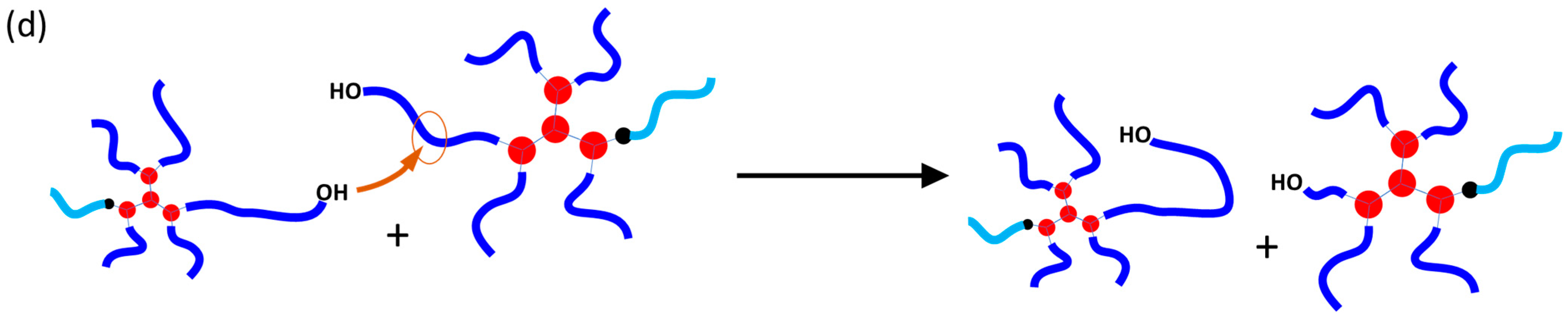
2.2. Proving the Covalent Incorporation of the HPPA Initiator into the PCL Structure
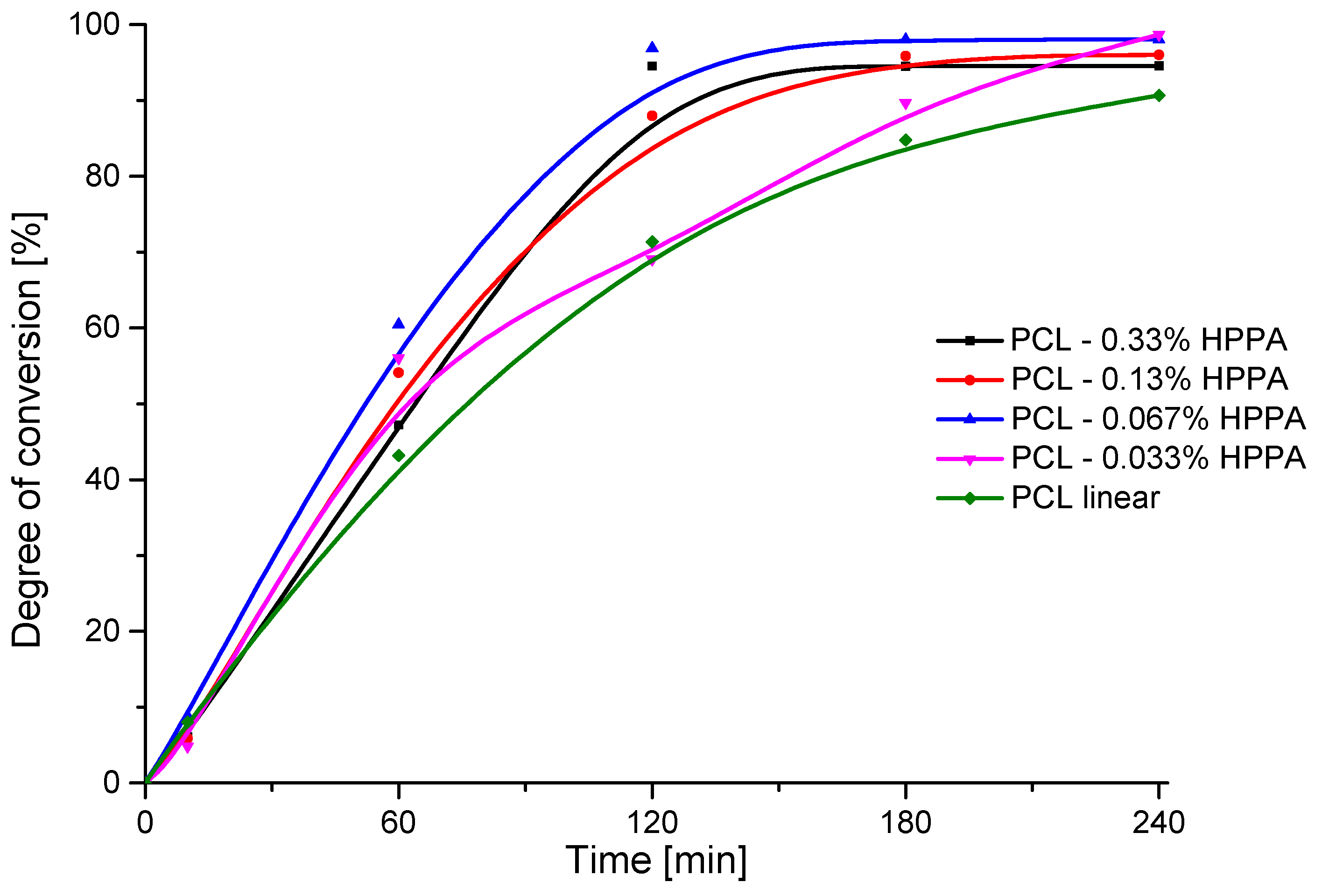
2.2.1. Kinetics of the HPPA-Initiated Polymerization
2.2.2. Core-Shell Structure and Nitrogen Presence Evaluated by ToF—SIMS
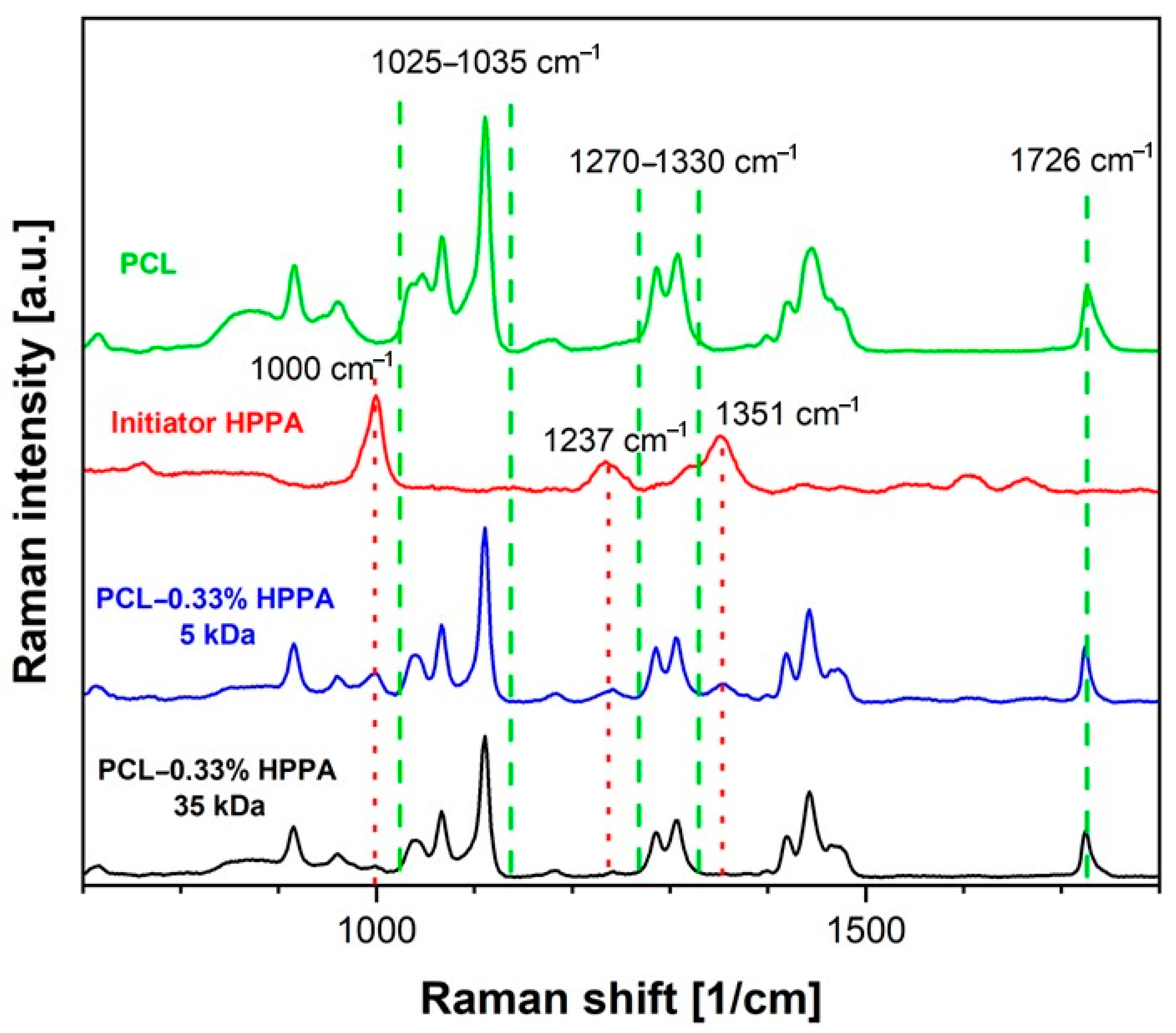
2.2.3. Raman Spectroscopy as a Method to Detect the HPPA Core
2.3. Shape and Size of the Highly Branched PCL Macromolecules
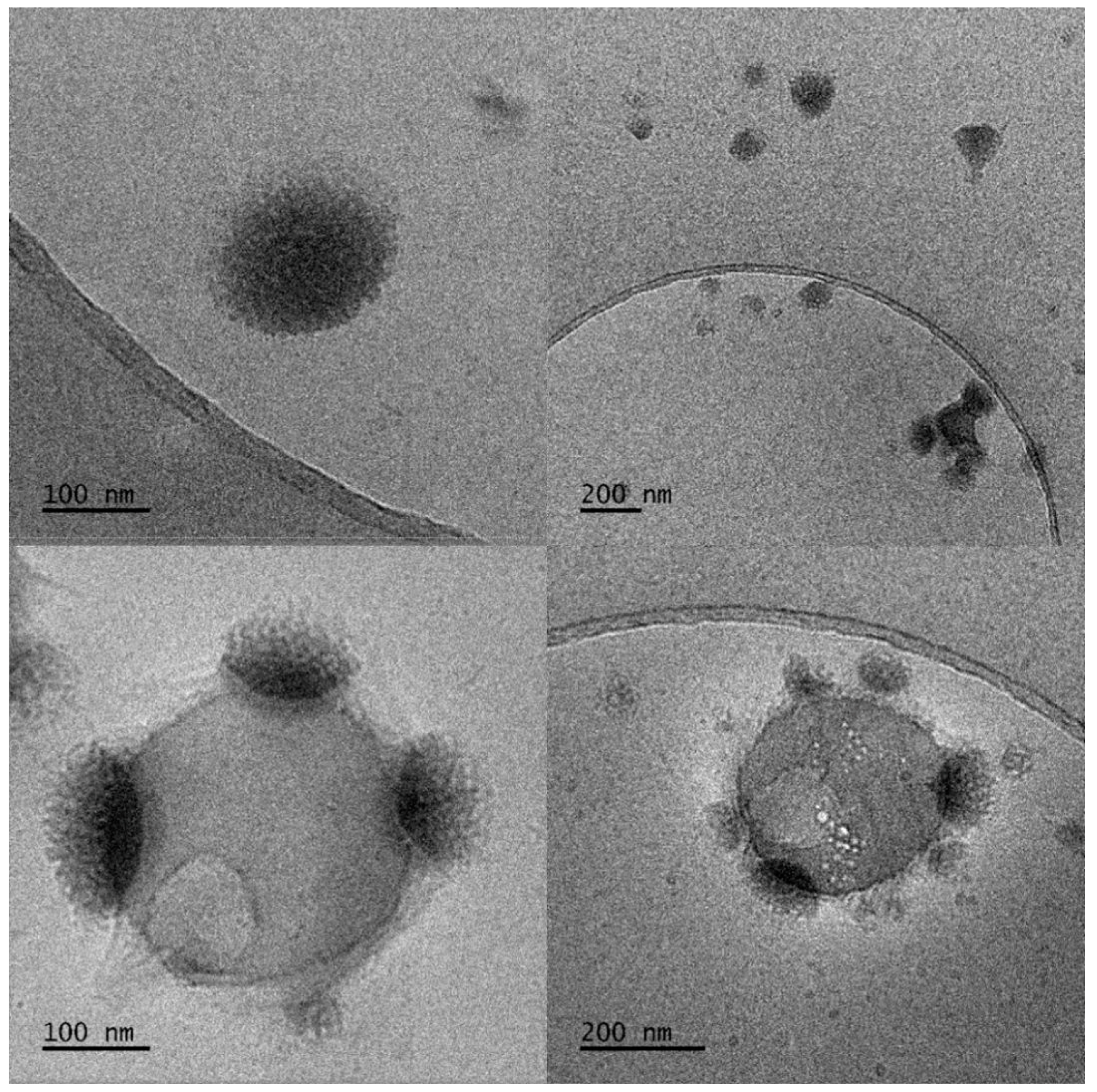
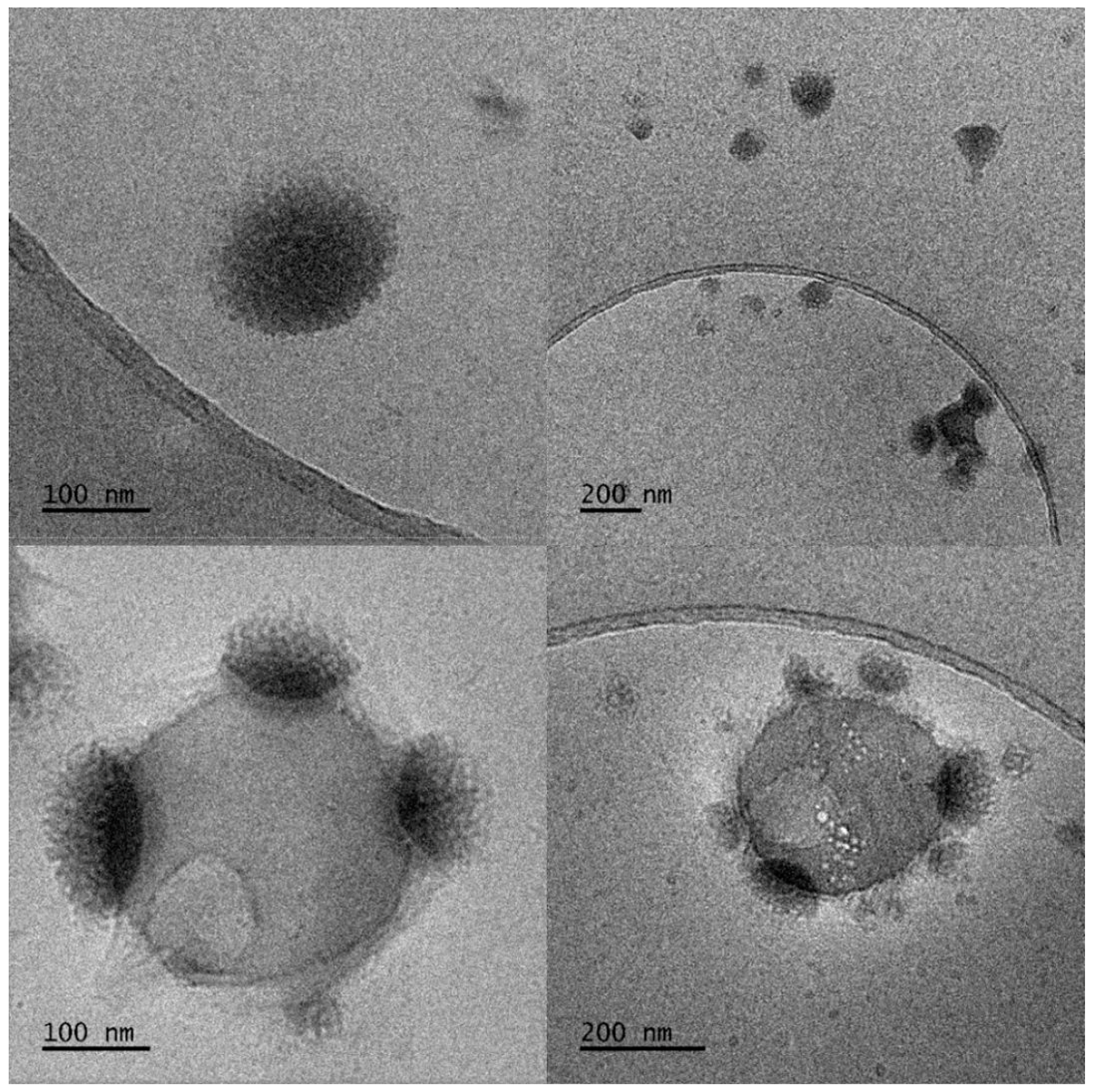
2.3.1. Observation by Cryogenic Transmission Electron Microscopy (Cryo-TEM)
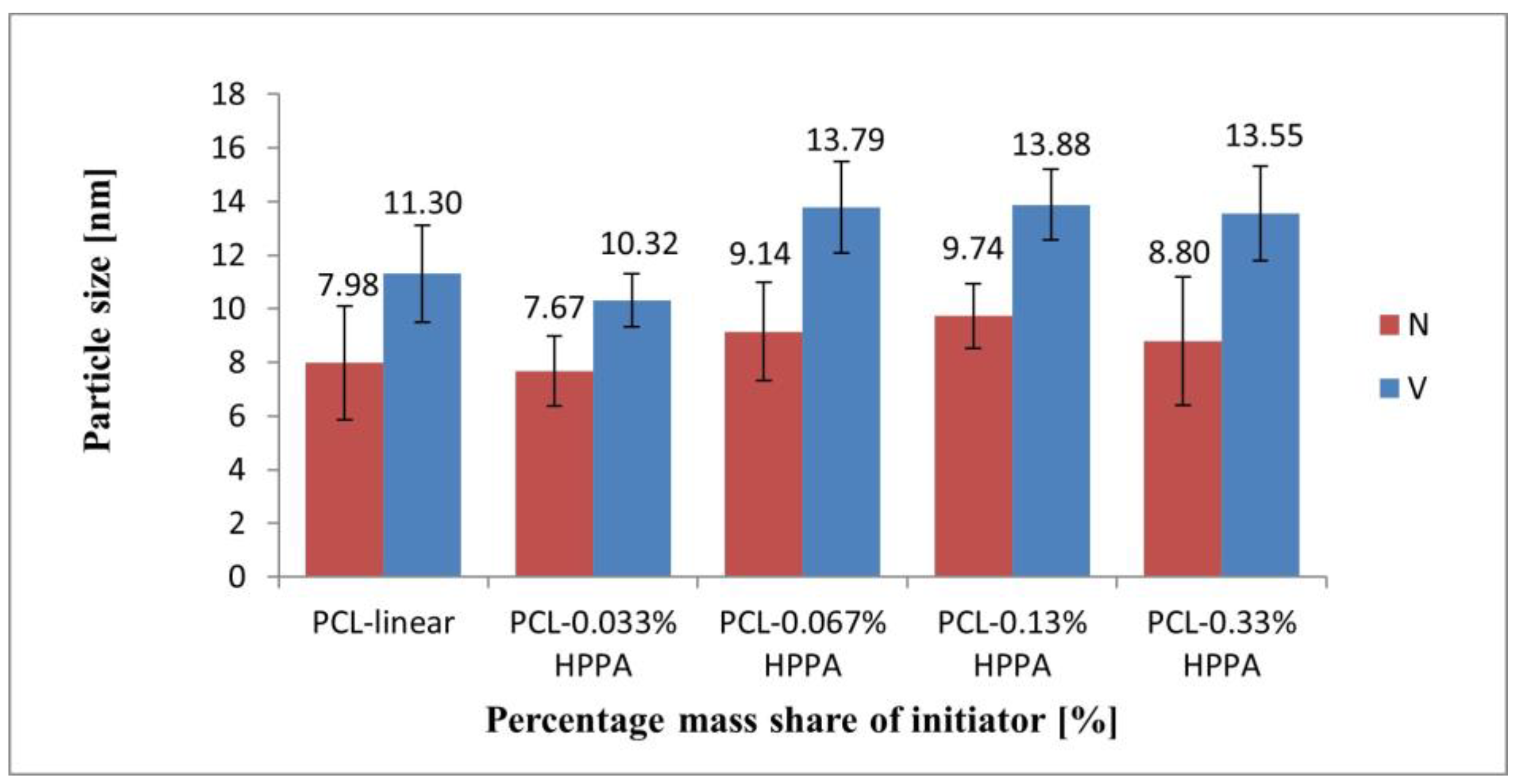
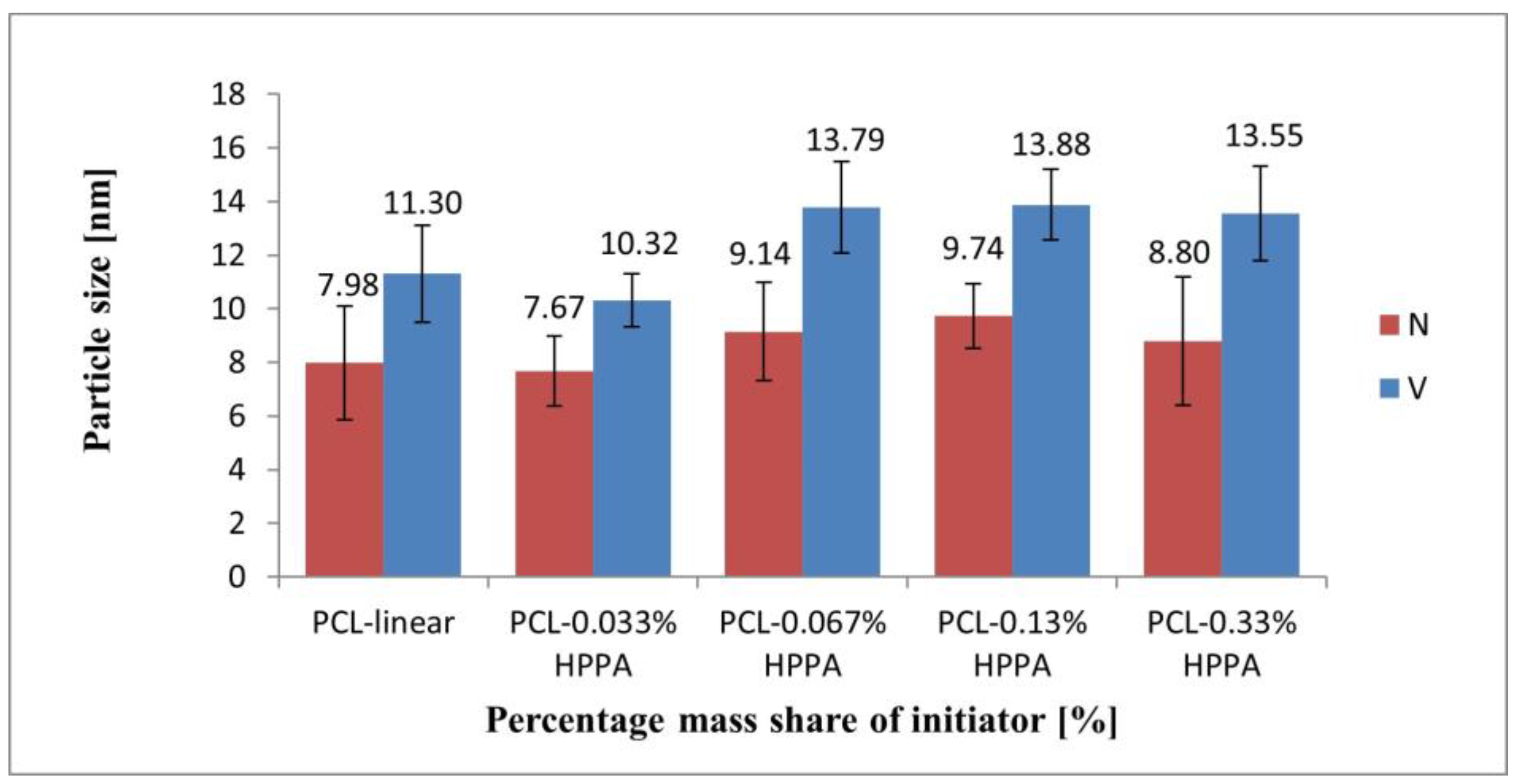
2.3.2. Size of Macromolecules as Observed by DLS
2.4. Phase Behavior of the Products
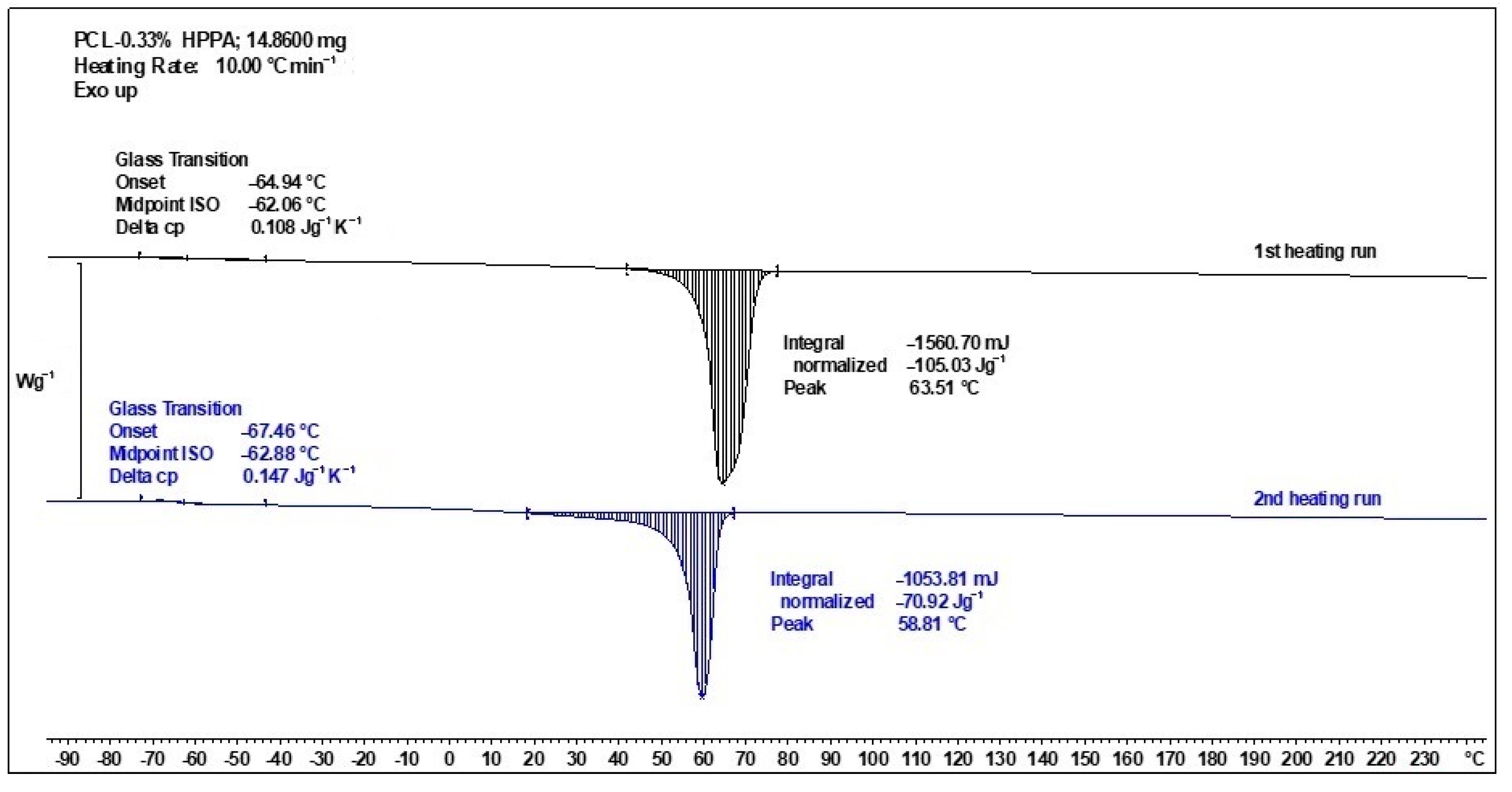
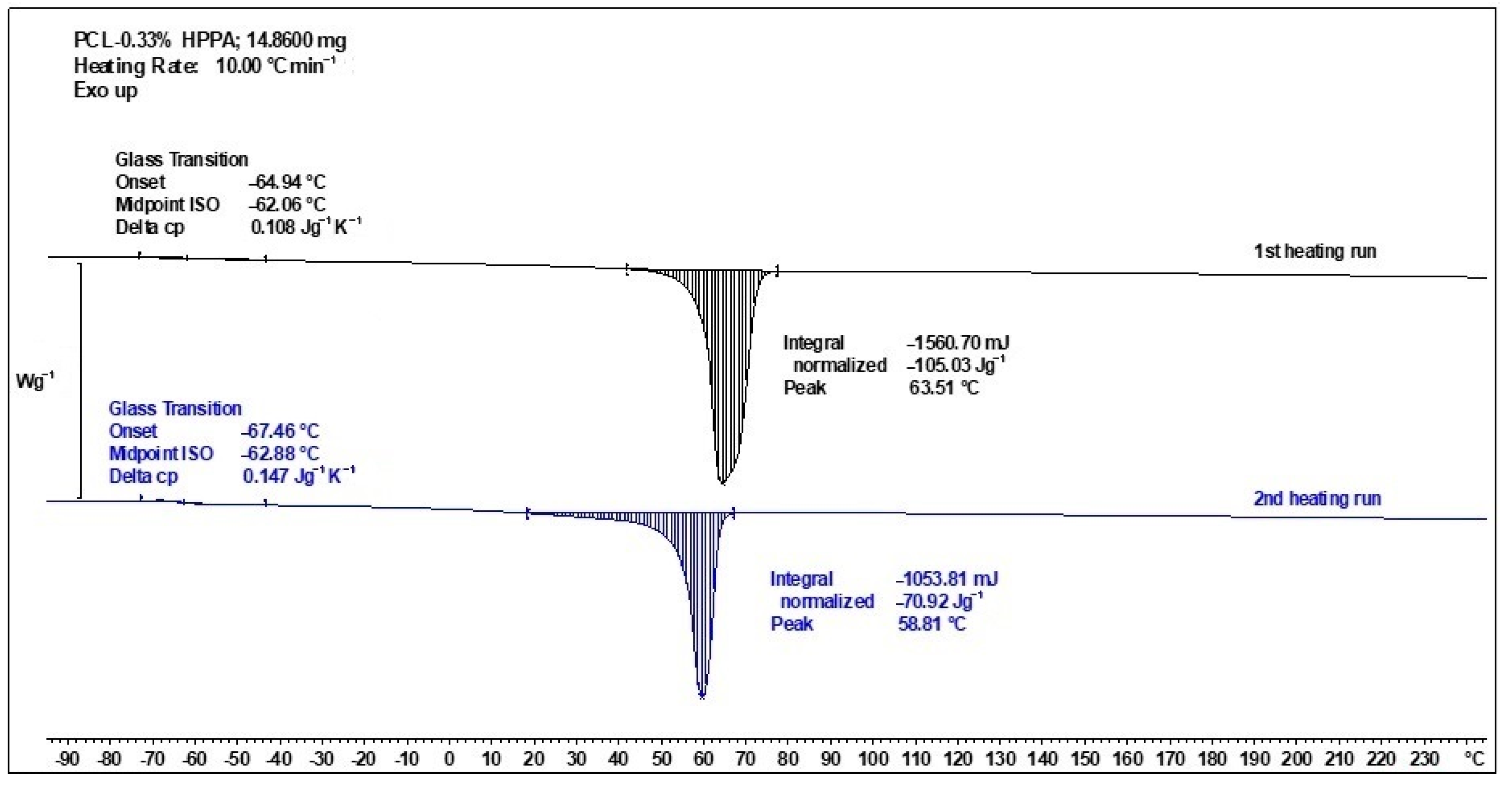
2.4.1. Thermal Transitions Observed by DSC
- Cr—degree of crystallinity
- ΔH—enthalpy of melting of the tested PCL
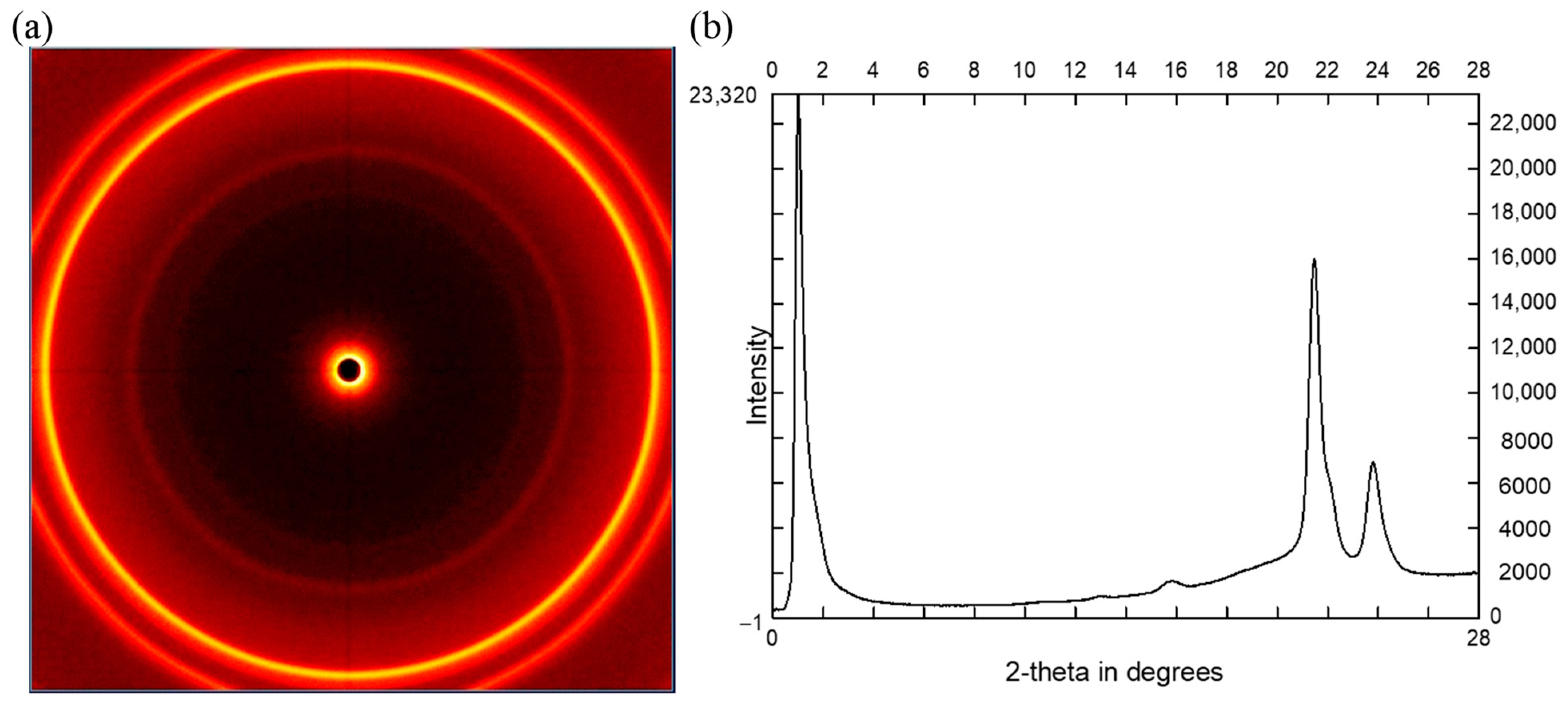
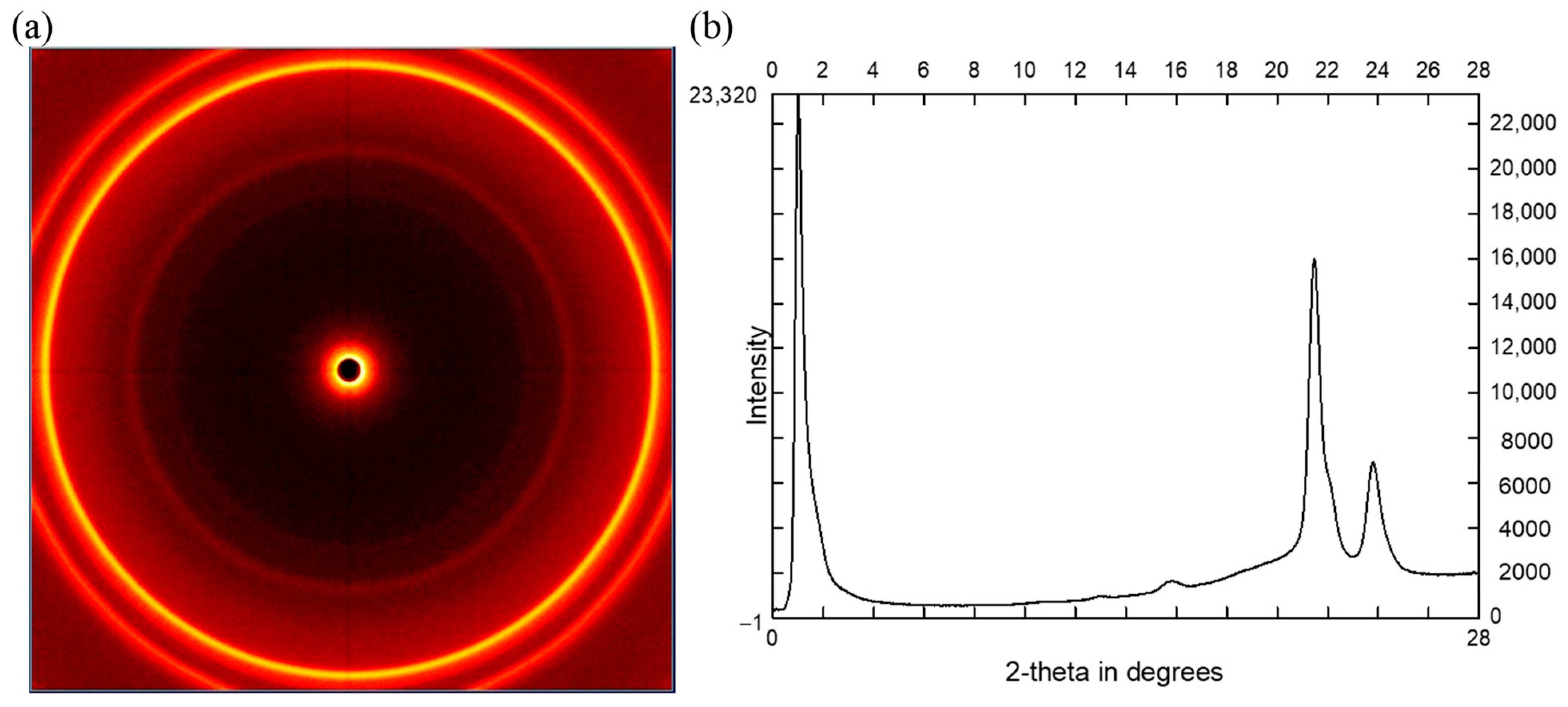
2.4.2. Crystallinity Observed by XRD
2.4.3. Mechanical Properties—Tensile Strength
3. Materials and Methods
3.1. Starting Materials
3.2. Synthesis of PCL
3.3. Characterization
3.3.1. 1H Nuclear Magnetic Resonance—1H NMR Spectroscopy
3.3.2. Carbon-13 Nuclear Magnetic Resonance—13C NMR
3.3.3. Kinetics Analysis by Means of 1H NMR
- P—the integration value of the signal coming from the polymer
- M—the integration value of the signal coming from the monomer
- p—degree of conversion
3.3.4. FT-IR Spectroscopy
3.3.5. Gel Permeation Chromatography—GPC
3.3.6. Dynamic Light Scattering—DLS
3.3.7. Differential Scanning Calorimetry—DSC
3.3.8. X-ray Diffraction—XRD
3.3.9. Cryogenic Transmission Electron Microscopy—Cryo-TEM
3.3.10. Time-of-Flight Secondary Ion Mass Spectrometry—ToF-SIMS
3.3.11. Raman Spectroscopy
3.3.12. Elemental Analysis
3.3.13. Mechanical Properties—Tensile Strength
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bhat, S.I.; Ahmadi, Y.; Ahmad, S. Recent advances in structural modifications of hyperbranched polymers and their applications. Ind. Eng. Chem. Res. 2018, 32, 10754–10785. [Google Scholar] [CrossRef]
- Wang, H.-B.; Chen, X.-S.; Pan, C.-Y. Synthesis and micellization of star-like hyperbranched polymer with poly(ethylene oxide) and poly(e-caprolactone) arms. J. Polym. Sci. Part A Polym. Chem. 2008, 46, 1388–1401. [Google Scholar] [CrossRef]
- Hölter, D.; Burgath, A.; Frey, H. Degree of branching in hyperbranched polymers. Acta Polymer. 1997, 48, 30–35. [Google Scholar] [CrossRef]
- Banaei, M.; Salami-Kalajahi, M. A “grafting to” approach to synthesize low cytotoxic poly(aminoamide)-dendrimer-grafted Fe3O4 magnetic nanoparticles. Adv. Polym. Technol. 2016, 37, 943–948. [Google Scholar] [CrossRef]
- Yang, X.; Kuang, Z.; Yang, X.; Hu, X.; Luo, P.; Lai, Q.; Zhang, B.; Zhang, X.; Wei, Y. Facile synthesis of curcumin-containing poly(amidoamine) dendrimers as pH-responsive delivery system for osteoporosis treatment. Colloids Surf. B 2023, 222, 113029. [Google Scholar] [CrossRef] [PubMed]
- Golshan, M.; Gheitarani, B.; Salami-Kalajahi, M.; Salami-Hosseini, M. Synthesis and characterization of fuorescence poly(amidoamine) dendrimer-based pigments. Sci. Rep. 2022, 12, 15180. [Google Scholar] [CrossRef] [PubMed]
- Neelgund, G.M.; Aguilar, S.F.; Kurkuri, M.D.; Rodrigues, D.F.; Ray, R.L. Elevated adsorption of lead and arsenic over silver nanoparticles deposited on poly(amidoamine) grafted carbon nanotubes. Nanomaterials 2022, 12, 3852. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Yan, D. Hyperbranched polymers: From synthesis to applications. Prog. Polym. Sci. 2004, 29, 183–275. [Google Scholar] [CrossRef]
- Cramail, H.; Testud, B.; Grau, E.; Taton, D.; Pintori, D. Hyperbranched polyesters by polycondensation of fatty acid-based abn-type monomers. Green Chem. 2017, 19, 259–269. [Google Scholar] [CrossRef]
- Istratov, V.V.; Polezhaev, A.V. Branched copolylactides: The effect of the synthesis method on their properties. J. Phys. Conf. Ser. 2021, 1990, 012046. [Google Scholar] [CrossRef]
- Moradi, L.G.; Ganjaee Sari, M.; Ramezanzadeh, B. Polyester-amide hyperbranched polymer as an interfacial modifier for graphene oxide nanosheets: Mechanistic approach in an epoxy nanocomposite coating. Prog. Org. Coat. 2020, 142, 105573. [Google Scholar] [CrossRef]
- Korake, S.; Shaikh, A.; Salve, R.; Gajbhiye, K.R.; Gajbhiye, V.; Pawar, A. Biodegradable dendritic Boltorn™ nanoconstructs: A promising avenue for cancer theranostics. Int. J. Pharm. 2021, 594, 120177. [Google Scholar] [CrossRef]
- Lee, K.W.; Chung, J.W.; Kwak, S.-Y. Highly branched polycaprolactone/glycidol copolymeric green plasticizer by one-pot solvent-free polymerization. ACS Sustain. Chem. Eng. 2018, 6, 9006–9017. [Google Scholar] [CrossRef]
- Labet, M.; Thielemans, W. Synthesis of polycaprolactone: A review. Chem. Soc. Rev. 2009, 38, 3484–3504. [Google Scholar] [CrossRef]
- Nukala, S.G.; Kong, I.; Patel, V.I.; Kakarla, A.B.; Kong, W.; Buddrick, O. Development of Biodegradable Composites Using Polycaprolactone and Bamboo Powder. Polymers 2022, 14, 4169. [Google Scholar] [CrossRef]
- Paredes, C.; Martínez-Vazquez, F.J.; Pajares, A.; Miranda, P. Co-continuous calcium phosphate/polycaprolactone composite bone scaffolds fabricated by digital light processing and polymer melt suction. Ceramics Int. 2021, 47, 17726–17735. [Google Scholar] [CrossRef]
- Castro, J.I.; Astudillo, S.; Hernandez, J.H.M.; Saavedra, M.; Zapata, P.A.; Valencia-Llano, C.H.; Chaur, M.N.; Grande-Tovar, C.D. Synthesis, characterization, and optimization studies of polycaprolactone/polylactic acid/titanium dioxide nanoparticle/orange essential oil membranes for biomedical applications. Polymers 2023, 15, 135. [Google Scholar] [CrossRef]
- Walczak, M.; Lubczak, J. ε-caprolactone and pentaerythritol derived oligomer for rigid polyurethane foams preparation. J. Cell. Plast. 2022, 58, 757–775. [Google Scholar] [CrossRef]
- Lu, C.; Liu, L.; Guo, S.-R.; Zhang, Y.; Li, Z.; Gu, J. Micellization and gelation of aqueous solutions of star-shaped PEG–PCL block copolymers consisting of branched 4-arm poly(ethylene glycol) and polycaprolactone blocks. Eur. Polym. J. 2007, 43, 1857–1865. [Google Scholar] [CrossRef]
- Hedrick, J.L.; Magbitang, T.; Connor, E.F.; Glauser, T.; Volksen, W.; Hawker, C.J.; Lee, V.Y.; Miller, R.D. Application of complex macromolecular architectures for advanced microelectronic materials. Chem. Eur. J. 2002, 8, 3308–3319. [Google Scholar] [CrossRef] [PubMed]
- Zhan, C.; Li, S.; Cui, J.; Chen, Y. Multiarm star poly(ε-caprolactone) with hyperbranched polyamidoamine as core capable of selective accommodating cationic or anionic guests. Chin. J. Polym. Sci. 2015, 33, 920–930. [Google Scholar] [CrossRef]
- Ryczek, A.; Walczak, M.; Galina, H. Hyperbranched polyamides of 3,5-diaminobenzoic acid. Polimery 2014, 59, 682–686. [Google Scholar] [CrossRef]
- Gurlek, A.C.; Sevinc, B.; Bayrak, E.; Erisken, C. Synthesis and characterization of polycaprolactone for anterior cruciate ligament regeneration. Mater. Sci. Eng. C 2017, 71, 820–826. [Google Scholar] [CrossRef] [PubMed]
- Lipik, V.T.; Widjaja, L.K.; Liow, S.S.; Abadie, M.J.M.; Venkatraman, S.S. Effects of transesterification and degradation on properties and structure of polycaprolactoneepolylactide copolymers. Polym. Degrad. Stab. 2010, 95, 2596–2602. [Google Scholar] [CrossRef]
- Yan, D.; Zhou, Z.; Müller, A.H.E. Molecular Weight Distribution of Hyperbranched Polymers Generated by Self-Condensing Vinyl Polymerization in Presence of a Multifunctional Initiator. Macromolecules 1999, 32, 245–250. [Google Scholar] [CrossRef]
- Voit, B.I.; Lederer, A. Hyperbranched and highly branched polymer architectures-synthetic strategies and major characterization aspects. Chem. Rev. 2009, 109, 5924–5973. [Google Scholar] [CrossRef] [PubMed]
- Awsiuk, K.; Budkowski, A.; Marzec, M.M.; Petrou, P.; Rysz, J.; Bernasik, A. Effects of polythiophene surface structure on adsorption and conformation of bovine serum albumin: A multivariate and multitechnique study. Langmuir 2014, 30, 13925–13933. [Google Scholar] [CrossRef] [PubMed]
- Baranowska-Korczyc, A.; Warowicka, A.; Jasiurkowska-Delaporte, M.; Grześkowiak, B.; Jarek, M.; Maciejewska, B.M.; Jurga-Stopac, J.; Jurga, S. Antimicrobial electrospun poly(ε-caprolactone) scaffolds for gingival fibroblast growth. RSC Adv. 2016, 6, 19647. [Google Scholar] [CrossRef]
- Pezzotti, G. Raman spectroscopy in cell biology and microbiology. J. Raman Spectrosc. 2021, 52, 2348. [Google Scholar] [CrossRef]
- Chen, G.-M.; Zou, T.-M.; Chen, L.; Huang, Y.-P. Crystallization properties of polycaprolactone induced by different hydroxyapatite nano-particles. Asian J. Chem. 2010, 22, 5902–5912. [Google Scholar] [CrossRef]
- Tomaszewski, P.E. Scherrer formula—The century of erroneous practices. Wiad. Chem. 2013, 67, 9–10. [Google Scholar]
- ISO 527-1:2020; Plastics—Determination of Tensile Properties. ISO: Geneva, Switzerland, 2020.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | C [%] | H [%] | N [%] |
|---|---|---|---|
| PCL-linear | 59.83 | 8.230 | 0 |
| PCL-0.33% HPPA—35 kDa | 61.76 | 8.351 | 0.26 |
| PCL-0.33% HPPA—5 kDa | 64.43 | 8.48 | 1.26 |
| HPPA-initiator | 55.33 | 5.247 | 18.25 |
| Sample | Mn [Dalton] | Mw [Dalton] | Mz [Dalton] | Mp [Dalton] | Mw/Mn |
|---|---|---|---|---|---|
| PCL-linear | 28,850 | 45,850 | 86,470 | 33,120 | 1.59 |
| PCL-0.033% HPPA | 37,110 | 67,030 | 126,700 | 45,370 | 1.80 |
| PCL-0.067% HPPA | 34,200 | 67,060 | 157,050 | 41,210 | 1.96 |
| PCL-0.13% HPPA | 36,630 | 88,440 | 270,400 | 42,430 | 2.41 |
| PCL-0.33% HPPA | 35,270 | 98,320 | 286,780 | 39,550 | 2.78 |
| Sample | Polydispersity Index PDI |
|---|---|
| PCL-linear | 0.49 |
| PCL-0.033% HPPA | 0.46 |
| PCL-0.067% HPPA | 0.49 |
| PCL-0.13% HPPA | 0.46 |
| PCL-0.33% HPPA | 0.49 |
| Polymer | Tg [°C] | Tm [°C] | Cr % DSC 1 | Cr % SAXS |
|---|---|---|---|---|
| PCL linear | −59.8 | 63.8 | 73.9 | 74.5 |
| PCL-0.033% HPPA | −61.3 | 60.8 | 79.4 | 73.0 |
| PCL-0.067% HPPA | −62.1 | 60.1 | 77.6 | 72.8 |
| PCL-0.13% HPPA | −65.6 | 64.0 | 78.4 | 72.0 |
| PCL-0.33% HPPA | −62.06 | 63.5 | 77.5 | 75.5 |
| Sample | Emod | Fmax | Elongation at Break | FBr |
|---|---|---|---|---|
| MPa | MPa | % | MPa | |
| PCL-linear | 331.0 | 21.9 | 338.8 | 21.2 |
| PCL-0.033% HPPA | 341.2 | 19.4 | 424.5 | 15.4 |
| PCL-0.067% HPPA | 425.2 | 21.4 | 371.5 | 15.3 |
| PCL-0.13% HPPA | 446.2 | 25.2 | 360.6 | 22.1 |
| PCL-0.33% HPPA | 485.7 | 21.9 | 317.5 | 15.0 |
| Sample | Time [Hours] | Initiator—HPPA Percentage Mass Share [%] | Monomer -Caprolactone CL [mL] | Catalyst Sn(Oct)2 [mL] |
|---|---|---|---|---|
| PCL-linear | 4 | 0 | 21 | 0.03 |
| PCL-0.033% HPPA | 0.033 | |||
| PCL-0.067% HPPA | 0.067 | |||
| PCL-0.13% HPPA | 0.13 | |||
| PCL-0.33% HPPA | 0.33 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zioło, A.; Mossety-Leszczak, B.; Walczak, M.; Strachota, B.; Strachota, A.; Awsiuk, K.; Janiszewska, N.; Raczkowska, J. Synthesis and Morphology Characteristics of New Highly Branched Polycaprolactone PCL. Molecules 2024, 29, 991. https://doi.org/10.3390/molecules29050991
Zioło A, Mossety-Leszczak B, Walczak M, Strachota B, Strachota A, Awsiuk K, Janiszewska N, Raczkowska J. Synthesis and Morphology Characteristics of New Highly Branched Polycaprolactone PCL. Molecules. 2024; 29(5):991. https://doi.org/10.3390/molecules29050991
Chicago/Turabian StyleZioło, Aleksandra, Beata Mossety-Leszczak, Małgorzata Walczak, Beata Strachota, Adam Strachota, Kamil Awsiuk, Natalia Janiszewska, and Joanna Raczkowska. 2024. "Synthesis and Morphology Characteristics of New Highly Branched Polycaprolactone PCL" Molecules 29, no. 5: 991. https://doi.org/10.3390/molecules29050991
APA StyleZioło, A., Mossety-Leszczak, B., Walczak, M., Strachota, B., Strachota, A., Awsiuk, K., Janiszewska, N., & Raczkowska, J. (2024). Synthesis and Morphology Characteristics of New Highly Branched Polycaprolactone PCL. Molecules, 29(5), 991. https://doi.org/10.3390/molecules29050991