
Synthesis and Photolysis Properties of a New Chloroquine Photoaffinity Probe

Abstract
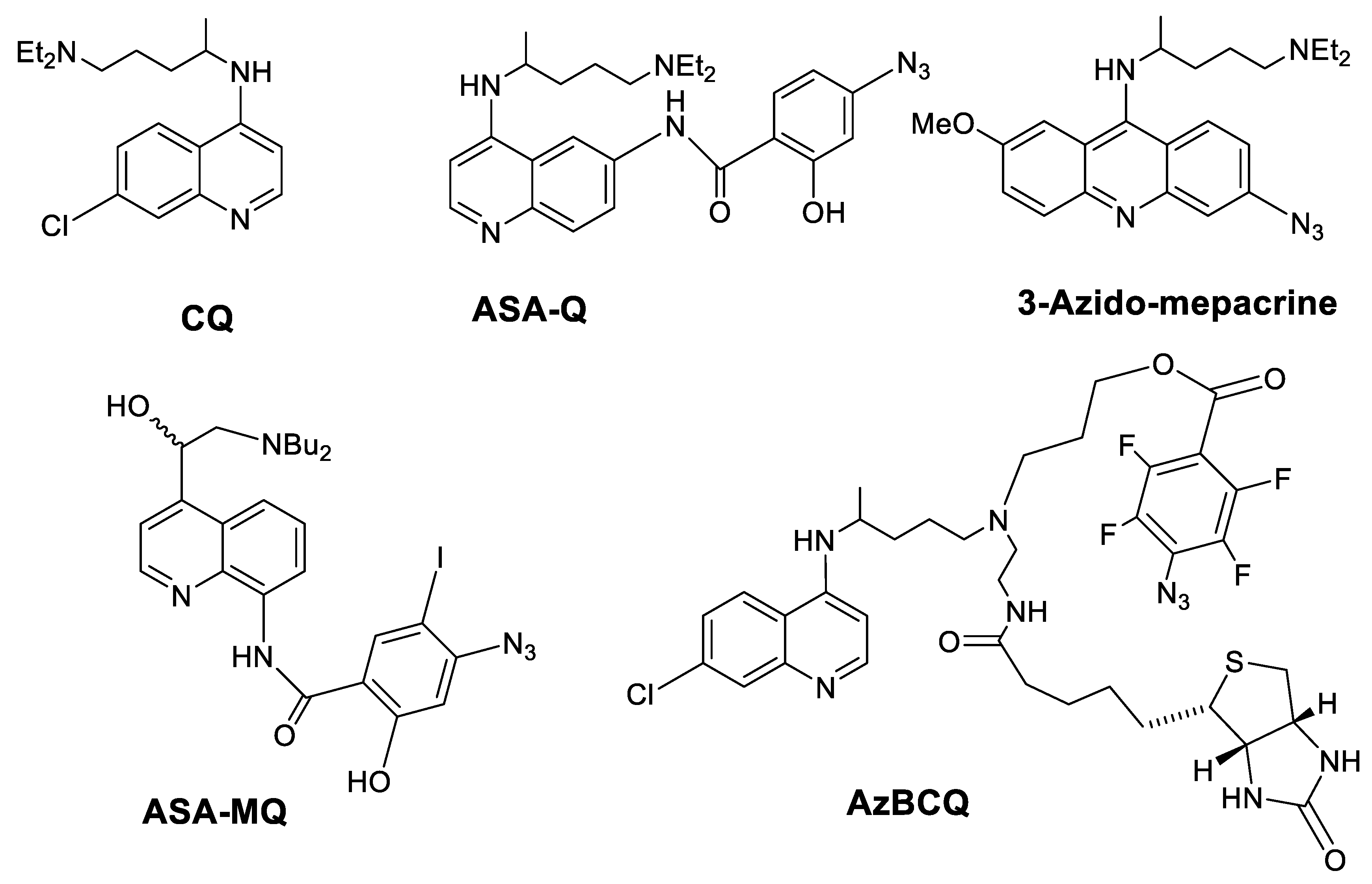
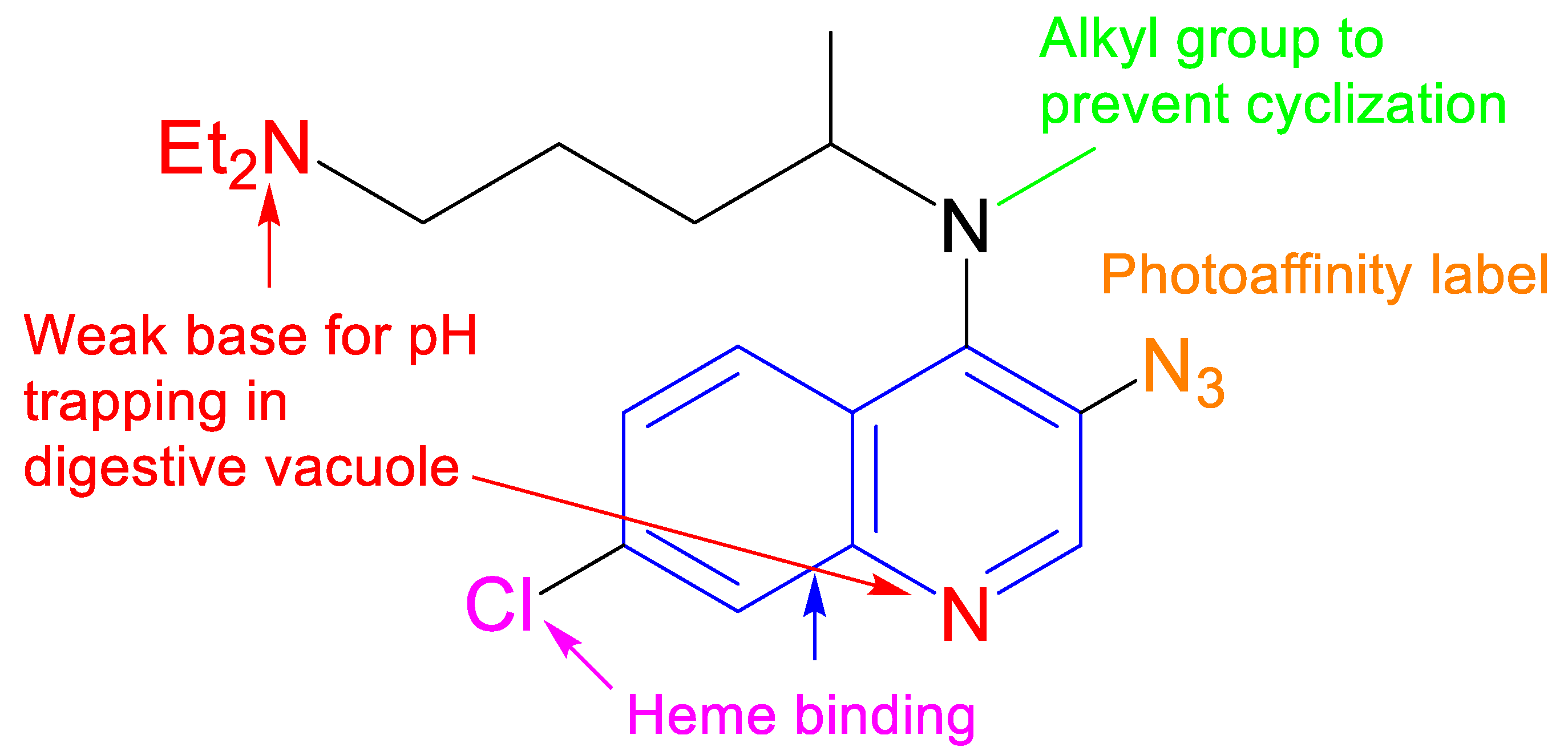
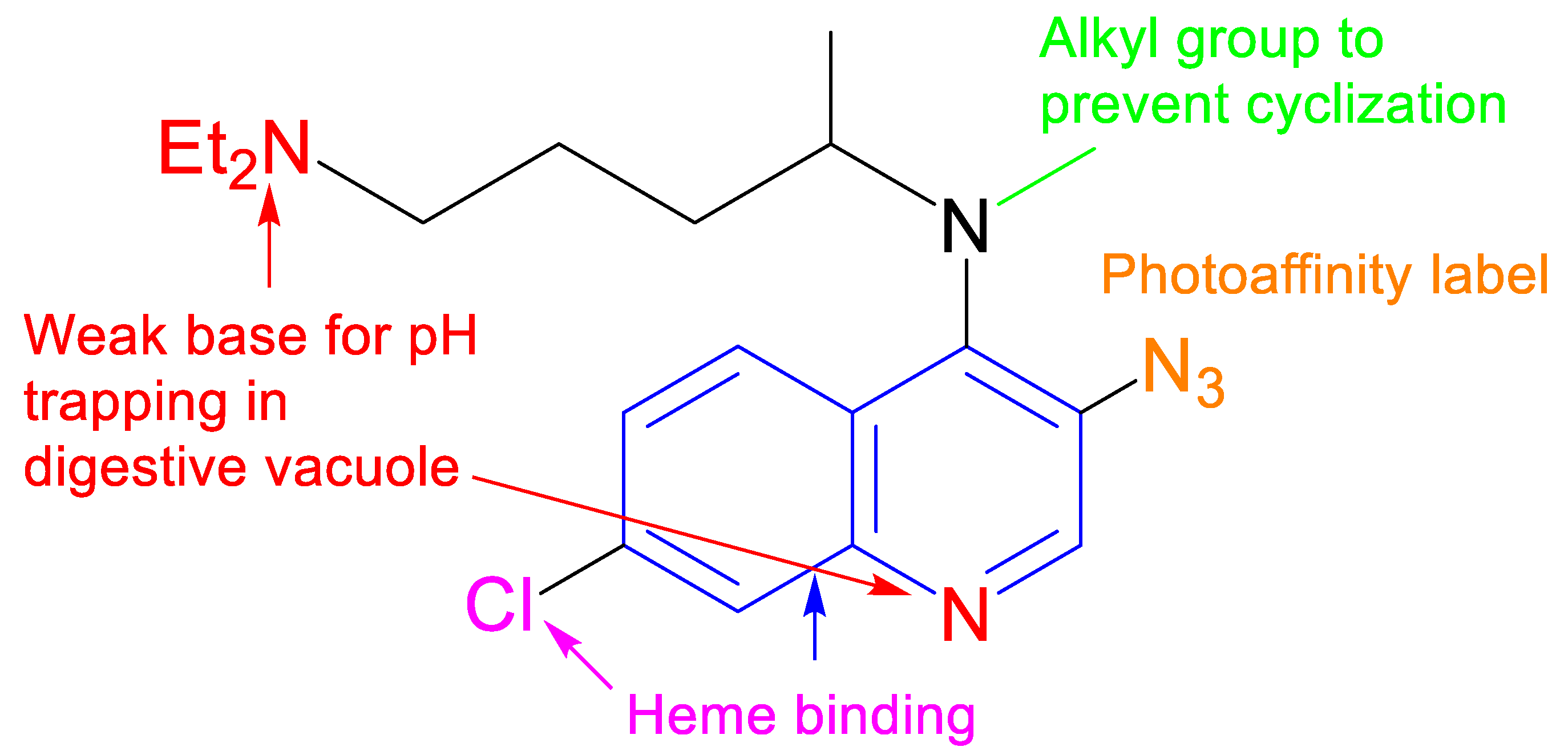
1. Introduction
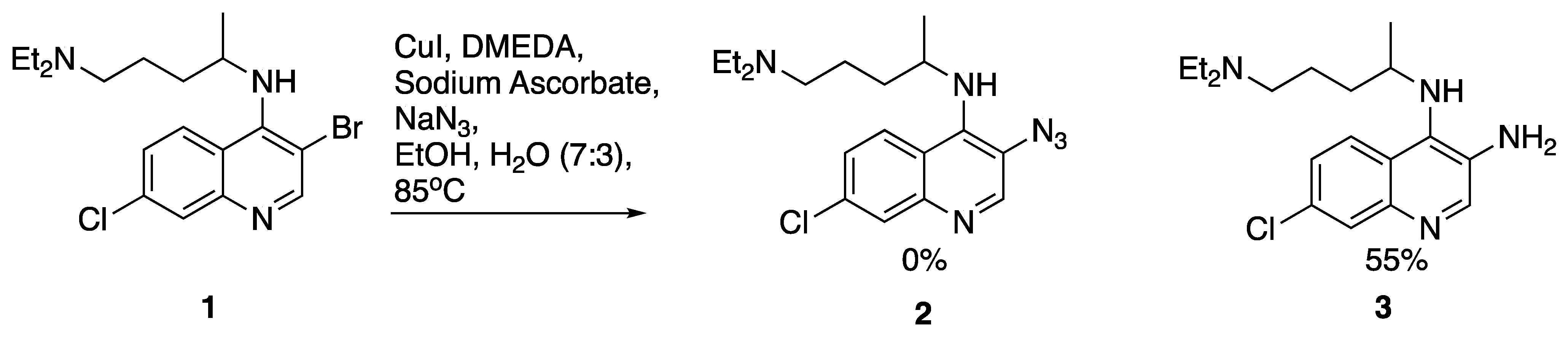
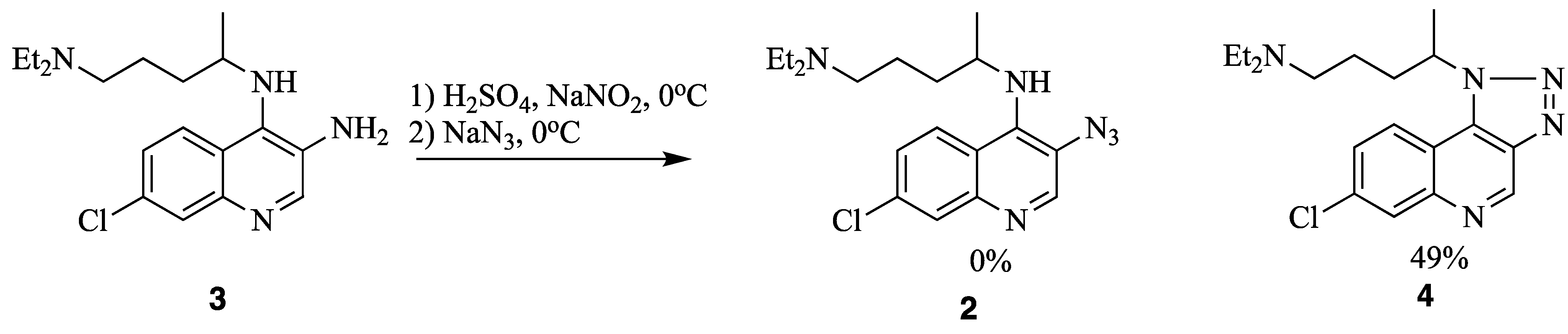
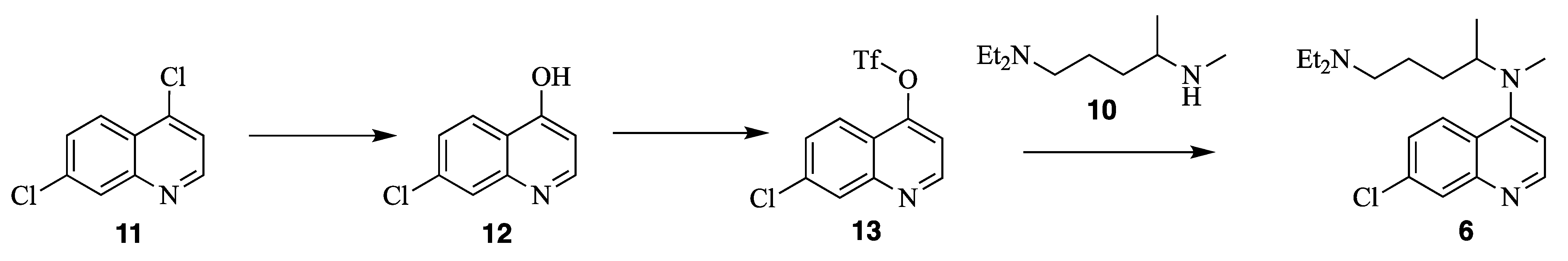
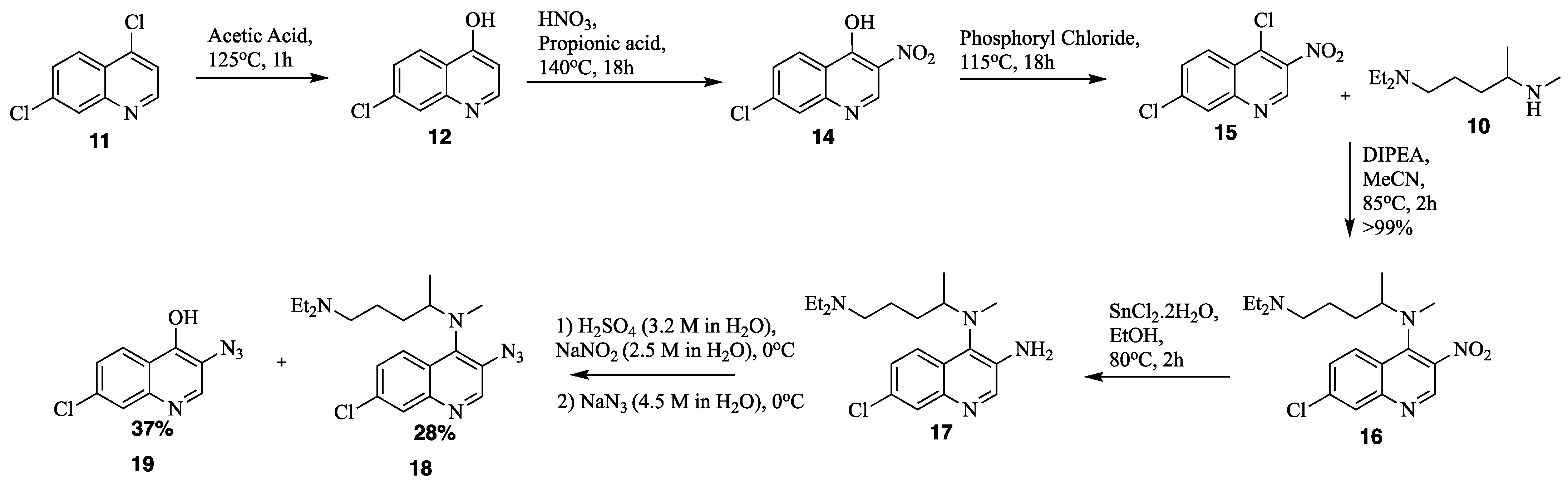
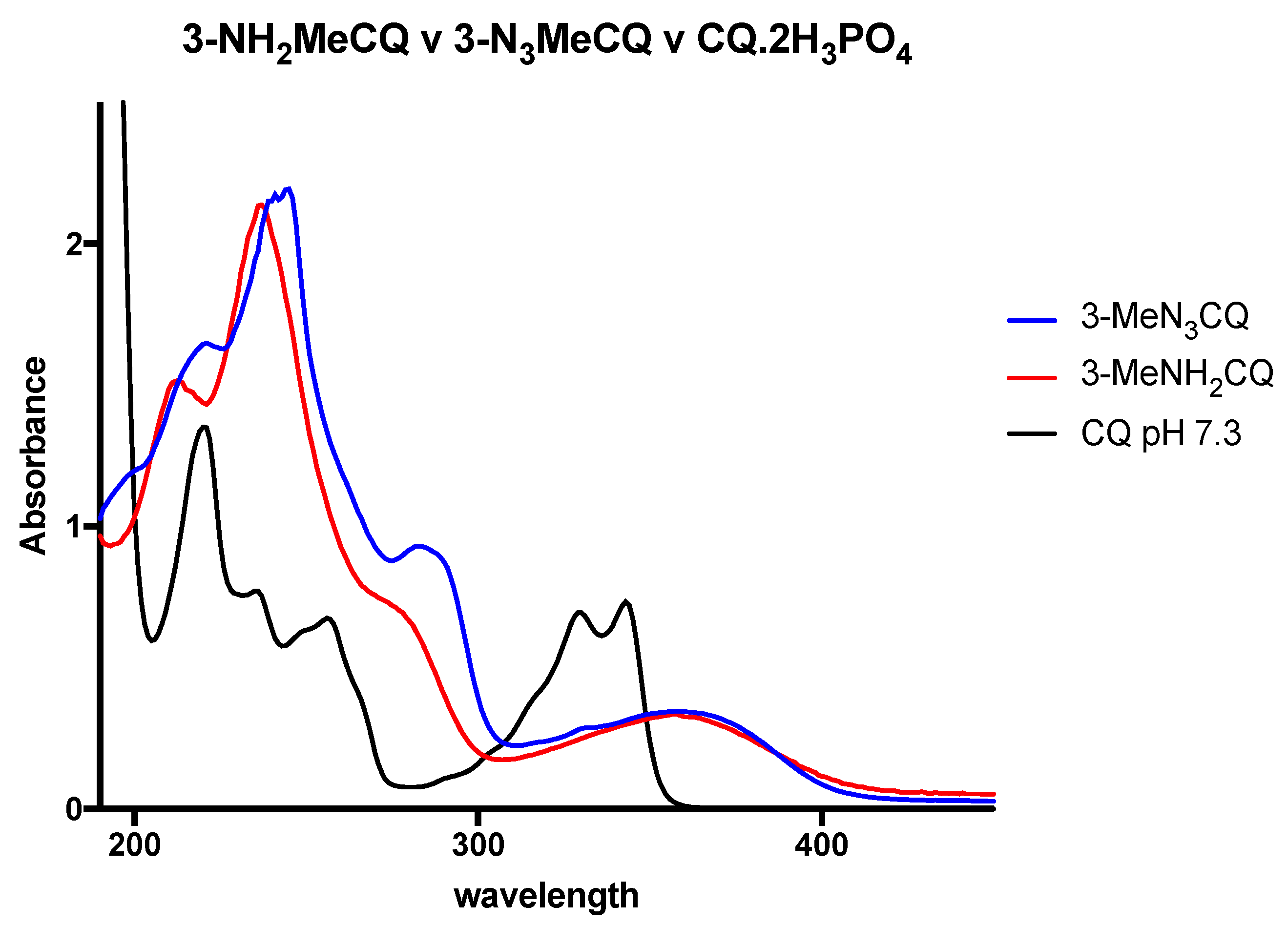
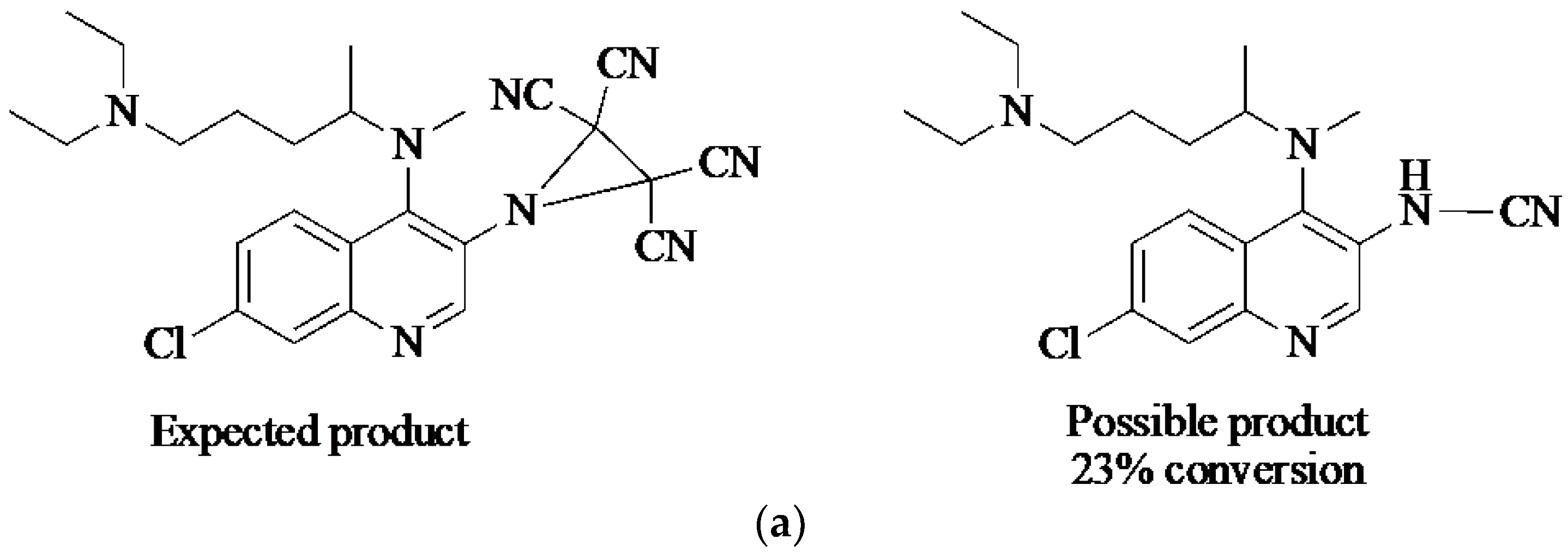
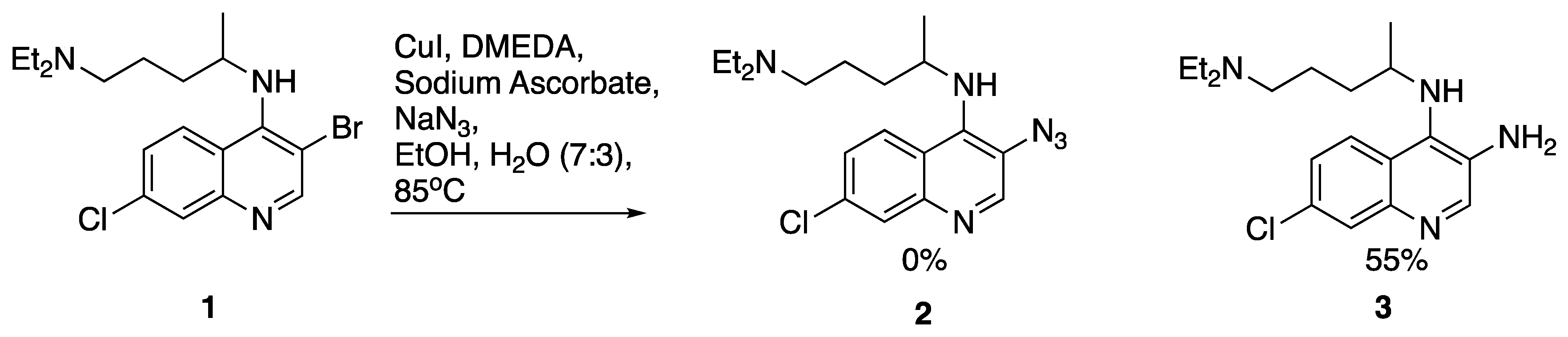
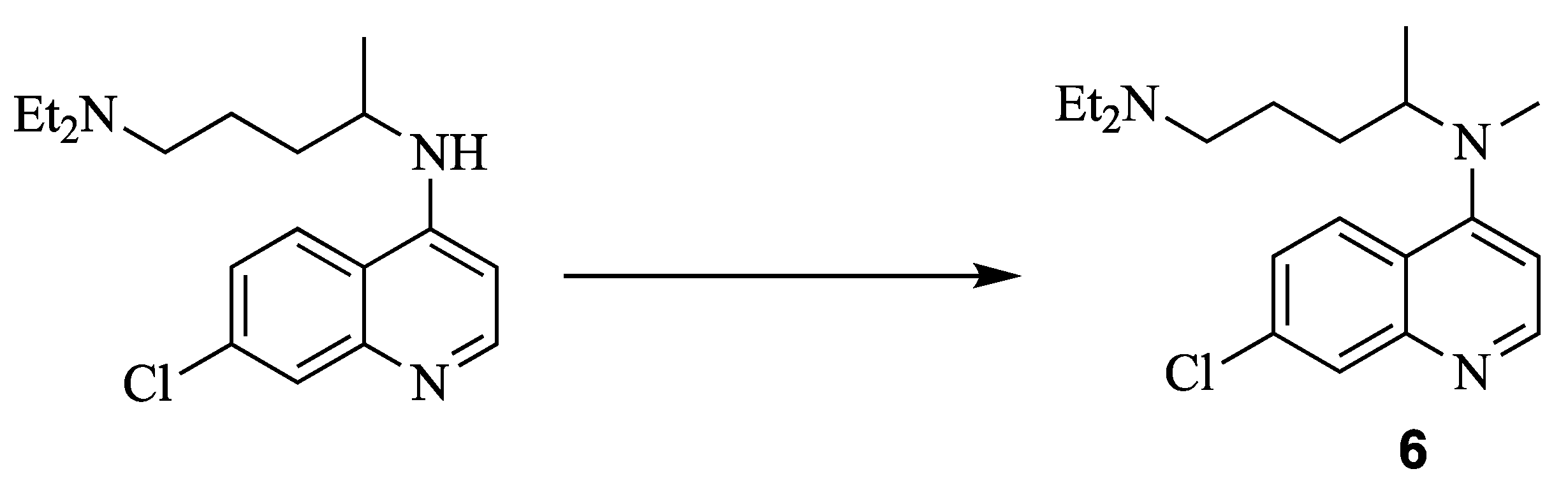
2. Results and Discussion
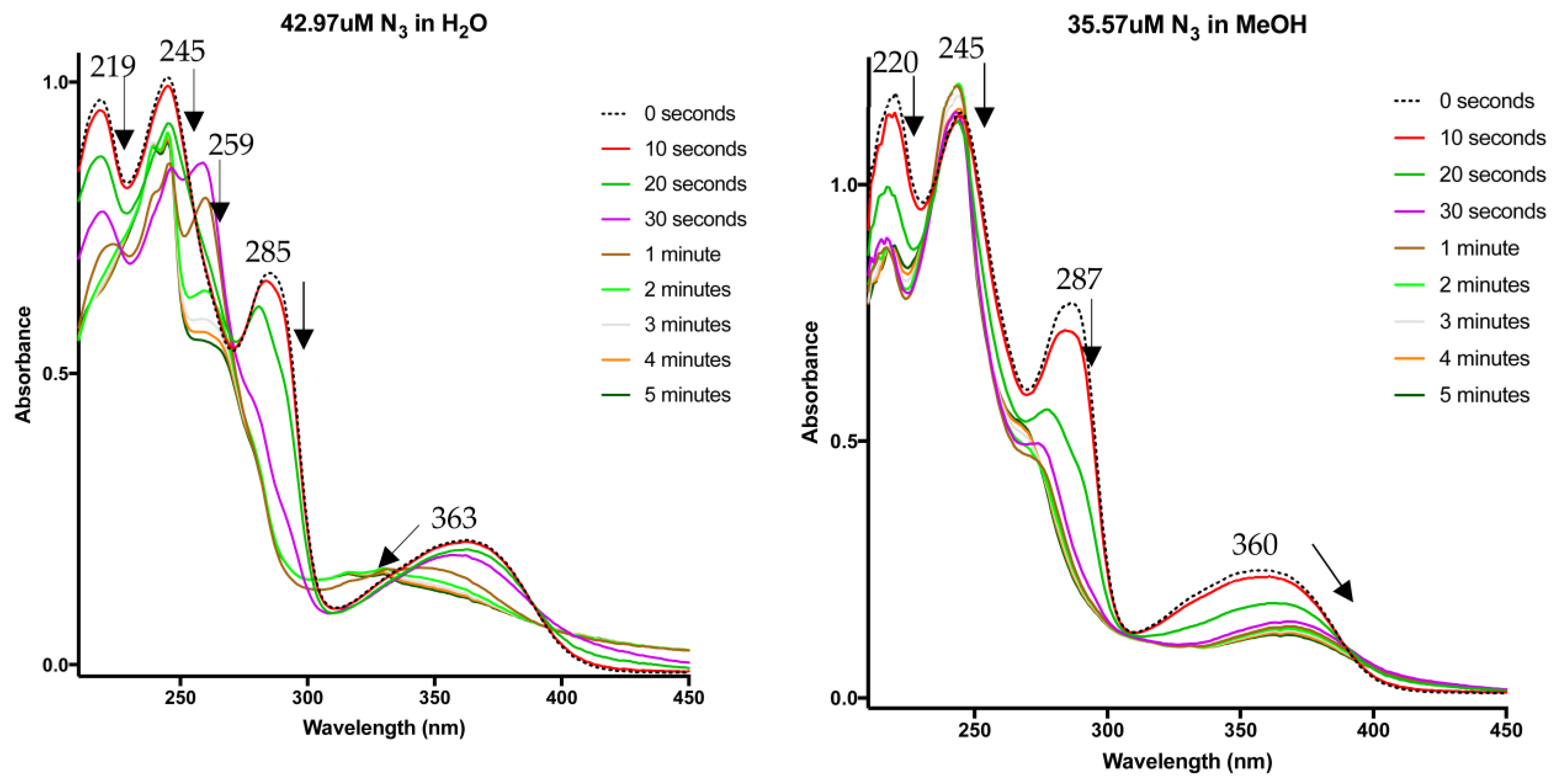
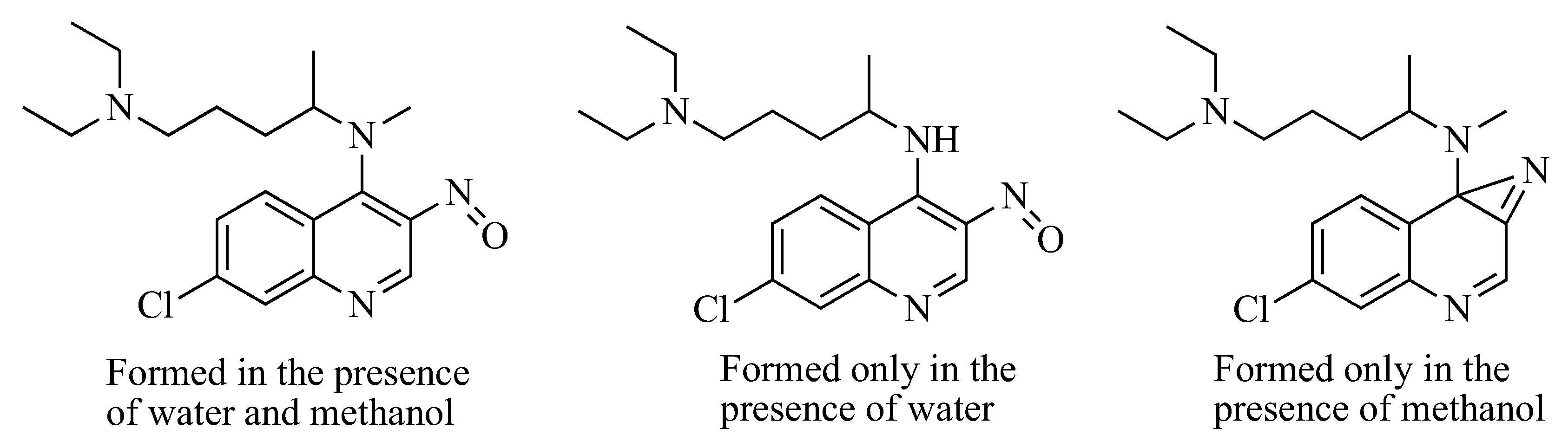
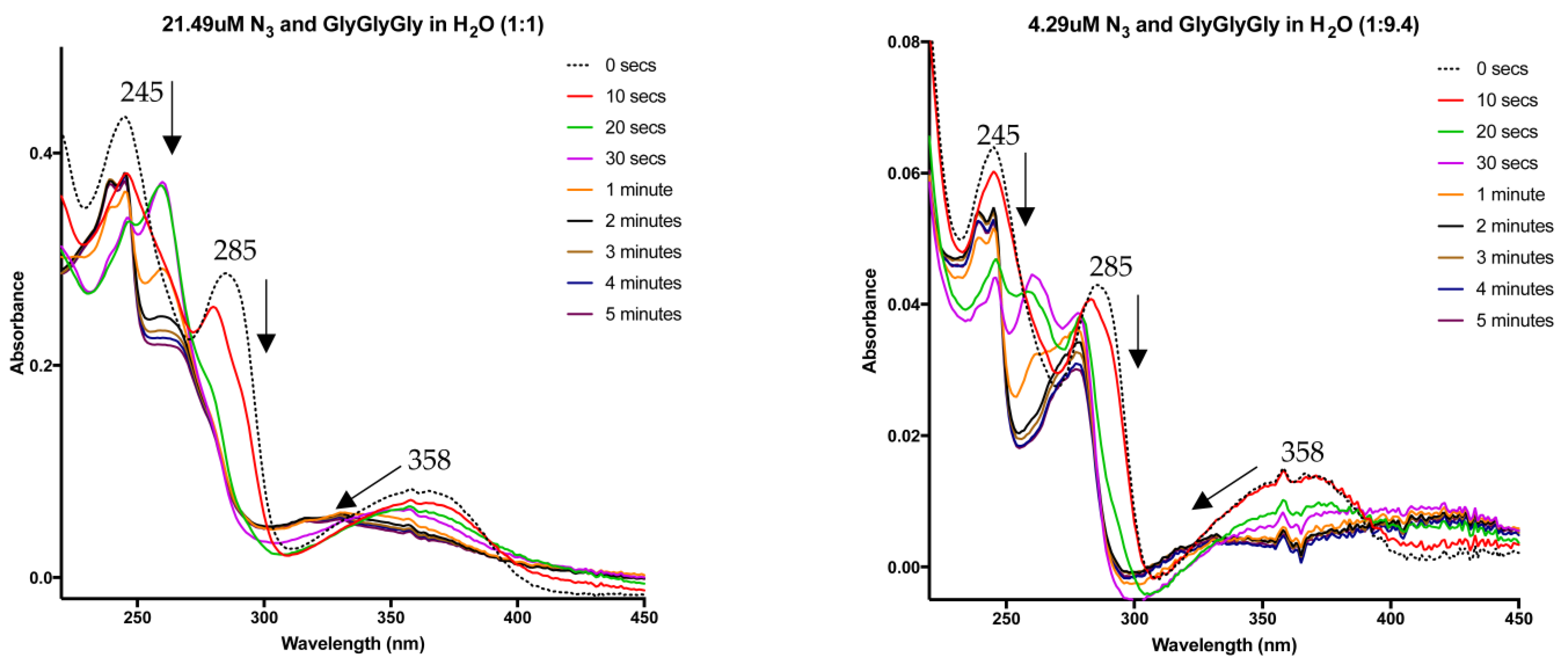
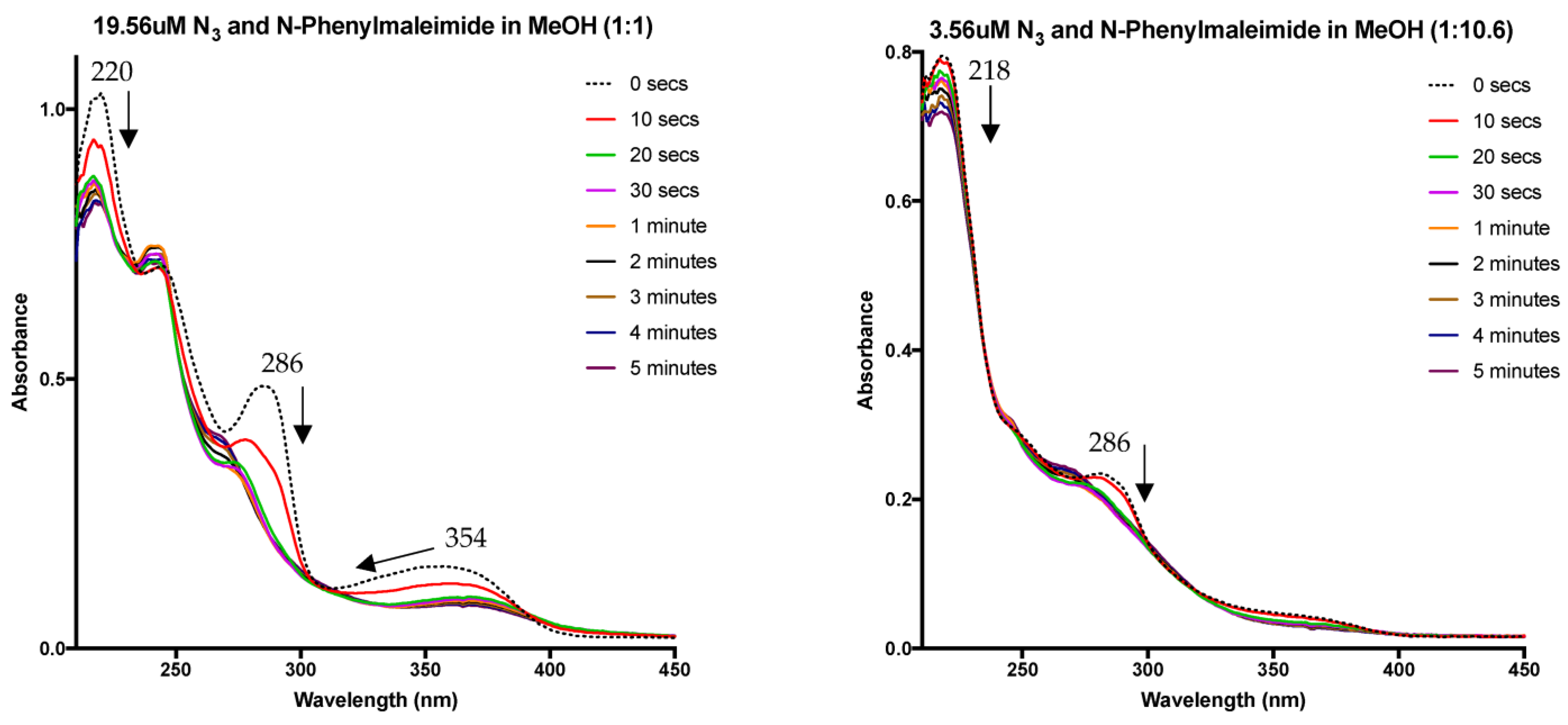
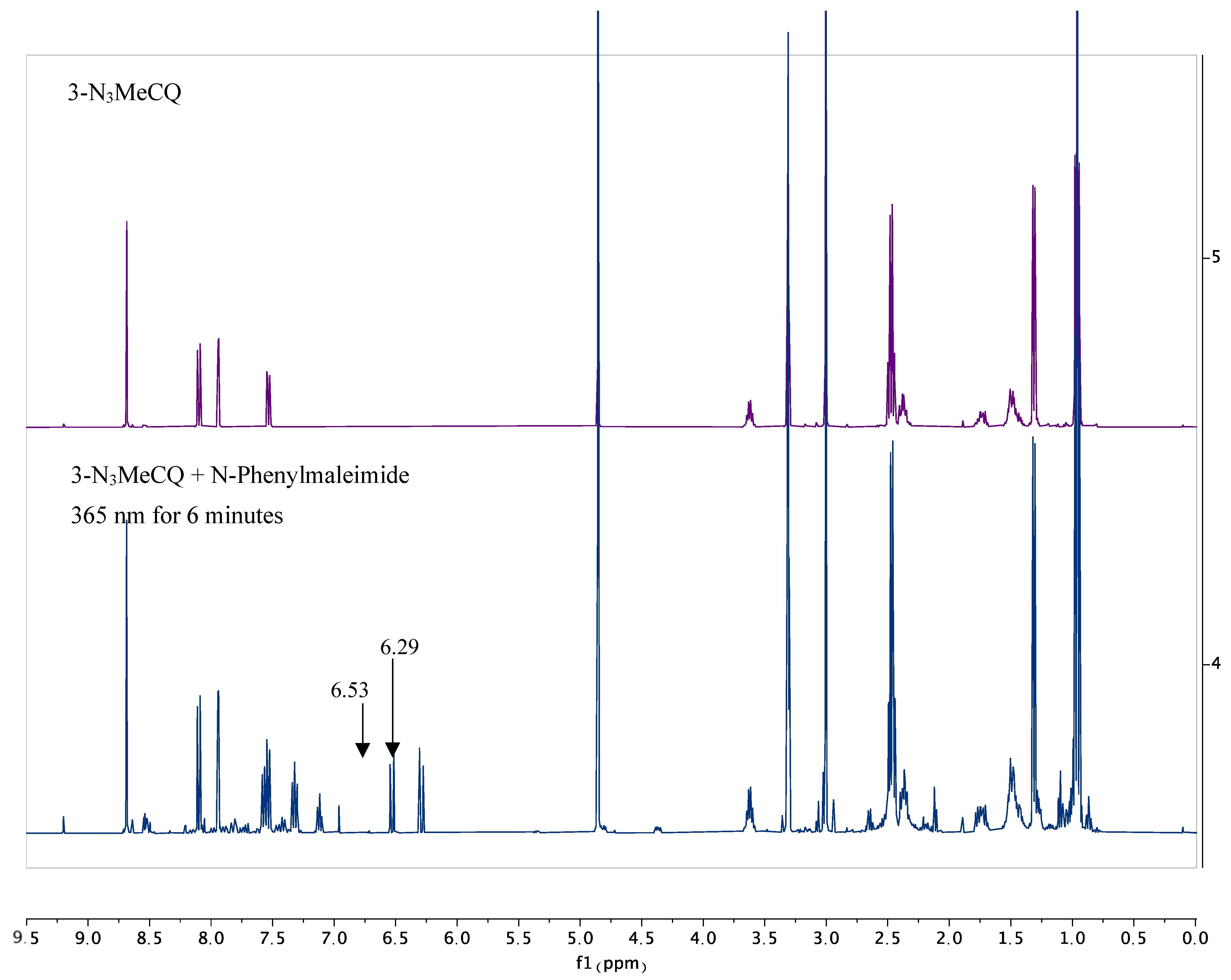
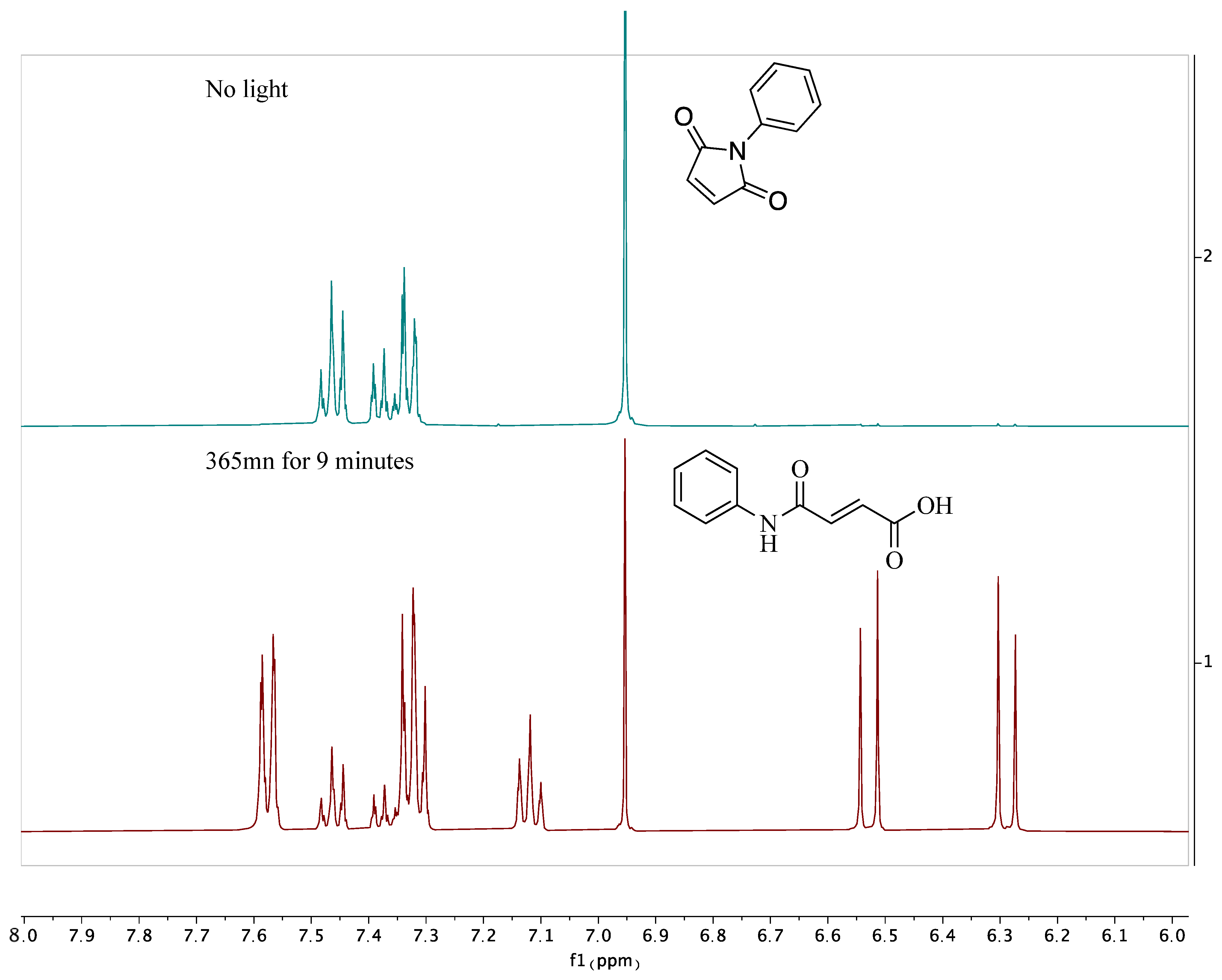
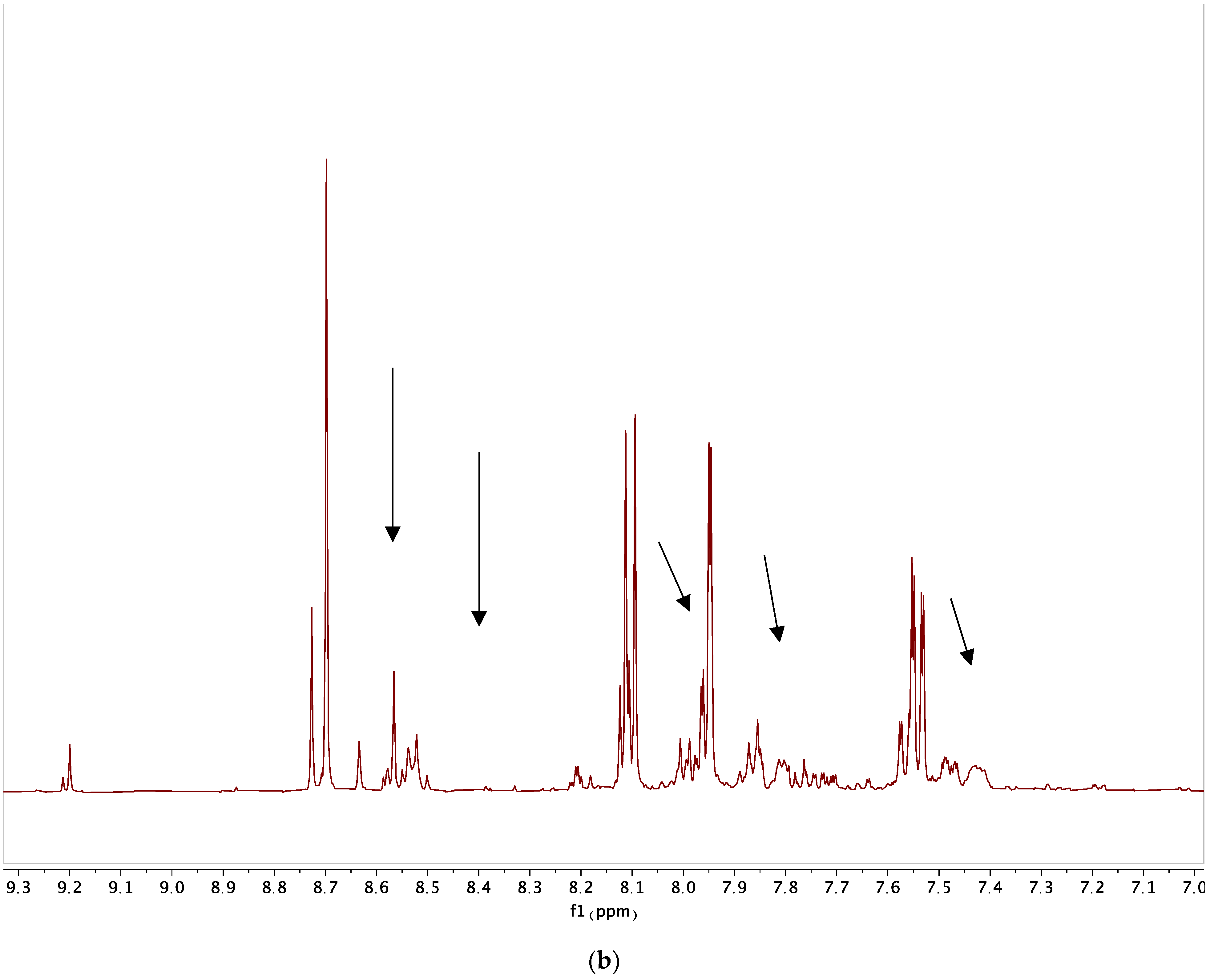
3. Photoaffinity Properties
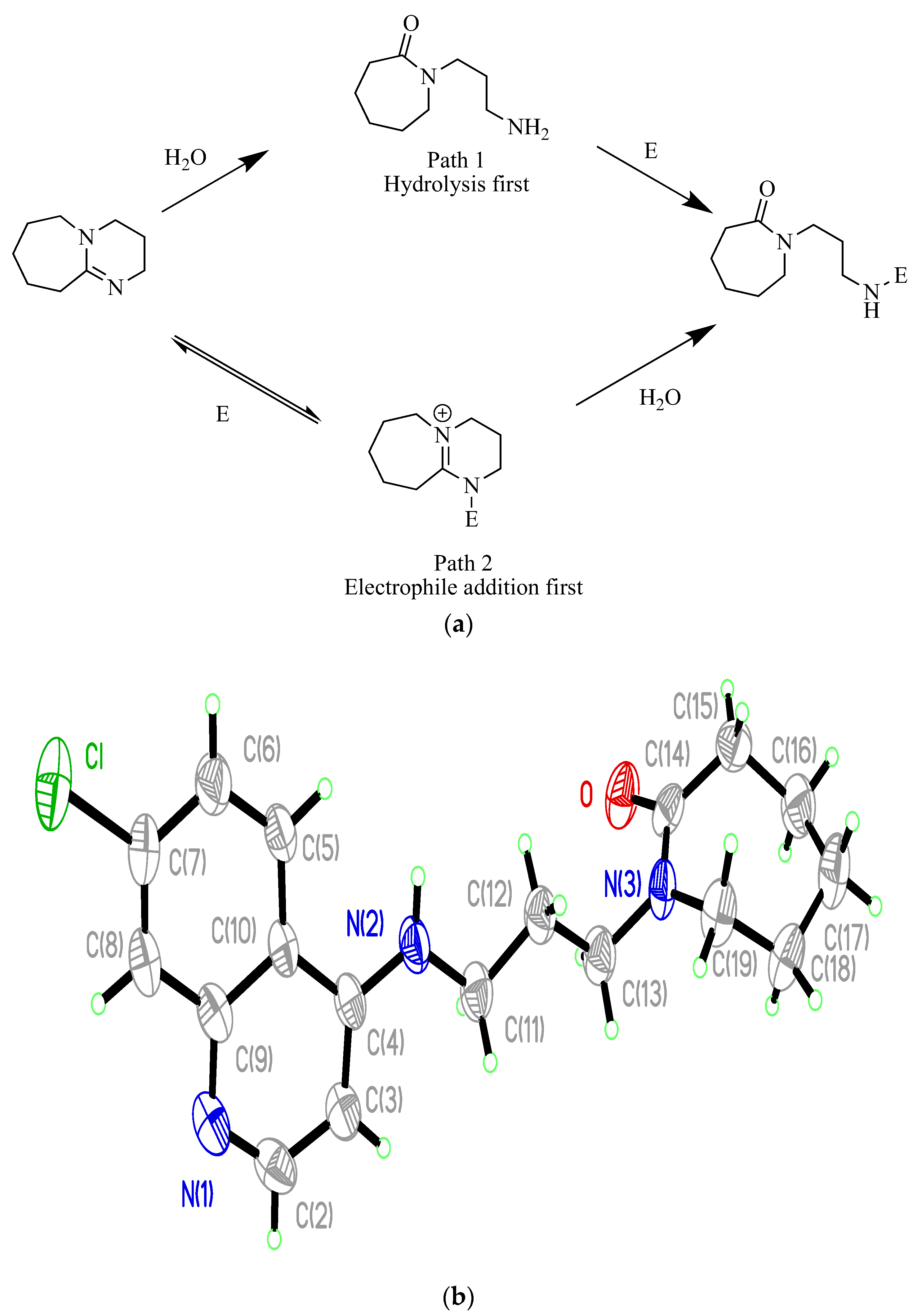
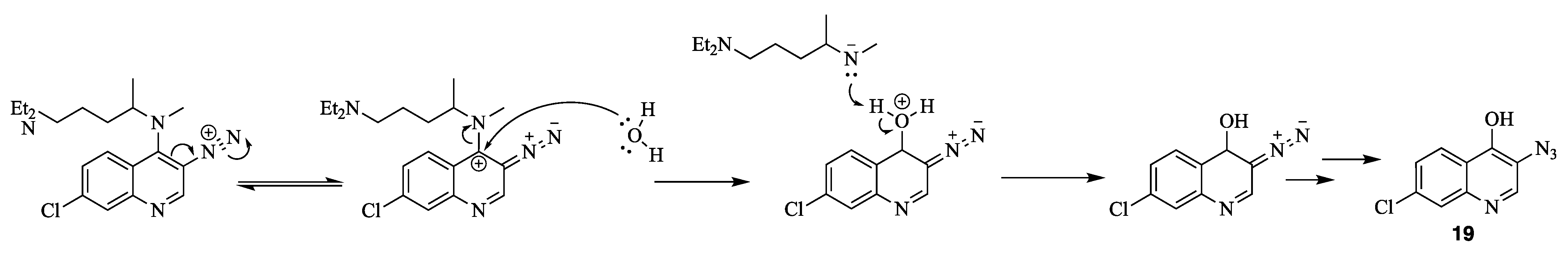
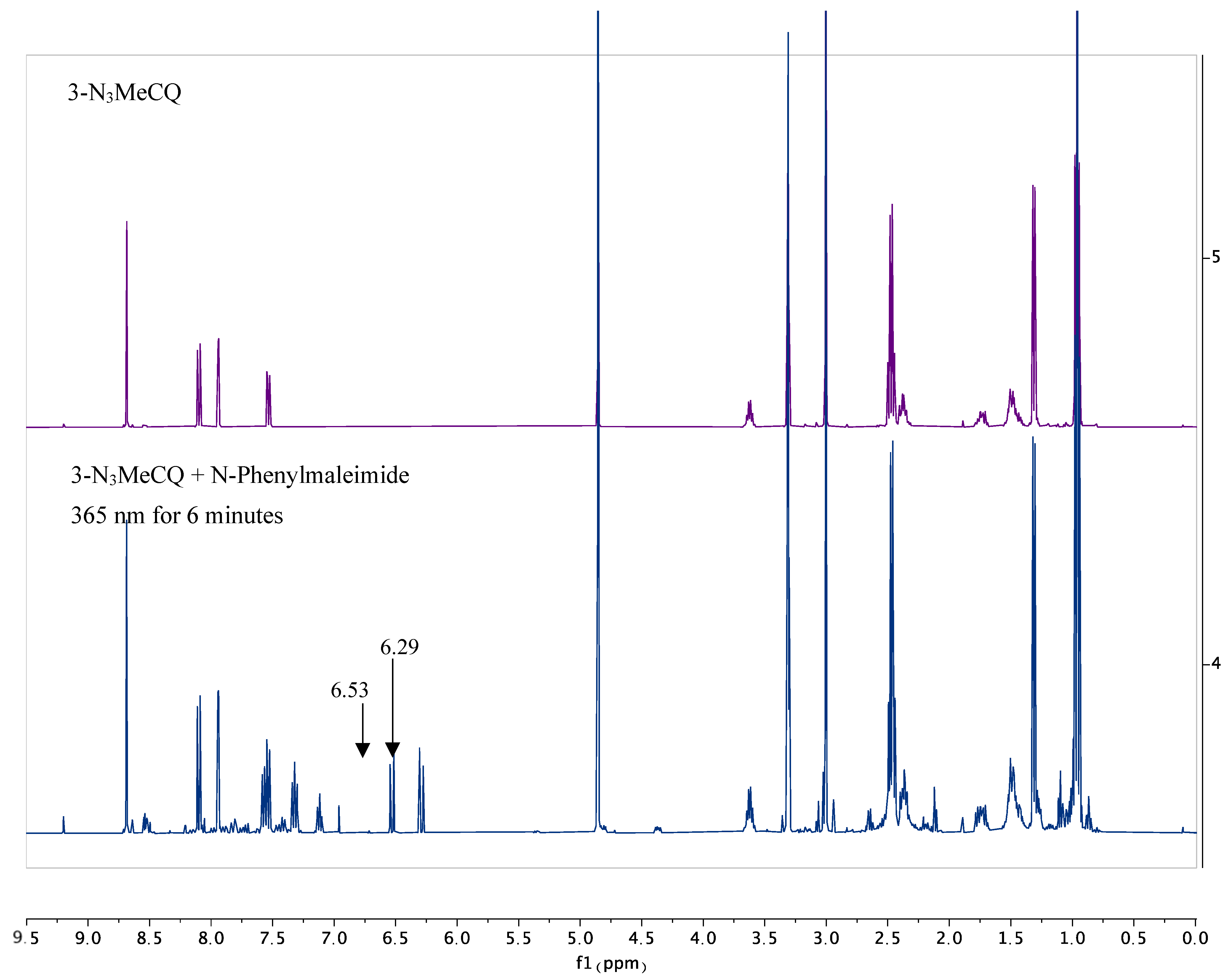
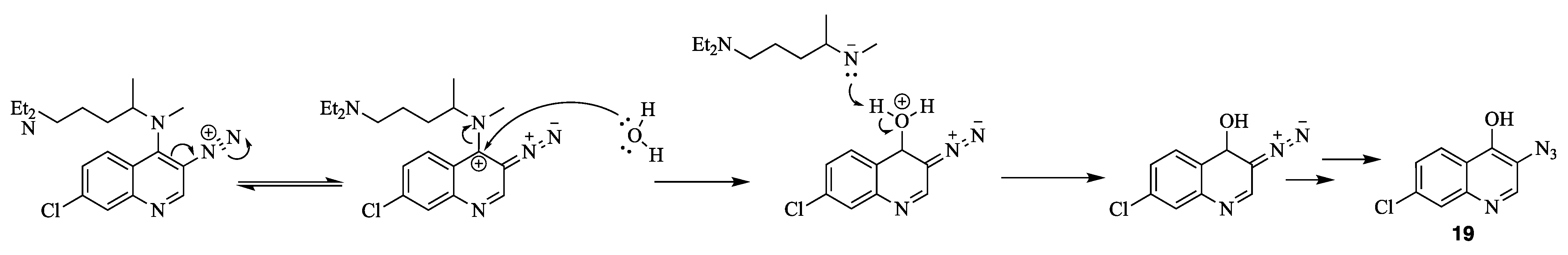
4. Nitrene Insertion
5. Conclusions
6. Experimental
6.1. General Information
6.2. Experimental Procedure
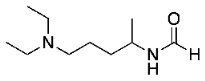 N-(5-(diethylamino)pentan-2-yl)formamide (9)
N-(5-(diethylamino)pentan-2-yl)formamide (9)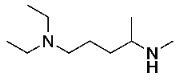 N1,N1-diethyl-N4-methylpentane-1,4-diamine (10)
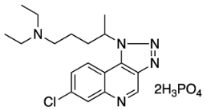
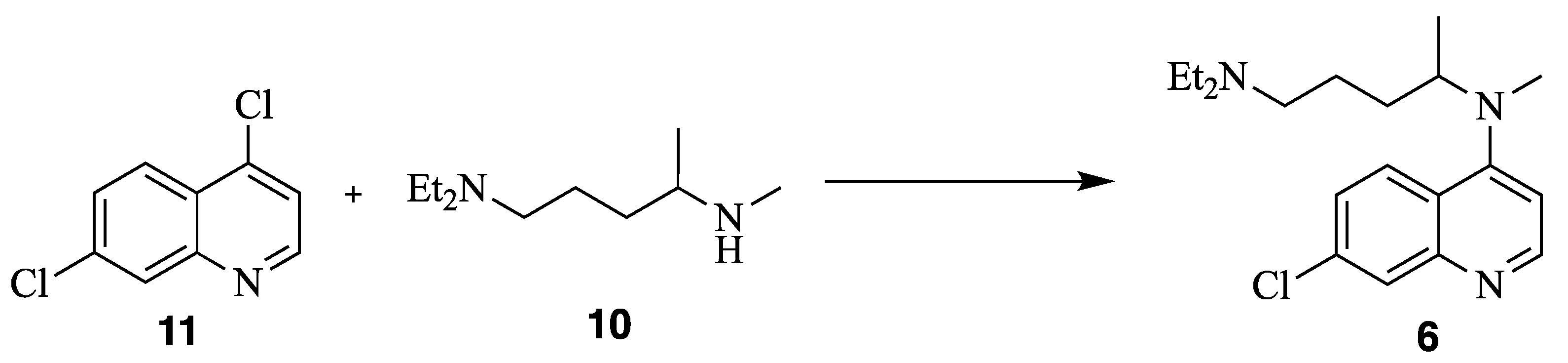
N1,N1-diethyl-N4-methylpentane-1,4-diamine (10)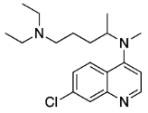 N4-(7-chloroquinolin-4-yl)-N1,N1-diethyl-N4-methylpentane-1,4-diamine (6)
N4-(7-chloroquinolin-4-yl)-N1,N1-diethyl-N4-methylpentane-1,4-diamine (6)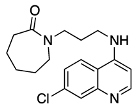 1-(3-((7-chloroquinolin-4-yl)amino)propyl)azepan-2-one
1-(3-((7-chloroquinolin-4-yl)amino)propyl)azepan-2-one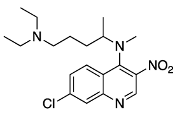 N4-(7-chloro-3-nitroquinolin-4-yl)-N1,N1-diethyl-N4-methylpentane-1,4-diamine (16)
N4-(7-chloro-3-nitroquinolin-4-yl)-N1,N1-diethyl-N4-methylpentane-1,4-diamine (16)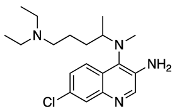 7-chloro-N4-(5-(diethylamino)pentan-2-yl)-N4-methylquinoline-3,4-diamine (17)
7-chloro-N4-(5-(diethylamino)pentan-2-yl)-N4-methylquinoline-3,4-diamine (17)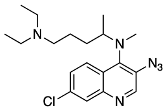 N4-(3-azido-7-chloroquinolin-4-yl)-N1,N1-diethyl-N4-methylpentane-1,4-diamine (18)
N4-(3-azido-7-chloroquinolin-4-yl)-N1,N1-diethyl-N4-methylpentane-1,4-diamine (18)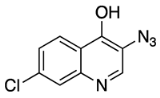 3-azido-7-chloroquinolin-4-ol (19)
3-azido-7-chloroquinolin-4-ol (19)6.3. X-ray Crystallography
6.4. UV–Vis Studies
6.5. General Procedure
6.6. Insertion Experiments
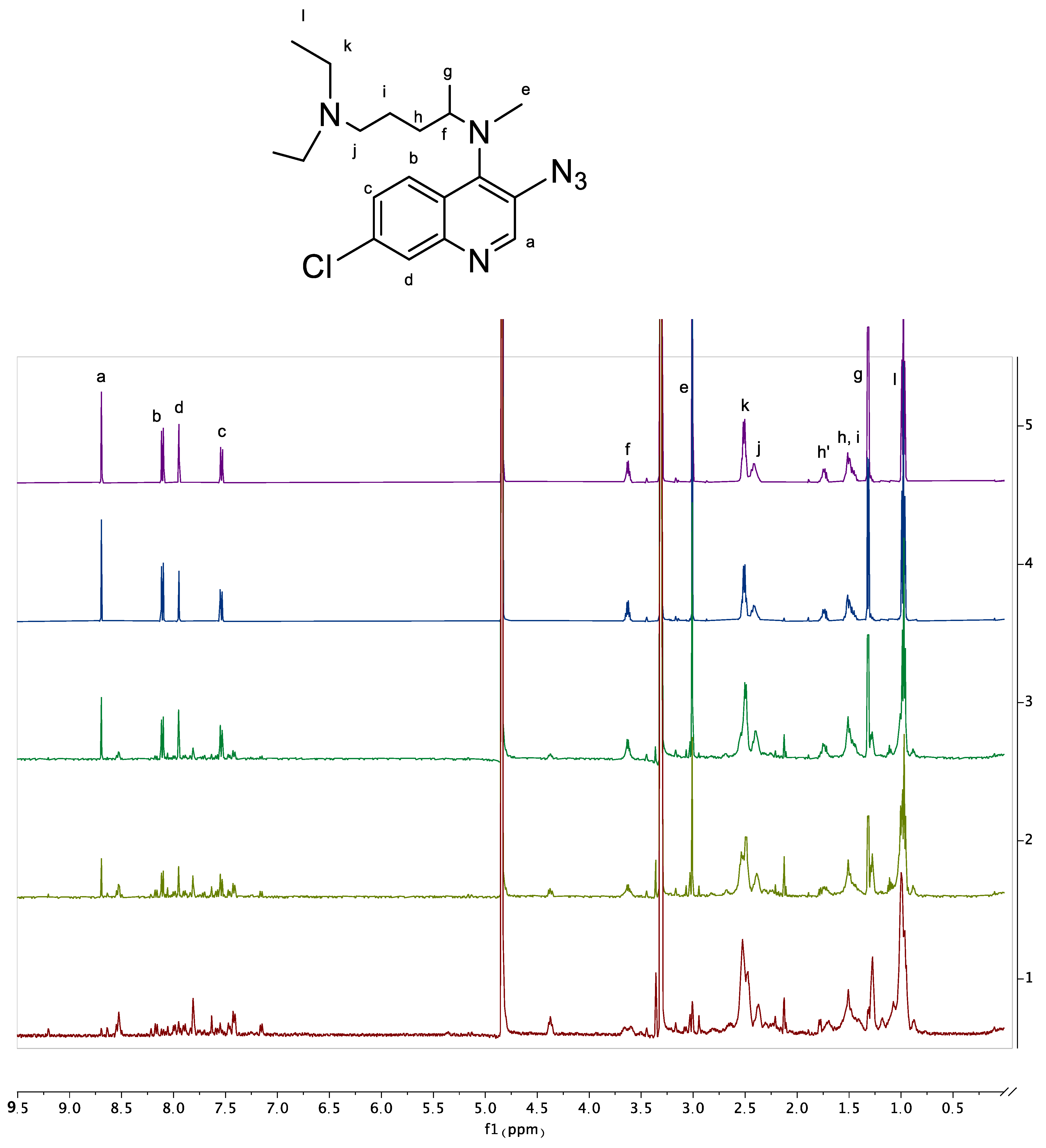
6.7. NMR Experiments
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Foley, M.; Deady, L.W.; Ng, K.; Cowman, A.F.; Tilley, L. Photoaffinity labeling of chloroquine-binding proteins in Plasmodium falciparum. J. Biol. Chem. 1994, 269, 6955–6961. [Google Scholar] [CrossRef]
- Mueller, D.M.; Hudson, R.A.; Lee, C.-P. Azide photoaffinity analogs for acridine dye binding sites. J. Am. Chem. Soc. 1981, 103, 1860–1862. [Google Scholar] [CrossRef]
- Desneves, J.; Thorn, G.; Berman, A.; Galatis, D.; La Greca, N.; Sinding, J.; Foley, M.; Deady, L.W.; Cowman, A.F.; Tilley, L. Photoaffinity labeling of mefloquine-binding proteins in human serum, uninfected erythrocytes and Plasmodium falciparum-infected erythrocytes. Mol. Biochem. Parasitol. 1996, 82, 181–194. [Google Scholar] [CrossRef]
- Lekostaj, J.K.; Natarajan, J.K.; Paguio, M.F.; Wolf, C.; Roepe, P.D. Photoaffinity Labeling of the Plasmodium falciparum Chloroquine Resistance Transporter with a Novel Perfluorophenylazido Chloroquine. Biochemistry 2008, 47, 10394–10406. [Google Scholar] [CrossRef] [PubMed]
- Edaye, S.; Tazoo, D.; Bohle, D.S.; Georges, E. 3-Iodo-4-aminoquinoline derivative sensitises resistant strains of Plasmodium falciparum to chloroquine. Int. J. Antimicrob. Agents 2016, 47, 482–485. [Google Scholar] [CrossRef] [PubMed]
- Edaye, S.; Tazoo, D.; Bohle, D.S.; Georges, E. 3-Halo Chloroquine Derivatives Overcome Plasmodium falciparum Chloroquine Resistance Transporter-Mediated Drug Resistance in P. falciparum. Antimicrob. Agents Chemother. 2015, 59, 7891–7893. [Google Scholar] [CrossRef] [PubMed]
- Ribas, X.; Güell, I. Cu(I)/Cu(III) catalytic cycle involved in Ullmann-type cross-coupling reactions. Pure Appl. Chem. 2014, 86, 345–360. [Google Scholar] [CrossRef]
- Andersen, J.; Bolvig, S.; Liang, X. Efficient One-Pot Synthesis of 1-Aryl 1,2,3-Triazoles from Aryl Halides and Terminal Alkynes in the Presence of Sodium Azide. Synlett 2005, 2005, 2941–2947. [Google Scholar] [CrossRef]
- Lanke, S.R.; Bhanage, B.M. Copper bis(2,2,6,6-Tetramethyl-3,5-heptanedionate)–Catalyzed Coupling of Sodium Azide with Aryl Iodides/Boronic Acids to Aryl Azides or Aryl Amines. Synth. Commun. 2014, 44, 399–407. [Google Scholar] [CrossRef]
- Zhu, W.; Ma, D. Synthesis of aryl azides and vinyl azides via proline-promoted CuI-catalyzed coupling reactions. Chem. Commun. 2004, 7, 888–889. [Google Scholar] [CrossRef]
- Markiewicz, J.T.; Wiest, O.; Helquist, P. Synthesis of Primary Aryl Amines Through a Copper-Assisted Aromatic Substitution Reaction with Sodium Azide. J. Org. Chem. 2010, 75, 4887–4890. [Google Scholar] [CrossRef]
- Messaoudi, S.; Brion, J.-D.; Alami, M. An Expeditious Copper-Catalyzed Access to 3-Aminoquinolinones, 3-Aminocoumarins and Anilines using Sodium Azide. Adv. Synth. Catal. 2010, 352, 1677–1687. [Google Scholar] [CrossRef]
- Goriya, Y.; Ramana, C.V. The [Cu]-catalyzed SNAR reactions: Direct amination of electron deficient aryl halides with sodium azide and the synthesis of arylthioethers under Cu(II)–ascorbate redox system. Tetrahedron 2010, 66, 7642–7650. [Google Scholar] [CrossRef]
- Qin, Z.; Kastrati, I.; Chandrasena, R.E.P.; Liu, H.; Yao, P.; Petukhov, P.A.; Bolton, J.L.; Thatcher, G.R.J. Benzothiophene selective estrogen receptor modulators with modulated oxidative activity and receptor affinity. J. Med. Chem. 2007, 50, 2682–2692. [Google Scholar] [CrossRef]
- Gehringer, M.; Forster, M.; Pfaffenrot, E.; Bauer, S.M.; Laufer, S.A. Novel Hinge-Binding Motifs for Janus Kinase 3 Inhibitors: A Comprehensive Structure-Activity Relationship Study on Tofacitinib Bioisosteres. ChemMedChem 2014, 9, 2516–2527. [Google Scholar] [CrossRef]
- Kaila, N.; Green, N.; Li, H.-Q.; Hu, Y.; Janz, K.; Gavrin, L.K.; Thomason, J.; Tam, S.; Powell, D.; Cuozzo, J.; et al. Identification of a novel class of selective Tpl2 kinase inhibitors: 4-Alkylamino-[1,7]naphthyridine-3-carbonitriles. Bioorg. Med. Chem. 2007, 15, 6425–6442. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Kundu, D.; Hajra, A.; Majee, A. Formylation without catalyst and solvent at 80 °C. Tetrahedron Lett. 2010, 51, 2896–2899. [Google Scholar] [CrossRef]
- Kim, J.-G.; Jang, D. Facile and Highly Efficient N-Formylation of Amines Using a Catalytic Amount of Iodine under Solvent-Free Conditions. Synlett 2010, 2010, 2093–2096. [Google Scholar] [CrossRef]
- Shastri, L.A.; Shastri, S.L.; Bathula, C.D.; Basanagouda, M.; Kulkarni, M.V. Mild, Simple, and Efficient Method for N -Formylation of Secondary Amines via Reimer–Tiemann Reaction. Synth. Commun. 2011, 41, 476–484. [Google Scholar] [CrossRef]
- Joseph, S.; Das, P.; Srivastava, B.; Nizar, H.; Prasad, M. A convenient procedure for N-formylation of amines. Tetrahedron Lett. 2013, 54, 929–931. [Google Scholar] [CrossRef]
- Collet, J.W.; Ackermans, K.; Lambregts, J.; Maes, B.U.; Orru, R.V.; Ruijter, E. Modular Three-Component Synthesis of 4-Aminoquinolines via an Imidoylative Sonogashira/Cyclization Cascade. J. Org. Chem. 2018, 83, 854–861. [Google Scholar] [CrossRef] [PubMed]
- Pajkert, R.; Böttcher, T.; Ponomarenko, M.; Bremer, M.; Röschenthaler, G.-V. Synthesis and characterization of novel carbene complexes of phosphorus(V) fluorides with potential liquid-crystalline properties. Tetrahedron 2013, 69, 8943–8951. [Google Scholar] [CrossRef]
- Sánchez-Martín, R.; Campos, J.M.; Conejo-García, A.; Cruz-López, O.; Báñez-Coronel, M.; Rodríguez-González, A.; Gallo, M.A.; Lacal, J.C.; Espinosa, A. Symmetrical bis-quinolinium compounds: New human choline kinase inhibitors with antiproliferative activity against the HT-29 cell line. J. Med. Chem. 2005, 48, 3354–3363. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, T.; Guedes, R.C.; dos Santos, D.J.; Carrasco, M.; Gut, J.; Rosenthal, P.J.; Moreira, R.; Lopes, F. Design, synthesis and structure–activity relationships of (1H-pyridin-4-ylidene)amines as potential antimalarials. Bioorg. Med. Chem. Lett. 2009, 19, 3476–3480. [Google Scholar] [CrossRef]
- Natarajan, J.K.; Alumasa, J.N.; Yearick, K.; Ekoue-Kovi, K.A.; Casabianca, L.B.; De Dios, A.C.; Wolf, C.; Roepe, P.D. 4-N-, 4-S-, and 4-O-chloroquine analogues: Influence of side chain length and quinolyl nitrogen pKa on activity vs chloroquine resistant malaria. J. Med. Chem. 2008, 51, 3466–3479. [Google Scholar] [CrossRef]
- Shi, M.; Shen, Y. A Novel Reaction of 1,8-Diazabicyclo[5.4.0]undec-7-ene (DBU) or 1,5-Diazabicyclo[4.3.0]non-5-ene (DBN) with Benzyl Halides in the Presence of Water. Helv. Chim. Acta 2002, 85, 1355. [Google Scholar] [CrossRef]
- Kraft, A. Branched non-covalent complexes between carboxylic acids and two tris(amidines). J. Chem. Soc. Perkin Trans. 1999, 1, 705–714. [Google Scholar] [CrossRef]
- Hyde, A.M.; Calabria, R.; Arvary, R.; Wang, X.; Klapars, A. Investigating the Underappreciated Hydrolytic Instability of 1,8-Diazabicyclo[5.4.0]undec-7-ene and Related Unsaturated Nitrogenous Bases. Org. Process Res. Dev. 2019, 23, 1860–1871. [Google Scholar] [CrossRef]
- Sather, A.C.; Lee, H.G.; De La Rosa, V.Y.; Yang, Y.; Müller, P.; Buchwald, S.L. A Fluorinated Ligand Enables Room-Temperature and Regioselective Pd-Catalyzed Fluorination of Aryl Triflates and Bromides. J. Am. Chem. Soc. 2015, 137, 13433–13438. [Google Scholar] [CrossRef]
- Sinha, M.; Dola, V.R.; Soni, A.; Agarwal, P.; Srivastava, K.; Haq, W.; Puri, S.K.; Katti, S.B. Synthesis of chiral chloroquine and its analogues as antimalarial agents. Bioorg. Med. Chem. 2014, 22, 5950–5960. [Google Scholar] [CrossRef]
- Johnson, W.S.; Buell, B.G. A New Synthesis of Chloroquine. J. Am. Chem. Soc. 1952, 74, 4513–4516. [Google Scholar] [CrossRef]
- Gorlitzer, K.; Kramer, C.; Meyer, H.; Walter, R.D.; Jomaa, H.; Wiesner, J. Pyrido[3,2-b]indol-4-yl-amine—Synthese und Prufung auf Wirksamkeit gegen Malaria. Die Pharm.—An Int. J. Pharm. Sci. 2004, 59, 243–250. [Google Scholar]
- Liu, Y.; Zhang, Z.; Wu, A.; Yang, X.; Zhu, Y.; Zhao, N. A Novel Process for Antimalarial Drug Pyronaridine Tetraphosphate. Org. Process Res. Dev. 2014, 18, 349–353. [Google Scholar] [CrossRef]
- Elslager, E.F.; Gold, E.H.; Tendick, F.H.; Werbel, L.M.; Worth, D.F. Amodiaquine N-oxides and other 7-chloro-4-aminoquinoline n-oxides. J. Heterocycl. Chem. 1964, 1, 6–12. [Google Scholar] [CrossRef]
- Wang, X.; Pan, W.-J.; Cai, Y.-H.; Xie, X.-Y.; Huang, C.-Y.; Li, J.-Y.; Chen, W.-N.; He, M.-H. Microwave-Assisted Efficient Synthesis of 4-Substituted Amino-2-methylquinolines Catalyzed by p-Toluenesulfonic Acid. Heterocycles 2016, 92, 1864. [Google Scholar] [CrossRef]
- Bayley, H.; Staros, J.V. Photoaffinity Labeling and Related Techniques. In Azides and Nitrenes; Elsevier: Amsterdam, The Netherlands, 1984; pp. 433–490. [Google Scholar] [CrossRef]
- Vezmar, M.; Georges, E. Direct binding of chloroquine to the multidrug resistance protein (MRP). Biochem. Pharmacol. 1998, 56, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Baruah, H.; Puthenveetil, S.; Choi, Y.-A.; Shah, S.; Ting, A.Y. An Engineered Aryl Azide Ligase for Site-Specific Mapping of Protein-Protein Interactions through Photo-Cross-Linking. Angew. Chem. Int. Ed. 2008, 47, 7018–7021. [Google Scholar] [CrossRef]
- Berman, A.; Shearing, L.N.; Ng, K.F.; Jinsart, W.; Foley, M.; Tilley, L. Photoaffinity labelling of Plasmodium falciparum proteins involved in phospholipid transport. Mol. Biochem. Parasitol. 1994, 67, 235–243. [Google Scholar] [CrossRef]
- Wang, J.; Burdzinski, G.; Platz, M.S. Ultrafast Time-Resolved Studies of the Photochemistry of Aryl Azides. In Nitrenes and Nitrenium Ions; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; pp. 1–31. [Google Scholar] [CrossRef]
- Vyas, S.; Winter, A.H.; Hadad, C.M. Theory and Computation in the Study of Nitrenes and their Excited-State Photoprecursors. In Nitrenes and Nitrenium Ions; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; pp. 33–76. [Google Scholar] [CrossRef]
- Bräse, S.; Banert, K. Organic Azides. Organic Azides: Syntheses and Applications; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2009. [Google Scholar] [CrossRef]
- Zott, H.; Heusinger, H. Photolysis of maleic anhydride and maleimides in solution studied by electron spin resonance. Radiat. Phys. Chem. 1977, 10, 297–302. [Google Scholar] [CrossRef]
- Murata, S.; Abe, S.; Tomioka, H. Photochemical Reactions of Mesityl Azide with Tetracyanoethylene: Competitive Trapping of Singlet Nitrene and Didehydroazepine. J. Org. Chem. 1997, 62, 3055–3061. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SADABS, TWINABS; Siemens Industrial Automation, Inc.: Madison, WI, USA, 1996. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Base | Solvent | Methylating Agent | Temperature (°C) | Yield (%) |
|---|---|---|---|---|
| NaH | DMF | MeI | −20 to 0 | 23 |
| NaH | DMF | MeI | Room temp. | 15 |
| NaH | DMF | MeI | −20 | 9 |
| NaH | DMF | MeI | −20 | N.D. |
| NaH | MeCN | MeI | −20 | - |

| Formylating Agent | Solvent | Temperature (°C) | Catalyst | Yield (%) |
|---|---|---|---|---|
| Formic acid | - | 70–84 | - | - |
| Formic acid | - | 70 | I2 | - |
| Formamide | THF | RT–70 | NaOMe | - |
| Sodium ethoxide | CHCl3 | 55 | - | - |

| Formylating Agent | Solvent | Temperature (°C) | Yield (%) |
|---|---|---|---|
| Formic acid | - | 100 | 57 |
| Formic acid, acetic anhydride | - | 60 | - |
| Ethyl formate | - | 70 | 97 |
| Base | Solvent | Temperature (°C) | Yield (%) |
|---|---|---|---|
| - | - | 85 | - |
| - | - | 120–125 | 6 |
| - | - | 120 a | - |
| - | Ethylene glycol | 120 a | 6 |
| NEt3 | - | 125 | - |
| NEt3 | - | 85 a | - |
| NEt3 | EtOH | 0–40 | - |
| DBU | - | 80 a | 10 |
| DBU | - | 125 | 82 b |
| DIPEA | DMSO | 80 | - |
| DIPEA | DMSO | 120 | - |
| DIPEA | MeCN | 85 | - |
| tBuOK | DMSO | 80–120 | Decomp. |
| tBuOK | THF | RT–80 °C | - |
| c | 1,4-dioxane | 85 | 8 |
| Base | Solvent | Temperature (°C) | Yield (%) |
|---|---|---|---|
| - | - | 80–125 | 24 |
| DIPEA | DMSO | 80–125 | trace |
| DIPEA | Acetonitrile | RT–40 | 15 |
| DIPEA | - | 125 | 26 |
| NEt3 | - | 80–125 | 22 |
| NEt3 | EtOH | 0–40 | - |
| Acid | Solvent | Temperature (°C) | Yield (%) |
|---|---|---|---|
| Phenol a | - | 125 | 14 b |
| Phenol | - | 125 c | 16 |
| p-toluenesulfonic acid | DMSO | 125 | - |
| p-toluenesulfonic acid | - | 120 c | 63 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kapuku, B.; Bohle, D.S. Synthesis and Photolysis Properties of a New Chloroquine Photoaffinity Probe. Molecules 2024, 29, 1084. https://doi.org/10.3390/molecules29051084
Kapuku B, Bohle DS. Synthesis and Photolysis Properties of a New Chloroquine Photoaffinity Probe. Molecules. 2024; 29(5):1084. https://doi.org/10.3390/molecules29051084
Chicago/Turabian StyleKapuku, Benita, and D. Scott Bohle. 2024. "Synthesis and Photolysis Properties of a New Chloroquine Photoaffinity Probe" Molecules 29, no. 5: 1084. https://doi.org/10.3390/molecules29051084
APA StyleKapuku, B., & Bohle, D. S. (2024). Synthesis and Photolysis Properties of a New Chloroquine Photoaffinity Probe. Molecules, 29(5), 1084. https://doi.org/10.3390/molecules29051084