




Probing Non-Covalent Interactions through Molecular Balances: A REG-IQA Study



Abstract
1. Introduction
2. Results and Discussion
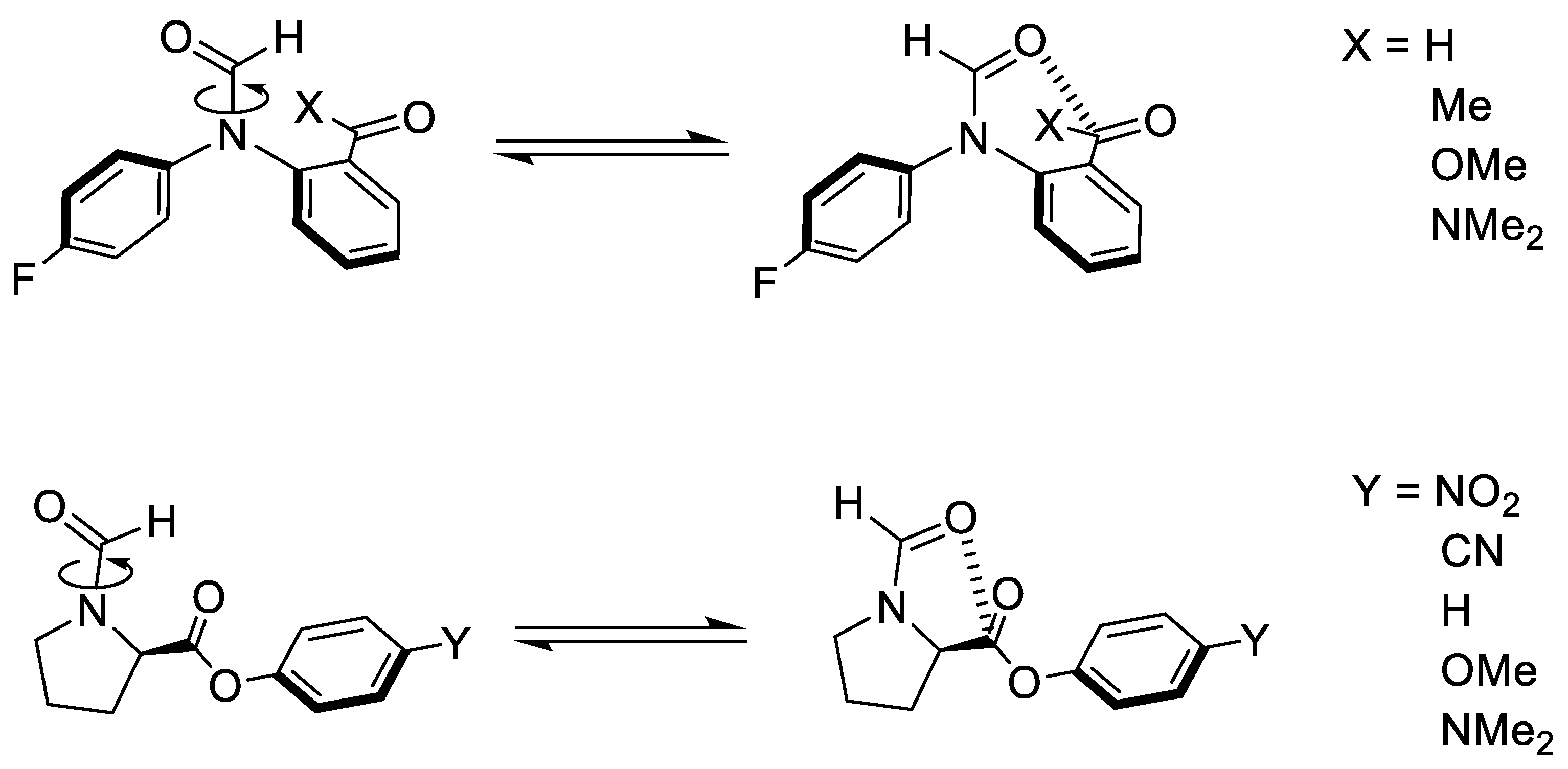
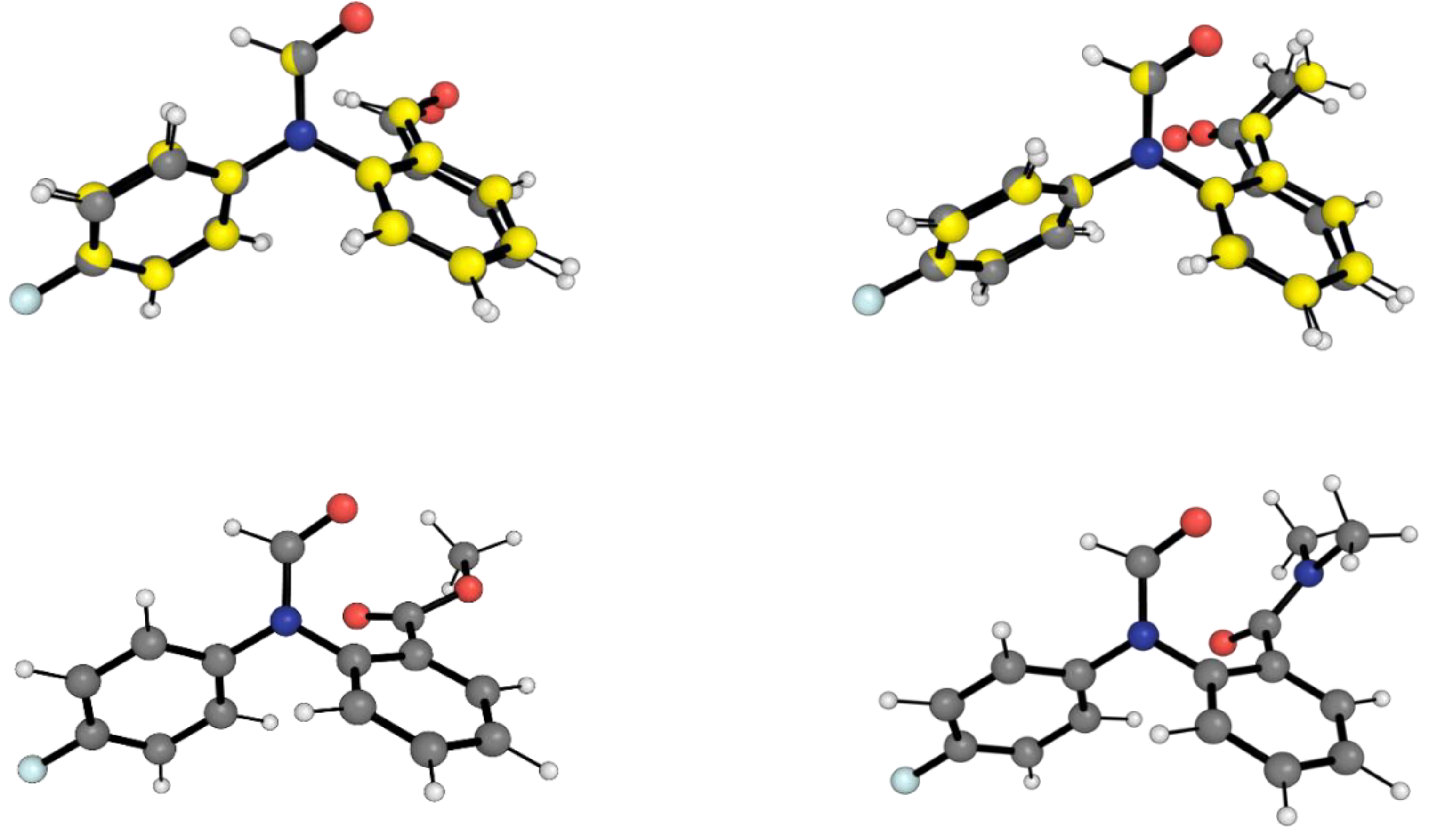
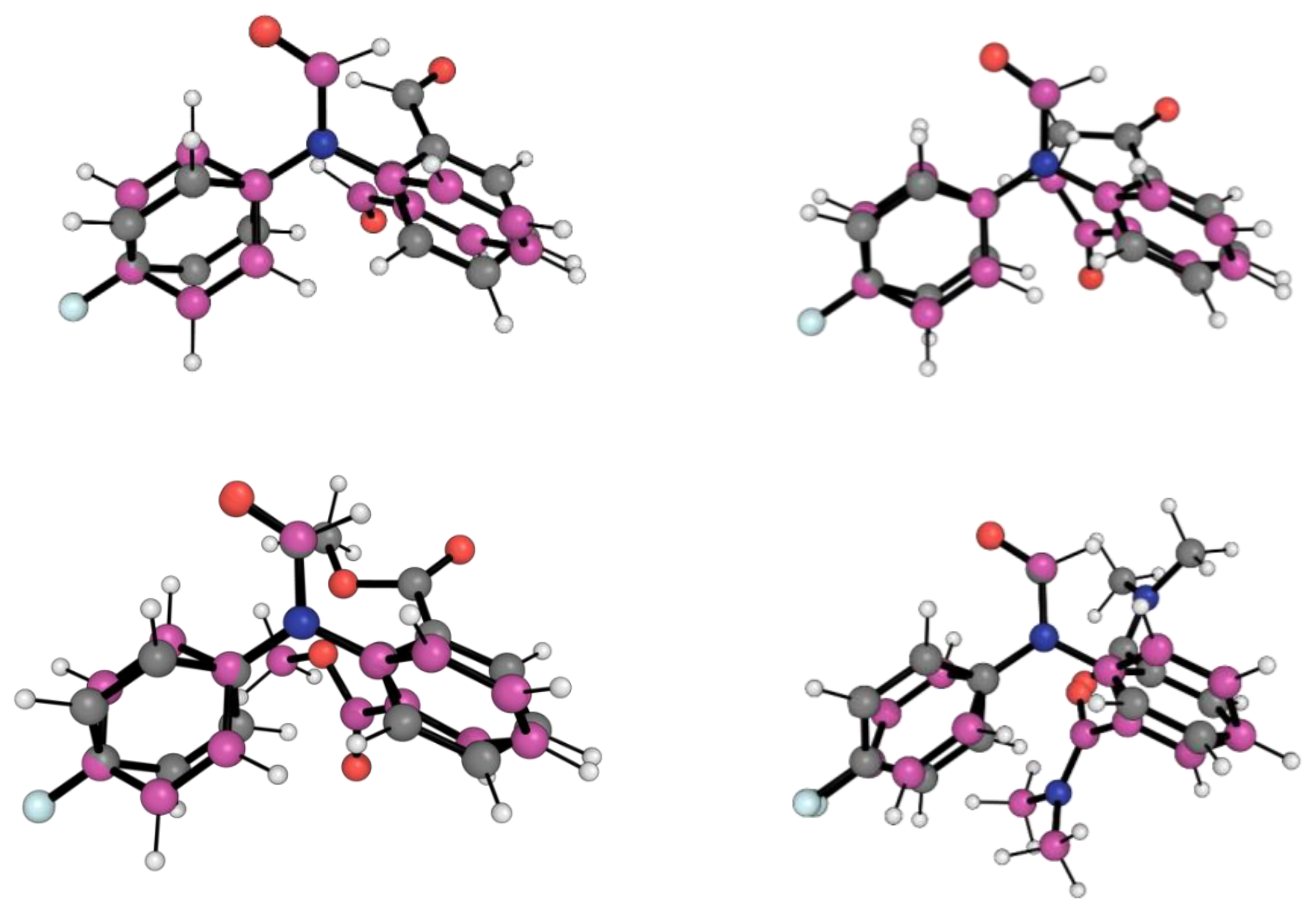
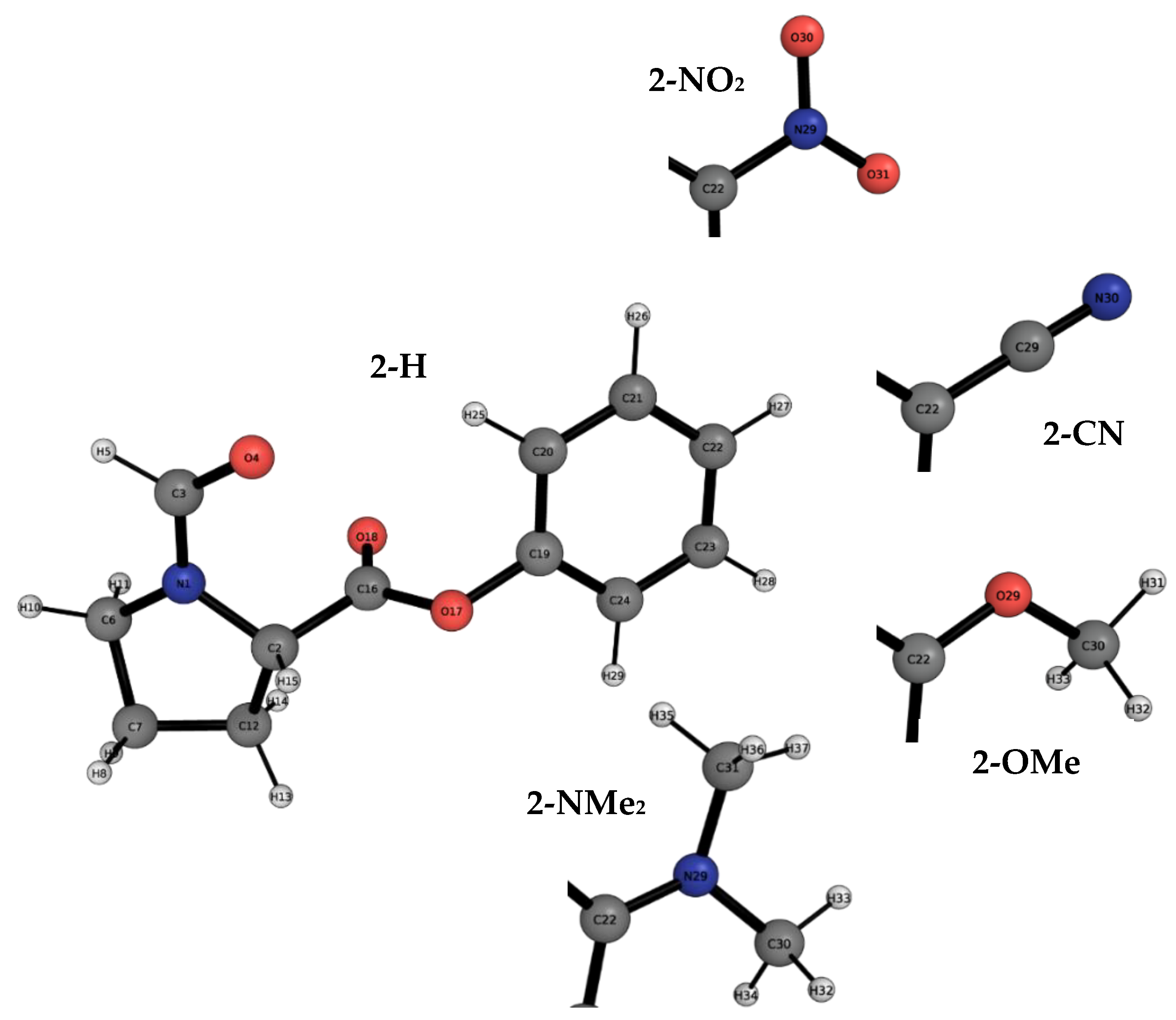
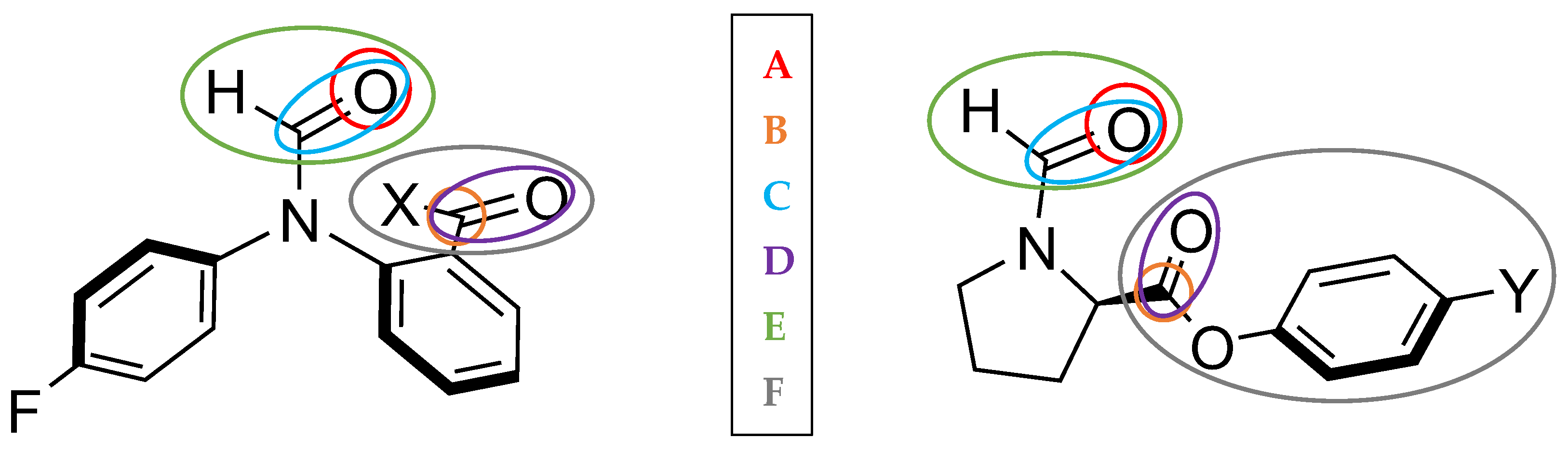
2.1. Conformational Landscape of 1-X
2.2. REG-IQA Analysis
3. Methods
3.1. Interacting Quantum Atoms (IQA)
3.2. Relative Energy Gradient (REG)
4. Computational Details
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kollman, P.A. Noncovalent interactions. Acc. Chem. Res. 1977, 10, 365–371. [Google Scholar] [CrossRef]
- Mueller-Dethlefs, K.; Hobza, P. Noncovalent Interactions: A challenge for experiment and theory. Chem. Rev. 2000, 100, 143–167. [Google Scholar] [CrossRef]
- London, F. The general theory of molecular forces. Trans. Faraday Soc. 1937, 33, 8b–26. [Google Scholar] [CrossRef]
- Dzyaloshinskii, I.E.; Lifshitz, E.M.; Pitaevskii, L.P. The general theory of van der Waals forces. Adv. Phys. 1961, 10, 165–209. [Google Scholar] [CrossRef]
- Hobza, P.; Müller-Dethlefs, K. Non-Covalent Interactions: Theory and Experiment; Royal Society of Chemistry: London, UK, 2010; Volume 2. [Google Scholar]
- Paliwal, S.; Geib, S.; Wilcox, C.S. Molecular torsion balance for weak molecular recognition forces. Effects of “tilted-T” edge-to-face aromatic interactions on conformational selection and solid-state structure. J. Am. Chem. Soc. 1994, 116, 4497–4498. [Google Scholar] [CrossRef]
- Cozzi, F.; Cinquini, M.; Annunziata, R.; Dwyer, T.; Siegel, J.S. Polar pi interactions between stacked aryls in 1, 8-diarylnaphthalenes. J. Am. Chem. Soc. 1992, 114, 5729–5733. [Google Scholar] [CrossRef]
- Cozzi, F.; Siegel, J.S. Interaction between stacked aryl groups in 1,8-diarylnaphthalenes: Dominance of polar/π over charge-transfer effects. Pure Appl. Chem. 1995, 67, 683–689. [Google Scholar] [CrossRef]
- Lin, B.; Karki, I.; Pellechia, P.J.; Shimizu, K.D. Electrostatically-gated molecular rotors. Chem. Commun. 2022, 58, 5869–5872. [Google Scholar] [CrossRef] [PubMed]
- Vik, E.C.; Li, P.; Madukwe, D.O.; Karki, I.; Tibbetts, G.S.; Shimizu, K.D. Analysis of the Orbital and Electrostatic Contributions to the Lone Pair–Aromatic Interaction Using Molecular Rotors. Org. Lett. 2021, 23, 8179–8182. [Google Scholar] [CrossRef]
- Li, P.; Vik, E.C.; Shimizu, K.D. N-Arylimide Molecular Balances: A Comprehensive Platform for Studying Aromatic Interactions in Solution. Accs. Chem. Res. 2020, 53, 2705–2714. [Google Scholar] [CrossRef]
- Vik, E.C.; Li, P.; Pellechia, P.J.; Shimizu, K.D. Transition-state stabilization by n → π* interactions measured using molecular rotors. J. Am. Chem. Soc. 2019, 141, 16579–16583. [Google Scholar] [CrossRef]
- Li, P.; Vik, E.C.; Maier, J.M.; Karki, I.; Strickland, S.M.; Umana, J.M.; Smith, M.D.; Pellechia, P.J.; Shimizu, K.D. Electrostatically driven CO−π aromatic interactions. J. Am. Chem. Soc. 2019, 141, 12513–12517. [Google Scholar] [CrossRef] [PubMed]
- Carroll, W.R.; Pellechia, P.; Shimizu, K.D. A Rigid Molecular Balance for Measuring Face-to-Face Arene−Arene Interactions. Org. Lett. 2008, 10, 3547–3550. [Google Scholar] [CrossRef]
- Hwang, J.; Li, P.; Carroll, W.R.; Smith, M.D.; Pellechia, P.J.; Shimizu, K.D. Additivity of Substituent Effects in Aromatic Stacking Interactions. J. Am. Chem. Soc. 2014, 136, 14060–14067. [Google Scholar] [CrossRef]
- Hwang, J.; Dial, B.E.; Li, P.; Kozik, M.E.; Smith, M.D.; Shimizu, K.D. How important are dispersion interactions to the strength of aromatic stacking interactions in solution? Chem. Sci. 2015, 6, 4358–4364. [Google Scholar] [CrossRef]
- Meredith, N.Y.; Borsley, S.; Smolyar, I.V.; Nichol, G.S.; Baker, C.M.; Ling, K.B.; Cockroft, S.L. Dissecting Solvent Effects on Hydrogen Bonding. Angew. Chem. 2022, 61, e202206604. [Google Scholar] [CrossRef] [PubMed]
- Elmi, A.; Cockroft, S.L. Quantifying interactions and solvent effects using molecular balances and model complexes. Acc. Chem. Res. 2020, 54, 92–103. [Google Scholar] [CrossRef]
- Muchowska, K.B.; Pascoe, D.J.; Borsley, S.; Smolyar, I.V.; Mati, I.K.; Adam, C.; Nichol, G.S.; Ling, K.B.; Cockroft, S.L. Reconciling electrostatic and n → π* orbital contributions in carbonyl interactions. Angew. Chem. 2020, 132, 14710–14716. [Google Scholar] [CrossRef]
- Yang, L.; Adam, C.; Nichol, G.S.; Cockroft, S.L. How much do van der Waals dispersion forces contribute to molecular recognition in solution? Nat. Chem. 2013, 5, 1006–1010. [Google Scholar] [CrossRef]
- Mati, I.K.; Adam, C.; Cockroft, S.L. Seeing through solvent effects using molecular balances. Chem. Sci. 2013, 4, 3965–3972. [Google Scholar] [CrossRef]
- Burns, R.J.; Mati, I.K.; Muchowska, K.B.; Adam, C.; Cockroft, S.L. Quantifying Through-Space Substituent Effects. Angew. Chem. Int. Ed. 2020, 132, 16860–16867. [Google Scholar] [CrossRef]
- Pascoe, D.J.; Ling, K.B.; Cockroft, S.L. The Origin of Chalcogen-Bonding Interactions. J. Am. Chem. Soc. 2017, 139, 15160–15167. [Google Scholar] [CrossRef]
- Yang, L.; Adam, C.; Cockroft, S.L. Quantifying Solvophobic Effects in Nonpolar Cohesive Interactions. J. Am. Chem. Soc. 2015, 137, 10084–10087. [Google Scholar] [CrossRef] [PubMed]
- Mati, I.K.; Cockroft, S.L. Molecular balances for quantifying non-covalent interactions. Chem. Soc. Rev. 2010, 39, 4195–4205. [Google Scholar] [CrossRef] [PubMed]
- Aliev, A.E.; Motherwell, M.B. Some Recent Advances in the Design and Use of Molecular Balances for the Experimental Quantification of Intramolecular Noncovalent Interactions of pi Systems. Chem. Eur. J. 2019, 25, 10516–10530. [Google Scholar] [CrossRef] [PubMed]
- Patkowski, K. Recent developments in symmetry-adapted perturbation theory. WIRES Comp. Mol. Sci. 2020, 10, e1452. [Google Scholar] [CrossRef]
- Szalewicz, K. Symmetry-adapted perturbation theory of intermolecular forces. WIRES Comp. Mol. Sci. 2012, 2, 254–272. [Google Scholar] [CrossRef]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. Natural bond orbital methods. WIRES Comp. Molec. Sci. 2012, 2, 1–42. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C. Natural Bond Orbitals and Extensions of Localized Bonding Concepts. Chem. Educ. Res. Pract. Eur. 2001, 2, 91–104. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Yang, L.; Brazier, J.B.; Hubbard, T.A.; Rogers, D.M.; Cockroft, S.L. Can Dispersion Forces Govern Aromatic Stacking in an Organic Solvent? Angew. Chem. Int. Ed. 2015, 55, 912–916. [Google Scholar] [CrossRef]
- Francisco, E.; Costales, A.; Menéndez-Herrero, M.; Martín Pendás, Á. Lewis Structures from Open Quantum Systems Natural Orbitals: Real Space Adaptive Natural Density Partitioning. J. Phys. Chem. A 2021, 125, 4013–4025. [Google Scholar] [CrossRef]
- Popelier, P.L.A. Non-covalent interactions from a Quantum Chemical Topology perspective. J. Mol. Model. 2022, 28, 276. [Google Scholar] [CrossRef]
- Thacker, J.C.R.; Popelier, P.L.A. The ANANKE Relative Energy Gradient (REG) Method to Automate IQA Analysis over Configurational Change. Theor. Chem. Acc. 2017, 136, 86. [Google Scholar] [CrossRef]
- Guevara-Vela, J.M.; Francisco, E.; Rocha-Rinza, T.; Martín Pendás, A. Interacting Quantum Atoms—A Review. Molecules 2020, 25, 4028. [Google Scholar] [CrossRef] [PubMed]
- Blanco, M.A.; Martín Pendás, A.; Francisco, E. Interacting Quantum Atoms: A Correlated Energy Decomposition Scheme Based on the Quantum Theory of Atoms in Molecules. J. Chem. Theor. Comp. 2005, 1, 1096–1109. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Atoms in Molecules. Acc. Chem. Res. 1985, 18, 9–15. [Google Scholar] [CrossRef]
- Wilson, A.L.; Popelier, P.L.A. Exponential Relationships capturing Atomistic Short-range Repulsion from the Interacting Quantum Atoms (IQA) Method. J. Phys. Chem. A 2016, 120, 9647–9659. [Google Scholar] [CrossRef] [PubMed]
- Gallegos, M.; Costales, A.; Martín Pendás, Á. Energetic Descriptors of Steric Hindrance in Real Space: An Improved IQA Picture. ChemPhysChem 2021, 22, 775–787. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Revilla, M.; Francisco, E.; Popelier, P.L.A.; Martín Pendás, Á. Domain-averaged Exchange Correlation Energies as a Physical Underpinning for Chemical Graphs. ChemPhysChem 2013, 14, 1211–1218. [Google Scholar] [CrossRef] [PubMed]
- Martín Pendás, Á.; Francisco, E.; Blanco, M.A.; Gatti, C. Bond Paths as Privileged Exchange Channels. Chem.—Eur. J. 2007, 13, 9362–9371. [Google Scholar] [CrossRef]
- Symons, B.C.B.; Williamson, D.J.; Brooks, C.M.; Wilson, A.L.; Popelier, P.L.A. Does the Intra-Atomic Deformation Energy of Interacting Quantum Atoms Represent Steric Energy? Chem. Open 2019, 8, 560–570. [Google Scholar]
- Falcioni, F.; Symons, B.C.B.; Popelier, P.L.A. REG-MULTI: Lifting the veil on electrostatic interactions. Adv. Quant. Chem. 2023, 88, 305–328. [Google Scholar]
- Triestram, L.; Falcioni, F.; Popelier, P.L.A. Interacting Quantum Atoms and Multipolar Electrostatic Study of XH···π Interactions. ACS Omega 2023, 8, 34844–34851. [Google Scholar] [CrossRef]
- Fischer, F.R.; Schweizer, W.B.; Diederich, F. Molecular Torsion Balances: Evidence for Favorable Orthogonal Dipolar Interactions Between Organic Fluorine and Amide Groups. Angew. Chem. Int. Ed. 2007, 46, 8270–8273. [Google Scholar] [CrossRef]
- Newberry, R.W.; VanVeller, B.; Guzei, I.A.; Raines, R.T. n → π* interactions of amides and thioamides: Implications for protein stability. J. Am. Chem. Soc. 2013, 135, 7843–7846. [Google Scholar] [CrossRef]
- Choudhary, A.; Kamer, K.J.; Raines, R.T. An n→ π* interaction in aspirin: Implications for structure and reactivity. J. Org. Chem. 2011, 76, 7933–7937. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, G.J.; Choudhary, A.; Raines, R.T.; Woolfson, D.N. n -> pi * interactions in proteins. Nat. Chem. Biol. 2010, 6, 615–620. [Google Scholar] [CrossRef] [PubMed]
- Hodges, J.A.; Raines, R.T. Energetics of an n → π* interaction that impacts protein structure. Org. Lett. 2006, 8, 4695–4697. [Google Scholar] [CrossRef]
- Hinderaker, M.P.; Raines, R.T. An electronic effect on protein structure. Protein Sci. 2003, 12, 1188–1194. [Google Scholar] [CrossRef]
- Newberry, R.W.; Raines, R.T. The n → π* Interaction. Acc. Chem. Res. 2017, 50, 1838–1846. [Google Scholar] [CrossRef] [PubMed]
- Paulini, R.; Mueller, K.; Diederich, F. Orthogonal Multipolar Interactions in Structural Chemistry and Biology. Angew. Chem. Int. Ed. 2005, 44, 1788–1805. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.A.; Schulte, M.M.; Weiss, J.; Zsolnai, L.; Jacobi, A.; Huttner, G.; Frenking, G.; Boehme, C.; Vyboishchikov, S.F. Transition metal coordinated Al(X)L-2 and Ga(X)L-2 fragments. J. Amer. Chem. Soc. 1998, 120, 1237–1248. [Google Scholar] [CrossRef]
- Hunter, C.A. Quantifying Intermolecular Interactions: Guidelines for the Molecular Recognition Toolbox. Angew. Chem. 2004, 43, 5310–5324. [Google Scholar]
- Alkorta, I.; Elguero, J.; Popelier, P.L. A relative energy gradient (REG) study of the nitrogen inversion in N-substituted aziridines. Chem. Phys. Lett. 2020, 758, 137927. [Google Scholar] [CrossRef]
- Kemnitz, C.R.; Loewen, M.J. “Amide resonance” correlates with a breadth of C–N rotation barriers. J. Am. Chem. Soc. 2007, 129, 2521–2528. [Google Scholar] [CrossRef]
- Clayden, J.; Greeves, N.; Warren, S. Organic Chemistry; Oxford University Press: Oxford, UK, 2012. [Google Scholar]
- Silva, A.F.; Duarte, L.J.; Popelier, P.L.A. Contributions of IQA electron correlation in understanding the chemical bond and non-covalent interactions. Struct. Chem. 2020, 31, 507–519. [Google Scholar] [CrossRef]
- Maxwell, P.; Martín Pendás, A.; Popelier, P.L.A. Extension of the Interacting Quantum Atoms (IQA) Approach to B3LYP Level Density Functional Theory. PhysChemChemPhys 2016, 18, 20986–21000. [Google Scholar] [CrossRef]
- Francisco, E.; Casals-Sainz, J.L.; Rocha-Rinza, T.; Martin Pendas, A. Partitioning the DFT Exchange-Correlation Energy in Line with the Interacting Quantum Atoms Approach. Theor. Chem. Accs. 2016, 135, 170. [Google Scholar] [CrossRef]
- Vincent, M.A.; Popelier, P.L.A. IQA analysis of the two-particle density matrix: Chemical insight and computational efficiency. Theor. Chem. Acc. 2023, 142, 119. [Google Scholar] [CrossRef]
- Stone, A.J. The Theory of Intermolecular Forces, 2nd ed.; Clarendon Press: Oxford, UK, 2013; Volume 32, p. 264. [Google Scholar]
- Frisch, M.J.; Scuseria, H.B.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; Li, X.; Caricato, M.; et al. GAUSSIAN16. 2016. Available online: https://gaussian.com/gaussian16/ (accessed on 31 January 2024).
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Density functional theory with London dispersion corrections. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 1, 211–228. [Google Scholar] [CrossRef]
- Schlegel, H.B. Optimization of Equilibrium Geometries and Transition Structures. J. Comp. Chem. 1982, 3, 214–218. [Google Scholar] [CrossRef]
- Keith, T.A.; AIMAll. TK Gristmill Software; AIMAll: Overland Park, KS, USA, 2019. [Google Scholar]
- Falcioni, F.; Duarte, L.J.; Popelier, P.L.A. REG.py (Version 0.1). Available online: https://github.com/popelier-group/REG (accessed on 31 January 2024).
- Schrödinger, L.L.C. The PyMOL Molecular Graphics System, Version 2.5; Schrödinger, LLC: New York, NY, USA, 2022. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Balance | ΔEexp | ΔELECC-BOTEC | ΔELECC-LEOC |
|---|---|---|---|
| 1-H | 3.1 | 0.3 | 1.2 |
| 1-Me | −4.5 | −6.1 | −5.7 |
| 1-OMe | −0.8 | −1.4 | 0.5 |
| 1-NMe2 | −5.3 | −12.6 | −4.4 |
| RMSE (kJ/mol) | 4.0 | 1.4 | |
| R2 | 0.78 | 0.84 |
| 1-H | 1-Me | ||||
| TERM | REG | R | TERM | REG | R |
| Eintra(n1) | −2.3 | −0.96 | Vcl(c4,c22) | −2.5 | −0.98 |
| Vcl(n1,o5) | −1.4 | −0.99 | Vcl(n1,o23) | −2.5 | −1.00 |
| Eintra(c4) | −1.1 | −0.98 | Vcl(o5,o23) | −2.3 | −0.98 |
| Vxc(c4,o5) | −0.8 | −0.98 | Eintra(n1) | −2.0 | −1.00 |
| Vcl(c4,c22) | −0.7 | −0.92 | Vcl(n1,o5) | −1.2 | −0.97 |
| Eintra(o5) | 0.5 | 0.97 | Vcl(n1,c2) | 1 | 0.99 |
| Vcl(o5,c22) | 0.7 | 0.82 | Vcl(n1,c22) | 1.8 | 1 |
| Vcl(n1,c2) | 0.8 | 0.88 | Vcl(o5,c22) | 2.1 | 0.97 |
| Vxc(n1,c4) | 1 | 0.96 | Vcl(c4,o23) | 3 | 0.99 |
| Vcl(n1,c4) | 4.7 | 0.99 | Vcl(n1,c4) | 3.6 | 0.97 |
| 1-OMe | 1-NMe2 | ||||
| TERM | REG | R | TERM | REG | R |
| Eintra(n1) | −2.1 | −0.98 | Vcl(c4,c22) | −3.2 | −0.99 |
| Vcl(c4,c22) | −1.2 | −0.98 | Vcl(o5,n24) | −2.6 | −0.99 |
| Vcl(n1,o5) | −1.2 | −0.99 | Vcl(o5,o23) | −2.5 | −0.99 |
| Eintra(c4) | −1.1 | −0.99 | Vcl(n1,o23) | −1.9 | −0.98 |
| Vcl(o5,o23) | −1.0 | −0.97 | Eintra(n1) | −1.6 | −0.95 |
| Vxc(n1,c4) | 0.7 | 0.95 | Vcl(h13,o23) | 1 | 0.91 |
| Vcl(n1,c2) | 0.8 | 0.92 | Vcl(c4,n24) | 1.7 | 0.98 |
| Vcl(o5,c22) | 1 | 0.89 | Vcl(c4,o23) | 3 | 0.99 |
| Vcl(c4,o23) | 1.5 | 0.99 | Vcl(o5,c22) | 3.3 | 0.99 |
| Vcl(n1,c4) | 4.1 | 1 | Vcl(n1,c4) | 3.9 | 0.97 |
| 2-H | |||||
| TERM | REG | R | |||
| Eintra(n1) | −4.1 | −0.99 | |||
| Vcl(c3,c16) | −2.2 | −0.98 | |||
| Eintra(c3) | −2.2 | −1.00 | |||
| Vcl(n1,o4) | −1.9 | −0.99 | |||
| Vcl(o4,o17) | −1.6 | −1.00 | |||
| Vcl(c3,o17) | 1.3 | 1 | |||
| Vcl(n1,c6) | 1.4 | 0.97 | |||
| Vcl(n1,c16) | 1.8 | 0.99 | |||
| Vcl(o4,c16) | 2.5 | 1 | |||
| Vcl(n1,c3) | 6.9 | 0.99 | |||
| 2-NO2 | 2-CN | ||||
| TERM | REG | R | TERM | REG | R |
| Eintra(n1) | −3.8 | −0.99 | Eintra(n1) | −3.8 | −0.98 |
| Vcl(c3,c16) | −2.3 | −0.98 | Vcl(c3,c16) | −2.3 | −0.99 |
| Eintra(c3) | −2.0 | −1.00 | Eintra(c3) | −2.0 | −1.00 |
| Vcl(n1,o4) | −1.8 | −0.99 | Vcl(n1,o4) | −1.8 | −0.99 |
| Vcl(o4,o17) | −1.7 | −1.00 | Vcl(o4,o17) | −1.6 | −1.00 |
| Vcl(n1,c6) | 1.3 | 0.97 | Vcl(c3,o17) | 1.2 | 1 |
| Vcl(c3,o17) | 1.3 | 1 | Vcl(n1,c6) | 1.4 | 0.97 |
| Vcl(n1,c16) | 1.7 | 0.99 | Vcl(n1,c16) | 1.7 | 0.99 |
| Vcl(o4,c16) | 2.7 | 1 | Vcl(o4,c16) | 2.6 | 1 |
| Vcl(n1,c3) | 6.4 | 0.99 | Vcl(n1,c3) | 6.4 | 0.99 |
| 2-OMe | 2-NMe2 | ||||
| TERM | REG | R | TERM | REG | R |
| Eintra(n1) | −4.1 | −0.99 | Eintra(n1) | −4.2 | −0.99 |
| Vcl(c3,c16) | −2.3 | −0.98 | Eintra(c3) | −2.3 | −1.00 |
| Eintra(c3) | −2.2 | −1.00 | Vcl(c3,c16) | −2.1 | −0.98 |
| Vcl(n1,o4) | −1.9 | −0.99 | Vcl(n1,o4) | −1.9 | −0.99 |
| Vcl(o4,o17) | −1.6 | −1.00 | Vcl(o4,o17) | −1.5 | −1.00 |
| Vcl(c3,o17) | 1.3 | 1 | Vcl(n1,c2) | 1.3 | 0.97 |
| Vcl(n1,c6) | 1.4 | 0.97 | Vcl(n1,c6) | 1.5 | 0.97 |
| Vcl(n1,c16) | 1.8 | 0.99 | Vcl(n1,c16) | 1.9 | 0.99 |
| Vcl(o4,c16) | 2.6 | 1 | Vcl(o4,c16) | 2.4 | 0.99 |
| Vcl(n1,c3) | 6.8 | 0.99 | Vcl(n1,c3) | 7.1 | 0.99 |
| 1-H | 1-Me | 1-OMe | 1-NMe2 | |||||
|---|---|---|---|---|---|---|---|---|
| Term | REG | REG Ratio | REG | REG Ratio | REG | REG Ratio | REG | REG Ratio |
| Vxc(A,B) | 0.05 | 99 | 0.21 | 17 | 0.06 | 66 | 0.05 | 82 |
| Vxc(C,D) | 0.07 | 64 | 0.33 | 11 | 0.19 | 21 | 0.10 | 37 |
| Vxc(E,F) | 0.10 | 46 | 0.06 | 59 | 0.18 | 23 | 0.27 | 14 |
| Vcl(A,B) | 0.67 | 7 | 2.07 | 2 | 1.05 | 4 | 3.28 | 1 |
| Vcl(C,D) | −0.05 | −91 | 0.28 | 13 | 0.28 | 15 | 0.55 | 7 |
| Vcl(E,F) | 0.02 | 220 | 0.30 | 12 | 0.21 | 20 | 0.32 | 12 |
| 2-NO2 | 2-CN | 2-H | 2-OMe | 2-NMe2 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Term | REG | REG Ratio | REG | REG Ratio | REG | REG Ratio | REG | REG Ratio | REG | REG Ratio |
| Vxc(A,B) | 0.22 | 30 | 0.21 | 31 | 0.19 | 36 | 0.18 | 37 | 0.17 | 42 |
| Vxc(C,D) | 0.19 | 34 | 0.18 | 35 | 0.12 | 55 | 0.13 | 53 | 0.09 | 83 |
| Vxc(E,F) | 0.31 | 21 | 0.27 | 24 | 0.22 | 31 | 0.17 | 41 | 0.09 | 76 |
| Vcl(A,B) | 2.67 | 2 | 2.64 | 2 | 2.55 | 3 | 2.57 | 3 | 2.42 | 3 |
| Vcl(C,D) | 0.00 | −2256 | 0.03 | 226 | −0.08 | −87 | −0.08 | −86 | −0.11 | −62 |
| Vcl(E,F) | −0.12 | −54 | −0.10 | −62 | −0.07 | −105 | −0.18 | −38 | −0.14 | −50 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Falcioni, F.; Bennett, S.; Stroer-Jarvis, P.; Popelier, P.L.A. Probing Non-Covalent Interactions through Molecular Balances: A REG-IQA Study. Molecules 2024, 29, 1043. https://doi.org/10.3390/molecules29051043
Falcioni F, Bennett S, Stroer-Jarvis P, Popelier PLA. Probing Non-Covalent Interactions through Molecular Balances: A REG-IQA Study. Molecules. 2024; 29(5):1043. https://doi.org/10.3390/molecules29051043
Chicago/Turabian StyleFalcioni, Fabio, Sophie Bennett, Pallas Stroer-Jarvis, and Paul L. A. Popelier. 2024. "Probing Non-Covalent Interactions through Molecular Balances: A REG-IQA Study" Molecules 29, no. 5: 1043. https://doi.org/10.3390/molecules29051043
APA StyleFalcioni, F., Bennett, S., Stroer-Jarvis, P., & Popelier, P. L. A. (2024). Probing Non-Covalent Interactions through Molecular Balances: A REG-IQA Study. Molecules, 29(5), 1043. https://doi.org/10.3390/molecules29051043