
An Efficient Approach to the Accurate Prediction of Mutational Effects in Antigen Binding to the MHC1



Abstract
1. Introduction
2. Results and Discussion
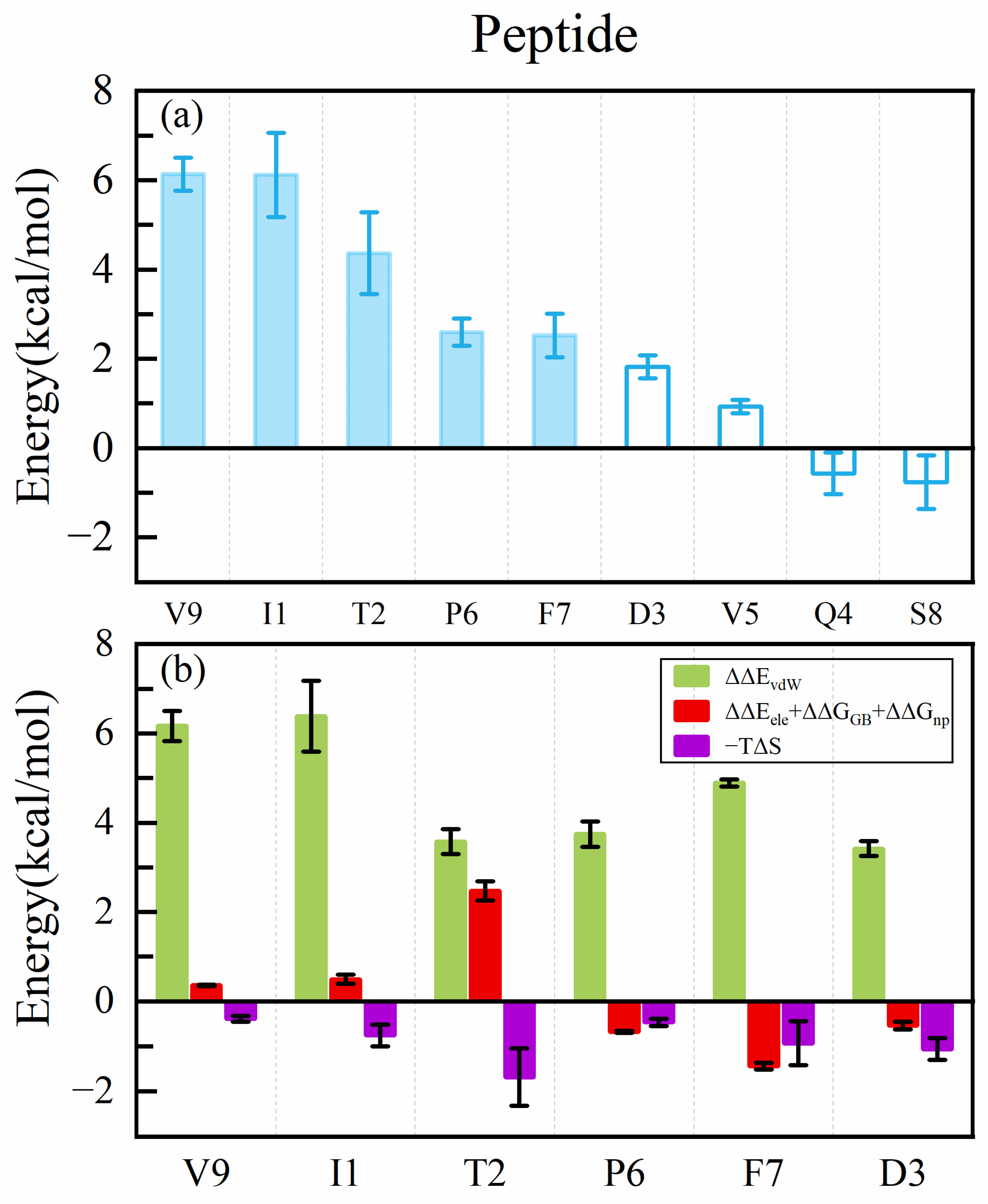
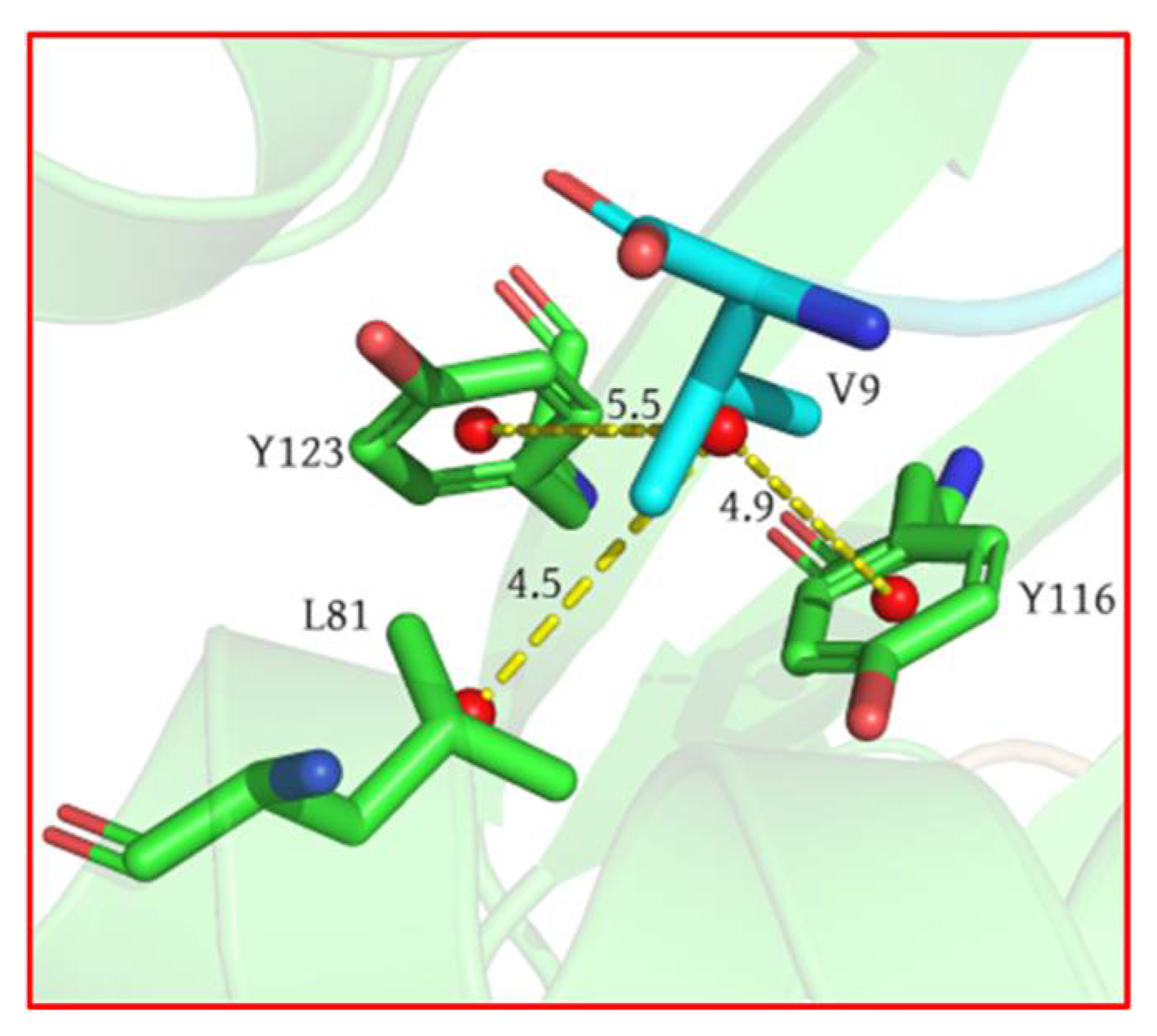
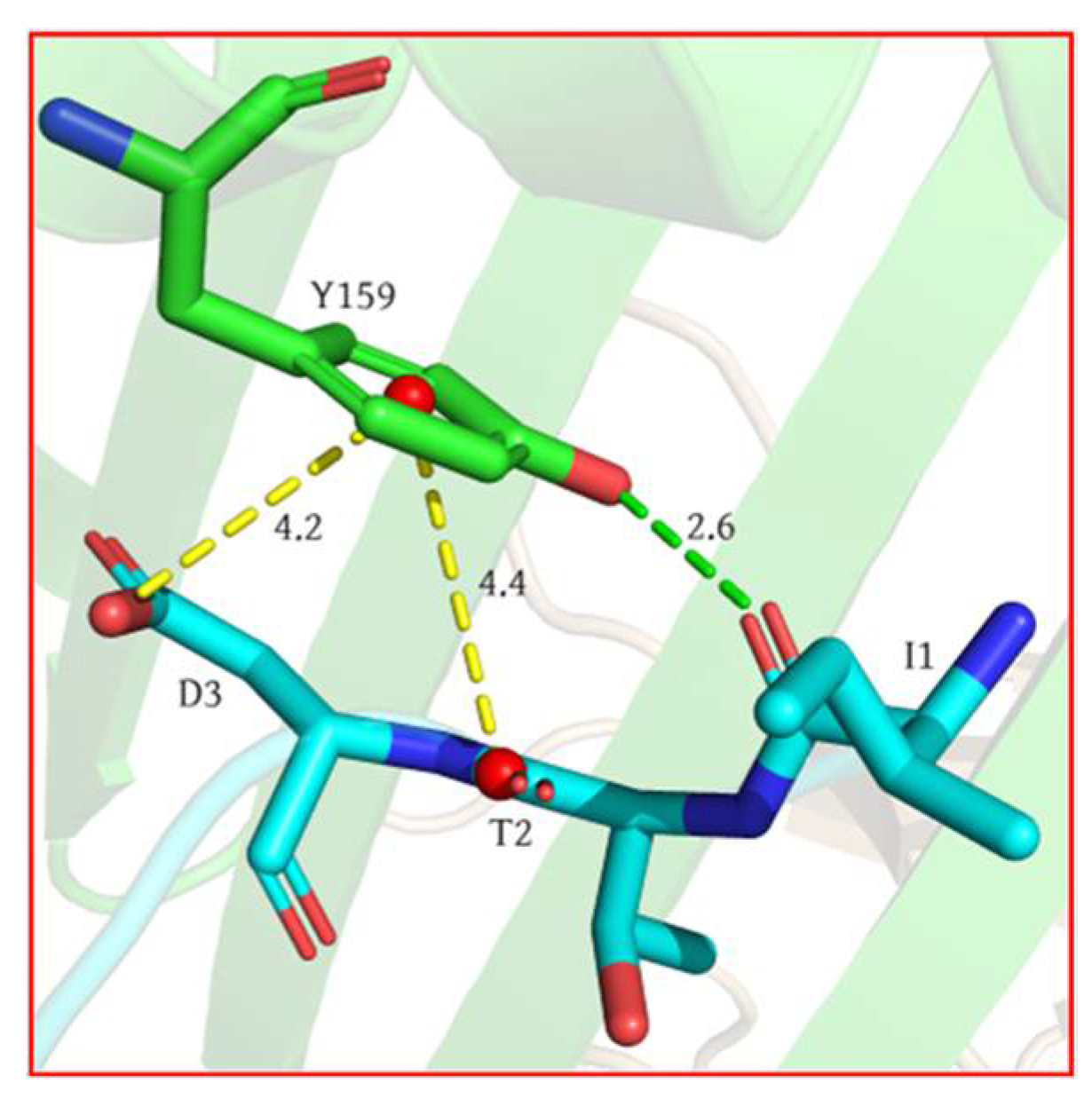
2.1. Analysis of Hotspot Residues on Peptide
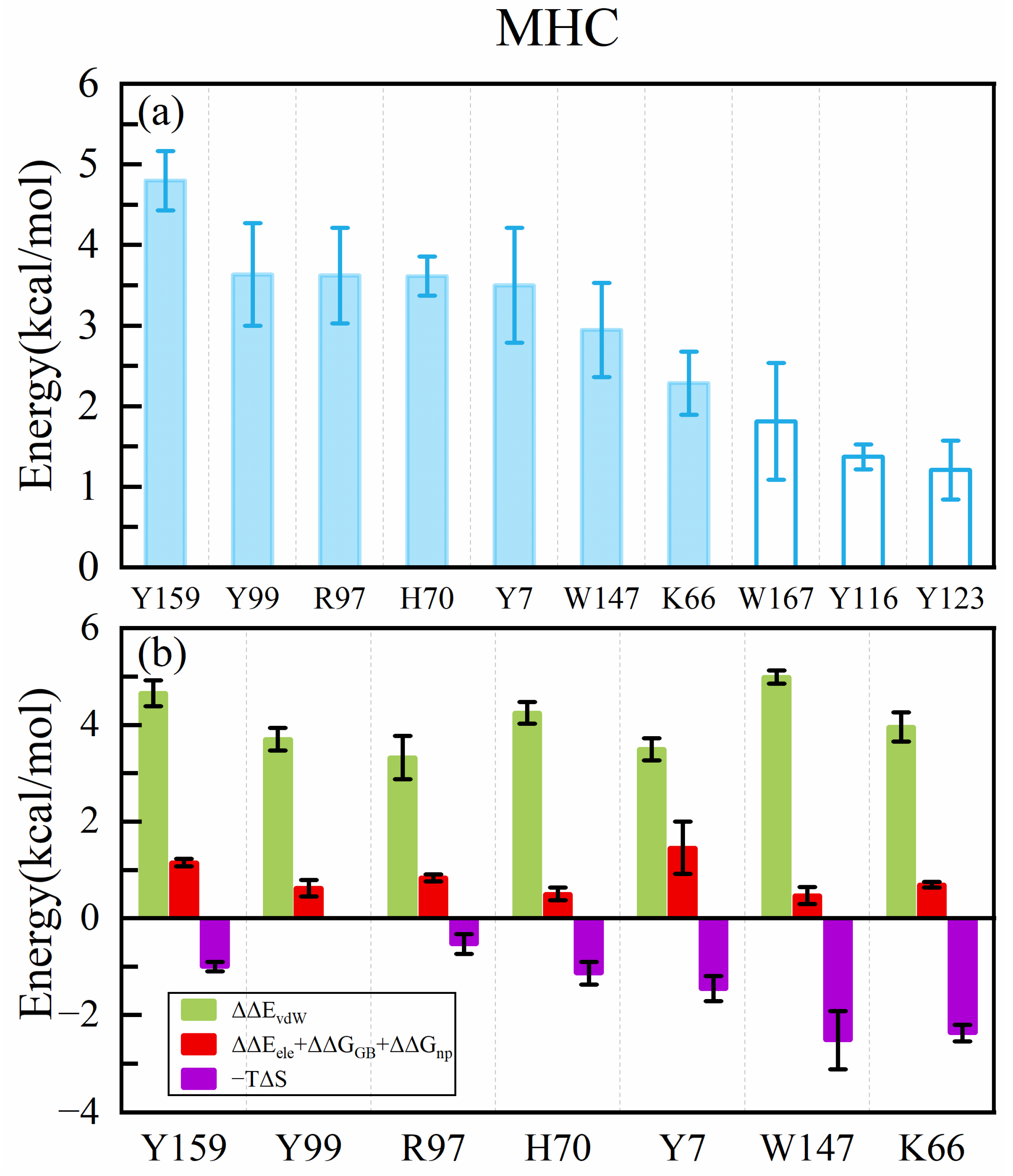
2.2. Analysis of Hotspot Residues on MHC1
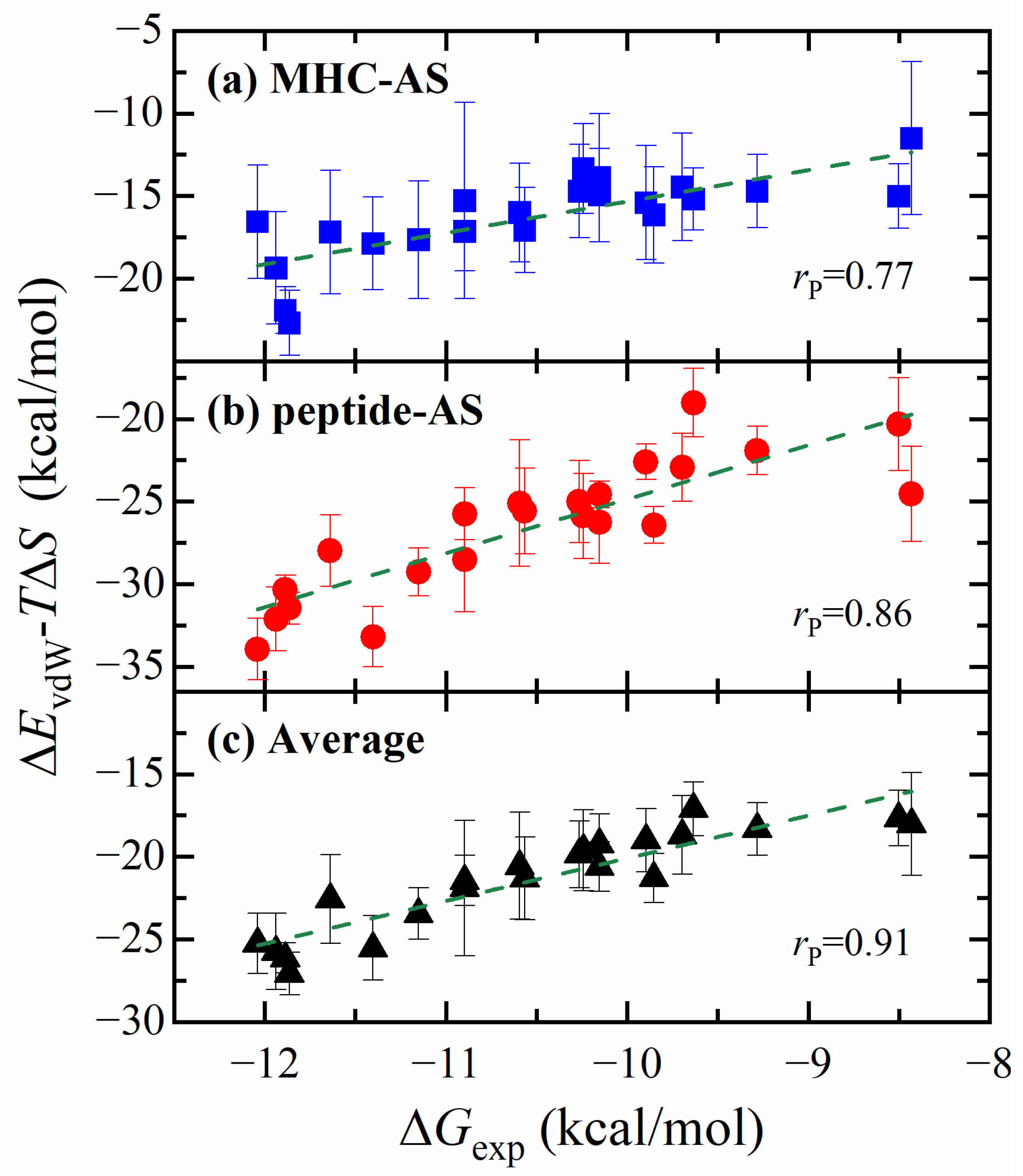
2.3. Free Energy Calculation Using ASGBIE Method
2.4. Comparison with Alchemical Method
3. Methods
3.1. MD Simulations
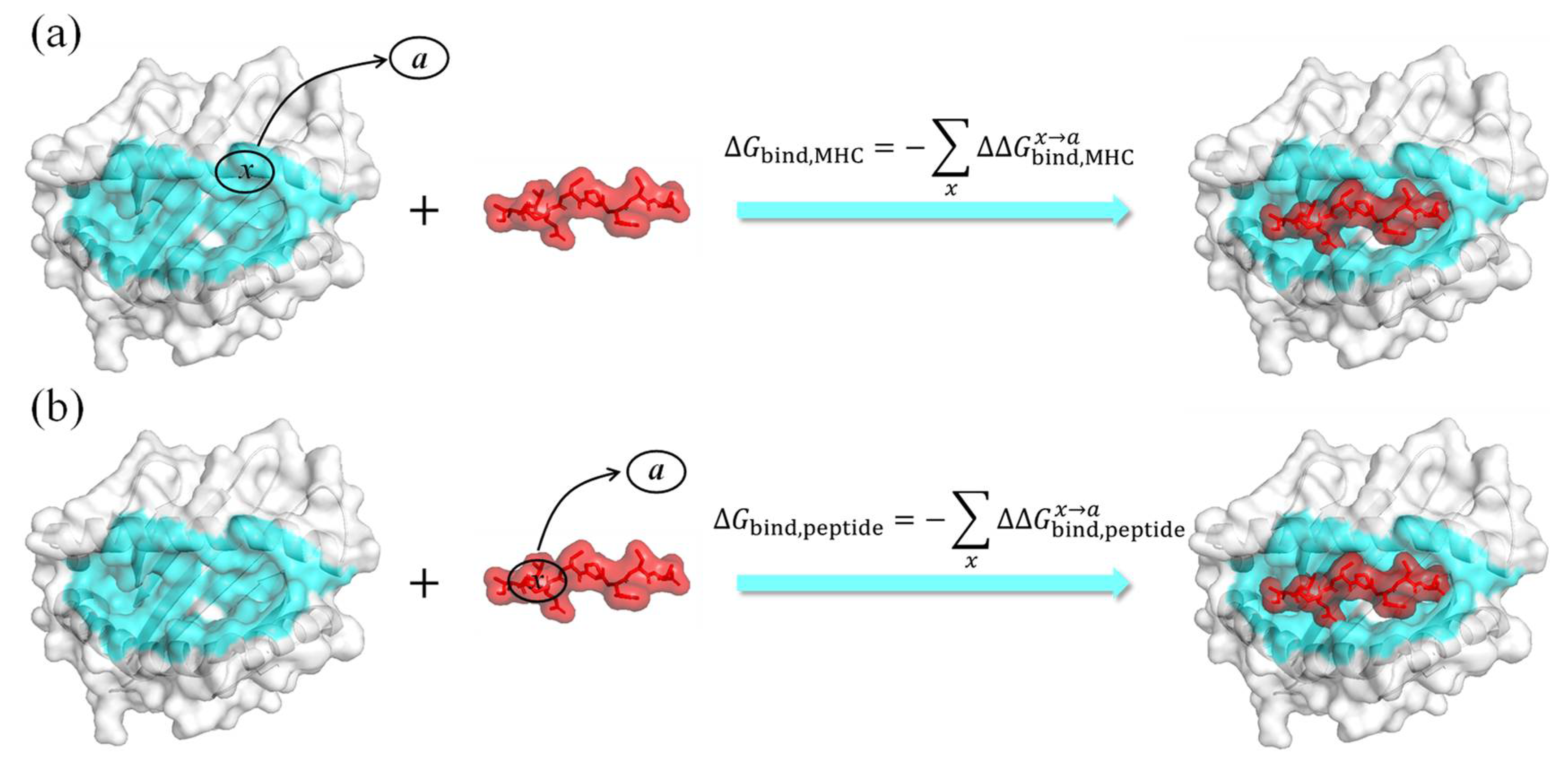
3.2. ASGBIE Method
3.2.1. Alanine Scanning
3.2.2. IE Method for Entropy Calculation
3.2.3. Optimized ASGBIE Method
4. Conclusions
- Our calculation showed that the two terminal residues of the peptide contributed the most to the peptide binding to the MHC1, with interactions dominated by the van der Waals interaction, followed by the no. 2 (threonine) residue. The antigen–MHC1 binding is dominated by hydrophobic interactions.
- The quantitative effect of the peptide (antigen) mutation on the antigen–MHC1 binding free energy is accurately predicted by the ASGBIE method with a good correlation to the experimental data. In particular, when electrostatic interactions (together with solvation) are neglected, the accuracy of the ASGBIE calculation is further improved. This finding suggests that for antigen–MHC1 bindings primarily governed by hydrophobic interactions, inaccuracies inherent in the point charge representation of electrostatic interactions within the force field and those in the implicit solvation model can detrimentally affect the accuracy of the results.
- The ASGBIE method is optimized by using an averaged version, i.e., averaging the summation over residues on both interacting partners. The accuracy of this optimized ASGBIE result is further improved. This enhancement stems from the complementary nature of the MHC–AS and Peptide–AS methods.
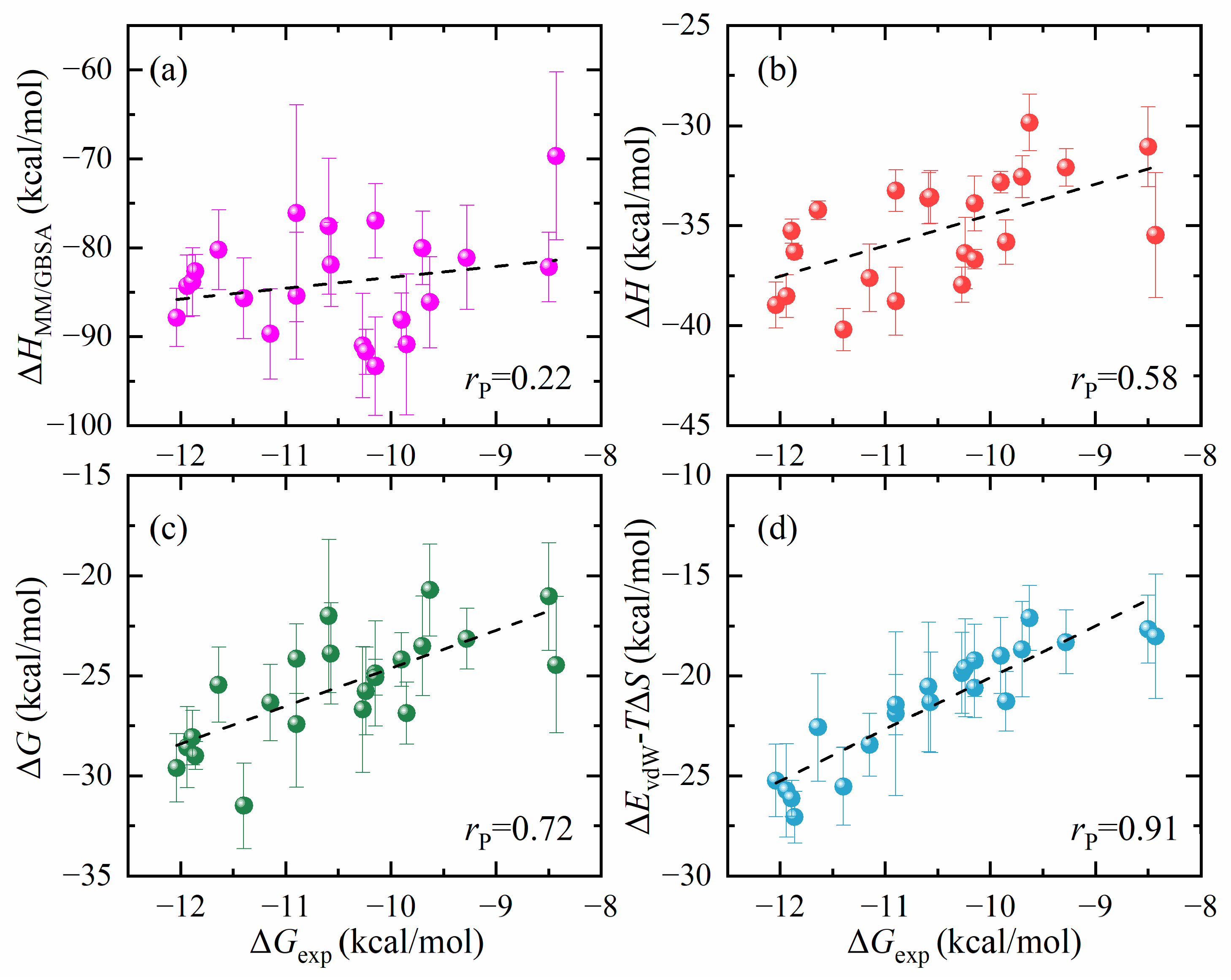
- The antigen–MHC1 binding energy calculation using the standard MM/GBSA method does not result in a good correlation with the experimental result. We also performed alchemical calculations of binding energy for comparison and the results are mixed. For peptide mutations that do not involve changes in electric charge, the alchemical calculation results in a very good correlation with the experimental result. However, for charge-changing mutations, the alchemical calculations result in significant errors. This discrepancy is a recognized limitation in alchemical methods involving a change of electric charge between the initial and the final states.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pishesha, N.; Harmand, T.J.; Ploegh, H.L. A guide to antigen processing and presentation. Nat. Rev. Immunol. 2022, 22, 751–764. [Google Scholar] [CrossRef] [PubMed]
- Neefjes, J.; Jongsma, M.L.M.; Paul, P.; Bakke, O. Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nat. Rev. Immunol. 2011, 11, 823–836. [Google Scholar] [PubMed]
- Rammensee, H.-G.; Friede, T.; Stevanović, S. MHC ligands and peptide motifs: First listing. Immunogenetics 1995, 41, 178–228. [Google Scholar] [CrossRef] [PubMed]
- Rudolph, M.G.; Stanfield, R.L.; Wilson, I.A. How TCRS bind MHCS, peptides, and coreceptors. Annu. Rev. Immunol. 2006, 24, 419–466. [Google Scholar] [CrossRef] [PubMed]
- Hoppes, R.; Oostvogels, R.; Luimstra, J.J.; Wals, K.; Toebes, M.; Bies, L.; Ekkebus, R.; Rijal, P.; Celie, P.H.N.; Huang, J.H.; et al. Altered Peptide Ligands Revisited: Vaccine Design through Chemically Modified HLA-A2–Restricted T Cell Epitopes. J. Immunol. 2014, 193, 4803–4813. [Google Scholar] [CrossRef]
- Tang, Y.; Lin, Z.; Ni, B.; Wei, J.; Han, J.; Wang, H.; Wu, Y. An altered peptide ligand for naïve cytotoxic T lymphocyte epitope of TRP-2(180–188) enhanced immunogenicity. Cancer Immunol. Immunother. 2007, 56, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Mei, S.; Li, F.; Leier, A.; Marquez-Lago, T.T.; Giam, K.; Croft, N.P.; Akutsu, T.; Smith, A.I.; Li, J.; Rossjohn, J.; et al. A comprehensive review and performance evaluation of bioinformatics tools for HLA class I peptide-binding prediction. Brief. Bioinf. 2020, 21, 1119–1135. [Google Scholar] [CrossRef]
- Liu, G.; Li, D.; Li, Z.; Qiu, S.; Li, W.; Chao, C.-C.; Yang, N.; Li, H.; Cheng, Z.; Song, X.; et al. PSSMHCpan: A novel PSSM-based software for predicting class I peptide-HLA binding affinity. GigaScience 2017, 6, 1–11. [Google Scholar] [CrossRef]
- Rammensee, H.G.; Bachmann, J.; Emmerich, N.P.N.; Bachor, O.A.; Stevanović, S. SYFPEITHI: Database for MHC ligands and peptide motifs. Immunogenetics 1999, 50, 213–219. [Google Scholar] [CrossRef]
- Jurtz, V.; Paul, S.; Andreatta, M.; Marcatili, P.; Peters, B.; Nielsen, M. NetMHCpan-4.0: Improved Peptide–MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J. Immunol. 2017, 199, 3360–3368. [Google Scholar] [CrossRef]
- O’Donnell, T.J.; Rubinsteyn, A.; Bonsack, M.; Riemer, A.B.; Laserson, U.; Hammerbacher, J. MHCflurry: Open-Source Class I MHC Binding Affinity Prediction. Cell Syst. 2018, 7, 129–132.e4. [Google Scholar] [CrossRef] [PubMed]
- Karosiene, E.; Lundegaard, C.; Lund, O.; Nielsen, M. NetMHCcons: A consensus method for the major histocompatibility complex class I predictions. Immunogenetics 2012, 64, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Ponomarenko, J.; Zhu, Z.; Tamang, D.; Wang, P.; Greenbaum, J.; Lundegaard, C.; Sette, A.; Lund, O.; Bourne, P.E.; et al. Immune epitope database analysis resource. Nucleic Acids Res. 2012, 40, W525–W530. [Google Scholar] [CrossRef]
- Kirkwood, J.G. Statistical Mechanics of Fluid Mixtures. J. Chem. Phys. 1935, 3, 300–313. [Google Scholar] [CrossRef]
- Zwanzig, R.W. High-Temperature Equation of State by a Perturbation Method. I. Nonpolar Gases. J. Chem. Phys. 1954, 22, 1420–1426. [Google Scholar] [CrossRef]
- Bennett, C.H. Efficient estimation of free energy differences from Monte Carlo data. J. Comput. Phys. 1976, 22, 245–268. [Google Scholar] [CrossRef]
- Shirts, M.R.; Chodera, J.D. Statistically optimal analysis of samples from multiple equilibrium states. J. Chem. Phys. 2008, 129, 124105. [Google Scholar] [CrossRef] [PubMed]
- de Ruiter, A.; Oostenbrink, C. Advances in the calculation of binding free energies. Curr. Opin. Struct. Biol. 2020, 61, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Song, L.F.; Merz, K.M., Jr. Evolution of Alchemical Free Energy Methods in Drug Discovery. J. Chem. Inf. Model. 2020, 60, 5308–5318. [Google Scholar] [CrossRef]
- Kollman, P.A.; Massova, I.; Reyes, C.; Kuhn, B.; Huo, S.; Chong, L.; Lee, M.; Lee, T.; Duan, Y.; Wang, W.; et al. Calculating Structures and Free Energies of Complex Molecules: Combining Molecular Mechanics and Continuum Models. Acc. Chem. Res. 2000, 33, 889–897. [Google Scholar] [CrossRef]
- Srinivasan, J.; Miller, J.; Kollman, P.A.; Case, D.A. Continuum Solvent Studies of the Stability of RNA Hairpin Loops and Helices. J. Biomol. Struct. Dyn. 1998, 16, 671–682. [Google Scholar] [CrossRef]
- Liu, X.; Peng, L.; Zhou, Y.; Zhang, Y.; Zhang, J.Z.H. Computational Alanine Scanning with Interaction Entropy for Protein–Ligand Binding Free Energies. J. Chem. Theory Comput. 2018, 14, 1772–1780. [Google Scholar] [CrossRef]
- Yan, Y.; Yang, M.; Ji, C.G.; Zhang, J.Z.H. Interaction Entropy for Computational Alanine Scanning. J. Chem. Inf. Model. 2017, 57, 1112–1122. [Google Scholar] [CrossRef]
- Duan, L.; Liu, X.; Zhang, J.Z.H. Interaction Entropy: A New Paradigm for Highly Efficient and Reliable Computation of Protein–Ligand Binding Free Energy. J. Am. Chem. Soc. 2016, 138, 5722–5728. [Google Scholar] [CrossRef]
- Clackson, T.; Wells, J.A. A Hot Spot of Binding Energy in a Hormone-Receptor Interface. Science 1995, 267, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Cukuroglu, E.; Engin, H.B.; Gursoy, A.; Keskin, O. Hot spots in protein–protein interfaces: Towards drug discovery. Prog. Biophys. Biophys. Chem. 2014, 116, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Qi, Y.; Gao, Y.; Zhang, J.Z.H. A method for efficient calculation of thermal stability of proteins upon point mutations. Phys. Chem. Chem. Phys. 2020, 22, 8461–8466. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Bao, J.; Yang, Y.; Dong, S.; Zhang, L.; Qi, Y.; Zhang, J.Z.H. Study of SHMT2 Inhibitors and Their Binding Mechanism by Computational Alanine Scanning. J. Chem. Inf. Model. 2019, 59, 3871–3878. [Google Scholar] [CrossRef] [PubMed]
- Song, L.F.; Lee, T.-S.; Zhu, C.; York, D.M.; Merz, K.M., Jr. Using AMBER18 for Relative Free Energy Calculations. J. Chem. Inf. Model. 2019, 59, 3128–3135. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, M.R.; Salgaller, M.L.; Southwood, S.; Robbins, P.F.; Sette, A.; Rosenberg, S.A.; Kawakami, Y. Improved induction of melanoma-reactive CTL with peptides from the melanoma antigen gp100 modified at HLA-A*0201-binding residues. J. Immunol. 1996, 157, 2539–2548. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Price, D.J.; Brooks, C.L., III. A modified TIP3P water potential for simulation with Ewald summation. J. Chem. Phys. 2004, 121, 10096–10103. [Google Scholar] [CrossRef] [PubMed]
- Pastor, R.W.; Brooks, B.R.; Szabo, A. An analysis of the accuracy of Langevin and molecular dynamics algorithms. Mol. Phys. 1988, 65, 1409–1419. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Massova, I.; Kollman, P.A. Combined molecular mechanical and continuum solvent approach (MM-PBSA/GBSA) to predict ligand binding. Perspect. Drug Discov. Des. 2000, 18, 113–135. [Google Scholar] [CrossRef]
- Li, Z.; Bao, J.; Qi, Y.; Zhang, J.Z.H. Computational approaches to studying methylated H4K20 recognition by DNA repair factor 53BP1. Phys. Chem. Chem. Phys. 2020, 22, 6136–6144. [Google Scholar] [CrossRef]
- Sun, Z.; Yan, Y.N.; Yang, M.; Zhang, J.Z.H. Interaction entropy for protein-protein binding. J. Chem. Phys. 2017, 146, 124124. [Google Scholar] [CrossRef]
- Knapp, B.; Ospina, L.; Deane, C.M. Avoiding False Positive Conclusions in Molecular Simulation: The Importance of Replicas. J. Chem. Theory Comput. 2018, 14, 6127–6138. [Google Scholar] [CrossRef]
- Hünenberger, P.H.; McCammon, J.A. Ewald artifacts in computer simulations of ionic solvation and ion–ion interaction: A continuum electrostatics study. J. Chem. Phys. 1999, 110, 1856–1872. [Google Scholar] [CrossRef]
- Rocklin, G.J.; Mobley, D.L.; Dill, K.A.; Hünenberger, P.H. Calculating the binding free energies of charged species based on explicit-solvent simulations employing lattice-sum methods: An accurate correction scheme for electrostatic finite-size effects. J. Chem. Phys. 2013, 139, 184103. [Google Scholar] [CrossRef] [PubMed]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. The J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Chen, W.; Deng, Y.; Russell, E.; Wu, Y.; Abel, R.; Wang, L. Accurate Calculation of Relative Binding Free Energies between Ligands with Different Net Charges. J. Chem. Theory Comput. 2018, 14, 6346–6358. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Energy Component | Pearson Correlation Coefficient | |
|---|---|---|
| MHC-AS | Peptide-AS | |
| ΔHMM/GBSA | 0.22 | 0.22 |
| ΔEvdW | 0.73 | 0.77 |
| ΔH | 0.30 | 0.58 |
| ΔG | 0.46 | 0.72 |
| ΔEvdW-TΔS | 0.77 | 0.86 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, M.; Zhao, F.; Yu, L.; Liu, J.; Wang, J.; Zhang, J.Z.H. An Efficient Approach to the Accurate Prediction of Mutational Effects in Antigen Binding to the MHC1. Molecules 2024, 29, 881. https://doi.org/10.3390/molecules29040881
Zhou M, Zhao F, Yu L, Liu J, Wang J, Zhang JZH. An Efficient Approach to the Accurate Prediction of Mutational Effects in Antigen Binding to the MHC1. Molecules. 2024; 29(4):881. https://doi.org/10.3390/molecules29040881
Chicago/Turabian StyleZhou, Mengchen, Fanyu Zhao, Lan Yu, Jinfeng Liu, Jian Wang, and John Z. H. Zhang. 2024. "An Efficient Approach to the Accurate Prediction of Mutational Effects in Antigen Binding to the MHC1" Molecules 29, no. 4: 881. https://doi.org/10.3390/molecules29040881
APA StyleZhou, M., Zhao, F., Yu, L., Liu, J., Wang, J., & Zhang, J. Z. H. (2024). An Efficient Approach to the Accurate Prediction of Mutational Effects in Antigen Binding to the MHC1. Molecules, 29(4), 881. https://doi.org/10.3390/molecules29040881