Abstract
Cancer is a complicated, multifaceted disease that can impact any organ in the body. Various chemotherapeutic agents have a low selectivity and are very toxic when used alone or in combination with others. Resistance is one of the most important hurdles that develop due to the use of many anticancer therapeutics. As a result, treating cancer requires a target-specific palliative care strategy. Remarkable scientific discoveries have shed light on several of the molecular mechanisms underlying cancer, resulting in the development of various targeted anticancer agents. One of the most important heterocyclic motifs is quinazoline, which has a wide range of biological uses and chemical reactivities. Newer, more sophisticated medications with quinazoline structures have been found in the last few years, and great strides have been made in creating effective protocols for building these pharmacologically active scaffolds. A new class of chemotherapeutic agents known as quinazoline-based derivatives possessing anticancer properties consists of several well-known compounds that block different protein kinases and other molecular targets. This review highlights recent updates (2021–2024) on various quinazoline-based derivatives acting against different protein kinases as anticancer chemotherapeutics. It also provides guidance for the design and synthesis of novel quinazoline analogues that could serve as lead compounds.
1. Introduction
Cancer is widely recognized as a serious public health issue and is universally acknowledged as the second-greatest cause of death in the United States [1]. According to the World Health Organization (WHO), the word “cancer” refers to a broad category of illnesses that can impact any area of the body [2]. Other terms used to characterize cancer are neoplasms and malignant tumors [3]. The fast growth of abnormal cells that eventually spread to other organs after initially residing in one area is one of the primary characteristics of cancer. This phenomenon is known as metastasis [4]. Widespread metastases are the main reason behind cancer-related deaths [4].
Recent WHO statistics indicate that 10 million deaths worldwide are expected to be related to cancer [5]. The International Agency for Research on Cancer (IARC) estimates that one in five people will develop cancer in their lifetime. One of the most important public health issues of the twenty-first century is cancer prevention [5]. Recent evidence suggests that effective primary prevention techniques could avoid at least 40% of cancer cases, and early tumor diagnosis could further reduce cancer-related death.
Protein kinases constitute one of the biggest protein families. By manipulating the location, activity, and functionality of many proteins via multisite phosphorylation, they regulate a broad spectrum of cellular processes [6]. Numerous critical cancer processes, such as tumor growth, metastasis, neovascularization, and chemotherapy resistance, have been shown to be significantly impacted by them. Protein kinases catalyze the transfer of a phosphate group from ATP to the hydroxy group of an amino acid residue. In cellular and molecular process, protein kinases are indispensable. As a result, they play a crucial part in the growth, dissemination, and survival of tumor cells in humans. Hence, this class of enzymes has drawn significant attention as a potential therapeutic target, with multiple kinase suppressors now receiving FDA approval for different cancer indications [7,8].
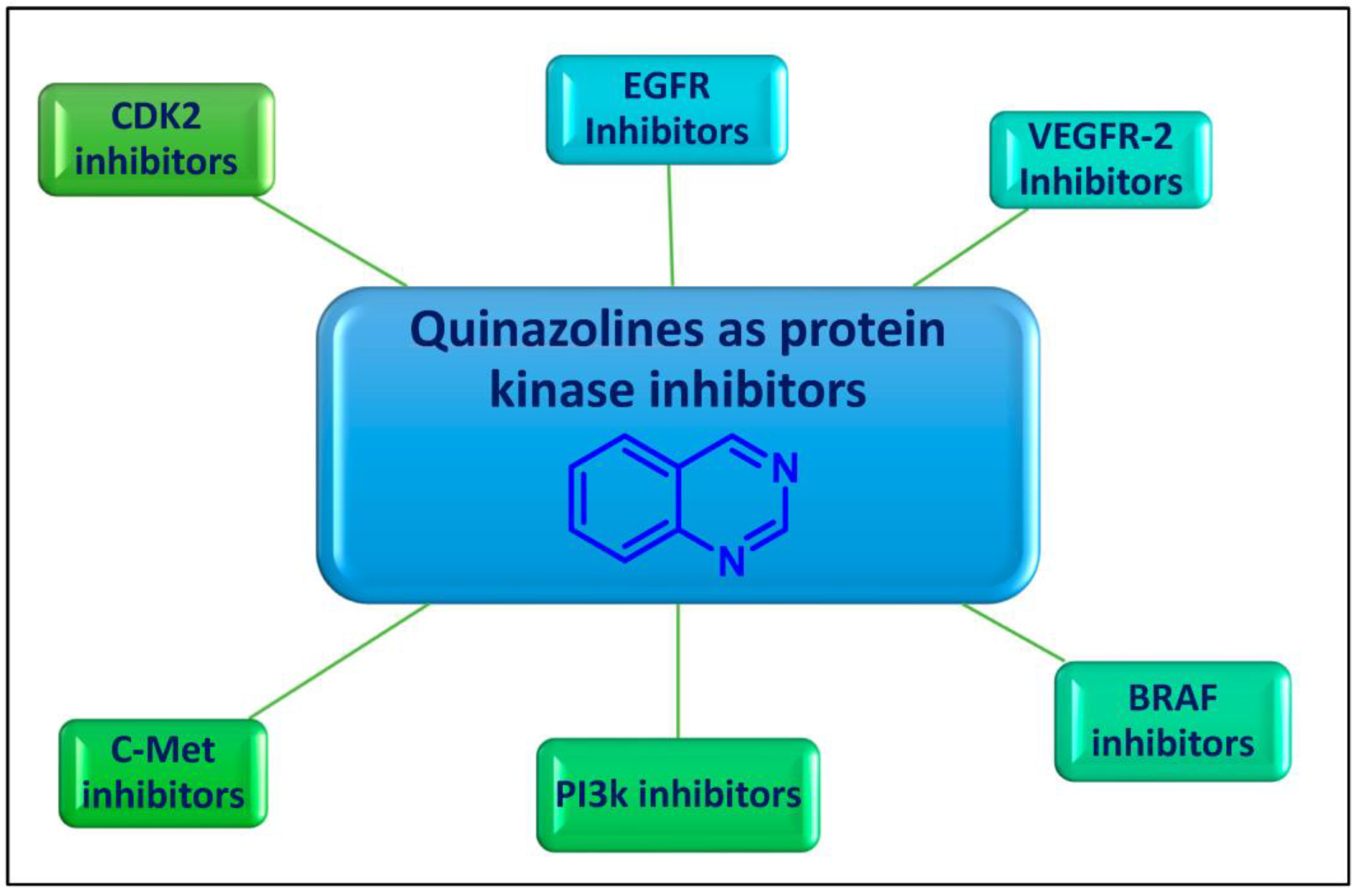
Quinazoline is an aromatic heterocyclic scaffold consisting of fused pyrimidine and a benzene ring. The quinazoline motif is considered to be one of the most important heterocyclic ring systems in pharmaceutical chemistry, present in many compounds and endowed with tremendous biological activities [9,10,11,12]. The quinazoline nucleus and its derivatives were reported to have significant potential as promising enzyme inhibitors (Figure 1).
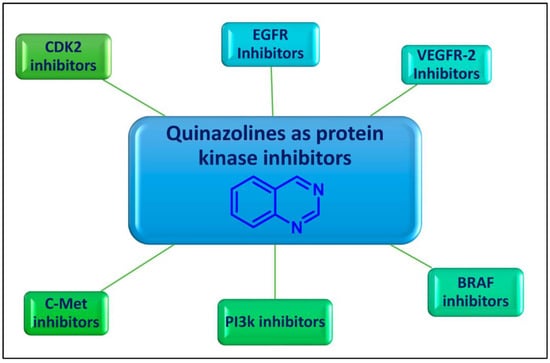
Figure 1.
Protein kinase inhibitory activity of quinazolines for cancer therapy.
2. Quinazolines as Protein Kinases Inhibitors
Over the years, diverse quinazolines have been discovered to act as single or multi-target protein kinase inhibitors. Epidermal growth factor receptor (EGFR), vascular endothelial growth factor receptor-2 (VEGFR-2), B rapidly accelerated fibrosarcoma (BRAF), phosphoinositide 3-kinase (PI3k), and mesenchymal–epithelial transition factor (c-Met) are among the protein kinases that are suppressed by quinazolines (Figure 1). FDA-approved medications based on quinazoline scaffolds and the latest developments in quinazolines as protein kinase suppressors for the treatment of cancer are the main topics of this review article.
2.1. Epidermal Growth Factor Receptor (EGFR) Inhibitors
One member of the ErbB family of the transmembrane tyrosine kinase receptors is the epidermal growth factor receptor (EGFR) [13]. It functions as a crucial mediator in cell proliferation, survival, adhesion, migration, and differentiation via downstream signaling transmission through auto-phosphorylation of several tyrosine residues following EGFR dimerization [13]. This receptor is activated when ligands, such as EGF or TGFa, bind to the extracellular domain, which sets off a signal transduction cascade that leads to cell proliferation, metastasis, and resistance to apoptosis in many types of cancers. Numerous malignancies, including those of the breast, ovary, colon, and non-small cell lung cancers (NSCLC), are linked to overexpression of EGFR [14].
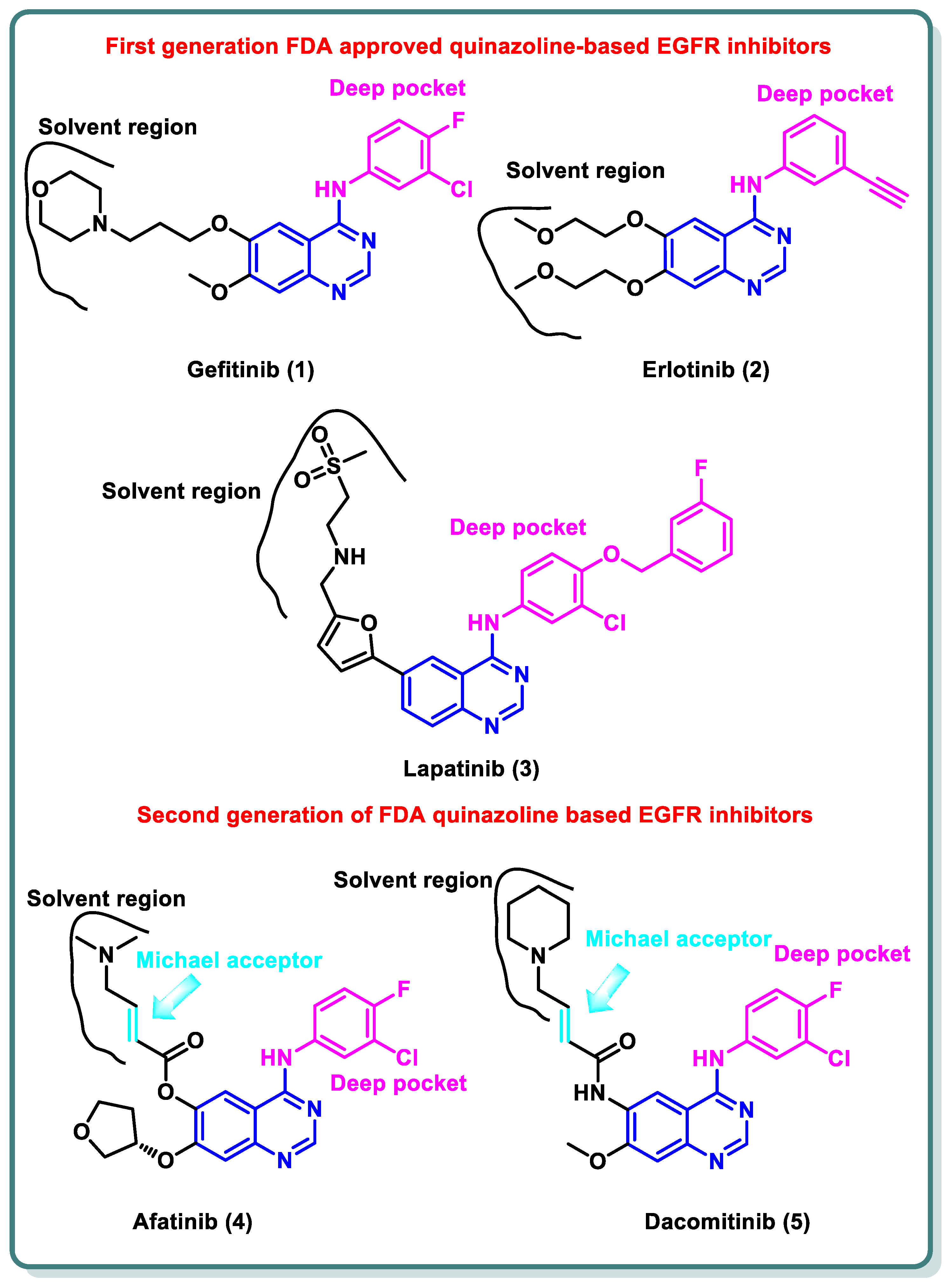
A variety of quinazolines have been discovered in recent years to inhibit EGFR [15]. It is suggested that the 4-anilinoquinazoline scaffold interacts with ATP at the EGFR binding site [16]. Gefitinib (1) and erlotinib (2), the first-generation EGFR inhibitors, have received FDA approval for the treatment of non-small cell lung cancer (NSCLC) [17].
Additionally, lapatinib (3) has received FDA approval for the management of certain cases of postmenopausal women’s breast cancer [18]. Despite the advancements made, many non-small cell lung cancers (NSCLCs) develop EGFR binding site mutations that promote tumor formation and carcinogenesis. The protein databank incorporates the crystal structure of EGFR co-crystalized with gefitinib (1), erlotinib (2), and lapatinib (3) [19,20]. These three inhibitors share the same binding mode to EGFR in which the quinazoline moiety occupies the ATP binding site and is stabilized by the ability of N1 to perform hydrogen bonding with the NH of Met793 in the hinge region, while N3 is involved in hydrogen bonding to Thr854 through a water molecule. The substituent at the 4-position is directed to the back of the ATP binding site and is involved in hydrophobic interactions with the protein, while the substitution at position 6 (and 7) is directed toward the solvent interface (Figure 2) [21].

Figure 2.
Structures of first- and second-generation FDA-approved quinazoline-based EGFR inhibitors.
Despite the advancements made, many NSCLCs develop EGFR binding site mutations that promote tumor formation and carcinogenesis. About 40% of NSCLC patients have an activating mutation of L858R, which raises the affinity for ATP and reduces the affinity for first-generation TKIs, resulting in resistance [22]. In contrast, about 50% of lung adenocarcinomas in patients who have become resistant to gefitinib (1) and erlotinib (2) exhibit the treatment resistance mutation T790M [23]. It entails changing threonine 790 at the gate region to a bulkier methionine, which prevents inhibitors from binding to the EGFR binding site. Hence, various structural alterations were made to the previous drugs to provide a range of ATP-competitive irreversible EGFR inhibitors that were recognized as second-generation, irreversible EGFR inhibitors [24]. Afatinib (4) and dacomitinib (5) occupy the ATP binding site of EGFR in the same manner as gefinitib (1); however, they are classified as irreversible EGFR inhibitors, as they have dimethylaminocrotonamide and 4-piperidin-1-yl-but-2-enamide, moieties, respectively, at the sixth position. These are capable of forming a covalent bond with the Cys797 present EGFR’s ATP binding site by acting as a Michael acceptor (Figure 2) [25].
Additionally, different research groups have recently reported diverse scaffolds based on the quinazoline moiety as EGFR inhibitors beside other protein kinases. For example, in 2024, Han Zhang and his coworkers designed and synthesized novel 4-phenoxyquinazoline compounds as dual EGFR/c-Met suppressors to treat non-small cell lung cancer [26] (Figure 3). The most promising candidate is derivative 6, which inhibits EGFR, EGFRL858R/T790M, and c-Met kinases at IC50 values of 64.8, 305.4, and 137.4 nM, respectively. It has significant anti-proliferative activity (IC50 = 2.27–3.35 µM), similar to that of afatinib, towards five types of cancer cell lines. Analysis of the cell cycle revealed that derivative 6 has the capacity to induce arrest at G2/M. In vivo findings in xenograft models demonstrated that derivative 6 might both cause apoptosis and suppress tumor development. Altogether, the results showed that derivative 6 is a novel compound with dual inhibitory activity against both EGFR and c-Met, and it can be considered a potential therapeutic medication to treat a variety of tumors (Figure 3) [26].

Figure 3.
Design strategy of quinazoline derivative 6 as a dual EGFR and cMet inhibitor.
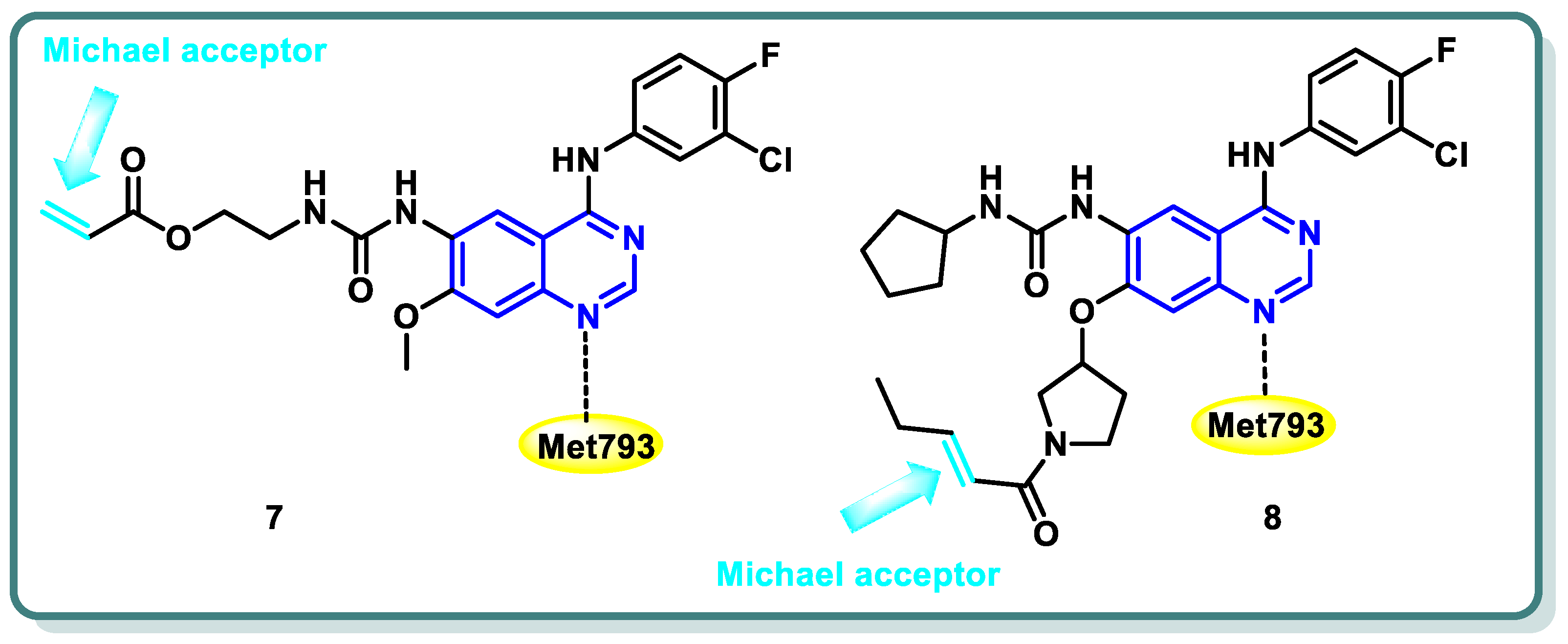
Diverse series of quinazolines were designed as EGFRL858R/T790M inhibitors in 2022 by Wenhui Gan and coworkers [27]. Urea and thiourea moieties were incorporated in one series (compound 7 is a representative example, Figure 4), whereas a Michael receptor active warhead was introduced in another series (compound 8 is a representative example, Figure 4). Most of the designed and synthesized candidates demonstrated potent anti-proliferative impacts against different cell lines, including A549 and H1975 cancer cells. Additionally, they demonstrated moderate to outstanding kinase inhibitory efficacy against EGFRWT and EGFRL858R/T790M. Urea derivative 7 revealed the most promising EGFRWT and EGFRL858R/T790M inhibitory activities (IC50 = 0.8 and 2.7 nM, respectively). Furthermore, both derivatives 7 and 8 strongly caused apoptosis of A549 cells. A cell cycle analysis showed that arrest of the cell cycle of A549 cells occurred at the S phase in derivative 7 and the G1 phase in derivative 8 [27] (Figure 4).

Figure 4.
Structures of quinazoline derivatives 7 and 8.
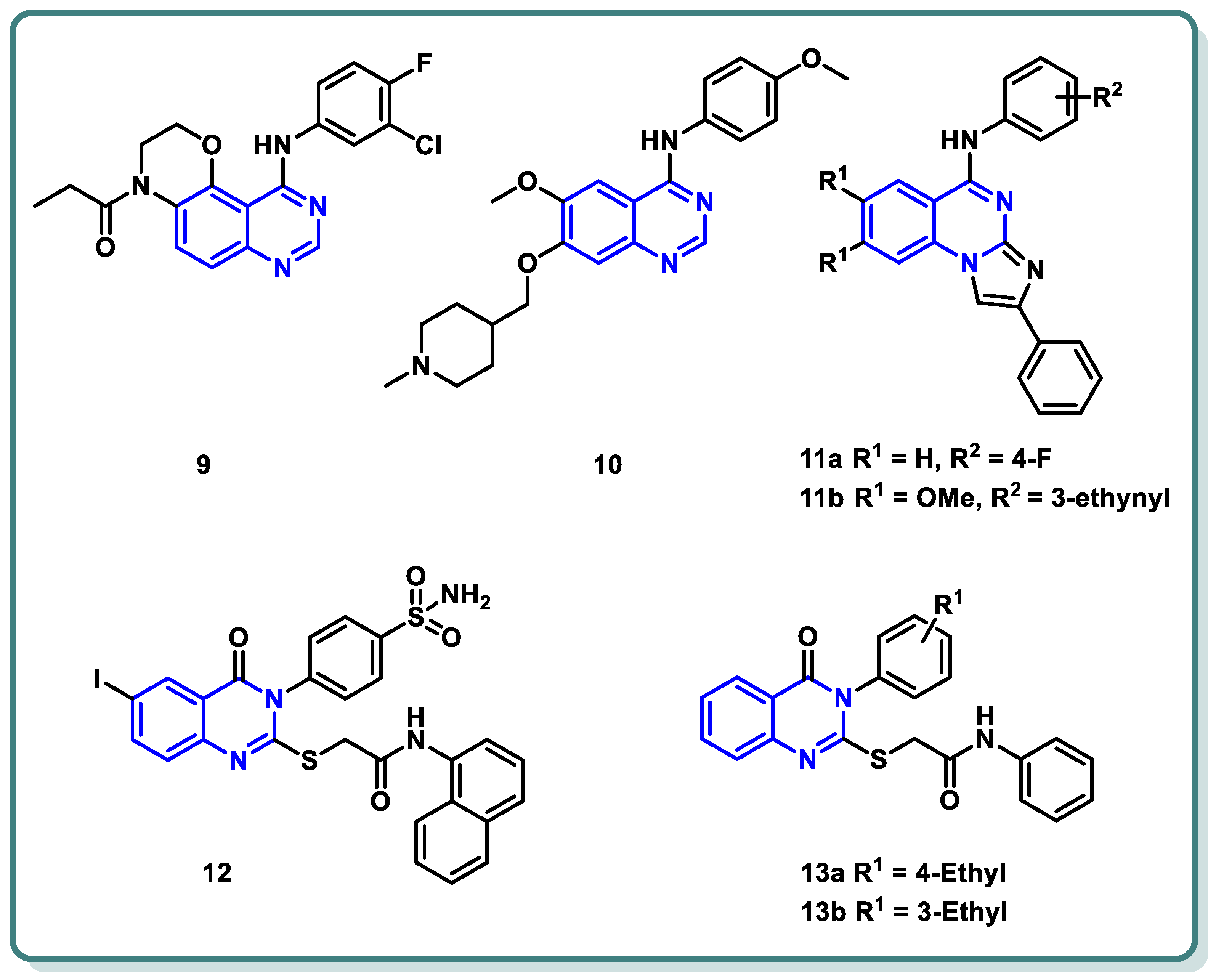
Moreover, a new family of 3,4-dihydro-2H-[1,4]oxazino [2,3-f]quinazoline derivatives that target EGFR was described by Qin et al. (Figure 5) [28]. With an IC50 ≤ 937.7 nM, the synthesized candidates showed strong EGFR inhibitory efficacy. Compound 9 (Figure 5) exhibited strong anti-proliferative properties against NCI-H1563 and H1975 cancer cell lines (IC50 = 25.69 nM). Furthermore, it produced an in vitro safety profile against the normal cell line, resembling 16HBE cells [28].
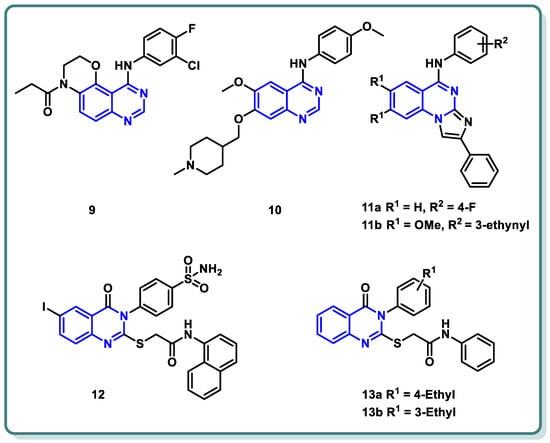
Figure 5.
Chemical structures of various substituted quinazolines-based compounds 9–13 as EGFR inhibitors.
A series of 4-anilinoquinazoline analogues that are expected to be accommodated in the hinge region and allosteric pocket of EGFRC797S was designed by Dou Dou [29]. When screening the designed and synthesized quinazolines regarding their inhibitory activity against EGFRC797S, derivative 10 (Figure 5) displayed a potent activity, with an IC50 = 0.128 µM. A potential anti-proliferative impact against BaF3-EGFRL858R/T790M/C797S and BaF3-EGFR19del/T790M/C797S was recorded for derivative 10 (IC50 = 0.75 and 0.09 μM, respectively). Additionally, derivative 10 demonstrated dose-dependent inhibition of EGFR and its downstream signaling cascades in BaF3-EGFR19del/T790M/C797S cells. Interestingly, quinazoline 10 (Figure 5) demonstrated in vivo inhibition of tumor growth in a BaF3-EGFR19del/T790M/C797S xenograft model (30 mg/kg, TGI = 67.95%) [29].
In 2023, a class of imidazo [1,2-a]quinazolines was presented by Hasenvand et al. as containing potent EGFR inhibitors [30]. When compared to erlotinib, the reference drug, imidazoquinazolines, 11a,b (Figure 5) revealed significant anticancer activity against PC3, HepG2, HeLa, and MDA-MB-231, with IC50 values in the micro-molar range. Additional analyses showed that they could cause cell growth inhibition at the G0 phase of the cell cycle and induce apoptotic cell death. Two derivatives, 11a,b were shown to have suppression activity and selectivity toward EGFR (as seen by their respective EGFR-IC50 = 82.0 µM and 12.3 µM, respectively). Furthermore, 11a,b were shown via Western blot analysis to decrease extracellular signal-regulated kinase (ERK1/2) as well as EGFR phosphorylation. The degree of B-Actin phosphorylation, however, remained unchanged [30] (Figure 5).
In addition, Ghorab et al. introduced a series of novel quinazoline sulfonamide derivatives in 2023 [31]. Using the MTT assay, the compounds’ growth inhibitory activities against four cancer cell lines, namely HepG2, MCF-7, HCT116, and A549, were assessed. Based on the obtained findings, the compounds showing the most significant potencies were examined as EGFRT790M and VEGFR-2 inhibitors. With an IC50 = 0.0977 µM against MCF-7, the quinazoline sulfonamide derivative 12 (Figure 5) exhibited the most noticeable cytotoxic effect and the most inhibitory activity against EGFRT790M and VEGFR-2 (IC50 = 0.0728 and 0.0523 µM, respectively). In MCF-7 cells, compound 12 induced apoptosis and cell cycle stopping at the G2/M phase. Following an 8 Gy gamma irradiation dose, the radiosensitizing evaluation of compound 12 demonstrated its noteworthy capacity to sensitize the cancer cells to the impact of radiation. According to molecular docking simulation studies, compound 12 was predicted to occupy the VEGF and EGF receptors’ ATP-binding site and block their functions [31] (Figure 5).
The same research group designed and synthesized a class of N-substituted-2-((4-oxo-3-phenyl-3,4-dihydroquinazolin-2-yl)thio)acetamides [32]. Some of the synthesized candidates demonstrated potent cytotoxic activity on a HepG2 cell line with an IC50 reaching 1.11 µM. The quinazolines 13a and 13b inhibited EGFR (IC50 = 73.23 and 58.26 µM, respectively) in reference to erlotinib (IC50 = 9.79 µM) [32] (Figure 5).
2.2. Vascular Endothelia Growth Factor Receptor (VEGFR) Inhibitors
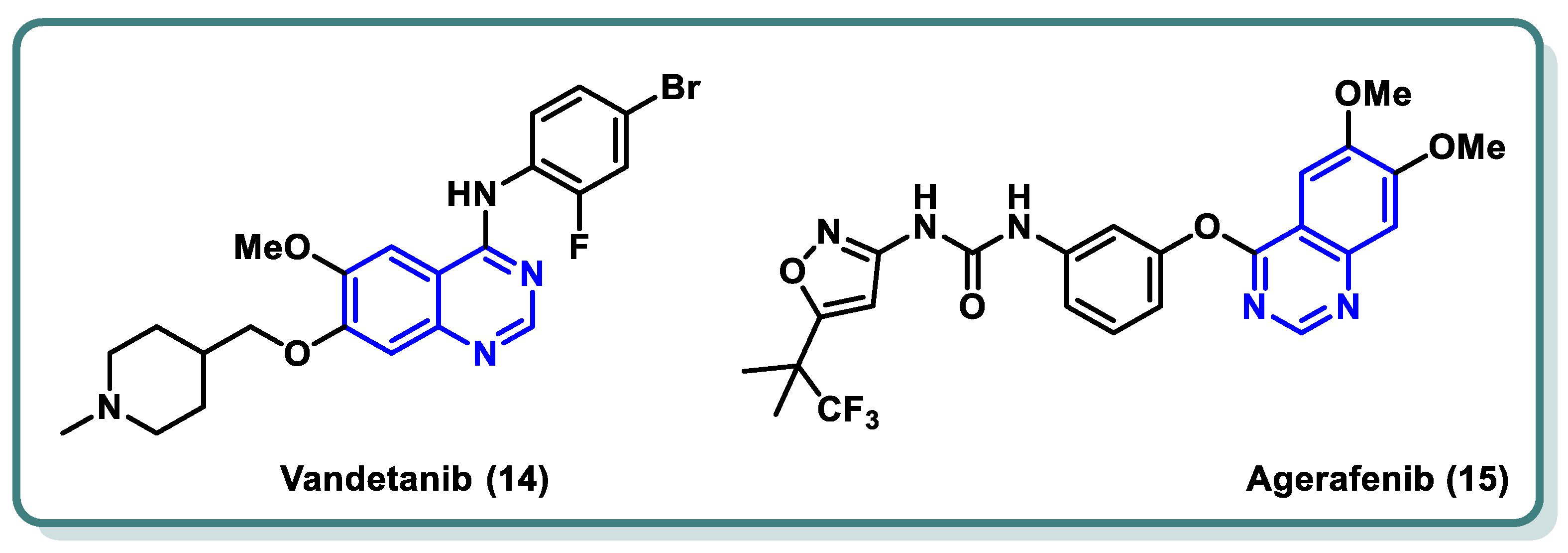
In both normal and pathological circumstances, the process of angiogenesis, or the formation of new blood vessels, is mediated by the tyrosine kinase system known as vascular endothelial growth factor (VEGF)/vascular endothelial growth factor receptor (VEGFR) [33,34,35]. When a solid tumor experiences hypoxia, massive levels of VEGF-A are released. By attaching itself to VEGFR-2, VEGF-A activates the receptor by starting a phosphorylation cascade. This then starts a signaling pathway downstream that eventually results in angiogenesis. When compared to healthy tissues, VEGFR-2 is the most expressed of the three VEGF receptors in a variety of cancer forms, including malignant melanoma, breast cancer, hepatocellular carcinoma, colon cancer, and so on [36,37,38]. Therefore, blocking VEGFR-2 is thought to be an effective way to prevent angiogenesis [38,39]. This ultimately leads to the cancer cells’ sources of nutrition and oxygen being blocked, which in turn prevents the cancer cells from surviving, proliferating, metastasizing, and dying off [40]. Vandetanib (14) is an example of a quinazoline derivative that acts as a VEGFR and EGFR inhibitor, and it has recently received a license from the FDA for the management of advanced medullary thyroid carcinoma [41,42]. Moreover, agerafenib (15) is a potential anti-tumor agent and potent mutlikinase (VEGFR-2, PDGFR, BRAFWT, and BRAFV600E) inhibitor [43] (Figure 6).

Figure 6.
FDA-approved drugs as VEGFR-2 inhibitors for cancer treatment.
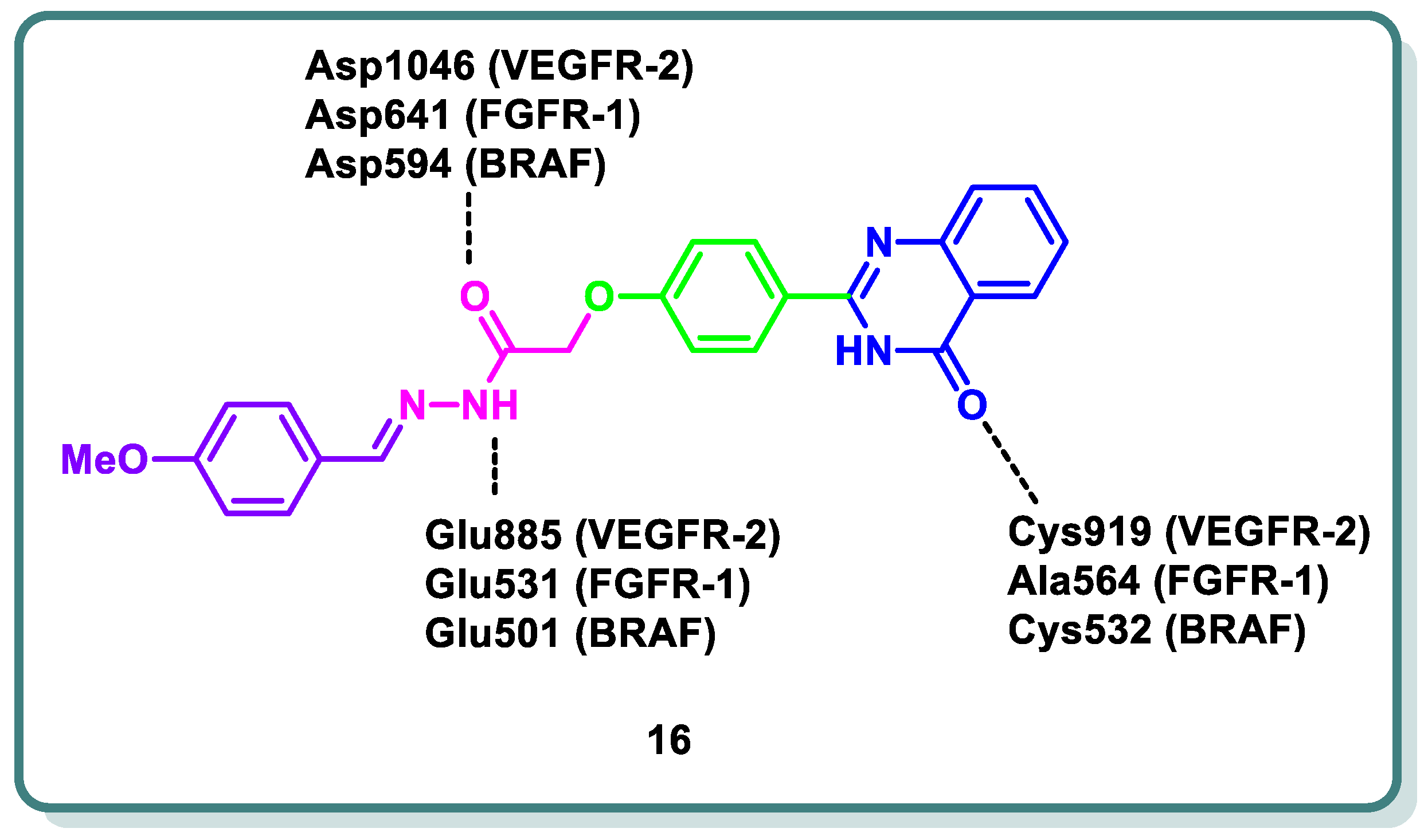
Abd El-Karim et al. recently designed and discovered a novel class of quinazolinone N-acetohydrazides as significant inhibitors of VEGFR-2 alongside other kinases [44]. Compound 16 was the most potent candidate (Figure 7). It demonstrated IC50 values of 0.29, 0.35, 0.47, and 0.30 μM on VEGRF-2, FGFR-1, BRAF, and BRAFV600E, respectively. Also, compound 16 was the most potent candidate (Figure 7). It demonstrated IC50 values of 0.29, 0.35, 0.47, and 0.30 μM on VEGRF-2, FGFR-1, BRAF, and BRAFV600E, respectively. Additionally, compound 16 revealed growth inhibition potential against NCI-60 cancer cell lines (GI50 = 1.64 μM). In addition, the same study showed that compound 16 led to cell cycle arrest in the MDA-MB-231 cell line at the G2/M phase alongside an apoptotic effect, as observed based on the elevation in the percentage of cells in the early and late apoptosis phases from 0.05 and 0.99% in control cells to 0.44% and 2.43%, respectively. The newly quinazolines exhibit analogous binding mechanisms to the protein kinase active sites under study, as demonstrated by in silico molecular docking simulations. This mechanism represents the settlement of the quinazolinone scaffold within the hinge region of the target kinases, where the C=O forms a hydrogen bond with the important amino acids. Meanwhile, the essential amino acids in the gate area interact through hydrogen bonding with the N-acetohydrazide linker and the peripheral phenyl moiety occupies the allosteric hydrophobic back pocket, producing a number of hydrophobic interactions (Figure 7) [44].

Figure 7.
Design strategy of compound 16 as a VEGFR-2, FGFR-1, and BRAF inhibitor for cancer treatment.
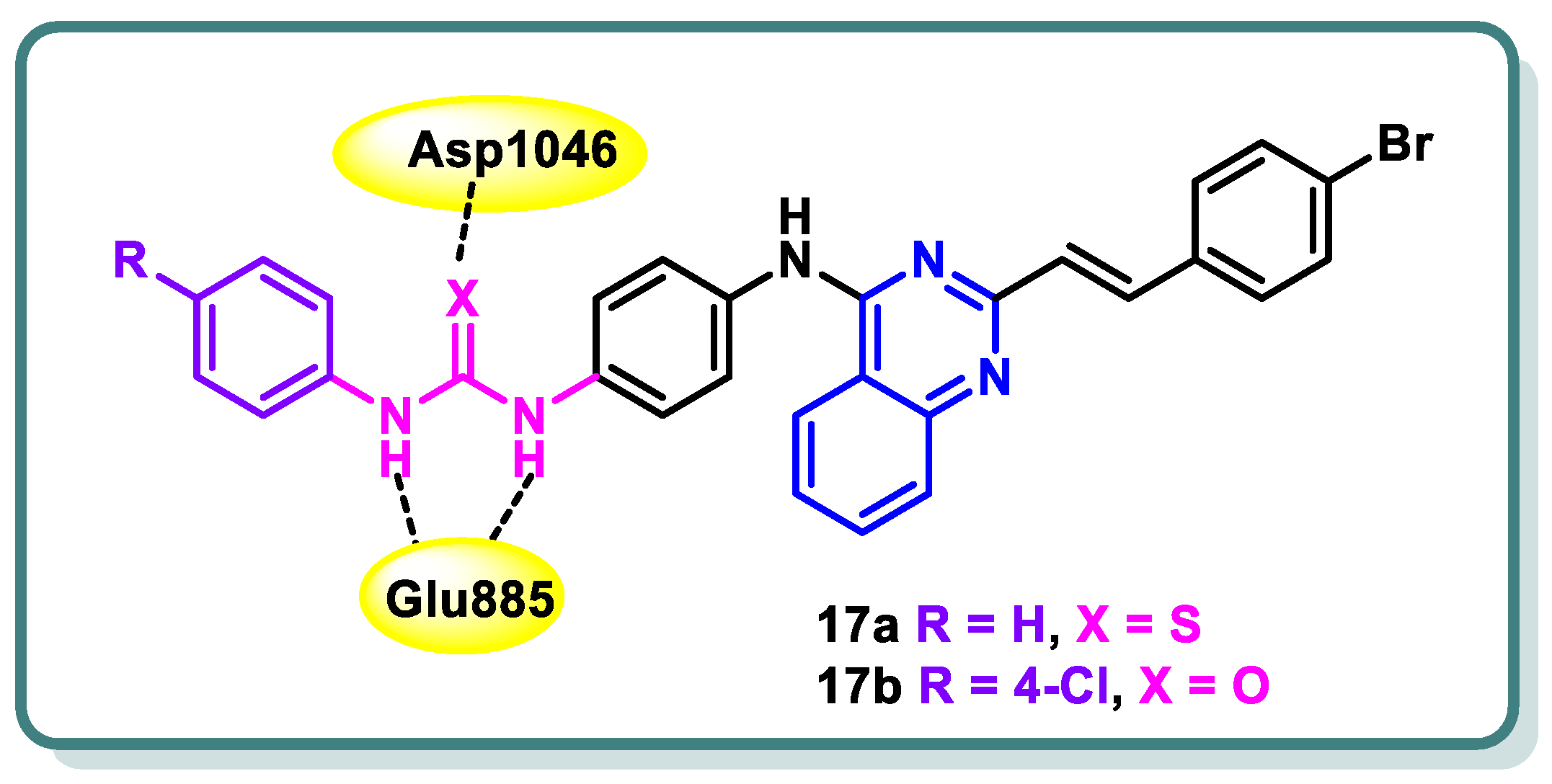
Hamdi and colleagues [45] have successfully synthesized a novel series of 4-anilino-2-vinylquinazolines. The evaluation encompassed the assessment of in vitro anticancer activity, suppression of VEGFR-2, and activation of apoptosis. Among the products that underwent testing, compounds 17a and 17b (Figure 8) demonstrated significant potencies as anticancer agents against HCT-116, HepG-2, and MCF-7 cancer cell lines. Their effectiveness, with IC50 values ranging from approximately 4.92 to 11.31 μM, was found to be equivalent to that of sorafenib, which exhibited IC50 values ranging from approximately 5.47 to 9.18 μM. In addition, compound 17a demonstrated a significant suppression impact against VEGFR-2, with an IC50 value of 60.27 nM, which is similar to the inhibitory activity of sorafenib (IC50 = 55.43 nM). On the other hand, compound 17b showed a moderate suppression effect against VEGFR-2 (IC50 = 93.5 nM). Compound 17a exhibited a notable level of apoptosis-inducing activity, amounting to 36.24%, when tested on MCF-7 cancer cells. This efficacy is equivalent to the apoptotic impact demonstrated by sorafenib, which amounted to 32.46%. Furthermore, compound 17a was found to stop the cell cycle at the G1/S phase. The anticancer and VEGFR-2 inhibitory effects were elucidated using molecular modeling investigations, which demonstrated that compound 17a possesses the crucial element necessary for interacting with the DFG-binding domain, a critical factor in VEGFR-2 inhibition. Furthermore, compound 17a exhibited molecular fields that were comparable to those observed by sorafenib (Figure 8) [45].

Figure 8.
Design of quinazolines 17a and 17b as VEGFR-2 inhibitors.
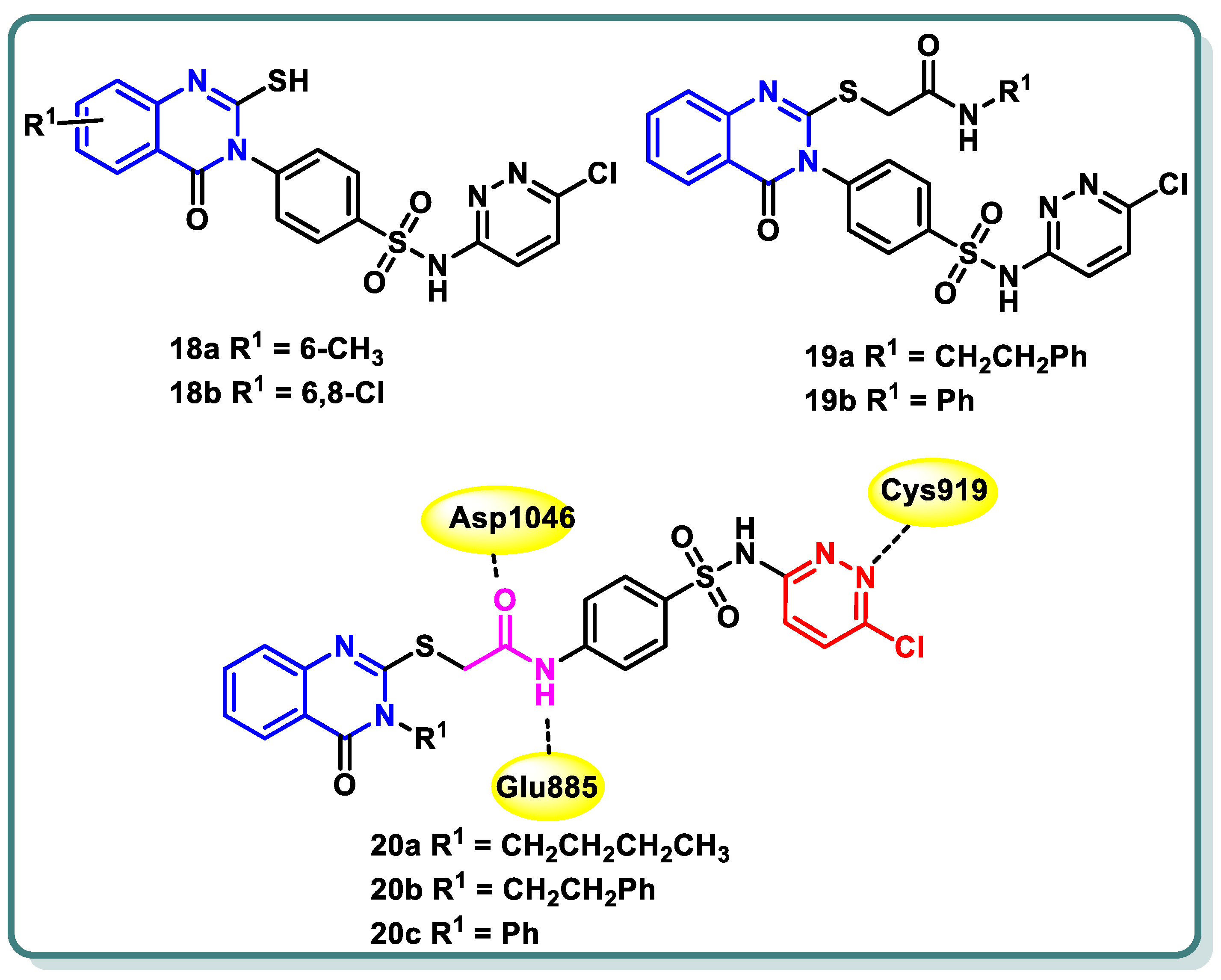
In their study, Zahran et al. [46] documented the development and creation of innovative sulfachloropyridazine-bearing quinazoline-4(3H)-one derivatives. The cytotoxic activity of all the produced compounds was evaluated in vitro at a dose of 10 μM against a panel of 60 human cancer cell lines. Compounds 18a, 18b, 19a, 19b, 20a, 20b, and 20c (Figure 9), which exhibited the highest levels of activity, were chosen for the purpose of assessing their capacity to suppress VEGFR-2 in comparison with sorafenib and vandetanib as standard medicines. Thioacetamide derivative 20b exhibited the highest level of inhibitory action against VEGFR-2, with an IC50 value of 66 nM. Additionally, the assessment of the antiangiogenic characteristics necessary for inhibiting wound healing in HUVEC cells was carried out for 20b, which exhibited a closure percentage of 58.52%, which is similar to that of sorafenib (46.67%). The analogue 20b exhibited moderate suppression activity against PDGFR (IC50 = 180 ± 0.009 nM), EGFR (IC50 = 98 ± 0.004 nM), and FGFR-1 (IC50 = 82 ± 0.004 nM) as well. The Annexin V-FITC/PI test showed that 20b notably enhanced early apoptosis. Additionally, it led to an elevation in the levels of active caspase-3 and Bax while simultaneously decreasing the levels of Bcl-2 in UO-31 cells compared to the untreated control group. The molecular docking analysis revealed that the highly active compounds exhibited a favorable binding conformation inside the active site of VEGFR-2, suggesting their potential role in exerting anticancer effects (Figure 9) [46].

Figure 9.
Structures of sulfachloropyridazine-bearing quinazoline-4(3H)-one derivatives 18a,b, 19a,b and 20a–c.
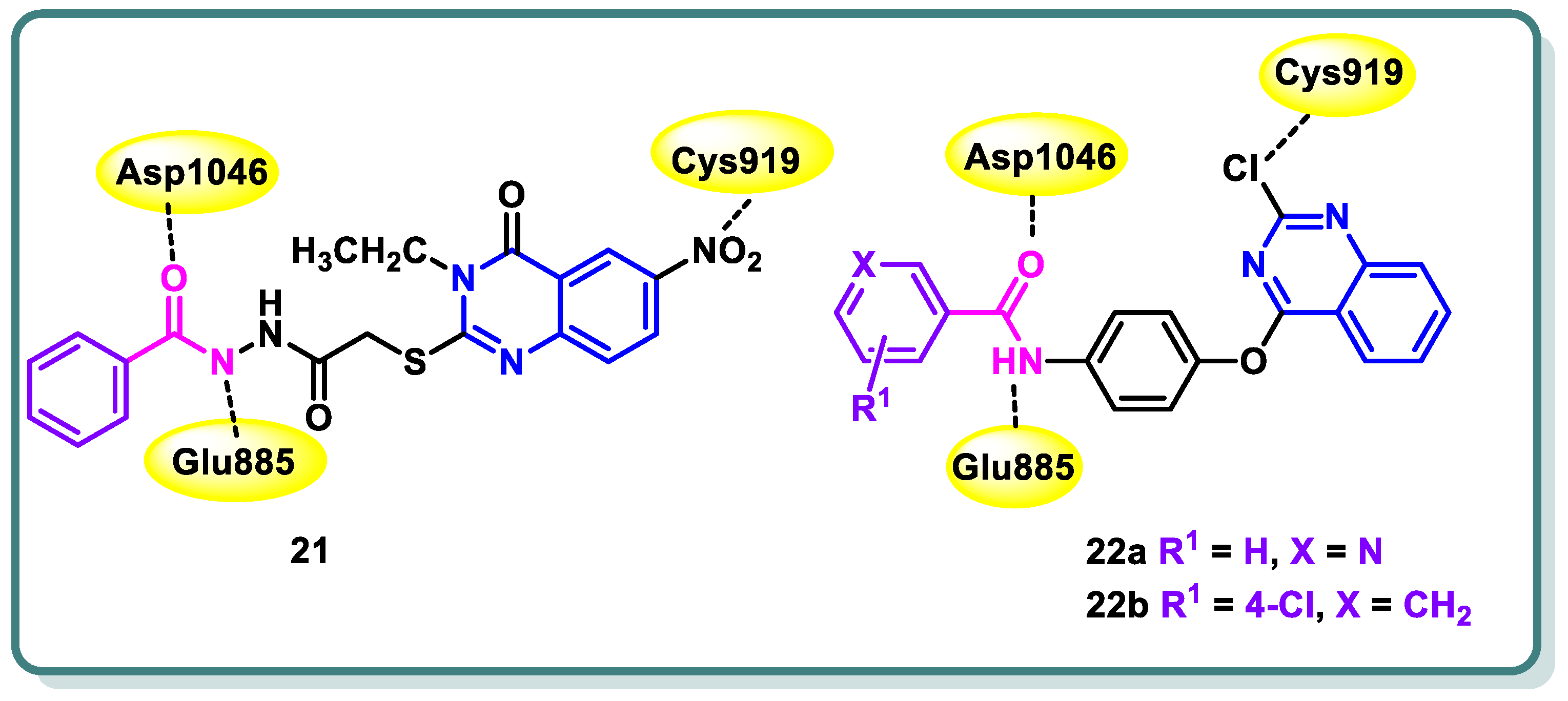
Additionally, in 2021, Abdallah and coworkers [47] reported the design, synthesis, and biological assessment of nineteen derivatives of quinazolin-4-one. These compounds were investigated for their potential anticancer action, taking into consideration the pharmacophoric properties of VEGFR-2 kinase inhibitors. The thioacetyl-benzohydrazide compound 21 exhibited the highest potency among the tested candidates. It demonstrated an IC50 value of 4.6 ± 0.06 μM against VEGFR-2 kinase. Furthermore, it demonstrated IC50 values of 17.23 ± 1.5, 26.10 ± 2.2, and 30.85 ± 2.3 μg/mL against HepG2, PC3, and MCF cell lines, respectively. Simultaneously, the experiment yielded an IC50 value of 145.93 ± 1.1 μg/mL when tested against the normal human lung fibroblasts cell line (WI-38), suggesting a favorable selectivity index. Additional examination of the HepG2 cell cycle revealed that compound 21 possesses the capacity to trigger apoptosis and halt cell proliferation specifically at the G2/M stage (Figure 10). In addition, the docking investigations revealed that compound 21 possesses the capability to effectively connect to VEGFR-2 by forming three crucial hydrogen bonds with the critical residues Glu885, Asp1046, and Cys919, as depicted in Figure 10. In conclusion, the study proposes that compound 21 exhibits potential as a primary candidate for the advancement of efficacious anticancer medicines that specifically target VEGFR-2 [47].

Figure 10.
Quinazolines 21 and 22 showing their expected interactions with the VEGFR-2 active site.
One year later, the same research group synthesized 17 novel compounds by utilizing the quinazoline, quinoxaline, and benzene nuclei [48]. The strategy of their work was to align with the known pharmacophoric characteristics of type II VEGFR-2 inhibitors. The biological findings indicated that the quinazoline derivatives 22a and 22b (Figure 10) exhibited notable potential as anticancer medicines against the hepatocellular carcinoma cell line (HepG2) that specifically target VEGFR-2 kinase. In the VEGFR-2 inhibitory experiment, compounds 22a and 22b displayed IC50 values of 60.00 and 86.36 nM, respectively, whereas sorafenib, the positive control, displayed an IC50 value of 54.00 nM. Simultaneously, the IC50 values acquired for the two contenders against HepG2 were 24.10 and 17.39 mM, respectively. Upon further investigation, compound 22a was found to possess significant potential as an inducer of apoptosis in HepG2 cells. Compound 22a increased the rate of apoptosis from 1.20% to 12.46% by upregulating the expression levels of caspase-3, BAX, and P53 and reducing the level of Bcl-2 (Figure 10) [48].
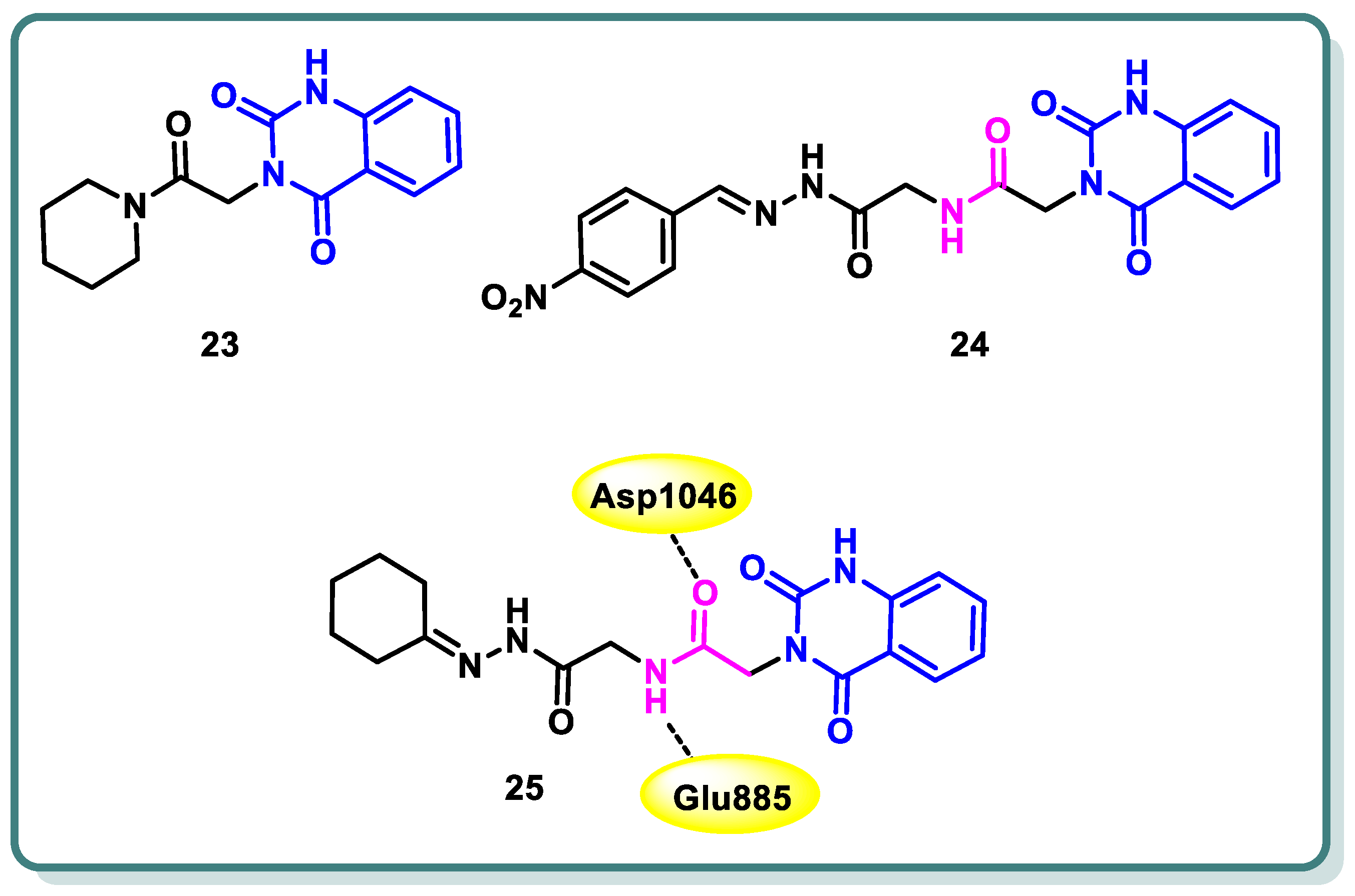
In a study conducted by Hassan et al. [49], the authors developed a series of 3-substituted quinazoline-2,4(1H,3H)-diones using novel synthetic methodologies with the aim of creating dual inhibitors of c-Met and VEGFR-2 tyrosine kinases. The antiproliferative efficacy of the target compounds was evaluated using the HCT-116 colon cancer cell line, which overexpresses c-Met/VEGFR-2. Among the tested compounds 23, 24, and 25 (Figure 11), a significant inhibition was observed for both c-Met and VEGFR-2 tyrosine kinases, with IC50 values ranging from 0.052 to 0.084 µM. The molecular docking study revealed that compound 25 displayed hydrogen bonding with Glu885 and Asp1046 (Figure 11) [49].
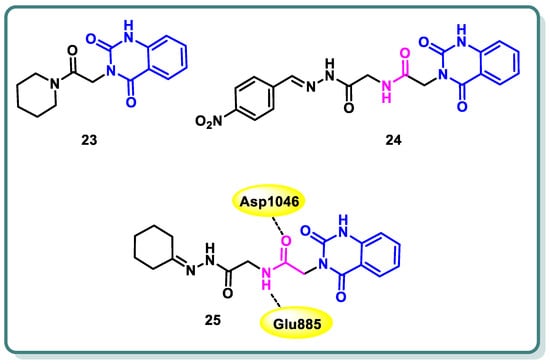
Figure 11.
Structures of quinazolines 23, 24, and 25.
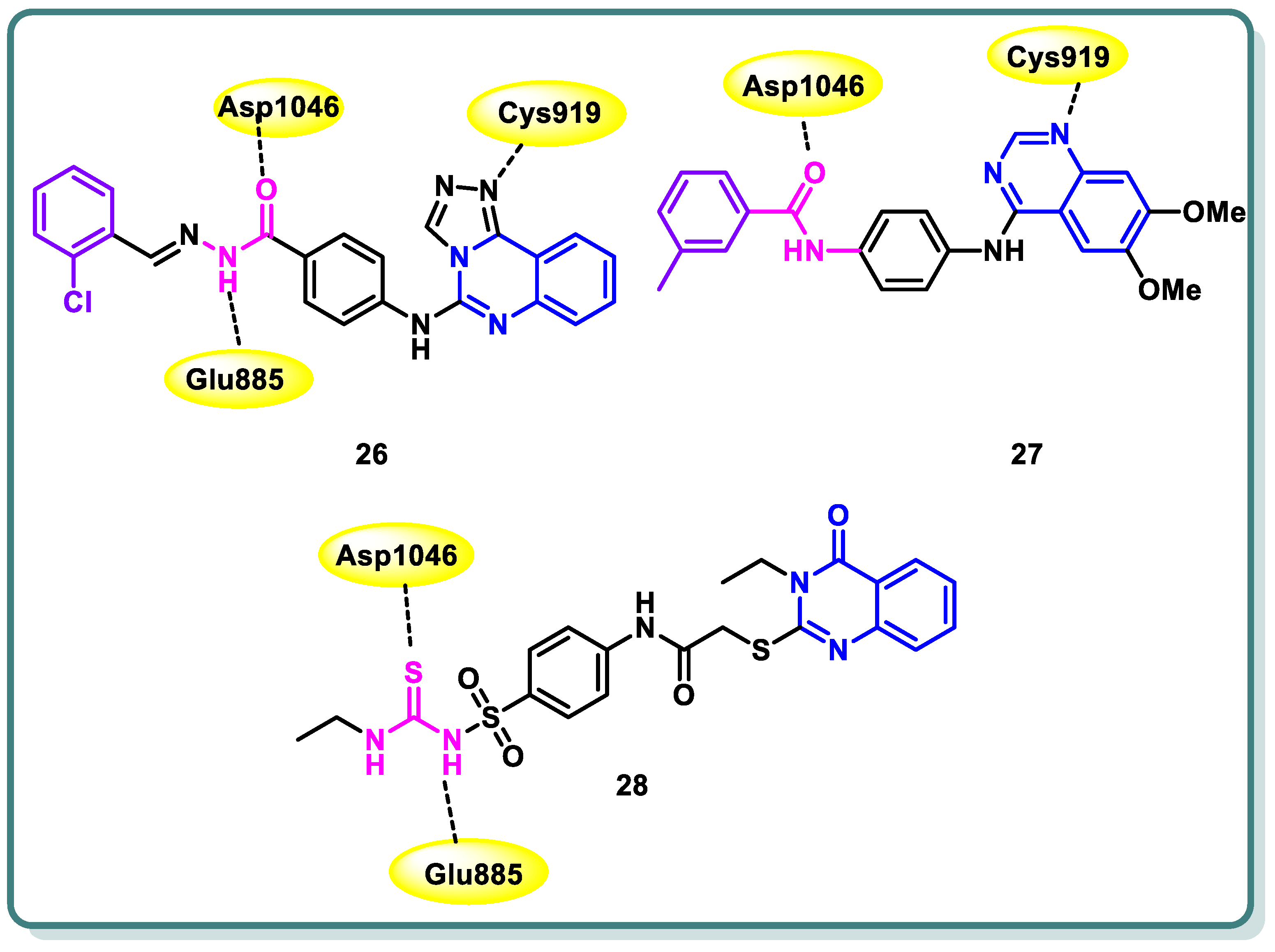
Furthermore, Azab and colleagues [50] designed and synthesized new compounds belonging to the [1,2,4]triazolo [4,3-c]quinazoline class. These compounds were strategically intended to possess the crucial pharmacophoric characteristics necessary for inhibiting VEGFR-2. The aim of this study was to assess the antiproliferative impacts of the synthesized compounds on two tumor cell lines, namely HepG2 and HCT-116. Sorafenib was employed as a positive control in this investigation. Compound 26 demonstrated significant potential antiproliferative activity (IC50 = 4.88 and 5.21 μM, respectively). Furthermore, the results demonstrated that it had the most potent inhibitory effect against VEGFR-2, as indicated by an IC50 value of 53.81 nM, surpassing that of sorafenib (IC50 = 44.34 nM). The cell cycle analysis’s findings showed that compound 26 can cause cell cycle stoppage in HepG2 cells, impacting both the S and G2/M stages. Furthermore, the administration of this substance resulted in a tenfold augmentation in the number of apoptotic cells as compared to the control group. In addition, the administration of this chemical resulted in a significant 3.35-fold upregulation of BAX expression levels, accompanied by a modest 1.25-fold down regulation of Bcl-2 expression levels. The ratio of BAX to Bcl-2 was determined to be 4.57, suggesting a favorable apoptotic outcome. Furthermore, the results demonstrated a notable elevation in the expression of caspase-3 (4.12- fold) in comparison to the control cells (Figure 12) [50].

Figure 12.
Structures of quinazoline derivatives 26–28.
Wang et al. reported the design and synthesis of new 6,7-dimethoxy-4-anilinoquinazoline derivatives and assessed them as potent VEGFR-2 inhibitors [51]. Among the target derivatives, methylbenzamide compound 27 showed the most significant VEGFR-2 kinase inhibitory activity (IC50 = 0.016 ± 0.002 μM), better than sorafenib (IC50 = 0.021 ± 0.004 μM). Furthermore, it showed the highest antiproliferative effect against Hep-G2 and MCF-7, with IC50 values in the low-micromolar range (Figure 12). In addition, the target compound was docked into the ATP binding site of VEGFR-2 (PDB: 3B8Q) using the Discovery Studio 4.5/CDOCKER protocol. Figure 12 illustrates the interaction mechanism between 14b and VEGFR-2. It demonstrates that the incorporation into the ATP-binding site of VEGFR-2 was through various interactions, including hydrogen bonds, π–cation interactions, π–anion interactions, π–π stacked interactions, alkyl interactions, and van der Waals interactions with the amino acid residues of VEGFR-2.
A novel set of quinazolin-4 (3H)-ones was also conceptualized, synthesized with the intention of functioning as inhibitors of VEGFR-2 [52], and subsequently assessed for their efficacy in impeding the proliferation of hepatocellular carcinoma, specifically targeting the HepG-2 cell line. Compound 28 exhibited greater potency compared to doxorubicin and 78% of the sorafenib activity. Compound 28 displayed an IC50 value of 4.33 ± 0.2 μg/mL, while doxorubicin showed an IC50 value of 4.50 ± 0.2 μg/mL, and sorafenib exhibited an IC50 value of 3.40 ± 0.25 μg/mL. In addition, compound 28 significantly inhibited VEGFR-2, with IC50 values of 3.1 ± 0.04 μM compared to the IC50 value of sorafenib, which was found to be 2.4 ± 0.05 μM. Moreover, compound 28 was found to cause apoptosis and halt the progression of the cell cycle at the G2/M phase. Furthermore, the in vivo anticancer studies demonstrated that compounds 28 exhibited a substantial reduction in tumor growth. The immuno-histochemical analysis of activated caspase-3 expression indicated a progressive rise in the levels of cleaved caspase-3 protein over time following the administration of compound 28 to HepG-2 cells. In addition, the fibroblastic proliferative index test demonstrated that compound 28 exhibited the ability to mitigate liver fibrosis. The findings obtained from the biological screening were further validated through docking studies (Figure 12) [52].
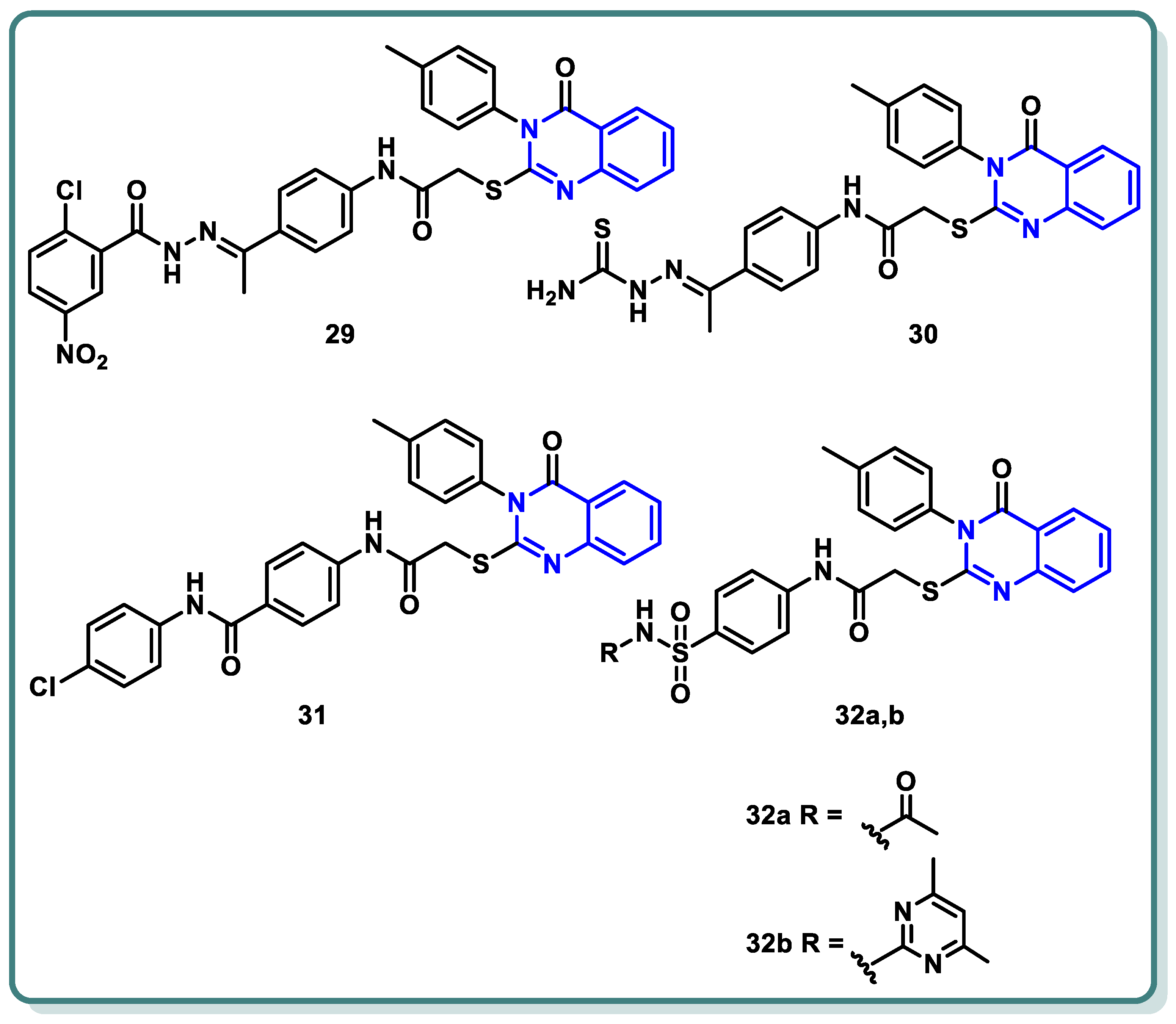
A new series of different quinazoline derivatives of potential cytotoxic and VEGFR-2 inhibitory activities were designed and synthesized by El-Adl et al. [53]. Compounds 29, 30, 31, 32a, and 32b produced significant impacts. They were found to be mostly more active than the standard or almost the same, with IC50 results varying between 1.68 ± 0.06 and 9.89 ± 0.39 μM compared to doxorubicin of IC50 values, which varied from 8.28 ± 0.33 to 9.63 ± 0.38 μM, and with sorafenib of IC50 values ranging from 7.31 ± 0.29 to 9.40 ± 0.37 μM against HepG2, HCT-116, and MCF-7 cells, respectively. An in vitro VEGFR-2 enzyme assay showed that the latter five compounds, 29, 30, 31, 32a, and 32b significantly suppressed VEGFR-2 (IC50 = 0.290 ± 0.05, 0.517 ± 0.07, 0.380 ± 0.04, 0.377 ± 0.04, and 0.415 ± 0.03 μg/mL, respectively) compared to sorafenib, with an IC50 value of 0.588 μg/mL (Figure 13) [53].

Figure 13.
Structures of quinazolines 29–32.
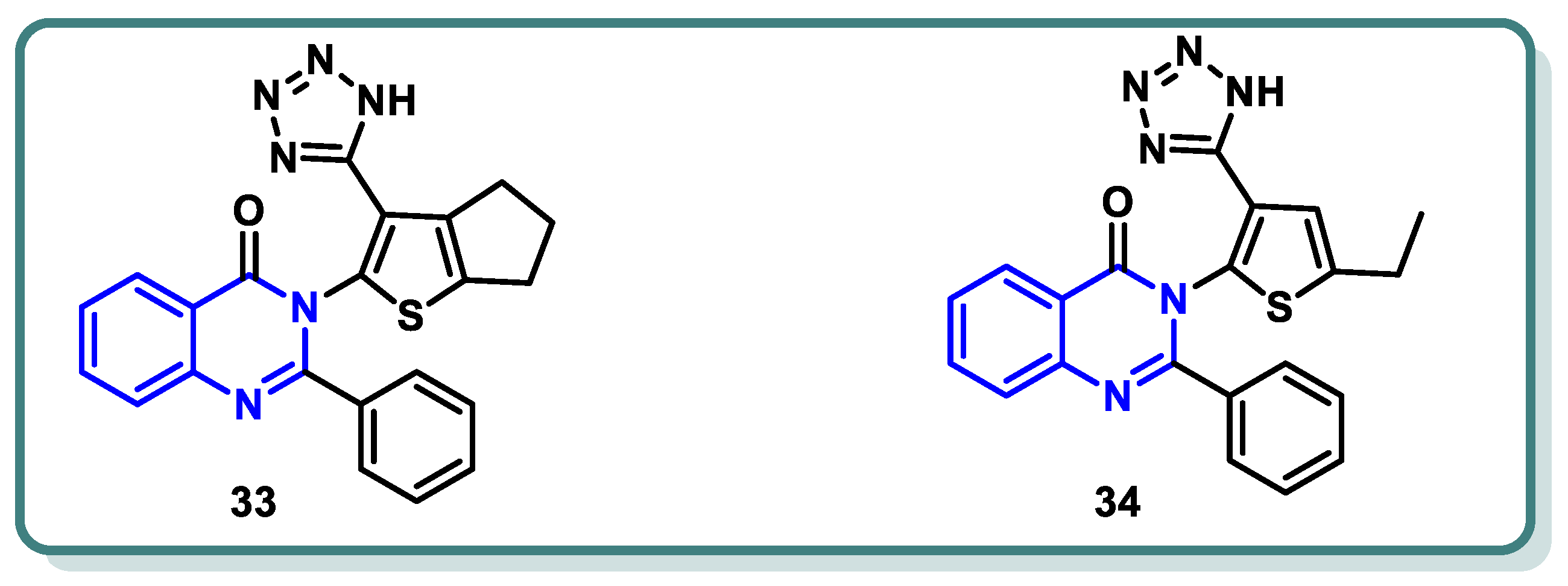
In a recent study conducted by Elrayess et al. [54], novel derivatives of 2-phenylquinazolin-4(3H)-one were developed. Their cytotoxic effects were assessed in situ against MCF-7 and HepG2 cells, revealing promising results. Derivatives 33 and 34 (Figure 14) exhibited notable antiproliferative effects against MCF-7 (IC50 = 1.35 μM) and HepG2 cells (IC50 = 3.24 μM), which were equivalent to those of sorafenib (IC50 values of 3.04 and 2.93 μM, respectively). When comparing compounds 33 and 34 to sorafenib as VEGFR-2 inhibitors, they exhibited better activities, with IC50 values of 13, 67, and 30 nM, respectively. Compound 33 boosted cell cycle arrest at the S-phase by 55.11 times by inducing apoptosis in breast cancer cells. This compound downregulated the antiapoptotic gene Bcl-2 and elevated the apoptotic-related genes caspases 3, 8, 9, P53, Bax, and others. Molecular docking studies revealed that compound 33 exhibited a considerable binding affinity to both the DFG loop and the allosteric region of VEGFR-2 (Figure 14).

Figure 14.
Structures of VEGFR-2 inhibitors 33 and 34.
2.3. Rapidly Accelerated Fibrosarcoma (RAF) Inhibitors
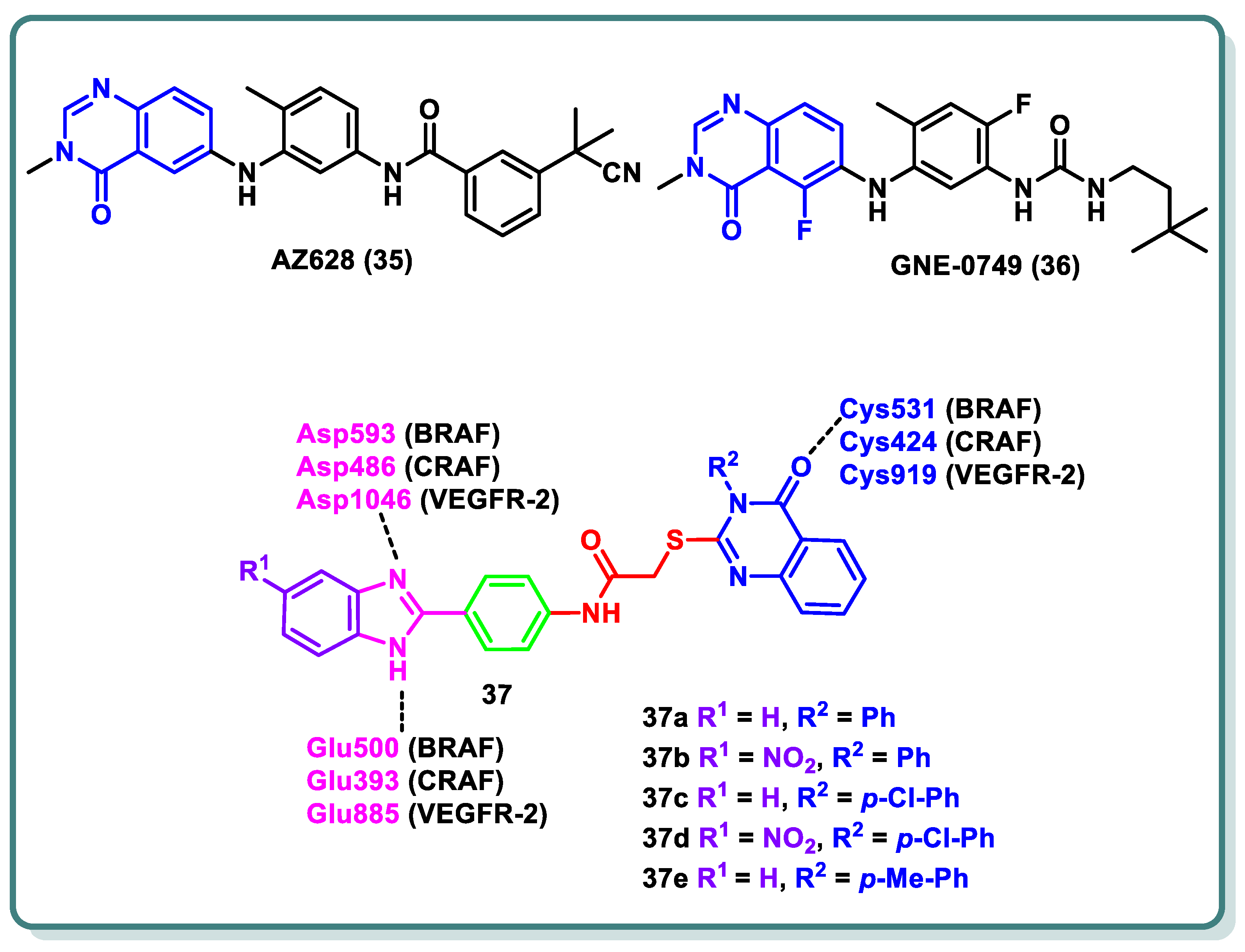
The three isoforms of rapidly accelerated fibrosarcoma (RAF) kinases, CRAF (RAF-1), BRAF, and ARAF, are serine/threonine protein kinases that are a component of the MAPK signaling cascade, a critical promoter involved in cellular development, differentiation, and proliferation [55]. Compared to ARAF and CRAF, BRAF is the most significant RAF kinase and an essential MEK1/2 activator [55]. Numerous forms of melanoma are commonly linked to BRAF mutations [41]. The most prevalent BRAF mutation is known as BRAF (V600E), in which a glutamate residue is placed in place of a valine residue at position 600 [41]. CRAF is activated via interactions with BRAF in a RAS-dependent process, which represents the complexity of the signaling pathway. The quinazolinone nucleus is a privileged scaffold with various derivatives acting as RAF inhibitors. While GNE-0749 (36), a fluoroquinazolinone urea derivative, acts as a highly selective pan-RAF suppressor in K-Ras mutant animals in pre-clinical research [56], AZ628 (35) is a quinazolinone derivative that acts as a BRAF (wt), BRAF (V600E), CRAF, and VEGFR-2 multi-kinase inhibitor [57].
In 2022, Ali and coauthors designed and synthesized of a series of 2-arylbenzimidazole-thioquinazolin-4(3H)-ones conjugates 37 (Figure 15) [58]. Dual potency on BRAFV600E and CRAF was found for 37a–d (IC50 values 0.002–0.37 µM). Additionally, multi-kinase inhibitory activity was observed for the quinazolinone derivative 37e (IC50 = 6.10, 6.70, 2.50, 10.80, 0.03, 0.13, and 0.12 µM a on VEGFR-2, BRAFWT, BRAFV600E, CRAF, PDGFR-β, FLT-3, and c-KIT, respectively). The synthesized compounds’ growth inhibition percentage (GI%) was determined at 10 µM after they were assessed in vitro against a panel of anticancer cell lines at NCI–US. Among the compounds chosen for screening in the five-dose trial, compounds 37c,d exhibited GI50 values as high as 1.1 µM. Moreover, a cell cycle analysis was carried out in an A-375 melanoma cell line for compound 37c, which arrested the cell cycle at the G2/M phase by 7.89%, as well as induction of the late apoptosis phase by 4.14%, respectively. Molecular docking experiments were conducted on BRAF, CRAF, and VEGFR-2 to further investigate the binding mechanisms for the compounds and their interactions via the important amino acids, namely BRAF (Glu500, Cys531, and Asp593), CRAF (Glu393, Cys424, and Asp486), and VEGFR-2 (Glu885, Cys919, and Asp1046) (Figure 15) [58].
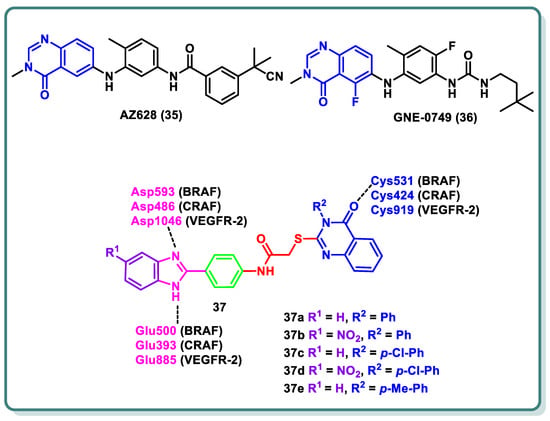
Figure 15.
Structures of various quinazolines as RAF-inhibiting candidates.
2.4. Phosphoinositide 3-Kinase (PI3K) Inhibitors
Phosphoinositide 3-kinase, or PI3K, is an intracellular lipid kinase that is involved in the regulation of cell proliferation, migration, differentiation, and death. It is part of the PI3K/AKT/mTOR signaling pathway [59].
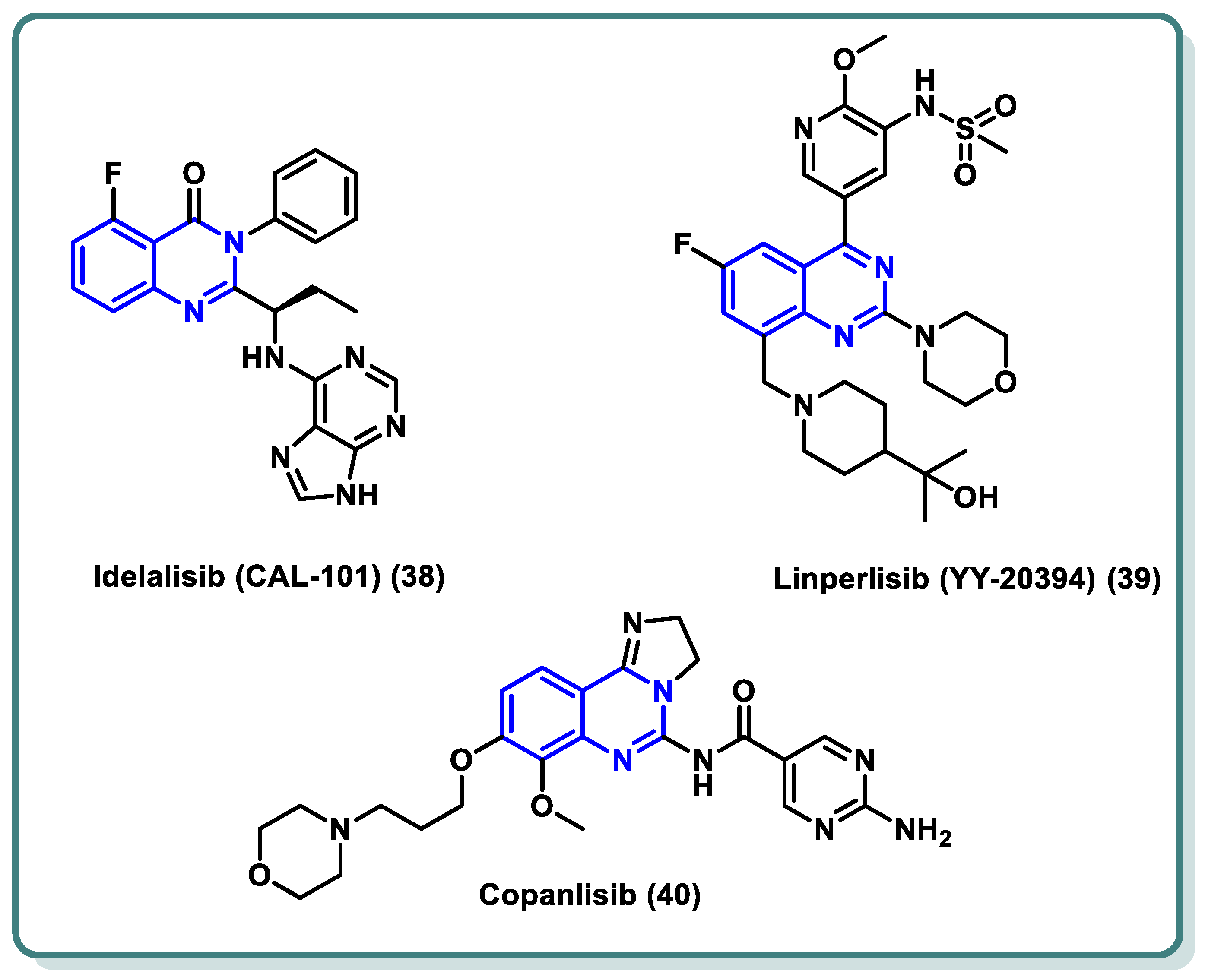
PI3Ks can be divided into three categories based on their structural characteristics and substrate selectivity, with class I PI3K receiving the most research attention. G protein-coupled receptor (GPCR) or receptor tyrosine kinase (RTK) signaling is mediated by class I PI3K [60,61]. The active class I PI3K is catalyzed by signals from the RTK and GPCR to phosphorylate PIP2 to PIP3. This lipid product participates in the phosphorylation of protein kinase B (PKB, Akt) and functions as a second messenger to regulate cellular processes. Class I PI3K is further classified into PI3Kα, PI3Kβ, PI3Kδ, and PI3Kγ [62]. Research has demonstrated a strong correlation between PI3K dysregulation and the emergence and progression of a wide range of tumors [63]. Specifically, PI3Kδ is a significant subtype of type I PI3K that primarily regulates B cell receptor (BCR) signaling [64]. As a result, PI3Kδ is now a useful target for the treatment of B cell-related illnesses as well as hematologic malignancies. In recent years, a number of quinazoline-based PI3Kδ inhibitors have been approved by the FDA in the past decade for the treatment of malignant tumors, asthma, immunodeficiency, and autoimmune diseases. As an illustration, idelalisib (38) (Figure 16), the first PI3Kδ inhibitor licensed for therapeutic use, was used to treat relapsed follicular lymphoma (FL), small lymphocytic lymphoma (SLL), and chronic lymphocytic leukaemia (CLL) [65]. Idelalisib occupies the ATP binding site of PI3Kδ, where the purine moiety is involved in hydrogen bonding through N3 and N9 with the amino acid residues Val-828 and Glu-826, respectively. Additionally, N7 of the purine moiety forms a water-mediated network with Asp911 in conjunction with N1 of the quinazoline moiety. Moreover, the inhibitor showed additional hydrophobic interaction through its ethyl group with the target protein [66]. Furthermore, linperlisib (39) (Figure 16) was successively approved for treating CLL, SLL, FL, and APDS (activated PI3Kδ syndrome) [67]. Also, Copanlisib (40) has been indicated for various cancers [68,69].

Figure 16.
Structures of different PI3K inhibitors 38–40 based on the quinazoline nucleus.
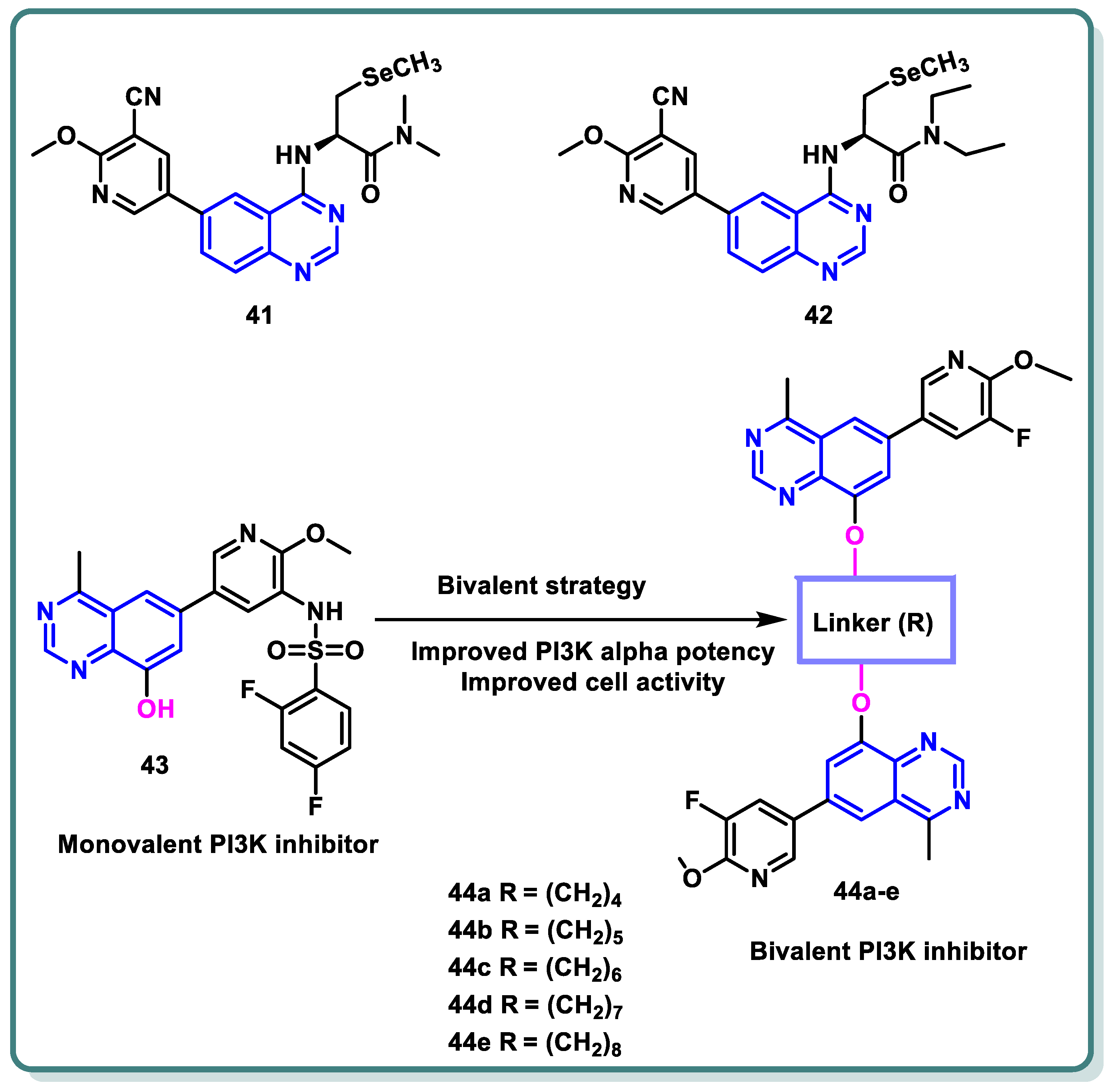
In a recent study by Gao et al., a series of selenium-containing PI3Kδ inhibitors based on quinazoline and pyrido [3,2-d]pyrimidine skeletons was designed and synthesized. Following the assessment of δ-kinase activities, the obtained data demonstrated that 41 and 42 tailed with a line-chain amine produced potent suppression activity against PI3Kδ (IC50 = 1.13 and 2.52 nM, respectively) [70]. Ab in vitro anticancer assay against the human malignant B cell line SU-DHL-6 demonstrated the promising activity of these compounds compared with idelalisib as a positive control (Figure 17).

Figure 17.
Structure of different quinazoline-based derivatives as PI3K inhibitors.
Furthermore, in order to create a number of bivalent PI3K inhibitors, Xia and colleagues selected a 4-methylquinazoline scaffold (Figure 17). To be more precise, legend 43 was chosen as the monovalent counterpart that might generate three important interactions with the PI3Kα target: a charged interaction between Lys802 and the deprotonated sulfonamide, a water bridge via pyridine nitrogen and Tyr836 and Asp810, and a hydrogen bond between the nitrogen atom in the quinazoline scaffold at position 3 and Val851. The quinazoline scaffold’s position 8 was appropriate for a range of functional groups that occupied the solvent-exposed region [71]. As a result, several bivalent PI3K inhibitors were created by adding linkers of various kinds and lengths to the quinazoline scaffold’s 8 locations (Figure 17), keeping the major structural groups attached to PI3Kα’s ATP-binding pocket. A variety of scaffold substituents (R) were investigated to identify the ideal “monovalent counterpart.” By blocking the PI3K pathway, these bivalent PI3K inhibitors are anticipated to exhibit greater PI3K potencies and offer a novel approach to the search for new cancer treatments. Different lengths of alkylene linkers were bonded as hydrophobic segments to the phenolic group in position 8 of the 4-methyl quinazoline scaffold using ether bonds. It was demonstrated that compound 44a, bearing a 4- methylene linker suppressed PI3Kα (IC50 value of 1166 nM), was roughly 2-fold more significant than compound 44b, bearing a 5-methylene linker (IC50 = 2722 nM). Compound 44c, bearing 6-methylene linker, showed a 2-fold potency more than that of compound 44b (IC50 = 1282 nM). In addition, compounds 44d and 44e, which have 7- and 8-methylene linkers, exhibited lower PI3K potencies than compound 44c. Their IC50 values were 2137 nM and 4247 nM, respectively. With an additional chain length extension from the 9-methylene linker to 12-methylene linker, the inhibitory effects on PI3Kα appeared to be diminished (IC50 > 10,000 nM).
2.5. Cyclin-Dependent Kinase (CDK) Inhibitors
Cyclin-dependent kinases (CDKs) are a family of protein kinases with central roles in orchestrating the eukaryotic cell cycle. Among the 13 mammalian CDKs, CDK1, CDK2, CDK3, CDK4, and CDK6 hold particular significance for their control of cell cycle progression through targeted phosphorylation of protein substrates [72]. Dysregulation of these kinases, leading to uncontrolled cell proliferation, is a hallmark of cancer [73]. CDK2 [74,75,76,77,78,79] occupies a pivotal position in this regulatory network. Its influence extends beyond cell cycle transitions and G1/S and G2 phase progression to encompass DNA repair [80], gene transcription, and epigenetic modifications in tumors [81,82,83]. Abnormal expression of cyclins, the regulatory subunits essential for CDK2 activity, further underscores its oncogenic potential, particularly through overexpression of cyclin A and/or E in various cancers [84,85].
Recognizing CDK2’s multifaceted role in cancer development, researchers have focused on its inhibition as a therapeutic strategy. Potent CDK2 inhibitors, such as dinaciclib [86], milciclib [80], and roniciclib [87], are currently undergoing clinical evaluation, targeting this kinase specifically or in combination with others. However, the presence of adverse effects observed in clinical trials underscores the need for further refinement and discovery of more selective and efficacious CDK2 inhibitors [88].
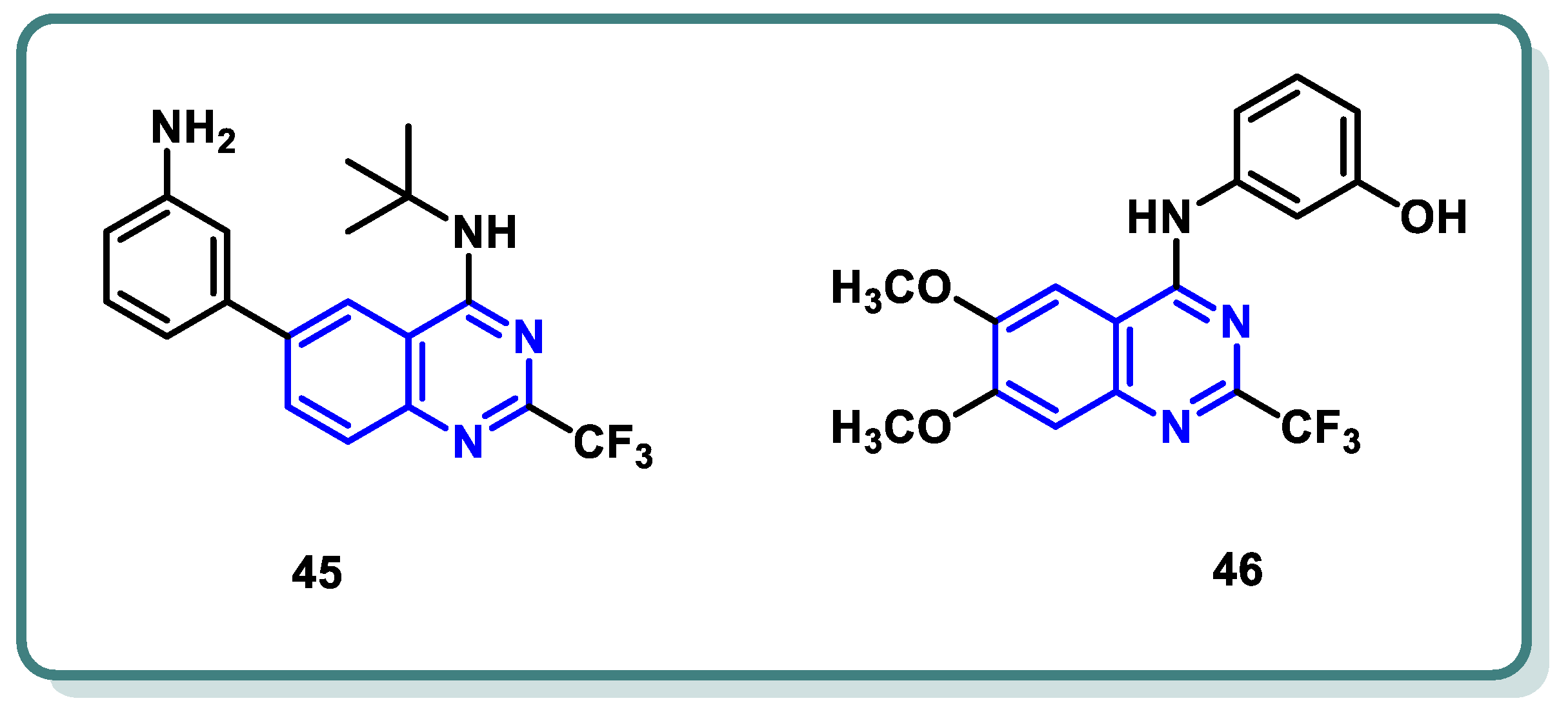
Our analysis of the literature revealed a diverse landscape of CDK2 inhibitors, encompassing scaffolds like pyrazolopyrimidines (dinaciclib), purines (fadraciclib [89], roscovitine [90]), and quinazolines (compounds 45 [91] and 46 [92]) (Figure 18).

Figure 18.
Reported quinazoline-based CDK2 inhibitors.
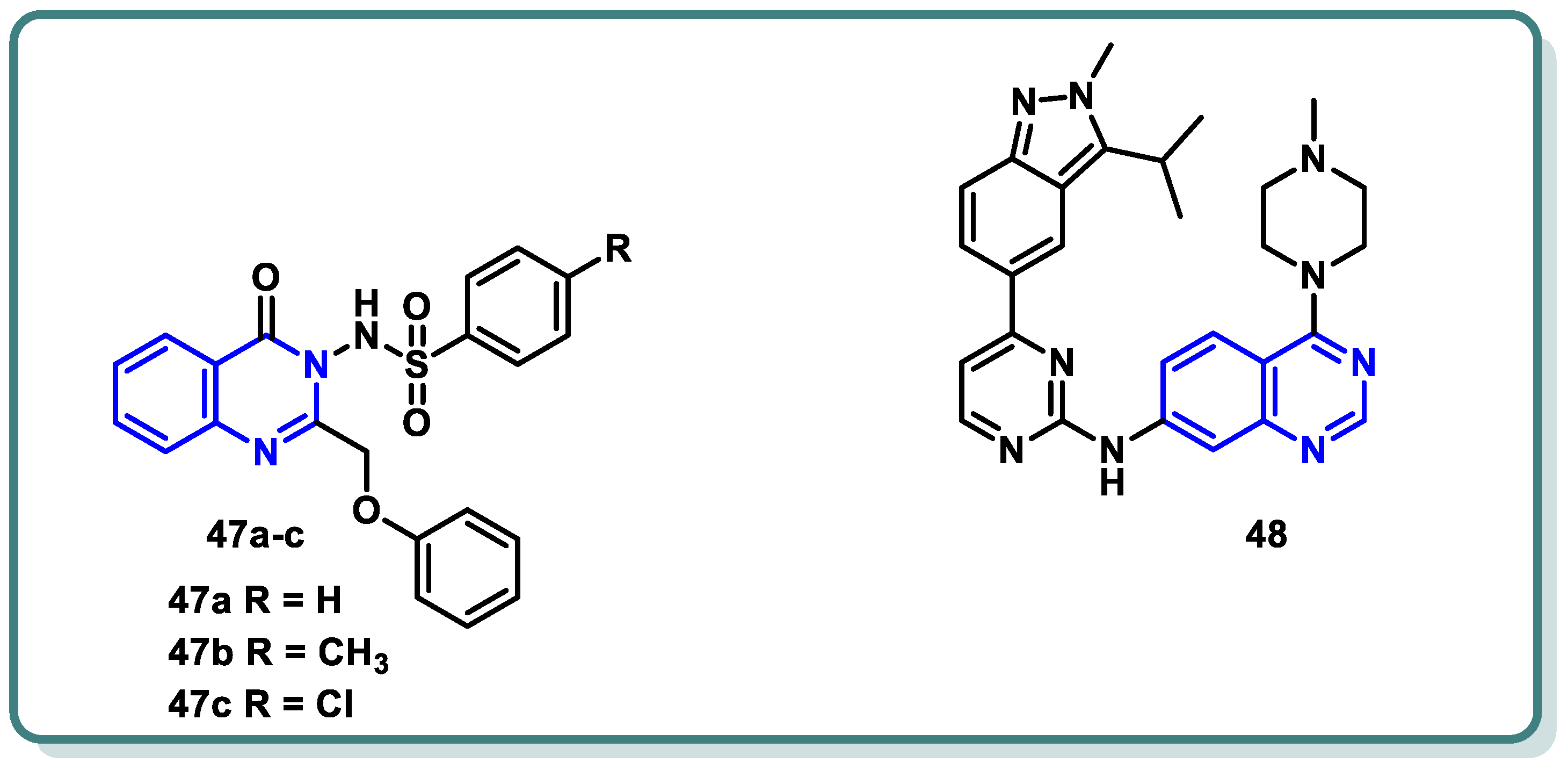
Recently, Mohammed et al. [93] strategically chose a substitution pattern at the 2 and 3 positions of the quinazolinone ring to create distinct electronic circumstances (Figure 19). This variation was intended to impact the lipophilicity, thereby influencing the activity of the target molecules. The objective behind the synthesis these analogues were to develop active CDK2 suppressors with enhanced efficacy against cancer cells characterized by overexpression of CDK2. In this study, quinazolin-4(3H)-one derivatives 47 were synthesized as CDK2 inhibitors, showing potent antiproliferative activity against melanoma (MDA-MB-435) and glioblastoma (SNB-75) cell lines. Compound 47c demonstrated a superior potency, with an IC50 of 3.03 µM against MDA-MB-435, surpassing the reference roscovitine [90]. Additionally, 47c induced cell cycle arrest and apoptosis, highlighting CDK2 as a potential target. CDK2 inhibitory assays confirmed the potency of 47c (IC50 = 0.63 µM). Additionally, computational ADME studies indicated that the most active compound 47c possessed promising pharmacokinetic properties. These findings endorse the potential of compound 47c as a valuable lead candidate in the quest for promising anti-melanoma agents through CDK2 inhibition.

Figure 19.
Quinazolines 47 and 48 as CDK inhibitors.
On the other hand, investigations by Huang et al. [94] revealed that compound 48 exhibits robust multi-targeted inhibition of cyclin-dependent kinases (CDKs), encompassing CDK2, CDK1, CDK4, CDK8, and CDK9. These results led to G1 arrest and pro-apoptotic signaling in hematologic malignancies. The inclusive approach of compound 48 addressed the potential resistance mechanisms, broadening its impact across a spectrum of vulnerable cancer cells. While compound 48 showed promise in preclinical studies, further research is essential to fully unravelling the potential of compound 48, paving the way for transformative clinical trials in the complex landscape of hematologic cancers.
3. Conclusions
Research on new cancer treatments has progressed from a time of harmful, non-selective medications to safer, selective ones. The main shortcomings of chemotherapeutic drugs are their lack of selectivity and the toxicity that follows. Accordingly, there is significant evidence to support the improvement of targeted therapy with site-specific action in the management of this fatal illness. Extensive work in the drug discovery field and understanding of cancer biology led to massive exploration of quinazoline pharmacophore-based derivatives as a kinase-targeted cancer therapy. Several quinazoline-based drugs acting as kinase inhibitors have been licensed by the FDA and are available for clinical use for the treatment of advanced cancer diseases. The majority of the alterations that were made to the quinazoline skeleton at positions 4, 6, and 7 in order to create anticancer molecules produced substances with extremely strong biological activities against cancer. Additional structural alterations to the quinazoline nucleus may result in novel derivatives with a greater efficacy than already prescribed medications. The literature review above highlights the significance of quinazoline nucleus structural modifications and resulting molecular changes for cancer therapy. This study provides scientists working towards improvements in novel target-specific therapeutic candidates for cancer treatment with fresh insights and an enhanced comprehension.
Author Contributions
Conceptualization, H.T.A.-M., M.M.A., N.S.A., S.S.A.E.-K. and S.H.A.; data curation, H.T.A.-M., M.M.A., N.S.A., S.S.A.E.-K. and S.H.A.; methodology, H.T.A.-M., M.M.A., N.S.A., S.S.A.E.-K. and S.H.A.; supervision, H.T.A.-M., M.M.A., N.S.A., S.S.A.E.-K. and S.H.A.; writing—original draft, H.T.A.-M., M.M.A., N.S.A., S.S.A.E.-K. and S.H.A.; writing—review and editing, H.T.A.-M., M.M.A., N.S.A., S.S.A.E.-K. and S.H.A. All authors have read and agreed to the published version of the manuscript.
Funding
Funding for this research was provided by the Robert A. Welch Foundation, grant number L-0002, and Prairie View University Faculty, grant number 552509-00018.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article references.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- de Martel, C.; Georges, D.; Bray, F.; Ferlay, J.; Clifford, G.M. Global burden of cancer attributable to infections in 2018: A worldwide incidence analysis. Lancet Glob. Health 2020, 8, e180–e190. [Google Scholar] [CrossRef]
- Singh, D.; Vaccarella, S.; Gini, A.; De Paula Silva, N.; Steliarova-Foucher, E.; Bray, F. Global patterns of Hodgkin lymphoma incidence and mortality in 2020 and a prediction of the future burden in 2040. Int. J. Cancer 2022, 150, 1941–1947. [Google Scholar] [CrossRef]
- Shenoy, G.P.; Pal, R.; Purwarga Matada, G.S.; Singh, E.; Raghavendra, N.M.; Dhiwar, P.S. Discoidin domain receptor inhibitors as anticancer agents: A systematic review on recent development of DDRs inhibitors, their resistance and structure activity relationship. Bioorg. Chem. 2023, 130, 106215. [Google Scholar] [CrossRef]
- Giaquinto, A.N.; Miller, K.D.; Tossas, K.Y.; Winn, R.A.; Jemal, A.; Siegel, R.L. Cancer statistics for African American/Black People 2022. CA Cancer J. Clin. 2022, 72, 202–229. [Google Scholar] [CrossRef]
- Cohen, P. The role of protein phosphorylation in human health and disease. The Sir Hans Krebs Medal Lecture. Eur. J. Biochem. 2001, 268, 5001–5010. [Google Scholar] [CrossRef]
- Cohen, P. Protein kinases—The major drug targets of the twenty-first century? Nat. Rev. Drug Discov. 2002, 1, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Bhullar, K.S.; Lagaron, N.O.; McGowan, E.M.; Parmar, I.; Jha, A.; Hubbard, B.P.; Rupasinghe, H.P.V. Kinase-targeted cancer therapies: Progress, challenges and future directions. Mol. Cancer 2018, 17, 48. [Google Scholar] [CrossRef] [PubMed]
- Faisal, M.; Saeed, A. Chemical Insights into the Synthetic Chemistry of Quinazolines: Recent Advances. Front. Chem. 2020, 8, 594717. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Mohsen, H.T.; Petreni, A.; Supuran, C.T. Investigation of the carbonic anhydrase inhibitory activity of benzenesulfonamides incorporating substituted fused-pyrimidine tails. Arch. Pharm. 2022, 355, e2200274. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Mohsen, H.T.; Omar, M.A.; Petreni, A.; Supuran, C.T. Novel 2-substituted thioquinazoline-benzenesulfonamide derivatives as carbonic anhydrase inhibitors with potential anticancer activity. Arch. Pharm. 2022, 355, e2200180. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Mohsen, H.T.; El Kerdawy, A.M.; Petreni, A.; Supuran, C.T. Novel benzenesulfonamide-thiouracil conjugates with a flexible N-ethyl acetamide linker as selective CA IX and CA XII inhibitors. Arch. Pharm. 2023, 356, e2200434. [Google Scholar] [CrossRef] [PubMed]
- Wieduwilt, M.J.; Moasser, M.M. The epidermal growth factor receptor family: Biology driving targeted therapeutics. Cell. Mol. Life Sci. CMLS 2008, 65, 1566–1584. [Google Scholar] [CrossRef] [PubMed]
- Uribe, M.L.; Marrocco, I.; Yarden, Y. EGFR in Cancer: Signaling Mechanisms, Drugs, and Acquired Resistance. Cancers 2021, 13, 2748. [Google Scholar] [CrossRef]
- Ranson, M. Epidermal growth factor receptor tyrosine kinase inhibitors. Br. J. Cancer 2004, 90, 2250–2255. [Google Scholar] [CrossRef] [PubMed]
- Pal, R.; Teli, G.; Matada, G.S.P.; Dhiwar, P.S. Designing strategies, structural activity relationship and biological activity of recently developed nitrogen containing heterocyclic compounds as epidermal growth factor receptor tyrosinase inhibitors. J. Mol. Struct. 2023, 1291, 136021. [Google Scholar] [CrossRef]
- Thomas, P.; Vincent, B.; George, C.; Joshua, J.M.; Pavithran, K.; Vijayan, M. A comparative study on erlotinib & gefitinib therapy in non-small cell lung carcinoma patients. Indian J. Med. Res. 2019, 150, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Ge, R.; Sang, D.; Luo, T.; Li, W.; Ji, X.; Yuan, P.; Wang, B. Real-world data of lapatinib and treatment after lapatinib in patients with previously treated HER2-positive metastatic breast cancer: A multicenter, retrospective study. Cancer Med. 2020, 9, 2981–2988. [Google Scholar] [CrossRef]
- Wood, E.R.; Truesdale, A.T.; McDonald, O.B.; Yuan, D.; Hassell, A.; Dickerson, S.H.; Ellis, B.; Pennisi, C.; Horne, E.; Lackey, K.; et al. A unique structure for epidermal growth factor receptor bound to GW572016 (Lapatinib): Relationships among protein conformation, inhibitor off-rate, and receptor activity in tumor cells. Cancer Res. 2004, 64, 6652–6659. [Google Scholar] [CrossRef]
- Stamos, J.; Sliwkowski, M.X.; Eigenbrot, C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 2002, 277, 46265–46272. [Google Scholar] [CrossRef]
- Amelia, T.; Kartasasmita, R.E.; Ohwada, T.; Tjahjono, D.H. Structural Insight and Development of EGFR Tyrosine Kinase Inhibitors. Molecules 2022, 27, 819. [Google Scholar] [CrossRef]
- Sharma, S.V.; Bell, D.W.; Settleman, J.; Haber, D.A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 2007, 7, 169–181. [Google Scholar] [CrossRef]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2, e73. [Google Scholar] [CrossRef]
- Wang, S.; Li, J. Second-generation EGFR and ErbB tyrosine kinase inhibitors as first-line treatments for non-small cell lung cancer. OncoTargets Ther. 2019, 12, 6535–6548. [Google Scholar] [CrossRef]
- Li, H.S.; Wang, S.Z.; Xu, H.Y.; Yan, X.; Zhang, J.Y.; Lei, S.Y.; Li, T.; Hao, X.Z.; Zhang, T.; Yang, G.J.; et al. Afatinib and Dacomitinib Efficacy, Safety, Progression Patterns, and Resistance Mechanisms in Patients with Non-Small Cell Lung Cancer Carrying Uncommon EGFR Mutations: A Comparative Cohort Study in China (AFANDA Study). Cancers 2022, 14, 5307. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gan, W.; Fan, D.; Zheng, P.; Lv, Q.; Pan, Q.; Zhu, W. Novel quinazoline-based dual EGFR/c-Met inhibitors overcoming drug resistance for the treatment of NSCLC: Design, synthesis and anti-tumor activity. Bioorg. Chem. 2024, 142, 106938. [Google Scholar] [CrossRef] [PubMed]
- Gan, W.; Wang, C.; Pan, Q.; Li, Y.; Guo, Y.; Fan, D.; Peng, Y.; Rao, Z.; Xu, S.; Zheng, P.; et al. Discovery of novel 4-arylamino-quinazoline derivatives as EGFR(L858R/T790M) inhibitors with the potential to inhibit the non-small cell lung cancers. Bioorg. Chem. 2022, 127, 105994. [Google Scholar] [CrossRef]
- Qin, X.; Liu, P.; Li, Y.; Hu, L.; Liao, Y.; Cao, T.; Yang, L. Design, synthesis and biological evaluation of novel 3,4-dihydro-2H-[1,4]oxazino [2,3-f]quinazolin derivatives as EGFR-TKIs. Bioorg. Med. Chem. Lett. 2023, 80, 129104. [Google Scholar] [CrossRef] [PubMed]
- Dou, D.; Wang, J.; Qiao, Y.; Wumaier, G.; Sha, W.; Li, W.; Mei, W.; Yang, T.; Zhang, C.; He, H.; et al. Discovery and optimization of 4-anilinoquinazoline derivatives spanning ATP binding site and allosteric site as effective EGFR-C797S inhibitors. Eur. J. Med. Chem. 2022, 244, 114856. [Google Scholar] [CrossRef] [PubMed]
- Hasanvand, Z.; Oghabi Bakhshaiesh, T.; Peytam, F.; Firoozpour, L.; Hosseinzadeh, E.; Motahari, R.; Moghimi, S.; Nazeri, E.; Toolabi, M.; Momeni, F.; et al. Imidazo[1,2-a]quinazolines as novel, potent EGFR-TK inhibitors: Design, synthesis, bioactivity evaluation, and in silico studies. Bioorg. Chem. 2023, 133, 106383. [Google Scholar] [CrossRef]
- Ghorab, M.M.; Soliman, A.M.; El-Adl, K.; Hanafy, N.S. New quinazoline sulfonamide derivatives as potential anticancer agents: Identifying a promising hit with dual EGFR/VEGFR-2 inhibitory and radiosensitizing activity. Bioorg. Chem. 2023, 140, 106791. [Google Scholar] [CrossRef]
- Ghorab, W.M.; El-Sebaey, S.A.; Ghorab, M.M. Design, synthesis and Molecular modeling study of certain EGFRinhibitors with a quinazolinone scaffold as anti-hepatocellular carcinoma and Radio-sensitizers. Bioorg. Chem. 2023, 131, 106310. [Google Scholar] [CrossRef]
- Abhinand, C.S.; Raju, R.; Soumya, S.J.; Arya, P.S.; Sudhakaran, P.R. VEGF-A/VEGFR2 signaling network in endothelial cells relevant to angiogenesis. J. Cell Commun. Signal 2016, 10, 347–354. [Google Scholar] [CrossRef]
- Adams, R.H.; Alitalo, K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat. Rev. Mol. Cell Biol. 2007, 8, 464–478. [Google Scholar] [CrossRef]
- Modi, S.J.; Kulkarni, V.M. Vascular Endothelial Growth Factor Receptor (VEGFR-2)/KDR Inhibitors: Medicinal Chemistry Perspective. Med. Drug Discov. 2019, 2, 100009. [Google Scholar] [CrossRef]
- Hicklin, D.J.; Ellis, L.M. Role of the vascular endothelial growth factor pathway in tumor growth and angiogenesis. J. Clin. Oncol. 2005, 23, 1011–1027. [Google Scholar] [CrossRef]
- Muto, J.; Shirabe, K.; Sugimachi, K.; Maehara, Y. Review of angiogenesis in hepatocellular carcinoma. Hepatol. Res. 2015, 45, 1–9. [Google Scholar] [CrossRef]
- Abdel-Mohsen, H.T.; Girgis, A.S.; Mahmoud, A.E.E.; Ali, M.M.; El Diwani, H.I. New 2,4-disubstituted-2-thiopyrimidines as VEGFR-2 inhibitors: Design, synthesis, and biological evaluation. Arch. Pharm. 2019, 352, e1900089. [Google Scholar] [CrossRef]
- Abdel-Mohsen, H.; El Kerdawy, A. Design, Synthesis, Molecular Docking Studies and in Silico Prediction of ADME Properties of New 5-Nitrobenzimidazole/thiopyrimidine Hybrids as Anti-angiogenic Agents Targeting Hepatocellular Carcinoma. Egypt. J. Chem. 2023, 67, 437–446. [Google Scholar] [CrossRef]
- Rajabi, M.; Mousa, S.A. The Role of Angiogenesis in Cancer Treatment. Biomedicines 2017, 5, 34. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Kirkwood, J.M.; Grob, J.J.; Simeone, E.; Grimaldi, A.M.; Maio, M.; Palmieri, G.; Testori, A.; Marincola, F.M.; Mozzillo, N. The role of BRAF V600 mutation in melanoma. J. Transl. Med. 2012, 10, 85. [Google Scholar] [CrossRef] [PubMed]
- Ramos, H.E.; Hecht, F.; Berdelou, A.; Borget, I.; Leboulleux, S.; Baudin, E.; Schlumberger, M. Long-term follow-up and safety of vandetanib for advanced medullary thyroid cancer. Endocrine 2021, 71, 434–442. [Google Scholar] [CrossRef]
- Li, H.; Yu, Y.; Zhao, Y.; Wu, D.; Yu, X.; Lu, J.; Chen, Z.; Zhang, H.; Hu, Y.; Zhai, Y.; et al. Small molecule inhibitor agerafenib effectively suppresses neuroblastoma tumor growth in mouse models via inhibiting ERK MAPK signaling. Cancer Lett. 2019, 457, 129–141. [Google Scholar] [CrossRef]
- Abd El-Karim, S.S.; Syam, Y.M.; El Kerdawy, A.M.; Abdel-Mohsen, H.T. Rational design and synthesis of novel quinazolinone N-acetohydrazides as type II multi-kinase inhibitors and potential anticancer agents. Bioorg. Chem. 2024, 142, 106920. [Google Scholar] [CrossRef]
- Hamdi, A.; El-Shafey, H.W.; Othman, D.I.A.; El-Azab, A.S.; AlSaif, N.A.; Abdel-Aziz, A.A.M. Design, synthesis, antitumor, and VEGFR-2 inhibition activities of novel 4-anilino-2-vinyl-quinazolines: Molecular modeling studies. Bioorg. Chem. 2022, 122, 105710. [Google Scholar] [CrossRef] [PubMed]
- Zahran, S.S.; Ragab, F.A.; El-Gazzar, M.G.; Soliman, A.M.; Mahmoud, W.R.; Ghorab, M.M. Antiproliferative, antiangiogenic and apoptotic effect of new hybrids of quinazoline-4(3H)-ones and sulfachloropyridazine. Eur. J. Med. Chem. 2023, 245, 114912. [Google Scholar] [CrossRef] [PubMed]
- Abdallah, A.E.; Eissa, S.I.; Al Ward, M.M.S.; Mabrouk, R.R.; Mehany, A.B.M.; El-Zahabi, M.A. Design, synthesis and molecular modeling of new quinazolin-4(3H)-one based VEGFR-2 kinase inhibitors for potential anticancer evaluation. Bioorg. Chem. 2021, 109, 104695. [Google Scholar] [CrossRef]
- Abdallah, A.E.; Mabrouk, R.R.; Al Ward, M.M.S.; Eissa, S.I.; Elkaeed, E.B.; Mehany, A.B.M.; Abo-Saif, M.A.; El-Feky, O.A.; Alesawy, M.S.; El-Zahabi, M.A. Synthesis, biological evaluation, and molecular docking of new series of antitumor and apoptosis inducers designed as VEGFR-2 inhibitors. J. Enzym. Inhib. Med. Chem. 2022, 37, 573–591. [Google Scholar] [CrossRef]
- Hassan, A.; Mubarak, F.A.F.; Shehadi, I.A.; Mosallam, A.M.; Temairk, H.; Badr, M.; Abdelmonsef, A.H. Design and biological evaluation of 3-substituted quinazoline-2,4(1H,3H)-dione derivatives as dual c-Met/VEGFR-2-TK inhibitors. J. Enzym. Inhib. Med. Chem. 2023, 38, 2189578. [Google Scholar] [CrossRef]
- Azab, A.E.; Alesawy, M.S.; Eldehna, W.M.; Elwan, A.; Eissa, I.H. New [1,2,4]triazolo[4,3-c]quinazoline derivatives as vascular endothelial growth factor receptor-2 inhibitors and apoptosis inducers: Design, synthesis, docking, and antiproliferative evaluation. Arch. Pharm. 2022, 355, e2200133. [Google Scholar] [CrossRef]
- Wang, R.; Liu, H.; You, Y.Y.; Wang, X.Y.; Lv, B.B.; Cao, L.Q.; Xue, J.Y.; Xu, Y.G.; Shi, L. Discovery of novel VEGFR-2 inhibitors embedding 6,7-dimethoxyquinazoline and diarylamide fragments. Bioorg. Med. Chem. Lett. 2021, 36, 127788. [Google Scholar] [CrossRef]
- Eissa, I.H.; Ibrahim, M.K.; Metwaly, A.M.; Belal, A.; Mehany, A.B.M.; Abdelhady, A.A.; Elhendawy, M.A.; Radwan, M.M.; ElSohly, M.A.; Mahdy, H.A. Design, molecular docking, in vitro, and in vivo studies of new quinazolin-4(3H)-ones as VEGFR-2 inhibitors with potential activity against hepatocellular carcinoma. Bioorg. Chem. 2021, 107, 104532. [Google Scholar] [CrossRef]
- El-Adl, K.; El-Helby, A.-G.A.; Ayyad, R.R.; Mahdy, H.A.; Khalifa, M.M.; Elnagar, H.A.; Mehany, A.B.M.; Metwaly, A.M.; Elhendawy, M.A.; Radwan, M.M.; et al. Design, synthesis, and anti-proliferative evaluation of new quinazolin-4(3H)-ones as potential VEGFR-2 inhibitors. Bioorg. Med. Chem. 2021, 29, 115872. [Google Scholar] [CrossRef]
- Elrayess, R.; Elgawish, M.S.; Nafie, M.S.; Ghareb, N.; Yassen, A.S.A. 2-Phenylquinazolin-4(3H)-one scaffold as newly designed, synthesized VEGFR-2 allosteric inhibitors with potent cytotoxicity through apoptosis. Arch. Pharm. 2023, 356, e2200654. [Google Scholar] [CrossRef]
- Hussain, M.R.; Baig, M.; Mohamoud, H.S.; Ulhaq, Z.; Hoessli, D.C.; Khogeer, G.S.; Al-Sayed, R.R.; Al-Aama, J.Y. BRAF gene: From human cancers to developmental syndromes. Saudi J. Biol. Sci. 2015, 22, 359–373. [Google Scholar] [CrossRef]
- Khazak, V.; Astsaturov, I.; Serebriiskii, I.G.; Golemis, E.A. Selective Raf inhibition in cancer therapy. Expert Opin. Ther. Targets 2007, 11, 1587–1609. [Google Scholar] [CrossRef]
- Huestis, M.P.; Dela Cruz, D.; DiPasquale, A.G.; Durk, M.R.; Eigenbrot, C.; Gibbons, P.; Gobbi, A.; Hunsaker, T.L.; La, H.; Leung, D.H.; et al. Targeting KRAS Mutant Cancers via Combination Treatment: Discovery of a 5-Fluoro-4-(3H)-quinazolinone Aryl Urea pan-RAF Kinase Inhibitor. J. Med. Chem. 2021, 64, 3940–3955. [Google Scholar] [CrossRef]
- Ali, I.H.; Abdel-Mohsen, H.T.; Mounier, M.M.; Abo-elfadl, M.T.; El Kerdawy, A.M.; Ghannam, I.A.Y. Design, synthesis and anticancer activity of novel 2-arylbenzimidazole/2-thiopyrimidines and 2-thioquinazolin-4(3H)-ones conjugates as targeted RAF and VEGFR-2 kinases inhibitors. Bioorg. Chem. 2022, 126, 105883. [Google Scholar] [CrossRef]
- Burke, J.E.; Williams, R.L. Synergy in activating class I PI3Ks. Trends Biochem. Sci. 2015, 40, 88–100. [Google Scholar] [CrossRef]
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644. [Google Scholar] [CrossRef]
- Zhao, W.; Qiu, Y.; Kong, D. Class I phosphatidylinositol 3-kinase inhibitors for cancer therapy. Acta Pharm. Sin. B 2017, 7, 27–37. [Google Scholar] [CrossRef]
- Janku, F.; Yap, T.A.; Meric-Bernstam, F. Targeting the PI3K pathway in cancer: Are we making headway? Nat. Rev. Clin. Oncol. 2018, 15, 273–291. [Google Scholar] [CrossRef] [PubMed]
- Cantley, L.C. The phosphoinositide 3-kinase pathway. Science 2002, 296, 1655–1657. [Google Scholar] [CrossRef]
- Puri, K.D.; Di Paolo, J.A.; Gold, M.R. B-cell receptor signaling inhibitors for treatment of autoimmune inflammatory diseases and B-cell malignancies. Int. Rev. Immunol. 2013, 32, 397–427. [Google Scholar] [CrossRef] [PubMed]
- Cheah, C.Y.; Fowler, N.H. Idelalisib in the management of lymphoma. Blood 2016, 128, 331–336. [Google Scholar] [CrossRef]
- Somoza, J.R.; Koditek, D.; Villasenor, A.G.; Novikov, N.; Wong, M.H.; Liclican, A.; Xing, W.; Lagpacan, L.; Wang, R.; Schultz, B.E.; et al. Structural, biochemical, and biophysical characterization of idelalisib binding to phosphoinositide 3-kinase delta. J. Biol. Chem. 2015, 290, 8439–8446. [Google Scholar] [CrossRef]
- Wang, T.; Sun, X.; Qiu, L.; Su, H.; Cao, J.; Li, Z.; Song, Y.; Zhang, L.; Li, D.; Wu, H.; et al. The Oral PI3Kδ Inhibitor Linperlisib for the Treatment of Relapsed and/or Refractory Follicular Lymphoma: A Phase II, Single-Arm, Open-Label Clinical Trial. Clin. Cancer Res. 2023, 29, 1440–1449. [Google Scholar] [CrossRef] [PubMed]
- Patnaik, A.; Appleman, L.J.; Tolcher, A.W.; Papadopoulos, K.P.; Beeram, M.; Rasco, D.W.; Weiss, G.J.; Sachdev, J.C.; Chadha, M.; Fulk, M.; et al. First-in-human phase I study of copanlisib (BAY 80-6946), an intravenous pan-class I phosphatidylinositol 3-kinase inhibitor, in patients with advanced solid tumors and non-Hodgkin’s lymphomas. Ann. Oncol. 2016, 27, 1928–1940. [Google Scholar] [CrossRef]
- Dreyling, M.; Morschhauser, F.; Bouabdallah, K.; Bron, D.; Cunningham, D.; Assouline, S.E.; Verhoef, G.; Linton, K.; Thieblemont, C.; Vitolo, U.; et al. Phase II study of copanlisib, a PI3K inhibitor, in relapsed or refractory, indolent or aggressive lymphoma. Ann. Oncol. 2017, 28, 2169–2178. [Google Scholar] [CrossRef]
- Gao, L.; Chuai, H.; Ma, M.; Zhang, S.Q.; Zhang, J.; Li, J.; Wang, Y.; Xin, M. Design, synthesis and bioactivity evaluation of selenium-containing PI3Kδ inhibitors. Bioorg. Chem. 2023, 140, 106815. [Google Scholar] [CrossRef]
- Xia, L.; Jiang, L.; Du, T.; Lin, S.; Xiong, T.; Peng, S.; Tian, H.; Zhang, K.; Wu, D.; Sheng, L.; et al. Design, synthesis, and biological evaluation of novel bivalent PI3K inhibitors for the potential treatment of cancer. Bioorg. Chem. 2023, 140, 106814. [Google Scholar] [CrossRef]
- Fischer, P.M.; Endicott, J.; Meijer, L. Cyclin-dependent kinase inhibitors. Prog. Cell Cycle Res. 2003, 5, 235–248. [Google Scholar]
- Dictor, M.; Ehinger, M.; Mertens, F.; Akervall, J.; Wennerberg, J. Abnormal cell cycle regulation in malignancy. Am. J. Clin. Pathol. 1999, 112, S40–S52. [Google Scholar] [PubMed]
- Wu, J.; Chen, Y.; Li, R.; Guan, Y.; Chen, M.; Yin, H.; Yang, X.; Jin, M.; Huang, B.; Ding, X.; et al. Synergistic anticancer effect by targeting CDK2 and EGFR–ERK signaling. J. Cell Biol. 2023, 223, e202203005. [Google Scholar] [CrossRef]
- Wu, T.; Yang, Z.; Chen, W.; Jiang, M.; Xiao, Z.; Su, X.; Jiao, Z.; Yu, Y.; Chen, S.; Song, M.; et al. miR-30e-5p-mediated FOXD1 promotes cell proliferation by blocking cellular senescence and apoptosis through p21/CDK2/Rb signaling in head and neck carcinoma. Cell Death Discov. 2023, 9, 295. [Google Scholar] [CrossRef]
- Wang, X.; Shi, W.; Wang, X.; Lu, J.-J.; He, P.; Zhang, H.; Chen, X. Nifuroxazide boosts the anticancer efficacy of palbociclib-induced senescence by dual inhibition of STAT3 and CDK2 in triple-negative breast cancer. Cell Death Discov. 2023, 9, 355. [Google Scholar] [CrossRef] [PubMed]
- Sultan, R.; Ahmed, A.; Wei, L.; Saeed, H.; Islam, M.; Ishaq, M. The anticancer potential of chemical constituents of Moringa oleifera targeting CDK-2 inhibition in estrogen receptor positive breast cancer using in-silico and in vitro approches. BMC Complement. Med. Ther. 2023, 23, 396. [Google Scholar] [CrossRef]
- Gomha, S.M.; Zaki, M.E.A.; Maliwal, D.; Pissurlenkar, R.R.S.; Ibrahim, M.S.; Fathalla, M.; Hussein, A.M. Synthesis, in-silico studies, and biological evaluation of some novel 3-thiazolyl-indoles as CDK2–inhibitors. Results Chem. 2023, 6, 101209. [Google Scholar] [CrossRef]
- Wang, L.; Shao, X.; Zhong, T.; Wu, Y.; Xu, A.; Sun, X.; Gao, H.; Liu, Y.; Lan, T.; Tong, Y.; et al. Discovery of a first-in-class CDK2 selective degrader for AML differentiation therapy. Nat. Chem. Biol. 2021, 17, 567–575. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, L.; Hei, R.; Li, X.; Cai, H.; Wu, X.; Zheng, Q.; Cai, C. CDK inhibitors in cancer therapy, an overview of recent development. Am. J. Cancer Res. 2021, 11, 1913–1935. [Google Scholar]
- Ma, T.; Van Tine, B.A.; Wei, Y.; Garrett, M.D.; Nelson, D.; Adams, P.D.; Wang, J.; Qin, J.; Chow, L.T.; Harper, J.W. Cell cycle-regulated phosphorylation of p220(NPAT) by cyclin E/Cdk2 in Cajal bodies promotes histone gene transcription. Genes Dev. 2000, 14, 2298–2313. [Google Scholar] [CrossRef] [PubMed]
- Flores, O.; Wang, Z.; Knudsen, K.E.; Burnstein, K.L. Nuclear targeting of cyclin-dependent kinase 2 reveals essential roles of cyclin-dependent kinase 2 localization and cyclin E in vitamin D-mediated growth inhibition. Endocrinology 2010, 151, 896–908. [Google Scholar] [CrossRef] [PubMed]
- De Boer, L.; Oakes, V.; Beamish, H.; Giles, N.; Stevens, F.; Somodevilla-Torres, M.; Desouza, C.; Gabrielli, B. Cyclin A/cdk2 coordinates centrosomal and nuclear mitotic events. Oncogene 2008, 27, 4261–4268. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.C.; Clurman, B.E. Cyclin E in normal and neoplastic cell cycles. Oncogene 2005, 24, 2776–2786. [Google Scholar] [CrossRef] [PubMed]
- Spruck, C.H.; Won, K.A.; Reed, S.I. Deregulated cyclin E induces chromosome instability. Nature 1999, 401, 297–300. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.K.; LaPlant, B.; Chng, W.J.; Zonder, J.; Callander, N.; Fonseca, R.; Fruth, B.; Roy, V.; Erlichman, C.; Stewart, A.K. Dinaciclib, a novel CDK inhibitor, demonstrates encouraging single-agent activity in patients with relapsed multiple myeloma. Blood 2015, 125, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.F.; Lin, J.D.; Hsueh, C.; Chou, T.C.; Wong, R.J. Potent effects of roniciclib alone and with sorafenib against well-differentiated thyroid cancer. Endocr. Relat. Cancer 2018, 25, 853–864. [Google Scholar] [CrossRef]
- Tadesse, S.; Caldon, E.C.; Tilley, W.; Wang, S. Cyclin-Dependent Kinase 2 Inhibitors in Cancer Therapy: An Update. J. Med. Chem. 2019, 62, 4233–4251. [Google Scholar] [CrossRef]
- Frame, S.; Saladino, C.; MacKay, C.; Atrash, B.; Sheldrake, P.; McDonald, E.; Clarke, P.A.; Workman, P.; Blake, D.; Zheleva, D. Fadraciclib (CYC065), a novel CDK inhibitor, targets key pro-survival and oncogenic pathways in cancer. PLoS ONE 2020, 15, e0234103. [Google Scholar] [CrossRef]
- Le Tourneau, C.; Faivre, S.; Laurence, V.; Delbaldo, C.; Vera, K.; Girre, V.; Chiao, J.; Armour, S.; Frame, S.; Green, S.R.; et al. Phase I evaluation of seliciclib (R-roscovitine), a novel oral cyclin-dependent kinase inhibitor, in patients with advanced malignancies. Eur. J. Cancer 2010, 46, 3243–3250. [Google Scholar] [CrossRef]
- Sielecki, T.M.; Johnson, T.L.; Liu, J.; Muckelbauer, J.K.; Grafstrom, R.H.; Cox, S.; Boylan, J.; Burton, C.R.; Chen, H.; Smallwood, A.; et al. Quinazolines as cyclin dependent kinase inhibitors. Bioorg. Med. Chem. Lett. 2001, 11, 1157–1160. [Google Scholar] [CrossRef] [PubMed]
- Shewchuk, L.; Hassell, A.; Wisely, B.; Rocque, W.; Holmes, W.; Veal, J.; Kuyper, L.F. Binding mode of the 4-anilinoquinazoline class of protein kinase inhibitor: X-ray crystallographic studies of 4-anilinoquinazolines bound to cyclin-dependent kinase 2 and p38 kinase. J. Med. Chem. 2000, 43, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, E.R.; Elmasry, G.F. Development of newly synthesised quinazolinone-based CDK2 inhibitors with potent efficacy against melanoma. J. Enzym. Inhib. Med. Chem. 2022, 37, 686–700. [Google Scholar] [CrossRef]
- Huang, J.; Wang, X.; Dong, R.; Liu, X.; Li, H.; Zhang, T.; Xu, J.; Liu, C.; Zhang, Y.; Hou, S.; et al. Discovery of N-(4-(3-isopropyl-2-methyl-2H-indazol-5-yl)pyrimidin-2-yl)-4-(4-methylpiperazin-1-yl)quinazolin-7-amine as a Novel, Potent, and Oral Cyclin-Dependent Kinase Inhibitor against Haematological Malignancies. J. Med. Chem. 2021, 64, 12548–12571. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).