
Cyclodextrin-Enabled Enantioselective Complexation Study of Cathinone Analogs



Abstract
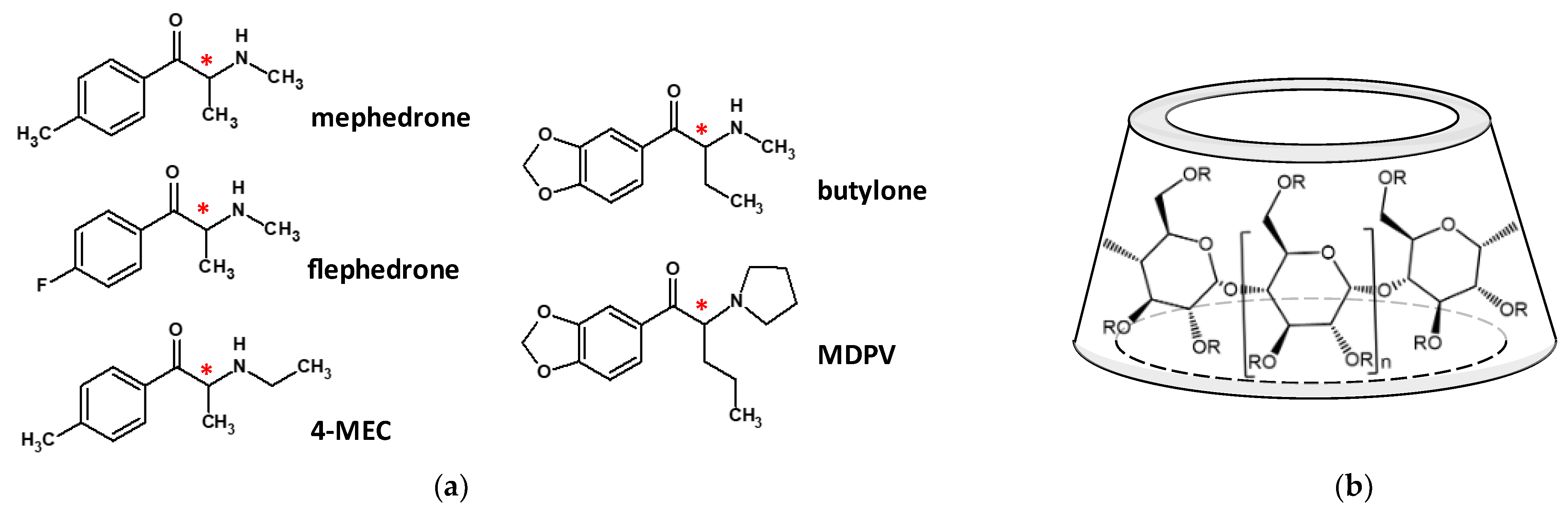
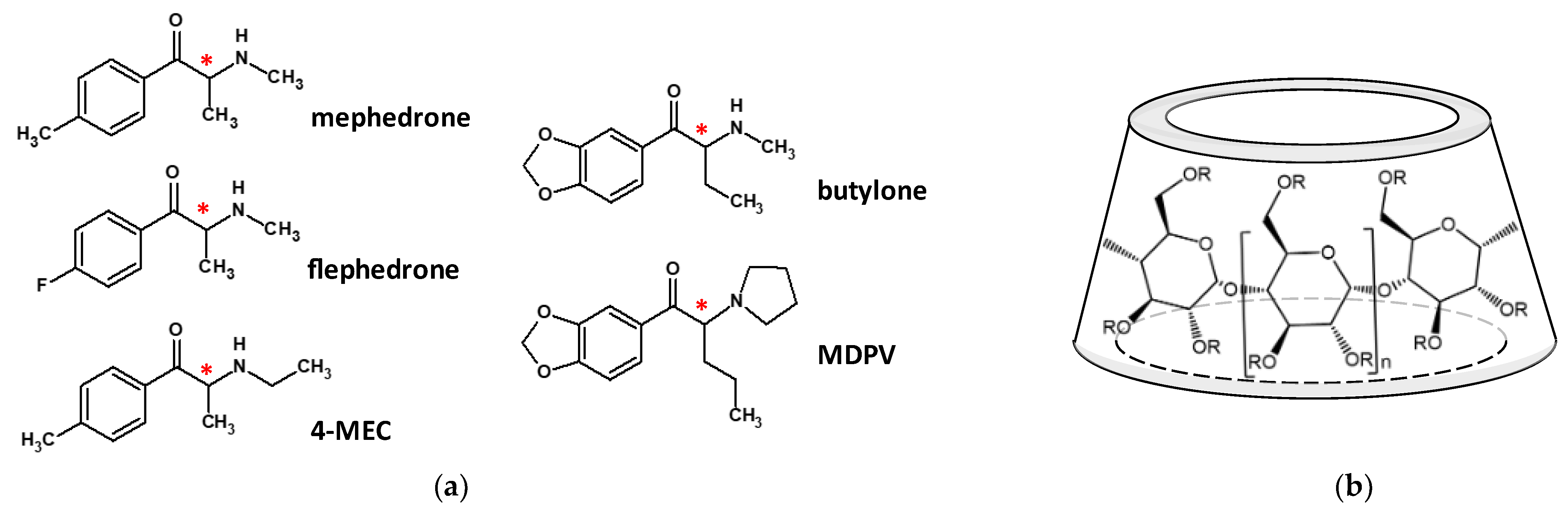
1. Introduction
2. Results and Discussion
2.1. Capillary Electrophoresis
2.1.1. pKa Determination of the Cathinone Derivatives by CE-pH Titration
2.1.2. Determination of the Cathinone–CD Complex Stabilities by ACE
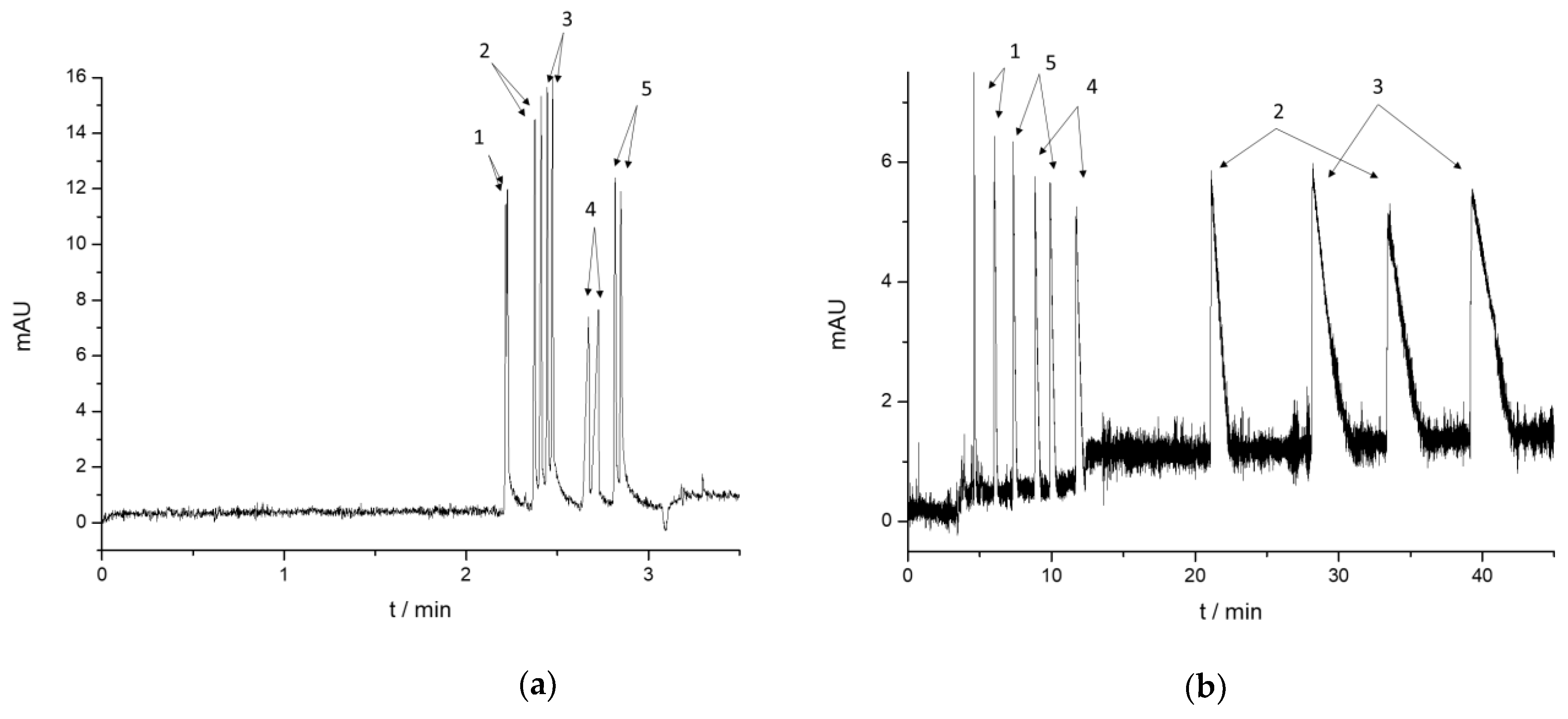
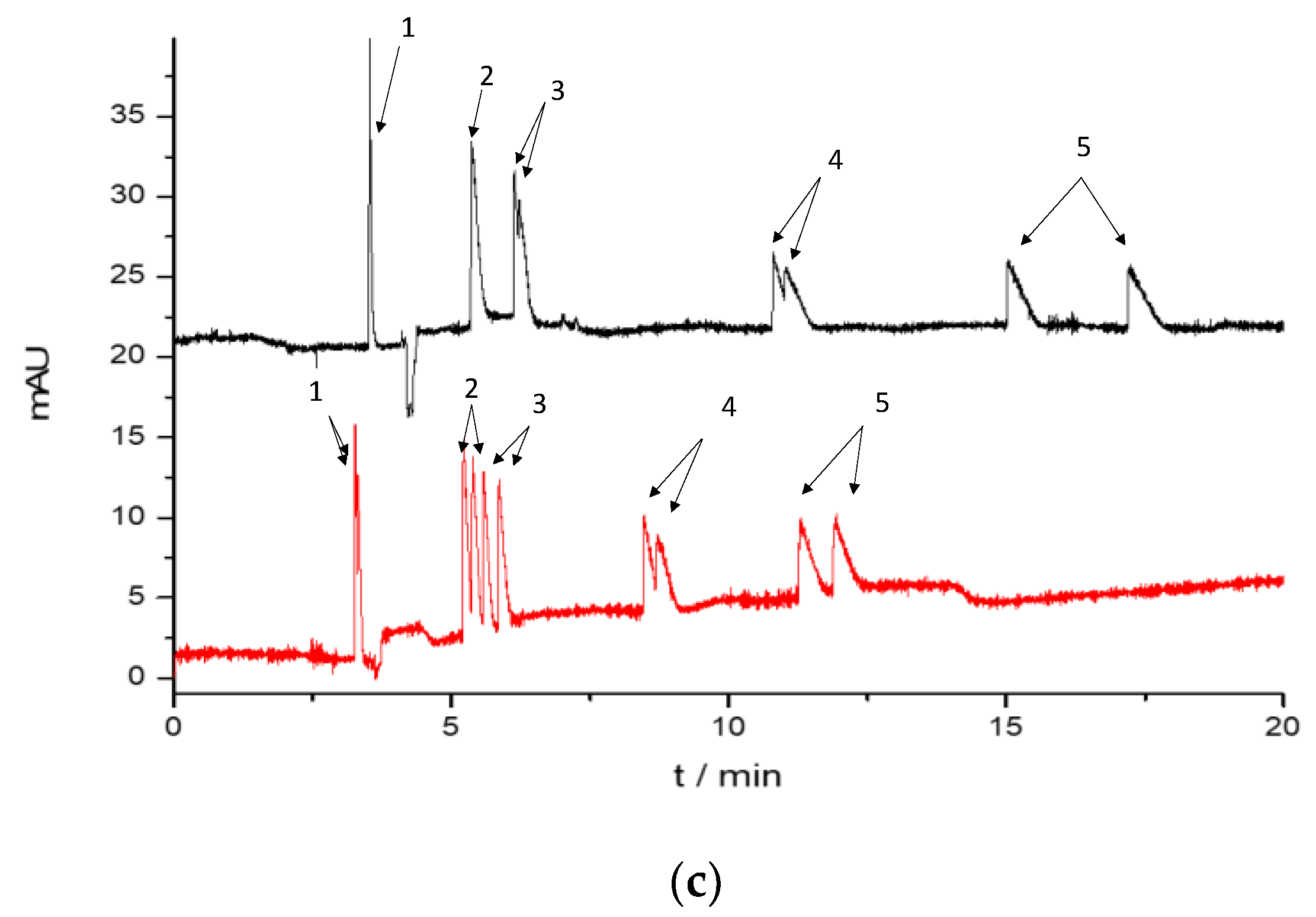
2.1.3. Enantioseparation of the Cathinone Derivatives
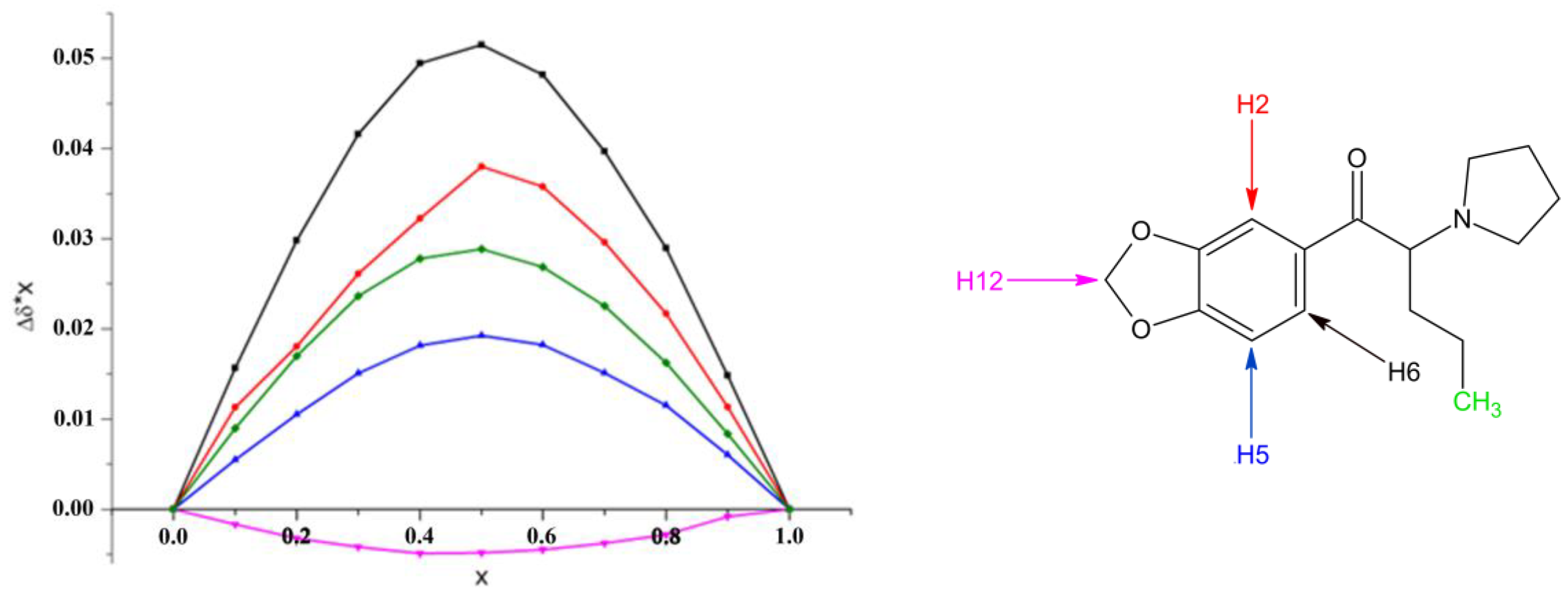
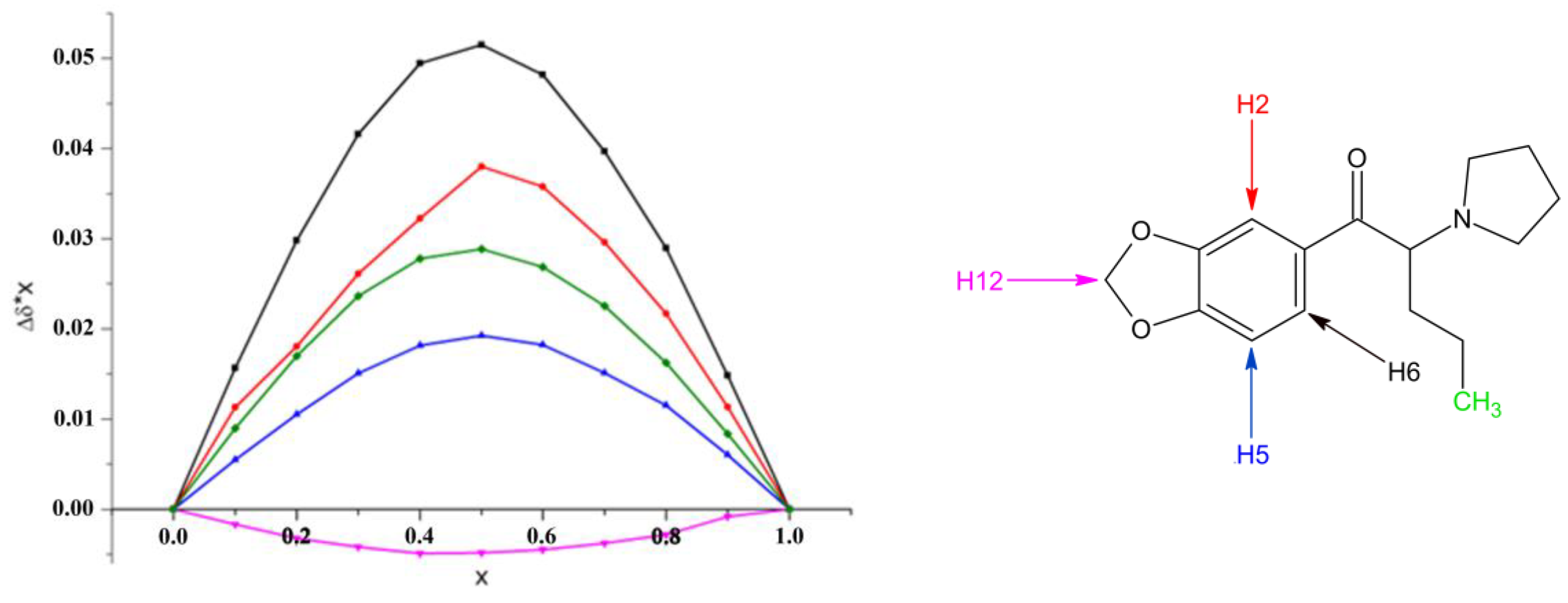
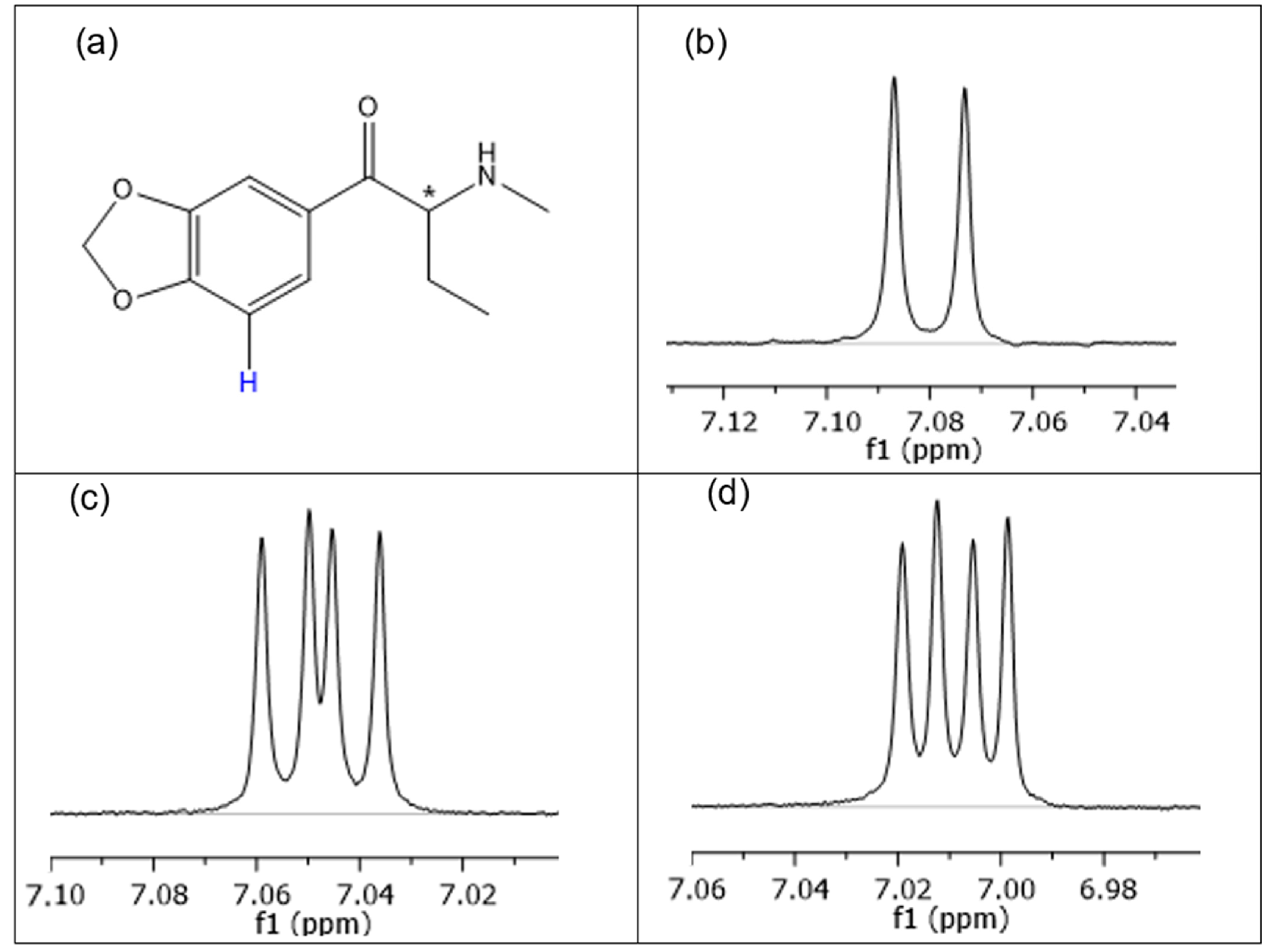
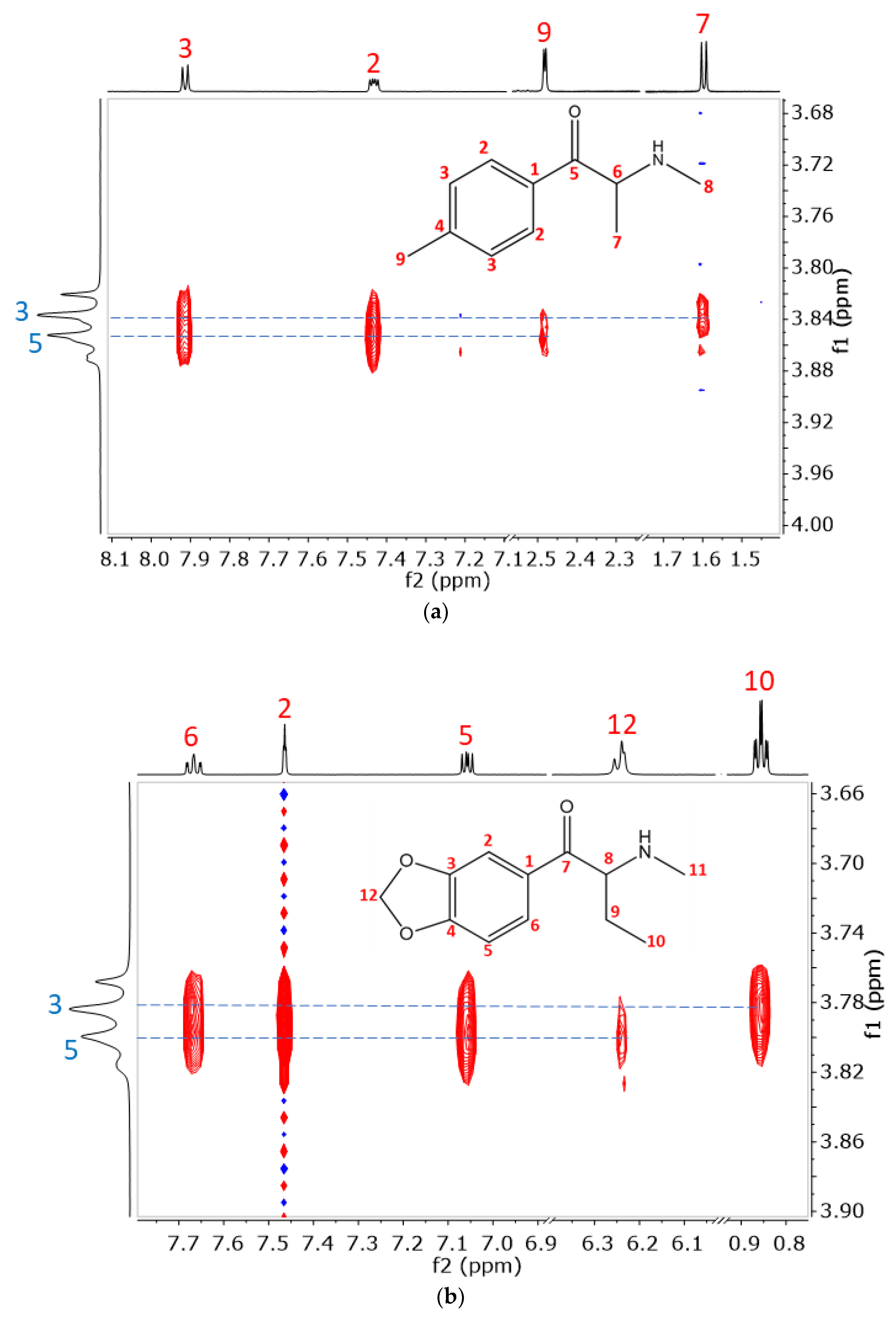
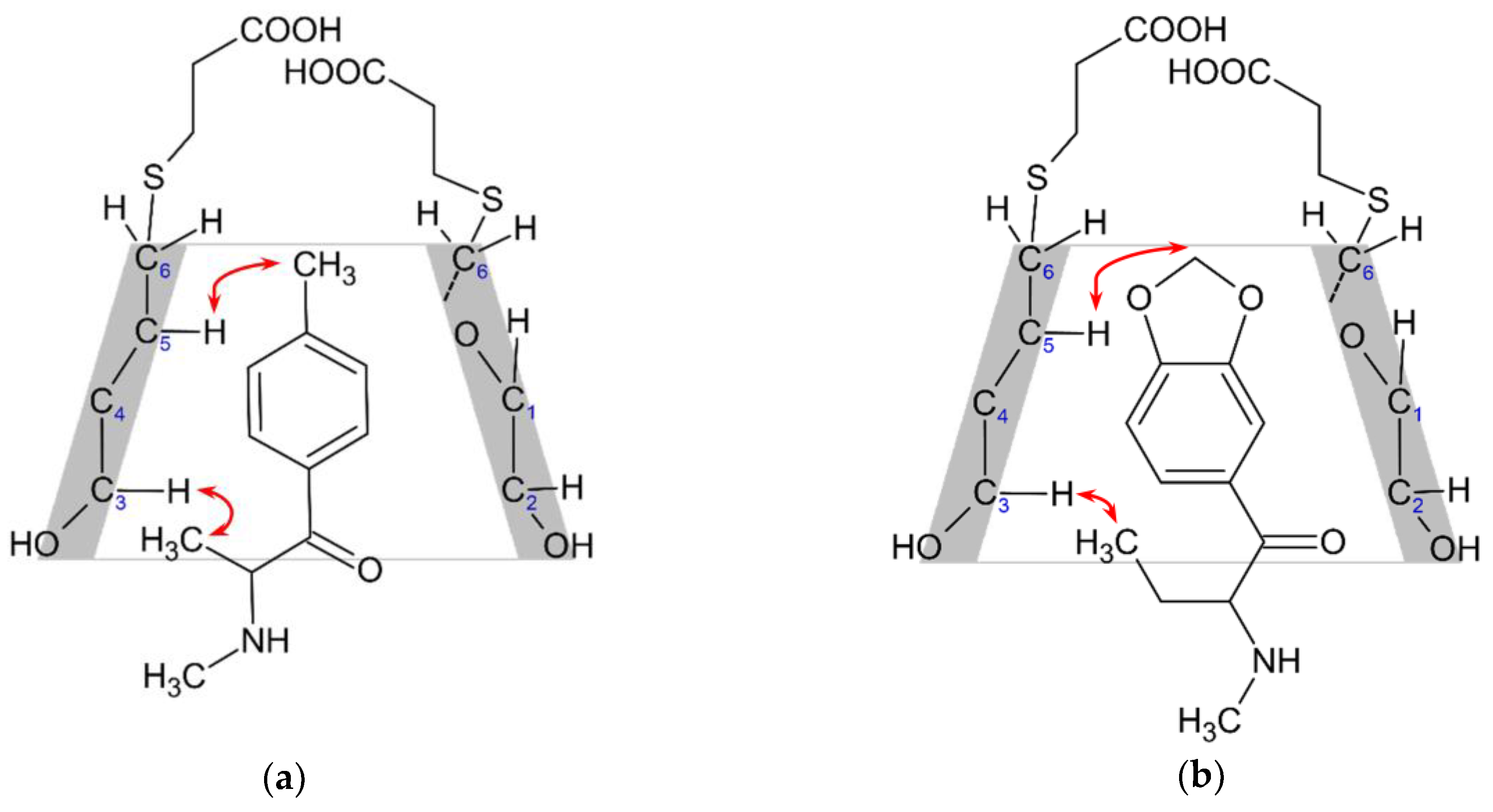
2.2. Structural Studies of the Complexes by NMR
3. Materials and Methods
3.1. Materials
3.2. Capillary Electrophoresis
3.2.1. pKa Determination of the Cathinone Derivatives
3.2.2. Determination of the Cathinone–CD Complex Stabilities by ACE
3.3. NMR Experiments
3.4. Potentiometry
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dal Cason, T.A.; Young, R.; Glennon, R.A. Cathinone: An Investigation of Several N-Alkyl and Methylenedioxy-Substituted Analogs. Pharmacol. Biochem. Behav. 1997, 58, 1109–1116. [Google Scholar] [CrossRef]
- Yin, H.; Zhang, X.; Wei, J.; Lu, S.; Bardelang, D.; Wang, R. Recent advances in supramolecular antidotes. Theranostics 2021, 11, 1513–1526. [Google Scholar] [CrossRef] [PubMed]
- Mayer, B.P.; Kennedy, D.J.; Lau, E.Y.; Valdez, C.A. Evaluation of polyanionic cyclodextrins as high affinity binding scaffolds for fentanyl. Sci. Rep. 2023, 13, 2680. [Google Scholar] [CrossRef] [PubMed]
- Lurie, I.S.; Klein, R.F.X.; Dal Cason, T.A.; LeBelle, M.J.; Brenneisen, R.; Weinberger, R.E. Chiral Resolution of Cationic Drugs of Forensic Interest by Capillary Electrophoresis with Mixtures of Neutral and Anionic Cyclodextrins. Anal. Chem. 1994, 66, 4019–4026. [Google Scholar] [CrossRef] [PubMed]
- Mohr, S.; Pilaj, S.; Schmid, M.G. Chiral separation of cathinone derivatives used as recreational drugs by cyclodextrin-modified capillary electrophoresis. Electrophoresis 2012, 33, 1624–1630. [Google Scholar] [CrossRef] [PubMed]
- Taschwer, M.; Weiß, J.A.; Kunert, O.; Schmid, M.G. Analysis and characterization of the novel psychoactive drug 4-chloromethcathinone (clephedrone). Forensic Sci. Int. 2014, 244, e56–e59. [Google Scholar] [CrossRef] [PubMed]
- Hubner, E.-M.; Steinkellner, P.; Schmid, M.G. Comparative studies on enantioseparation of New Psychoactive Substances using cyclodextrin-assisted capillary electrophoresis with UV detection. J. Pharm. Biopharm. Res. 2021, 3, 187–205. [Google Scholar] [CrossRef]
- Taschwer, M.; Hofer, M.G.; Schmid, M.G. Enantioseparation of benzofurys and other novel psychoactive compounds by CE and sulfobutylether β-cyclodextrin as chiral selector added to the BGE. Electrophoresis 2014, 35, 2793–2799. [Google Scholar] [CrossRef] [PubMed]
- Merola, G.; Fu, H.; Tagliaro, F.; Macchia, T.; Mccord, B.R. Chiral separation of 12 cathinone analogs by cyclodextrin-assisted capillary electrophoresis with UV and mass spectrometry detection. Electrophoresis 2014, 35, 3231–3241. [Google Scholar] [CrossRef]
- Fejős, I.; Varga, E.; Benkovics, G.; Malanga, M.; Sohajda, T.; Szemán, J.; Béni, S. Characterization of a single-isomer carboxymethyl-beta-cyclodextrin in chiral capillary electrophoresis. Electrophoresis 2017, 38, 1869–1877. [Google Scholar] [CrossRef]
- Nowak, P.M.; Olesek, K.; Woźniakiewicz, M.; Kościelniak, P. Simultaneous enantioseparation of methcathinone and two isomeric methylmethcathinones using capillary electrophoresis assisted by 2-hydroxyethyl-β-cyclodextrin. Electrophoresis 2018, 39, 2406–2409. [Google Scholar] [CrossRef]
- Hägele, J.S.; Hubner, E.M.; Schmid, M.G. Chiral separation of cathinone derivatives using β-cyclodextrin-assisted capillary electrophoresis–Comparison of four different β-cyclodextrin derivatives used as chiral selectors. Electrophoresis 2019, 40, 1787–1794. [Google Scholar] [CrossRef]
- Li, L.; Lurie, I.S. Regioisomeric and enantiomeric analyses of 24 designer cathinones and phenethylamines using ultra high performance liquid chromatography and capillary electrophoresis with added cyclodextrins. Forensic Sci. Int. 2015, 254, 148–157. [Google Scholar] [CrossRef]
- Ioannou, K.A.; Christou, A.; Stavrou, I.J.; Schmid, M.G.; Kapnissi-Christodoulou, C.P. Evaluation of cyclodextrin- and cyclofructan-based chiral selectors for the enantioseparation of psychoactive substances in capillary electrophoresis. Electrophoresis 2022, 43, 2392–2401. [Google Scholar] [CrossRef]
- Moini, M.; Rollman, C.M. Compatibility of highly sulfated cyclodextrin with electrospray ionization at low nanoliter/minute flow rates and its application to capillary electrophoresis/electrospray ionization mass spectrometric analysis of cathinone derivatives and their optical i. Rapid Commun. Mass Spectrom. 2015, 29, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Alcaraz, A.; Borrull, F.; Aguilar, C.; Calull, M. Enantioselective determination of cathinones in urine by high pressure in-line SPE–CE. Electrophoresis 2019, 40, 1762–1770. [Google Scholar] [CrossRef] [PubMed]
- Baciu, T.; Borrull, F.; Calull, M.; Aguilar, C. Enantioselective determination of cathinone derivatives in human hair by capillary electrophoresis combined in-line with solid-phase extraction. Electrophoresis 2016, 37, 2352–2362. [Google Scholar] [CrossRef] [PubMed]
- Taschwer, M.; Seidl, Y.; Mohr, S.; Schmid, M.G. Chiral separation of cathinone and amphetamine derivatives by HPLC/UV using sulfated ß-cyclodextrin as chiral mobile phase additive. Chirality 2014, 26, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Gregg, R.A.; Baumann, M.H.; Partilla, J.S.; Bonano, J.S.; Vouga, A.; Tallarida, C.S.; Velvadapu, V.; Smith, G.R.; Peet, M.M.; Reitz, A.B.; et al. Stereochemistry of mephedrone neuropharmacology: Enantiomer-specific behavioural and neurochemical effects in rats. Br. J. Pharmacol. 2015, 172, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Olabi, M.; Stein, M.; Wätzig, H. Affinity capillary electrophoresis for studying interactions in life sciences. Methods 2018, 146, 76–92. [Google Scholar] [CrossRef]
- Dubský, P.; Dvořák, M.; Ansorge, M. Affinity capillary electrophoresis: The theory of electromigration. Anal. Bioanal. Chem. 2016, 408, 8623–8641. [Google Scholar] [CrossRef] [PubMed]
- Busch, M.H.A.; Kraak, J.C.; Poppe, H. Principles and limitations of methods available for the determination of binding constants with affinity capillary electrophoresis. J. Chromatogr. A 1997, 777, 329–353. [Google Scholar] [CrossRef]
- Wang, Y.; Adeoye, D.I.; Ogunkunle, E.O.; Wei, I.A.; Filla, R.T.; Roper, M.G. Affinity Capillary Electrophoresis: A Critical Review of the Literature from 2018 to 2020. Anal. Chem. 2021, 93, 295–310. [Google Scholar] [CrossRef] [PubMed]
- Várnai, B.; Malanga, M.; Sohajda, T.; Béni, S. Molecular interactions in remdesivir-cyclodextrin systems. J. Pharm. Biomed. Anal. 2022, 209, 114482. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, S.K.; Kumar, G. NMR and molecular modelling studies on the interaction of fluconazole with β-cyclodextrin. Chem. Cent. J. 2009, 3, 9. [Google Scholar] [CrossRef]
- Nowak, P.M.; Woźniakiewicz, M.; Mitoraj, M.; Sagan, F.; Kościelniak, P. Thermodynamics of acid-base dissociation of several cathinones and 1-phenylethylamine, studied by an accurate capillary electrophoresis method free from the Joule heating impact. J. Chromatogr. A 2018, 1539, 78–86. [Google Scholar] [CrossRef]
- Dubský, P.; Ördögová, M.; Malý, M.; Riesová, M. CEval: All-in-one software for data processing and statistical evaluations in affinity capillary electrophoresis. J. Chromatogr. A 2016, 1445, 158–165. [Google Scholar] [CrossRef]
- Fejős, I.; Kalydi, E.; Kukk, E.L.; Seggio, M.; Malanga, M.; Béni, S. Single Isomer N-Heterocyclic Cyclodextrin Derivatives as Chiral Selectors in Capillary Electrophoresis. Molecules 2021, 26, 5271. [Google Scholar] [CrossRef]
- Ujj, D.; Kalydi, E.; Malanga, M.; Varga, E.; Sohajda, T.; Béni, S.; Benkovics, G. Sugammadex analogue cyclodextrins as chiral selectors for enantioseparation of cathinone derivatives by capillary electrophoresis. J. Chromatogr. A 2022, 1683, 463506. [Google Scholar] [CrossRef]
- Řezanka, P.; Macková, D.; Jurok, R.; Himl, M.; Kuchař, M. Enantioseparation and Determination of Mephedrone and Its Metabolites by Capillary Electrophoresis Using Cyclodextrins as Chiral Selectors. Molecules 2020, 25, 2879. [Google Scholar] [CrossRef] [PubMed]
- Job, P. Formation and stability of inorganic complexes in solution. Ann. Chim. 1928, 9, 113–203. [Google Scholar]
- Chankvetadze, B. Combined approach using capillary electrophoresis and NMR spectroscopy for an understanding of enantioselective recognition mechanisms by cyclodextrins. Chem. Soc. Rev. 2004, 33, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Benkovics, G.; Fejős, I.; Darcsi, A.; Varga, E.; Malanga, M.; Fenyvesi, É.; Sohajda, T.; Szente, L.; Béni, S.; Szemán, J. Single-isomer carboxymethyl-γ-cyclodextrin as chiral resolving agent for capillary electrophoresis. J. Chromatogr. A 2016, 1467, 445–453. [Google Scholar] [CrossRef]
- Bálint, M.; Darcsi, A.; Benkovics, G.; Varga, E.; Malanga, M.; Béni, S. Synthesis of the chiral selector heptakis(6-O-methyl)-β-cyclodextrin by phase-transfer catalysis and hydrazine-mediated transfer-hydrogenation. Electrophoresis 2019, 40, 1941–1950. [Google Scholar] [CrossRef] [PubMed]
- Wallingford, R.A.; Ewing, A. Capillary electrophoresis. Adv. Chromatogr. 1989, 29, 1–76. [Google Scholar] [CrossRef] [PubMed]
- Le Saux, T.; Varenne, A.; Gareil, P. Peak shape modeling by Haarhoff-Van der Linde function for the determination of correct migration times: A new insight into affinity capillary electrophoresis. Electrophoresis 2005, 26, 3094–3104. [Google Scholar] [CrossRef]
- Rundlett, K.L.; Armstrong, D.W. Examination of the origin, variation, and proper use of expressions for the estimation of association constants by capillary electrophoresis. J. Chromatogr. A 1996, 721, 173–186. [Google Scholar] [CrossRef]
- Bowser, M.T.; Chen, D.D.Y. Monte Carlo Simulation of Error Propagation in the Determination of Binding Constants from Rectangular Hyperbolae. 2. Effect of the Maximum-Response Range. J. Phys. Chem. A 1998, 103, 197–202. [Google Scholar] [CrossRef]
- Bowser, M.T.; Chen, D.D.Y. Monte Carlo Simulation of Error Propagation in the Determination of Binding Constants from Rectangular Hyperbolae. 1. Ligand Concentration Range and Binding Constant. J. Phys. Chem. A 1998, 102, 8063–8071. [Google Scholar] [CrossRef]
- Peng, X.; Bowser, M.T.; Britz-McKibbin, P.; Bebault, G.M.; Morris, J.R.; Chen, D.D.Y. Quantitative description of analyte migration behavior based on dynamic complexation in capillary electrophoresis with one or more additives. Electrophoresis 1997, 18, 706–716. [Google Scholar] [CrossRef]
- Østergaard, J.; Jensen, H.; Holm, R. Affinity capillary electrophoresis method for investigation of bile salts complexation with sulfobutyl ether-β-cyclodextrin. J. Sep. Sci. 2012, 35, 2764–2772. [Google Scholar] [CrossRef] [PubMed]
- Beneš, M.; Zusková, I.; Svobodová, J.; Gaš, B. Determination of stability constants of complexes of neutral analytes with charged cyclodextrins by affinity capillary electrophoresis. Electrophoresis 2012, 33, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Avdeef, A.; Bucher, J.J. Accurate Measurements of the Concentration of Hydrogen Ions with a Glass Electrode: Calibrations Using the Prideaux and Other Universal Buffer Solutions and a Computer-Controlled Automatic Titrator. Anal. Chem. 1978, 50, 2137–2142. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mephedrone | Flephedrone | 4-MEC | Butylone | MDPV | |
|---|---|---|---|---|---|
| CE-pH Titration | 8.61 ± 0.01 | 8.56 ± 0.01 | 8.81 ± 0.02 | 8.64 ± 0.01 | 9.00 ± 0.02 |
| Potentiometry | - | - | 8.85 ± 0.04 | - | 9.05 ± 0.04 |
| Cyclodextrin | Flephedrone | Mephedrone | 4-MEC | Butylone | MDPV | |
|---|---|---|---|---|---|---|
| Native CDs | β-CD | 350 ± 60 | 560 ± 50 | 390 ± 45 | 500 ± 50 | 1400 ± 135 |
| 430 ± 55 Rs 0.7 (7 mM) | 810 ± 155 Rs 0.8 (8 mM) | |||||
| Negative CDs | CM-β-CD | 190 ± 10 | 610 ± 30 | 620 ± 40 | 1500 ± 50 | 1900 ± 90 |
| 225 ± 10 Rs 1.3 (10 mM) | 615 ± 25 Rs 0.7 (3 mM) | 725 ± 45 Rs 0.6 (10 mM) | 1500 ± 60 Rs 0.4 (2 mM) | 2700 ± 150 Rs 3.3 (10 mM) | ||
| CE-β-CD | 150 ± 10 | 400 ± 30 | 590 ± 40 | 975 ± 95 | 1560 ± 120 | |
| 430 ± 25 Rs 0.8 (10 mM) | 2000 ± 155 Rs 1.6 (10 mM) | |||||
| SAX | 2000 ± 35 | 530 ± 45 | 5550 ± 650 | 2900 ± 200 | 2550 ± 145 2800 ± 75 Rs 1.3 (5 mM) | |
| SBX | 575 ± 35 610 ± 45 Rs 1.0 (3 mM) | 5000 ± 500 | 8000 ± 340 | 8250 ± 400 8750 ± 820 Rs 0.9 (3 mM) | 9650 ± 900 12,830 ± 900 Rs 1.6 (3 mM) | |
| SGX | 315 ± 20 | 825 ± 65 1000 ± 100 Rs 2.5 (4 mM) | 975 ± 90 960 ± 95 Rs 1.3 (4 mM) | 1700 ± 110 | 1500 ± 145 2000 ± 185 Rs 2.2 (4 mM) | |
| Succ-β-CD | 1600 ± 230 | 1900 ± 200 | 4500 ± 680 | 5200 ± 645 | 5600 ± 175 | |
| (DS~6) | 6200 ± 870 Rs 1.8 (4 mM) | |||||
| Succ-β-CD | 7200 ± 1000 | 2750 ± 300 | 8900 ± 920 | 13,500 ± 1350 | 12,100 ± 2150 | |
| (DS~4) | 8200 ± 880 Rs 1.1 (2 mM) | 30,500 ± 5300 Rs 1.3 (1.5 mM) | 10,600 ± 35 Rs 2.5 (2 mM) | |||
| Phos-β-CD | 330 ± 20 | 1900 ± 200 | 1200 ± 85 | 1400 ± 130 | 870 ± 60 | |
| 440 ± 35 Rs 4.0 (10 mM) | 2100 ± 230 Rs 1.8 (10 mM) | 1400 ± 120 Rs 1.4 (8 mM) | 1600 ± 140 Rs 1.7 (10 mM) | 1200 ± 85 Rs 2.6 (3 mM) | ||
| SBE-β-CD (DS~4) | 175 ± 25 | 300 ± 60 | 560 ± 60 | 1600 ± 280 | 3400 ± 45 | |
| 325 ± 60 Rs 0.7 (5 mM) | 550 ± 40 Rs 0.8 (5 mM) | |||||
| SBE-β-CD (DS~6.5) | 200 ± 20 | 500 ± 20 | 560 ± 35 | 1200 ± 60 | 2300 ± 140 | |
| 200 ± 12 Rs 1.0 (8 mM) | 500 ± 40 Rs 0.6 (8 mM) | 660 ± 35 Rs 1.4 (8 mM) | 1300 ± 60 Rs 0.7 (8 mM) | 2550 ± 200 Rs 1.1 (8 mM) | ||
| SP-β-CD (DS~2) | 140 ± 12 | 390 ± 20 | 440 ± 25 | 710 ± 45 | 1100 ± 60 | |
| 1220 ± 50 Rs 0.6 (0.8 mM) | ||||||
| SP-β-CD (DS~4) | 120 ± 12 | 500 ± 65 | 620 ± 65 | 1350 ± 130 | 3400 ± 60 | |
| 450 ± 65 Rs 0.5 (7 mM) | ||||||
| S-β-CD | 860 ± 100 | 2000 ± 110 | 1450 ± 190 | 2160 ± 260 | 2700 ± 250 | |
| 2300 ± 85 Rs 4.8 (2 mM) | 1350 ± 75 Rs 5.0 (3 mM) | |||||
| HS-β-CD | 580 ± 14 | 1400 ± 175 | n.d. | n.d. | n.d. | |
| 470 ± 65 Rs 0.7 (3 mM) | 1800 ± 25 Rs 6.2 (4 mM) | |||||
| HDAS-β-CD | 110 ± 37 | 450 ± 60 | 560 ± 40 | 725 ± 20 | 360 ± 55 | |
| 360 ± 75 Rs 2.8 (5 mM) | 730 ± 40 Rs 8.6 (5 mM) | 830 ± 45 Rs 7.9 (5 mM) | 340 ± 30 Rs 2.3 (5 mM) | |||
| HDMS-β-CD | n.d. | n.d. | 1670 ± 400 | 1750 ± 800 | 915 ± 265 |
| Cyclodextrin | Concentration (mM) | Flephedrone | Mephedrone | 4-MEC | Butylone | MDPV | |
|---|---|---|---|---|---|---|---|
| Negative CDs | CM-α-CD | 1 | 0.5 | 0.6 | 0.4 | 0.3 | 1.7 |
| 5 | 0.9 | 1.7 | 1.5 | 0.5 | 2.9 | ||
| 10 | 1.2 | 0.8 | 0.6 | 0.8 | 5.5 | ||
| CM-β-CD | 1 | 0.4 | 0 | 0.3 | 0.6 | 1.6 | |
| 5 | 0.8 | 0.1 | 0.4 | 0.3 | 2.1 | ||
| 10 | 0.8 | 0.4 | 0.5 | 0.4 | 3.5 | ||
| CM-γ-CD | 1 | 0 | 1.1 | 0.7 | 1.2 | 0.9 | |
| 5 | 0.9 | 2.6 | 2.1 | 1.6 | 1.9 | ||
| 10 | 0.9 | 2.2 | 1.7 | 1.9 | 0.5 | ||
| CE-β-CD | 1 | 0 | 0.1 | 0.2 | 0 | 0 | |
| 5 | 0 | 0 | 0 | 0 | 1.5 | ||
| 10 | 0 | 0.2 | 0 | 0 | 1.1 | ||
| SAX | 1 | 0 | 0 | 0 | 0 | 0.9 | |
| 5 | 0 | n.d. | n.d. | 0 | 1.1 | ||
| 10 | 0 | n.d. | n.d. | 0 | 1.8 | ||
| SBX | 1 | 0 | 0.4 | 0.5 | 0 | 0 | |
| 5 | 0.5 | 0 | n.d. | 0.7 | 2.7 | ||
| 10 | 0.6 | 0 | 0.8 | 0.9 | 1.9 | ||
| Succ-β-CD (DS~4) | 1 5 10 | 0 0.9 1.2 | 0 0 0 | 0.8 n.d. n.d. | 2.4 2.5 2.6 | n.d. 0.7 0.7 | |
| SBE-α-CD | 1 | 0.3 | 1.5 | 0.9 | 0.4 | n.d | |
| 5 | 0.4 | 1.8 | 1.6 | 0.9 | 0.5 | ||
| 10 | 0.5 | 2.3 | 2.2 | 1.5 | 0.8 | ||
| SBE-β-CD | 1 | 0.3 | 0.4 | 0.5 | 0.5 | 0.7 | |
| (DS~6.5) | 5 | 0.5 | 0.7 | 1.2 | 0.4 | 1.0 | |
| 10 | 0.4 | 0.8 | 1.5 | 0.6 | 1.4 | ||
| 6-(SB)7-β-CD | 1 5 10 | 0 0 0 | 0 0.2 0.2 | 0 0.7 0.6 | n.d. 0.4 0.6 | 1.8 2.6 n.d. | |
| S-β-CD | 1 | 0.1 | 1.6 | 1.9 | 0.5 | 0.6 | |
| 5 | 0.6 | 3.1 | 4.2 | n.d. | n.d. | ||
| 10 | 0.9 | n.d. | n.d. | n.d. | n.d. | ||
| S-γ-CD | 1 | 0 | 0.7 | 0.5 | 0.6 | 0 | |
| 5 | 0 | 1.1 | 0.6 | 0.7 | 1.1 | ||
| 10 | 0 | 1.0 | 0 | 1.4 | 1.5 | ||
| HS-β-CD | 1 | 3.1 | 2.9 | 2.4 | 3.4 | 3.4 | |
| 5 | 8.1 | 9.2 | 8.7 | 9.2 | 11.7 | ||
| 10 | n.d. | n.d. | n.d. | n.d. | n.d. | ||
| HDAS-β-CD | 1 | 5.1 | 2.1 | 2.7 | 3.7 | 1.4 | |
| 5 | 13.1 | 6.2 | 7.3 | 9.5 | 6.1 | ||
| 10 | 8.0 | 8.4 | 7.7 | 11.4 | 8.5 | ||
| HDMS-β-CD | 1 | 0.8 | 0.2 | 0.3 | 0.8 | 0.5 | |
| 5 | 1.7 | 1.6 | 1.5 | 2.7 | 1.9 | ||
| 10 | 2.6 | 2.3 | 2.1 | 4.0 | 2.9 | ||
| ODMS-γ-CD | 1 | 0.8 | 2.2 | 1.5 | 1.1 | 0 | |
| 5 | 2.2 | 5.4 | 4.0 | 2.9 | 1.5 | ||
| 10 | 3.3 | 7.5 | 5.8 | 4.4 | 2.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dohárszky, A.; Kalydi, E.; Völgyi, G.; Béni, S.; Fejős, I. Cyclodextrin-Enabled Enantioselective Complexation Study of Cathinone Analogs. Molecules 2024, 29, 876. https://doi.org/10.3390/molecules29040876
Dohárszky A, Kalydi E, Völgyi G, Béni S, Fejős I. Cyclodextrin-Enabled Enantioselective Complexation Study of Cathinone Analogs. Molecules. 2024; 29(4):876. https://doi.org/10.3390/molecules29040876
Chicago/Turabian StyleDohárszky, András, Eszter Kalydi, Gergely Völgyi, Szabolcs Béni, and Ida Fejős. 2024. "Cyclodextrin-Enabled Enantioselective Complexation Study of Cathinone Analogs" Molecules 29, no. 4: 876. https://doi.org/10.3390/molecules29040876
APA StyleDohárszky, A., Kalydi, E., Völgyi, G., Béni, S., & Fejős, I. (2024). Cyclodextrin-Enabled Enantioselective Complexation Study of Cathinone Analogs. Molecules, 29(4), 876. https://doi.org/10.3390/molecules29040876