

Recent Progress in the Synthesis of Benzoxazin-4-Ones, Applications in N-Directed Ortho-Functionalizations, and Biological Significance


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
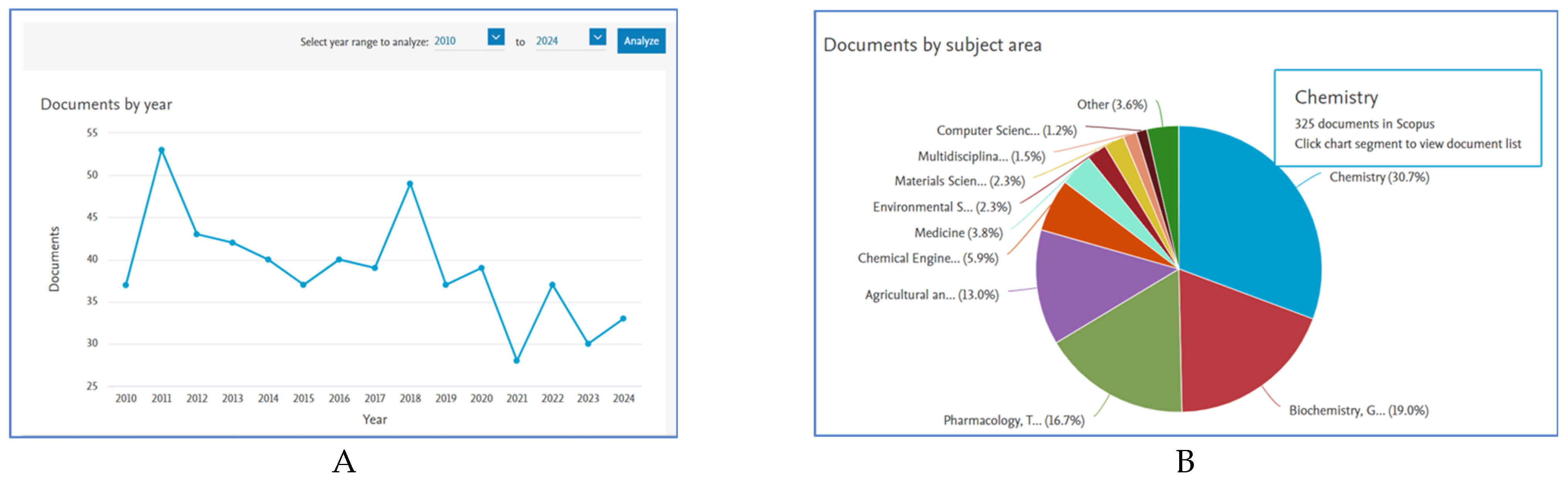
1. Introduction
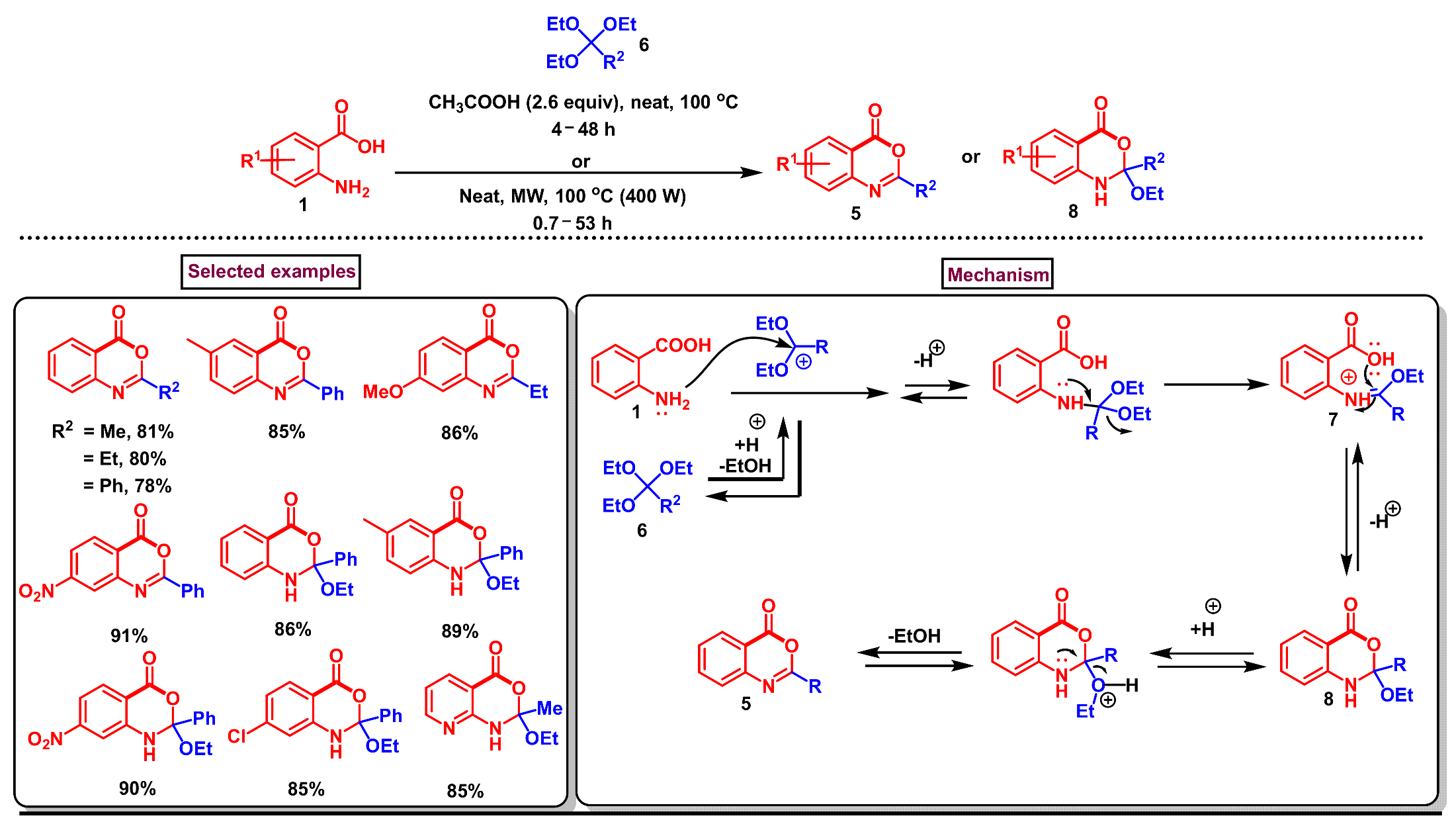
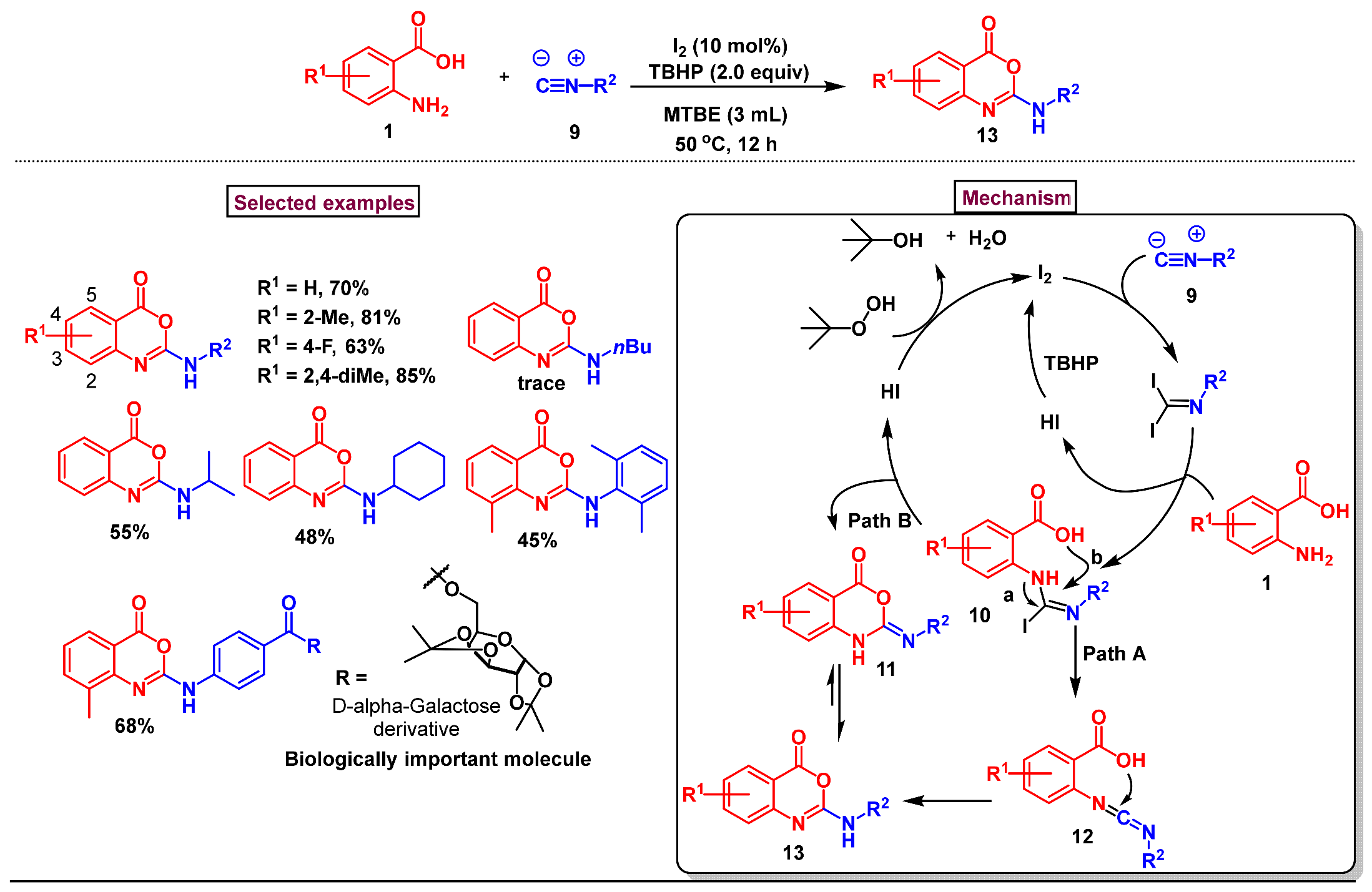
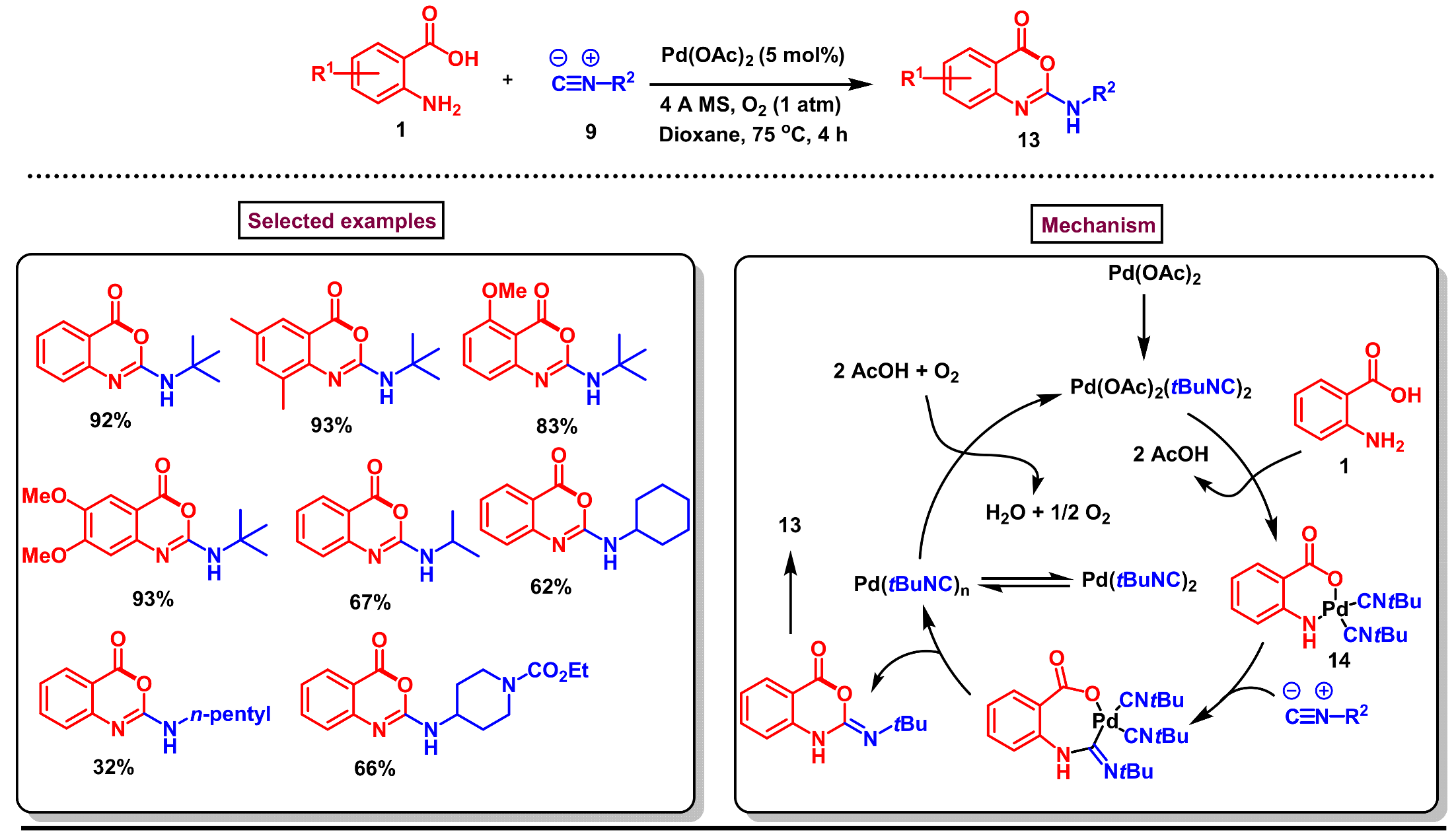
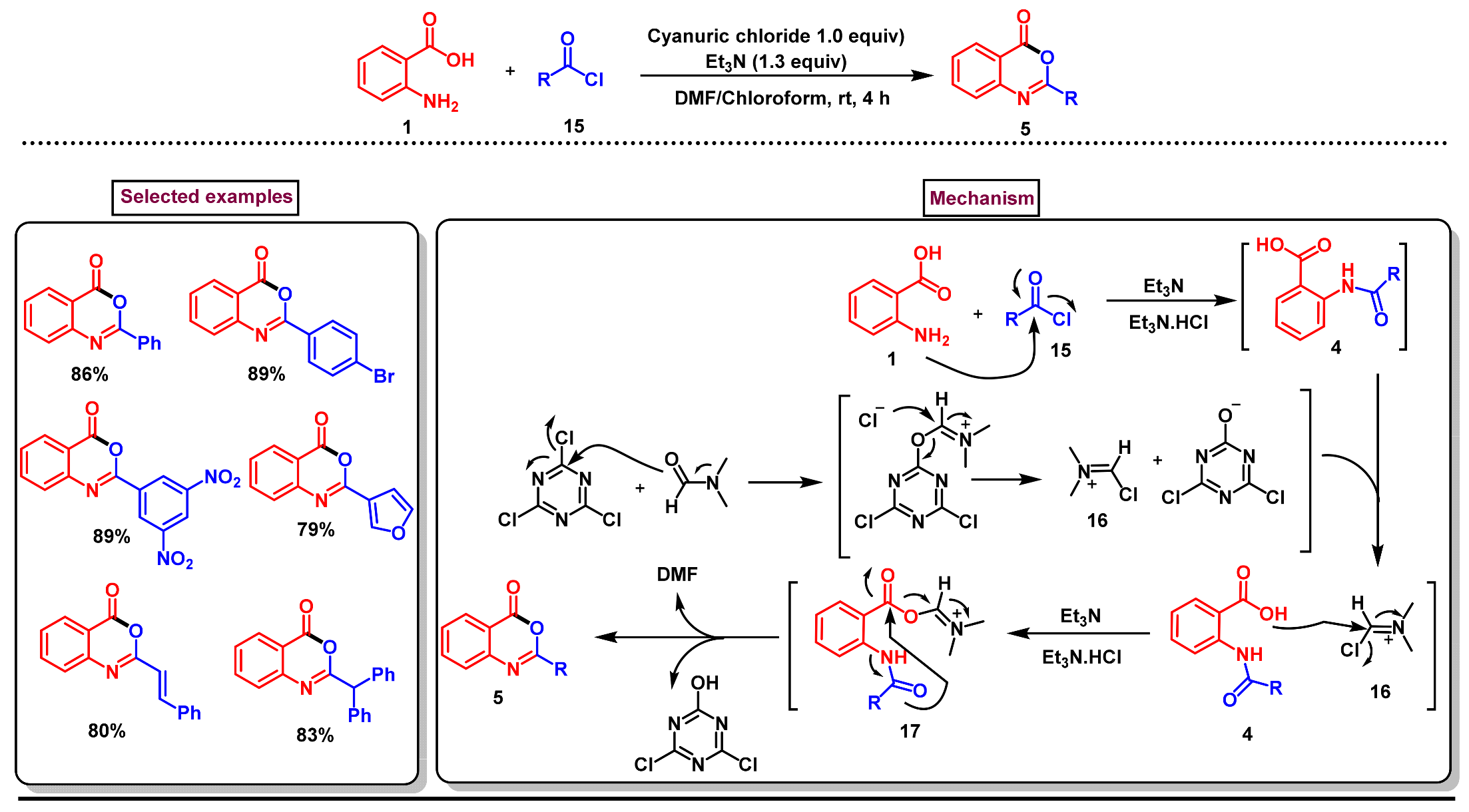
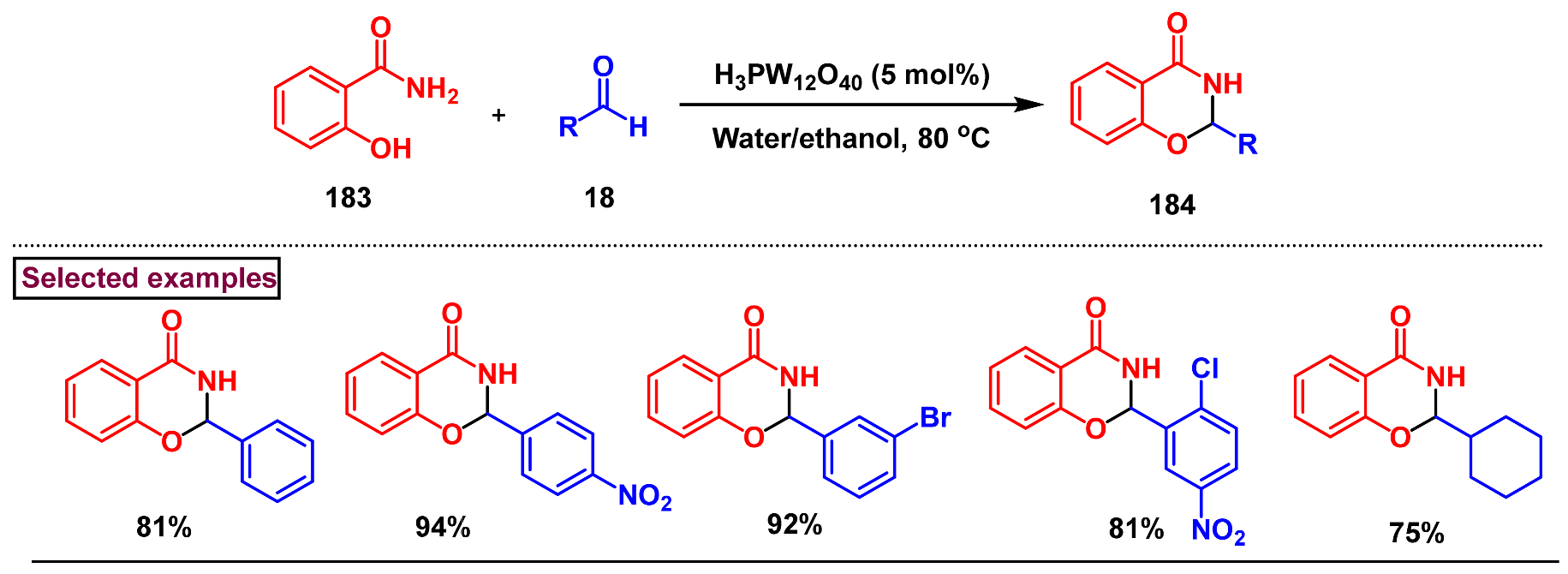
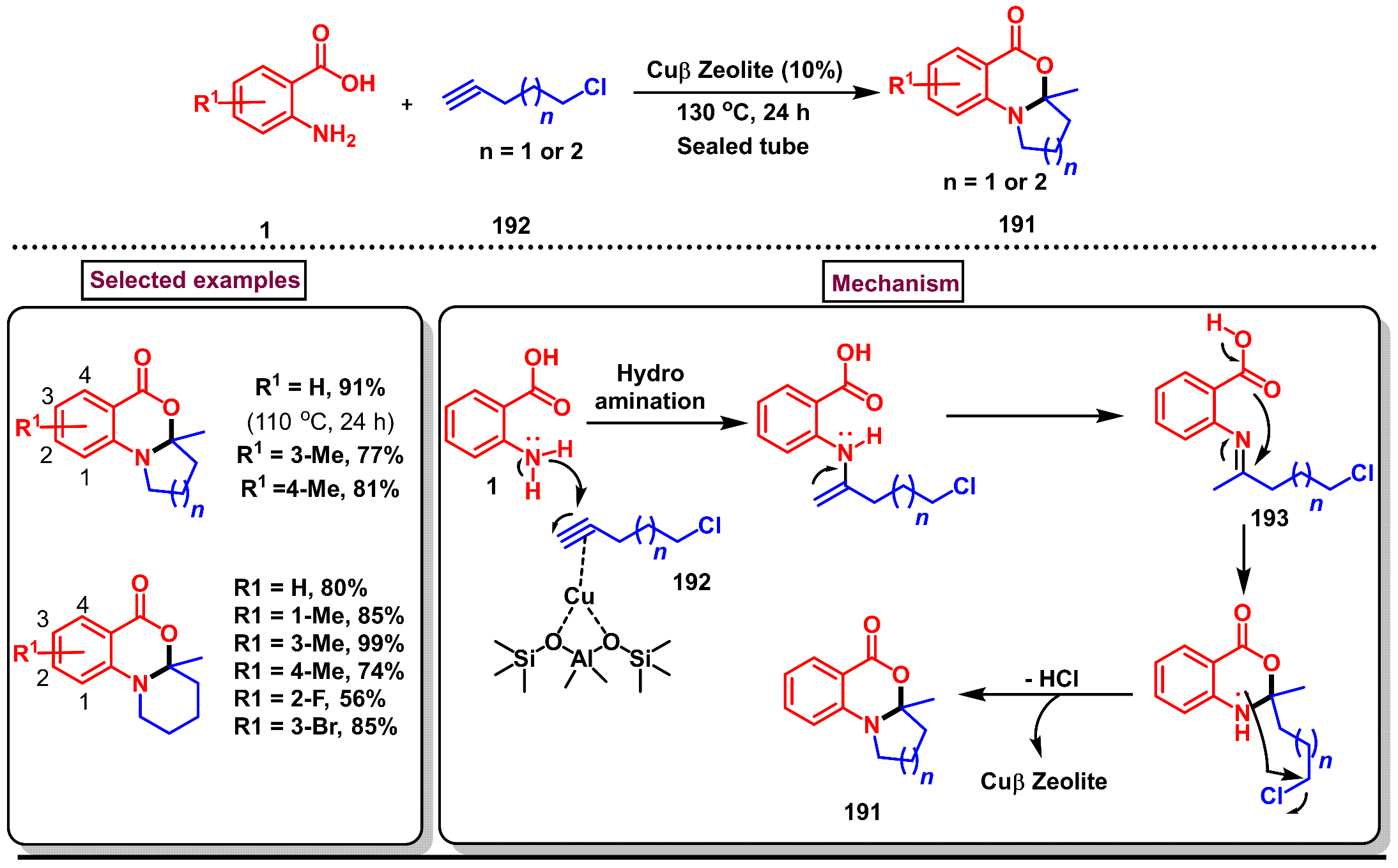
2. Preparation of Benzo [1,3]-Oxazin-4-One Derivatives from Anthranilic Acids
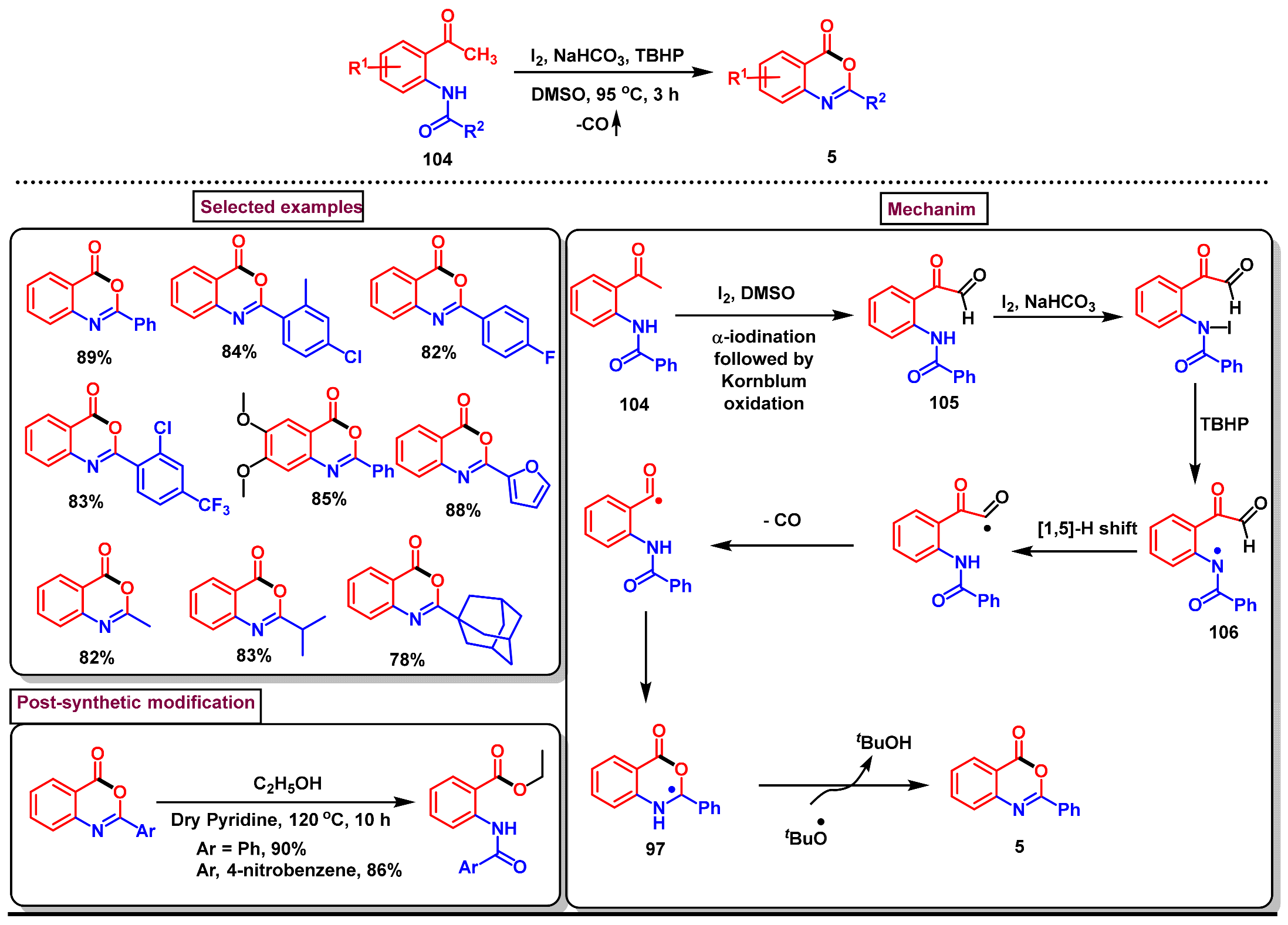
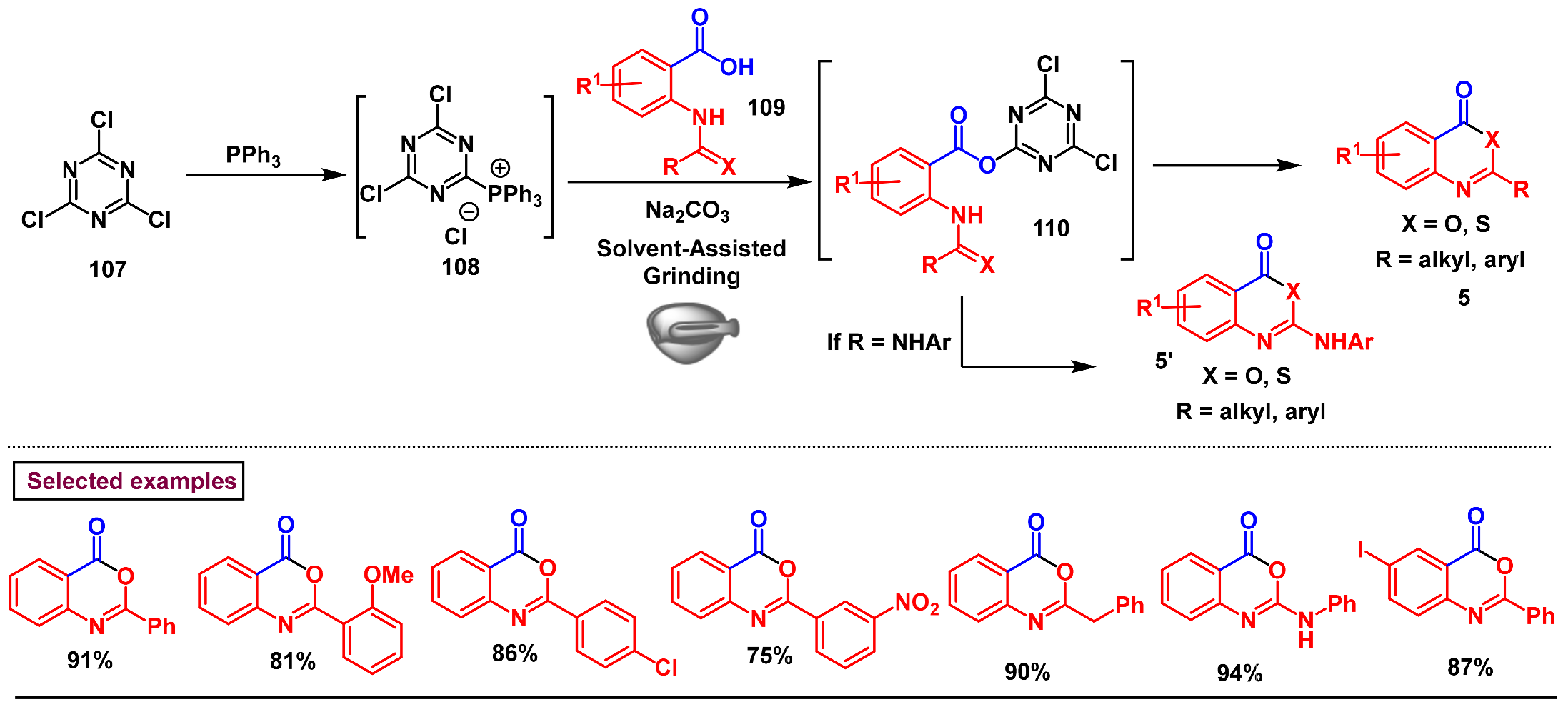
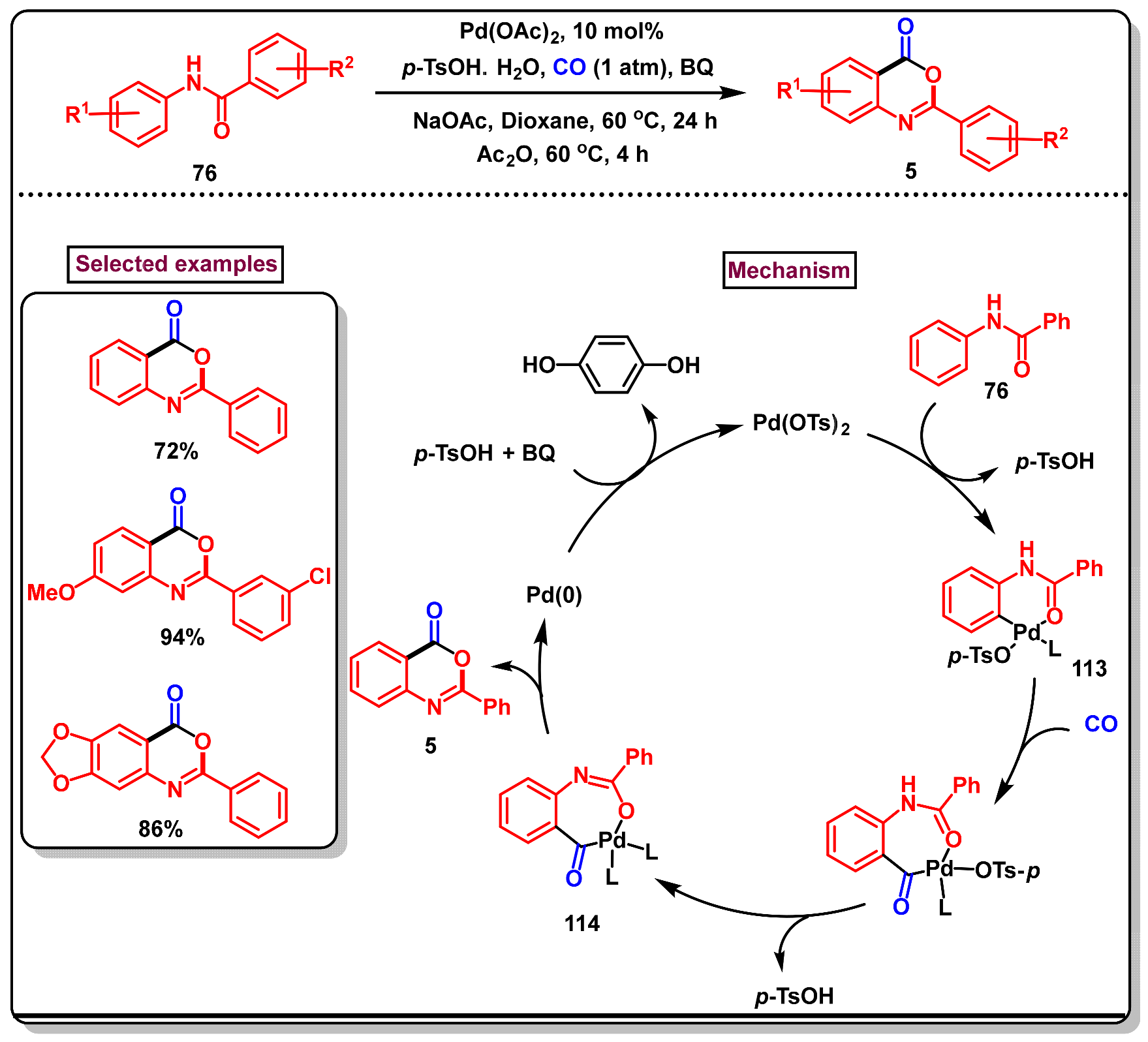
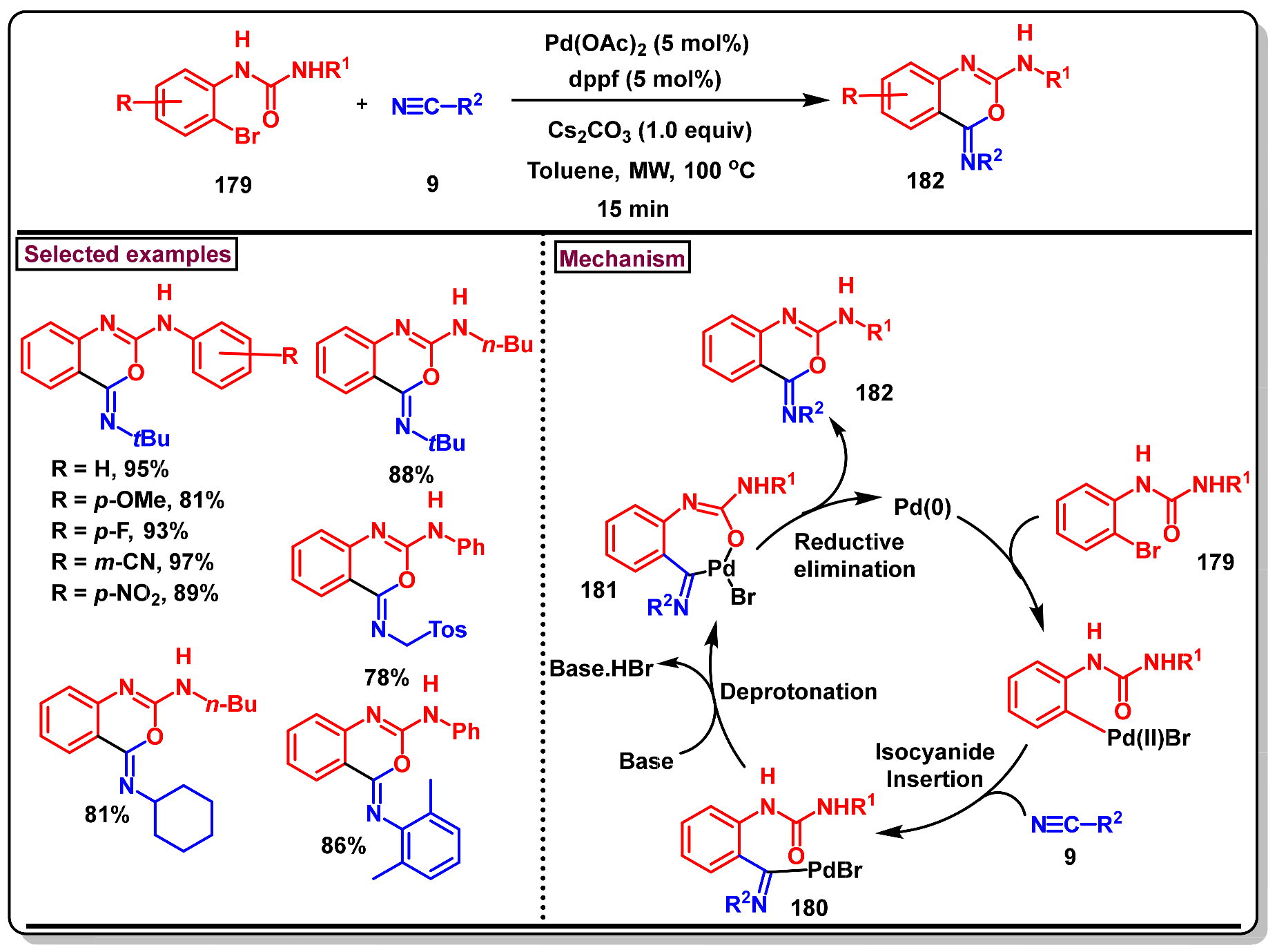
3. Preparation of Benzo[d][1,3]-Oxazin-4-Ones from Functionalized Amides
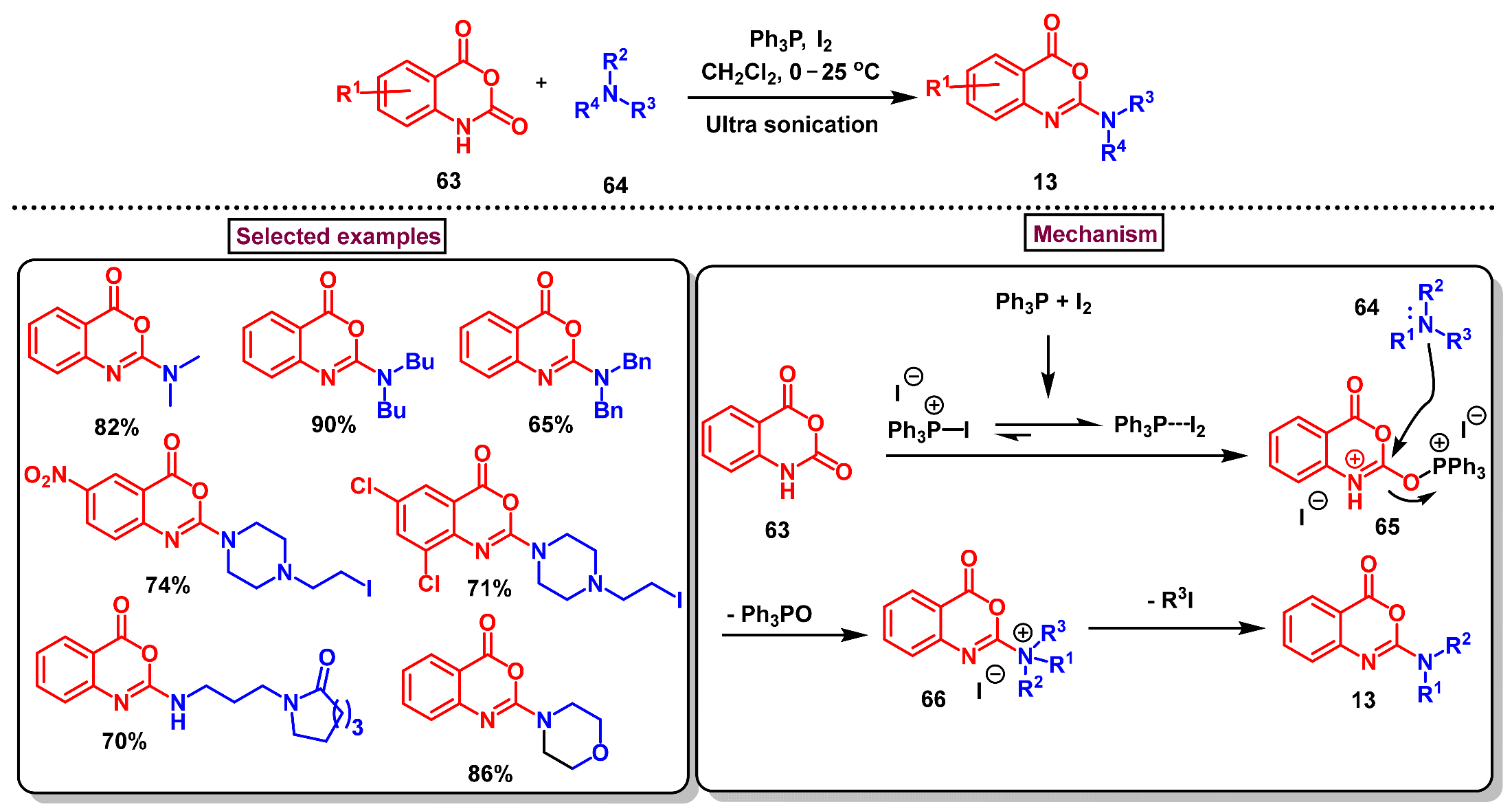
4. Preparation of Benzo[d][1,3]-Oxazin-4-Ones from Isatoic
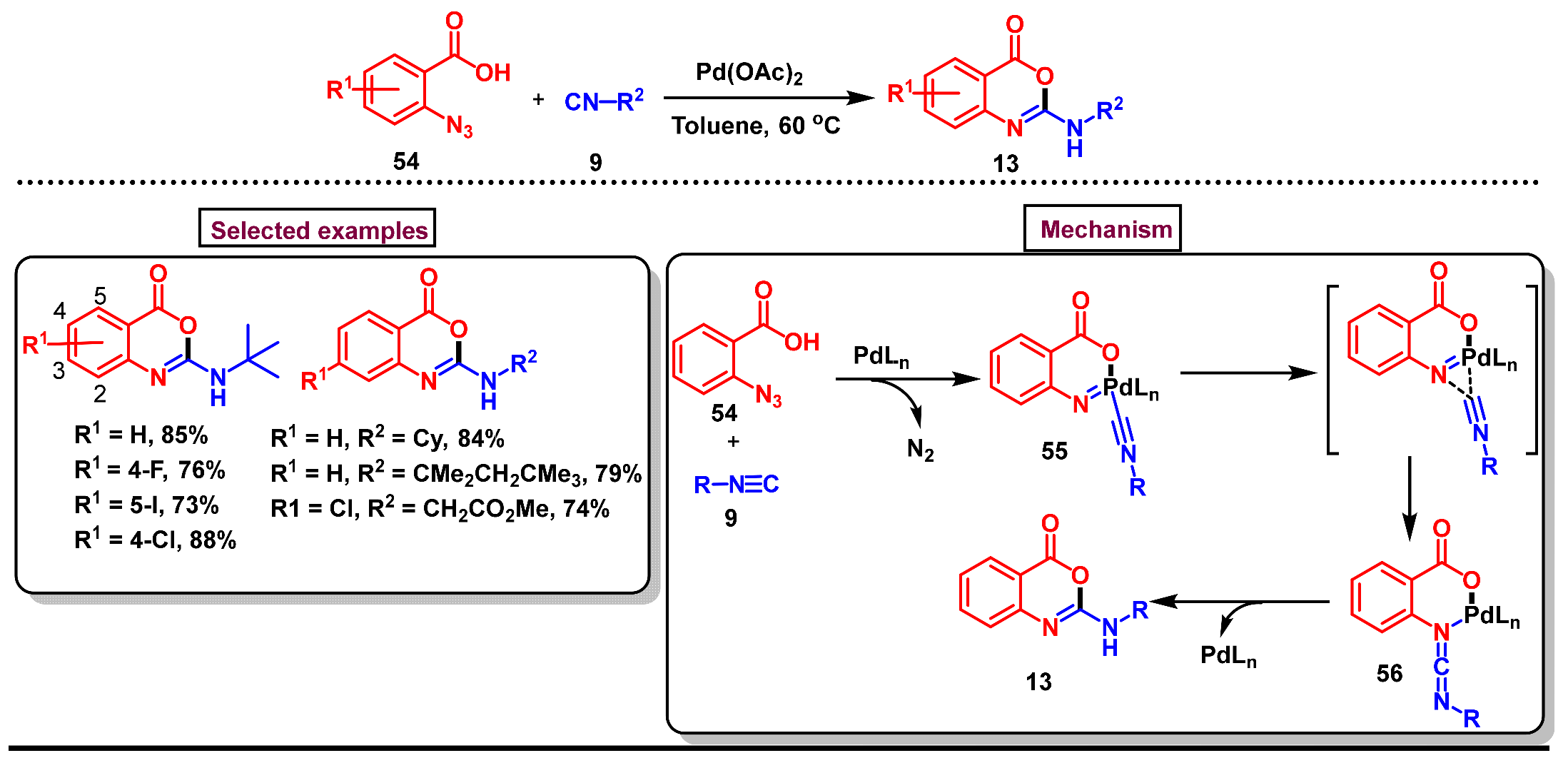
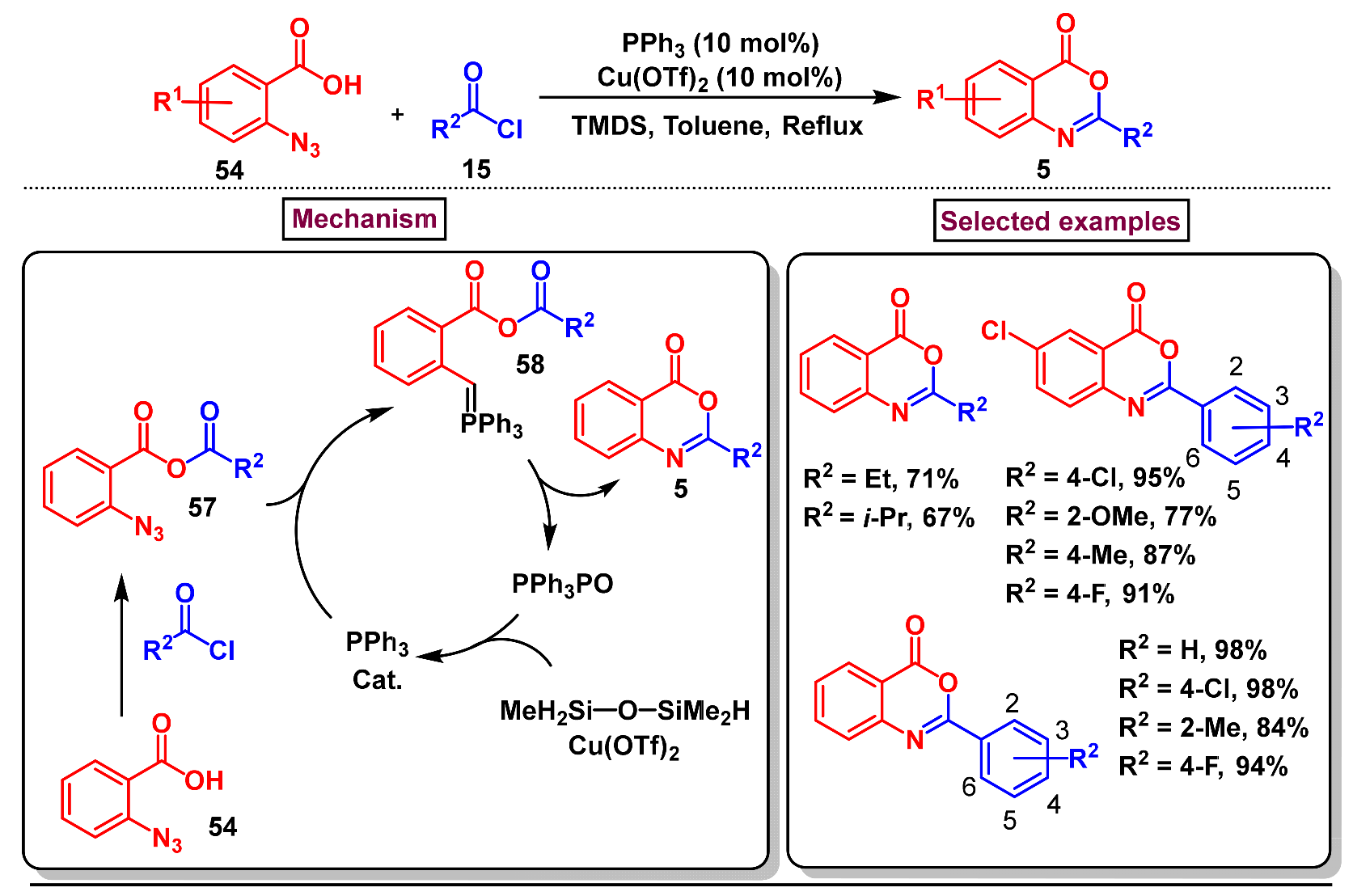
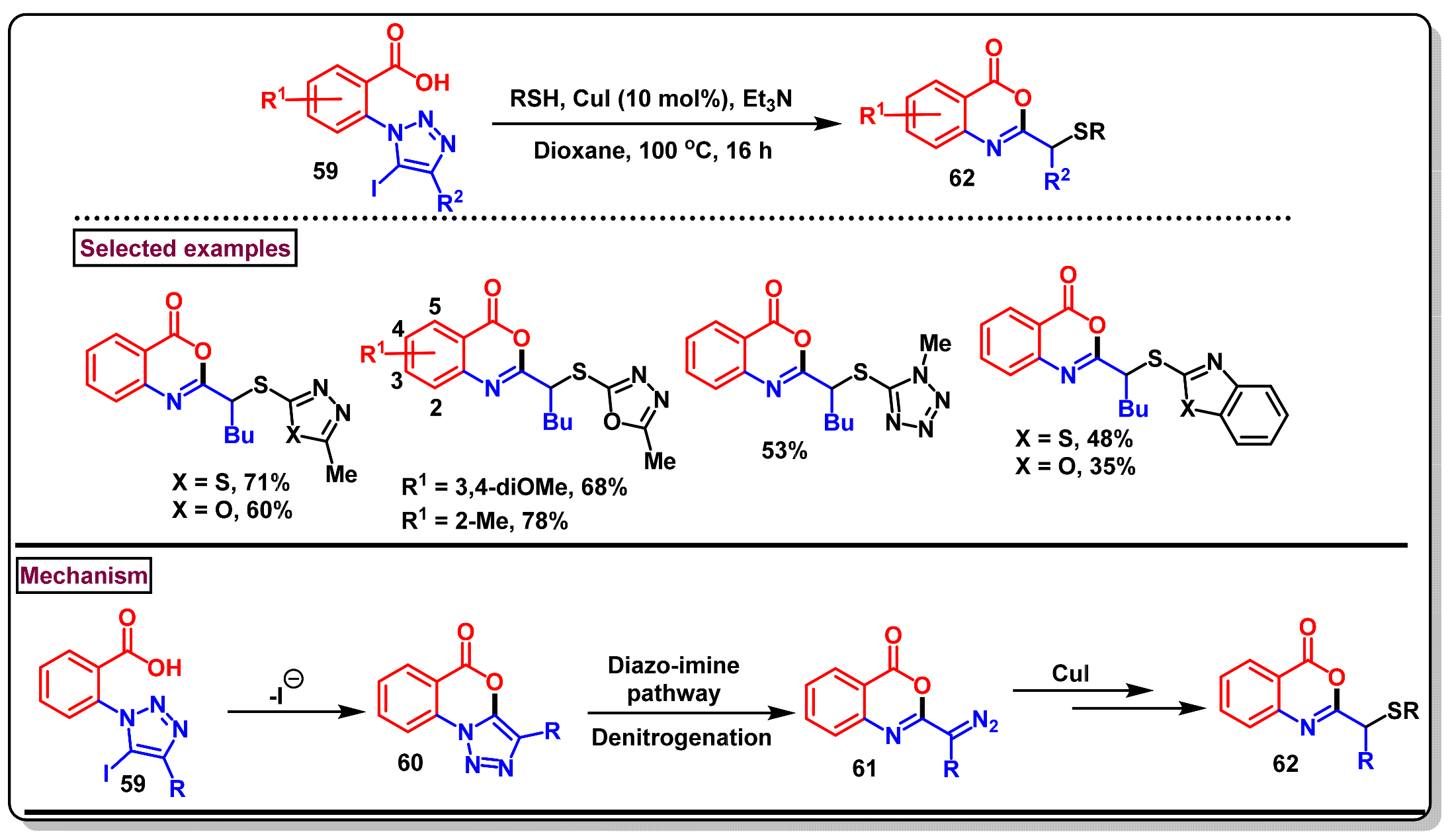
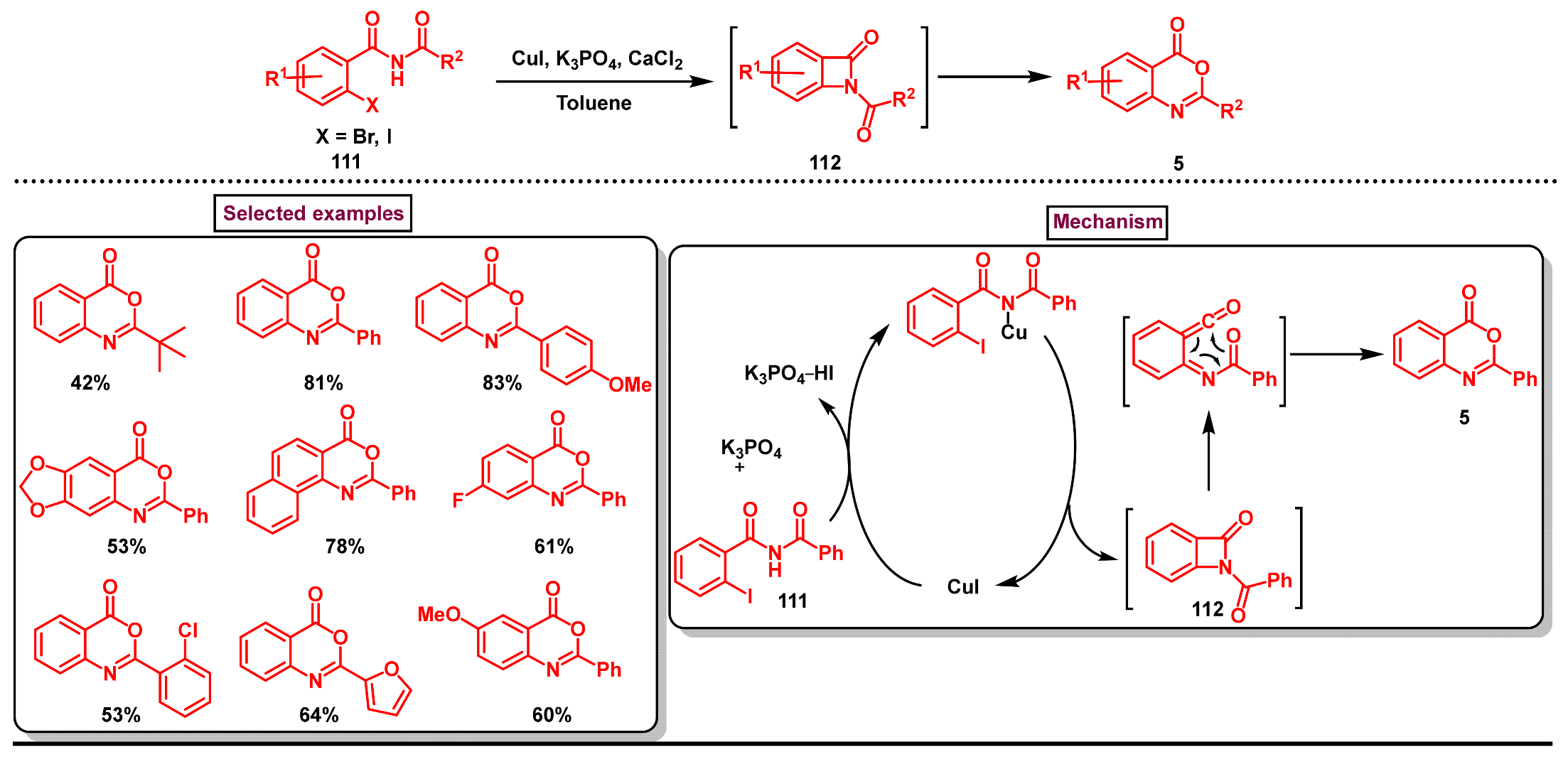
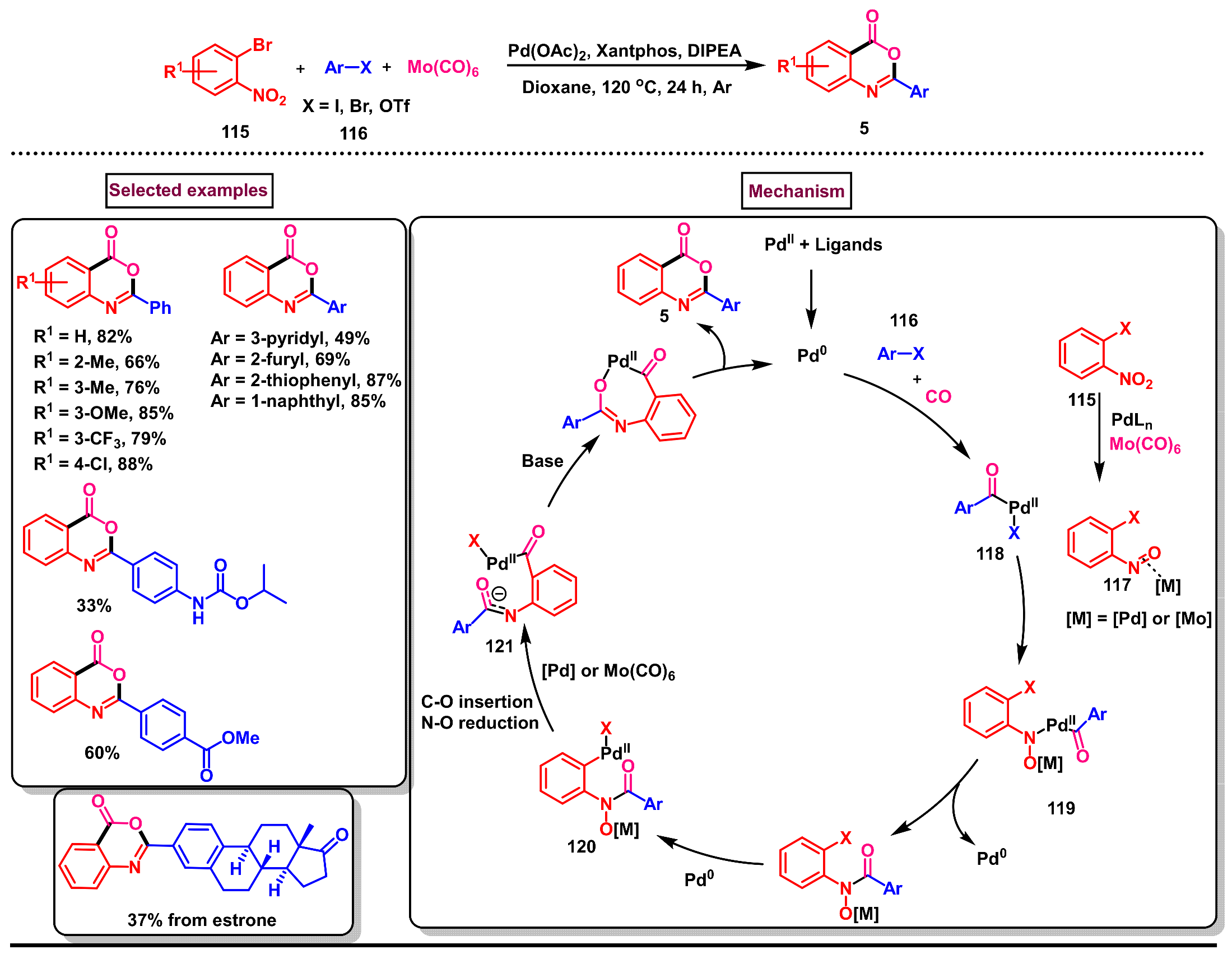
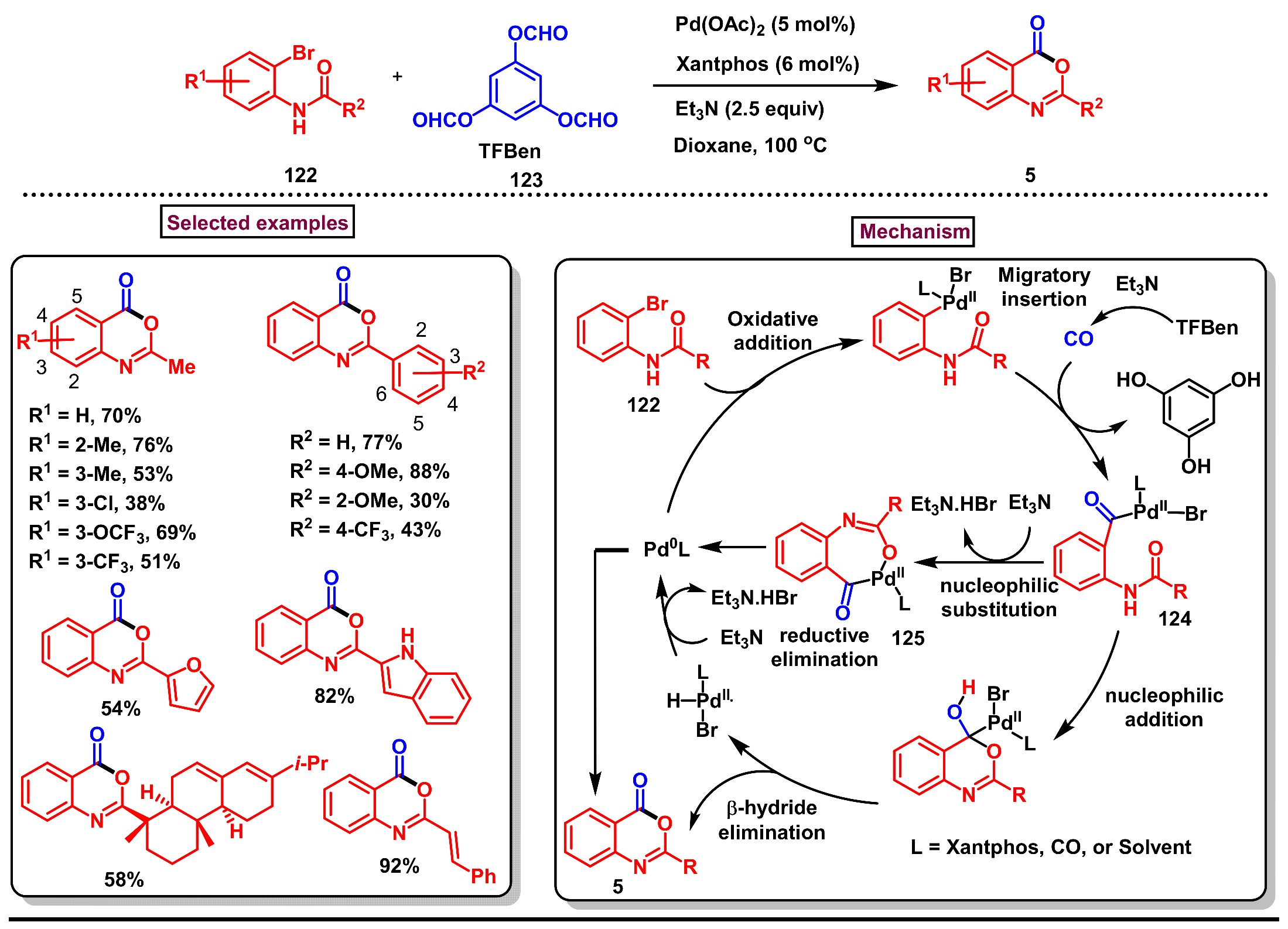
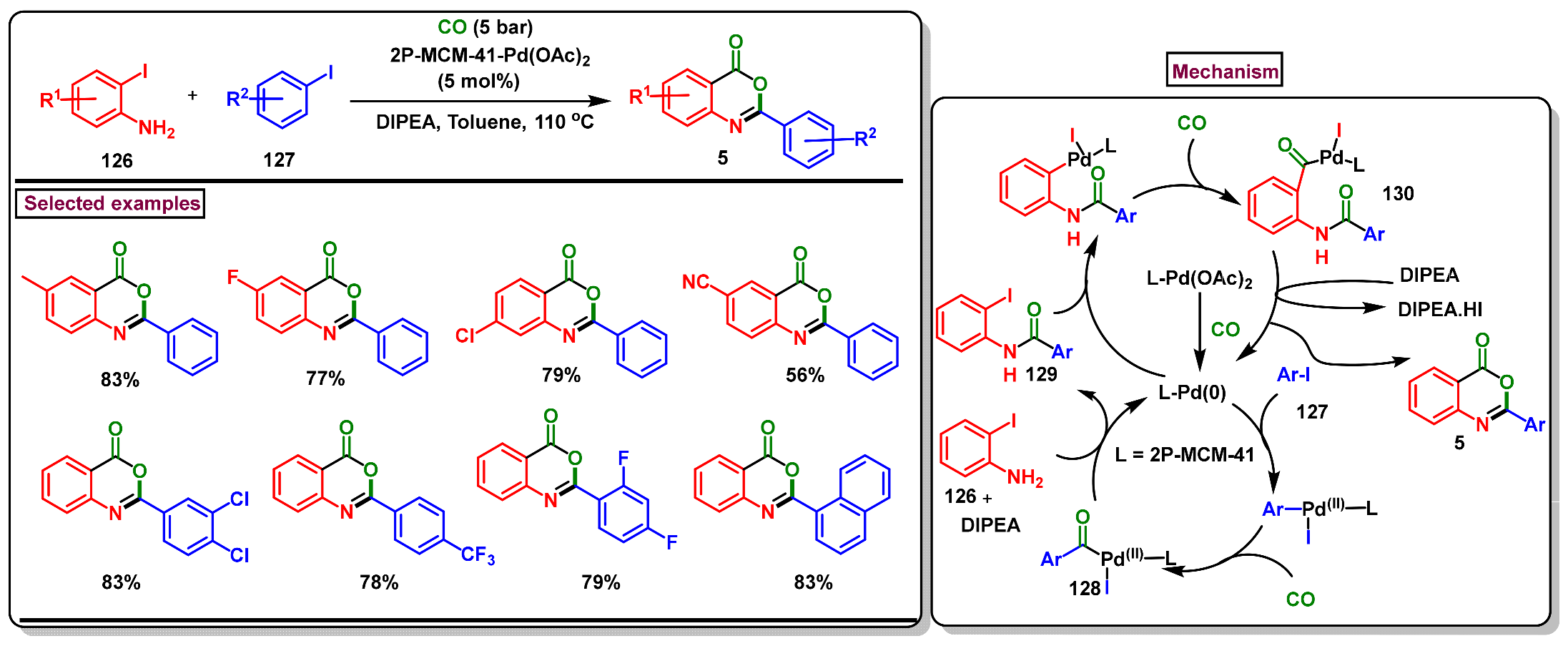
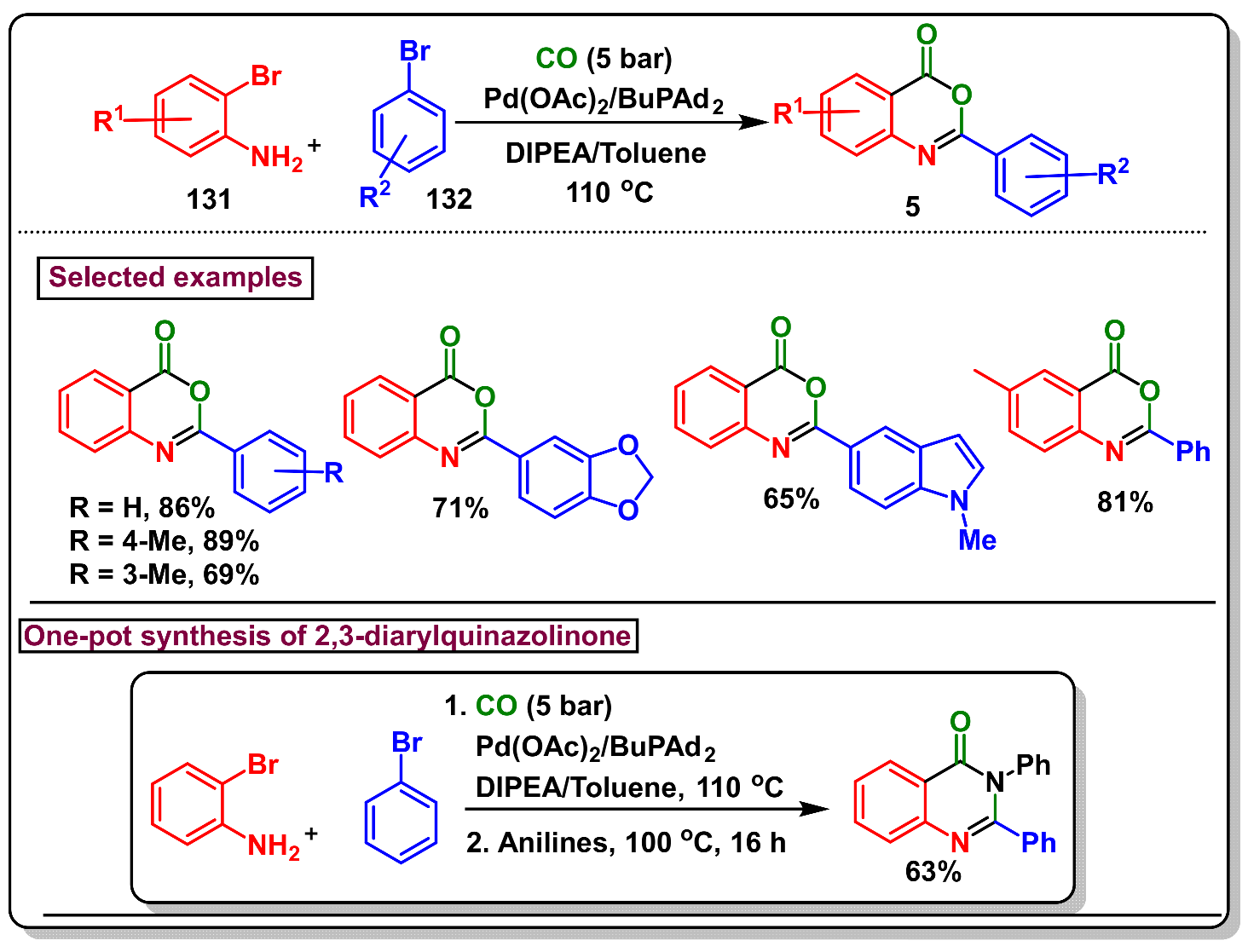
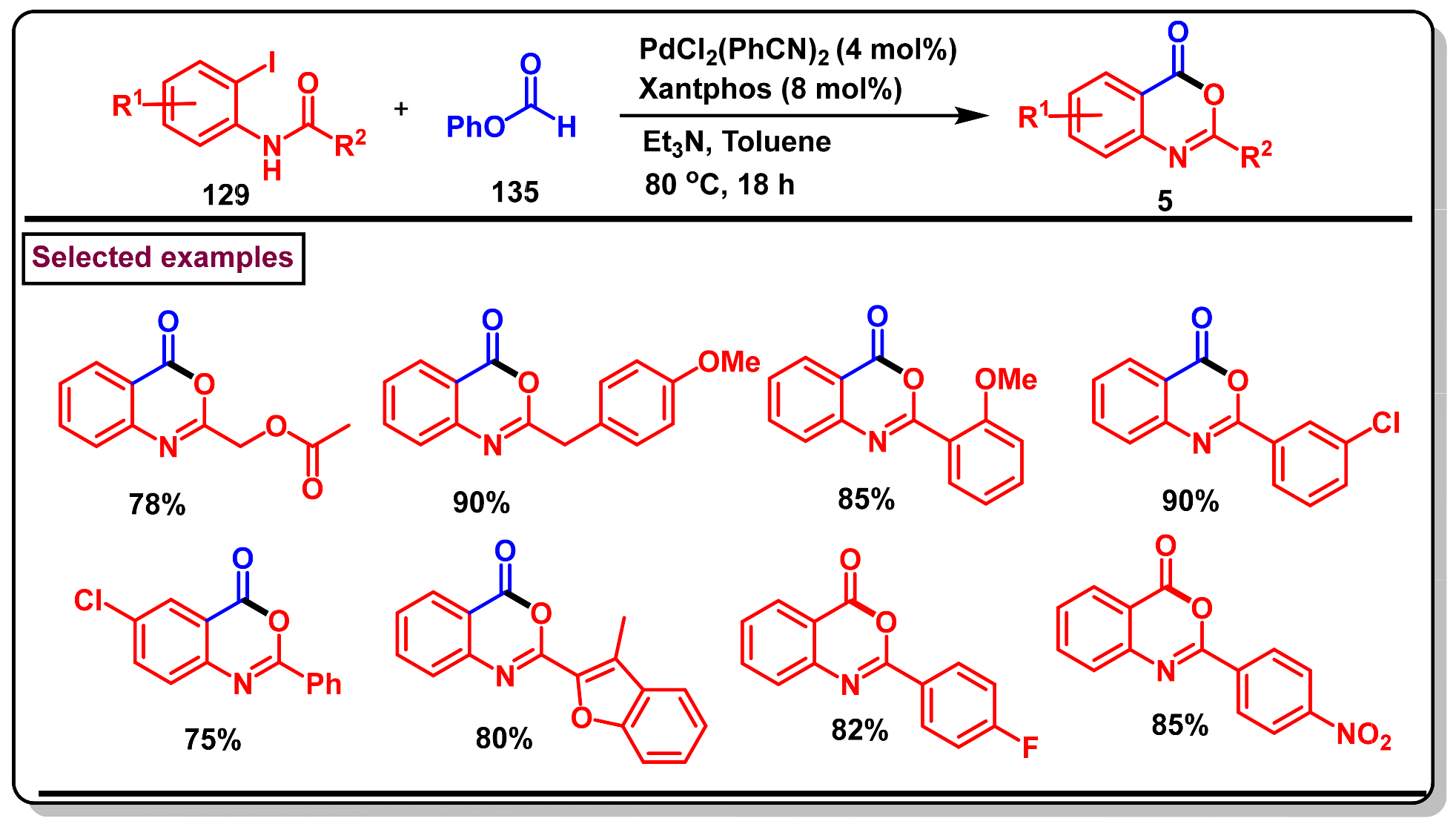
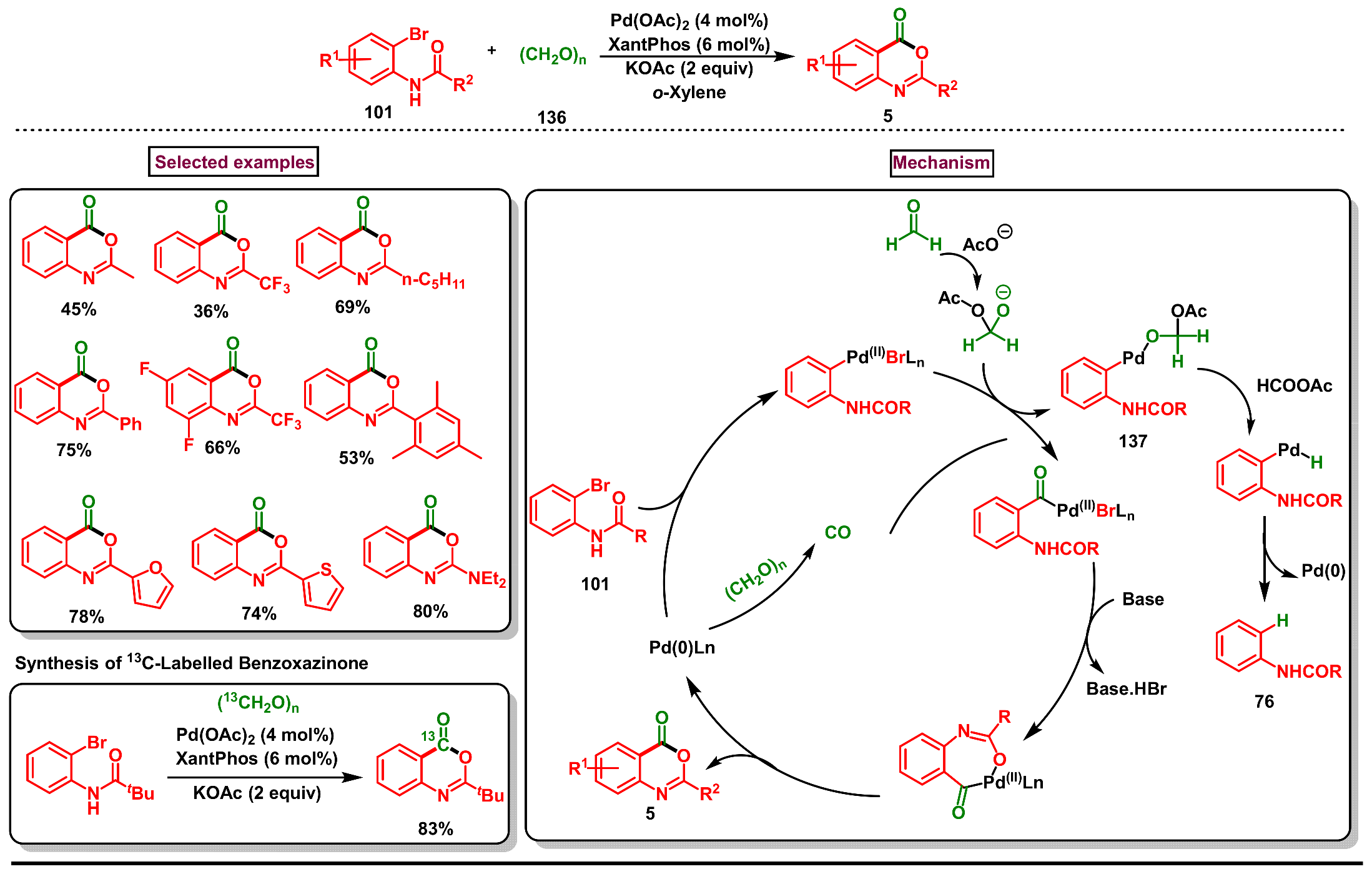
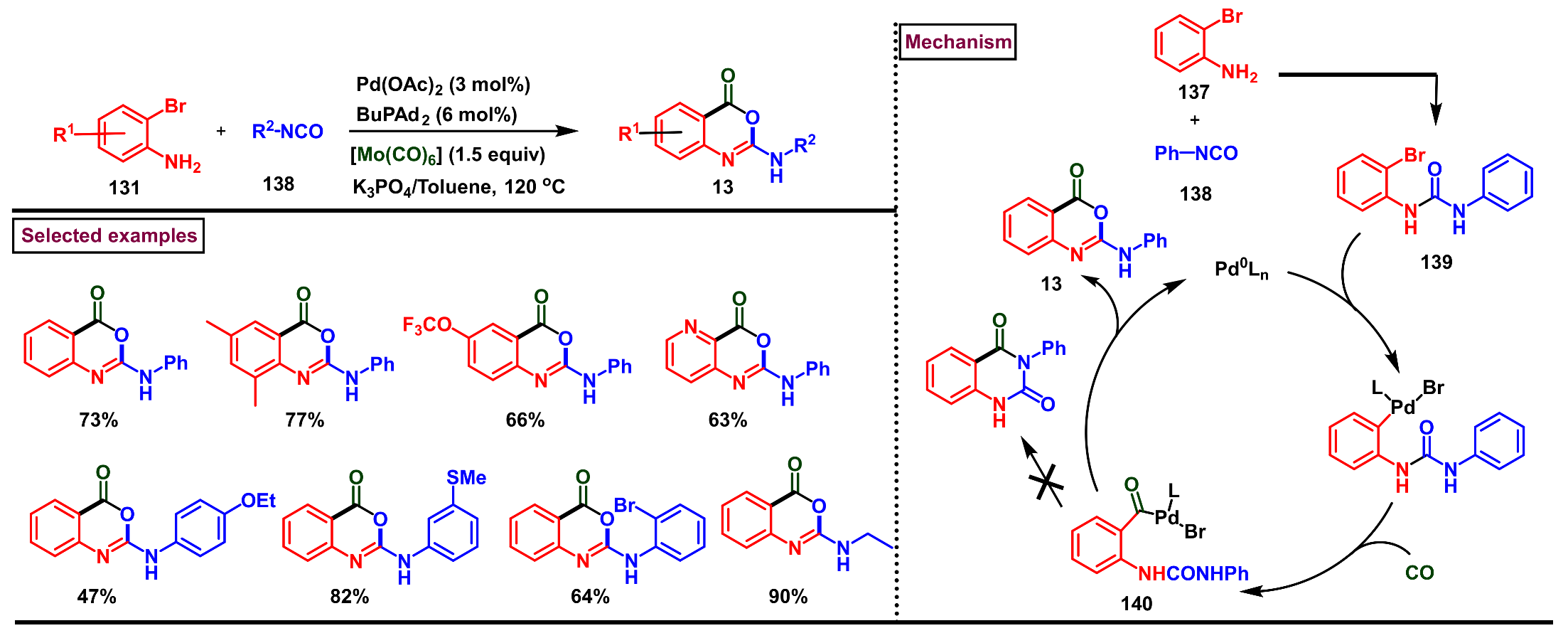
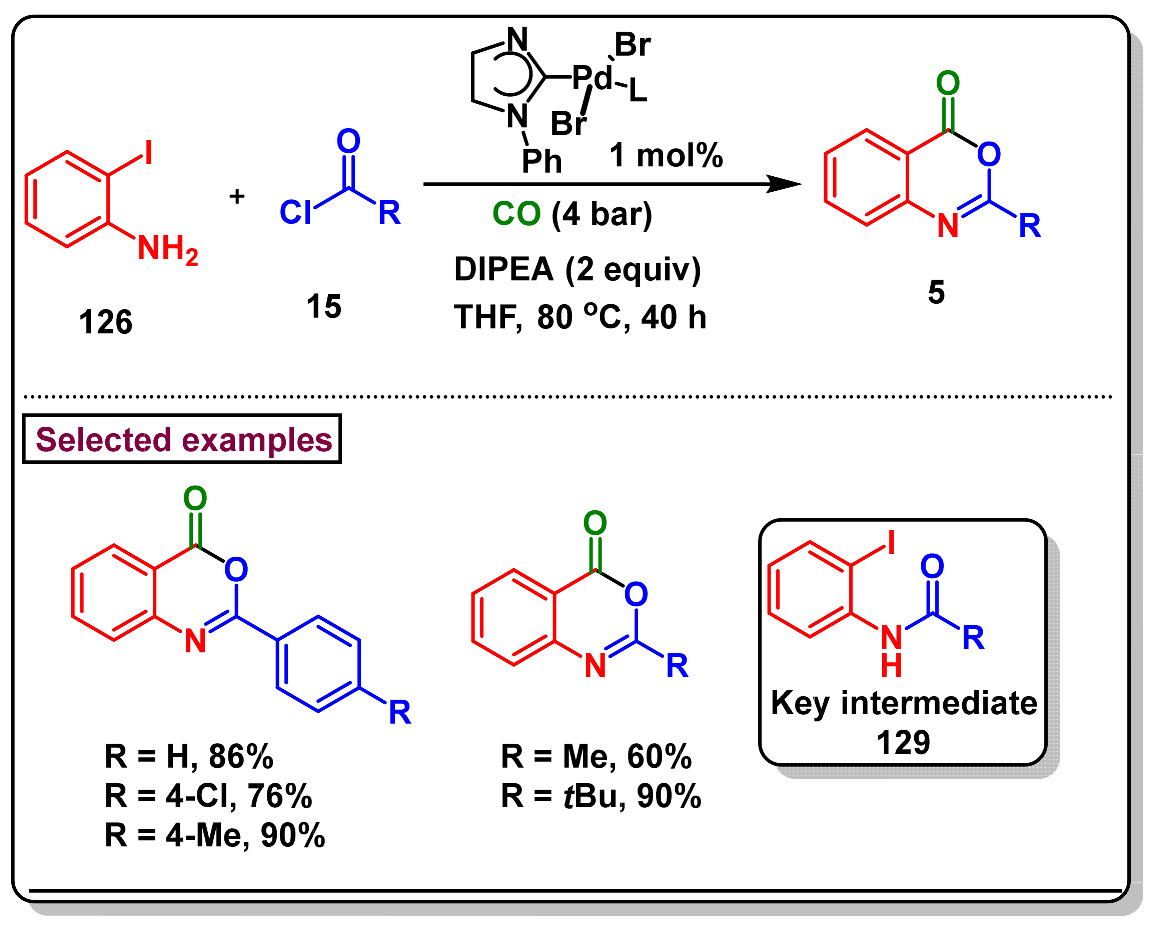
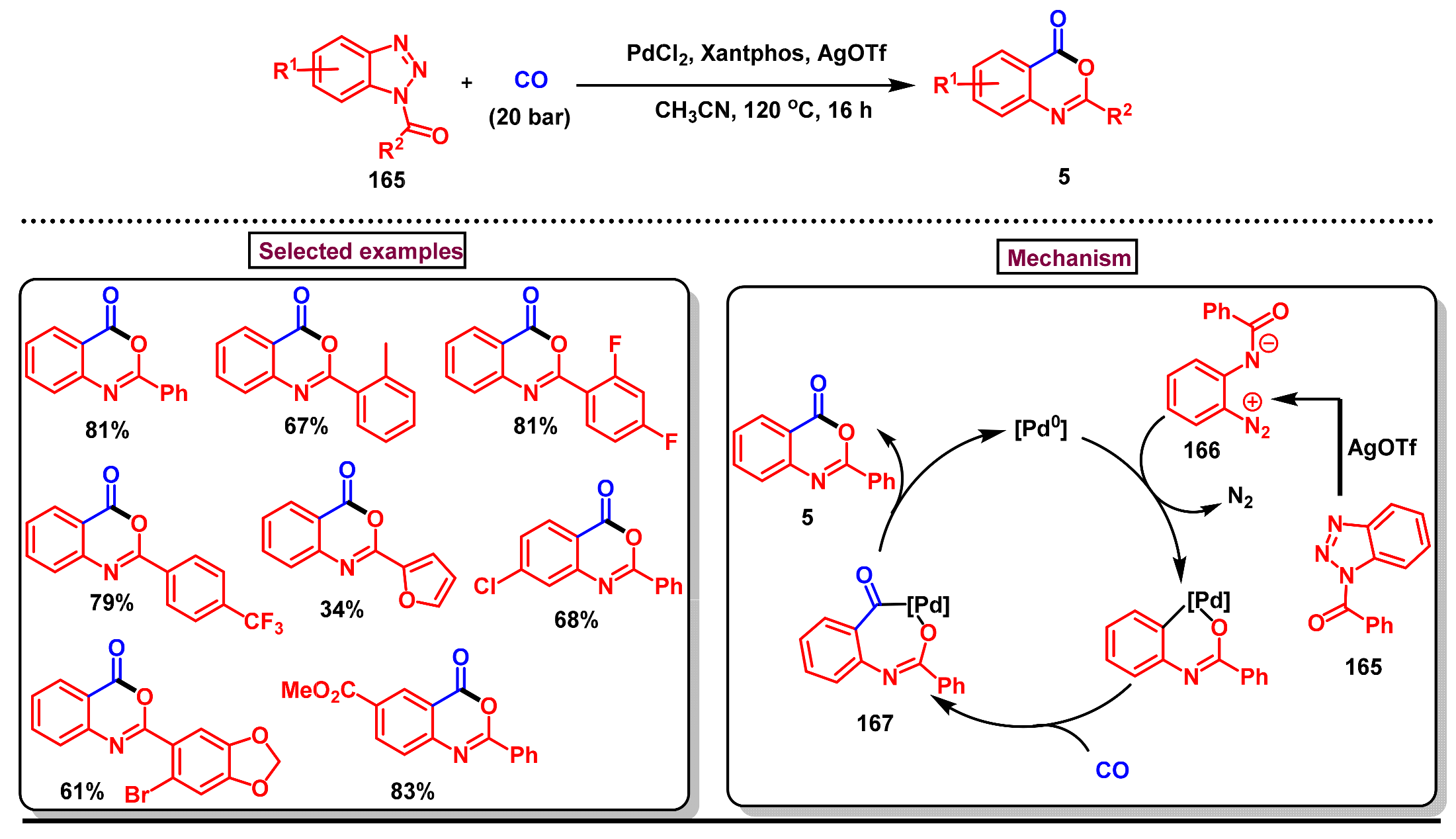
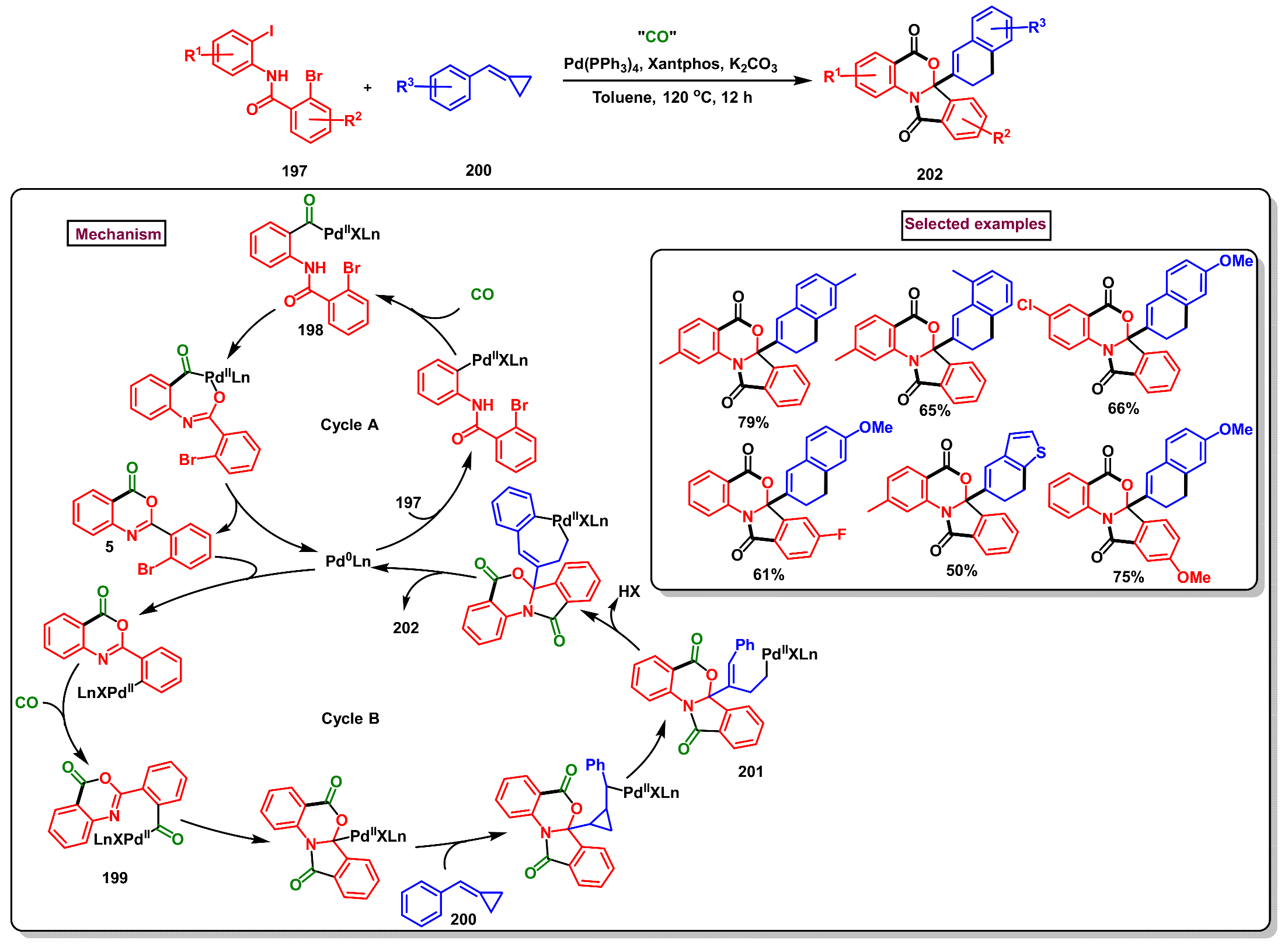
5. Preparation of Benzo[d][1,3]-Oxazin-4-Ones from Ortho-Aryl Halides
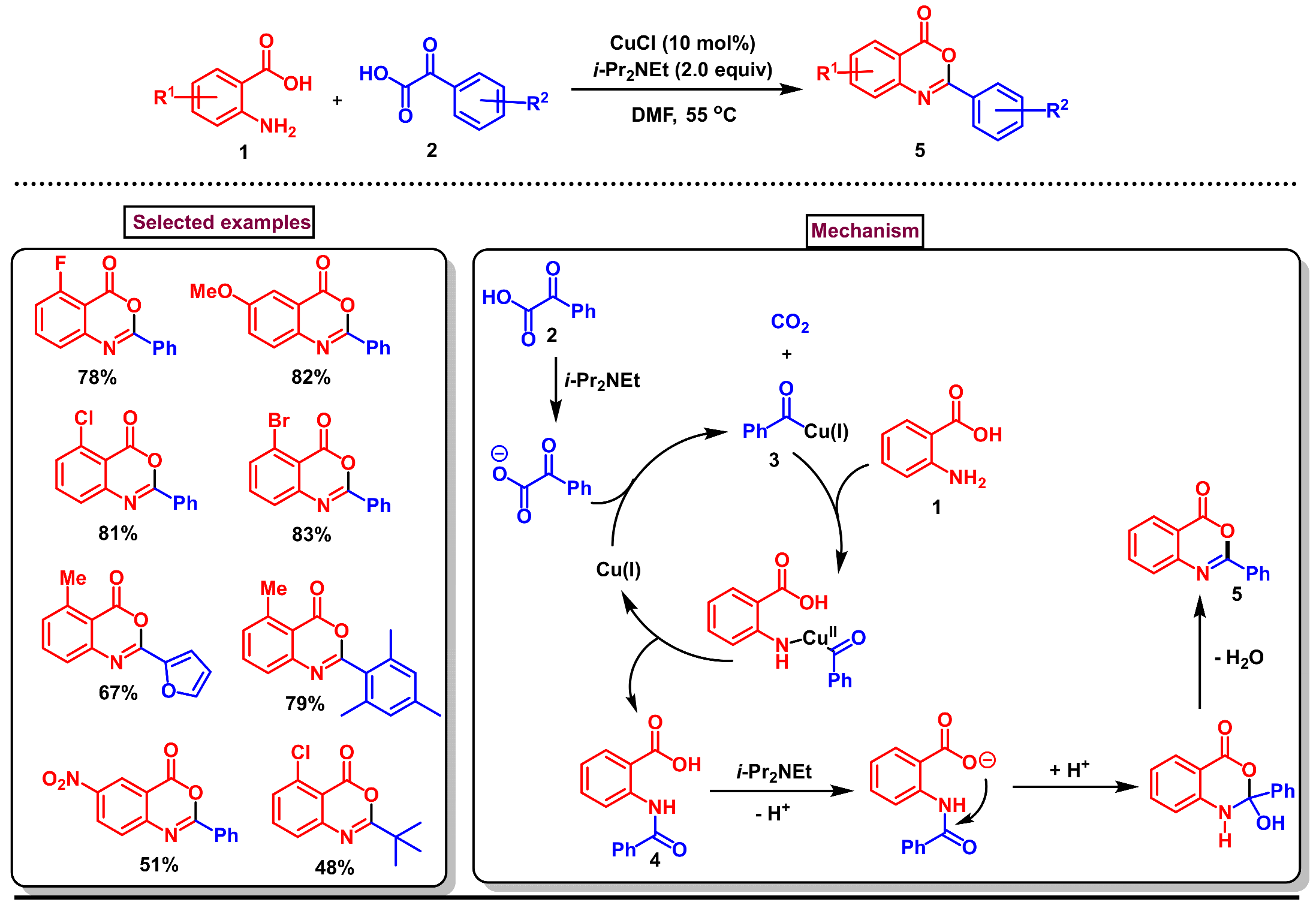
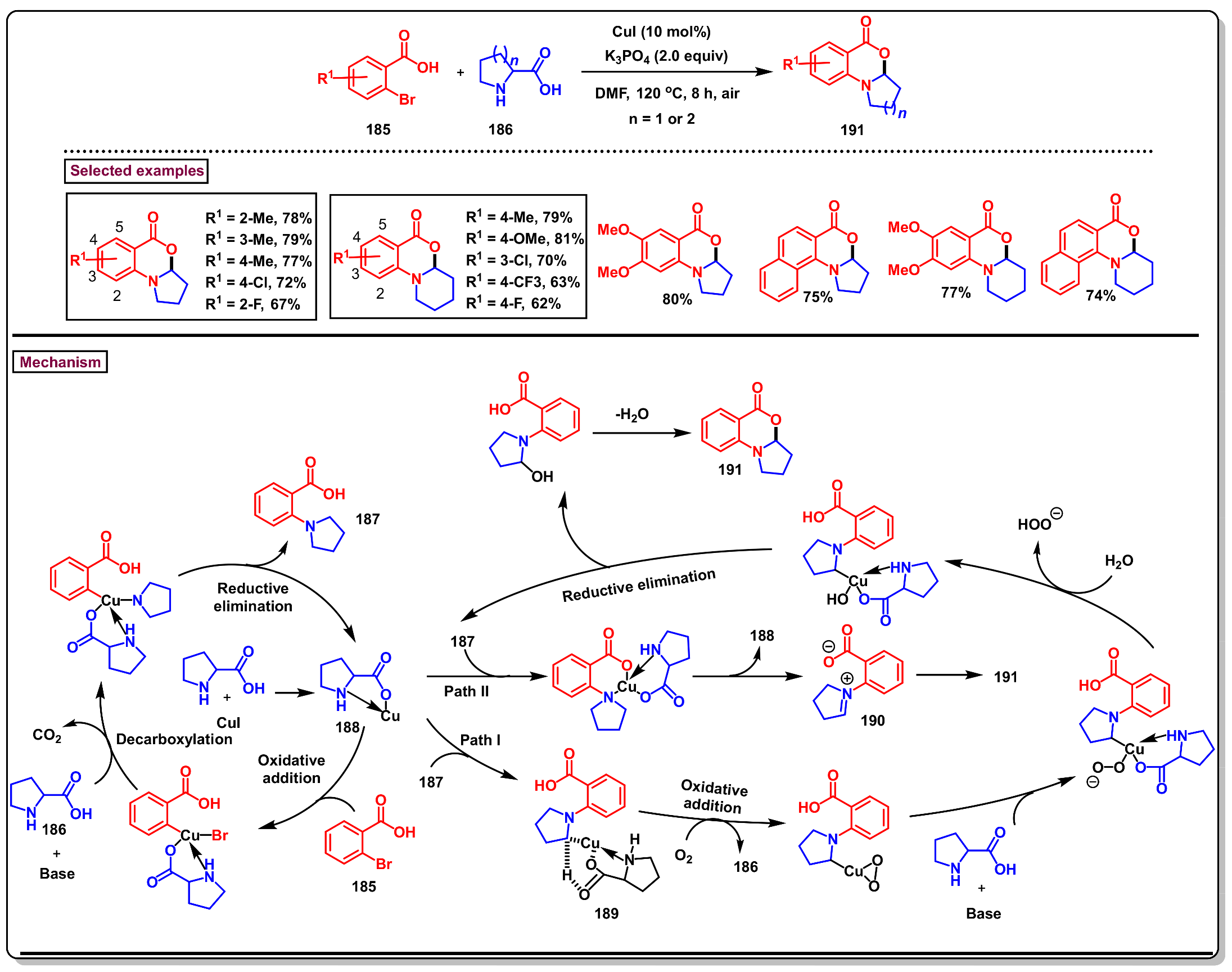
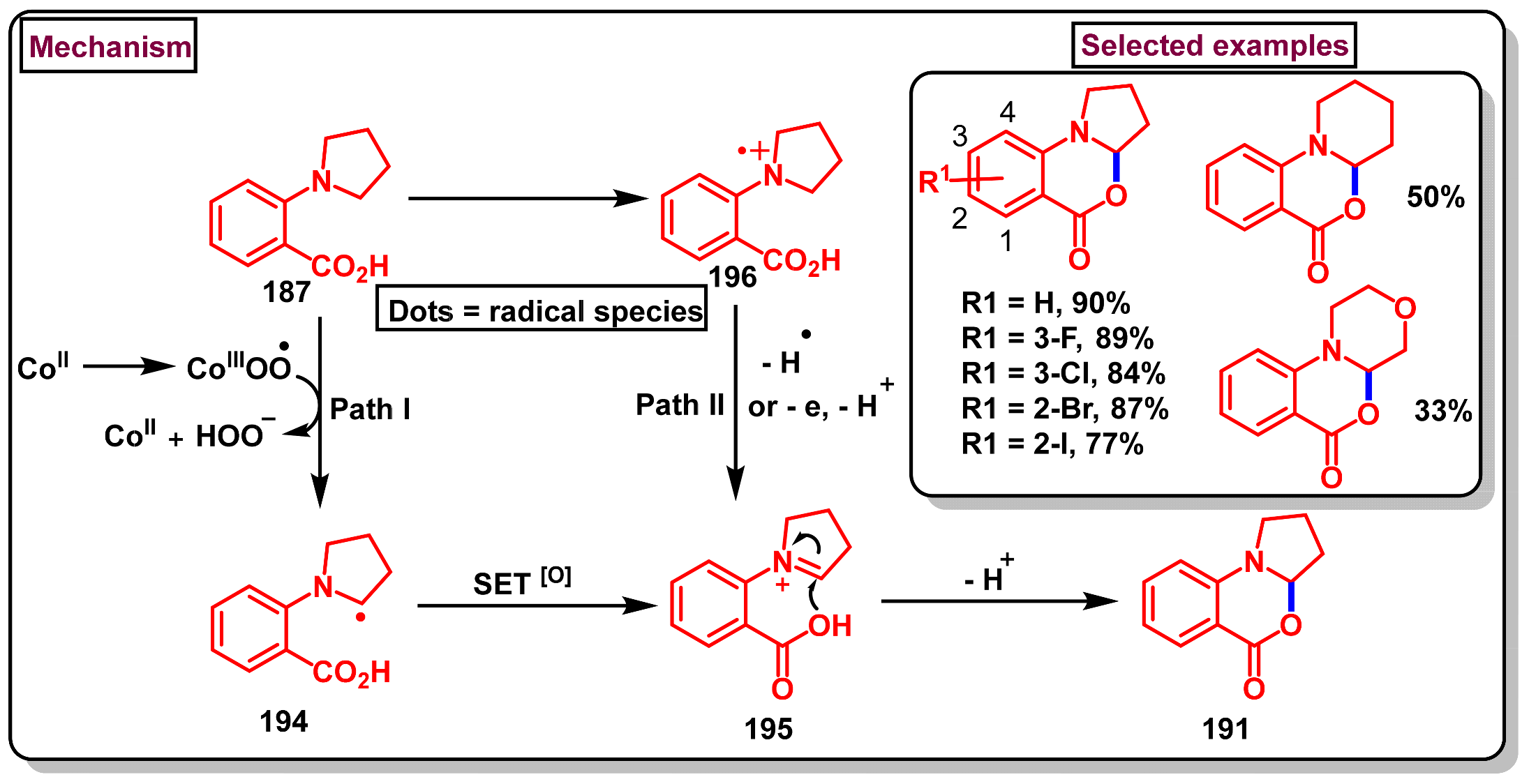
6. Preparation of Benzo[d][1,3]-Oxazin-4-Ones from Benzoic Acid Derivatives
7. Preparation of Benzo[d][1,3]-Oxazin-4-Ones from Indoles and Isatins
- From Functionalized Isatins
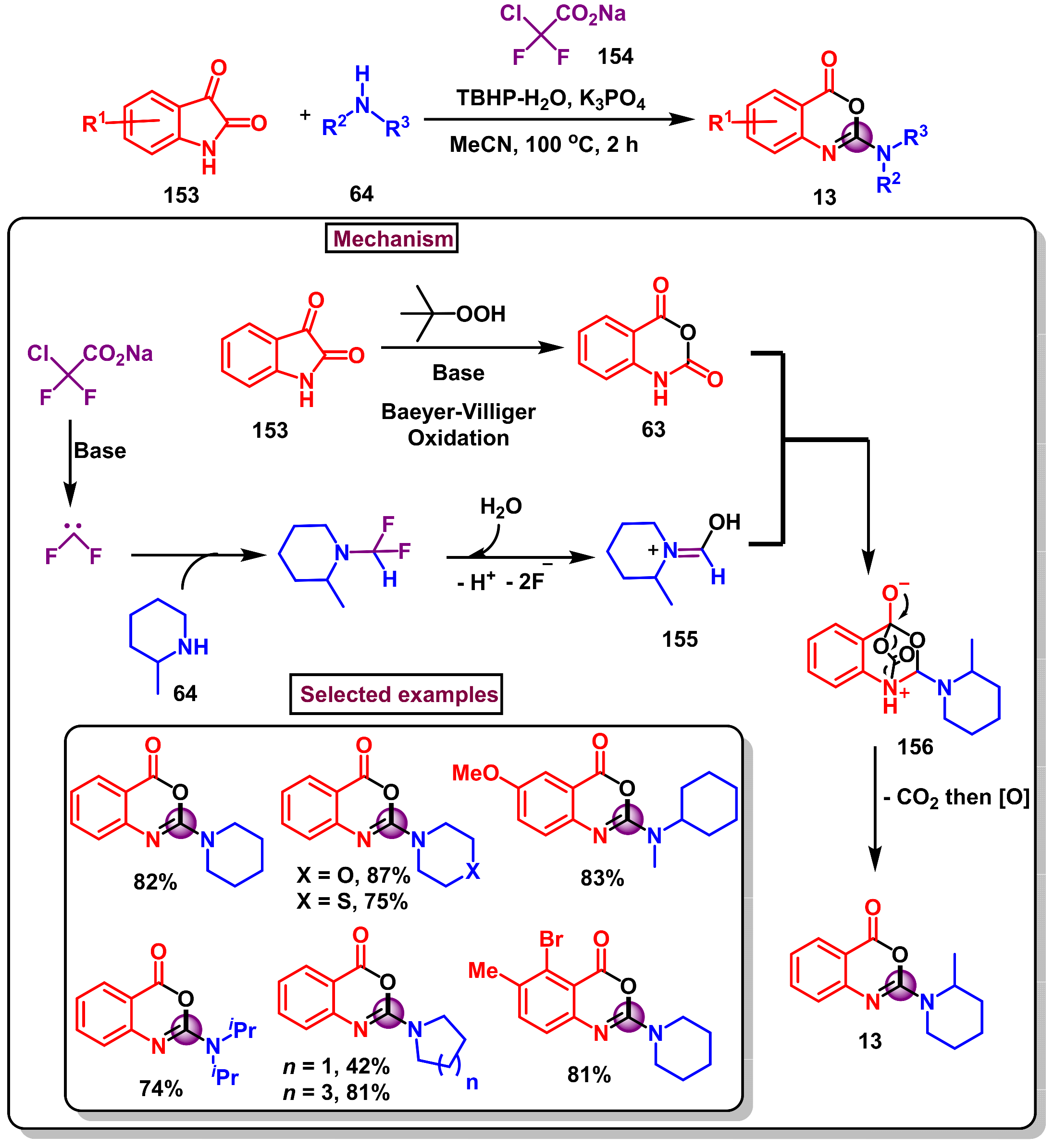
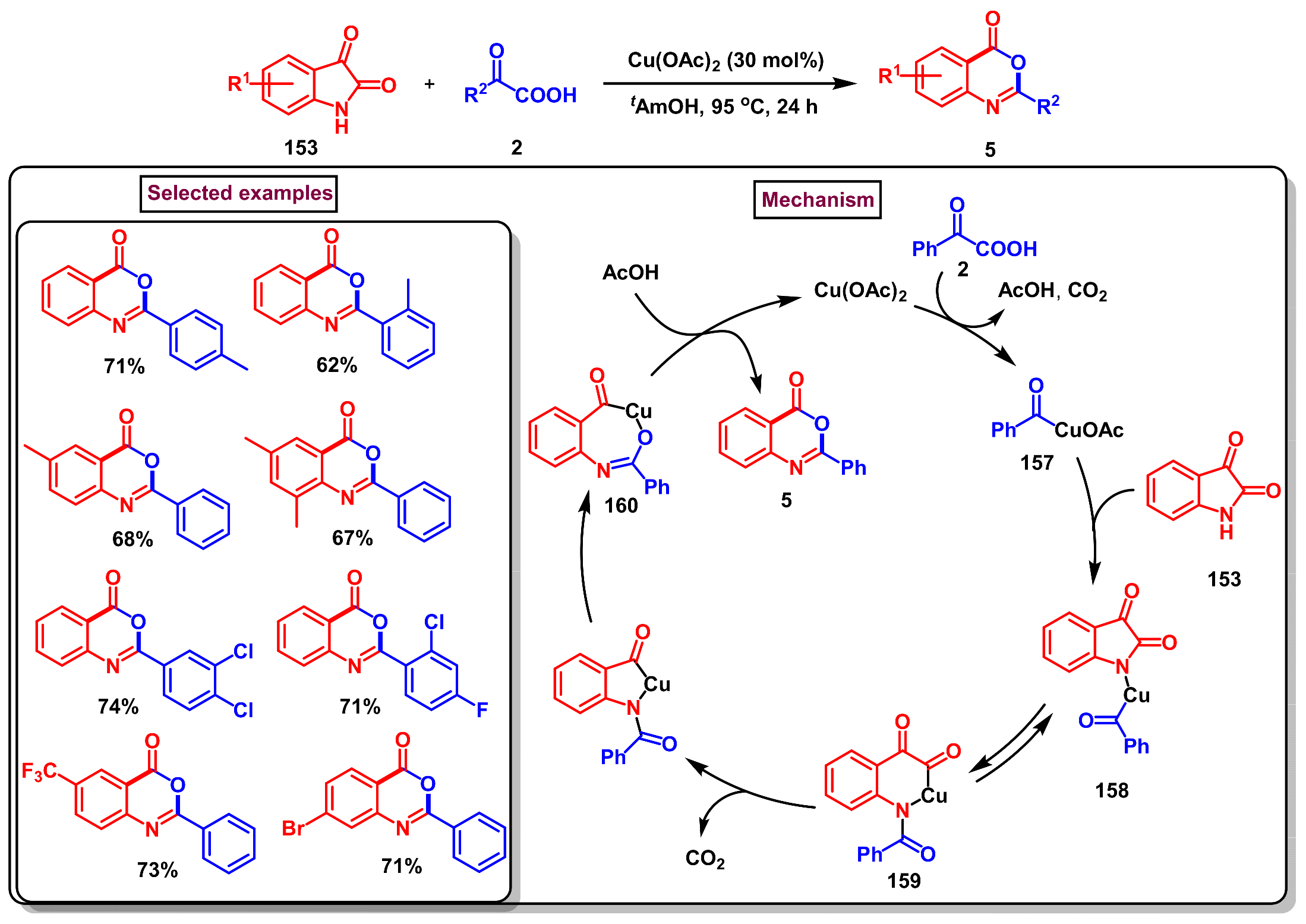
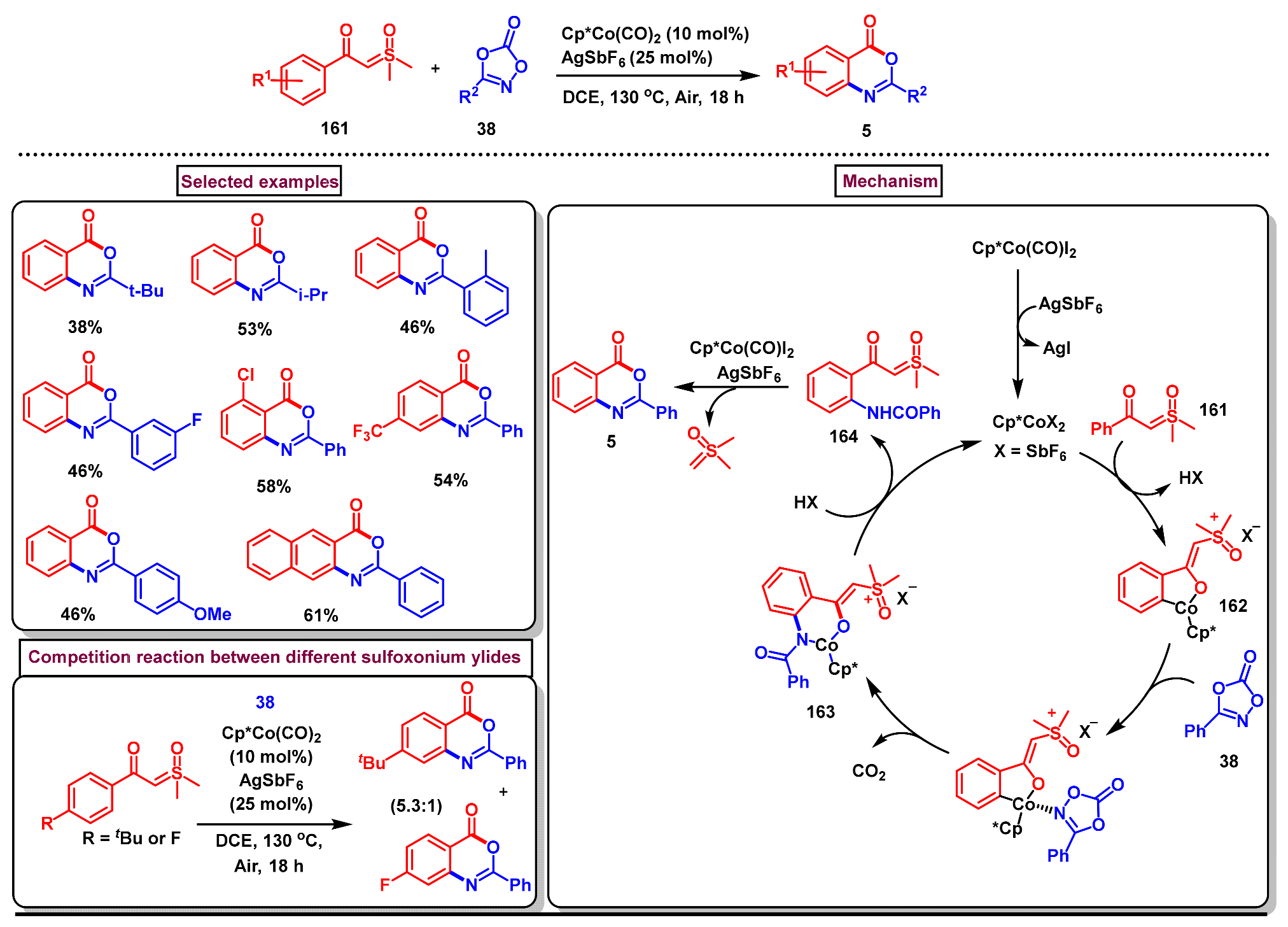
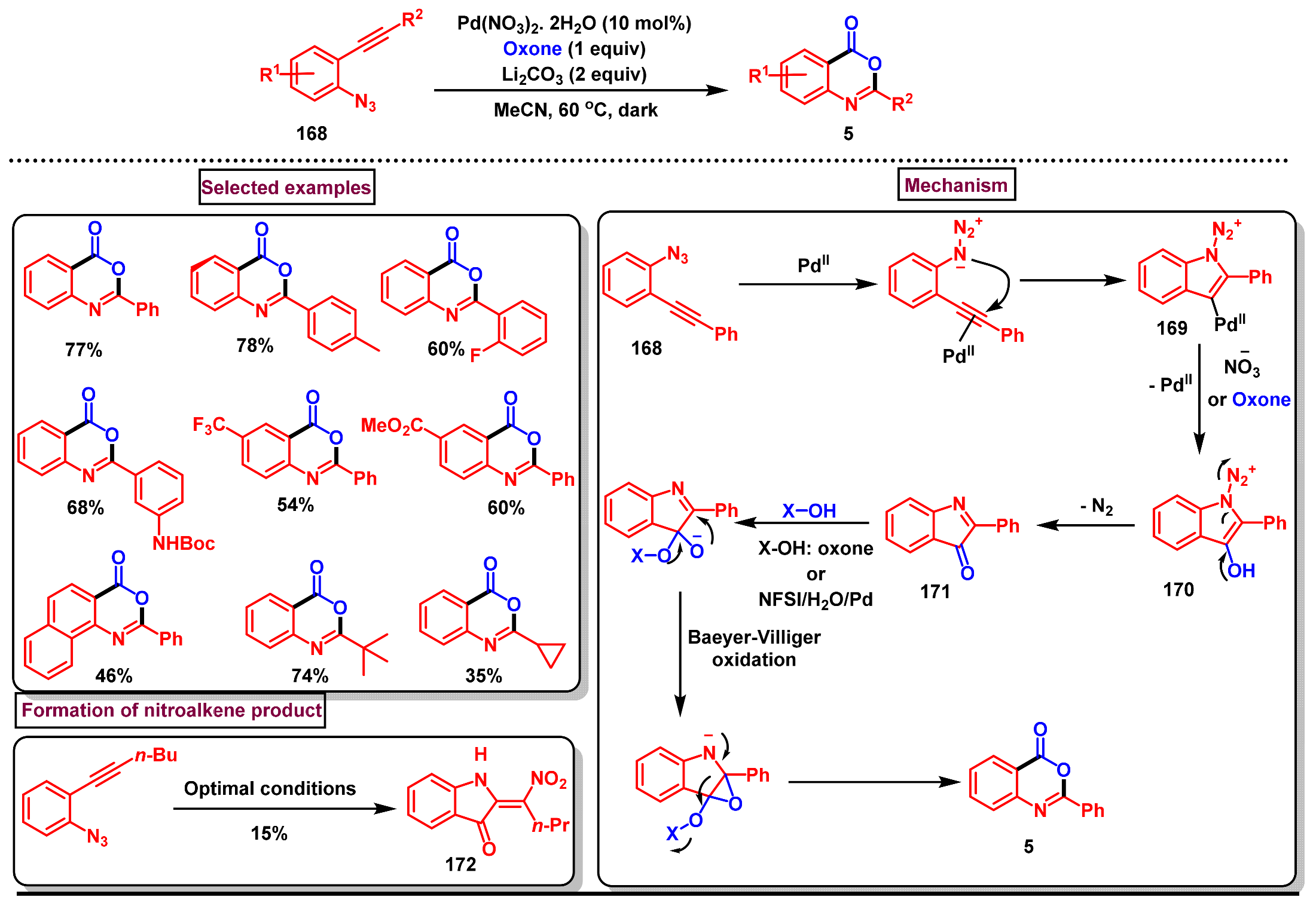
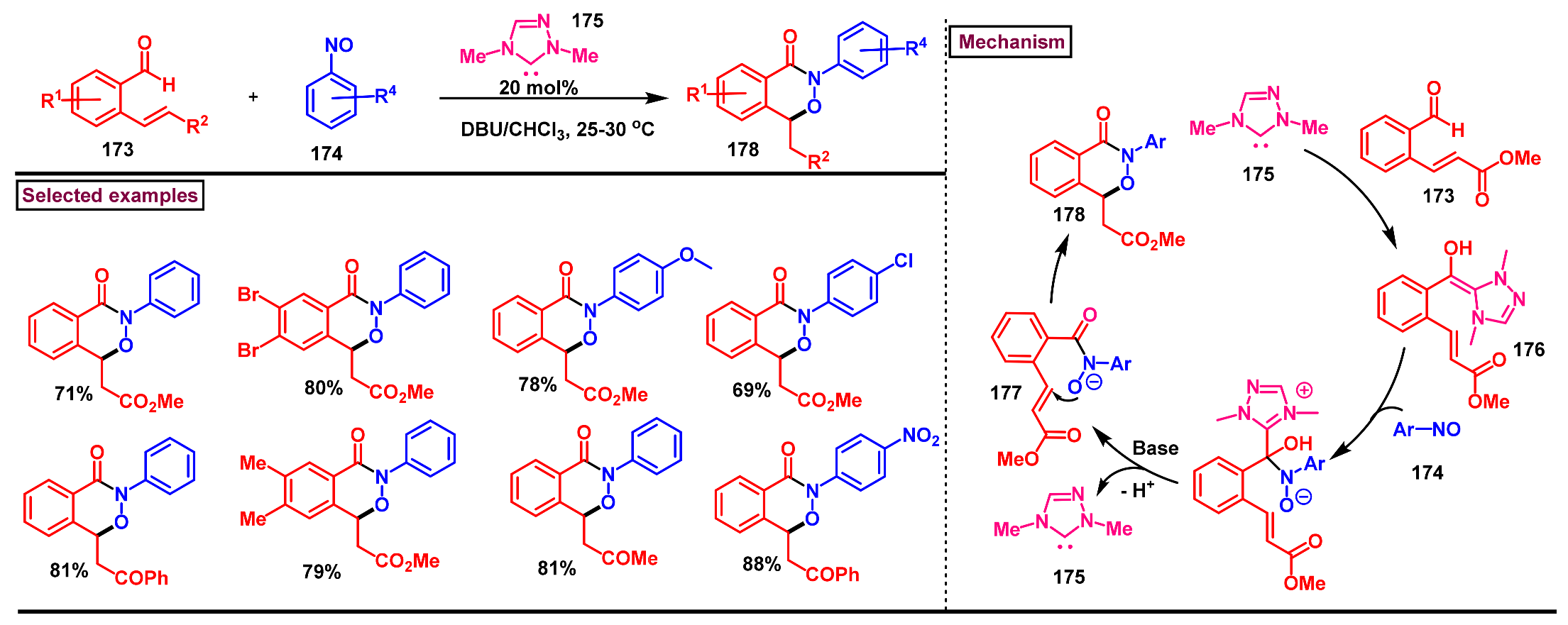
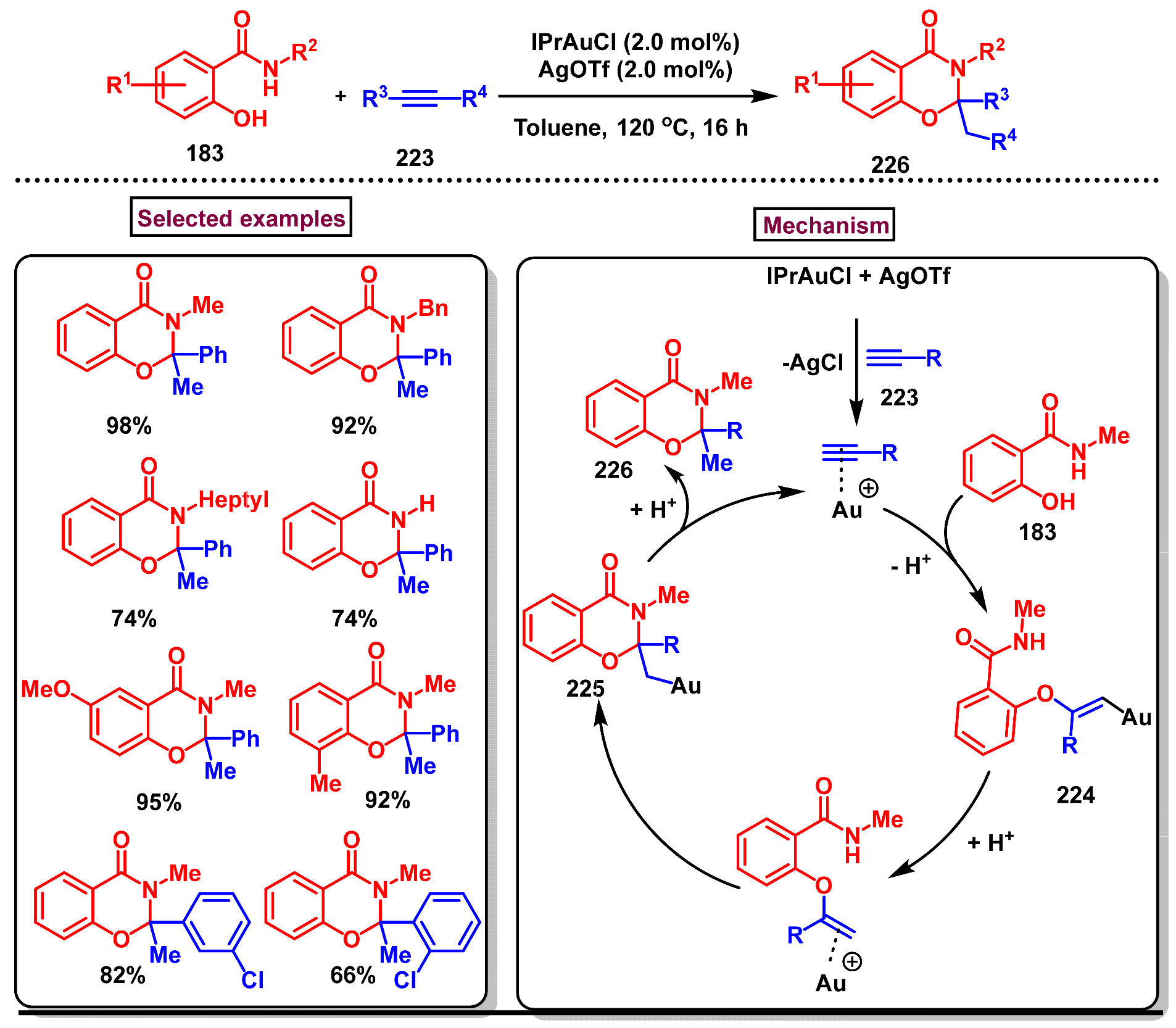
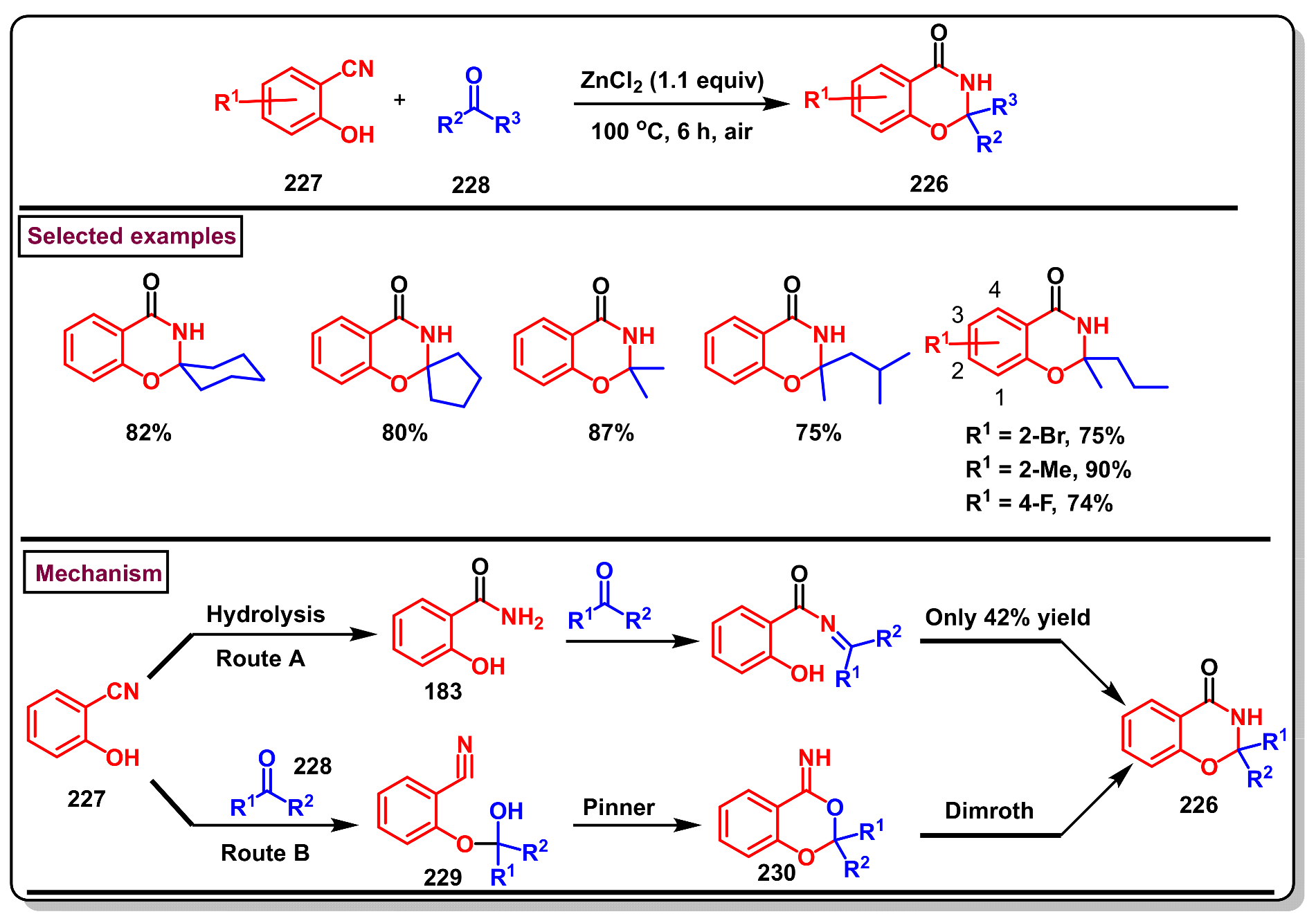
8. Preparation of Benzo[d][1,3]-Oxazin-4-Ones via Miscellaneous Procedures
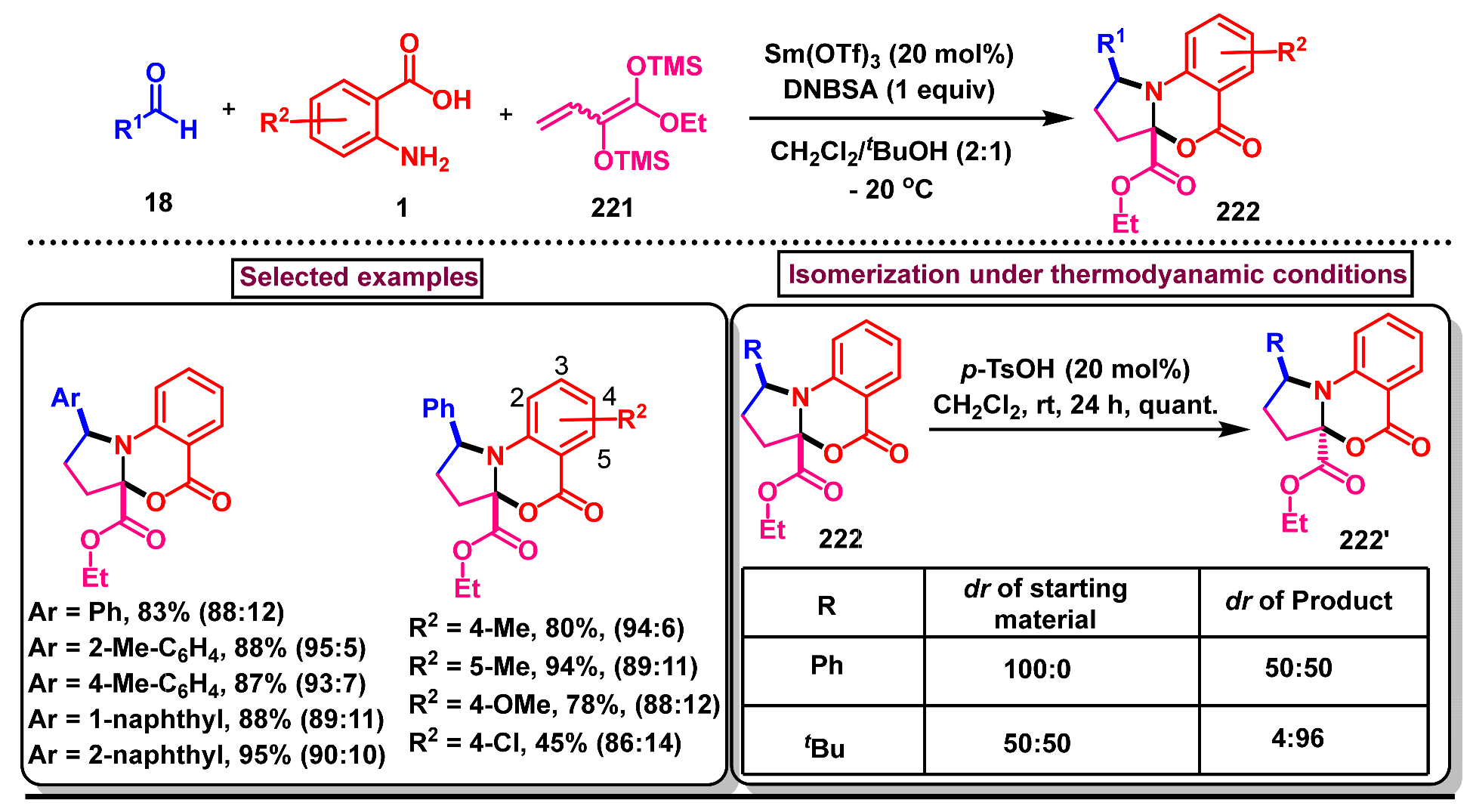
9. Preparation of Fused Benzo[d][1,3]-Oxazin-4-One Derivatives
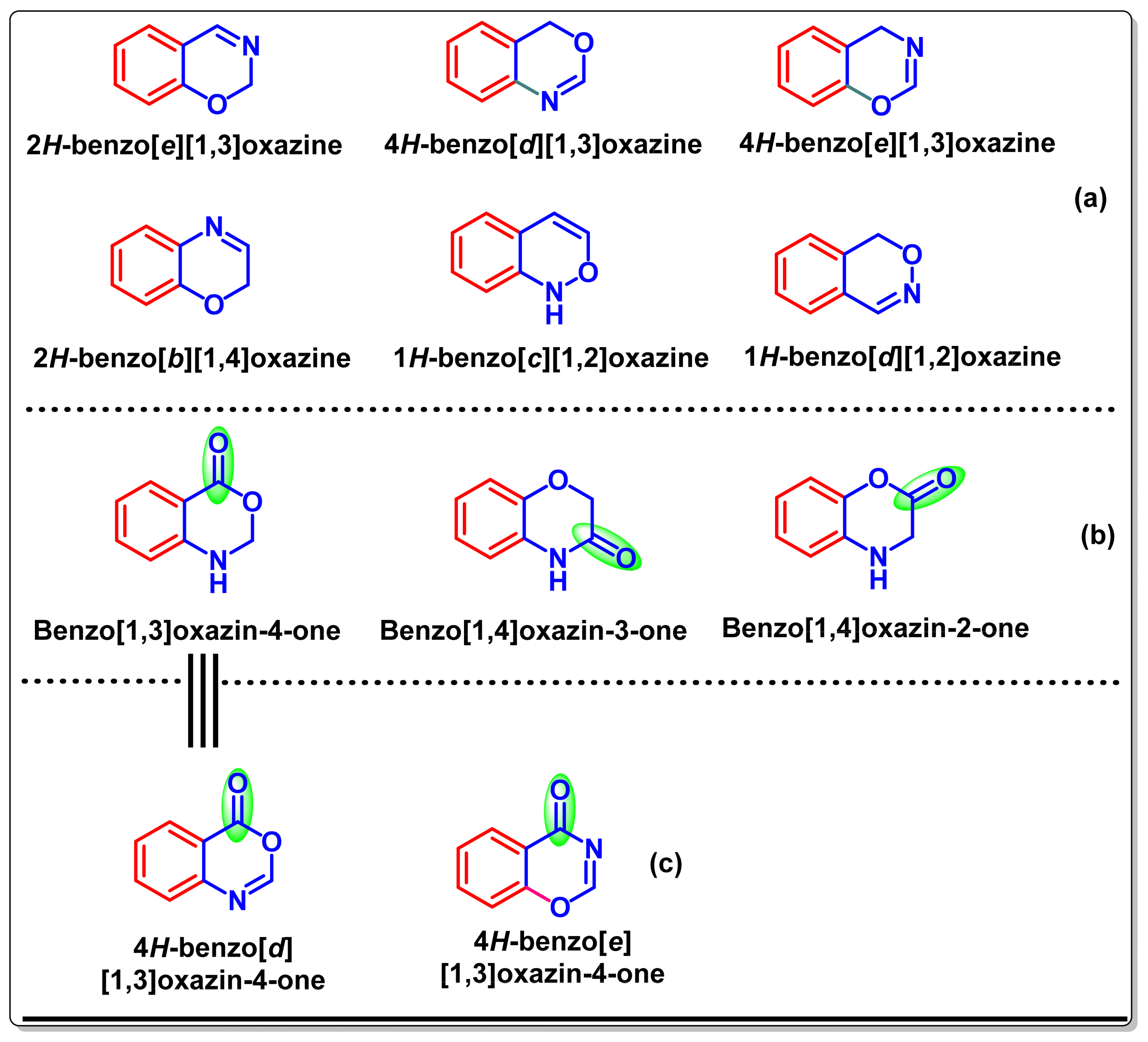
10. Preparation of Benzo[e][1,3]-Oxazin-4-One Derivatives
11. Applications in N-Directed Ortho-Functionalizations Via C-H Activation
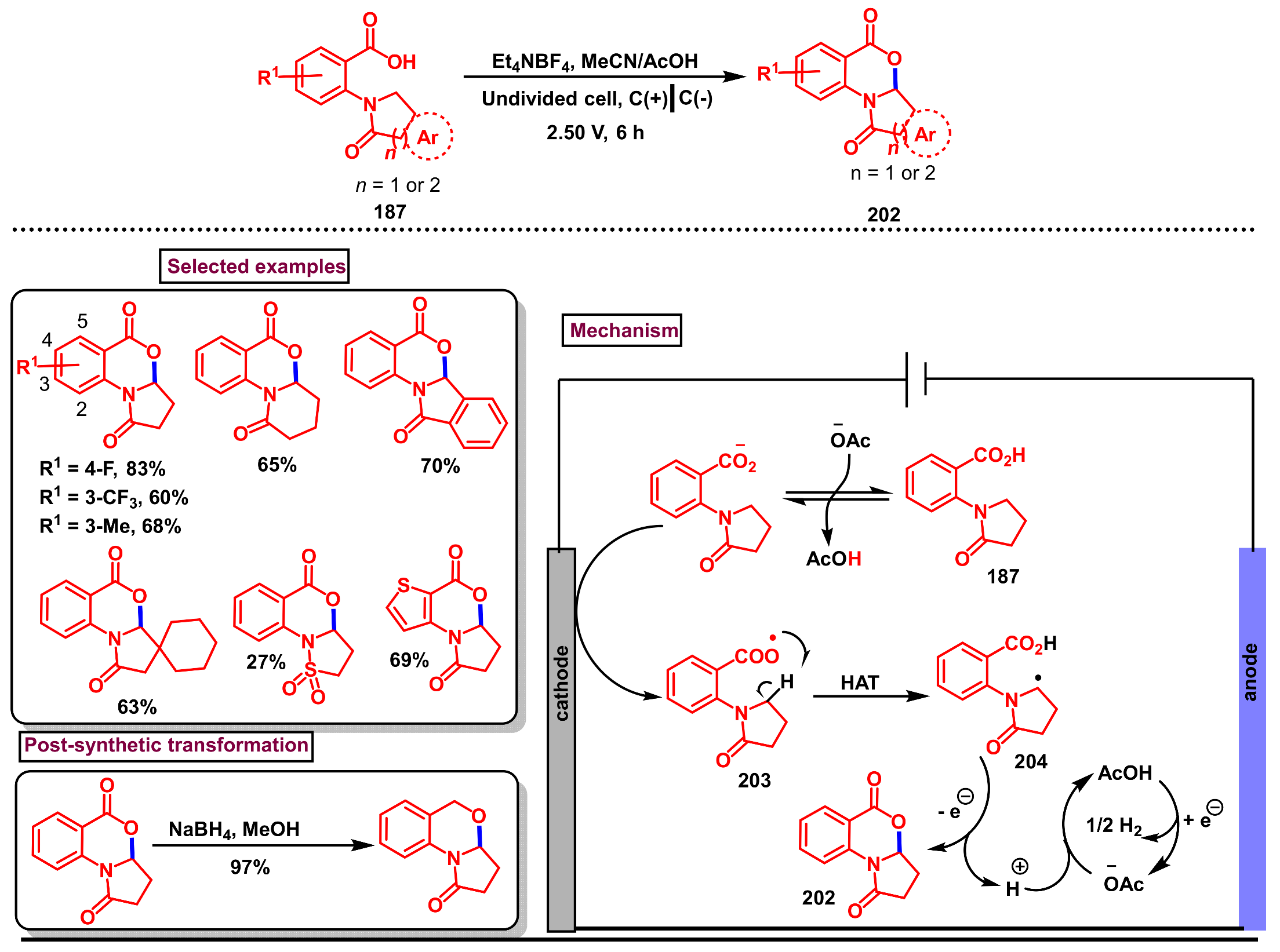
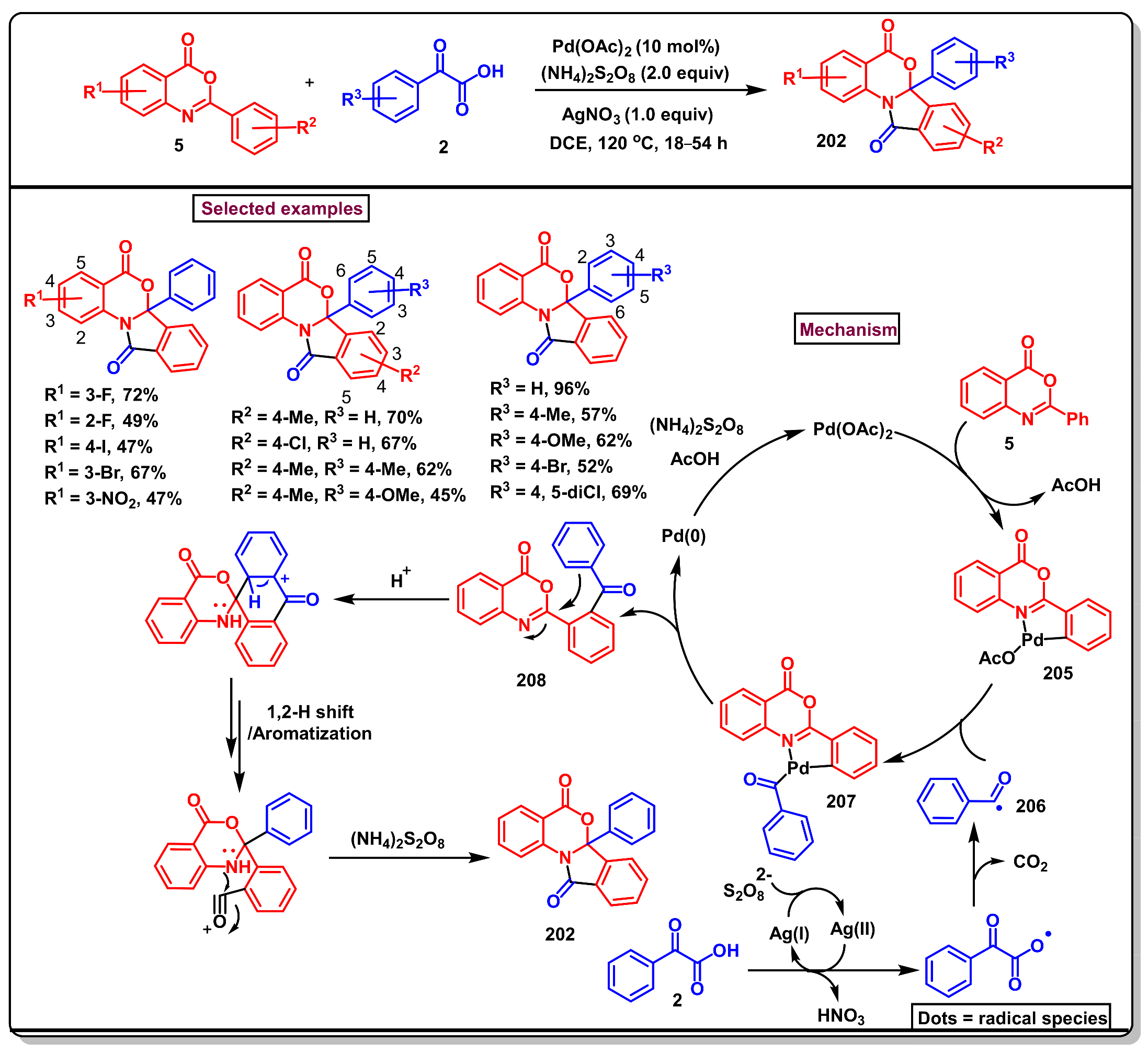
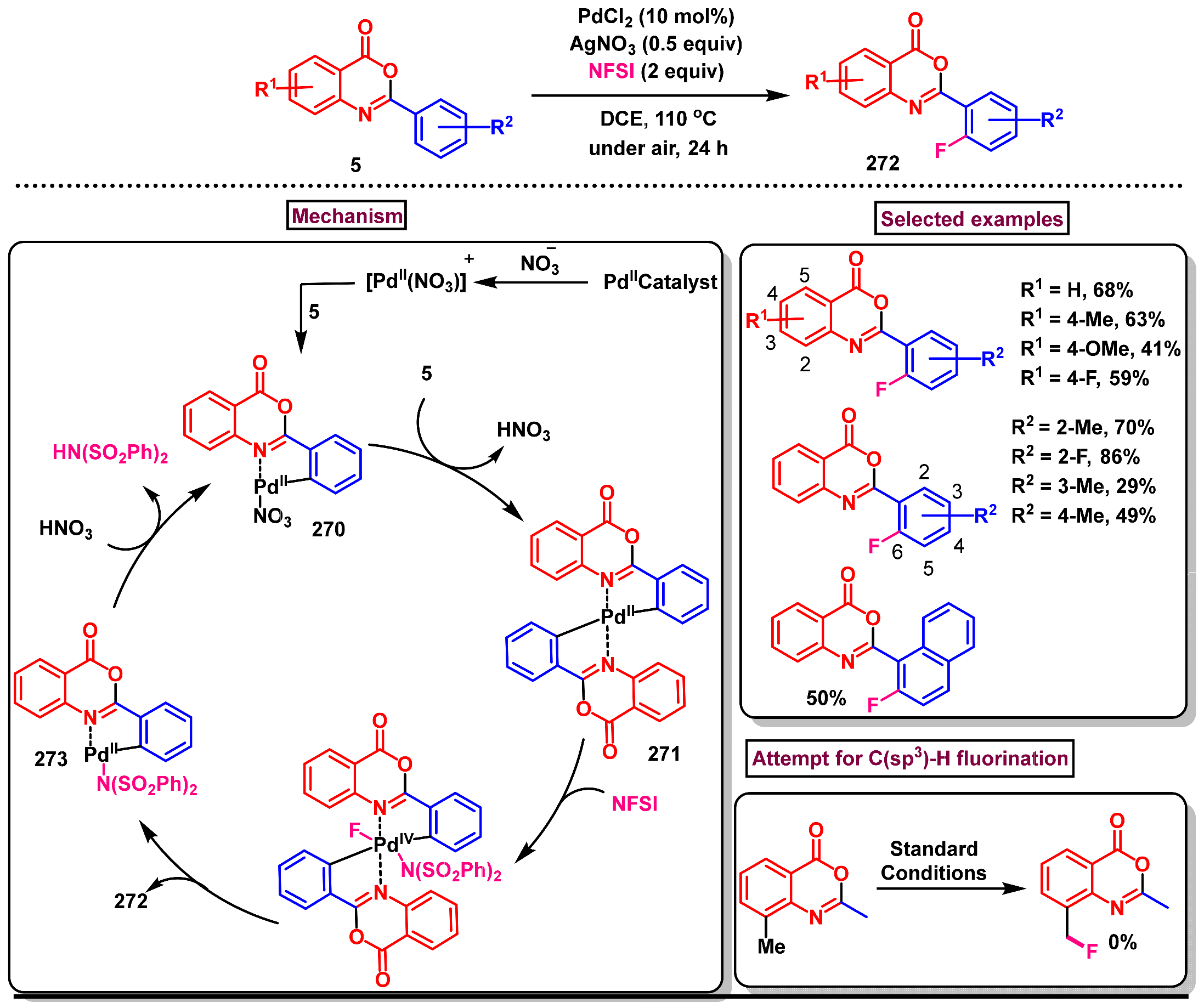
11.1. Pd-Catalyzed C(sp2)–H Monofluorination of 2-Arylbenzo[d]oxazin-4-one
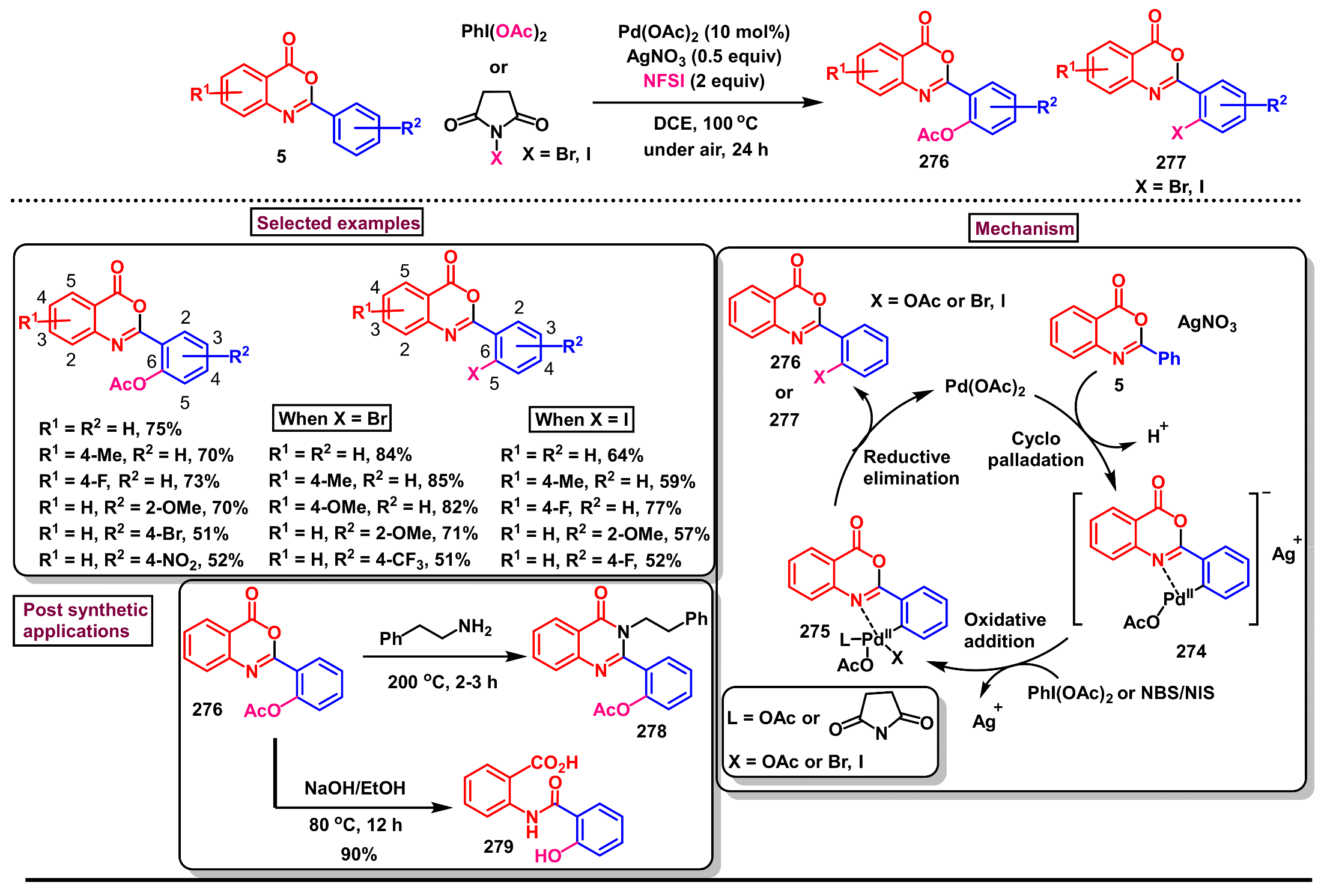
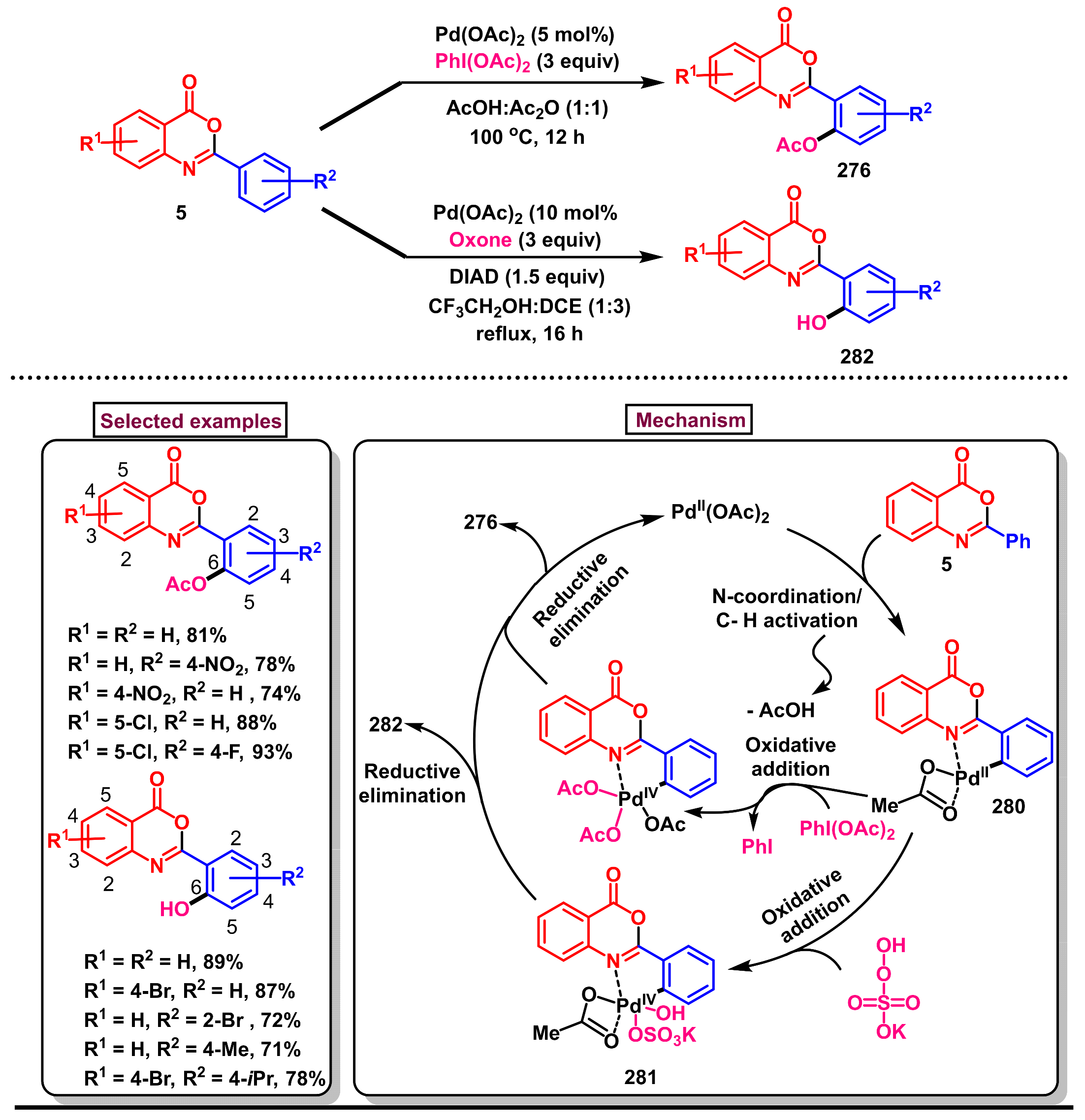
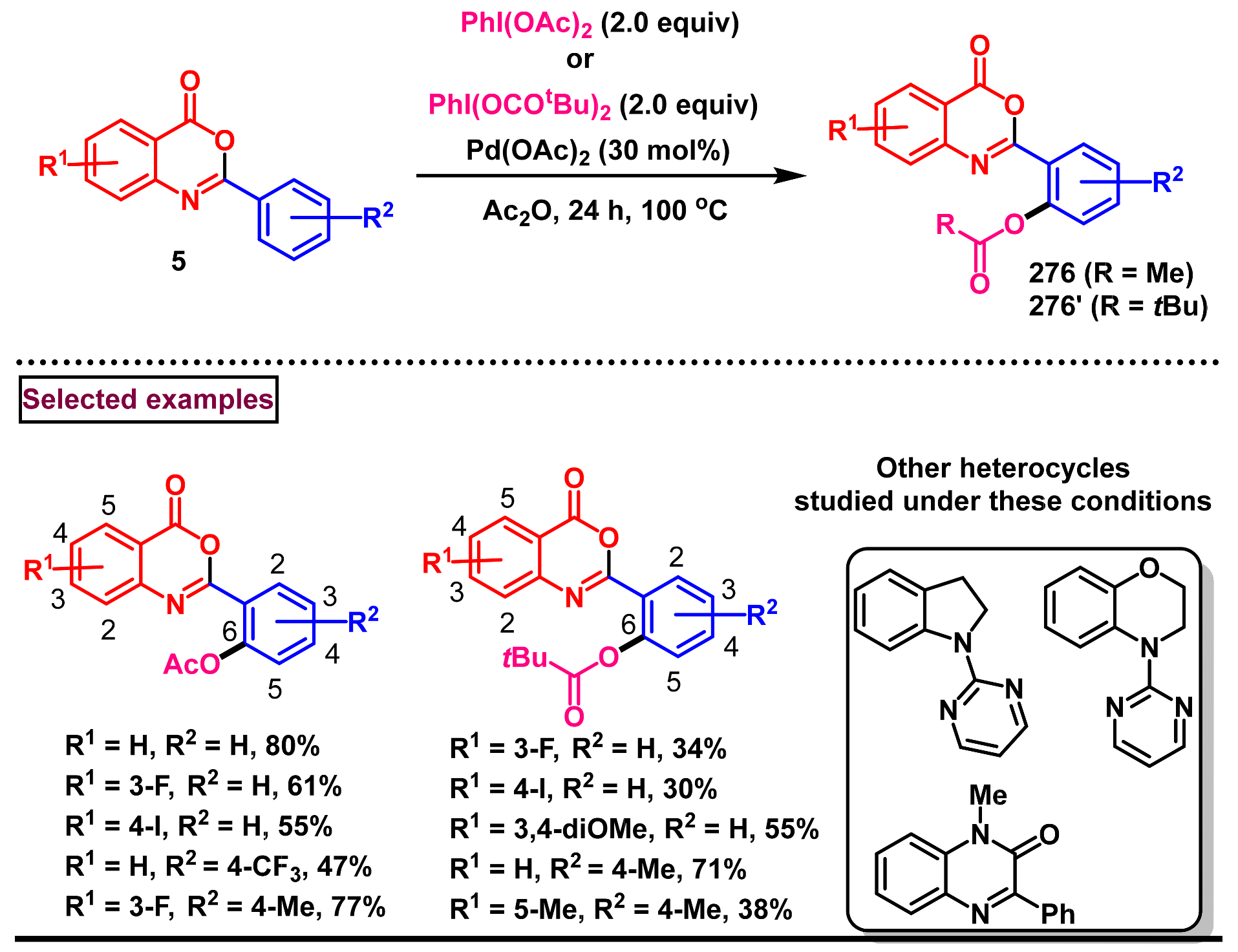
11.2. Pd-Catalyzed Selective Acetoxylation, Halogenation and Hydroxylation of 2-Arylbenzo[d]oxazin-4-ones
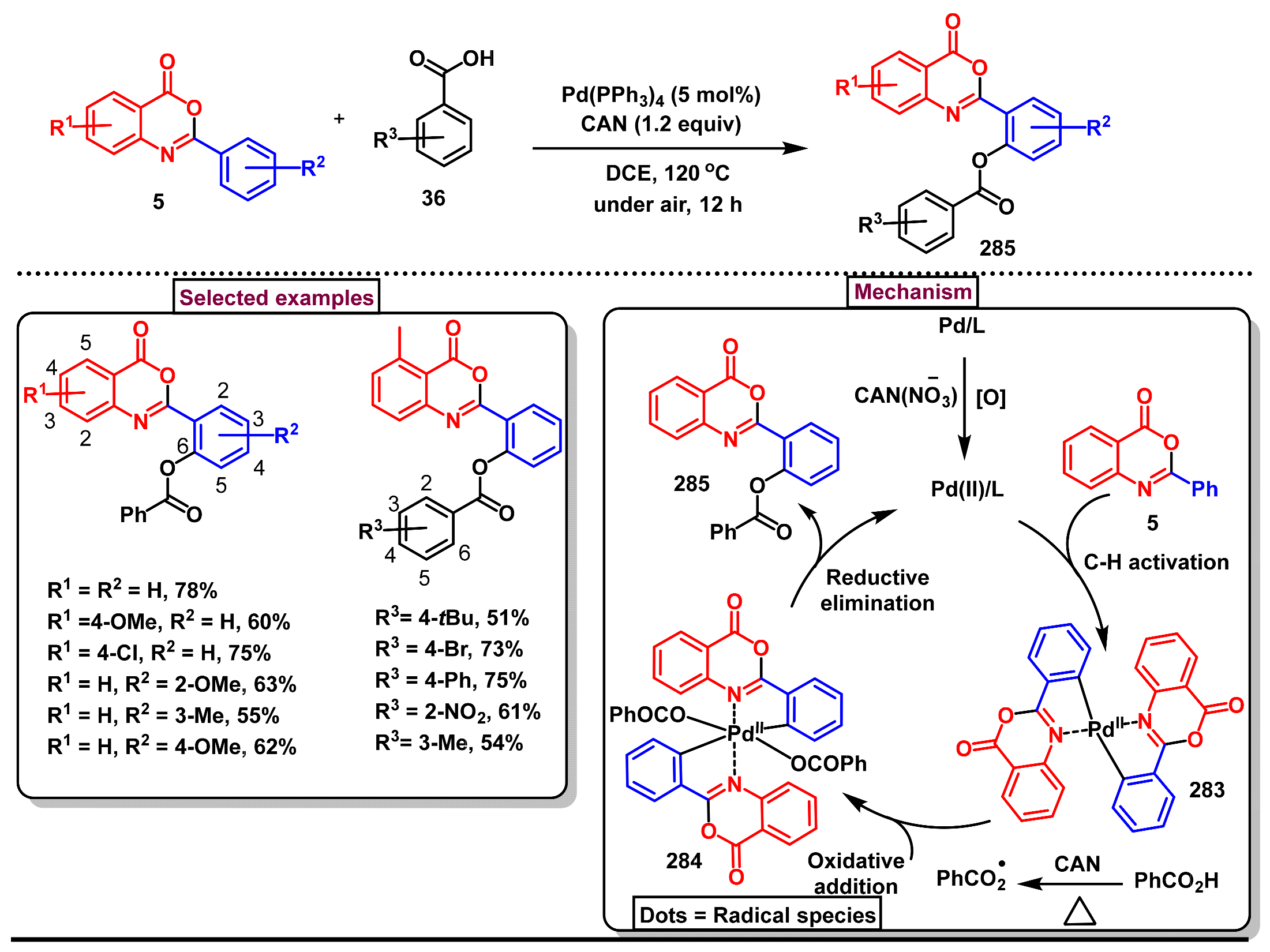
11.3. Pd-Catalyzed Selective Benzoxylation of 2-Arylbenzo[d]oxazin-4-ones
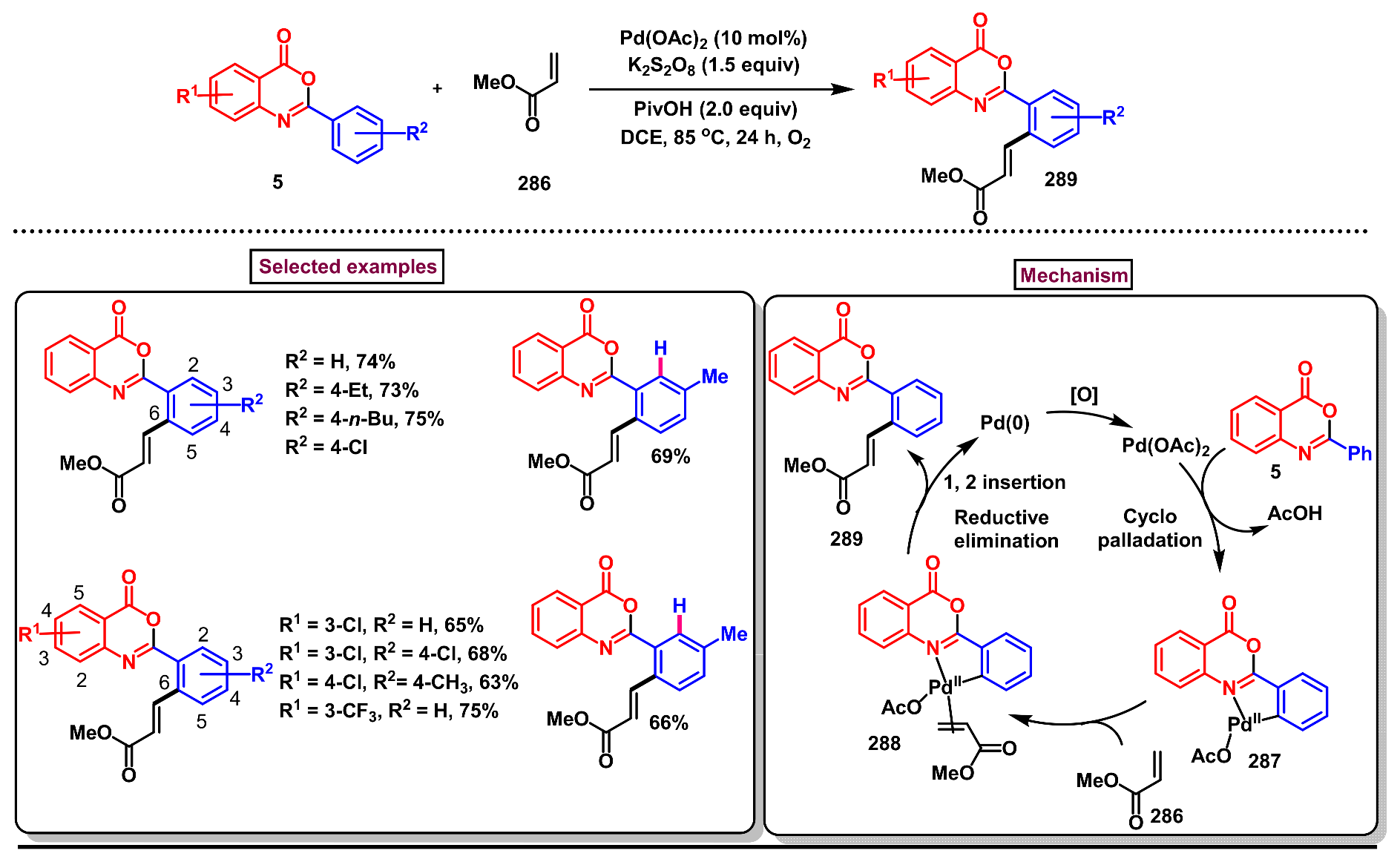
11.4. Pd-Catalyzed Selective Olefination of 2-Arylbenzo[d]oxazin-4-ones
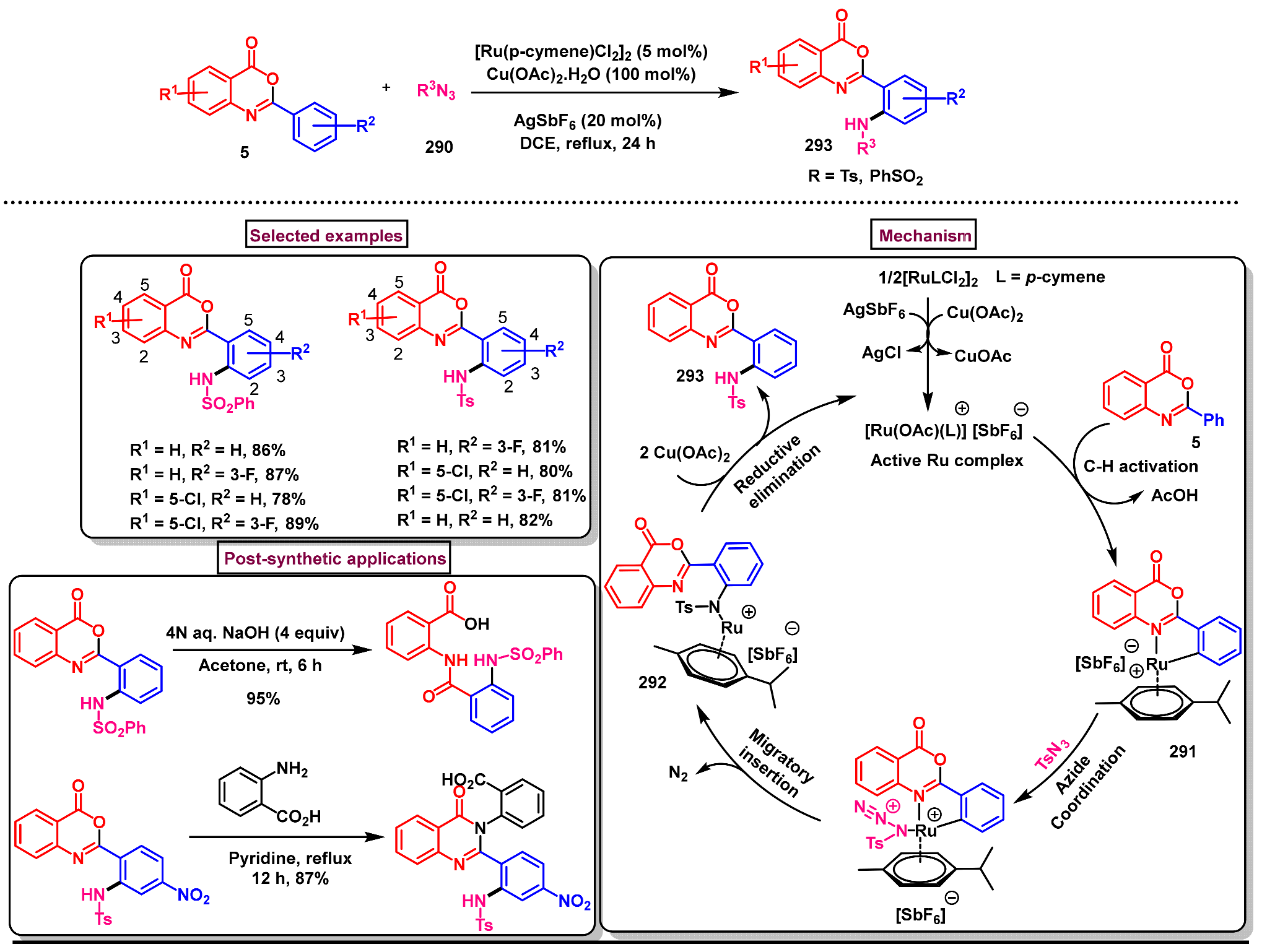
11.5. Pd-Catalyzed Selective Amidation of 2-Arylbenzo[d]oxazin-4-ones
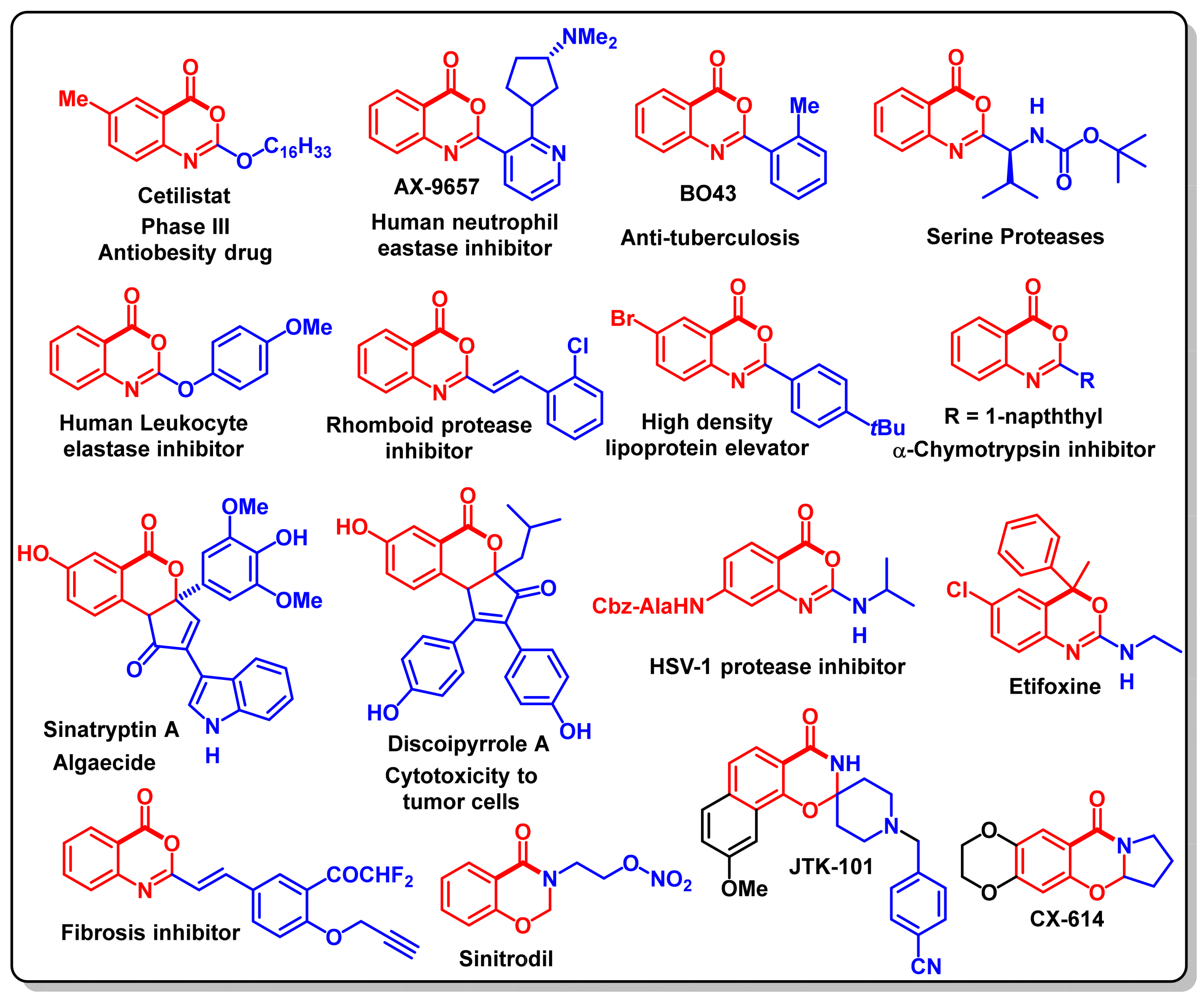
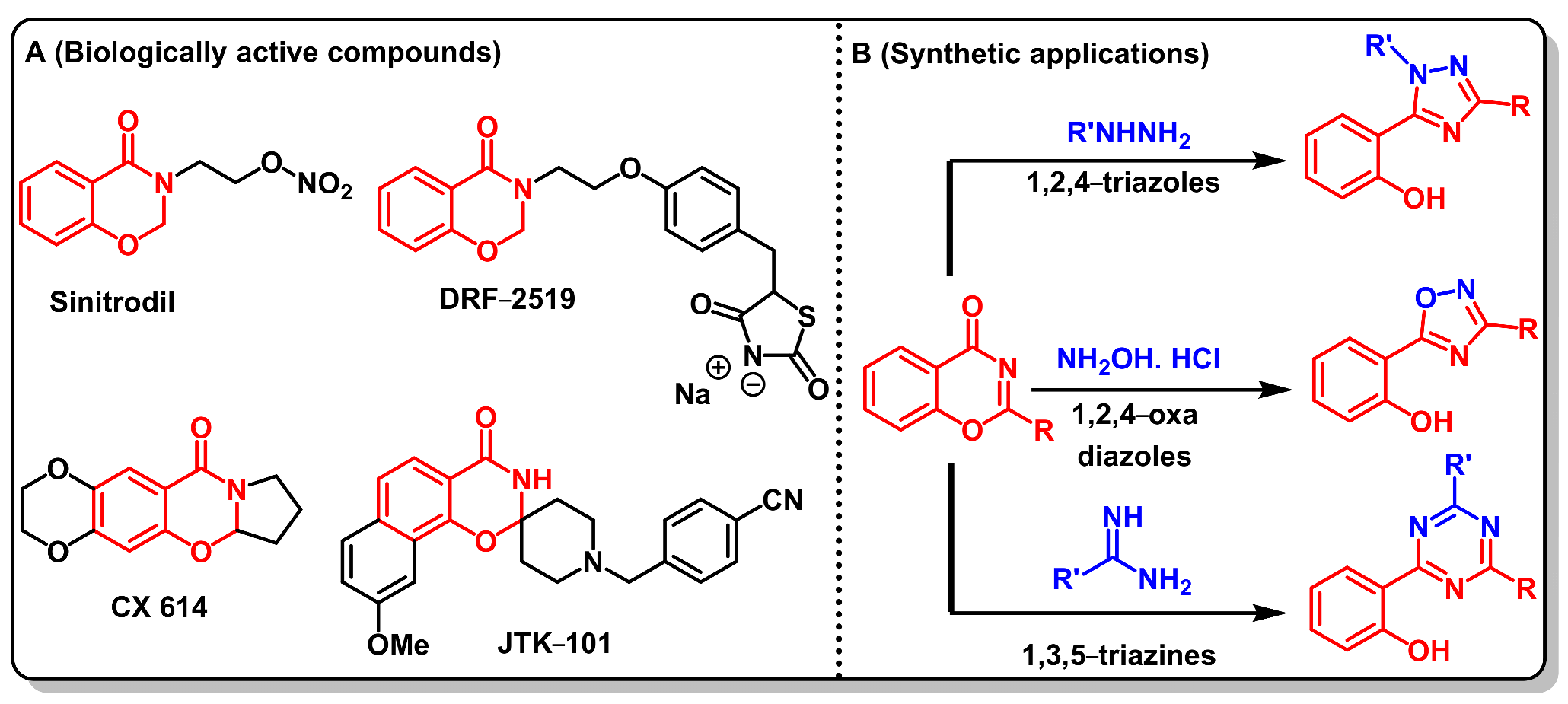
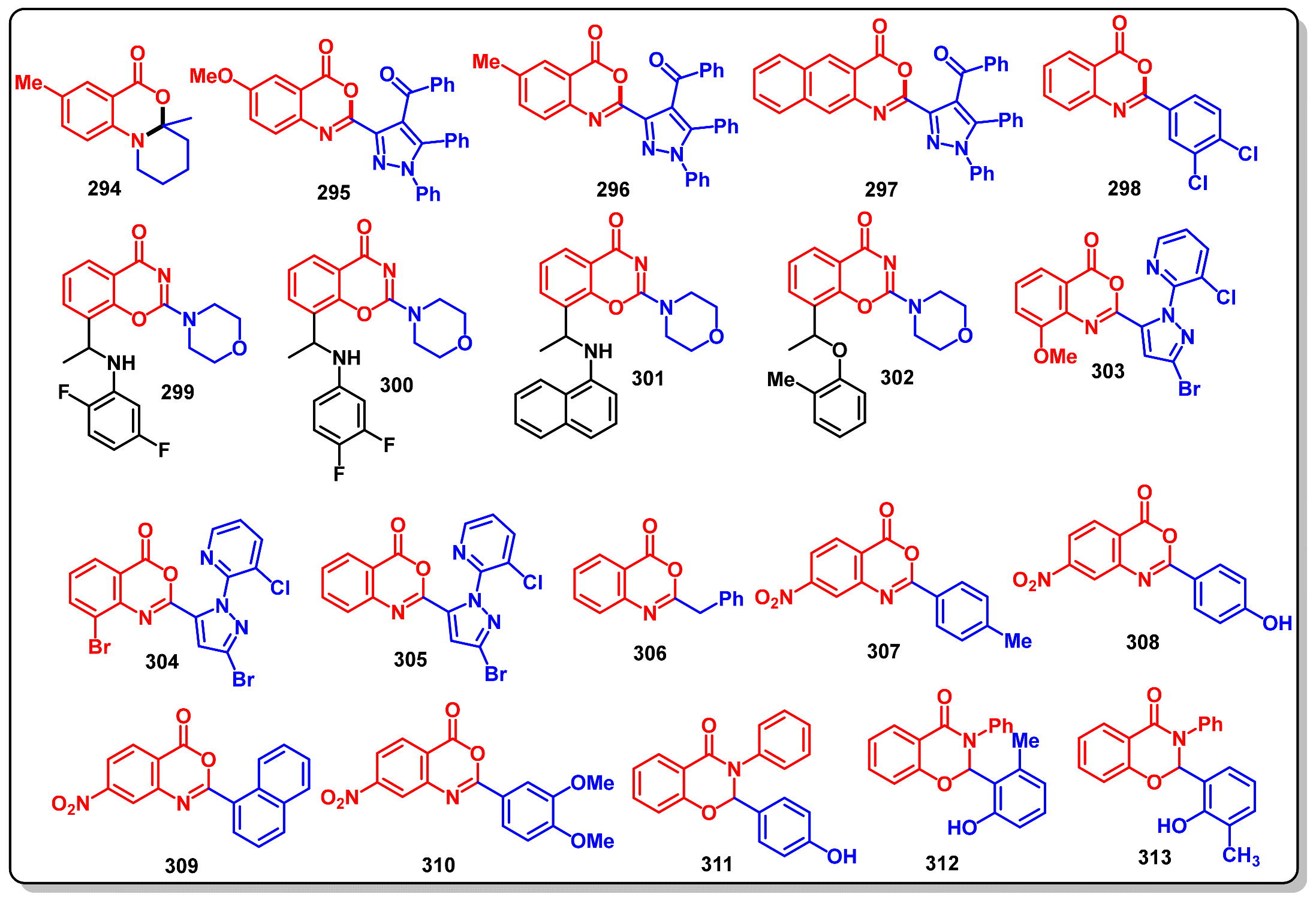
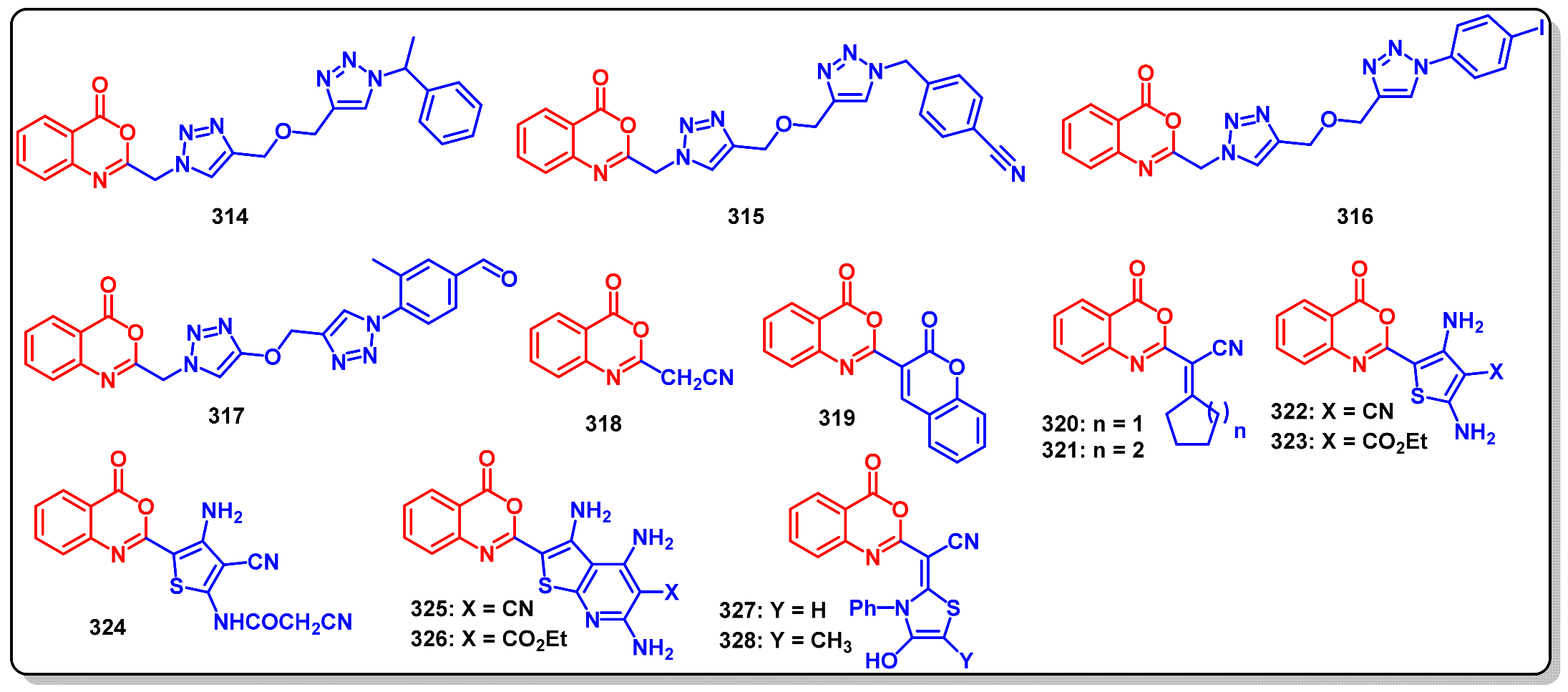
12. Biological Significance of Benzo[d]-Oxazin-4-Ones
Anticancer Activity of Benzo [1,3]oxazin-4-ones
13. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wei, L.; Min, L.-J.; Han, L.; Liu, X.-H. Recent advances on synthesis of 1,4-benzoxazines and its derivatives. Curr. Org. Chem. 2021, 25, 2840–2855. [Google Scholar] [CrossRef]
- Chatterjee, I.; Ali, K.; Panda, G. A Synthetic overview of benzoxazines and benzooxazepines as anticancer agents. ChemMedChem 2023, 18, e202200617. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Tan, Y.; Chen, H.; Wan, Y. Benzoxazine: A privileged scaffold in medicinal chemistry. Curr. Med. Chem. 2023, 30, 372–389. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, A.; Tokarczyk, M.; Weisbrodt, M.; Gziut, K. Adhesive films based on benzoxazine resins and the photoreactive epoxyacrylate Copolymer. Materials 2022, 15, 1839. [Google Scholar] [CrossRef] [PubMed]
- Higginson, C.J.; Malollari, K.G.; Xu, Y.; Kelleghan, A.V.; Ricapito, N.G.; Messersmith, P.B. Bioinspired design provides high-strength benzoxazine structural adhesives. Angew. Chem. Int. Ed. 2019, 58, 12271–12279. [Google Scholar] [CrossRef] [PubMed]
- Klfout, H.A.; Asiri, A.M.; Alamry, K.A.; Hussein, M.A. Recent advances in bio-based polybenzoxazines as an interesting adhesive coating. RSC Adv. 2023, 13, 19817–19835. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Awadasseid, A.; Narva, S.; Cao, S.; Tanaka, Y.; Wu, Y.; Fu, W.; Zhao, X.; Wei, C.; Zhang, W. Anti-cancer activity of benzoxazinone derivatives via targeting c-Myc G-quadruplex structure. Life Sci. 2020, 258, 118252. [Google Scholar] [CrossRef] [PubMed]
- Gholami, H.R.; Asghari, S.; Mohseni, M. Synthesis, characterization, and evaluation of antibacterial and antioxidant activities of novel benzoxazinones and benzoxathiinones. J. Heterocycl. Chem. 2019, 56, 1505–1513. [Google Scholar] [CrossRef]
- Abood, N.A.; Schretzman, L.A.; Flynn, D.L.; Houseman, K.A.; Wittwer, A.J.; Dilworth, V.M.; Hippenmeyer, P.J.; Holwerda, B.C. Inhibition of human cytomegalovirus protease by benzoxazinones and evidence of antiviral activity in cell culture. Bioorg. Med. Chem. Lett. 1997, 7, 2105–2108. [Google Scholar] [CrossRef]
- Tang, C.; Guo, W.; Yang, S.; Hu, X.; Chen, X.; Wang, X. Design, synthesis and antifungal activity of novel 1,4-benzoxazin-3-one derivatives containing an acylhydrazone moiety. Front Chem. 2023, 11, 1233443. [Google Scholar] [CrossRef]
- Kikuchi, S.; Horoiwa, S.; Kasahara, R.; Hariguchi, N.; Matsumoto, M.; Oshima, Y. Synthesis of febrifugine derivatives and development of an effective and safe tetrahydroquinazoline-type antimalarial. Eur. J. Med. Chem. 2014, 76, 10–19. [Google Scholar] [CrossRef] [PubMed]
- De Simone, A.; Tumiatti, V.; Andrisano, V.; Milelli, A. Glycogen Synthase Kinase 3β: A New Gold Rush in Anti-Alzheimer’s disease multitarget drug discovery? J. Med. Chem. 2021, 64, 26–41. [Google Scholar] [CrossRef] [PubMed]
- Wilder, P., Jr.; Winston, A. An infrared investigation of the stereochemistry of some tricyclic lactones. J. Am. Chem. Soc. 1955, 77, 5598. [Google Scholar] [CrossRef]
- Shehzad, M.T.; Imran, A.; Njateng, G.S.S.; Hameed, A.; Islam, M.; Al-Rashida, M.; Uroos, M.; Asari, A.; Shafiq, Z.; Iqbal, J. Benzoxazinone-thiosemicarbazones as antidiabetic leads via aldose reductase inhibition: Synthesis, biological screening and molecular docking study. Bioorg. Chem. 2019, 87, 857–866. [Google Scholar] [CrossRef]
- Bari, A.; Khan, Z.A.; Shahzad, S.A.; Raza Naqvi, S.A.; Khan, S.A.; Amjad, H.; Iqbal, A.; Yar, M. Design and syntheses of 7-nitro-2-aryl-4H-benzo[d][1,3]oxazin-4-ones as potent anticancer and antioxidant agents. J. Mol. Struct. 2020, 1214, 128252. [Google Scholar] [CrossRef]
- Torrens, A.; Mas, J.; Port, A.; Castrillo, J.A.; Sanfeliu, O.; Guitart, X.; Dordal, A.; Romero, G.; Fisas, M.A.; Sánchez, E.; et al. Synthesis of new benzoxazinone derivatives as neuropeptide Y5 antagonists for the treatment of obesity. J. Med. Chem. 2005, 48, 2080–2092. [Google Scholar] [CrossRef]
- Sengupta, S.; Reddy, J.R.; Rajesh, N.; Jaiswal, A.; Mabalirajan, U.; Palakodety, R.K.; Mukherjee, P.; Bandyopadhyay, A. Novel benzoxazinone derivative as potent human neutrophil elastase inhibitor: Potential implications in lung injury. Eur. J. Pharmacol. 2022, 931, 175187. [Google Scholar] [CrossRef]
- Gilmore, J.L.; Hays, S.J.; Caprathe, B.W.; Lee, C.; Emmerling, M.R.; Michael, W.; Jaen, J.C. Synthesis and evaluation of 2-aryl-4H-3,1-benzoxazin-4-ones as C1r serine protease inhibitors. Bioorg. Med. Chem. Lett. 1996, 6, 679–682. [Google Scholar] [CrossRef]
- Bryson, A.; De la Motte, S.; Dunk, C. Reduction of dietary fat absorption by the novel gastrointestinal lipase inhibitor cetilistat in healthy volunteers. Br. J. Clin. Pharmacol. 2009, 67, 309–315. [Google Scholar] [CrossRef]
- Padwal, R. Cetilistat, a new lipase inhibitor for the treatment of obesity. Curr. Opin. Investig. Drugs. 2008, 9, 414–421. [Google Scholar]
- Von Nussbaum, F.; Li, V.M. Netutrophil elastase inhibitors for the treatment of (cardio) pulmonary diseases: Into clinical testing with pre-adaptive pharmacophores. Chem. Lett. 2015, 25, 4370–4381. [Google Scholar] [CrossRef]
- Park, J.-D.; Li, Y.; Moon, K.; Han, E.J.; Lee, S.R.; Seyedsayamdost, M.R. Structural elucidation of cryptic algaecides in marine algal-bacterial symbioses by NMR spectroscopy and MicroED. Angew. Chem. Int. Ed. 2022, 61, e202114022. [Google Scholar] [CrossRef]
- Hu, Y.; Potts, M.B.; Colosimo, D.; Herrera-Herrera, M.L.; Legako, A.G.; Yousufuddin, M.; White, M.A.; MacMillan, J.B. Discoipyrroles A–D: Isolation, structure determination, and synthesis of potent migration inhibitors from Bacillus hunanensis. J. Am. Chem. Soc. 2013, 135, 13387–13392. [Google Scholar] [CrossRef] [PubMed]
- Dake, G.G.; Kaliappan, K.P. Total syntheses of Discoipyrroles A, B, and C, three marine natural products. J. Org. Chem. 2024, 89, 5825–5832. [Google Scholar] [CrossRef] [PubMed]
- Sahu, N.K.; Priyanka; Mahajan, A.T.; Sharma, V.; Suhas, K.P.; Tripathi, P.; Mathur, M.; Jain, M.; Chaudhary, S.C. ‘Cephalandole A’ analogues as a new class of antioxidant agents: Design, microwave-assisted synthesis, bioevaluation, SAR and in silico studies. J. Mol. Struct. 2024, 1303, 137445. [Google Scholar] [CrossRef]
- Luo, C.; Ai, J.; Ren, E.; Li, J.; Feng, C.; Li, X.; Luo, X. Research progress on evodiamine, a bioactive alkaloid of Evodiae fructus: Focus on its anti-cancer activity and bioavailability (Review). Exp Ther Med. 2021, 22, 1327. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Xiong, Y.; Peng, Y.; Zhang, X.; Li, S.; Peng, Y.; Peng, X.; Zhuo, L.; Jiang, W. Natural product evodiamine-inspired medicinal chemistry: Anticancer activity, structural optimization and structure-activity relationship. Eur. J. Med. Chem. 2023, 247, 115031. [Google Scholar] [CrossRef]
- Iwashita, K.; Hosokawa, Y.; Ihara, R.; Miamoto, T.; Otani, M.; Abe, J.; Asano, K.; Mercier, O.; Miyata, K.; Barlow, S. Flumioxazin, a PPO inhibitor: A weight-of-evidence consideration of its mode of action as a developmental toxicant in the rat and its relevance to humans. Toxicology 2022, 472, 153160. [Google Scholar] [CrossRef]
- El-Din, N.A.E.S. 3,4-dihydro-2H-1,3-benzoxazines and their oxo-derivatives-Chemistry and bioactivities. J. Serb. Chem. Soc. 2021, 86, 213–246. [Google Scholar] [CrossRef]
- El-Mekabaty, A. Chemistry of 4H-3,1-benzoxazin-4-ones. Int. J. Modern Org. Chem. 2013, 2, 81–121. [Google Scholar]
- Coppola, G.M. The chemistry of 4H-3,1-benzoxazin-4-ones. J. Heterocycl. Chem. 1999, 36, 563–588. [Google Scholar] [CrossRef]
- Zhang, R.-L.; Qiu, K.-X.; Gao, H.-F.; Yu, F.; Sun, Z.-W. Advancements in asymmetric catalytic approaches involving benzoxazinone derivatives. Adv. Synth. Catal. 2023, 365, 2767–2788. [Google Scholar] [CrossRef]
- Li, T.-R.; Wang, Y.-N.; Xiao, W.-J.; Lu, L.-Q. Transition-metal-catalyzed cyclization reactions using vinyl and ethynyl benzoxazinones as dipole precursors. Tetrahedron Lett. 2018, 59, 1521–1530. [Google Scholar] [CrossRef]
- El-Hashash, M.A.; El-Naggar, A.M.; El-Bordany, E.A.; Marzouk, M.I.; Nawar, T.M.S. 6-iodo-2-isopropyl-4H-3,1-benzoxazin-4-one as building block in heterocyclic synthesis. Synth. Commun. 2016, 46, 2009–2021. [Google Scholar] [CrossRef]
- Marzouk, M.I.; Farghaly, T.A.; El-Hashash, M.A.; Shaker, S.A.; Hussein, S.M. Transformation of benzoxazinone derivatives to some interesting heterocyclic compounds with expected biological activity. Heterocycles 2015, 91, 1399–1416. [Google Scholar] [CrossRef]
- Maizuru, N.; Inami, T.; Kurahashi, T.; Matsubara, S. Nickel-catalyzed cycloaddition of anthranilic acid derivatives to alkynes. Org. Lett. 2011, 13, 1206–1209. [Google Scholar] [CrossRef]
- Rajesh, N.; Manisha, B.; Ranjith, J.; Krishna, P.R. A metal-free tandem ring-opening/ring-closing strategy for the heterocyclic conversion of benzoxazin-4-ones to oxazolines. RSC Adv. 2016, 6, 6058–6064. [Google Scholar] [CrossRef]
- Banerjee, A.; Santra, S.K.; Mohanta, P.R.; Patel, B.K. Ruthenium (II)-catalyzed Regiosepcific C-H/O-H annulations of directing arenes via weak coordination. Org. Lett. 2015, 17, 5678–5681. [Google Scholar] [CrossRef] [PubMed]
- Baravkar, S.B.; Roy, A.; Gawade, R.L.; Puranik, V.G.; Sanjayan, G.J. Nucleophilic ring-opening of benzoxazinones by DBU: Some observations. Synth. Commun. 2014, 44, 2955–2960. [Google Scholar] [CrossRef]
- Bunk, C.; Komber, H.; Lang, M.; Fribiczer, N.; Geisler, M.; Formanek, P.; Jakisch, L.; Seiffert, S.; Voit, B.; Böhme, F. Amphiphilic tetra-PCL-b-PEG star block copolymers using benzoxazinone-based linking groups. Polym. Chem. 2023, 14, 1965–1977. [Google Scholar] [CrossRef]
- El-Badry, Y.; Mahamoud, K.H. Optical study of a static benzoxazinone derivative doped poly (vinyl) pyrrolidone-poly(vinyl) alcohol blend system. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2019, 219, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Nayak, M.K.; Kim, B.-H.; Kwon, J.E.; Park, S.; Seo, J.; Chung, J.W.; Park, S.Y. Gelation-induced enhanced fluorescence emission from organogels of salicylanilide-containing compounds exhibiting excited-state intramolecular proton transfer: Synthesis and self-assembly. Chem. Eur. J. 2010, 16, 7437–7447. [Google Scholar] [CrossRef] [PubMed]
- Macías, F.A.; Marín, D.; Oliveros-Bastidas, A.; Molinillo, J.M. Optimization of benzoxazinones as natural herbicide models by lipophilicity enhancement. J. Agric. Food Chem. 2006, 54, 9357–9365. [Google Scholar] [CrossRef] [PubMed]
- Youssef, Y.M.; El-Sayed, A.A.; Azab, M.E. Utility of benzoxazine-4-one and 3-aminoquinazolin-4-one derivatives as precursors for construction of potent insecticidal heterocycles. J. Heterocycl. Chem. 2019, 56, 2889–2901. [Google Scholar] [CrossRef]
- Heller, G.; Fiesselmann, G. Ueber die Einwirkung von Formaldehyd auf Anthranilsäure. Justus Liebigs Ann. Chem. 1902, 324, 118–137. [Google Scholar] [CrossRef]
- Yan, J.; Yang, Z.; Guo, J.; Xue, Q.; Chen, P. Efficient synthesis of 2-aryl-4H-benzo[d][1,3]oxazin-4-ones by copper-catalyzed decarboxylation of α-keto acids with anthranilic acids. Asian J. Org. Chem. 2024, 13, e202400157. [Google Scholar] [CrossRef]
- Annor-Gyamfi, J.K.; Bunce, R.A. 4H-Benzo[d][1,3]oxazin-4-ones and dihydro analogs from substituted anthranilic acids and ortho esters. Molecules 2019, 24, 3555. [Google Scholar] [CrossRef]
- Qiu, G.; Ding, Q.; Wu, J. Recent advances in isocyanide insertion chemistry. Chem. Soc. Rev. 2013, 42, 5257–5269. [Google Scholar] [CrossRef]
- Wang, H.-X.; Wei, T.-Q.; Xu, P.; Wang, S.-Y.; Ji, S.-J. I2/TBHP-mediated oxidative coupling of amino-based bisnucleophiles and isocyanides: Access to 2-aminobenzoxazinones, 2-aminobenzoxazines, and 2-aminoquinazolies under metal-free conditions. J. Org. Chem. 2018, 83, 13491–13497. [Google Scholar] [CrossRef]
- Vlaar, T.; Orru, R.V.A.; Maes, B.U.W.; Ruijter, E. Palladium-catalyzed synthesis of 2-aminobenzoxazinones by aerobic oxidative coupling of anthranilic acids and isocyanides. J. Org. Chem. 2013, 78, 10469–10475. [Google Scholar] [CrossRef]
- Shariat, M.; Samsudin, M.W.; Zakaria, Z. One-pot synthesis of 2-substituted 4H-3,1-benzoxazin-4-one derivatives under mild conditions using iminium cation from cyanuric chloride/dimethylformamide as a cyclizing agent. Chem. Cent. J. 2013, 7, 58. [Google Scholar] [CrossRef] [PubMed]
- Munusamy, S.; Muralidharan, V.P.; Iyer, S.K. Direct oxidative cascade cyclization of 2-aminobenzoic acid and arylaldehydes to aryl 4H-3,1-benzoxazin-4-ones with oxone. Tetrahedron Lett. 2017, 58, 520–523. [Google Scholar] [CrossRef]
- Azarifar, D.; Sheikh, D. An efficient and facile ultrasonic-accelerated one-pot synthesis of N-acetyl-2-aryl-1,2-dihydro-(4H)-3,1-benzoxazin-4-ones. Heteroat. Chem. 2011, 22, 106. [Google Scholar] [CrossRef]
- Grande, F.; Brizzi, A.; Garofalo, A.; Aiello, F. Active manganese dioxide promoted cyclization of ortho-(1H-pyrrol-1-yl)aryl and heteroaryl carboxylic acids to 5H-pyrrolo [1,2-a][1,3]benzoxazin-5-one derivatives. Tetrahedron 2013, 69, 9951–9956. [Google Scholar] [CrossRef]
- Abe, M.; Takano, M.; Mizukami, A.; Kimachi, T.; Inamoto, K. Gold-catalyzed heteroannulation of anthranilic acids with alkynes: Synthesis of 3,1-benzoxazin-4-ones. J. Org. Chem. 2024, 89, 12651–12657. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.-Y.; Zhang, Y.; Li, X.-C.; Zi, J.; Guo, X.-X. Pd-catalyzed intramolecular chemoselective C(sp2)-H and C(sp3)-H activation of N-alkyl-N-arylanthranilic acids. Org. Lett. 2019, 21, 989–992. [Google Scholar] [CrossRef]
- Liu, L.; Du, L.; Zhang-Negrerie, D.; Du, Y. NIS-Mediated intramolecular oxidative α-functionalization of tertiary amines: Transition metal-free synthesis of 1,2-dihydro-(4H)-3,1-benzoxazin-4-one derivatives. RSC Adv. 2015, 5, 29774–29781. [Google Scholar] [CrossRef]
- Zhang, N.; Cheng, R.; Zhang-Negrerie, D.; Du, Y.; Zhao, K. Hypervalent iodine-mediated oxygenation of N, N-diaryl tertiary amines: Intramolecular functionalization of sp3 C-H bonds adjacent to nitrogen. J. Org. Chem. 2014, 79, 10581–10587. [Google Scholar] [CrossRef]
- Li, J.; Zhang, S.; Lonka, M.R.; Zhang, J.; Zou, H. Rhodium(III)-catalyzed cascade reactions of benzoic acids with dioxazolones: Discovery of 2,5-substituted benzoxazinones as AIE molecules. Chem. Commun. 2019, 55, 11203–11206. [Google Scholar] [CrossRef]
- Jiang, J.; Cai, X.; Hu, Y.; Liu, X.; Chen, X.; Wang, S.-Y.; Zhang, Y.; Zhang, S. Thermo-promoted reactions of anthranils with carboxylic acids, amines, phenols, and malononitrile under catalyst-free conditions. J. Org. Chem. 2019, 84, 2022–2031. [Google Scholar] [CrossRef]
- Munusamy, S.; Venkatesan, S.; Sathiyanarayanan, K.I. Copeer catalyzed C-N bond formation/C-H activation: Synthesis of aryl 4H-3,1-benzoxazin-4-ones. Tetrahedron Lett. 2015, 56, 203–205. [Google Scholar] [CrossRef]
- Kobayashi, K.; Hashimoto, H.; Matsumoto, M.; Inouchi, H. A facile synthesis of 2-[1-(dialkylamino)alkyl]-4H-3,1-benzoxazin-4-ones by the reaction of 1,1-dimethylethyl 2-isocyanobenzoates with N, N-dialkyliminium iodides. Tetrahedron 2014, 70, 6398–6401. [Google Scholar] [CrossRef]
- Waseem, M.A.; Srivastava, A.; Srivastava, A.; Siddiqui, R.I.R. [bmIm]OH: An efficient basic catalyst for the synthesis of 4H-benzo[d][1,3]-oxazin-4-one derivatives in solvent-free conditions. Tetrahedron Lett. 2014, 55, 6072–6076. [Google Scholar] [CrossRef]
- Ansari, A.J.; Pathare, R.S.; Maurya, A.K.; Agnihotri, V.K.; Khan, S.; Roy, T.K.; Sawant, D.M.; Pardasani, R.T. Synthesis of diverse N-heterocycles via Pd-catalyzed tandem azide-isocyanide cross-coupling/cyclization: Mechanistic insight using experimental and theoritical studies. Adv. Synth. Catal. 2018, 360, 290–297. [Google Scholar] [CrossRef]
- Pedrood, K.; Montazer, M.N.; Larijani, B.; Mahdavi, M. Recent Advances in the Synthesis of Heterocycles by the Aza-Wittig Reaction. Synthesis 2021, 53, 2342–2366. [Google Scholar] [CrossRef]
- Pertejo, P.; Corres, N.; Torroba, T.; Garcia-Valverde, M. Reversal of diastereoselectivity in the synthesis of peptidomimetic 3-carboxamide-1,4-benzodiazepin-5-ones. Org. Lett. 2015, 17, 612–615. [Google Scholar] [CrossRef] [PubMed]
- Eguchi, S.; Matsushita, Y.; Yamashita, K. The aza-wittig reaction in heterocyclic synthesis. A review. Org. Prep. Proced. Int. 1992, 24, 209–243. [Google Scholar] [CrossRef]
- Wang, L.; Xie, Y.-B.; Huang, N.-Y.; Yan, J.-Y.; Hu, W.-M.; Liu, M.-G.; Ding, M.-W. Catalytic aza-Wittig reaction of acid anhydride for the synthesis of 4H-benzo[d][1,3]oxazin-4-ones and 4-benzylidene-2-aryloxazol-5(4H)-ones. ACS Catal. 2016, 6, 4010–4016. [Google Scholar] [CrossRef]
- Voloshkin, V.A.; Kotovshchikov, Y.N.; Latyshev, G.V.; Lukashev, N.V.; Beletskaya, I.P. Annulation-Triggered Denitrogenative Transformations of 2-(5-Iodo-1,2,3-triazolyl)benzoic acids. J. Org. Chem. 2022, 87, 7064–7065. [Google Scholar] [CrossRef]
- Abbas, S.Y.; El-Bayouki, K.A.M.; Basyouni, W.M. Utilization of isatoic anhydride in the syntheses of various types of quinazoline and quinazolinone derivatives. Synth. Commun. 2016, 46, 993–1035. [Google Scholar] [CrossRef]
- Tashrifi, Z.; Mohammadi-Khanaposhtani, M.; Biglar, M.; Larijani, B.; Madhavi, M. Isatoic anhydride: A fascinating and basic molecule for the synthesis of substituted quinazolinones and benzo di/triazepines. Curr. Org. Chem. 2019, 23, 1090–1130. [Google Scholar] [CrossRef]
- Shevkhgeimer, M.G.A. Synthesis of heterocyclic compounds based on isatoic anhydrides (2H-3,1-Benzoxazine-2,4-diones). Chem. Heterocycle. Compd. 2001, 37, 385–443. [Google Scholar] [CrossRef]
- Pattarawarapan, M.; Yamano, D.; Wiriya, N.; Hongsibsong, S.; Phakhodee, W. Direct access to 2-aminobenzoxazinones via Ph3P-I2 mediated deoxygenative amination of isatoic anhydrides with tertiary amines. Eur. J. Org. Chem. 2022, e202201069. [Google Scholar] [CrossRef]
- Yang, M.; Wang, J.; Lv, W.; Ba, D.; Cheng, G.; Wang, L. Synthesis of 2-alkenyl-4H-3,1-benzoxazin-4-ones through HFIP-mediated decarboxylative [4+2]-annulation of isatoic anhydrides with cyclopropenones under silver catalysis. Adv. Synth. Catal. 2021, 363, 1–7. [Google Scholar] [CrossRef]
- Pal, K.; Hoque, A.; Volla, C.M.R. Rh-catalyzed denitrogenative reaction of N-sulfonyl-1,2,3-triazoles with isatoic anhydrides and oxadiazolones. Chem. Eur. J. 2018, 24, 2558–2564. [Google Scholar] [CrossRef]
- Zhao, T.-F.; Xu, X.-L.; Sun, W.-Y.; Lu, Y. Construction of benzoxazinones from anilides and their derivatives. Org. Lett. 2023, 25, 4968–4973. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Kim, Y.; Chang, S. Transition metal-catalyzed C-H amination: Scope, mechanism, and applications. Chem. Rev. 2017, 117, 9247–9301. [Google Scholar] [CrossRef]
- Li, X.; Zhao, Q.; Shen, Y.; Ma, R. Rh(III)-catalyzed C-H diamidation and diamidation/intramolecular cyclization of N-iminopyridinium ylides with dioxazolones. J. Org. Chem. 2022, 87, 3468–3481. [Google Scholar] [CrossRef]
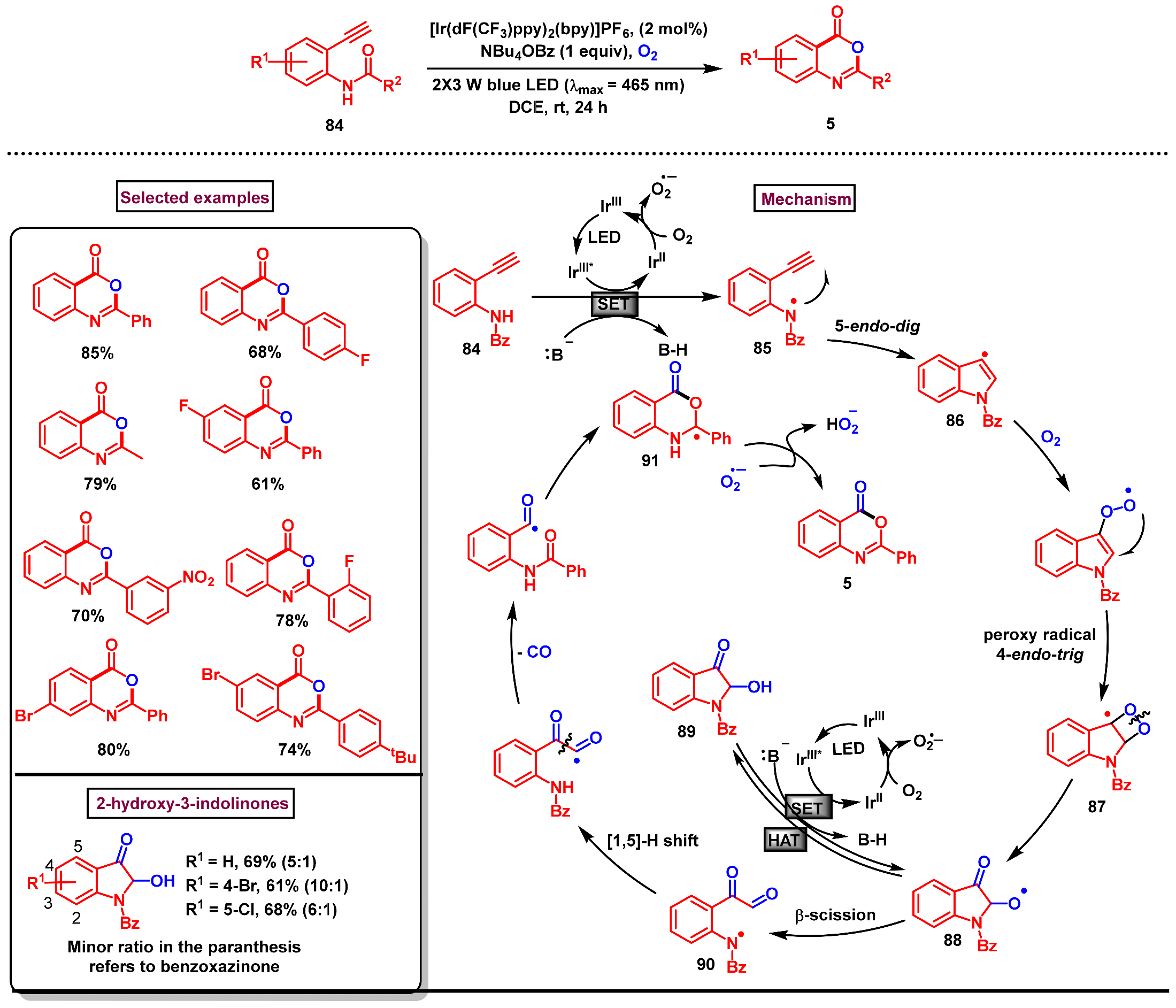
- Tan, T.-D.; Zhai, T.-Y.; Liu, B.-Y.; Li, L.; Qian, P.-C.; Sun, Q.; Zhou, J.-M.; Ye, L.-W. Controllable synthesis of benzoxazinones and 2-hydroxy-3-indolinones by visible-light promoted 5-endo-dig N-radical cyclization cascade. Cell Rep. Phys. Sci. 2021, 2, 100577. [Google Scholar] [CrossRef]
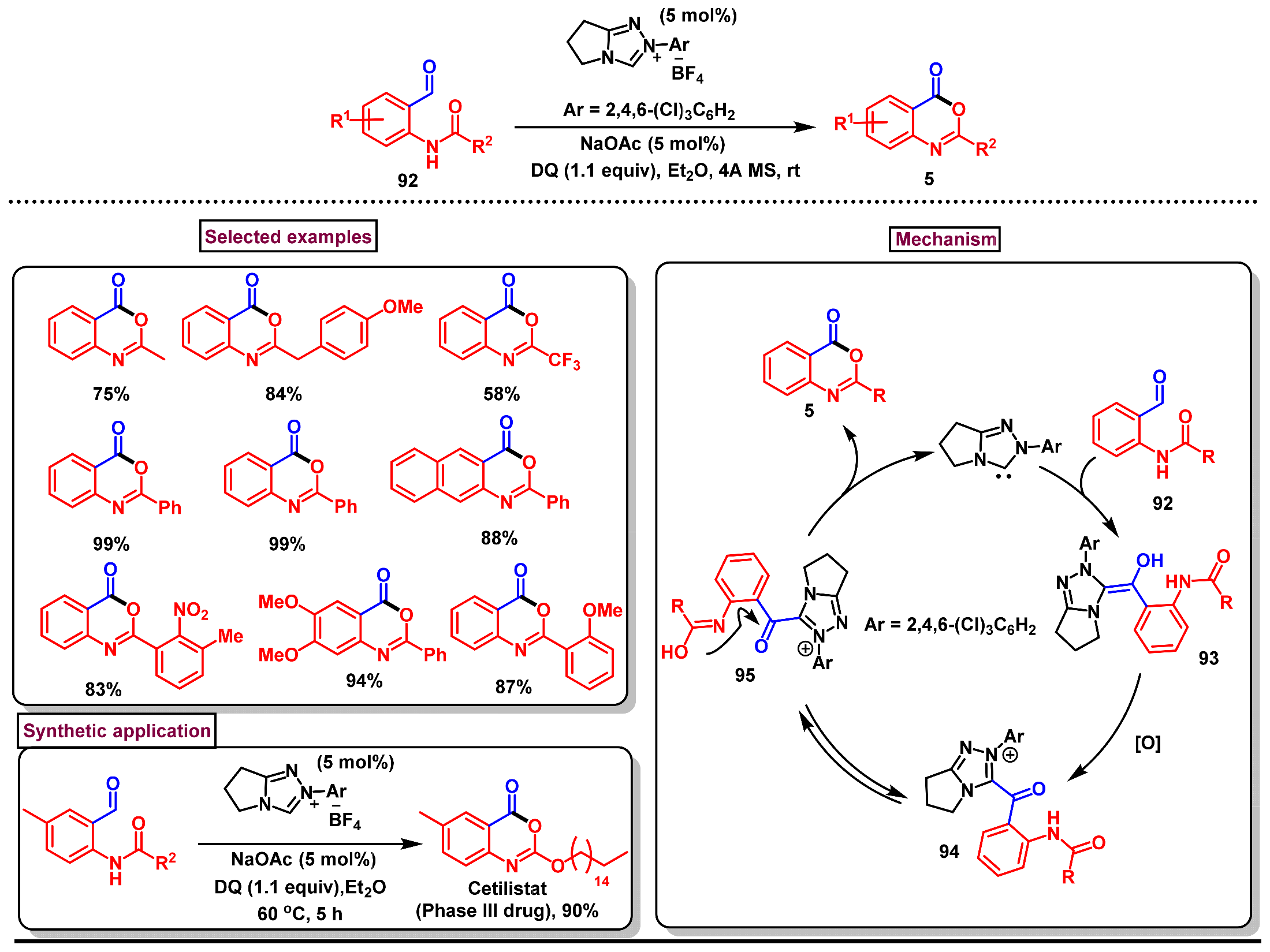
- Lang, M.; Wang, J. Carbene-catalyzed tandem isomerization/cyclization strategy: Efficient assembly of benzoxazinones. Org. Chem. Front. 2019, 6, 1367–1371. [Google Scholar] [CrossRef]
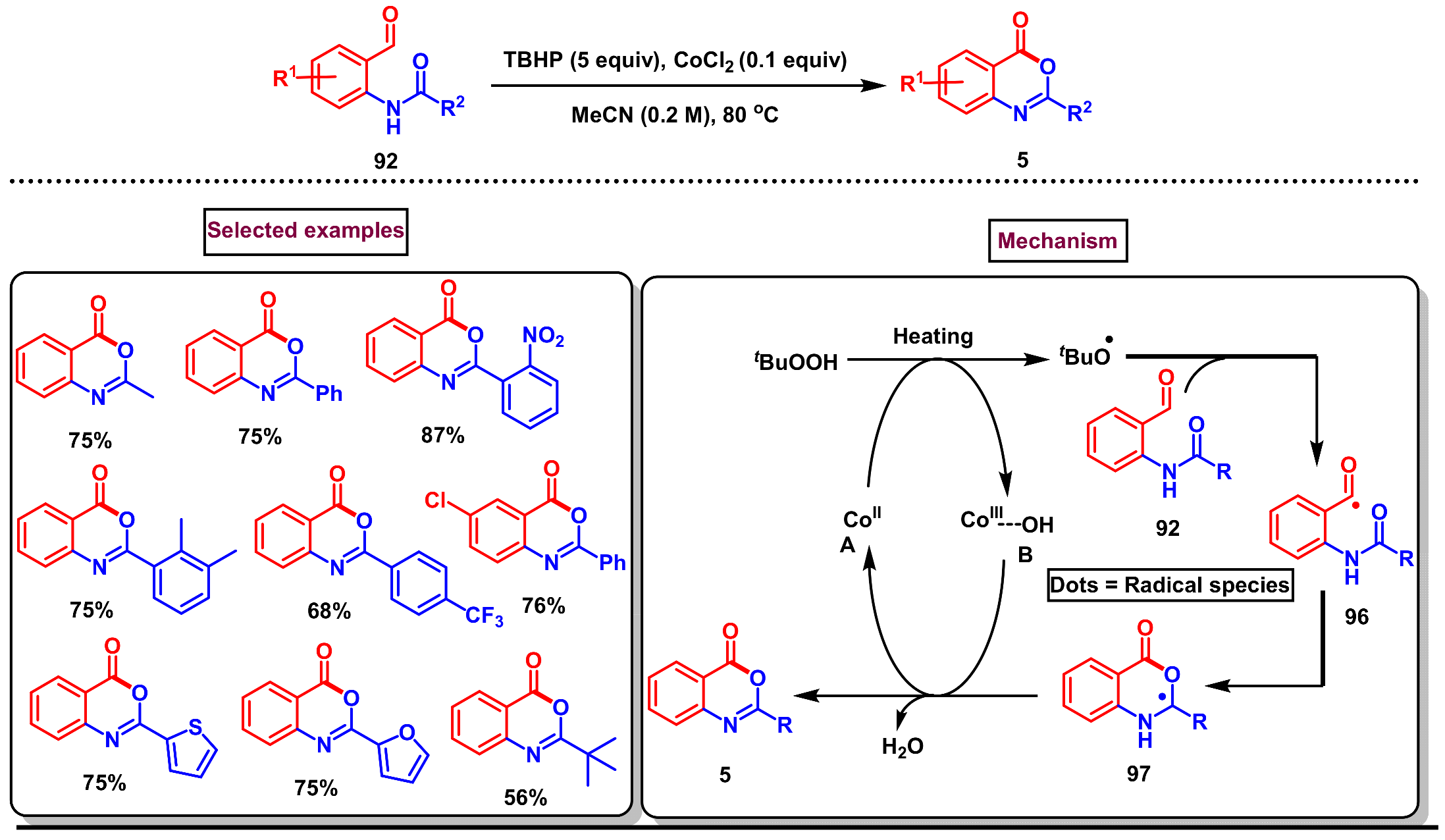
- Yu, J.; Zhang-Negrerie, D.; Du, Y. TBHP/CoCl2-mediated intramolecular oxidative cyclization of N-(2-formylphenyl)amides: An approach to the construction of 4H-3,1-benzoxazin-4-ones. Eur. J. Org. Chem. 2016, 2016, 562–568. [Google Scholar] [CrossRef]
- Bharathimohan, K.; Ponpandian, T.; Jafar, A.A. Silver mediated synthesis of 4H-benzoxazin-4-ones by intramolecular decarboxylative O-acylation reactions with α-oxocarboxylic acid. Eur. J. Org. Chem. 2017, 2017, 2806–2813. [Google Scholar] [CrossRef]
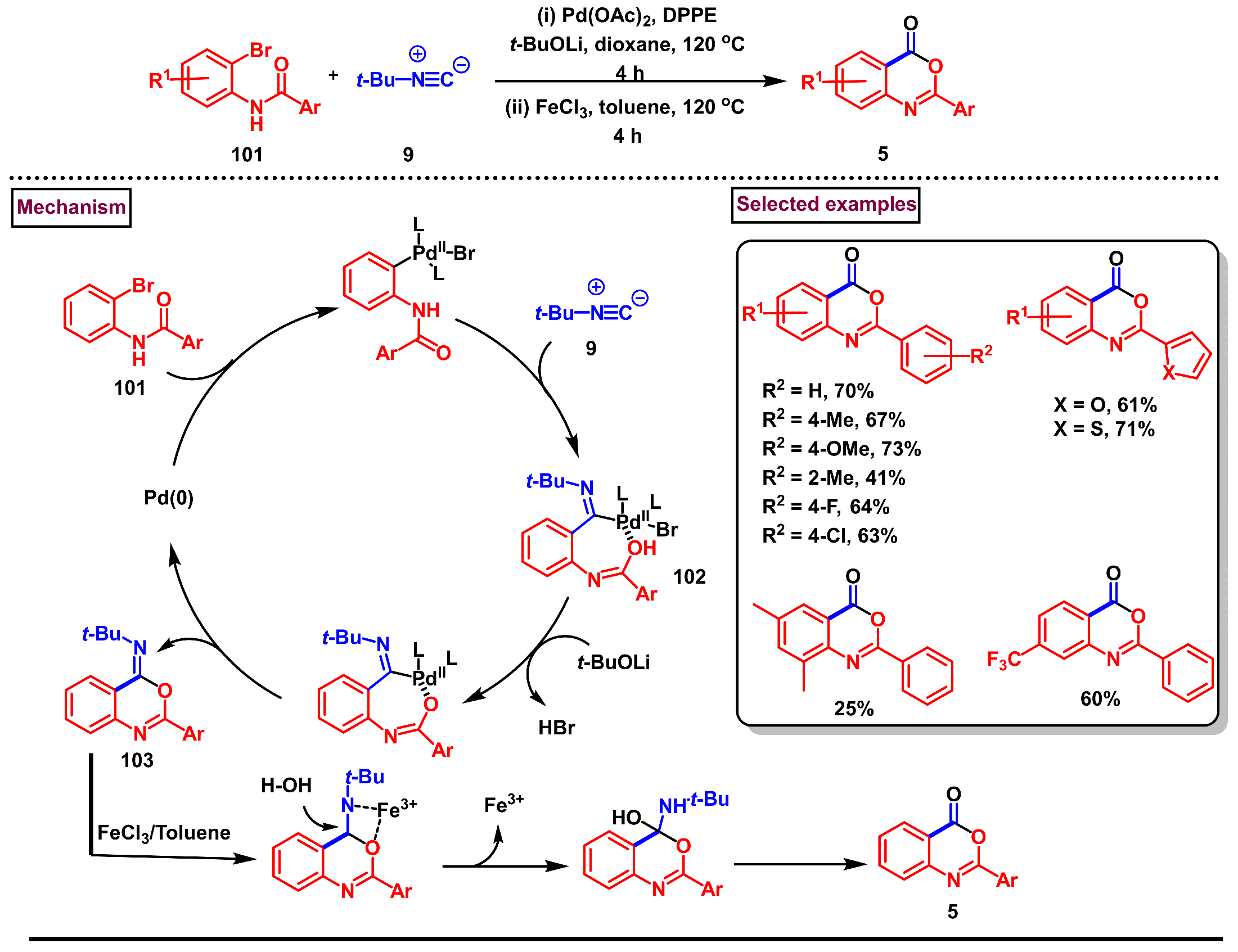
- Wang, J.-M.; Jiang, X.; Zhang, Y.; Zhu, Y.-M.; Shen, J.-K. Palladium-catalyzed synthesis of 4H-benzo[d][1,3]oxazin-4-ones and N-(2-cyanophenyl)benzamides via tert-butyl isocyanide insertion. Tetrahedron Lett. 2015, 56, 2349–2354. [Google Scholar] [CrossRef]
- Verma, A.; Kumar, S. Selective oxidative decarboxylative cleavage of unstrained C(sp3)-C(sp2) bond: Synthesis of substituted benzoxazinones. Org. Lett. 2016, 18, 4388–4391. [Google Scholar] [CrossRef] [PubMed]
- Pattarawarapan, M.; Wet-Osot, S.; Yamano, D.; Phakhodee, W. Mechanochemical synthesis of substituted 4H-3,1-benzoxazin-4-ones, 2-aminobenzoxazin-4-ones, and 2-amino-4H-3,1-benzothiazin-4-ones mediated by 2,4,6-trichloro-1,3,5-triazin and triphenylphosphine. Synlett 2017, 28, 589–592. [Google Scholar] [CrossRef]
- Ge, Z.-Y.; Xu, Q.-M.; Fei, X.-D.; Tang, T.; Zhu, Y.-M.; Ji, S.-J. Copper-catalyzed C-N bond formation/Rearrangement sequence: Synthesis of 4H-3,1-benzoxazin-4-ones. J. Org. Chem. 2013, 78, 4524–4529. [Google Scholar] [CrossRef]
- Giri, R.; Lam, J.K.; Yu, J.-Q. Synthetic applications of Pd(II)-catalyzed C-H carboxylation and mechanistic insights: Expedient routes to anthranilic acids, oxazolinones, and quinazolinones. J. Am. Chem. Soc. 2010, 132, 686–693. [Google Scholar] [CrossRef]
- Liu, X.; An, T.; Yin, Z.; Zhang, W. Palladium-catalyzed reductive double carbonylation of nitroarenes with aryl halides using Mo(CO)6 as a reductant and carbonyl source. Chem. Eur. J. 2023, 29, e202202880. [Google Scholar] [CrossRef]
- Zheng, Y.; Dong, M.; Qu, E.; Bai, J.; Wu, X.-F.; Li, W. Pd-catalyzed carbonylative synthesis of 4H-benzo[d][1,3]-oxazin-4-ones using benzene-1,3,5-triyl triformate as the CO source. Chem. Eur. J. 2021, 27, 16219–16224. [Google Scholar] [CrossRef]
- Xu, Z.; Huang, B.; Zhou, Z.; Cai, M. Recyclable heterogeneous palladium-catalyzed carbonylative cyclization of 2-iodoanilines with aryl iodides leading to 2-aryl-benzoxazinones. Synthesis 2020, 52, 581–590. [Google Scholar] [CrossRef]
- Wu, X.-F.; Schranck, J.; Neumann, H.; Beller, M. A convenient and general palladium-catalyzed carbonylative coupling for the synthesis of 2-arylbenzoxazinones. Chem. Eur. J. 2011, 17, 12246–12249. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yin, Z.; Wang, H.; Wu, X.-F. Pd/C-catalyzed carbonylative synthesis of 2-aminobenzoxazinones from 2-iodoaryl azides and amines. Org. Lett. 2019, 21, 3242–3246. [Google Scholar] [CrossRef] [PubMed]
- Chavan, S.P.; Bhanage, B.M. Carbonylative synthesis of phthalimides and benzoxazinones by using phenyl formate as a carbon monoxide source. Eur. J. Org. Chem. 2015, 2405–2410. [Google Scholar] [CrossRef]
- Li, W.; Wu, X.-F. Palladium-catalyzed carbonylative synthesis of benzoxazinones from N-(o-bromoaryl)amides using paraformaldehyde as the carbonyl source. J. Org. Chem. 2014, 79, 10410–10416. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.-F.; Sharif, M.; Shoaib, K.; Neumann, H.; Pews-Davtyan, A.; Langer, P.; Beller, M. A convenient palladium-catalyzed carbonylative synthesis of 2-aminobenzoxazinones from 2-bromoanilines and isocyanates. Chem. Eur. J. 2013, 19, 6230–6233. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.-F.; Neumann, H.; Beller, M. A general palladium-catalyzed carbonylative synthesis of 2-alkylbenzoxazinones from 2-bromoanilines and acid anhydrides. Chem. Eur. J. 2012, 18, 12599–12602. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Shi, L.; Han, Y.; Xia, C.; Huynh, H.V.; Li, F. Pd-carbene catalyzed carbonylation reactions of aryl iodides. Dalton Trans. 2011, 40, 7632–7638. [Google Scholar] [CrossRef]
- Salvadori, J.; Balducci, E.; Zaza, S.; Petricci, E.; Taddei, M. Microwave-assisted carbonylation and cyclocarbonylation of aryl iodides under ligand free heterogeneous catalysis. J. Org. Chem. 2010, 75, 1841–1847. [Google Scholar] [CrossRef]
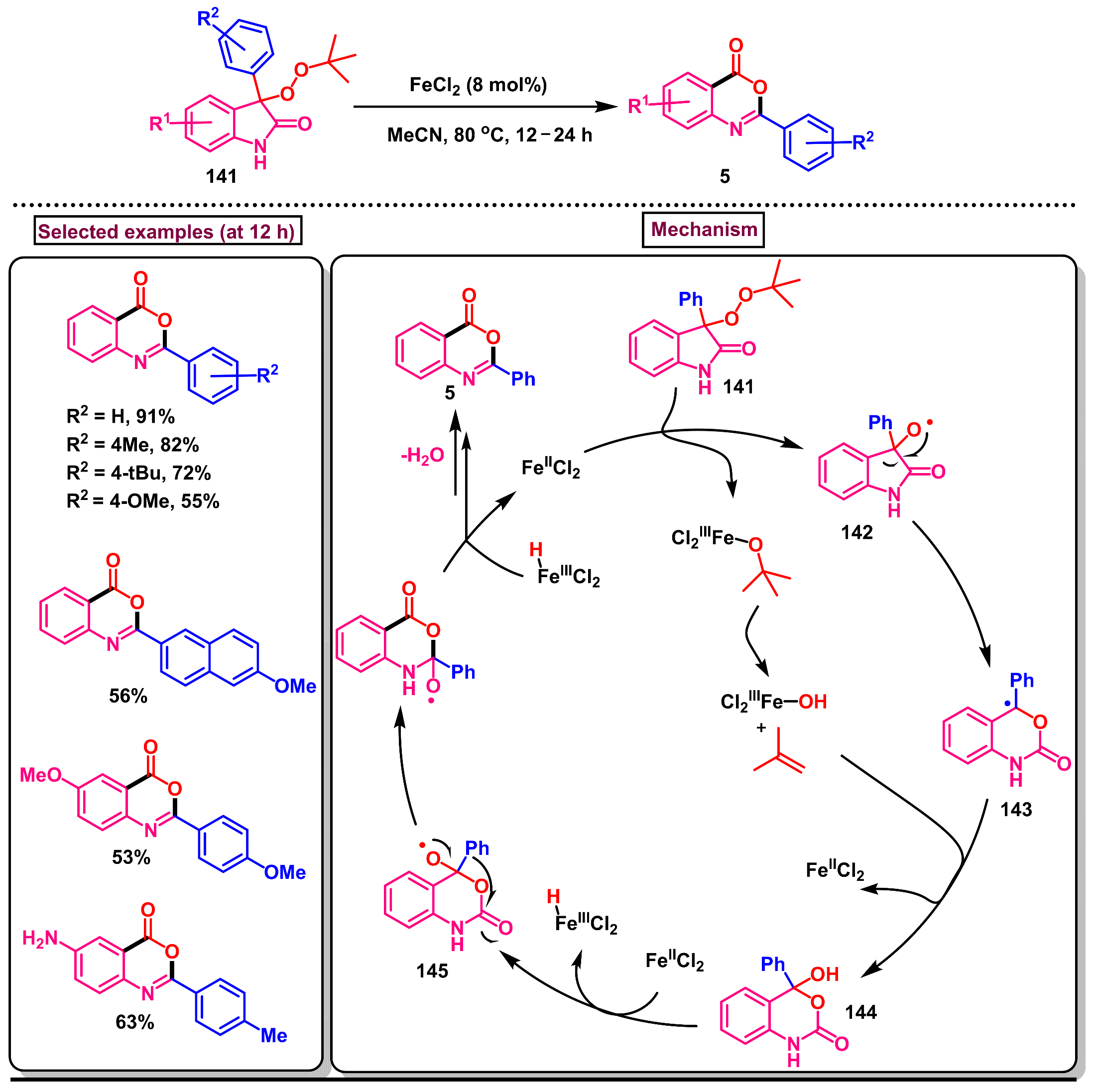
- Mohanta, N.; Samal, P.P.; Krishnamurthy, S.; Gnanaprakasam, B. FeCl2-catalyzed rearrangement of aryl peroxyoxindoles into 1,3-benzoazin-4-one. Adv. Synth. Catal. 2023, 365, 515–521. [Google Scholar] [CrossRef]
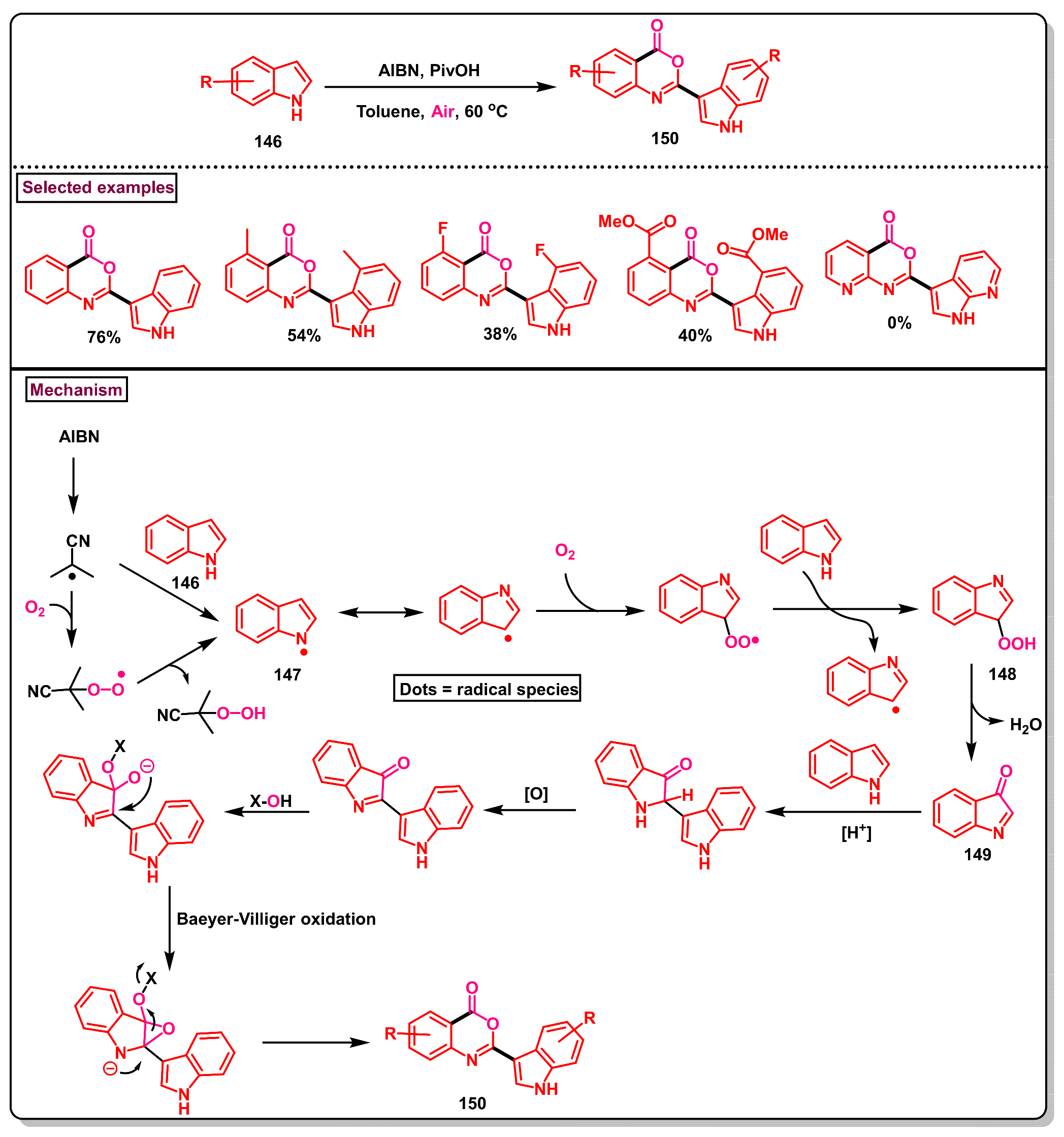
- Liu, X.-X.; Luo, X.-L.; Wu, Z.-Y.; Cui, X.-F.; Zhou, X.-Q.; He, Y.-Q.; Huang, G.-S. Oxidative re-cyclization of 1H-indoles for synthesis of 2-indolylbenzoxazinones via cleavage C2-C3 bond with AIBN under air. J. Org. Chem. 2017, 82, 2107–2113. [Google Scholar] [CrossRef] [PubMed]
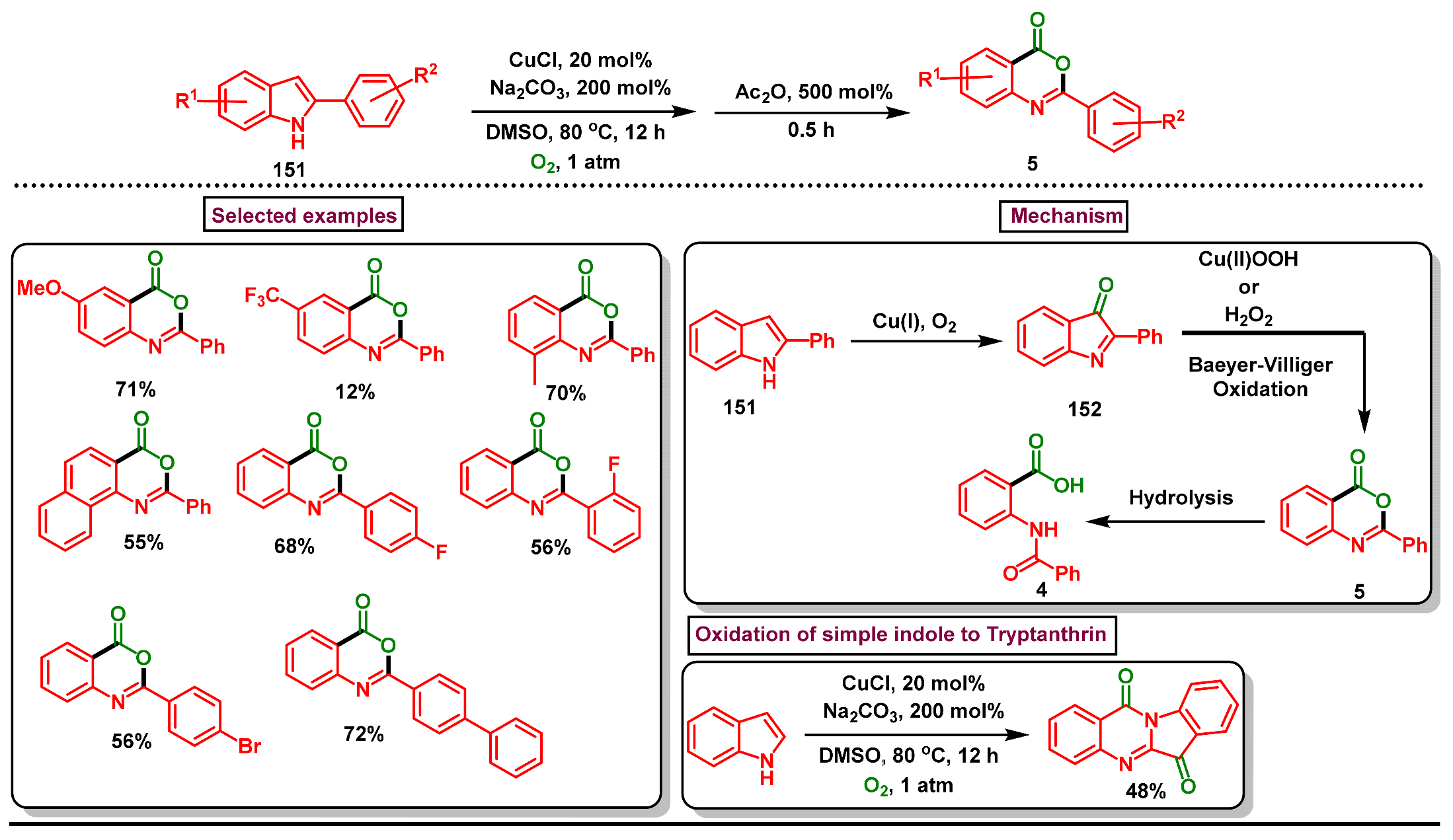
- Yamashita, M.; Iida, A. Copper-mediated oxidative tandem reactions with molecular oxygen: Synthesis of 2-arylbenzoxazinone derivatives from indoles. Tetrahedron Lett. 2014, 55, 2991–2993. [Google Scholar] [CrossRef]
- Wang, C.; Zhang, L.; Ren, A.; Lu, P.; Wang, Y. Cu-catalyzed synthesis of tryptanthrin derivatives from substituted indoles. Org. Lett. 2013, 15, 2982–2985. [Google Scholar] [CrossRef] [PubMed]
- Hirano, K.; Miura, M. Copper-mediated oxidative direct C–C (hetero)aromatic cross-coupling. Chem. Commun. 2012, 48, 10704–10714. [Google Scholar] [CrossRef] [PubMed]
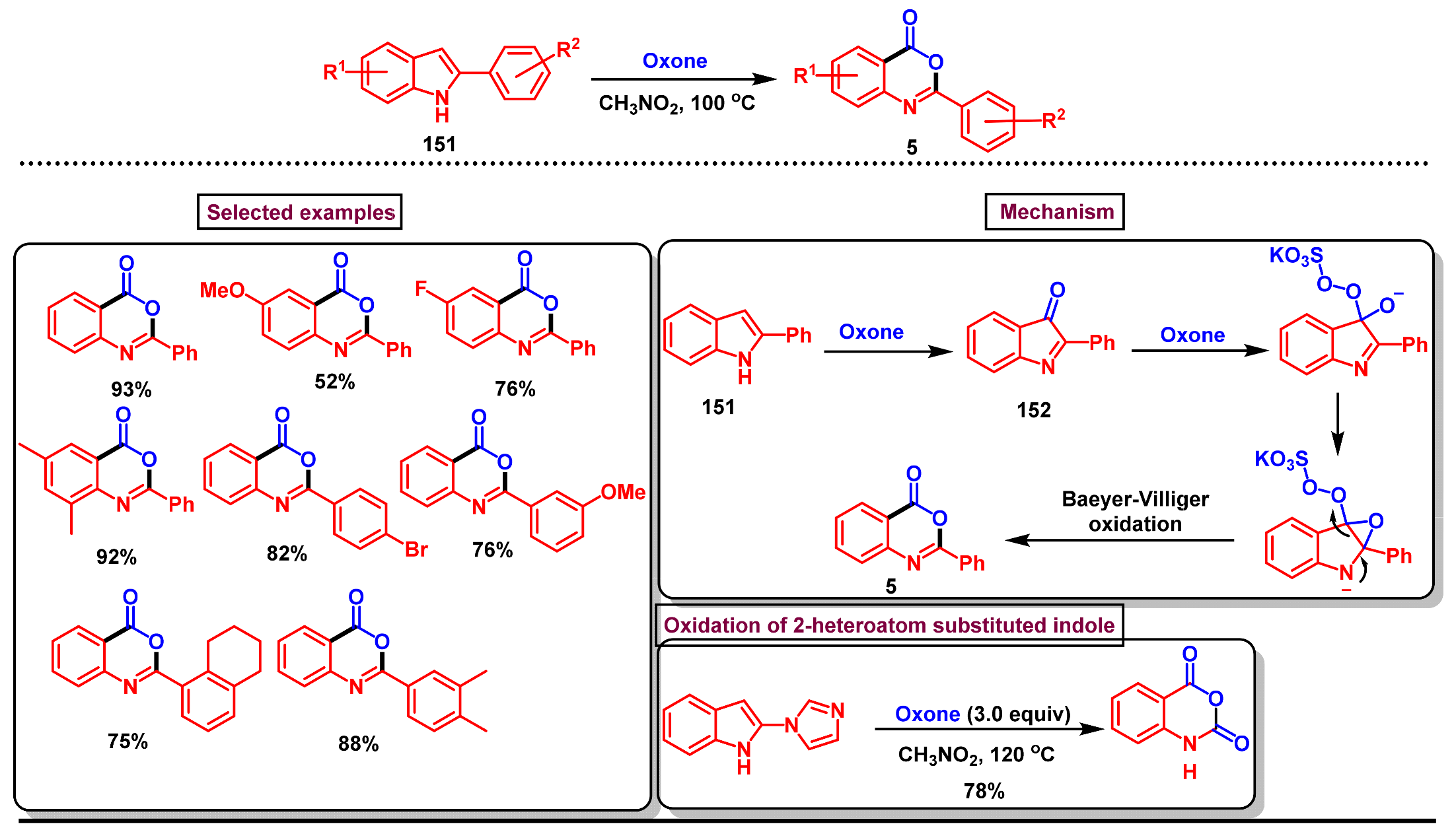
- Lian, X.-L.; Lei, H.; Quan, X.-J.; Ren, Z.-H.; Wang, Y.-Y.; Guan, Z.-H. Oxidation of 2-arylindoles for synthesis of 2-arylbenzoxazinones with oxone as the sole oxidant. Chem. Commun. 2013, 49, 8196–8198. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wang, Y.; Xu, C.; Zou, J.; Wy, Y.; Yin, G. TBHP-promoted multicomponent reaction to access 2-aminobenzoxazinones using sodium chlorodifluoroacetate as the C1 synthon. Org. Chem. Front. 2023, 10, 1988–1993. [Google Scholar] [CrossRef]
- Prakash, R.; Gogoi, S. Copper-catalyzed C-N, C-O coupling reaction of arylglyoxylic acids with isatins. Adv. Synth. Catal. 2016, 358, 3046–3049. [Google Scholar] [CrossRef]
- Yu, Y.; Xia, Z.; Wu, Q.; Liu, D.; Yu, L.; Xiao, Y.; Tan, Z.; Deng, W.; Zhu, G. Direct synthesis of benzoxazinones via Cp*Co(III)-catalyzed C-H activation and annulation of sulfoxonium ylides with dioxazolones. Chin. Chem. Lett. 2021, 32, 1263–1266. [Google Scholar] [CrossRef]
- Yin, Z.; Wang, Z.; Wu, X.-F. Silver and palladium cocatalyzed carbonylative activation of benzotriazoles to benzoxazinones under neutral conditions. Org. Lett. 2017, 19, 6232–6235. [Google Scholar] [CrossRef]
- An, T.; Liu, C.; Yin, Y.; Wu, X.-F.; Yin, Z. Palladium-catalyzed denitrogenative carbonylation of benzotriazoles with Cr(CO)6 as the carbonyl source. Organometallics 2022, 41, 1731–1737. [Google Scholar] [CrossRef]
- Liu, Q.; Chen, P.; Liu, G. Palladium-catalyzed C-C triple bond cleavage: Efficient synthesis of 4H-benzo[d][1,3]oxazin-4-ones. ACS Catal. 2013, 3, 178–181. [Google Scholar] [CrossRef]
- Sun, Z.-X.; Cheng, Y. N-Heterocyclic carbene-catalyzed cascade annulation reaction of O-vinylarylaldehydes with nitrosoarenes: One-step assembly of functionalized 2,3-benzoxazin-4-ones. Org. Biomol. Chem. 2012, 10, 4088–4094. [Google Scholar] [CrossRef] [PubMed]
- Pandey, G.; Batra, S. Microwave-assisted Palladium-Catalyzed Isonitrile Insertion in 2-Bromophenylureas for Efficient Synthesis of 4-Substituted Imino 4H-Benzo[d][1,3]-oxazin-2-amines. RSC Adv. 2015, 5, 28875–28878. [Google Scholar] [CrossRef]
- Tajbakhsh, M.; Hosseinzadeh, R.; Rezaee, P.; Tajbakhsh, M. H3PW12O40 Catalyzed Synthesis of Benzoxazine and Quinazoline in Aqueous Media. Chin. J. Catal. 2014, 35, 58–65. [Google Scholar] [CrossRef]
- Jiang, W.; Li, Z.-H.; Li, T.-J.; Liu, J.-Q.; Wang, X.-S. Copper-catalyzed decarboxylation cross-coupling cascade reaction for synthesis of fused dihydro-benzoxazinones. J. Org. Chem. 2024, 89, 7472–7477. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Paul, S. Solid acids: Green alternatives for acid catalysis. Catal. Today 2014, 236, 153–170. [Google Scholar] [CrossRef]
- Dasu, S.; Gajula, K.S.; Amrutham, V.; Boosa, M.; Madasu, R.; Lekkala, M.; Andugulapati, S.B.; Nama, N. A facile heterogeneous catalytic approach for one-pot synthesis of fused benzoxazinones using Cuβ zeolites and evaluation of biological activities. Tetrahedron 2024, 156, 133931. [Google Scholar] [CrossRef]
- Shang, X.-J.; Liu, Z.-Q. A Co(II)-catalyzed aerobic intramolecular C-O bond formation via selective (sp3) C-H bond activation: Facile access to dihydro-benzoxazinone derivatives. Tetrahedron Lett. 2015, 56, 482–484. [Google Scholar] [CrossRef]
- Möhrle, H.; Busch, M. Tricyclische Benzoxazinone. Arch Pharm. 1981, 314, 524–531. [Google Scholar] [CrossRef]
- Li, L.; Ji, M.-M.; Tang, Y.; Wang, W.-F.; Peng, J.-B. Palladium-catalyzed cascade carbonylation reaction: Synthesis of fused isoindolinones. Org. Lett. 2024, 26, 5625–5629. [Google Scholar] [CrossRef]
- Liu, C.; Liu, Y.; Yang, S.; Zheng, B.; Zhang, Y. Electrochemical lactonization enabled by unusal Shono-type oxidation from functionalzied benzoic acids. Org. Lett. 2024, 26, 1936–1940. [Google Scholar] [CrossRef] [PubMed]
- Lalji, R.S.K.; Kumar, P.; Gupta, M.; Parmar, V.S.; Singh, B.K. Palladium-catalyzed decarboxylative synthesis of 5H-benzo [4,5][1,3]oxazino [2,3-a]isoindole-5,11(6aH)-diones using 2-phenyl-4H-benzo [4,5][1,3]oxazin-4-ones and α-oxo carboxylic acids. Adv. Synth. Catal. 2020, 362, 552–560. [Google Scholar] [CrossRef]
- Kumar, P.; Gupta, M.; Bahadur, V.; Parmar, V.S.; Singh, B.K. Radical-induced, palladium-catalyzed C–H activation: An approach to functionalize 4H-Benzo[d][1,3]oxazin-4-one derivatives by using toluenes, aldehydes, and benzyl alcohols. Eur. J. Org. Chem. 2018, 2018, 1552–1558. [Google Scholar] [CrossRef]
- Patra, D.; Saha, A. C-H activation of 2-arylbenzoxazinones in aqueous medium: Synthesis of fused isoindolinones using a heterogeneous magnetic Pd-catalyst. Eur. J. Org. Chem. 2022, 2022, e202201042. [Google Scholar] [CrossRef]
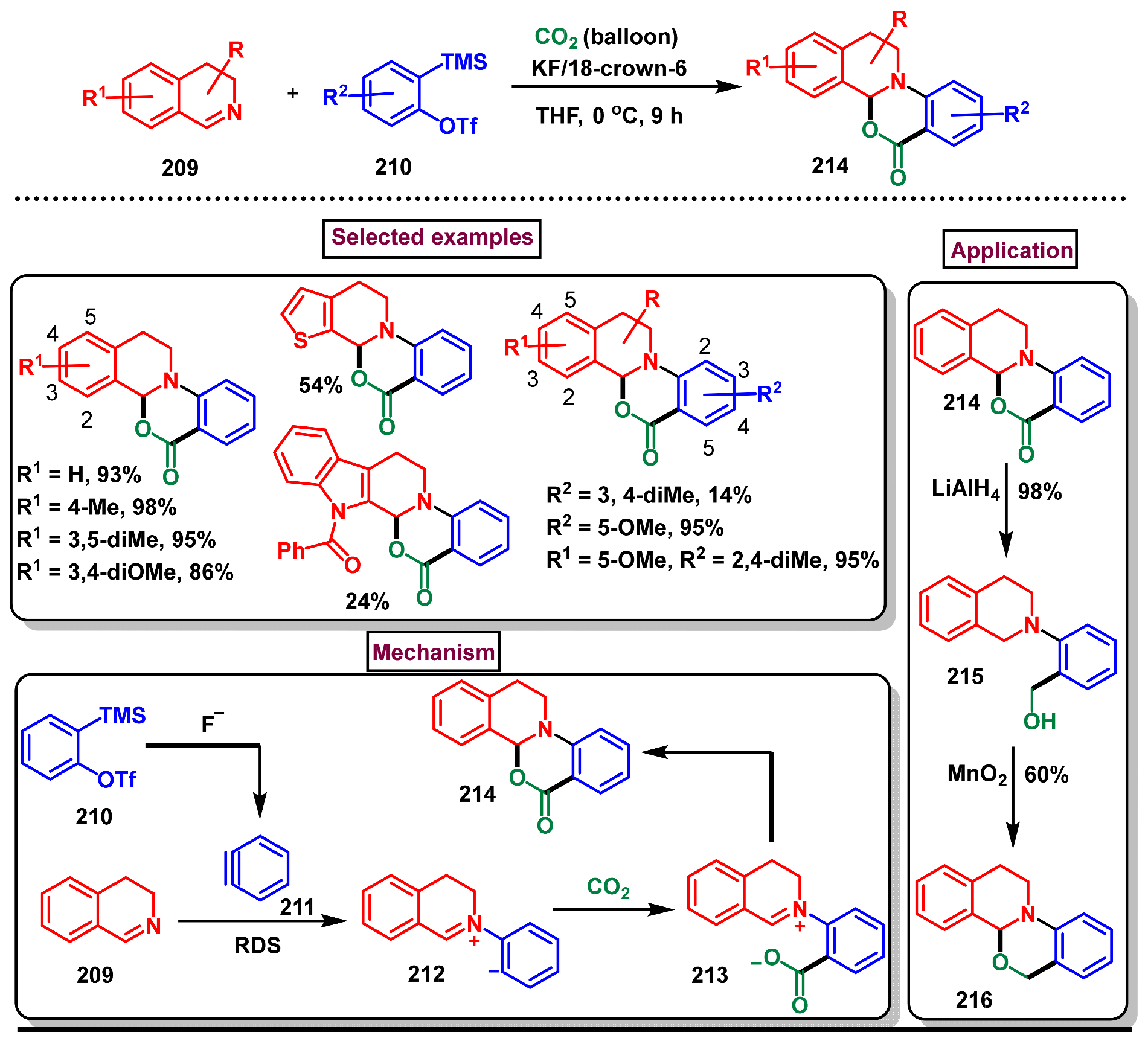
- Liu, S.; Zhang, K.; Meng, Y.; Xu, J.; Chen, N. Aryne and CO2-based formal [2+2+2] annulation to access tetrahydroisoquinoline-fused benzoxazinones. Org. Biomol. Chem. 2023, 21, 6892–6897. [Google Scholar] [CrossRef] [PubMed]
- Sharada, D.S.; Shinde, A.H.; Patel, S.M.; Vidyacharan, S. Scaffold diversity through a branching double annulation cascade strategy: An iminium induced one-pot synthesis of diverse fused tetrahydroisoquinoline (THIQ) scaffolds. J. Org. Chem. 2016, 81, 6463–6471. [Google Scholar] [CrossRef] [PubMed]
- To, W.-P.; Liu, Y.; Lau, T.-C.; Che, C.-M. A robust palladium (II)-porphyrin complex as catalyst for visible light-induced oxidative C-H functionalization. Chem. Eur. J. 2013, 19, 5654–5664. [Google Scholar] [CrossRef] [PubMed]
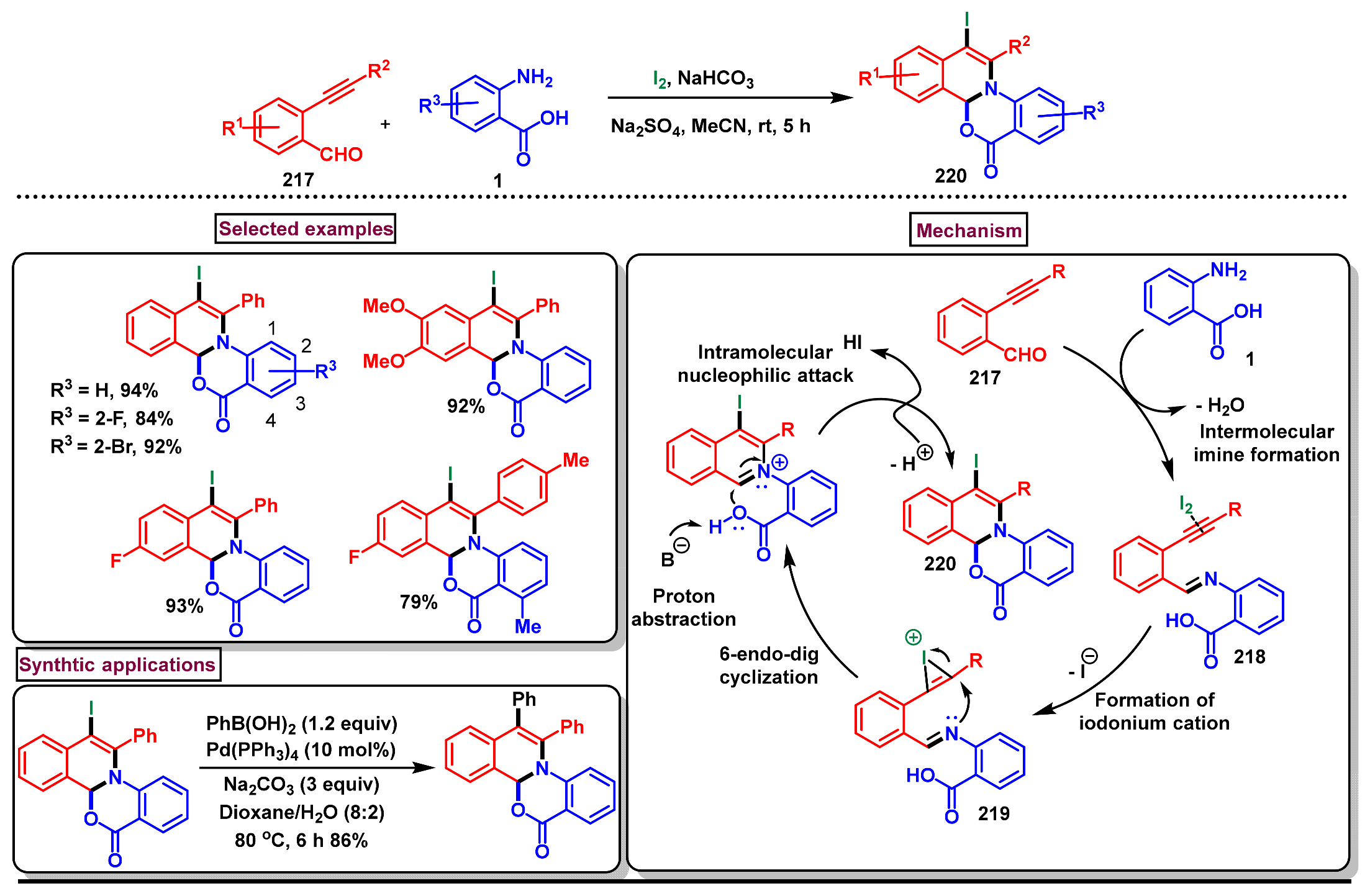
- Dighe, S.U.; Batra, S. Iodine-mediated electrophilic tandem cyclization of 2-alkynylbenzaldehydes with anthranilic acid leading to 1,2-dihydroisoquinoline-fused benzoxazinones. Tetrahedron 2013, 69, 9875–9885. [Google Scholar] [CrossRef]
- Boomhoff, M.; Schneider, C. A novel three-component [3+2] cycloannulation process for the rapid and highly stereoselective synthesis of pyrrolobenzoxazoles. Chem. Eur. J. 2012, 18, 4185–4189. [Google Scholar] [CrossRef] [PubMed]
- Boomhoff, M.; Yadav, A.K.; Appun, J.; Schneider, C. Modular, flexible, and stereoselective synthesis of pyrroloquinolines: Rapid assembly of complex heterocyclic scaffolds. Org. Lett. 2014, 16, 6236–6239. [Google Scholar] [CrossRef] [PubMed]
- Boomhoff, M.; Ukis, R.; Schneider, C. A highly stereocontrolled, one-pot approach towards pyrrolobenzoxazinones and pyrroloquinazolinones through a Lewis acid-catalyzed [3+2]-cycloannulation process. J. Org. Chem. 2015, 80, 8236–8244. [Google Scholar] [CrossRef]
- Minghetti, P.; Casiraghi, A.; Montanari, L.; Monzani, M.V. In vitro skin permeation of Sinitrodil, a member of a new class of nitrovasodilator drugs. Eur. J. Pharm. Sci. 1999, 7, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, R.; Misra, P.; Viramadithyan, R.K.; Premkumar, M.; Hiriyan, J.; Datla, S.R.; Damarla, R.K.B.; Suresh, J.; Rajagopalan, R. Antidiabetic and hypolipidemic potential of DRF 2519—A dual activator of PPAR-α and PPAR-γ. Eur. J. Pharmacol. 2004, 491, 195–206. [Google Scholar] [CrossRef]
- Arai, A.C.; Kessler, M.; Rogers, G.; Lynch, G. Effects of the potent ampakine CX614 on hippocampal and recombinant AMPA receptors: Interactions with Cyclothiazide and GYKI 52466. Mol. Pharmacol. 2000, 58, 802–813. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yamataka, K.; Okamoto, M.; Ikeda, S.; Baba, M. Potent and selective inhibition of Tat-dependent HIV-1 replication in chronically infected cells by a novel naphthalene derivative JTK-101. Antivir. Chem. Chemother. 2007, 18, 201–211. [Google Scholar] [CrossRef] [PubMed]
- Baykov, S.V.; Shetnev, A.A.; Semenov, A.V.; Baykova, S.O.; Boyarskiy, V.P. Room temperature synthesis of bioactive 1,2,4-oxadiazoles. Int. J. Mol. Sci. 2023, 24, 5406. [Google Scholar] [CrossRef]
- Le Falher, L.; Ayad, O.B.; Ziyaret, O.; Mamontov, A.; Botuha, C.; Thorimbert, S.; Slowinski, F. Access to pyridyl-substituted 1,3,5-triazines from 4H-Pyrido [1,3]oxazin-4-ones via a cyclocondensation process. J. Org. Chem. 2014, 79, 6579–6589. [Google Scholar] [CrossRef]
- Le Falher, L.; Ayad, O.B.; Ziyaret, O.; Botuha, C.; Thorimbert, S.; Slowinski, F. Preparation of Halogen-Containing 4H-Pyrido[e][1,3]oxazin-4-ones and Their Transformation into 2-Hydroxypyridinyl-Substituted 1,2,4-Oxadiazoles and 1,2,4-Triazoles. Eur. J. Org. Chem. 2015, 2015, 3830–3840. [Google Scholar] [CrossRef]
- Abe, M.; Kawamoto, M.; Inoue, M.; Kimachi, T.; Inamoto, K. Gold(I)-catalyzed heteroannulation of salicylic amides with alkynes: Synthesis of 1,3-Benzoxazin-4-one derivatives. Org. Lett. 2022, 24, 5684–5687. [Google Scholar] [CrossRef]
- Su, Z.; Chai, H.; Xu, J.; Li, J. ZnCl2-promoted domino reaction of 2-hydroxybenzonitriles with ketones for synthesis of 1,3-Benzoxazin-4-ones. RSC Adv. 2021, 11, 29906–29911. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Wang, Y.; Hua, R. Base-promoted chemodivergent formation of 1,4-Benzoxazepin-5(4H)-ones and 1,3-Benzoxazin-4(4H)-ones swtiched by solvents. Molecules 2019, 24, 3773. [Google Scholar] [CrossRef] [PubMed]
- Ogiwara, Y.; Suzuki, Y.; Sato, K.; Sakai, N. Construction of N-heterocyclic systems containing a fully substituted allylic carbon by palladium/phosphine catalysis. Org. Lett. 2018, 20, 6965–6969. [Google Scholar] [CrossRef] [PubMed]
- Modak, A.; Dutta, U.; Kancherla, R.; Maity, S.; Bhadra, M.; Mobin, S.M.; Maiti, D. Predictably selective (sp3)C-O bond formation through copper catalyzed dehydrogenative coupling: Facile synthesis of dihydro-oxazinone derivatives. Org. Lett. 2014, 16, 2602–2605. [Google Scholar] [CrossRef]
- Leas, D.A.; Wu, J.; Ezell, E.L.; Garrison, J.C.; Vennerstrom, J.L.; Dong, Y. Formation of 2-imino Benzo[e]-1,3-oxazin-4-ones from reactions of salicyclic acids and anilines with HATU: Mechanistic and synthetic studies. ACS Omega 2018, 3, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Prajapati, R.; Kumar, A.; Kandhikonda, R.; Kant, R.; Tadigoppula, N. Synthesis of 4H-1,3-benzoxazin-4-ones from 2,2-diazidobenzofuran-3(2H)-ones. Tetrahedron 2019, 75, 374–380. [Google Scholar] [CrossRef]
- Åkerbladh, L.; Chow, S.Y.; Odell, L.R.; Larhed, M. Synthesis of 4H-Benzo[e][1,3]oxazin-4-ones by a carbonylation-cyclization domino reaction of ortho-halophenols and cyanamide. ChemistryOpen 2017, 6, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zhang, X.; Wu, Y.; Cheng, M.; Zhang, D.; Li, G.; Lin, Z.; Zhang, X.; Huang, H. A facile and efficient synthesis of 4H-1,3-benzoxazin-4-one with electron-withdrawing group derivatives. Tetrahedron Lett. 2015, 56, 4683–4685. [Google Scholar] [CrossRef]
- Raw, S.A.; O’Kearney-McMullan, A.M.; Graham, M.A. Unexpected ring-expansion of 1,2-benzisoxazol-3-ones. Tetrahedron Lett. 2011, 52, 6775–6778. [Google Scholar] [CrossRef]
- Slowinski, F.; Ayad, O.B.; Ziyaret, O.; Botuha, C.; Le Falher, L.; Aouane, K.; Thorimbert, S. Expeditive access to 2-substituted 4H-pyrido [1,3]oxazin-4-ones via an intramolecular O-arylation. Org. Lett. 2013, 15, 3494–3497. [Google Scholar] [CrossRef] [PubMed]
- Docherty, J.H.; Lister, T.M.; Mcarthur, G.; Findlay, M.T.; Domingo-Legarda, P.; Kenyon, J.; Choudhary, S.; Larrosa, I. Transition-metal-catalyzed C–H bond activation for the formation of C–C bonds in complex molecules. Chem. Rev. 2023, 123, 7692–7760. [Google Scholar] [CrossRef]
- Dhawa, U.; Kaplaneries, N.; Ackermann, L. Green strategies for transition metal-catalyzed C–H activation in molecular syntheses. Org. Chem. Front. 2021, 8, 4886–4913. [Google Scholar] [CrossRef]
- Mohanty, S.R.; Prusty, N.; Nanda, T.; Mahulkar, P.S.; Ravikumar, P.C. Directing group-assisted selective C–H activation of six-membered N-heterocycles and benzo-fused N-heterocycles. Org. Chem. Front. 2024, 11, 540–575. [Google Scholar] [CrossRef]
- Zarkadoulas, A.; Zgouleta, I.; Tzouras, N.V.; Vougioukalakis, G.C. Traceless directing groups in sustainable metal-catalyzed C–H activation. Catalysts 2021, 11, 554. [Google Scholar] [CrossRef]
- Purser, S.; Morre, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef] [PubMed]
- McMurtrey, K.B.; Racowski, J.M.; Sanford, M.S. Pd-catalyzed C–H fluorination with nucleophilic fluoride. Org. Lett. 2012, 14, 4094–4097. [Google Scholar] [CrossRef] [PubMed]
- Dubost, E.; Fossey, C.; Cailly, T.; Rault, S.; Fabis, F. Selective ortho-Bromination of substituted benzaldoximes using Pd-catalyzed C–H activation: Application to the synthesis of substituted 2-Bromobenzaldehydes. J. Org. Chem. 2011, 76, 6414–6420. [Google Scholar] [CrossRef] [PubMed]
- Dabiri, M.; Lehi, N.F.; Movahed, S.K.; Khavasi, H.R. Pd-catalyzed regioselective C–H halogenation of quinazolinones and benzoxazinones. Org. Biomol. Chem. 2017, 15, 6264–6268. [Google Scholar] [CrossRef] [PubMed]
- Vu, H.M.; Li, X.-Q.; Chen, F.-W. Palladium-catalyzed C-H bond monofluorination of 2-arylbenzo[d]oxazinone using nitrate as crucial promoter. Synthesis 2019, 51, 1578–1584. [Google Scholar] [CrossRef]
- Vu, H.M.; Chen, F.-W.; Li, X.-Q. Acetoxylation and halogenation of 2-arylbenzoxazinones via palladium-catalyzed C(sp2)-H bond oxidation. ChemistrySelect 2019, 4, 9465–9469. [Google Scholar] [CrossRef]
- Bakthadoss, M.; Kumar, P.V.; Kumar, R.; Agarwal, V. Palladium catalyzed chemo- and site selective C-H acetoxylation and hydroxylation of oxobenzoxazine derivatives. Org. Biomol. Chem. 2019, 17, 4465–4469. [Google Scholar] [CrossRef]
- Gupta, M.; Kumar, S.; Kumar, P.; Singh, A.K.; Bahadur, V.; Singh, B.K. N-directed Pd-catalyzed direct ortho-acetoxylation and ortho-tert-butoxylation of 2-phenyl-4H-benzo[d][1,3]oxazin-4-ones via C-H activation. ChemistrySelect 2019, 4, 13992–13997. [Google Scholar] [CrossRef]
- Yong, J.-Y.; Vu, H.-M.; Li, X.-Q.; Gong, A.-J. Palladium (0)-catalyzed C(sp2)-H oxygeneation with carboxylic acids. Tetrahedron 2020, 76, 130969. [Google Scholar] [CrossRef]
- Deb, A.; Bag, S.; Kancherla, R.; Maiti, D. Palladium-catalyzed aryl C-H olefination with unactivated, aliphatic alkenes. J. Am. Chem. Soc. 2014, 136, 13602–13605. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.G.; Kitamura, T.; Fujiwara, Y. Catalytic functionalization of arenes and alkanes via C-H bond activation. Acc. Chem. Res. 2001, 34, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Lu, W. Towards ideal synthesis: Alkenylation of aryl C-H bonds by a Fujiwara-Moritani reaction. Chem. Eur. J. 2014, 20, 634–642. [Google Scholar] [CrossRef]
- Panja, S.; Maity, S.; Majhi, B.; Ranu, B.C. Palladium-catalyzed olefination of 4H-benzo[d][1,3]oxazin-4-one derivatives with activated alkenes via preferential cyclic imine-N-directed aryl C-H activation. Eur. J. Org. Chem. 2019, 2019, 5777–5786. [Google Scholar] [CrossRef]
- Bakthadoss, M.; Kumar, P.V.; Kumar, R.; Surendar, M.; Sharada, D.S. Ruthenium catalyzed chemo and site-selective C-H amidation of oxobenzoxazine derivatives with sulfonyl azides. New J. Chem. 2019, 43, 14190–14195. [Google Scholar] [CrossRef]
- Hsieh, P.-W.; Chang, F.-R.; Chang, C.-H.; Cheng, P.-W.; Chiang, L.-C.; Zeng, F.-L.; Lin, K.-H.; Wu, Y.-C. 2-Substituted benzoxazinone analogues as anti-human coronavirus (anti-HCoV) and ICAM-1 expression inhibition agents. Bioorg. Med. Chem. Lett. 2004, 14, 4751–4754. [Google Scholar] [CrossRef]
- Gütschow, M.; Neumann, U. Inhibition of cathepsin G by 4H-3,1-benzoxazin-4-ones. Bioorg. Med. Chem. 1997, 5, 1935–1942. [Google Scholar] [CrossRef]
- Krantz, A.; Spencer, R.W.; Tam, T.F.; Liak, T.J.; Copp, L.J.; Thomas, E.M.; Rafferty, S.P. Design and synthesis of 4H-3,1-benzoxazin-4-ones as potent alternate substrate inhibitors of human leukocyte elastase. J. Med. Chem. 1990, 33, 464–479. [Google Scholar] [CrossRef] [PubMed]
- Marasini, B.P.; Rahim, F.; Perveen, S.; Karim, A.; Khan, K.M.; Rahman, A.U.; Choudhary, M.I. Synthesis, structure-activity relationships studies of benzoxazinone derivatives as α-chymotrypsin inhibitors. Bioorg. Chem. 2017, 70, 210–221. [Google Scholar] [CrossRef]
- Zheng, B.-F.; Zuo, Y.; Huang, G.-Y.; Wang, Z.-Z.; Ma, J.-Y.; Wu, Q.-Y.; Yang, G.-F. Synthesis and Biological Activity Evaluation of Benzoxazinone-Pyrimidinedione Hybrids as Potent Protoporphyrinogen IX Oxidase Inhibitor. J. Agric. Food Chem. 2023, 71, 14221–14231. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Qiu, L.; Xie, X.; Ye, J.; Hu, A. Synthesis, crystal structure, DFT calculation, Hirshfeld suface analysis and antitumor activity of 4-(tert-butyl)-5-(1H-1,2,4-triazole-1-yl)thiazole derivatives containing benzoxazinone. J. Mol. Struct. 2024, 1310, 138047. [Google Scholar] [CrossRef]
- Benarjee, V.; Saritha, B.; Gangadhar, K.H.; Sailaja, B.B.V. Synthesis of some new 1,4-benzoxazine-pyrazoles in water as EGFR targeting anticancer agents. J. Mol. Struct. 2022, 1265, 133188. [Google Scholar] [CrossRef]
- Narsimha, S.; Battula, K.S.; Nukala, S.K.; Gondru, R.; Reddy, Y.N.; Nagavelli, V.R. One-pot synthesis of fused benzoxazino [1–3]triazolyl[4,5-c]quinolinone derivatives and their anticancer activity. RSC Adv. 2016, 6, 74332–74339. [Google Scholar] [CrossRef]
- Benarjee, V.; Saritha, B.; Gangadhar, K.H.; Sailaja, B.B.V. Synthesis of new 1,3,4-oxadiazole-1,4-benzoxazinone hybrids as tubulin polymerization inhibiting anticancer agents and their in silico studies. Tetrahedron 2022, 124, 132979. [Google Scholar] [CrossRef]
- Yang, J.; Barniol-Xicota, M.; Nguyen, M.T.N.; Ticha, A.; Strisovsky, K.; Verhelst, S.H.L. Benzoxazin-4-ones as novel, easily accessible inhibitors for rhomboid proteases. Bioorg. Med. Chem. Lett. 2018, 28, 1423–1427. [Google Scholar] [CrossRef] [PubMed]
- Goel, P.; Jumpertz, T.; Ticha, A.; Ogorek, I.; Mikles, D.C.; Hubalek, M.; Pietrzik, C.U.; Strisovsky, K.; Schmidt, B.; Weggen, S. Discovery and validation of 2-styryl substituted benzoxazin-4-ones as a novel scaffold for rhomboid protease inhibitors. Bioorg. Med. Chem. Lett. 2018, 28, 1417–1422. [Google Scholar] [CrossRef]
- Cil, O.; Anderson, M.O.; de Souza Goncalves, L.; Tan, J.-A.; Haggie, P.M.; Verkman, A.S. Small molecule inhibitors of intestinal epithelial anion exchanger SLC26A3 (DRA) with a luminal, extracellular site of action. Eur. J. Med. Chem. 2023, 249, 115149. [Google Scholar] [CrossRef]
- Szilagyi, K.; Hajdu, I.; Flachner, B.; Lorincz, Z.; Balczer, J.; Gal, P.; Zavodszky, P.; Pirli, C.; Balogh, B.; Mandity, I.M.; et al. Design and Selection of Novel C1s Inhibitors by In Silico and In Vitro Approaches. Molecules 2019, 24, 3641. [Google Scholar] [CrossRef] [PubMed]
- Koca, I.; Kamaci, V.; Özsoy, C.; Sert, Y.; Kani, I.; Tutar, L.; Tutar, Y. Pyrazolyl-Benzoxazinone Derivatives as Dual Hsp Inhibitors in Human Breast Cancer. ChemistrySelect 2022, 7, e202200359. [Google Scholar] [CrossRef]
- Kesuma, D.; Yuniarta, T.A.; Putra, G.S.; Sumari, S.; Sulistyowaty, M.I.; Anwari, F. In-silico, synthesis, structure elucidation and anticancer activity study of 2-(3,4-dichlorophenyl)-4H-benzo[d][1,3]oxazin-4-one. Pak. J. Pharm. Sci. 2022, 35, 1391–1398. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, E.U.R.; Porter, Z.J.; Jennings, I.G.; Al-Rawi, J.M.A.; Thompson, P.E.; Angove, M.J. Synthesis and biological evaluation of 4H-benzo[e][1,3]oxazin-4-ones analogues of TGX-221 as inhibitors of PI3Kβ. Bioorg. Med. Chem. 2022, 69, 116832. [Google Scholar] [CrossRef] [PubMed]
- Radwan, T.M.; El-Hashash, M.A.-A.; Wasfy, A.A.-H.F.; Abdallah, S.A. Antitumor, cytotoxic, and antioxidant evaluation of six heterocyclic compounds containing different heterocycle moieties. J. Heterocycl. Chem. 2020, 57, 1111–1122. [Google Scholar] [CrossRef]
- Radwan, T.M.; El-Hashash, M.A.-A.; Wasfy, A.A.-H.F.; Abdallah, S.A. Synthesis and Characteristics of Metastable 2-Benzyl-4H-3,1-benzoxazin-4-one as Anticancer Agent and its Comparison with other Heterocyclic Compounds. ChemistrySelect 2019, 4, 14056–14062. [Google Scholar] [CrossRef]
- Kesuma, D.; Putra, G.S.; Yuniarta, T.A.; Sulistyowaty, M.I.; Siswandono; Budiati, T. Synthesis of 2-phenyl-4H-benzo[d][1,3]oxazin-4-one and Their Biological Activity Against A549 Cancer Cell Line through Methionyl-tRNA Synthetase Inhibition Approach on in-silico Studies. J. Appl. Pharm. Sci. 2020, 11, 564–571. [Google Scholar]
- Kandale, A.; Ohlyan, R.; Kumar, G. Synthesis and in vitro antiproliferative activity of 2,3-aryl substituted 1,3-benzoxazin-4-one derivatives. Int. J. Pharm. Pharm. Sci. 2014, 6, 68–71. [Google Scholar]
- Avuthu, V.S.R.; Allaka, T.R.; Afzal, M.; Kishore, P.V.V.N.; Venkatesan, S.; Patel, P.R. Novel 1,2,3-triazole derivatives containing benzoxazinone scaffold: Synthesis, docking study, DFT analysis and biological evaluation. Results Chem. 2024, 11, 101800. [Google Scholar] [CrossRef]
- Mohareb, R.M.; Mohamed, A.A.; Ibrahim, R.A. Design, Synthesis, Molecular Docking and Biological Studies of 4H-Benzo[d][1,3]oxazin-4-one Derivatives and Their Uses in Heterocyclic Synthesis. ChemistrySelect 2022, 7, e202200325. [Google Scholar] [CrossRef]
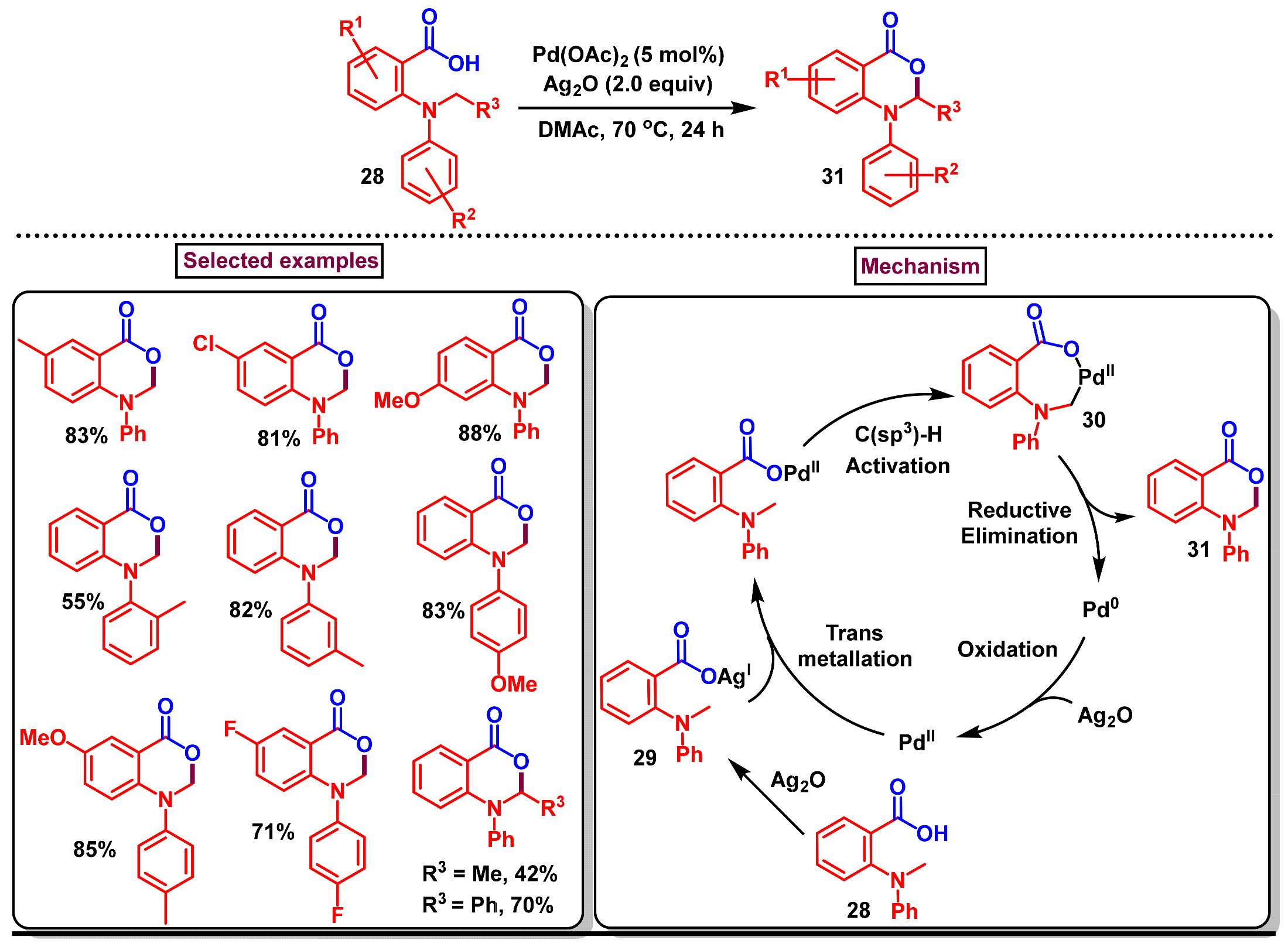
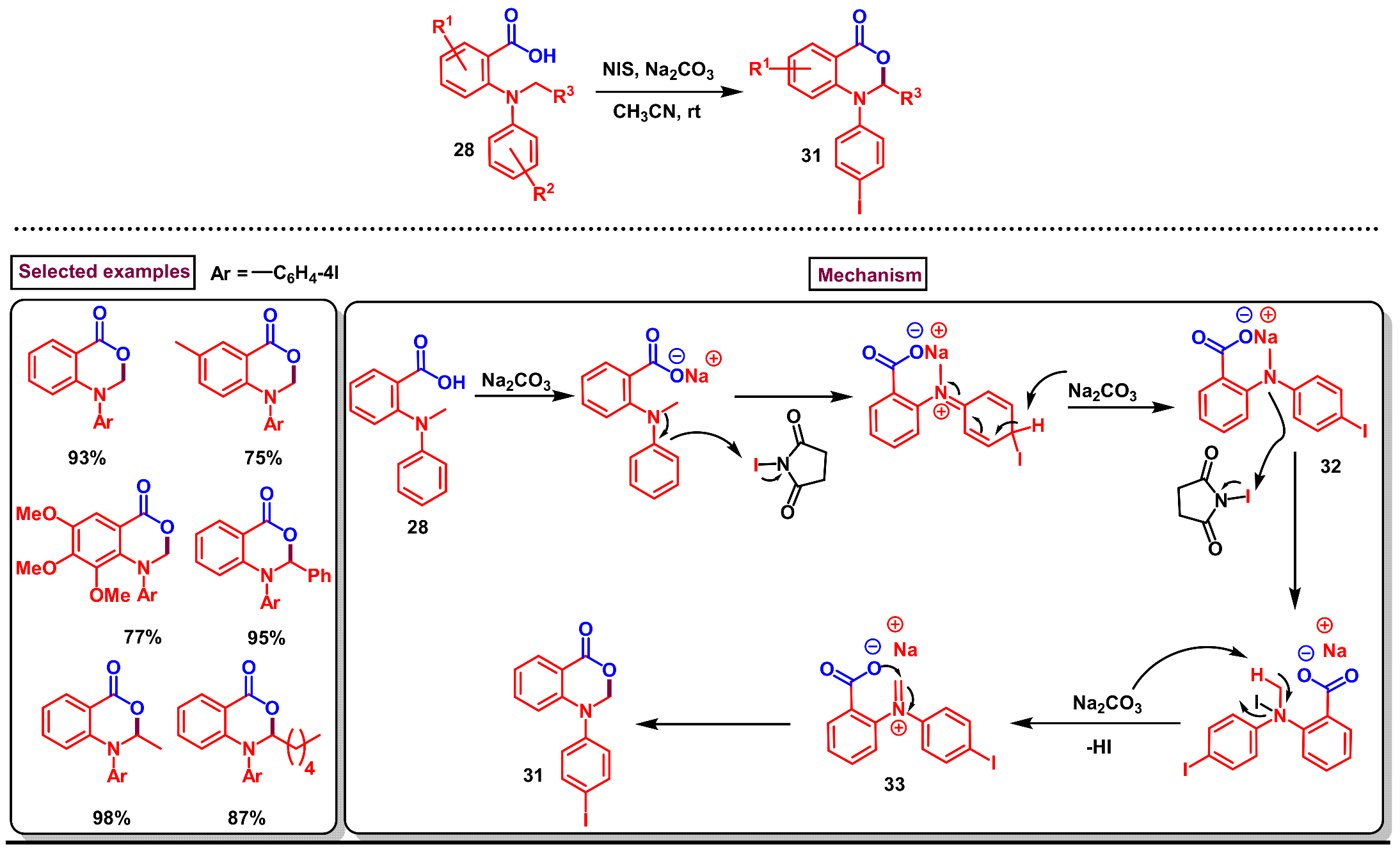
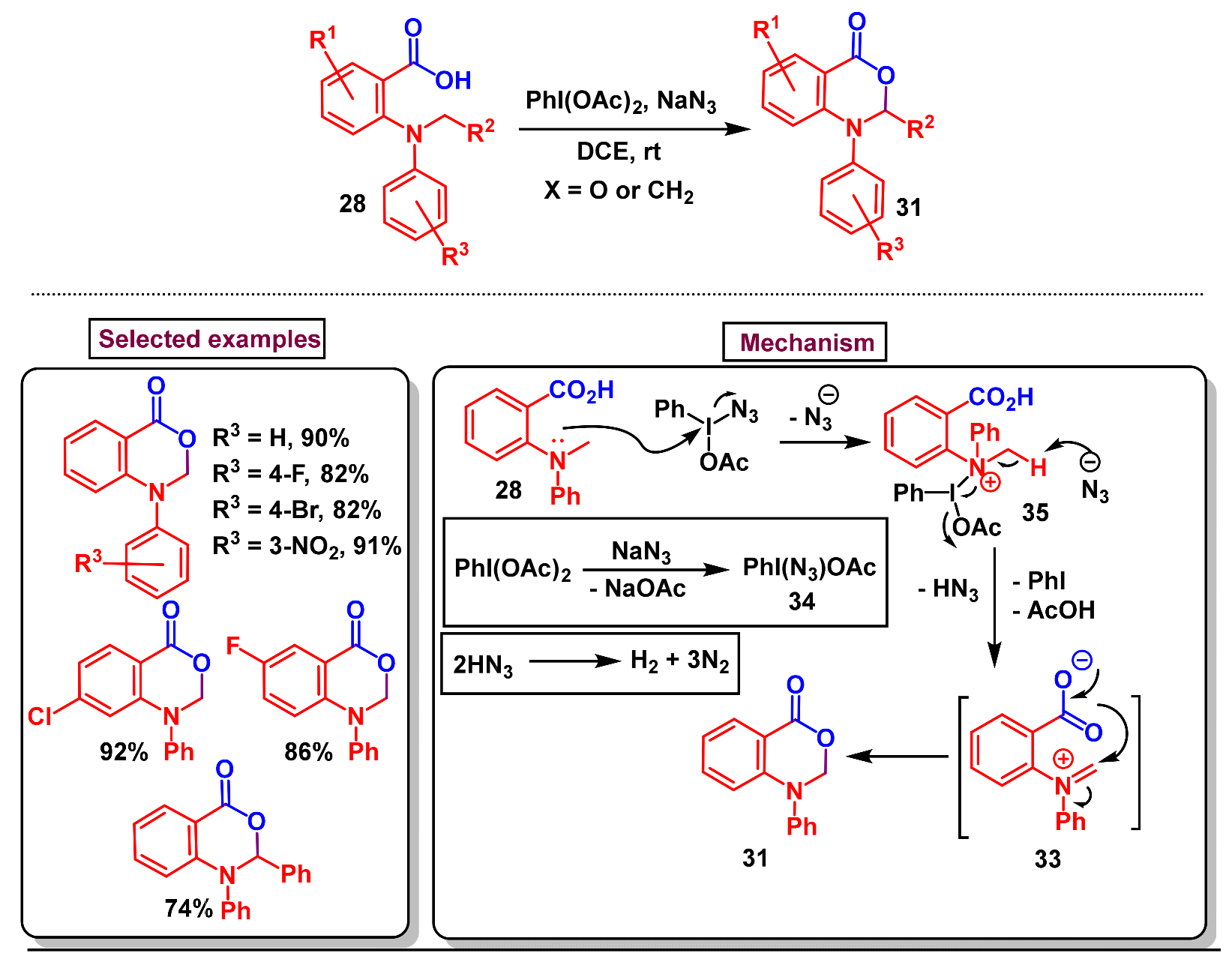
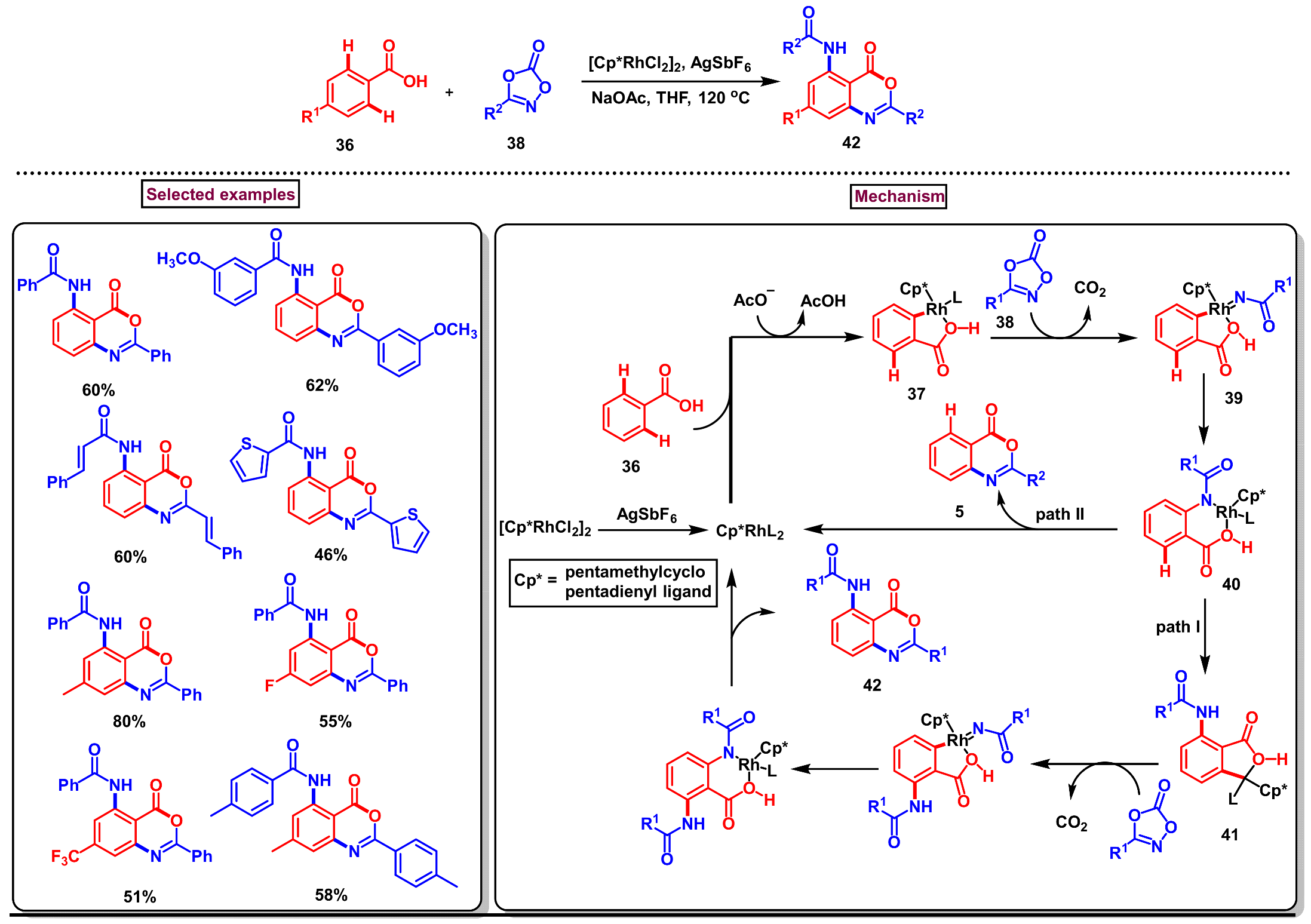
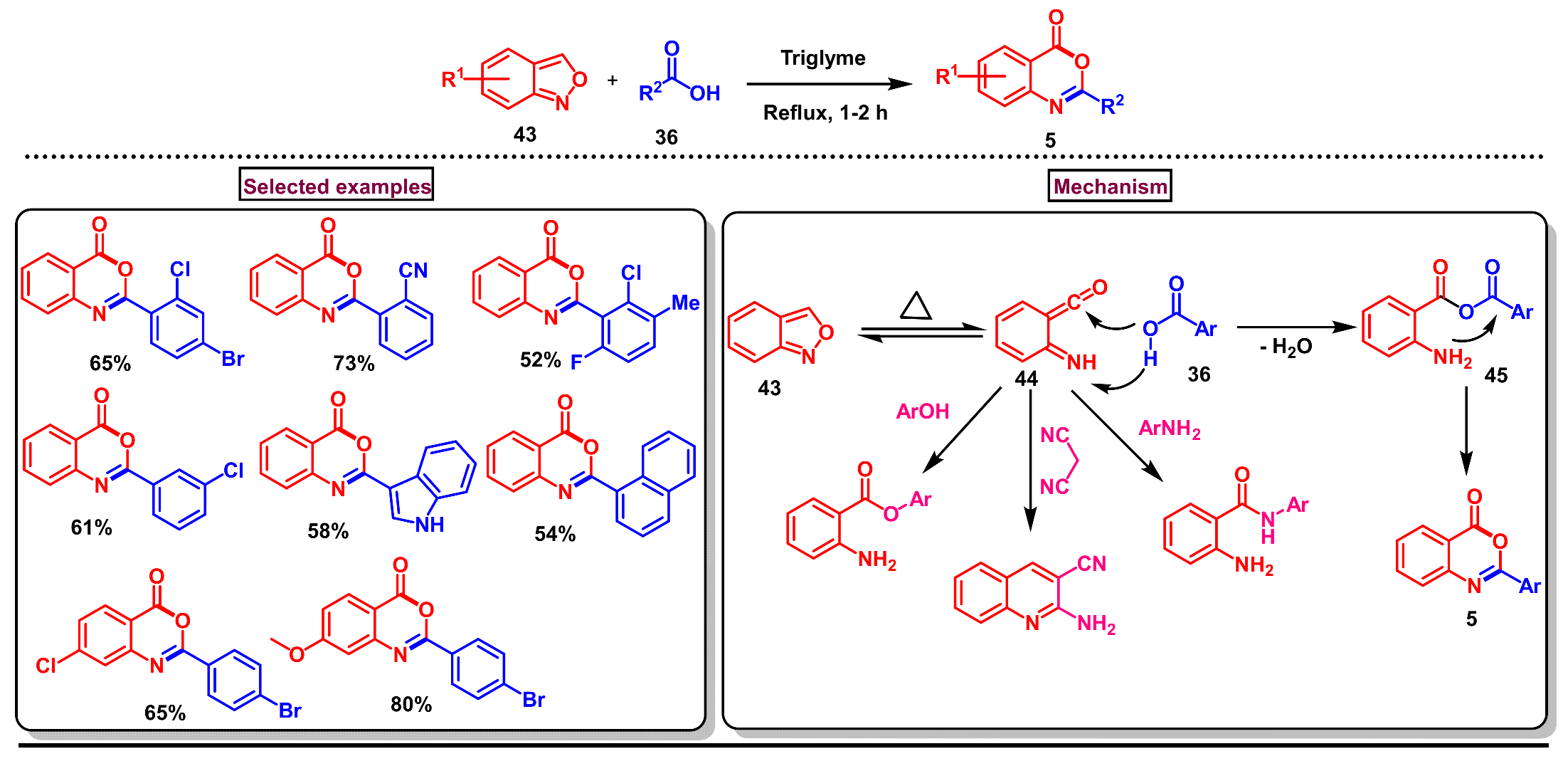
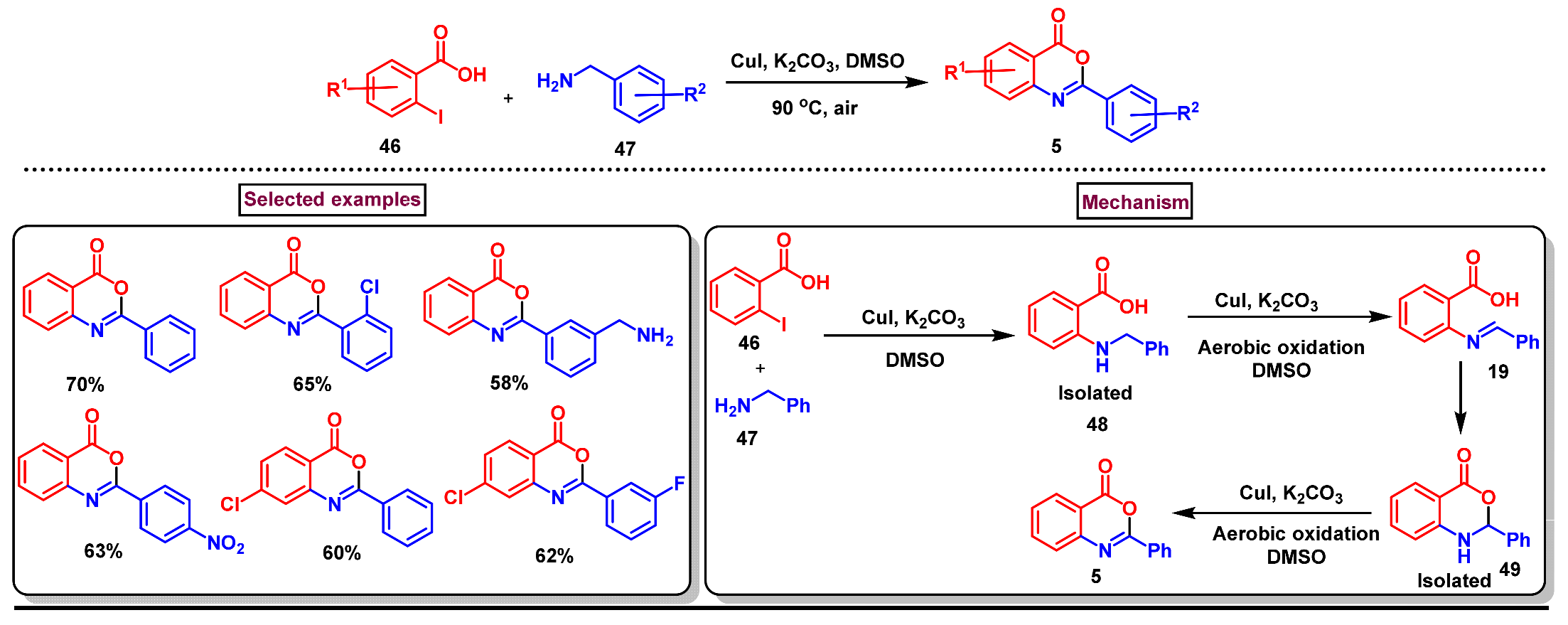
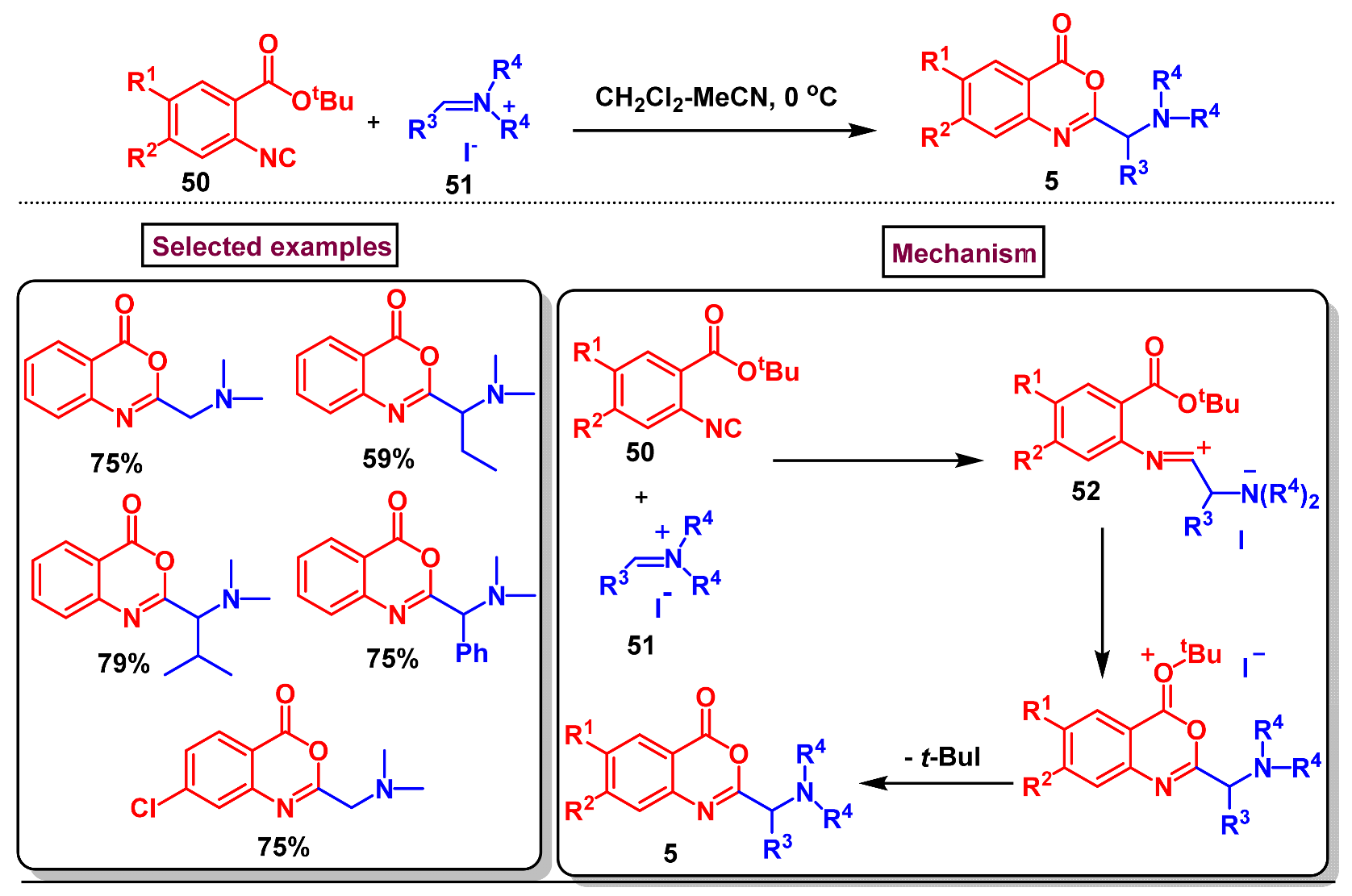
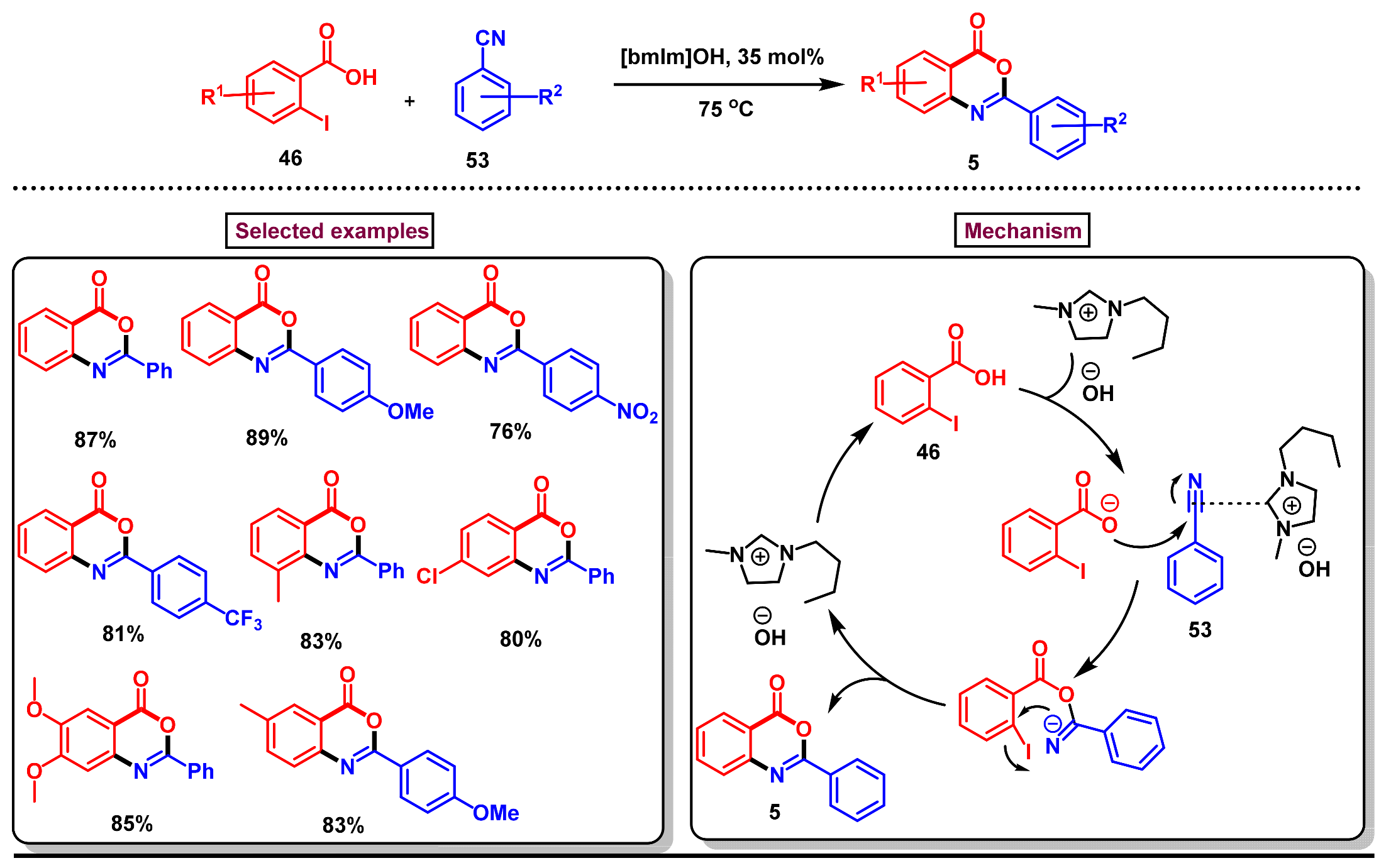


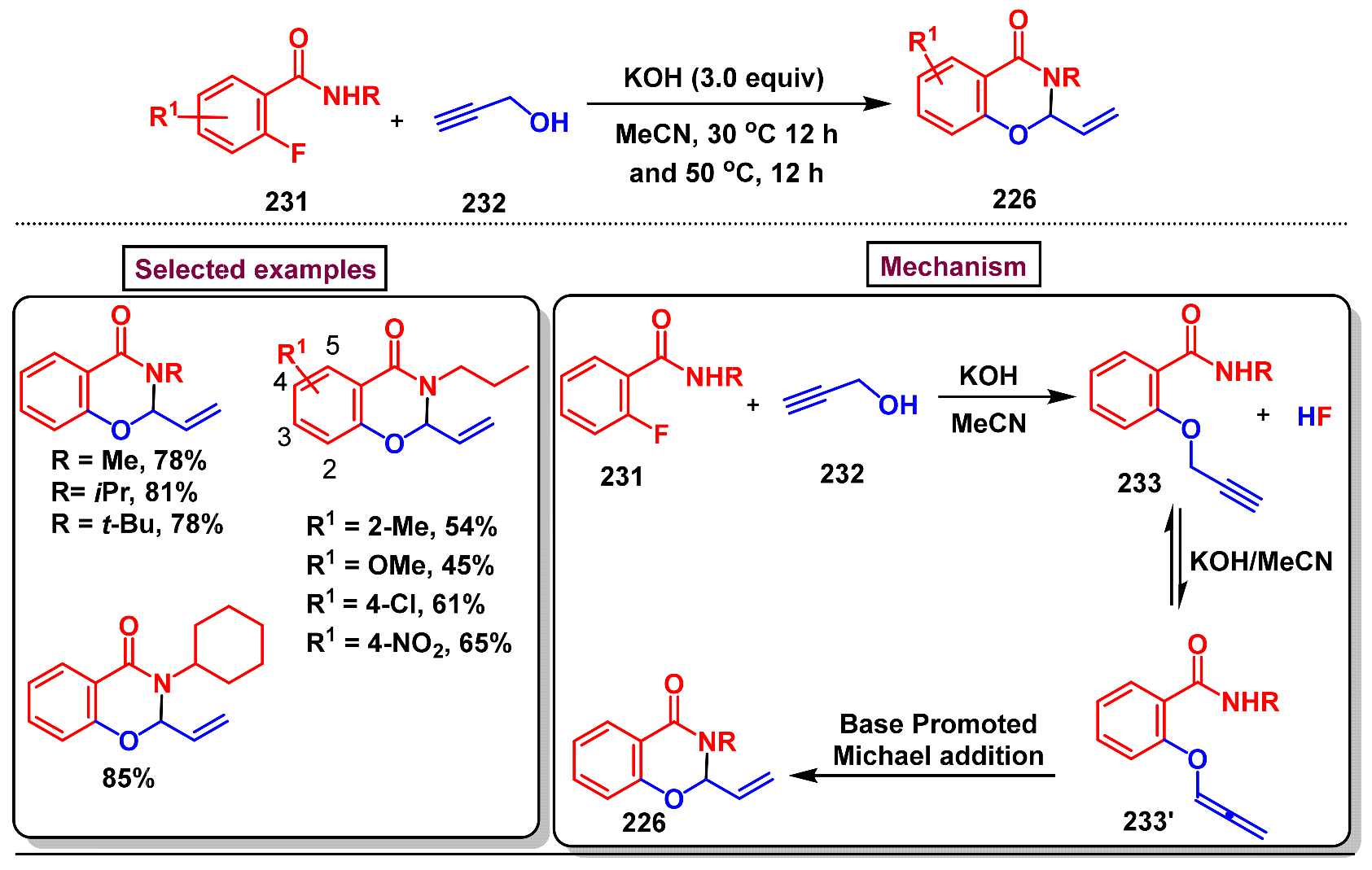
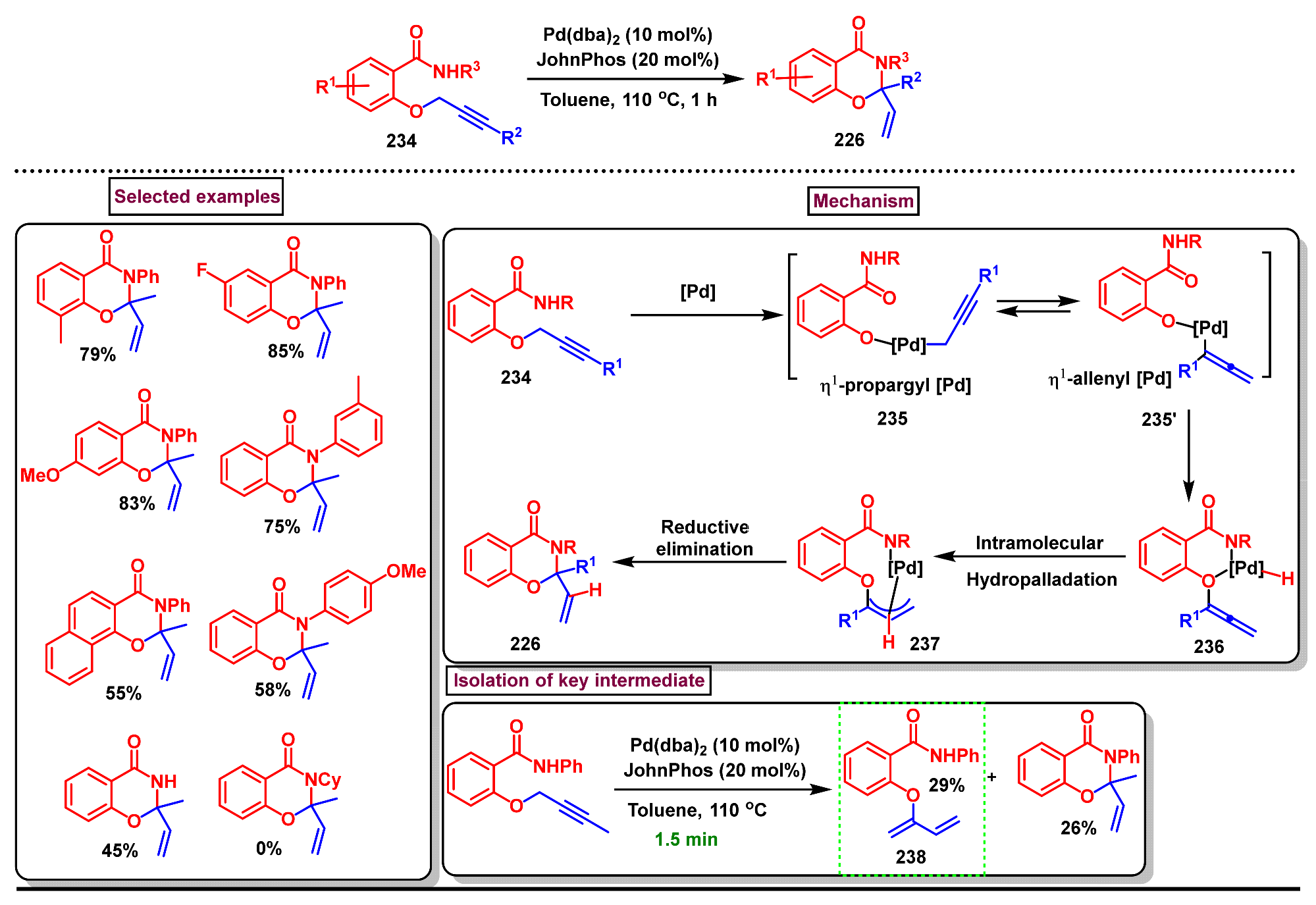
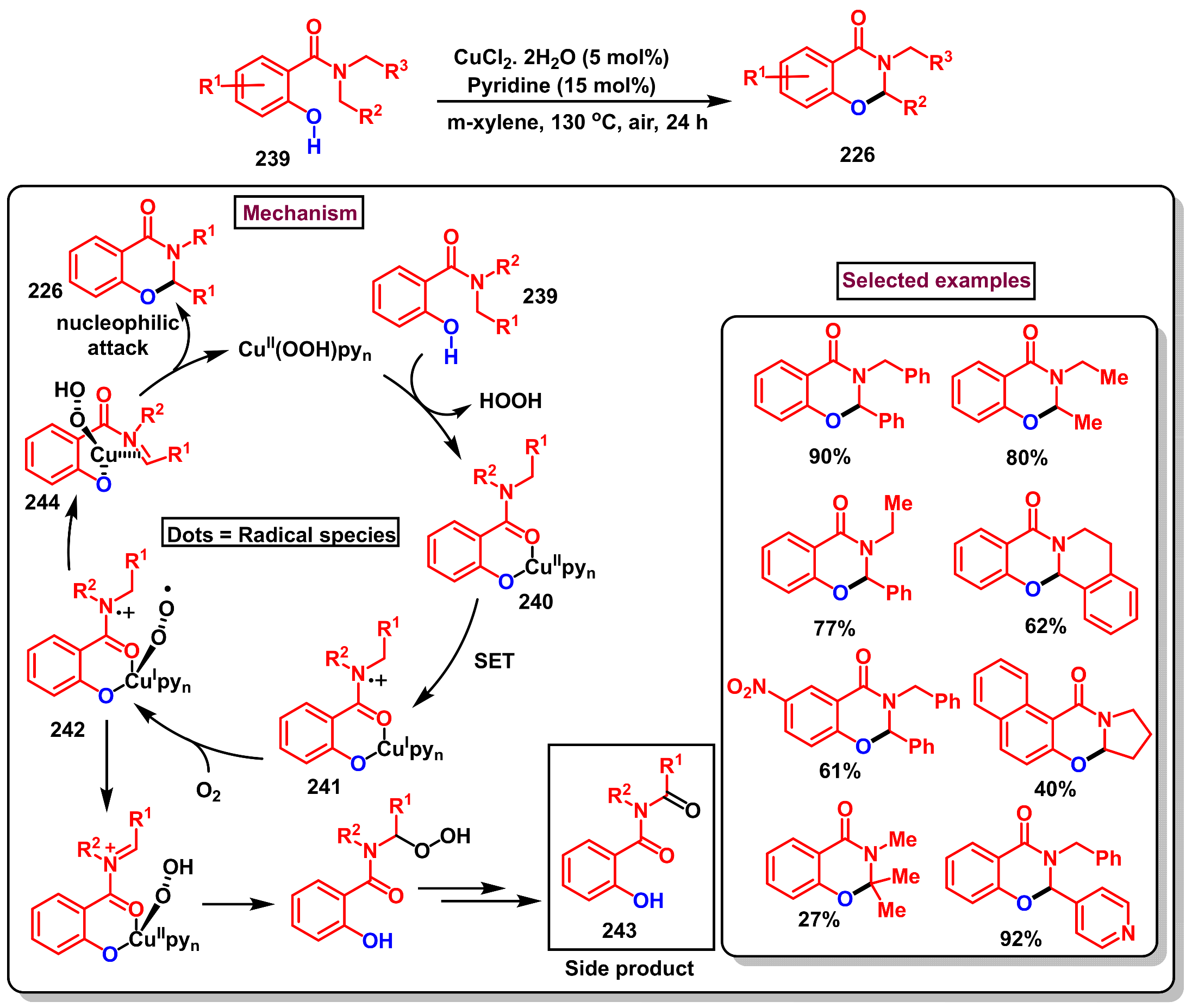
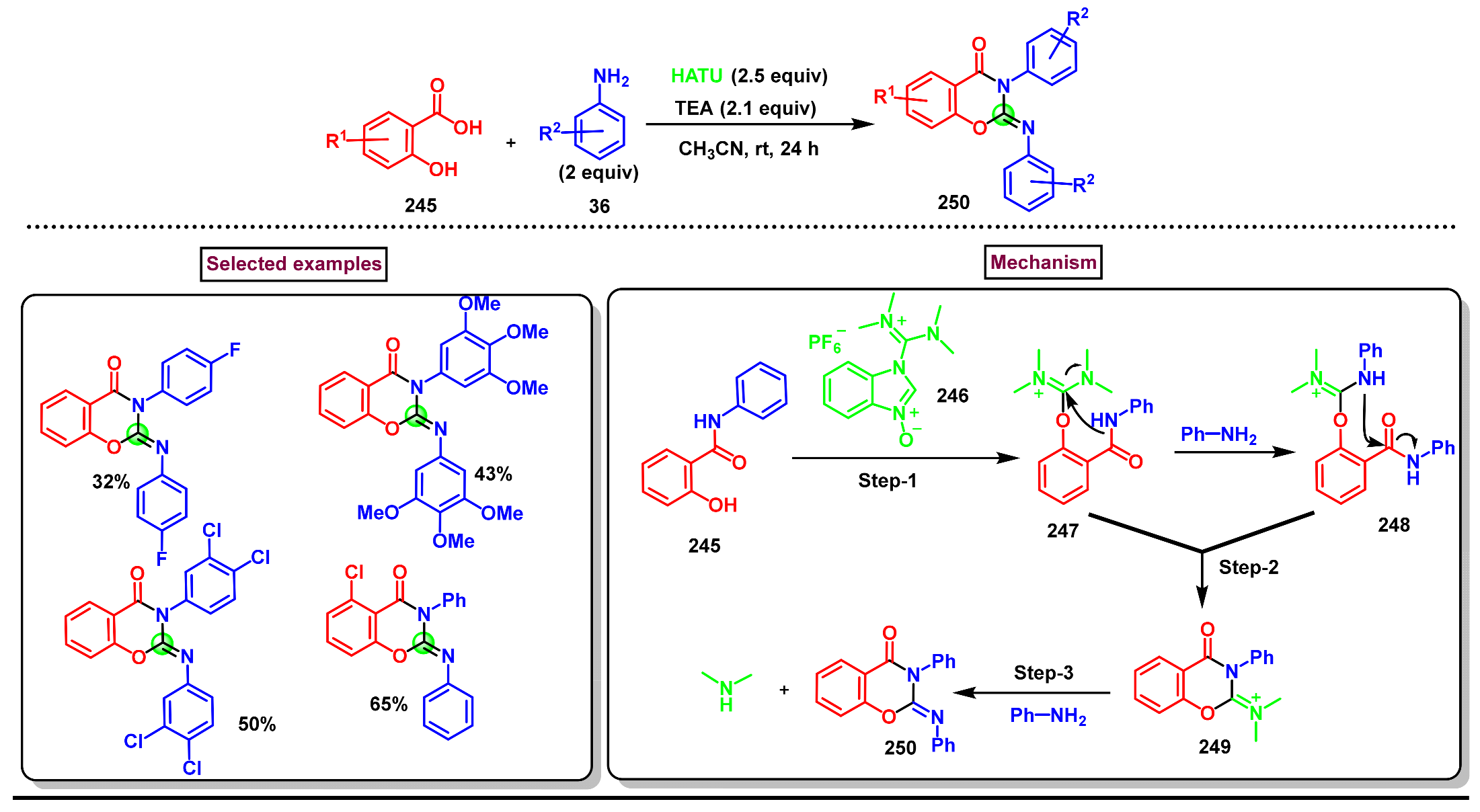
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moussa, Z.; Ramanathan, M.; Al-Masri, H.T.; Ahmed, S.A. Recent Progress in the Synthesis of Benzoxazin-4-Ones, Applications in N-Directed Ortho-Functionalizations, and Biological Significance. Molecules 2024, 29, 5710. https://doi.org/10.3390/molecules29235710
Moussa Z, Ramanathan M, Al-Masri HT, Ahmed SA. Recent Progress in the Synthesis of Benzoxazin-4-Ones, Applications in N-Directed Ortho-Functionalizations, and Biological Significance. Molecules. 2024; 29(23):5710. https://doi.org/10.3390/molecules29235710
Chicago/Turabian StyleMoussa, Ziad, Mani Ramanathan, Harbi Tomah Al-Masri, and Saleh A. Ahmed. 2024. "Recent Progress in the Synthesis of Benzoxazin-4-Ones, Applications in N-Directed Ortho-Functionalizations, and Biological Significance" Molecules 29, no. 23: 5710. https://doi.org/10.3390/molecules29235710
APA StyleMoussa, Z., Ramanathan, M., Al-Masri, H. T., & Ahmed, S. A. (2024). Recent Progress in the Synthesis of Benzoxazin-4-Ones, Applications in N-Directed Ortho-Functionalizations, and Biological Significance. Molecules, 29(23), 5710. https://doi.org/10.3390/molecules29235710