

Law and Order of Colloidal Tectonics: From Molecules to Self-Assembled Colloids


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
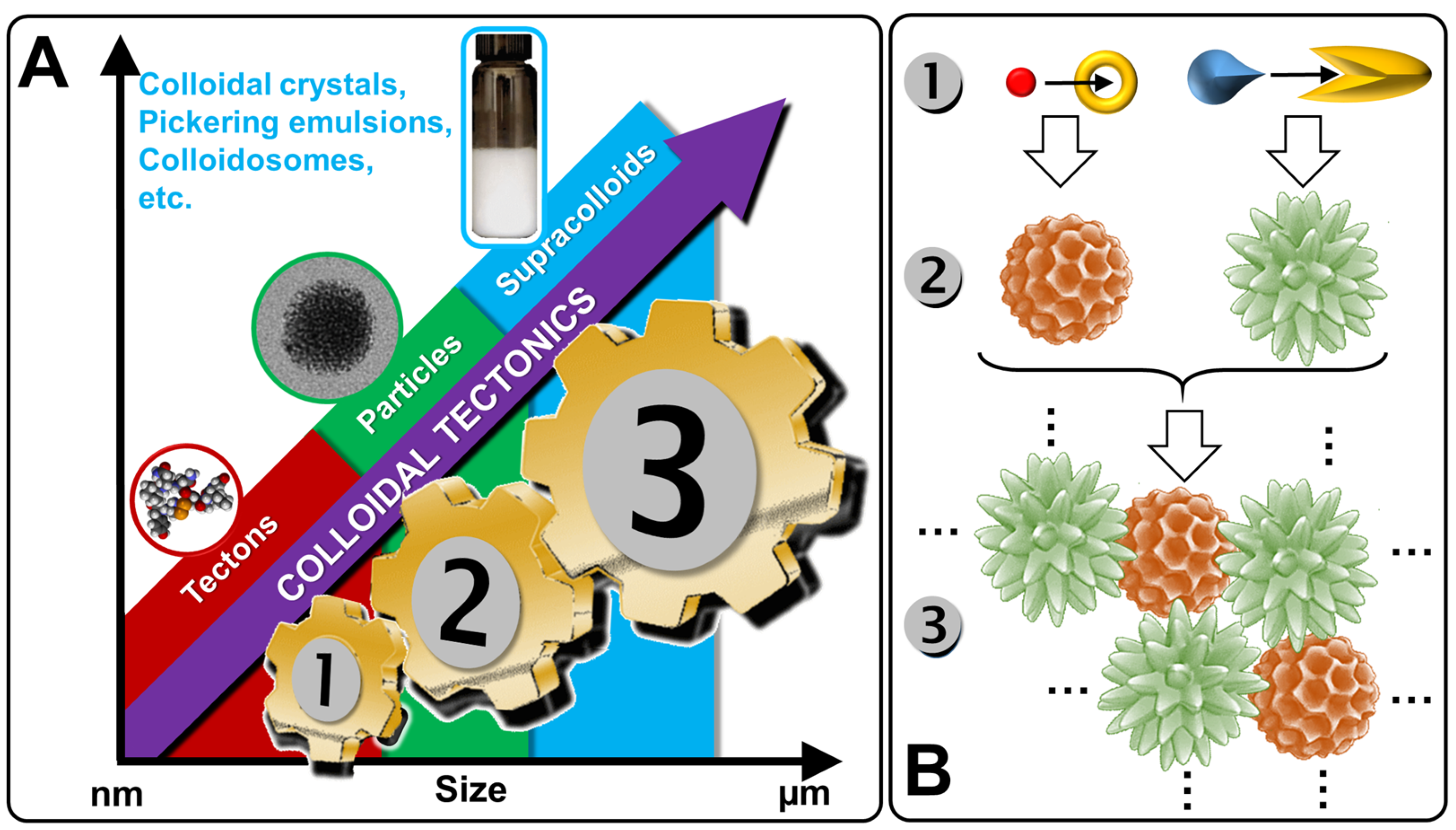
1. Introduction
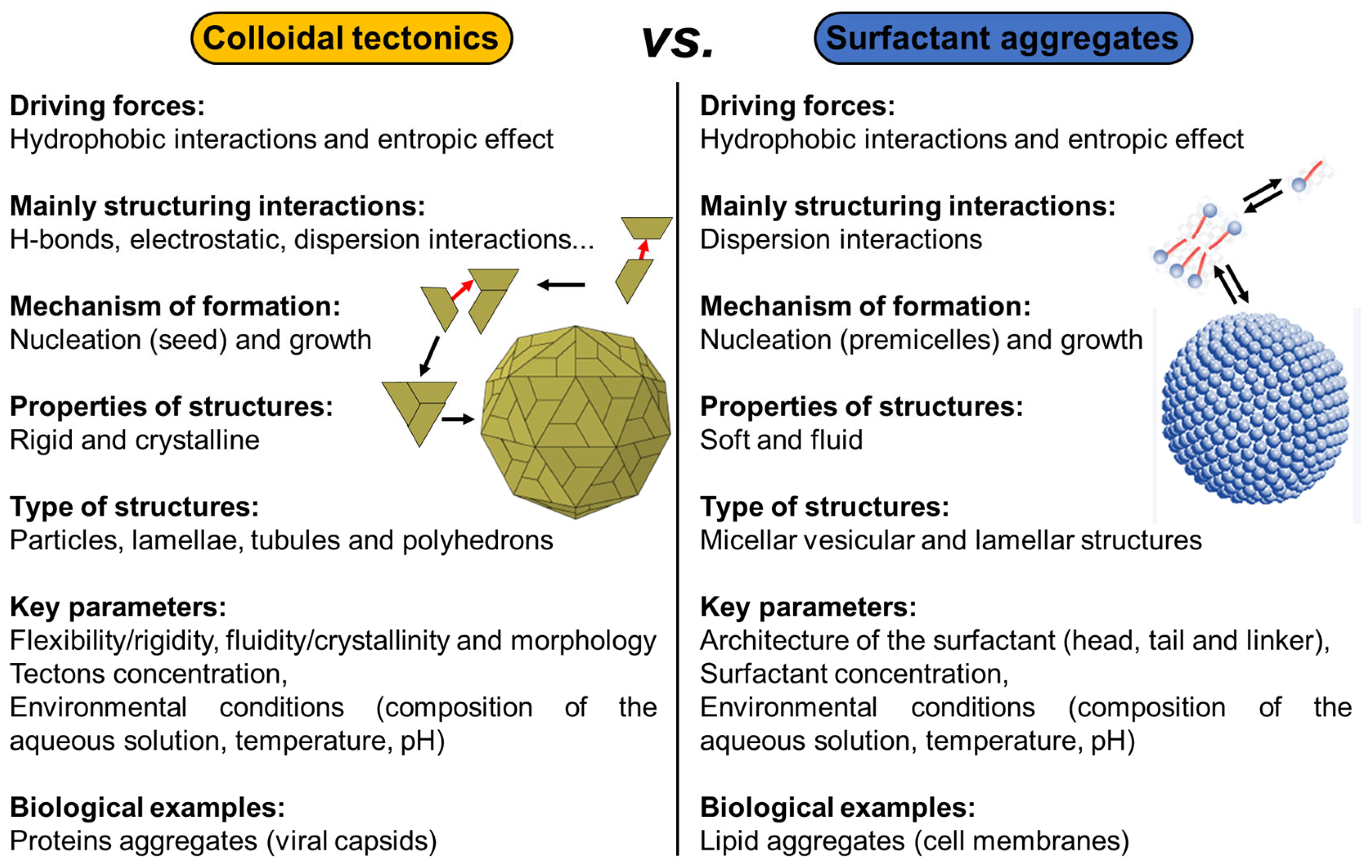
2. Colloidal Tectonics: A Bioinspired Concept
2.1. Milk Casein Particles
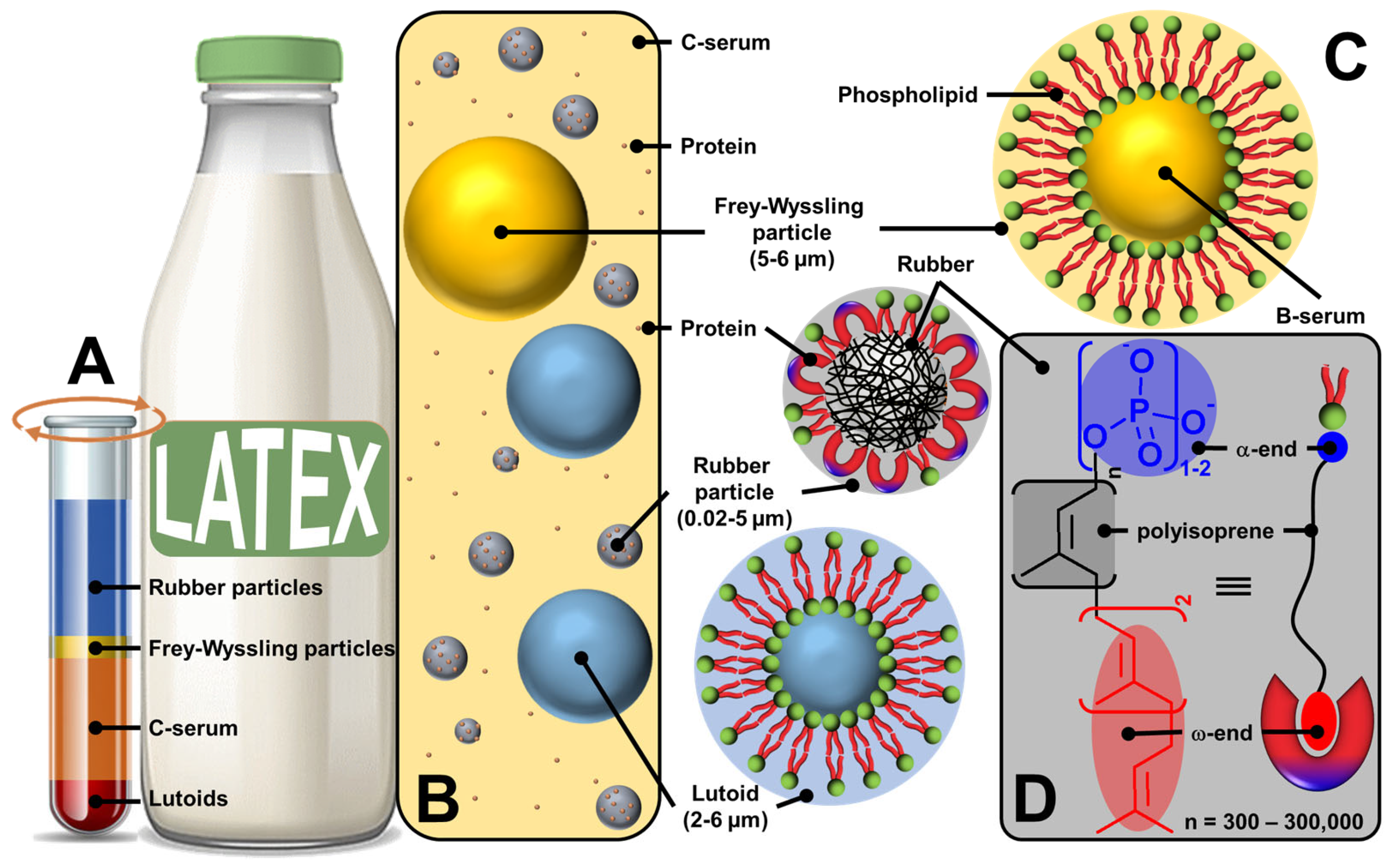
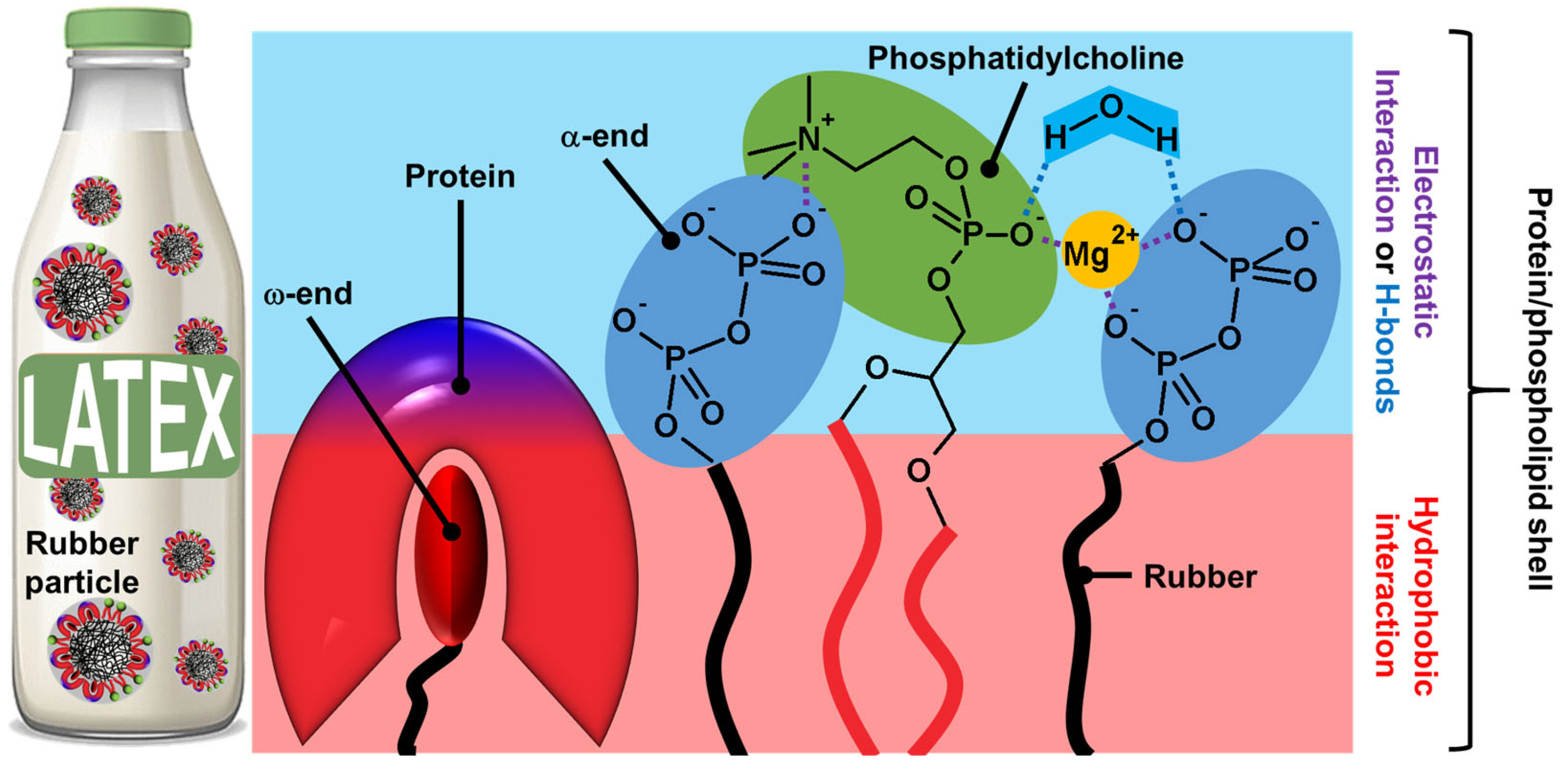
2.2. Plant Latex Particles
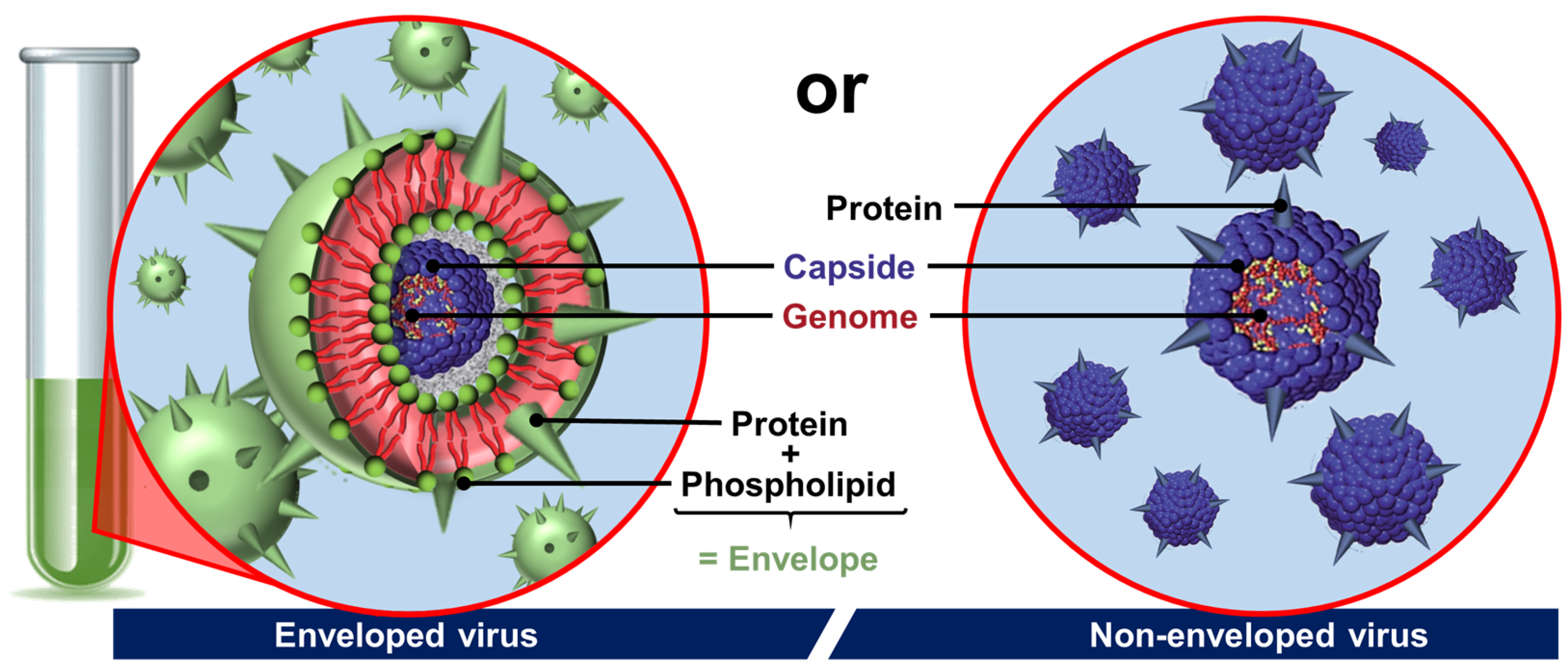
2.3. Viral Particles
2.4. Nature and Colloidal Tectonics
3. Design and Applications of Artificial Architectures Based on Colloidal Tectonics
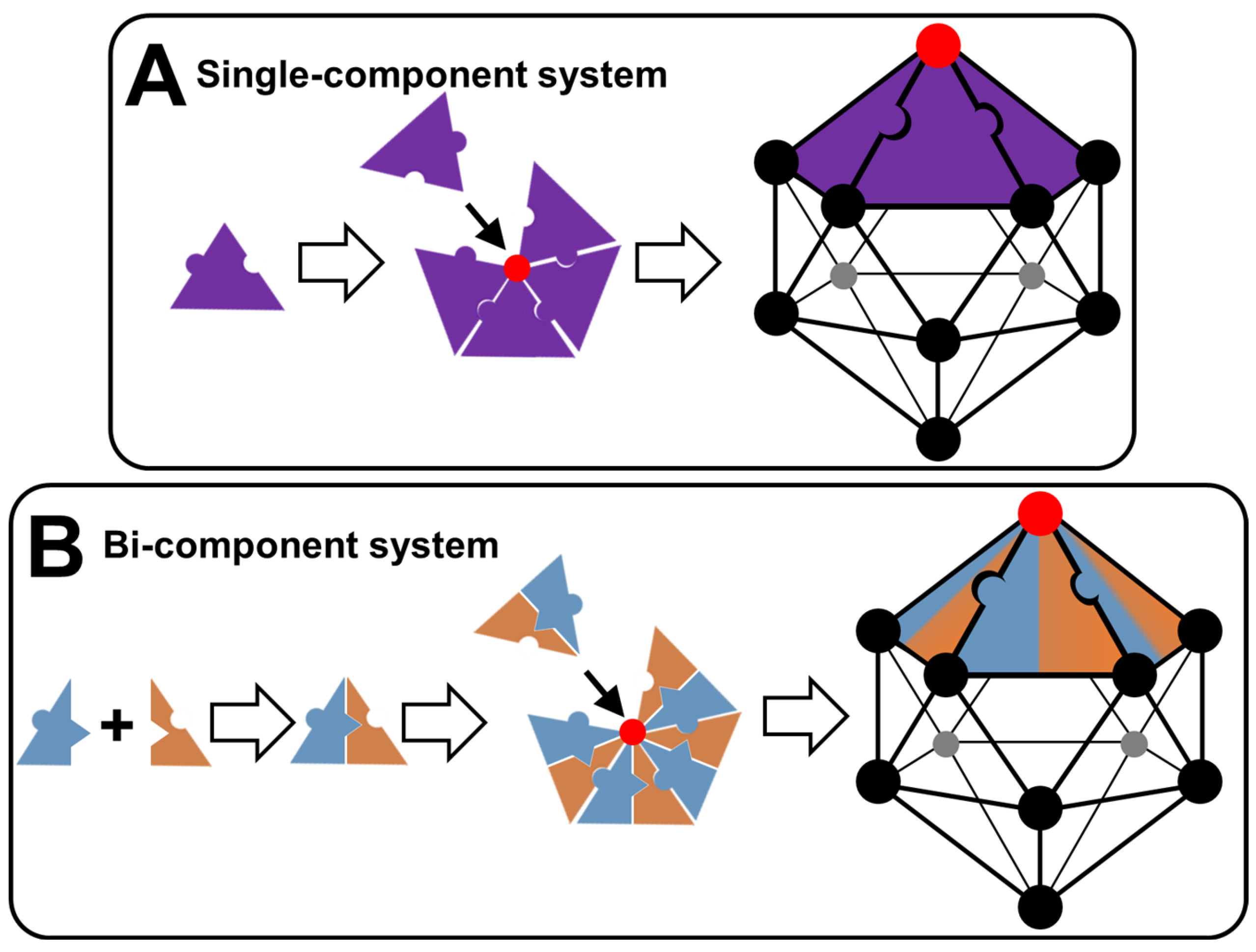
3.1. Single-Component Colloidal Systems
3.1.1. Homopolymers
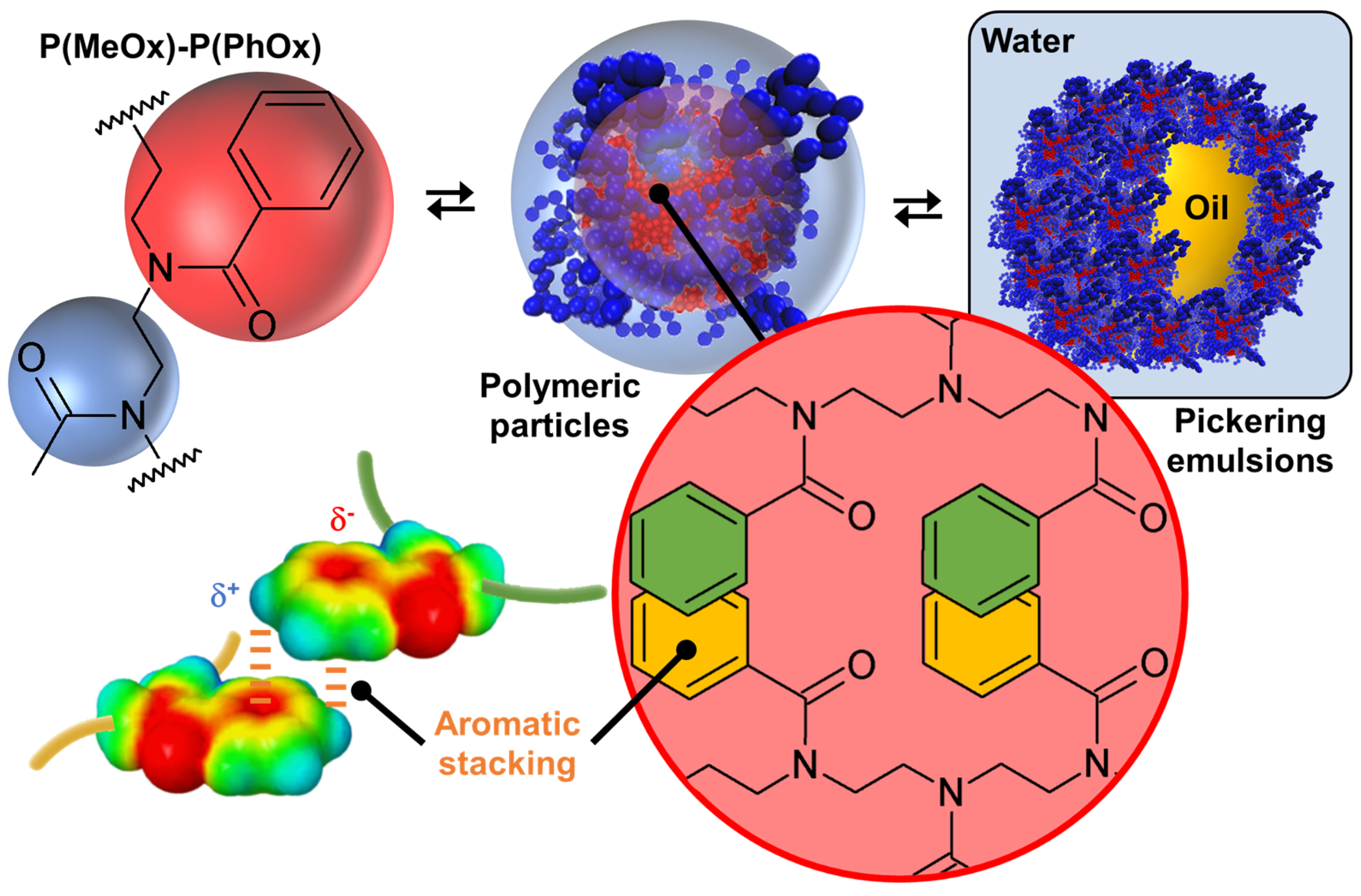
3.1.2. Copolymers
3.1.3. Surfactants
3.1.4. Other Systems
3.2. Multi-Component Colloidal Systems
3.2.1. Organic Architectures
3.2.2. Inorganic–Organic Hybrid Architectures
4. Conclusions and Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Wedlich-Söldner, R.; Betz, T. Self-organization: The fundament of cell biology. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2018, 373, 20170103. [Google Scholar] [CrossRef]
- Ruiz-Mirazo, K.; Briones, C.; de la Escosura, A. Prebiotic systems chemistry: New perspectives for the origins of life. Chem. Rev. 2014, 114, 285–366. [Google Scholar] [CrossRef] [PubMed]
- Hamley, I.W. Nanotechnology with soft materials. Angew. Chem. Int. Ed. 2003, 42, 1692–1712. [Google Scholar] [CrossRef] [PubMed]
- Sanger, F.; Tuppy, H. The amino-acid sequence in the phenylalanyl chain of insulin. I. The identification of lower peptides from partial hydrolysates. Biochem. J. 1951, 49, 463–481. [Google Scholar] [CrossRef] [PubMed]
- Pauling, L.; Corey, R.B.; Branson, H.R. The structure of proteins; two hydrogen-bonded helical configurations of the polypeptide chain. Proc. Natl. Acad. Sci. USA 1951, 37, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Wuyun, Q.; Chen, Y.; Shen, Y.; Cao, Y.; Hu, G.; Cui, W.; Gao, J.; Zheng, W. Recent progress of protein tertiary structure prediction. Molecules 2024, 29, 832. [Google Scholar] [CrossRef]
- Marciano, S.; Dey, D.; Listov, D.; Fleishman, S.J.; Sonn-Segev, A.; Mertens, H.; Busch, F.; Kim, Y.; Harvey, S.R.; Wysockid, V.H.; et al. Protein quaternary structures in solution are a mixture of multiple forms. Chem. Sci. 2022, 13, 11680–11695. [Google Scholar] [CrossRef]
- Cohen, R.D.; Pielak, G.J. Electrostatic Contributions to Protein Quinary Structure. J. Am. Chem. Soc. 2016, 138, 13139–13142. [Google Scholar] [CrossRef]
- Service, R.F. How Far Can We Push Chemical Self-Assembly? Science 2005, 309, 95. [Google Scholar] [CrossRef]
- Mattia, E.; Otto, S. Supramolecular systems chemistry. Nat. Nanotechnol. 2015, 10, 111–119. [Google Scholar] [CrossRef]
- Wang, H.; Feng, Z.; Xu, B. Bioinspired assembly of small molecules in cell milieu. Chem. Soc. Rev. 2017, 46, 2421–2436. [Google Scholar] [CrossRef] [PubMed]
- Simard, M.; Su, D.; Wuest, J.D. Use of hydrogen bonds to control molecular aggregation. Self-assembly of three-dimensional networks with large chambers. J. Am. Chem. Soc. 1991, 113, 4696–4698. [Google Scholar] [CrossRef]
- Su, D.; Wang, X.; Simard, M.; Wuest, J.D. Molecular tectonics. Supramol. Chem. 1995, 6, 171–178. [Google Scholar] [CrossRef]
- Leclercq, L.; Schmitzer, A.R. The liquid crystal state: An intermediate state to obtain crystal packing. J. Mol. Liq. 2014, 200, 283–288. [Google Scholar] [CrossRef]
- Leclercq, L. Get beyond limits: From colloidal tectonics concept to the engineering of eco-friendly catalytic systems. Front. Chem. 2018, 6, 168. [Google Scholar] [CrossRef]
- Leclercq, L.; Dechézelles, J.-F.; Rauwel, G.; Nardello-Rataj, V. In vitro study of versatile drug formulations based on α-cyclodextrin and polyethylene glycol using colloidal tectonics. J. Drug Deliv. Sci. Technol. 2020, 59, 101913. [Google Scholar] [CrossRef]
- Chai, C.; Oh, S.; Imm, J.-Y. Roles of milk fat globule membrane on fat digestion and infant nutrition. Food Sci. Anim. Resour. 2022, 42, 351–371. [Google Scholar] [CrossRef]
- Ballard, O.; Morrow, A.L. Human milk composition: Nutrients and bioactive factors. Pediatr. Clin. 2013, 60, 49–74. [Google Scholar] [CrossRef]
- Sadiq, U.; Gill, H.; Chandrapala, J. Casein micelles as an emerging delivery system for bioactive food components. Foods 2021, 10, 1965. [Google Scholar] [CrossRef]
- Gorissen, S.H.M.; Crombag, J.J.R.; Senden, J.M.G.; Huub Waterval, W.A.; Bierau, J.; Verdijk, L.B.; van Loon, L.J.C. Protein content and amino acid composition of commercially available plant-based protein isolates. Amino Acids 2018, 50, 1685–1695. [Google Scholar] [CrossRef]
- Uversky, V.N.; Dunker, A.K. Understanding Protein Non-Folding. Biochim. Biophys. Acta 2010, 1804, 1231–1264. [Google Scholar] [CrossRef] [PubMed]
- McMahon, D.J.; Oommen, B. Supramolecular Structure of the Casein Micelle. J. Dairy Sci. 2008, 91, 1709–1721. [Google Scholar] [CrossRef] [PubMed]
- Perinelli, D.R.; Cespi, M.; Lorusso, N.; Palmieri, G.P.; Bonacucina, G.; Blasi, P. Surfactant self-assembling and critical micelle concentration: One approach fits all? Langmuir 2020, 36, 5745–5753. [Google Scholar] [CrossRef]
- Huppertz, T.; Gazi, I.; Luyten, H.; Nieuwenhuijse, H.; Alting, A.; Schokker, E. Hydration of casein micelles and caseinates: Implications for casein micelle structure. Int. Dairy J. 2017, 74, 1–11. [Google Scholar] [CrossRef]
- Kamigaki, T.; Ito, Y.; Nishino, Y.; Miyazawa, A. Microstructural observation of casein micelles in milk by cryo-electron microscopy of vitreous sections (CEMOVIS). Microscopy 2018, 67, 164–170. [Google Scholar] [CrossRef]
- Møller, T.L.; Nielsen, S.B.; Pedersen, J.S.; Corredig, M. Structural and compositional characterization of Ca- and β-casein enriched casein micelles. Food Hydrocoll. 2024, 151, 109811. [Google Scholar] [CrossRef]
- Noble, R.W., Jr.; Waugh, D.F. Casein micelles. Formation and structure. I. J. Am. Chem. Soc. 1965, 87, 2236–2245. [Google Scholar] [CrossRef]
- Noble, R.W., Jr.; Waugh, D.F. Casein micelles. Formation and structure. II. J. Am. Chem. Soc. 1965, 87, 2246–2257. [Google Scholar] [CrossRef]
- Payens, T.A.J.; Schmidt, D.G. Boundary spreading of rapidly polymerizing αS1-casein B and C during sedimentation: Numerical solutions of the Lamm-Gilbert-Fujita equation. Arch. Biochem. Biophys. 1966, 115, 136–145. [Google Scholar] [CrossRef]
- Slattery, C.W.; Evard, R. A model for the formation and structure of casein micelles from subunits of variable composition. Biochim. Biophys. Acta 1973, 317, 529–538. [Google Scholar] [CrossRef]
- Schmidt, D.G. Colloidal aspects of casein. Neth. Milk Dairy J. 1980, 34, 42–64. [Google Scholar]
- Walstra, P. On the stability of casein micelles. J. Dairy Sci. 1990, 73, 1965–1979. [Google Scholar] [CrossRef]
- Walstra, P. Casein sub-micelles: Do they exist? Int. Dairy J. 1999, 9, 189–192. [Google Scholar] [CrossRef]
- Dalgleish, D.G.; Corredig, M. The structure of the casein micelle of milk and its changes during processing. Annu. Rev. Food Sci. Technol. 2012, 3, 449–467. [Google Scholar] [CrossRef]
- Horne, D.S. Casein micelle structure: Models and muddles. Curr. Opin. Colloid Interface Sci. 2006, 11, 148–153. [Google Scholar] [CrossRef]
- Casein, D.S. Interactions: Casting light on the black boxes, the structure in dairy products. Int. Dairy J. 1998, 8, 171–177. [Google Scholar] [CrossRef]
- Fox, P.F.; Brodkorb, A. The casein micelle: Historical aspects, current concepts and significance. Int. Dairy J. 2008, 18, 677–684. [Google Scholar] [CrossRef]
- Bouchoux, A.; Gésan-Guiziou, G.; Pérez, J.; Cabane, B. How to squeeze a sponge: Casein micelles under osmotic stress, a SAXS study. Biophys. J. 2010, 99, 3754–3762. [Google Scholar] [CrossRef]
- Fan, Z.; Fehér, B.; Hettinga, K.; Voets, I.K.; Bijl, E. Effect of temperature, pH and calcium phosphate concentration on the properties of reassembled casein micelles. Food Hydrocoll. 2024, 149, 109592. [Google Scholar] [CrossRef]
- Antuma, L.J.; Stadler, M.; Garamus, V.M.; Boom, R.M.; Keppler, J.K. Casein micelle formation as a calcium phosphate phase separation process: Preparation of artificial casein micelles through vacuum evaporation and membrane processes. Innov. Food Sci. Emerg. Technol. 2024, 92, 103582. [Google Scholar] [CrossRef]
- Zamora, A.; Ferragut, V.; Guamis, B.; Trujillo, A.J. Changes in the surface protein of the fat globules during ultra-high pressure homogenisation and conventional treatments of milk. Food Hydrocoll. 2012, 29, 135–143. [Google Scholar] [CrossRef]
- Rodríguez Urbina, J.C.; Osswald, T.A.; Estela Garcia, J.E.; Román, A.J. Environmentally safe preservation and stabilization of natural rubber latex in an acidic environment. SPE Polym. 2023, 4, 93–104. [Google Scholar] [CrossRef]
- Gracz-Bernaciak, J.; Mazur, O.; Nawrot, R. Functional studies of plant latex as a rich source of bioactive compounds: Focus on proteins and alkaloids. Int. J. Mol. Sci. 2021, 22, 12427. [Google Scholar] [CrossRef] [PubMed]
- Bauer, G.; Gorb, S.N.; Klein, M.-C.; Nellesen, A.; von Tapavicza, M.; Speck, T. Comparative study on plant latex particles and latex coagulation in Ficus benjamina, Campanula glomerata and three Euphorbia species. PLoS ONE 2014, 9, e113336. [Google Scholar] [CrossRef]
- Lahmar, I.; Nasri-Ayachi, M.B.; Belghith, K. Laticifer identification, rubber characterization, phenolic content, and antioxidant activity of Pergularia tomentosa latex extract. BioMed Res. Int. 2022, 2022, 7158905. [Google Scholar] [CrossRef]
- Konno, K. Plant latex and other exudates as plant defense systems: Roles of various defense chemicals and proteins contained therein. Phytochemistry 2011, 72, 1510–1530. [Google Scholar] [CrossRef]
- Nor, H.M.; Ebdon, J.R. Telechelic liquid natural rubber: A review. Prog. Polym. Sci. 1998, 23, 143–177. [Google Scholar] [CrossRef]
- Sakdapipanich, J.T. Structural characterization of natural rubber based on recent evidence from selective enzymatic treatments. J. Biosci. Bioeng. 2007, 103, 287–292. [Google Scholar] [CrossRef]
- Lam, K.L.; Yang, K.L.; Sunderasan, E.; Ong, M.T. Latex C-serum from Hevea brasiliensis induces non-apoptotic cell death in hepatocellular carcinoma cell line (HepG2). Cell Prolif. 2012, 45, 577–585. [Google Scholar] [CrossRef]
- Rochette, L.; Ghibu, S.; Richard, C.; Zeller, M.; Cottin, Y.; Vergely, C. Direct and indirect antioxidant properties of α-lipoic acid and therapeutic potential. Mol. Nutr. Food Res. 2013, 57, 114–125. [Google Scholar] [CrossRef]
- Rippel, M.M.; Leite, C.A.P.; Lee, L.T.; Galembeck, F. Formation of calcium crystallites in dry natural rubber particles. J. Colloid Int. Sci. 2005, 288, 449–456. [Google Scholar] [CrossRef]
- Buaksuntear, K.; Limarun, P.; Suethao, S.; Smitthipong, W. Non-covalent interaction on the self-healing of mechanical properties in supramolecular polymers. Int. J. Mol. Sci. 2022, 23, 6902. [Google Scholar] [CrossRef] [PubMed]
- Nawamawat, K.; Sakdapipanich, J.T.; Ho, C.C.; Ma, Y.; Song, J.; Vancso, J.G. Surface nanostructure of Hevea brasiliensis natural rubber latex particles. Colloids Surf. A Physicochem. Eng. Asp. 2011, 390, 157–166. [Google Scholar] [CrossRef]
- Wadeesirisak, K.; Castano, S.; Vaysse, L.; Bonfils, F.; Peruch, F.; Rattanaporn, K.; Liengprayoon, S.; Lecomte, S.; Bottier, C. Interactions of REF1 and SRPP1 rubber particle proteins from Hevea brasiliensis with synthetic phospholipids: Effect of charge and size of lipid headgroup. Biochem. Biophys. Res. Commun. 2023, 679, 205–214. [Google Scholar] [CrossRef]
- Dai, L.; Nie, Z.; Kang, G.; Li, Y.; Zeng, R. Identification and subcellular localization analysis of two rubber elongation factor isoforms on Hevea brasiliensis rubber particles. Plant. Physiol. Biochem. 2017, 111, 97–106. [Google Scholar] [CrossRef]
- Garcia, A.; Zou, H.; Hossain, K.R.; Xu, Q.H.; Buda, A.; Clarke, R.J. Polar interactions play an important role in the energetics of the main phase transition of phosphatidylcholine membranes. ACS Omega 2019, 4, 518–527. [Google Scholar] [CrossRef]
- De Gennes, P.G. Reptation of a polymer chain in the presence of fixed obstacles. J. Chem. Phys. 1971, 55, 572–579. [Google Scholar] [CrossRef]
- Sansatsadeekul, J.; Sakdapipanich, J.; Rojruthai, P. Characterization of associated proteins and phospholipids in natural rubber latex. J. Biosci. Bioeng. 2011, 111, 628–634. [Google Scholar] [CrossRef]
- Kumar, H.; Basavaraj, M.G. Plant latex as a versatile and sustainable emulsifier. Langmuir 2022, 38, 13217–13225. [Google Scholar] [CrossRef]
- Bruinsma, R.F.; Wuite, G.J.L.; Roos, W.H. Physics of viral dynamics. Nat. Rev. Phys. 2021, 3, 76–91. [Google Scholar] [CrossRef]
- Mateu, M.G. Assembly, stability and dynamics of virus capsids. Arch. Biochem. Biophys. 2013, 531, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Crick, F.H.; Watson, J.D. Structure of small viruses. Nature 1956, 177, 473–475. [Google Scholar] [CrossRef] [PubMed]
- Caspar, D.L.; Klug, A. Physical principles in the construction of regular viruses. Cold Spring Harb. Symp. Quant. Biol. 1962, 27, 1–24. [Google Scholar] [CrossRef]
- Rossmann, M.G.; Rao, V.B. Principles of virus structural organization. Viral Mol. Mach. 2012, 726, 17–47. [Google Scholar] [CrossRef]
- Hoh, F.; Uzest, M.; Drucker, M.; Plisson-Chastang, C.; Bron, P.; Blanc, S.; Dumas, C. Structural insights into the molecular mechanisms of cauliflower mosaic virus transmission by its insect vector. J. Virol. 2010, 84, 4706–4713. [Google Scholar] [CrossRef]
- Campbell, E.M.; Hope, T.J. HIV-1 capsid: The multifaceted key player in HIV-1 infection. Nat. Rev. Microbiol. 2015, 13, 471–483. [Google Scholar] [CrossRef]
- Lomonossoff, G.P.; Wege, C. TMV particles: The journey from fundamental studies to bionanotechnology applications. Adv. Virus Res. 2018, 102, 149–176. [Google Scholar] [CrossRef]
- Morozov, A.Y.; Bruinsma, R.F.; Rudnick, J. Assembly of viruses and the pseudo-law of mass action. J. Chem. Phys. 2009, 131, 155101. [Google Scholar] [CrossRef]
- Ting, C.L.; Wu, J.; Wang, Z.G. Thermodynamic basis for the genome to capsid charge relationship in viral encapsidation. Proc. Natl. Acad. Sci. USA 2011, 108, 16986–16991. [Google Scholar] [CrossRef]
- Zandi, R.; van der Schoot, P.; Reguera, D.; Kegel, W.; Reiss, H. Classical nucleation theory of virus capsids. Biophys. J. 2006, 90, 1939–1948. [Google Scholar] [CrossRef]
- Wilson, C.J.; Bommarius, A.S.; Champion, J.A.; Chernoff, Y.O.; Lynn, D.G.; Paravastu, A.K.; Liang, C.; Hsieh, M.-C.; Heemstra, J.M. Biomolecular assemblies: Moving from observation to predictive design. Chem. Rev. 2018, 118, 11519–11574. [Google Scholar] [CrossRef] [PubMed]
- Perlmutter, J.D.; Hagan, M.F. Mechanisms of virus assembly. Annu. Rev. Phys. Chem. 2015, 66, 217–239. [Google Scholar] [CrossRef] [PubMed]
- Niklasch, M.; Zimmermann, P.; Nassal, M. The Hepatitis B virus nucleocapsid—Dynamic compartment for infectious virus production and new antiviral target. Biomedicines 2021, 9, 1577. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.T.; Lin, Y.; Böker, A.; Su, L.; Carl, P.; Zettl, H.; He, J.; Sill, K.; Tangirala, R.; Emrick, T.; et al. Self-assembly and cross-linking of bionanoparticles at liquid-liquid interfaces. Angew. Chem. Int. Ed. 2005, 44, 2420–2426. [Google Scholar] [CrossRef]
- Kaur, G.; He, J.; Xu, J.; Pingali, S.V.; Jutz, G.; Böker, A.; Niu, Z.; Li, T.; Rawlinson, D.; Emrick, T.; et al. Interfacial Assembly of Turnip Yellow Mosaic Virus Nanoparticles. Langmuir 2009, 25, 5168–5176. [Google Scholar] [CrossRef]
- Wang, Z.; Gao, S.; Liu, X.; Tian, Y.; Wu, M.; Niu, Z. Programming self-assembly of tobacco mosaic virus coat proteins at Pickering emulsion interfaces for nanorod-constructed capsules. ACS Appl. Mater. Interfaces 2017, 9, 27383–27389. [Google Scholar] [CrossRef]
- Lee, A.G. How lipids affect the activities of integral membrane proteins. Biochim. Biophys. Acta-Biomembr. 2004, 1666, 62–87. [Google Scholar] [CrossRef]
- Pike, L.J. The challenge of lipid rafts. J. Lipid Res. 2008, 50, S323–S328. [Google Scholar] [CrossRef]
- Lombardo, D.; Kiselev, M.A.; Magazù, S.; Calandra, P. Amphiphiles self-assembly: Basic concepts and future perspectives of supramolecular approaches. Adv. Condens. Matter Phys. 2015, 2015, 151683. [Google Scholar] [CrossRef]
- Jagodzinski, F.; Clark, P.; Grant, J.; Liu, T.; Monastra, S.; Streinu, I. Rigidity analysis of protein biological assemblies and periodic crystal structures. BMC Bioinform. 2013, 14, S2. [Google Scholar] [CrossRef]
- Douyère, G.; Leclercq, L.; Nardello-Rataj, V. From polyethyleneimine hydrogels to Pickering-like smart “On/Off” emulgels switched by pH and temperature. J. Colloid Int. Sci. 2022, 628, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Douyère, G.; Leclercq, L.; Nardello-Rataj, V. Université de Lille, Lille 59000, France. 2024; unpublished work. [Google Scholar]
- Douyère, G.; Leclercq, L.; Nardello-Rataj, V. Cross-linked poly(4-vinylpyridine) particles for pH- and ionic strength-responsive “on–off” Pickering emulsions. Colloids Surf. A Physicochem. Eng. Asp. 2021, 631, 127705. [Google Scholar] [CrossRef]
- Bardoula, V.; Leclercq, L.; Hoogenboom, R.; Nardello-Rataj, V. Amphiphilic polymeric particles based on gradient and block copoly(2-oxazoline)s: Interplay between structure and Pickering emulsion properties. Colloids Surf. A Physicochem. Eng. Asp. 2024, 699, 134634. [Google Scholar] [CrossRef]
- Rivas, C.J.M.; Tarhini, M.; Badri, W.; Miladi, K.; Greige-Gerges, H.; Nazari, Q.A.; Galindo Rodríguez, S.A.; Román, R.A.; Fessi, H.; Elaissari, A. Nanoprecipitation process: From encapsulation to drug delivery. Int. J. Pharm. 2017, 532, 66–81. [Google Scholar] [CrossRef]
- Martinez, C.R.; Iverson, B.L. Rethinking the term “pi-stacking”. Chem. Sci. 2012, 3, 2191–2201. [Google Scholar] [CrossRef]
- Kohlan, T.B.; Atespare, A.E.; Yildiz, M.; Menceloglu, Y.Z.; Unal, S.; Dizman, B. Synthesis and structure–property relationship of amphiphilic poly(2-ethyl-co-2-(alkyl/aryl)-2-oxazoline) copolymers. ACS Omega 2022, 7, 40067–40077. [Google Scholar] [CrossRef]
- Valentin Bardoula, V.; Leclercq, L.; Hoogenboom, R.; Nardello-Rataj, V. Amphiphilic nonionic block and gradient copoly(2-oxazoline)s based on 2-methyl-2-oxazoline and 2-phenyl-2-oxazoline as efficient stabilizers for the formulation of tailor-made emulsions. J. Colloid Int. Sci. 2023, 632, 223–236. [Google Scholar] [CrossRef]
- Neuhaus, F.; Mueller, D.; Tanasescu, R.; Balog, S.; Ishikawa, T.; Brezesinski, G.; Zumbuehl, A. Vesicle origami: Cuboid phospholipid vesicles formed by template-free self-assembly. Angew. Chem. Int. Ed. 2017, 56, 6515–6518. [Google Scholar] [CrossRef]
- Tanasescu, R.; Lanz, M.A.; Mueller, D.; Tassler, S.; Ishikawa, T.; Reiter, R.; Brezesinski, G.; Zumbuehl, A. Vesicle origami and the influence of cholesterol on lipid packing. Langmuir 2016, 32, 4896–4903. [Google Scholar] [CrossRef]
- Weinberger, A.; Tanasescu, R.; Stefaniu, C.; Fedotenko, L.A.; Favarger, F.; Ishikawa, T.; Brezesinski, G.; Marques, C.M.; Zumbuehl, A. Bilayer Properties of 1,3-diamidophospholipids. Langmuir 2015, 31, 1879–1884. [Google Scholar] [CrossRef]
- Holme, M.N.; Fedotenko, I.A.; Abegg, D.; Althaus, J.; Babel, L.; Favarger, F.; Reiter, R.; Tanasescu, R.; Zaffalon, P.L.; Ziegler, A.; et al. Shear-stress sensitive lenticular vesicles for targeted drug delivery. Nat. Nanotechnol. 2012, 7, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Zumbuehl, A. Artificial phospholipids and their vesicles. Langmuir 2019, 35, 10223–10232. [Google Scholar] [CrossRef] [PubMed]
- Pardin, C.; Leclercq, L.; Schmitzer, A.R. N,N’-Methylenediimidazolium salts: From self-assembly to an efficient DNAse protection system. Chem. Eur. J. 2010, 16, 4686–4692. [Google Scholar] [CrossRef]
- Dong, K.; Zhang, S.; Wang, D.; Yao, X. Hydrogen bonds in imidazolium ionic liquids. J. Phys. Chem. A 2006, 110, 9775–9782. [Google Scholar] [CrossRef]
- Valéry, C.; Paternostre, M.; Robert, B.; Gulik-Krzywicki, T.; Narayanan, T.; Dedieu, J.-C.; Keller, G.; Torres, M.-L.; Cherif-Cheikh, R.; Calvo, P.; et al. Biomimetic organization: Octapeptide self-assembly into nanotubes of viral capsid-like dimension. Proc. Natl. Acad. Sci. USA 2004, 100, 10258–10262. [Google Scholar] [CrossRef]
- Hill, J.P.; Jin, W.; Kosaka, A.; Fukushima, T.; Ichihara, H.; Shimomura, T.; Ito, K.; Hashizume, T.; Ishii, N.; Aida, T. Self-assembled hexa-peri-hexabenzocoronene graphitic nanotube. Science 2004, 304, 1481–1483. [Google Scholar] [CrossRef]
- Leclercq, L. Interactions between cyclodextrins and cellular components: Towards greener medical applications? Beilstein J. Org. Chem. 2016, 12, 2644–2662. [Google Scholar] [CrossRef]
- Saenger, W. Crystal packing patterns of cyclodextrin inclusion complexes. J. Incl. Phenom. 1984, 2, 445–454. [Google Scholar] [CrossRef]
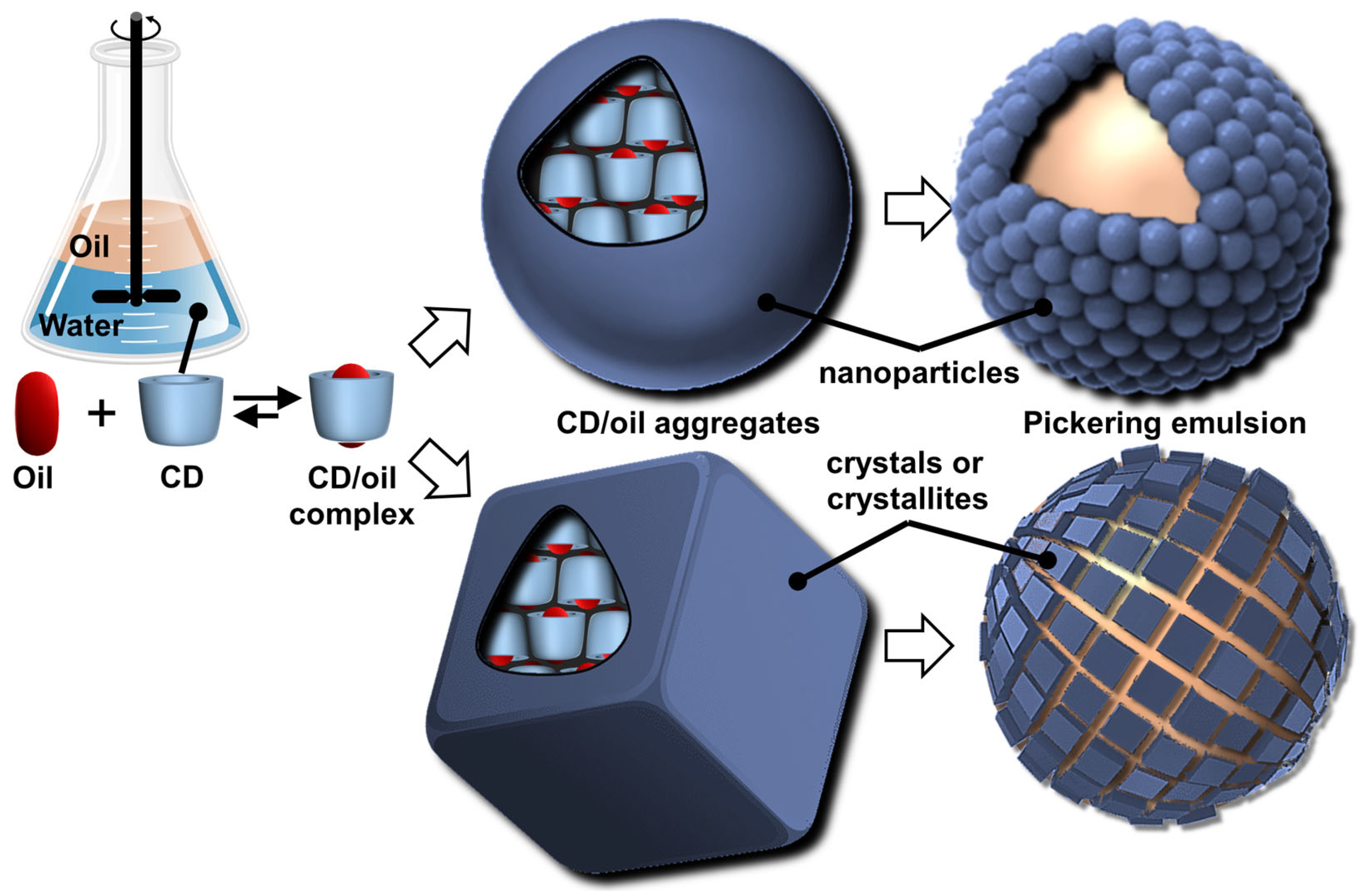
- Hashizaki, K.; Kageyama, T.; Inoue, M.; Taguchi, H.; Ueda, H.; Saito, Y. Study on preparation and formation mechanism of n-alkanol/water emulsion using α-cyclodextrin. Chem. Pharm. Bull. 2007, 55, 1620–1625. [Google Scholar] [CrossRef]
- Davarpanah, L.; Vahabzadeh, F. Formation of oil-in-water (O/W) pickering emulsions via complexation between β-cyclodextrin and selected organic solvents. Starch-Särke 2021, 64, 898–913. [Google Scholar] [CrossRef]
- Leclercq, L.; Tessier, J.; Douyère, G.; Nardello-Rataj, V.; Schmitzer, A.R. Phytochemical- and cyclodextrin-based Pickering emulsions: Natural potentiators of antibacterial, antifungal, and antibiofilm activity. Langmuir 2020, 36, 4317–4323. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, L.; Company, R.; Mühlbauer, A.; Mouret, A.; Aubry, J.-M.; Nardello-Rataj, V. Versatile eco-friendly Pickering emulsions based on substrate/native cyclodextrin complexes: A winning approach for solvent-free oxidations. ChemSusChem 2013, 6, 1533–1540. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Xu, H.-N. Faceted crystal growth of cyclodextrin-oil inclusion complexes. Carbohydr. Polym. 2024, 343, 122446. [Google Scholar] [CrossRef]
- Leclercq, L.; Tessier, J.; Nardello-Rataj, V.; Schmitzer, A.R. Highly active, entirely biobased antimicrobial Pickering emulsions. ChemMedChem 2021, 16, 2223–2230. [Google Scholar] [CrossRef]
- Mathapa, B.G.; Paunov, V.N. Cyclodextrin stabilised emulsions and cyclodextrinosomes. Phys. Chem. Chem. Phys. 2013, 15, 17903–17914. [Google Scholar] [CrossRef]
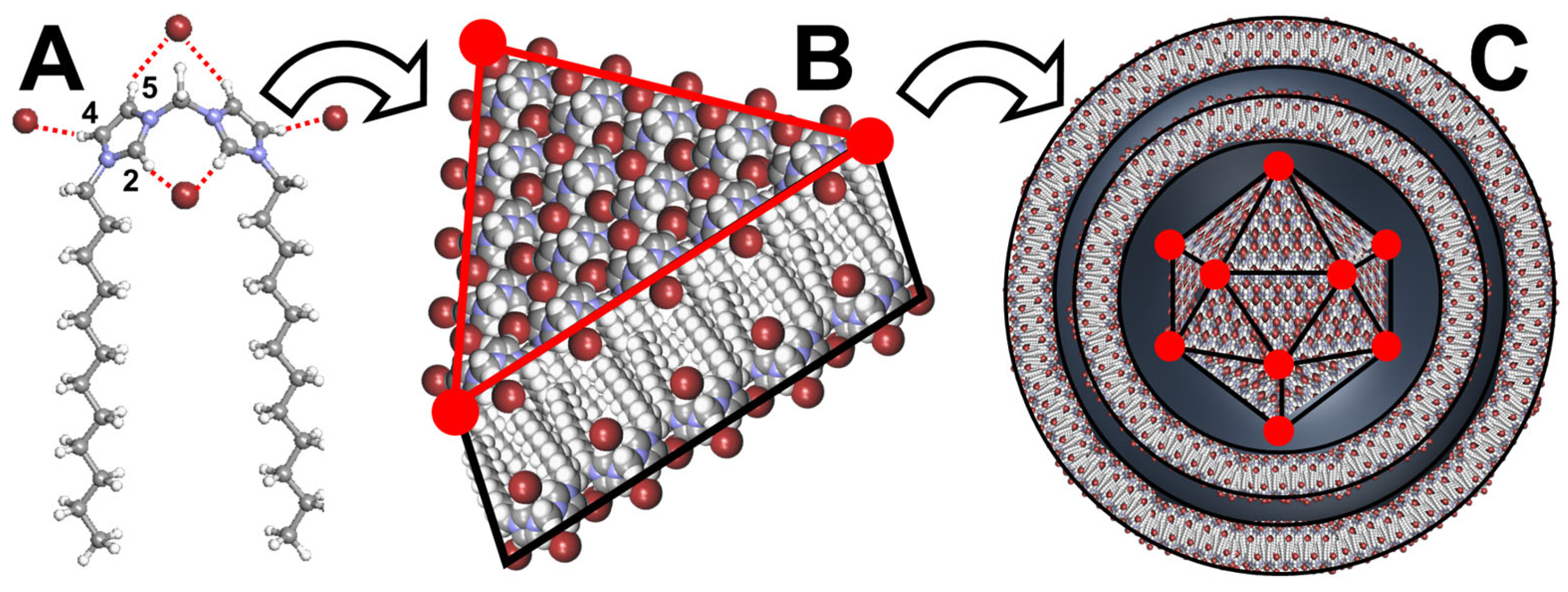
- Bochot, A.; Trichard, L.; Le Bas, G.; Alphandary, H.; Grossiord, J.-L.; Duchêne, D.; Fattal, E. α-Cyclodextrin/oil beads: An innovative self-assembling system. Int. J. Pharm. 2007, 339, 121–129. [Google Scholar] [CrossRef]
- Hamoudi, C.; Bochot, A. Oil-cyclodextrin based beads for oral delivery of poorly-soluble drugs. Curr. Top. Med. Chem. 2014, 14, 510–517. [Google Scholar] [CrossRef]
- Trichard, L.; Fatta, E.; Besnard, M.; Bochot, A. α-Cyclodextrin/oil beads as a new carrier for improving the oral bioavailability of lipophilic drugs. J. Control. Release 2007, 122, 47–53. [Google Scholar] [CrossRef]
- Trichard, L.; Delgado-Charro, M.B.; Guy, R.H.; Fattal, E.; Bochot, A. Novel beads made of α-cyclodextrin and oil for topical delivery of a lipophilic drug. Pharm. Res. 2008, 25, 435–440. [Google Scholar] [CrossRef]
- Trichard, L.; Chaminade, P.; Grossior, J.-L.; Le Bas, G.; Huang, N.; Durand, D.; Fattal, E.; Bochot, A. Beads made of α-cyclodextrin and vegetable oils: Oil composition and physicochemical properties influence bead feasibility and properties. J. Drug Deliv. Sci. Technol. 2011, 21, 189–194. [Google Scholar] [CrossRef]
- Potier, J.; Menuel, S.; Chambrier, M.-H.; Burylo, L.; Blach, J.-F.; Woisel, P.; Monflier, E.; Hapiot, F. Pickering emulsions based on supramolecular hydrogels: Application to higher olefins’ hydroformylation. ACS Catal. 2013, 3, 1618–1621. [Google Scholar] [CrossRef]
- Potier, J.; Menuel, S.; Monflier, E.; Hapiot, F. Synergetic Effect of randomly methylated β-cyclodextrin and a supramolecular hydrogel in Rh-catalyzed hydroformylation of higher olefins. ACS Catal. 2014, 4, 2342–2346. [Google Scholar] [CrossRef]
- Chevry, M.; Vanbésien, T.; Menuel, S.; Monflier, E.; Hapiot, F. Tetronics/cyclodextrin-based hydrogels as catalyst-containing media for the hydroformylation of higher olefins. Catal. Sci. Technol. 2017, 7, 114–123. [Google Scholar] [CrossRef]
- Lin, C.E.; Huang, H.C.; Chen, H.W. A capillary electrophoresis study on the influence of β-cyclodextrin on the critical micelle concentration of sodium dodecyl sulfate. J. Chromatogr. A 2001, 917, 297–310. [Google Scholar] [CrossRef]
- Jiang, L.; Peng, Y.; Yan, Y.; Deng, M.; Wang, Y.; Huang, J. “Annular ring” microtubes formed by SDS@2β-CD complexes in aqueous solution. Soft Matter 2010, 6, 1731–1736. [Google Scholar] [CrossRef]
- Jiang, L.; Peng, Y.; Yan, Y.; Huang, J. Aqueous self-assembly of SDS@2β-CD complexes: Lamellae and vesicles. Soft Matter 2011, 7, 1726–1731. [Google Scholar] [CrossRef]
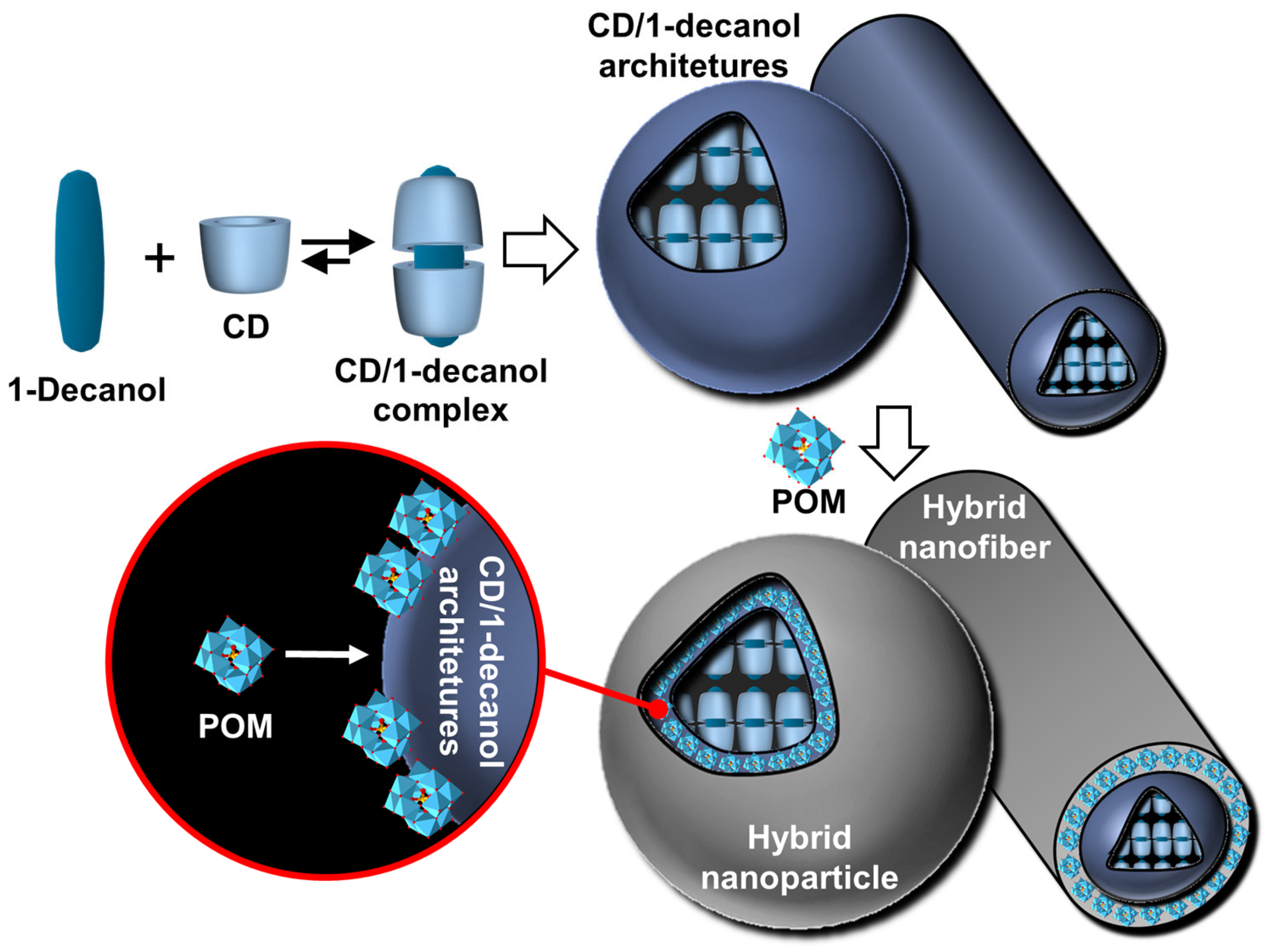
- Yang, S.; Yan, Y.; Huang, J.; Petukhov, A.V.; Kroon-Batenburg, L.M.J.; Drechsler, M.; Zhou, C.; Tu, M.; Granick, S.; Jiang, L. Giant capsids from lattice self-assembly of cyclodextrin complexes. Nat. Commun. 2017, 8, 15856. [Google Scholar] [CrossRef]
- dos Santos Silva Araújo, L.; Lazzara, G.; Chiappisi, L. Cyclodextrin/surfactant inclusion complexes: An integrated view of their thermodynamic and structural properties. Adv. Colloid Interface Sci. 2021, 289, 102375. [Google Scholar] [CrossRef]
- Liu, Z.; Zhao, Q.; Gao, S.; Yan, Y.; Xu, B.; Ma, C.; Huang, J. Multi-responsive self-assembly of polymeric-like building blocks nC12C6C12(Me)@nγ-CD: Vesicles, nanotubes and sheet crystal. Colloids Surf. A Physicochem. Eng. Asp. 2023, 676, 132269. [Google Scholar] [CrossRef]
- Wu, C.; Xie, Q.; Xu, W.; Tu, M.; Jiang, L. Lattice self-assembly of cyclodextrin complexes and beyond. Curr. Opin. Colloid Interface Sci. 2019, 39, 76–85. [Google Scholar] [CrossRef]
- Liu, K.; Ma, C.; Wu, T.; Qi, W.; Yan, Y.; Huang, J. Recent advances in assemblies of cyclodextrins and amphiphiles: Construction and regulation. Curr. Opin. Colloid Interface Sci. 2020, 45, 44–56. [Google Scholar] [CrossRef]
- Gu, T.; Huang, J.; Yan, Y. New opportunities for cyclodextrins in supramolecular assembly: Metal organic frameworks, crystalline self-assembly, and catalyzed assembly. Chem. Commun. 2023, 59, 14759–14775. [Google Scholar] [CrossRef] [PubMed]
- Landman, J.; Ouhajji, S.; Prévost, S.; Narayanan, T.; Groenewold, J.; Philipse, A.P.; Kegel, W.K.; Petukhov, A.V. Inward growth by nucleation: Multiscale self-assembly of ordered membranes. Sci. Adv. 2018, 4, eaat1817. [Google Scholar] [CrossRef] [PubMed]
- Dubois, M.; Demé, B.; Gulik-Krzywicki, T.; Dedieu, J.-C.; Vautrin, C.; Désert, S.; Perez, E.; Zemb, T. Self-assembly of regular hollow icosahedra in salt-free catanionic solutions. Nature 2001, 411, 672–675. [Google Scholar] [CrossRef]
- Dubois, M.; Lizunov, V.; Meister, A.; Gulik-Krzywicki, T.; Verbavatz, J.M.; Perez, E.; Zimmerberg, J.; Zemb, T. Shape control through molecular segregation in giant surfactant aggregates. Proc. Natl. Acad. Sci. USA 2004, 101, 15082. [Google Scholar] [CrossRef]
- Greenfield, M.A.; Palmer, L.C.; Vernizzi, G.; Olvera de la Cruz, M.; Stupp, S.I. Buckled membranes in mixed-valence ionic amphiphile vesicles. J. Am. Chem. Soc. 2009, 131, 12030–12031. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, G.; Song, A.; Wang, L.; Lin, M.; Dong, Z.; Yang, Z. Faceted fatty acid vesicles formed from single-tailed perfluorinated surfactants. Soft Matter 2015, 11, 7143–7150. [Google Scholar] [CrossRef]
- Bowick, M.J.; Sknepnek, R. Pathways to faceting of vesicles. Soft Matter 2013, 9, 8088–8095. [Google Scholar] [CrossRef]
- Guttman, S.; Ocko, B.M.; Deutsch, M.; Sloutskin, E. From faceted vesicles to liquid icoshedra: Where topology and crystallography meet. Curr. Opin. Colloid Interface Sci. 2016, 22, 35–40. [Google Scholar] [CrossRef]
- Shen, Y.; Ou-Yang, Z.; Hao, J.; Lin, H.; Jiang, L.; Liu, Z.; Gao, X. The mechanism for the transition from vesicles to punctured lamellae and faceted vesicles in cationic and anionic fluorinated surfactant mixture. Colloids Surf. A Physicochem. Eng. Asp. 2016, 500, 40–44. [Google Scholar] [CrossRef]
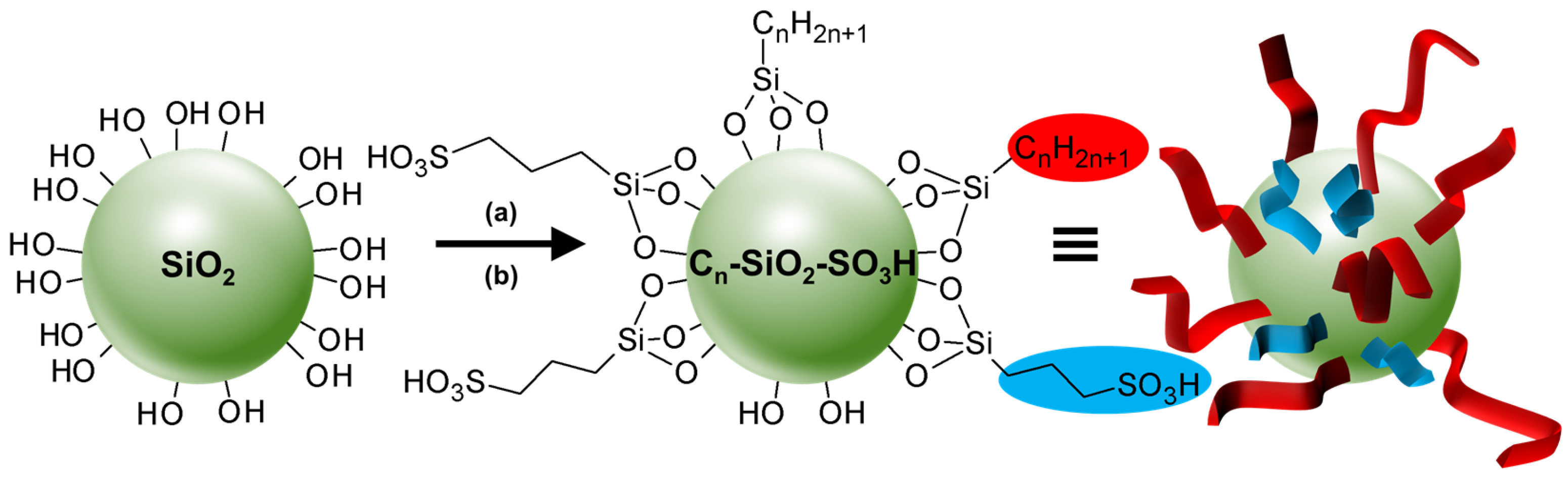
- Leclercq, L.; Mouret, A.; Proust, A.; Schmitt, V.; Bauduin, P.; Aubry, J.-M.; Nardello-Rataj, V. Pickering emulsion stabilized by catalytic polyoxometalate nanoparticles: A new effective medium for oxidation reactions. Chem. Eur. J. 2012, 8, 14352–14358. [Google Scholar] [CrossRef] [PubMed]
- Bonhomme, F.; Kanatzidis, M.G. Structurally characterized mesostructured hybrid surfactant−inorganic lamellar phases containing the adamantane [Ge4S10]4− anion: Synthesis and properties. Chem. Mater. 1998, 10, 1153–1159. [Google Scholar] [CrossRef]
- Leclercq, L.; Mouret, A.; Bauduin, P.; Nardello-Rataj, V. Supramolecular colloidosomes based on tri(dodecyltrimethylammonium) phosphotungstate: A bottom-up approach. Langmuir 2014, 30, 5386–5393. [Google Scholar] [CrossRef] [PubMed]
- Leclercq, L.; Mouret, A.; Renaudineau, S.; Schmitt, V.; Proust, A.; Nardello-Rataj, V. Self-assembled polyoxometalates nanoparticles as pickering emulsion stabilizers. J. Phys. Chem. B 2015, 119, 6326–6337. [Google Scholar] [CrossRef]
- Mouret, A.; Leclercq, L.; Mühlbauer, A.; Nardello-Rataj, V. Eco-friendly solvents and amphiphilic catalytic polyoxometalate nanoparticles: A winning combination for olefin epoxidation. Green Chem. 2014, 16, 269–278. [Google Scholar] [CrossRef]
- Wu, Y.; Shi, R.; Wu, Y.-L.; Holcroft, J.M.; Liu, Z.; Frasconi, M.; Wasielewski, M.R.; Li, H.; Stoddart, J.F. Complexation of polyoxometalates with cyclodextrins. J. Am. Chem. Soc. 2015, 137, 4111–4118. [Google Scholar] [CrossRef]
- Naskar, B.; Diat, O.; Nardello-Rataj, V.; Bauduin, P. Nanometer-size polyoxometalate anions adsorb strongly on neutral soft surfaces. J. Phys. Chem. C 2015, 119, 20985–20992. [Google Scholar] [CrossRef]
- Girard, L.; Naskar, B.; Dufrêche, J.-F.; Lai, J.; Diat, O.; Bauduin, P. A thermodynamic model of non-ionic surfactants’ micellization in the presence of polyoxometalates. J. Mol. Liq. 2019, 293, 111280. [Google Scholar] [CrossRef]
- Pacaud, B.; Leclercq, L.; Dechézelles, J.-F.; Nardello-Rataj, V. Hybrid core-shell nanoparticles by “plug and play” self-assembly. Chem. Eur. J. 2018, 24, 17672–17676. [Google Scholar] [CrossRef]
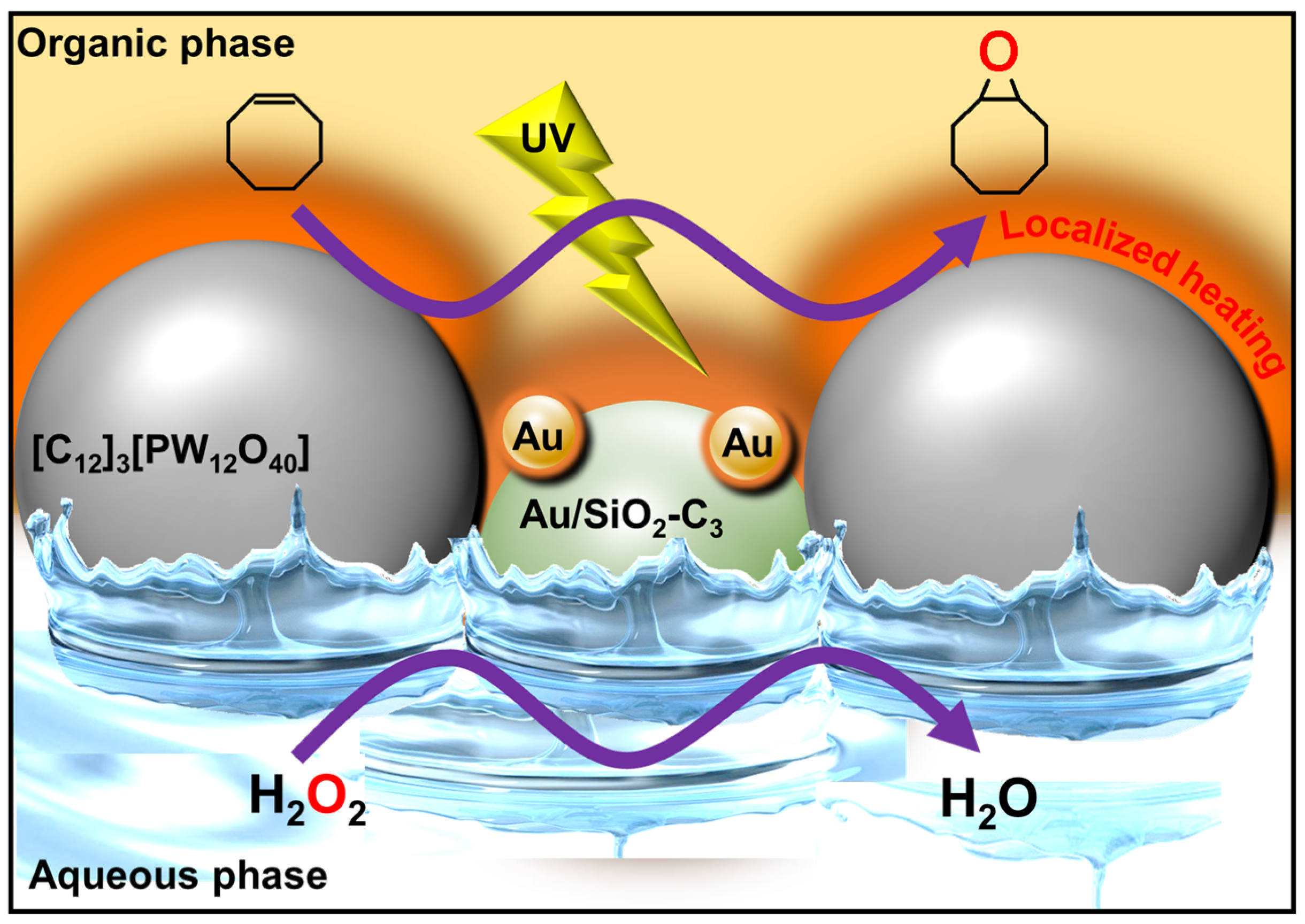
- Yang, B.; Leclercq, L.; Schmitt, V.; Pera-Titus, M.; Nardello-Rataj, V. Colloidal tectonics for tandem synergistic Pickering interfacial catalysis: Oxidative cleavage of cyclohexene oxide into adipic acid. Chem. Sci. 2019, 10, 501–507. [Google Scholar] [CrossRef]
- Feng, Y.; Dechezelles, J.-F.; D’Acremont, Q.; Courtade, E.; De Waele, V.; Pera-Titus, M.; Nardello-Rataj, V. Light-driven Pickering interfacial catalysis for the oxidation of alkenes at near-room temperature. Green Chem. 2023, 25, 1417–1423. [Google Scholar] [CrossRef]
- van Blaaderen, A. Colloidal molecules and beyond. Science 2003, 301, 470–471. [Google Scholar] [CrossRef] [PubMed]
- Manoharan, V.N.; Elsesser, M.T.; Pine, D.J. Dense packing and symmetry in small clusters of microspheres. Science 2003, 301, 483–487. [Google Scholar] [CrossRef]
- Kim, Y.-J.; Moon, J.-B.; Hwang, H.; Kim, Y.S.; Yi, G.-R. Advances in colloidal building blocks: Toward patchy colloidal clusters. Adv. Mater. 2023, 35, 2203045. [Google Scholar] [CrossRef]
- Cho, Y.-S.; Kim, S.-H.; Yi, G.-R.; Yang, S.-M. Self-organization of colloidal nanospheres inside emulsion droplets: Higher-order clusters, supraparticles, and supraballs. Colloids Surf. A Physicochem. Eng. Asp. 2009, 345, 237–245. [Google Scholar] [CrossRef]







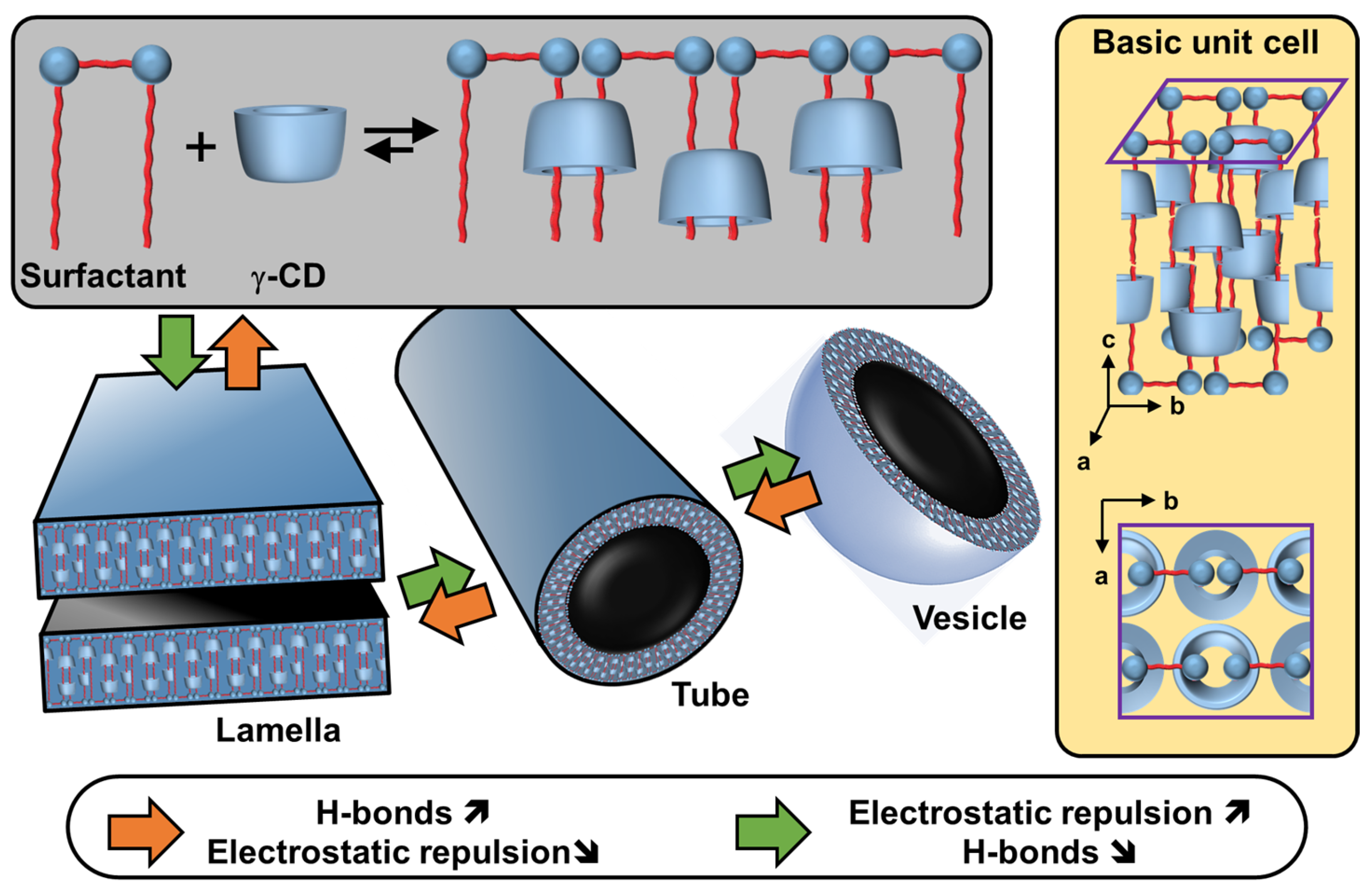
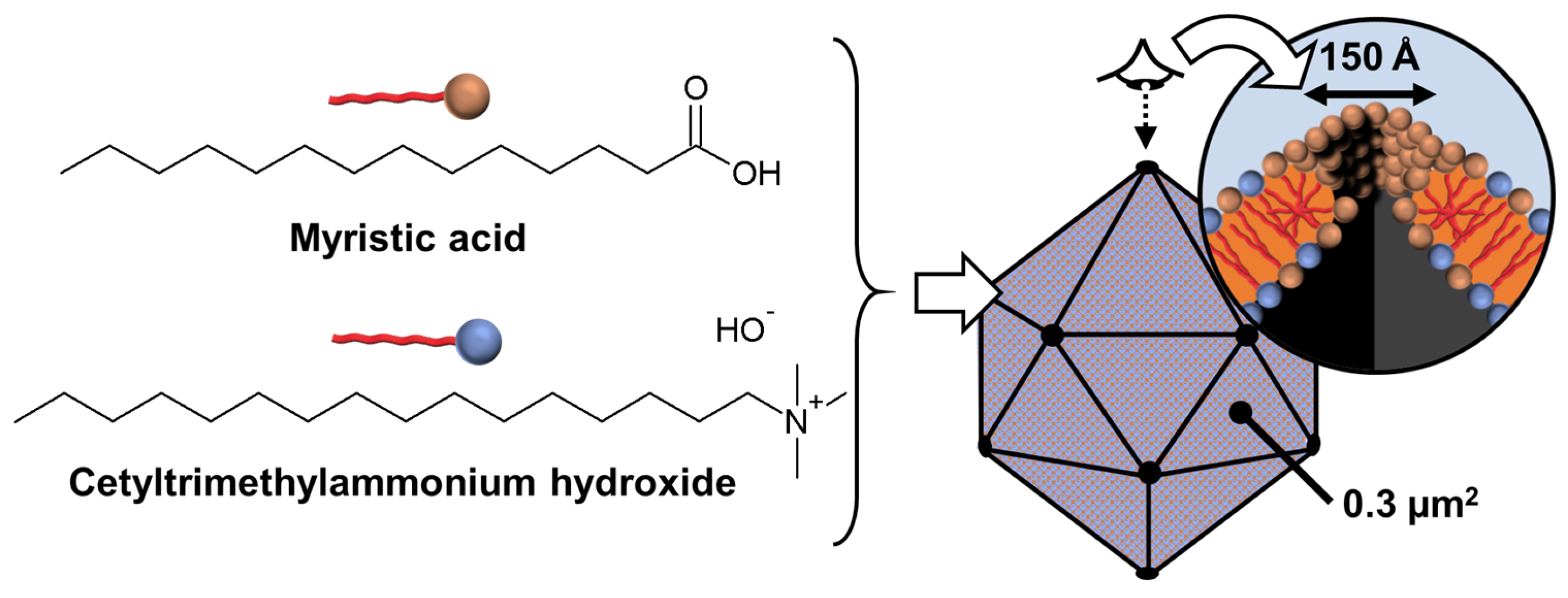
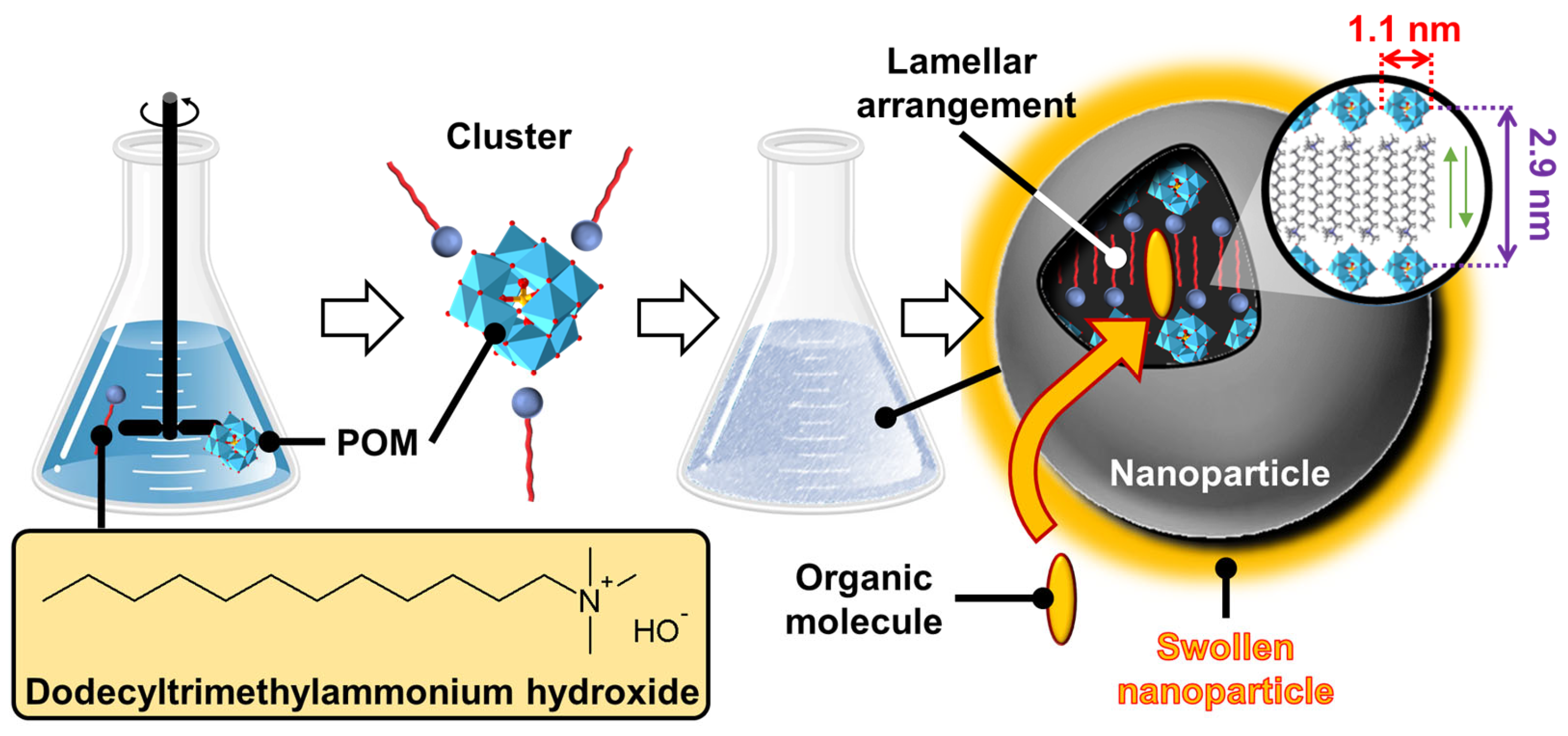
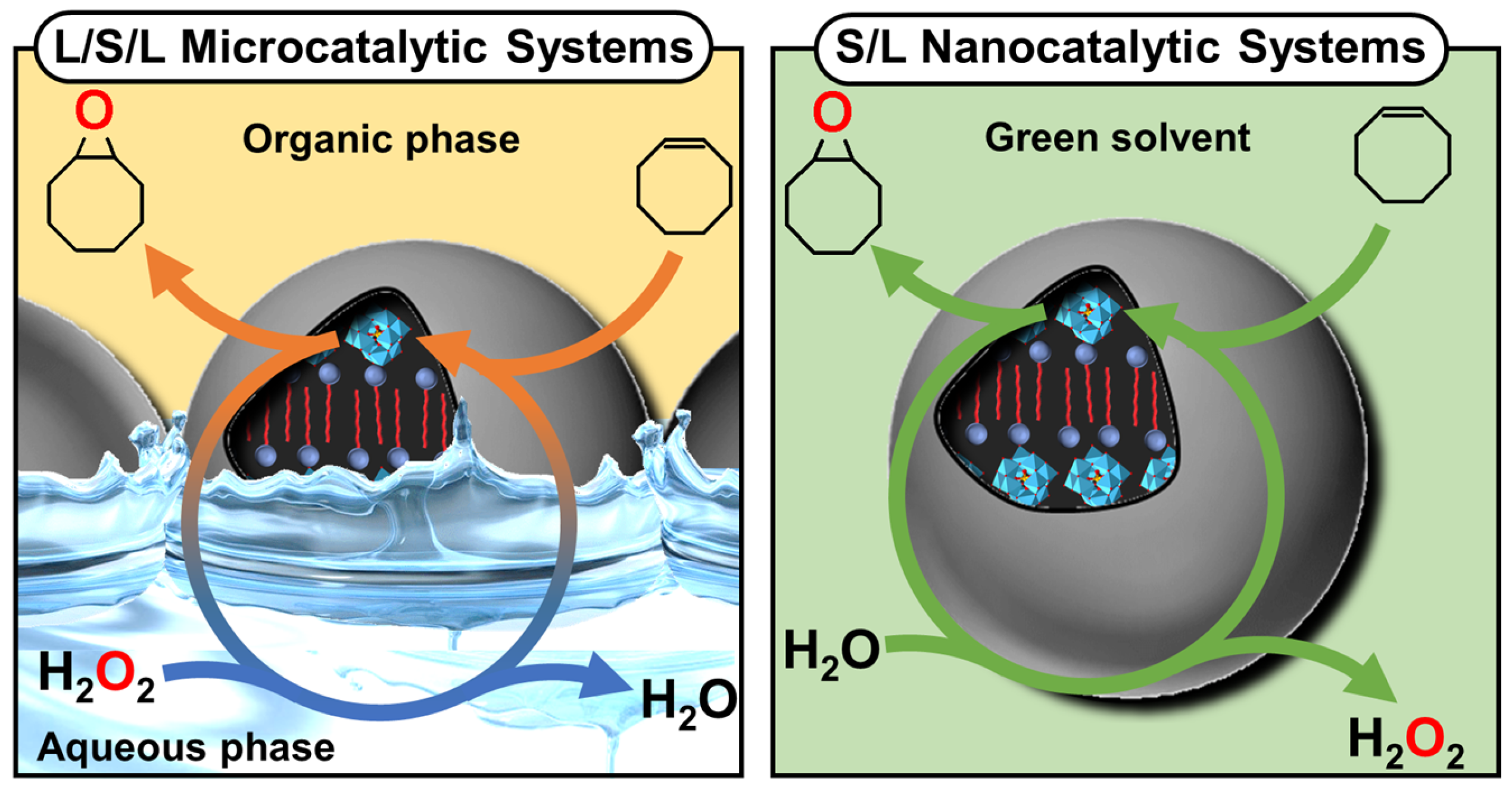

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leclercq, L. Law and Order of Colloidal Tectonics: From Molecules to Self-Assembled Colloids. Molecules 2024, 29, 5657. https://doi.org/10.3390/molecules29235657
Leclercq L. Law and Order of Colloidal Tectonics: From Molecules to Self-Assembled Colloids. Molecules. 2024; 29(23):5657. https://doi.org/10.3390/molecules29235657
Chicago/Turabian StyleLeclercq, Loïc. 2024. "Law and Order of Colloidal Tectonics: From Molecules to Self-Assembled Colloids" Molecules 29, no. 23: 5657. https://doi.org/10.3390/molecules29235657
APA StyleLeclercq, L. (2024). Law and Order of Colloidal Tectonics: From Molecules to Self-Assembled Colloids. Molecules, 29(23), 5657. https://doi.org/10.3390/molecules29235657