
Iron Porphyrin-Based Composites for Electrocatalytic Oxygen Reduction Reactions



Abstract

1. Introduction
2. Electrocatalytic Mechanism for ORRs
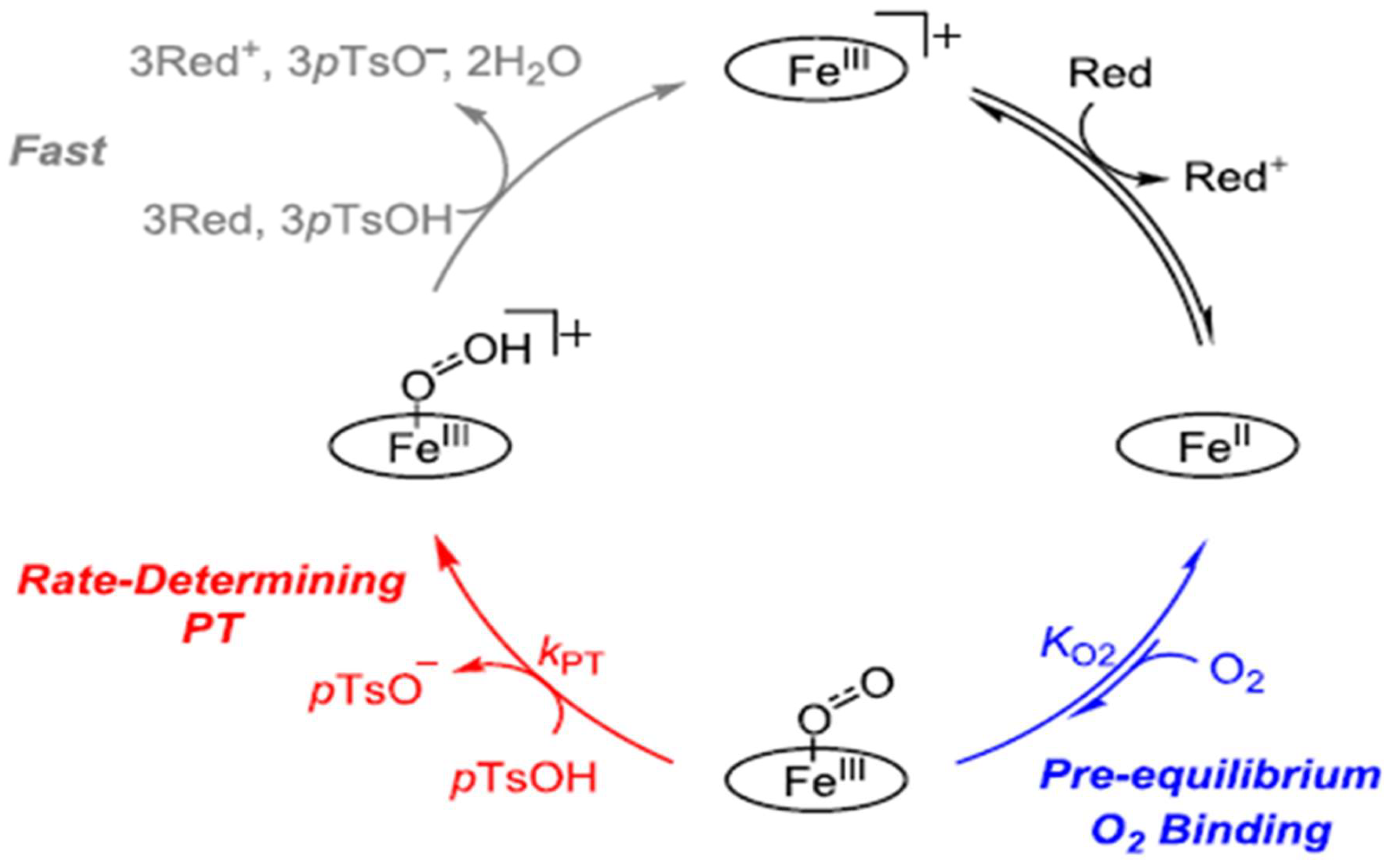
2.1. Evaluation of the ORR Mechanism
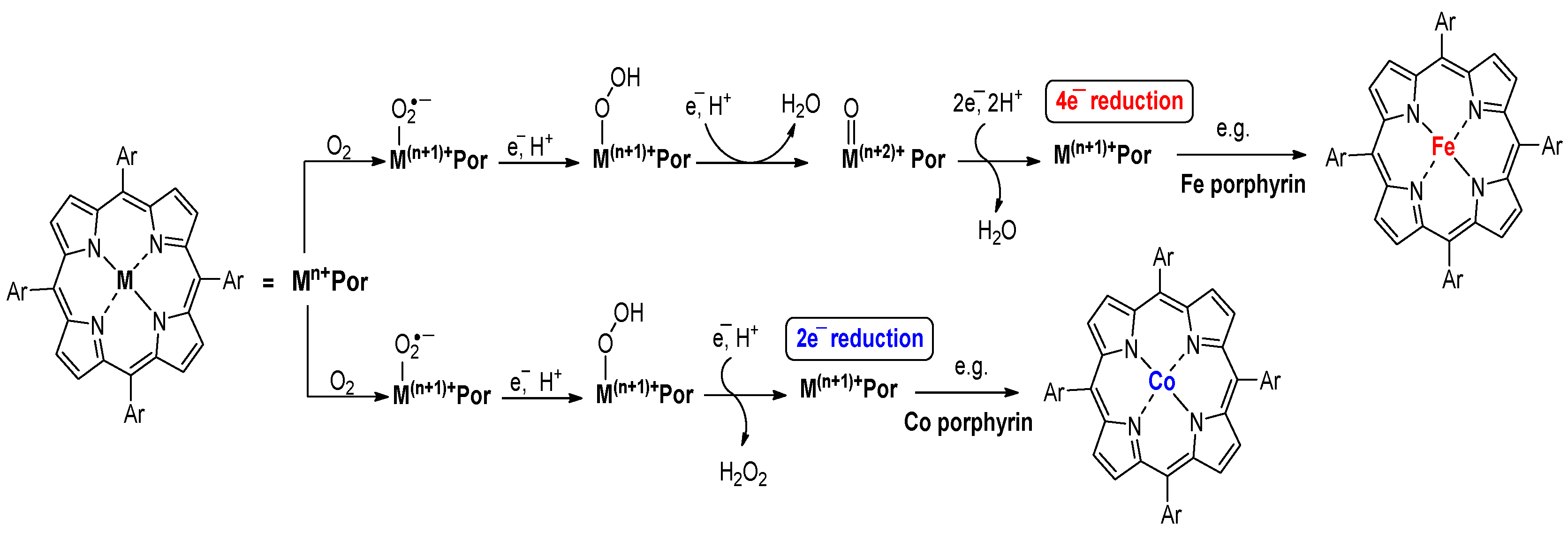
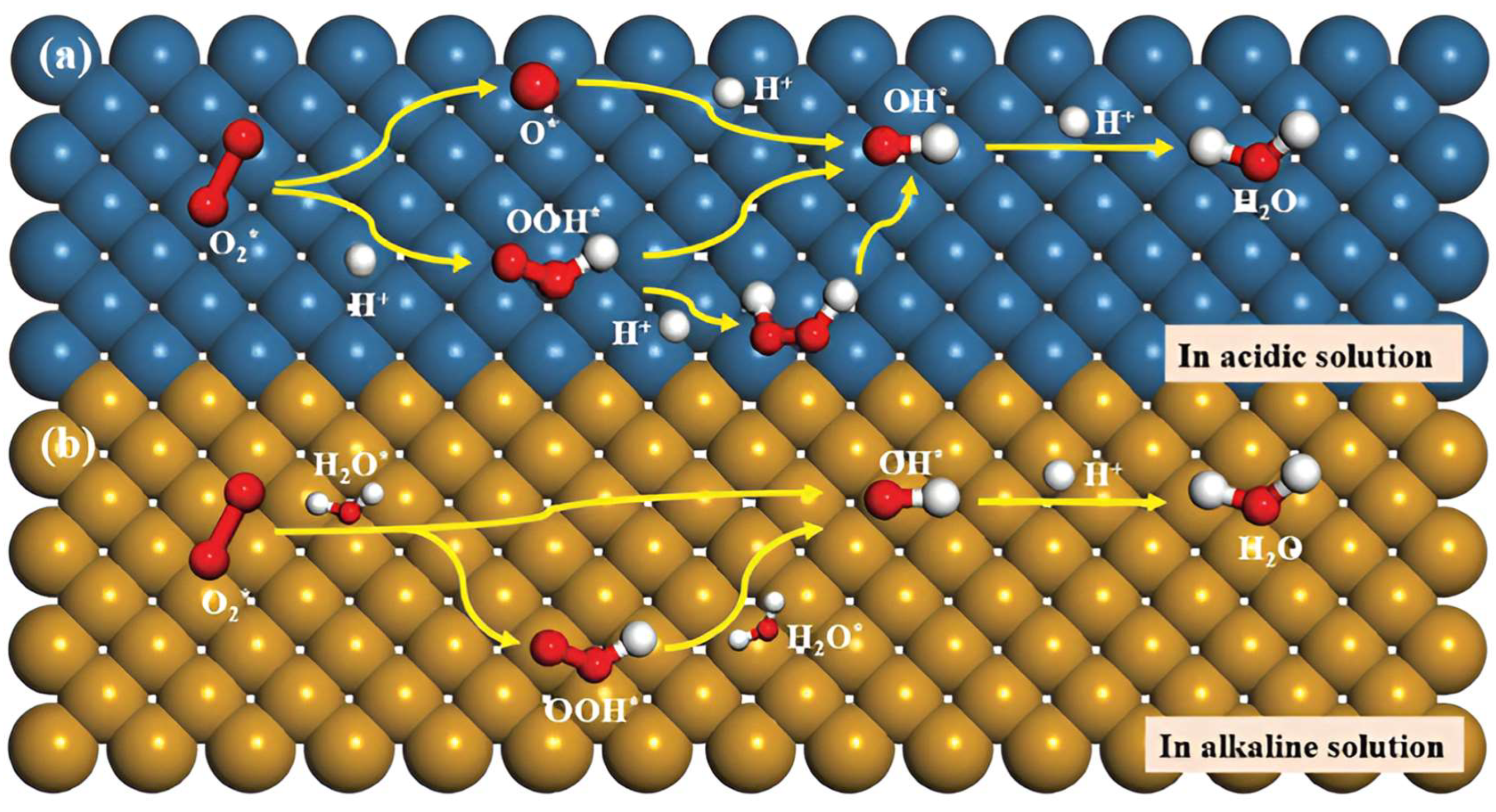
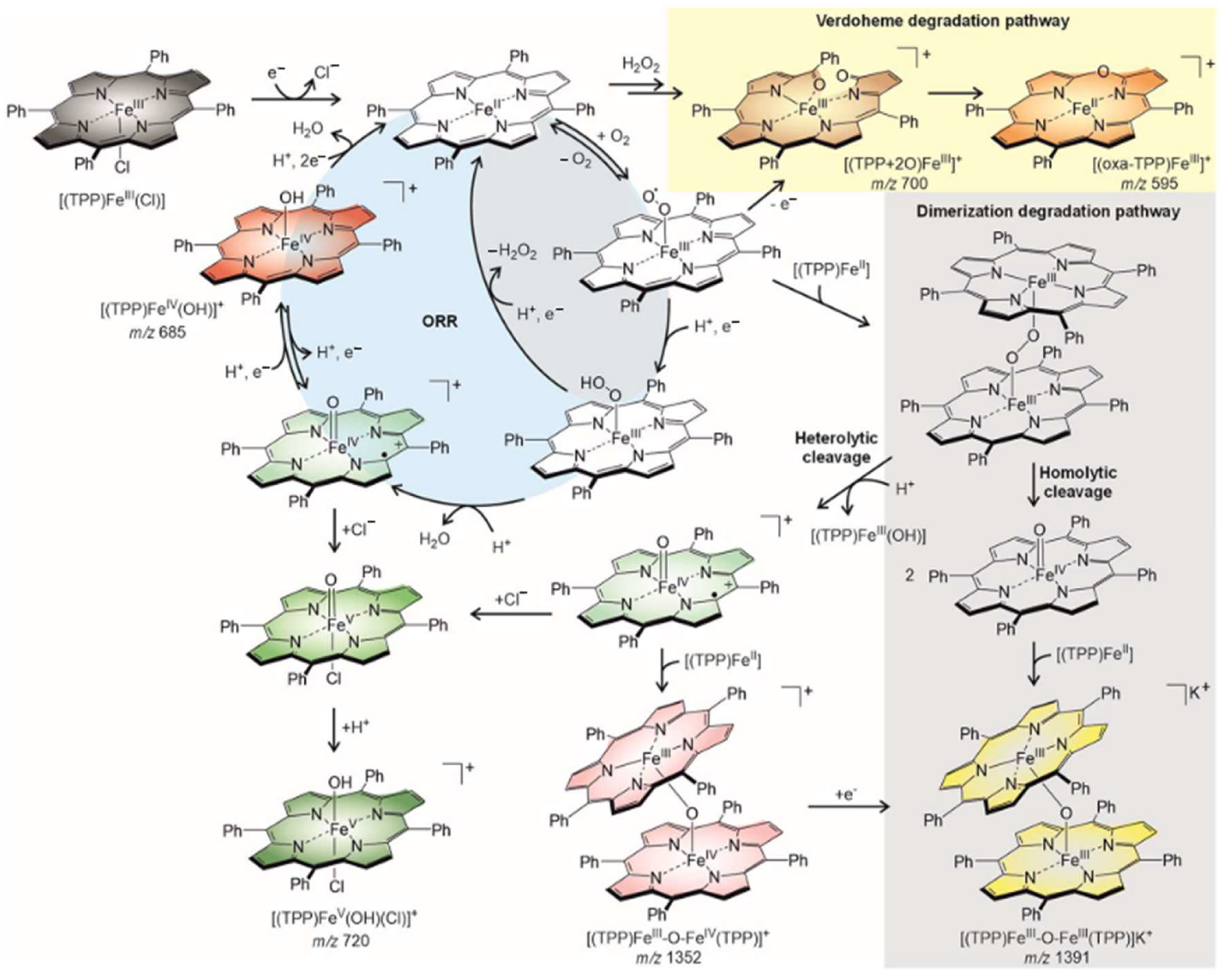
2.2. ORR Mechanism
3. Synthesis and Characterization of Iron Porphyrin-Based Composites
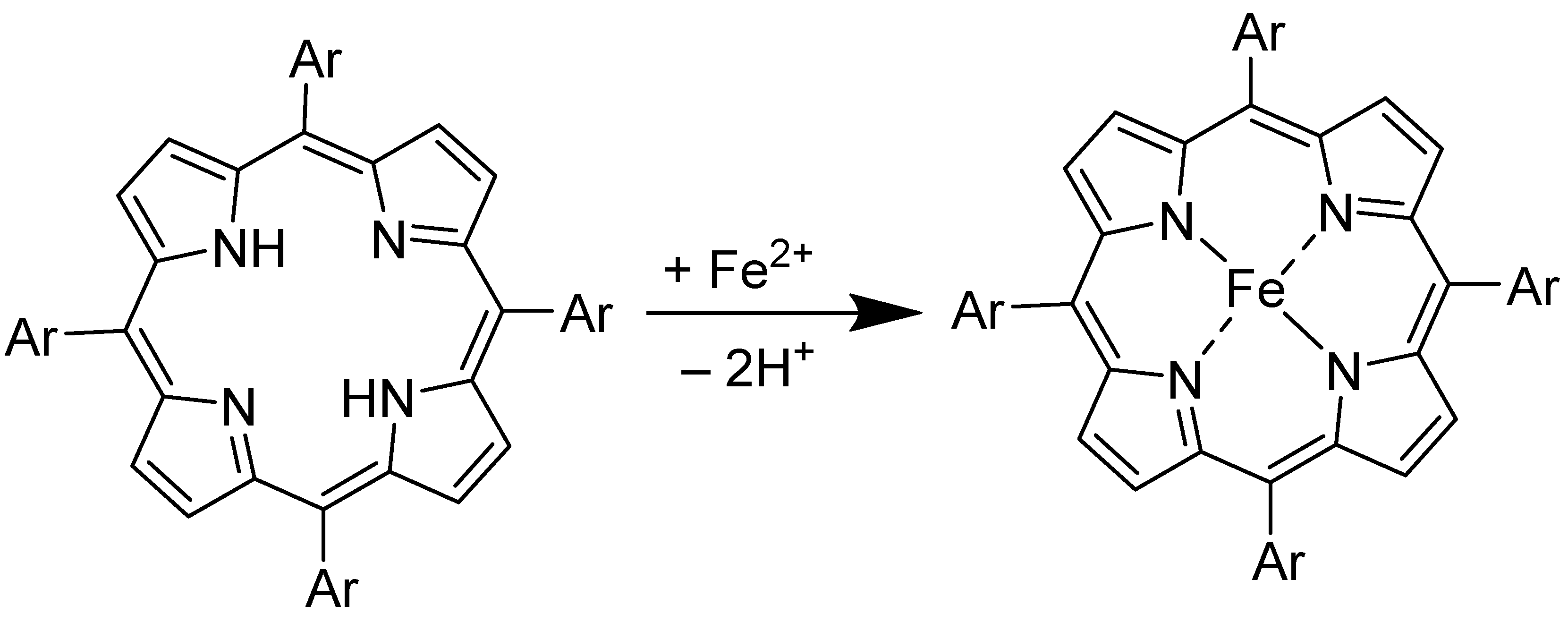
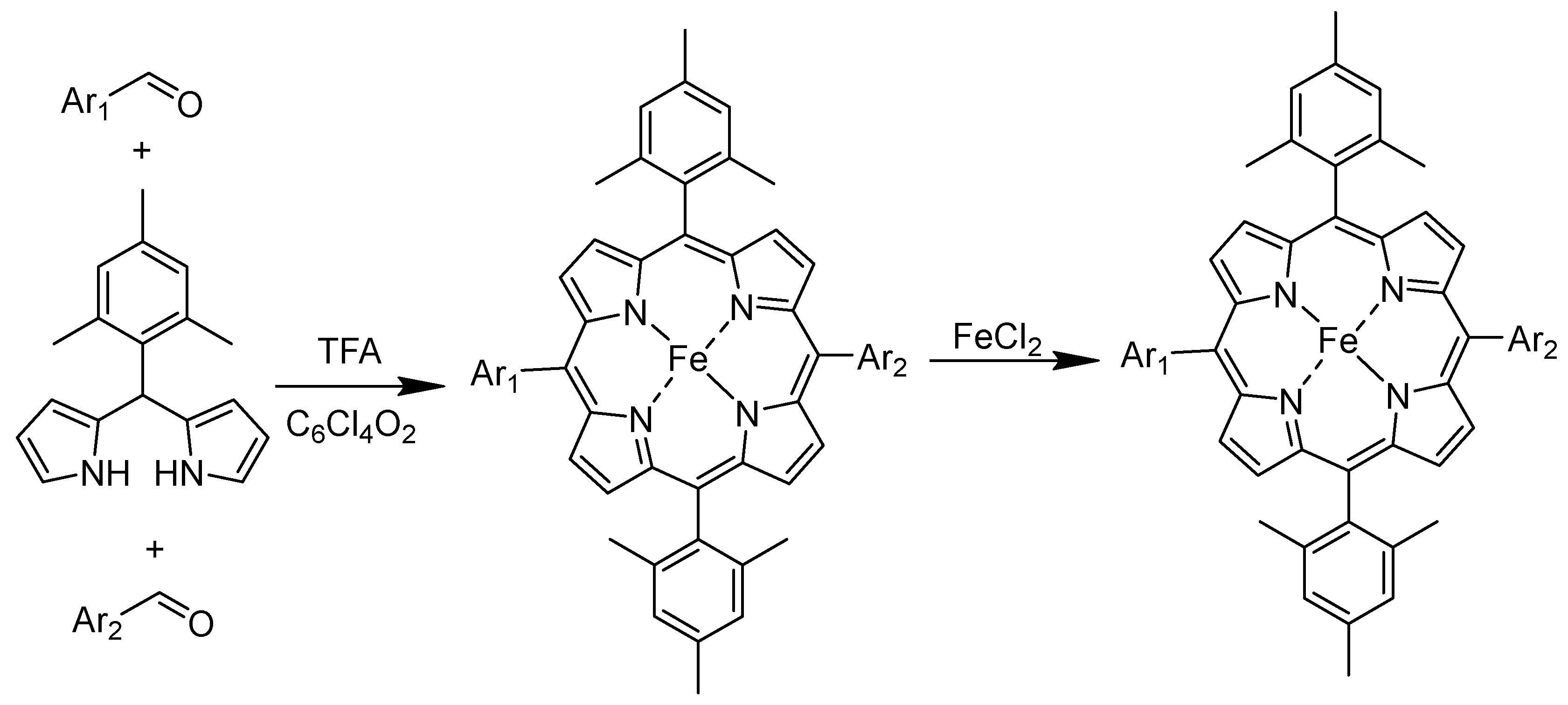
3.1. Synthesis
3.2. Characterization
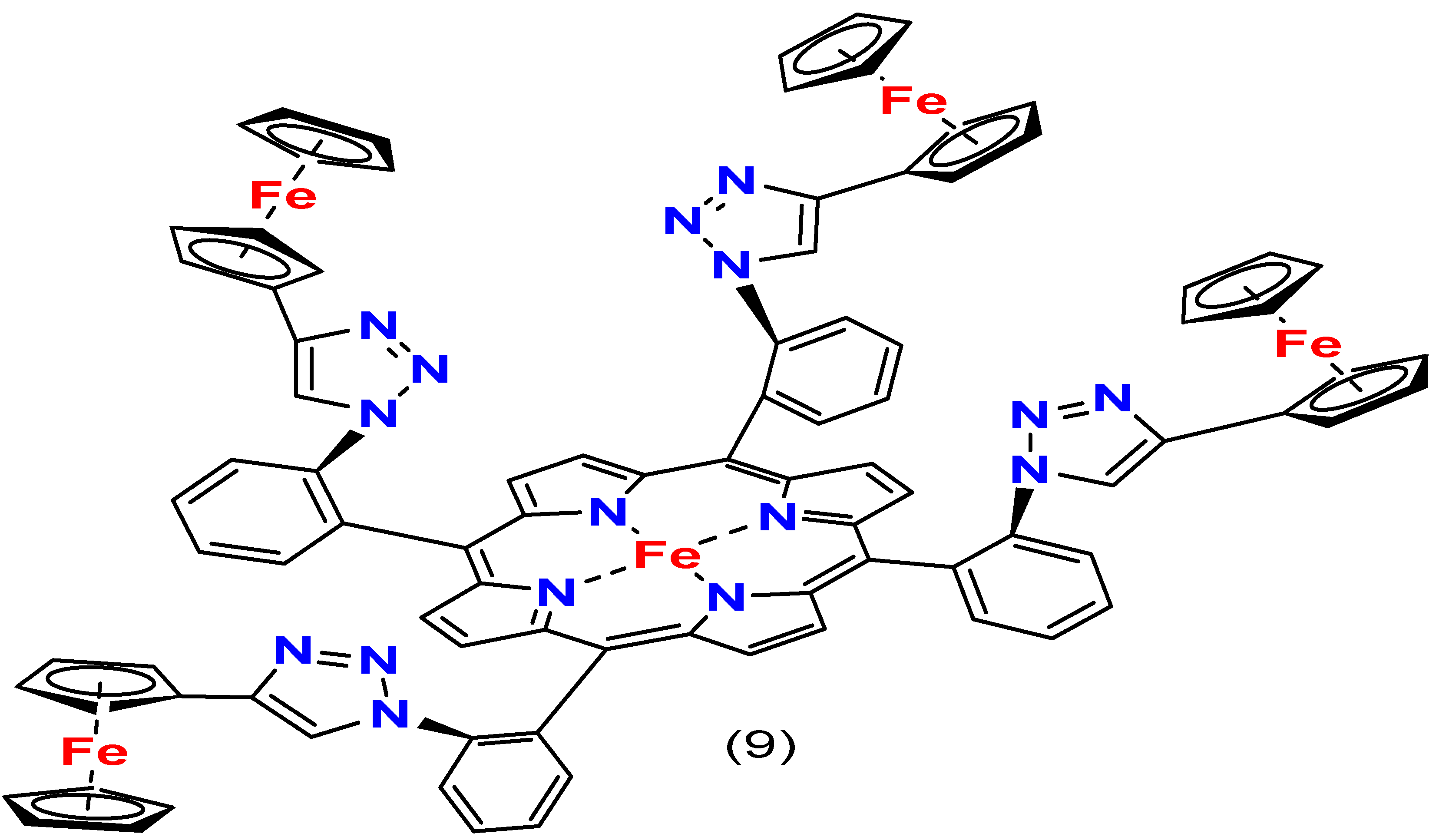
4. Effects of Iron Porphyrin Structure on ORR
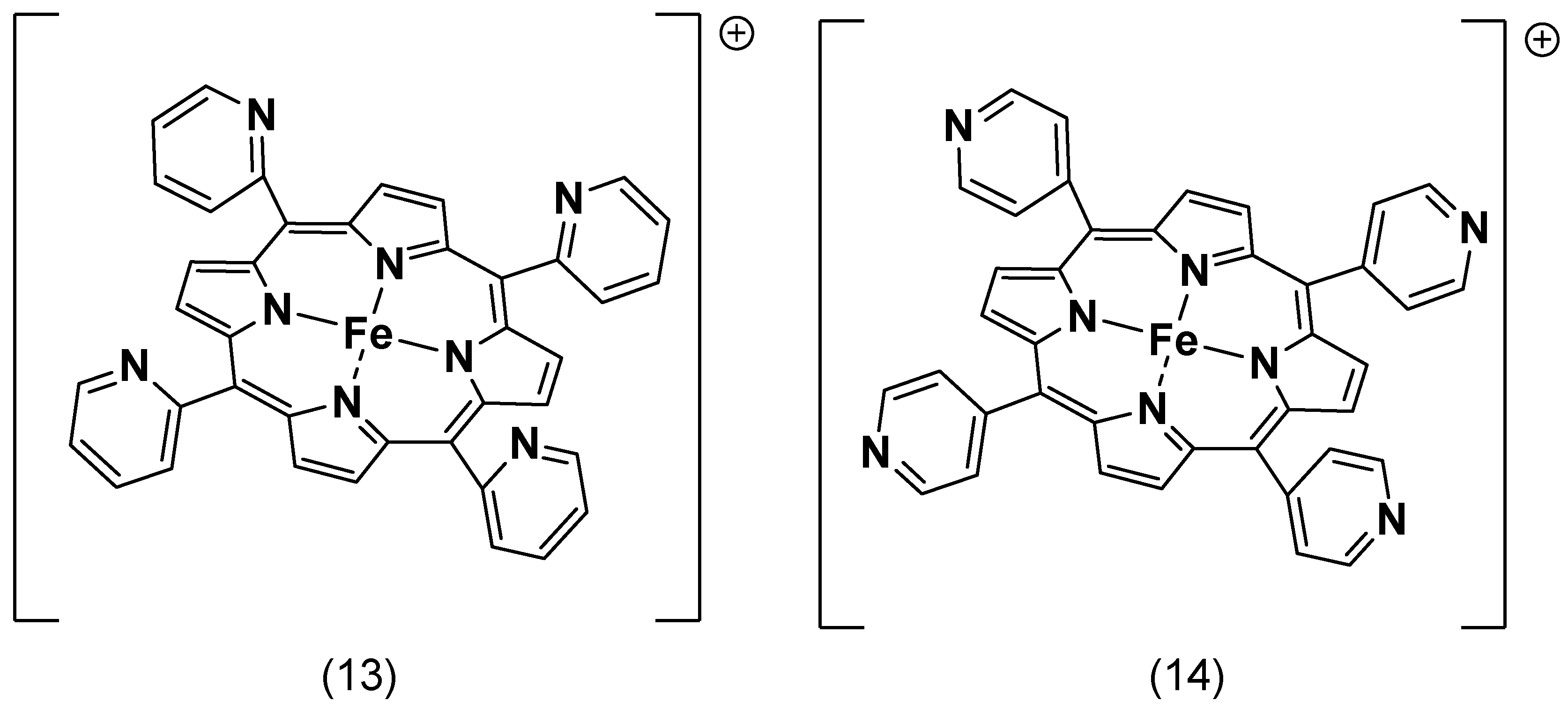
4.1. Axial Ligand Effects
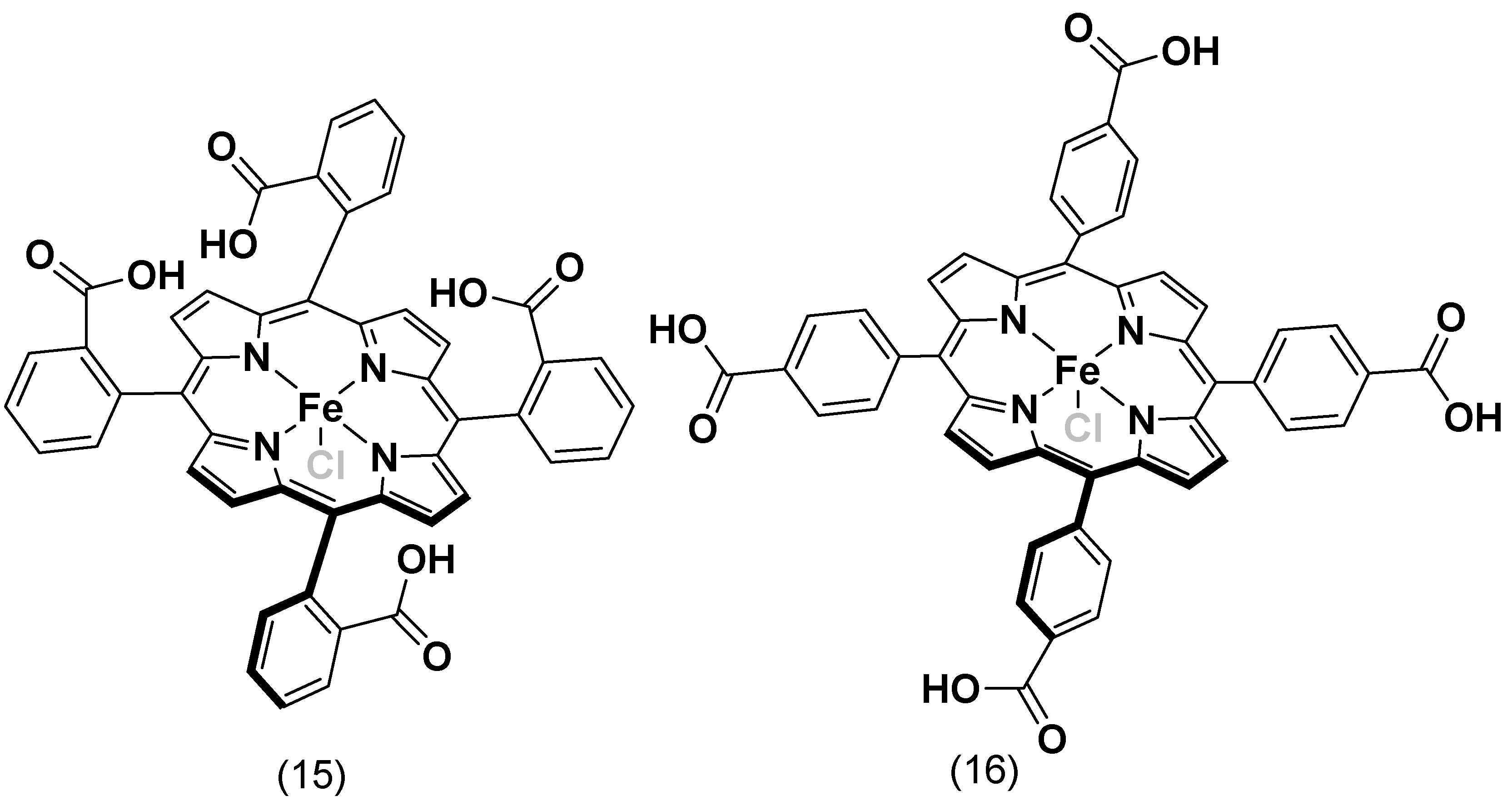
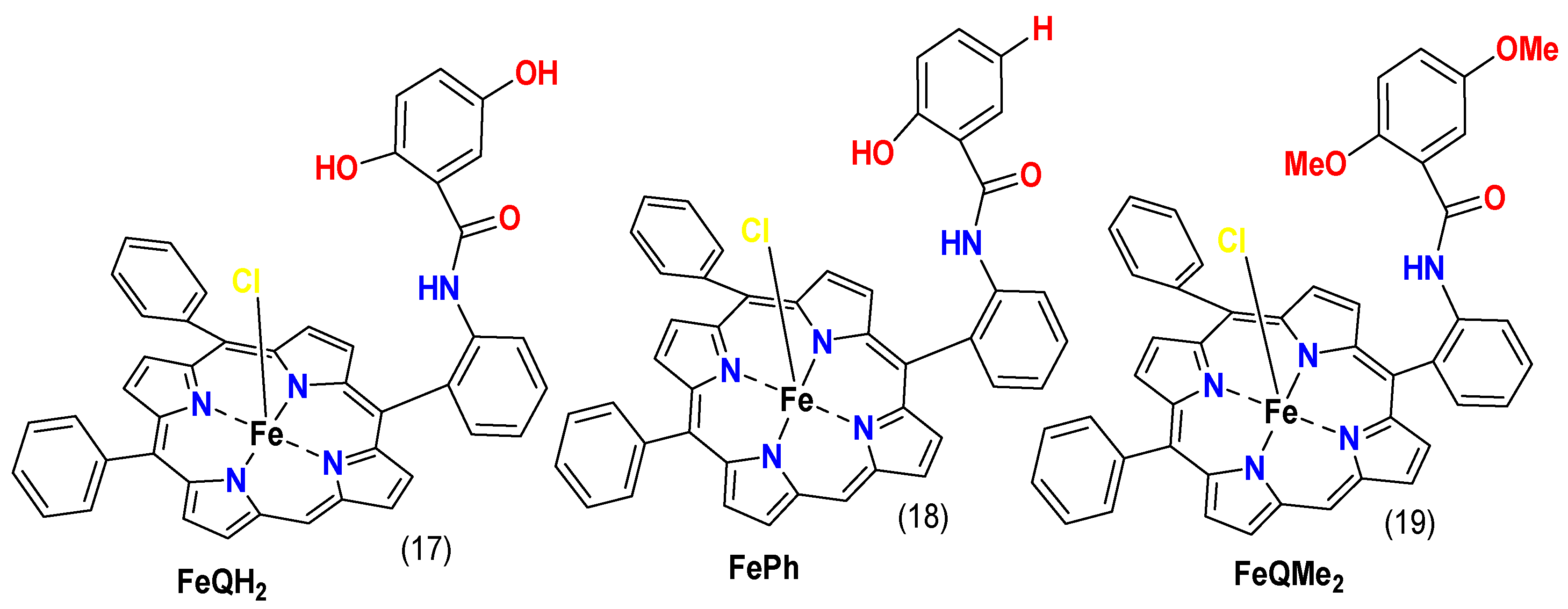
4.2. Substituents Effect
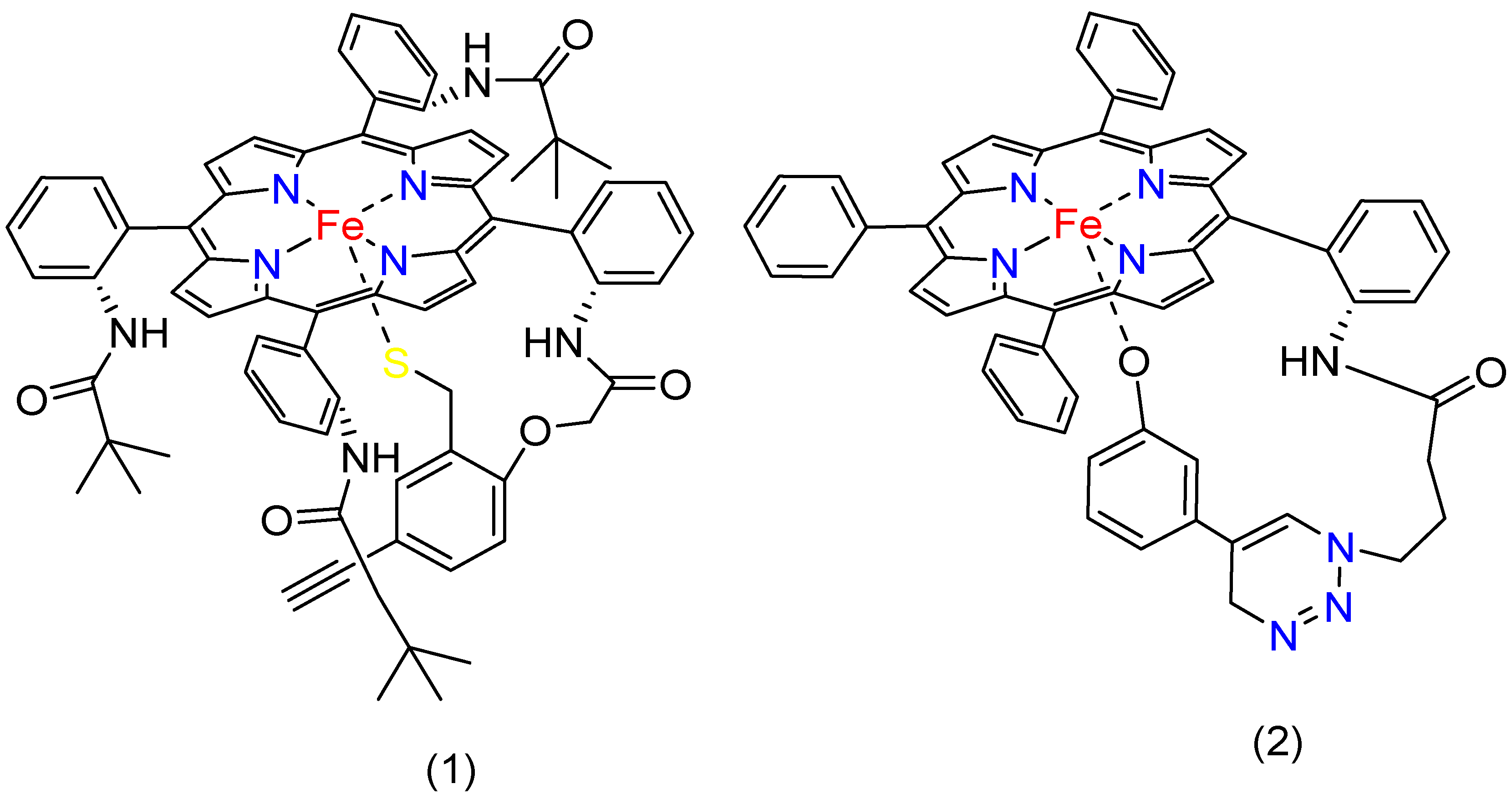
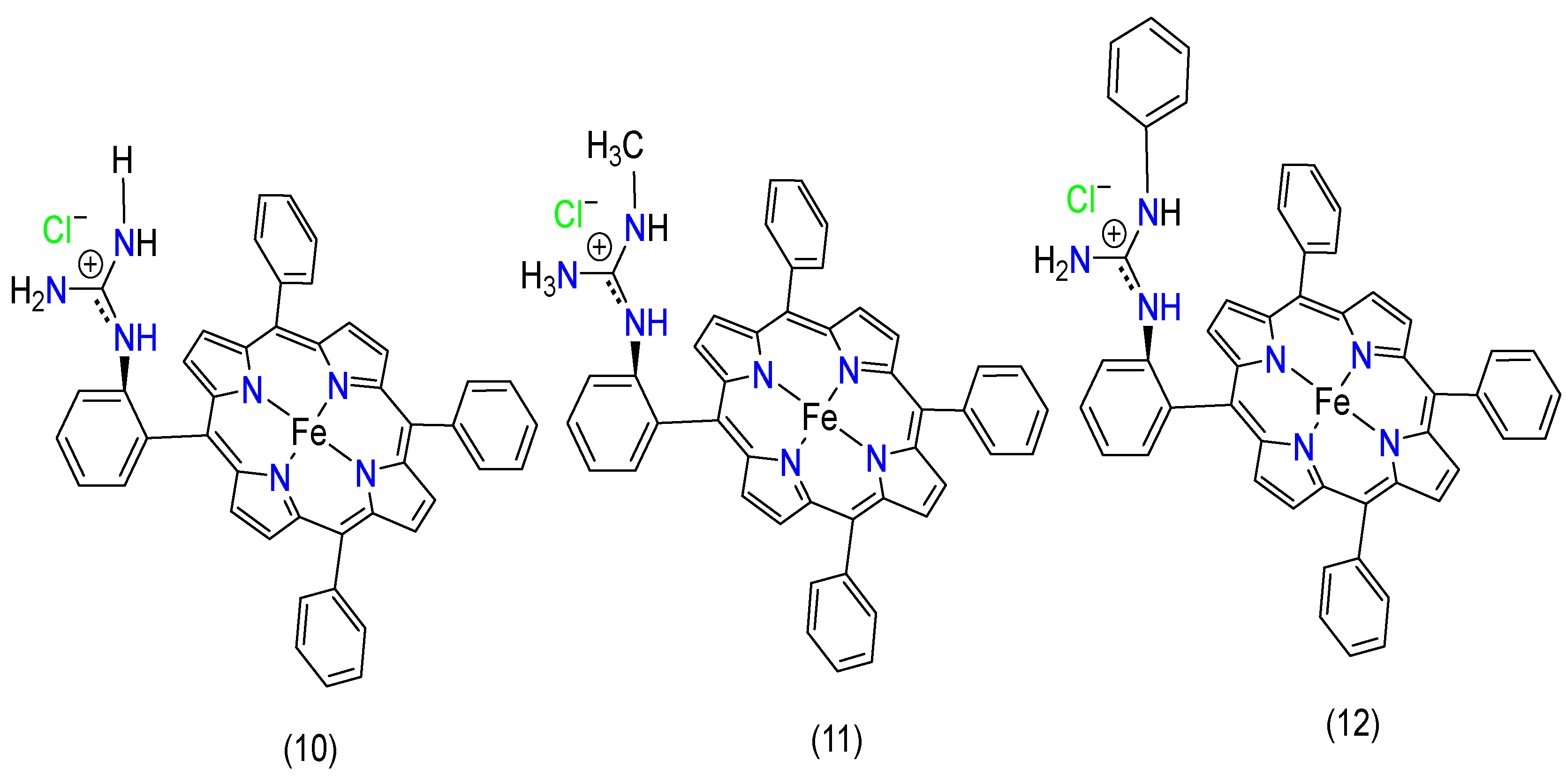
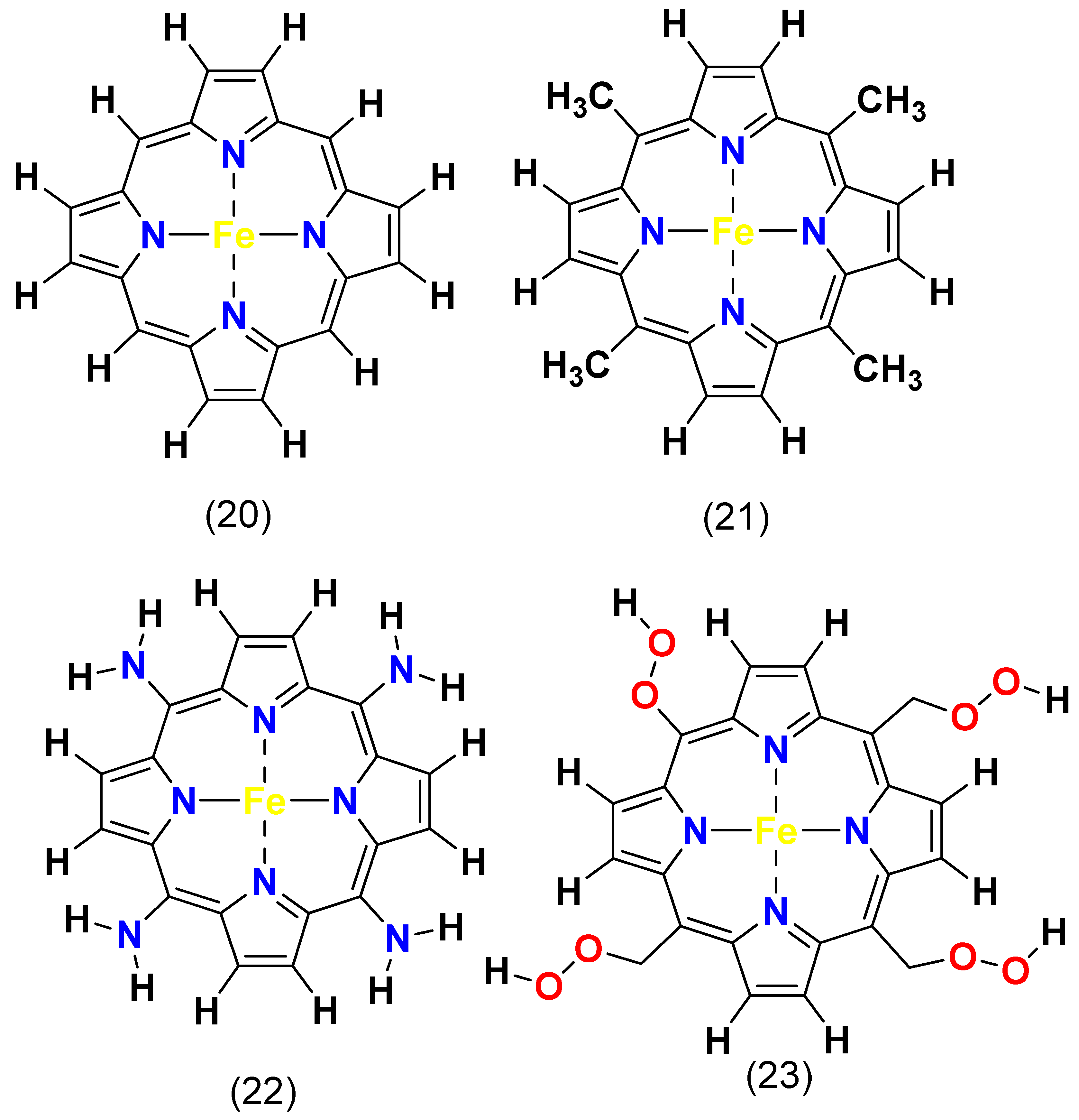
4.3. Functional Group Effect
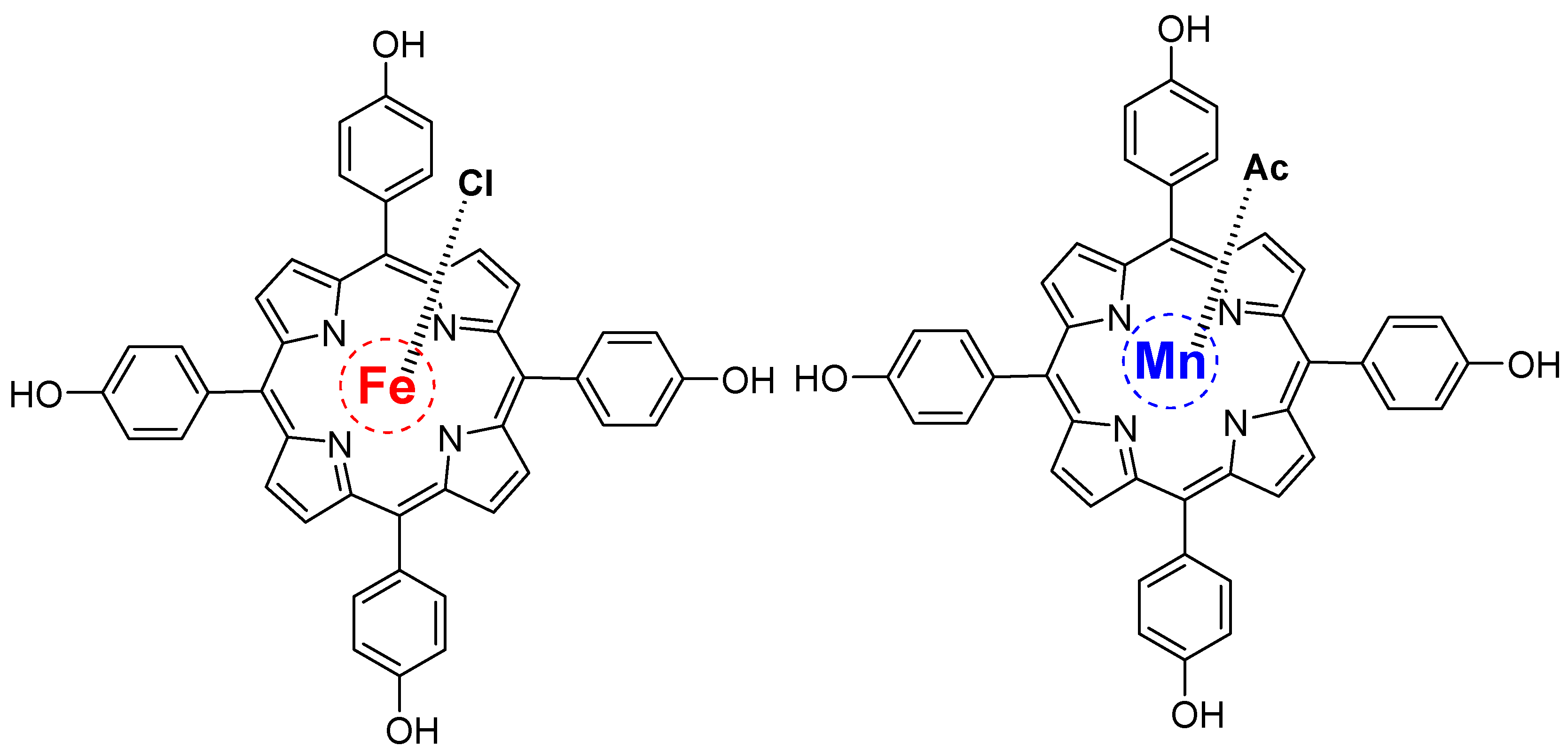
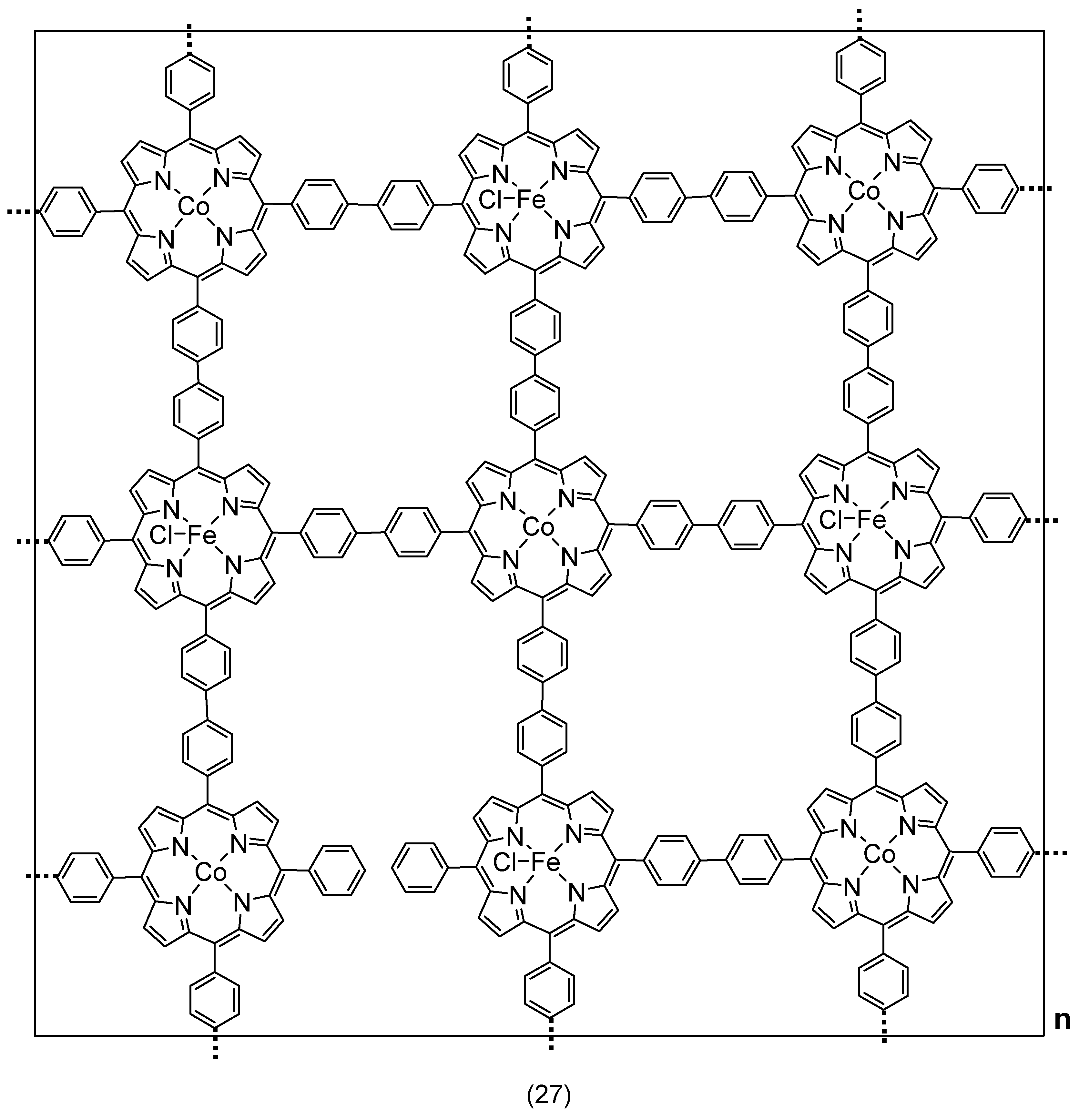
4.4. Iron–Metal Coupled Porphyrins
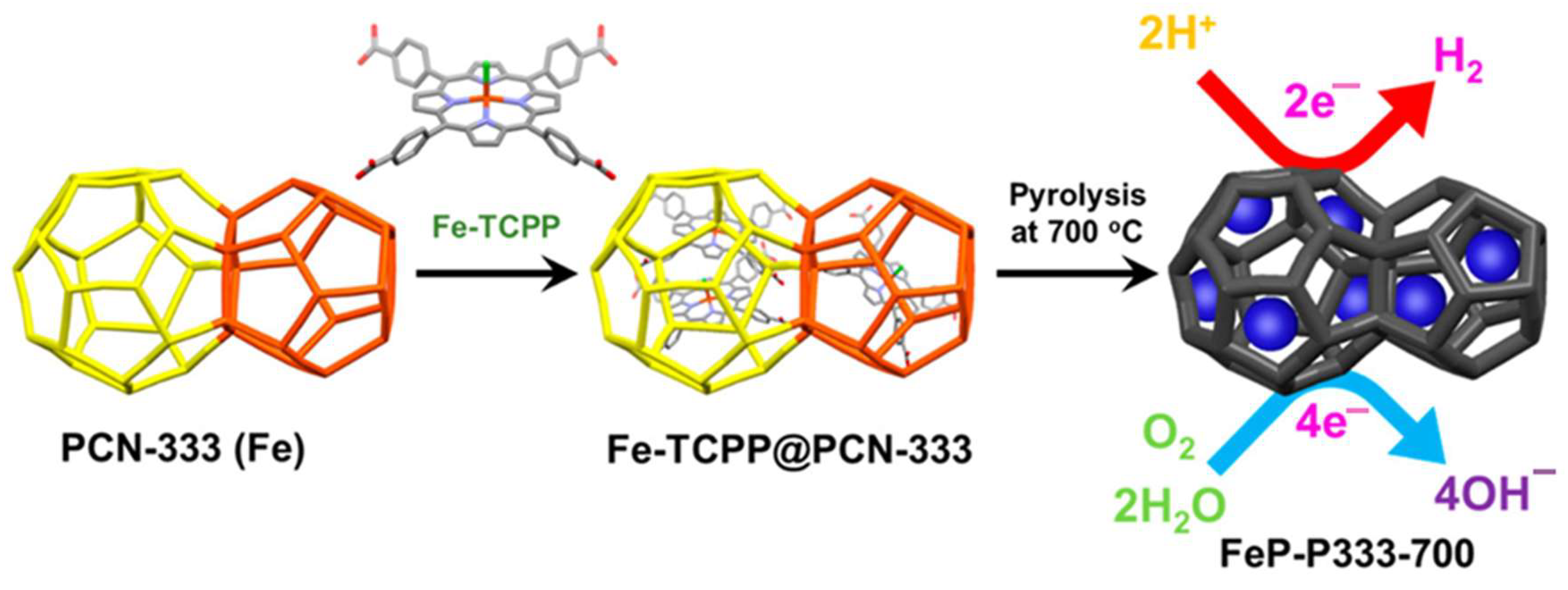
4.5. Iron Porphyrin-Based Metal–Organic Framework
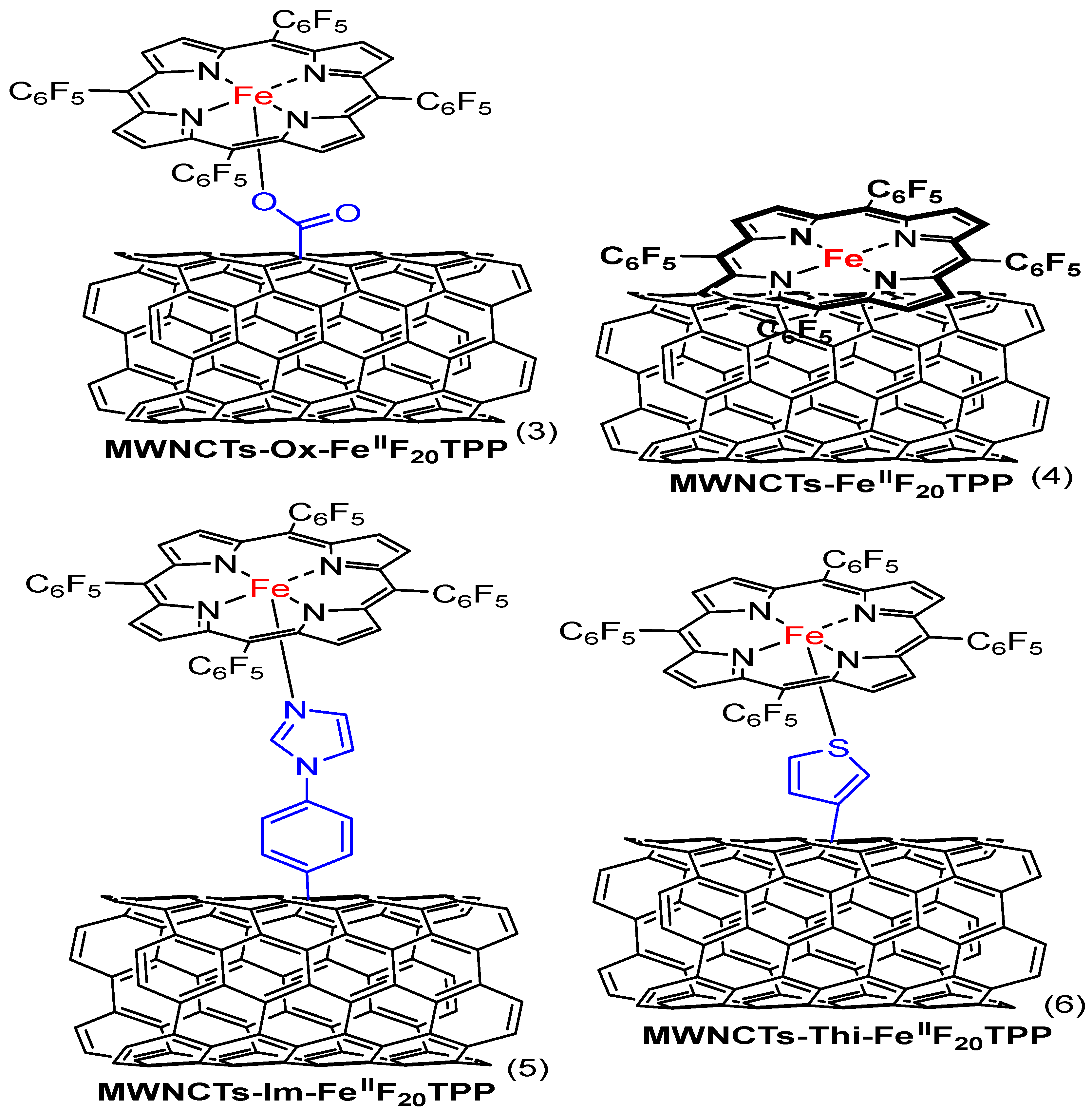
4.6. Heteroatom Doping Iron-Based Composites
5. Platinum-Based Catalyst vs. Iron Porphyrin-Based Composites
6. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Holechek, J.L.; Geli, H.M.E.; Sawalhah, M.N.; Valdez, R. A Global Assessment: Can Renewable Energy Replace Fossil Fuels by 2050? Sustainability 2022, 14, 4792. [Google Scholar] [CrossRef]
- Banham, D.; Ye, S.; Pei, K.; Ozaki, J.-I.; Kishimoto, T.; Imashiro, Y. A review of the stability and durability of non-precious metal catalysts for the oxygen reduction reaction in proton exchange membrane fuel cells. J. Power Sources 2015, 285, 334–348. [Google Scholar] [CrossRef]
- Bessagnet, B.; Bossioli, E.; Cholakian, A.; Vivanco, M.G.; Cuvelier, K.; Theobald, M.R.; Gil, V.; Menut, L.; de Meij, A.; Pisoni, E.; et al. Impact of air quality model settings for the evaluation of emission reduction strategies to curb air pollution. Environ. Res. 2024, 255, 119112. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Song, A.; Tian, H.; Liu, J.; Shao, G.; Liu, H.; Wang, G. Single-atom catalysts for high-energy rechargeable batteries. Chem. Sci. 2021, 12, 7656–7676. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, B.; Basu, S.; Jena, B.K. Transition metal-based single-atom catalysts (TM-SACs); rising materials for electrochemical CO2 reduction. J. Energy Chem. 2022, 70, 444–471. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, Y.; Pei, Z.; Wu, K.-H.; Tan, C.; Wang, H.; Wei, L.; Mahmood, A.; Yan, C.; Dong, J.; et al. Recent Progress of Carbon-Supported Single-Atom Catalysts for Energy Conversion and Storage. Matter 2020, 3, 1442–1476. [Google Scholar] [CrossRef]
- Cao, R.; Lee, J.-S.; Liu, M.; Cho, J. Recent Progress in Non-Precious Catalysts for Metal-Air Batteries. Adv. Energy Mater. 2012, 2, 816–829. [Google Scholar] [CrossRef]
- Lv, J.; Abbas, S.C.; Huang, Y.; Liu, Q.; Wi, M.; Dai, L.; Wang, Y. A photo-responsive bifunctional electrocatalyst for oxygen reduction and evolution reactions. Nano Energy 2017, 43, 130–137. [Google Scholar] [CrossRef]
- Shi, M.; Tong, X.; Li, W.; Fang, J.; Chen, L.; Ma, C.-A. Enhanced Electrocatalytic Oxygen Reduction on NiWOx Solid Solution with Induced Oxygen Defects. ACS Appl. Mater. Interfaces 2017, 9, 34990–35000. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Yasin, G.; Vashistha, V.K.; Das, D.K.; Rehman, M.U.; Iqbal, R.; Mo, Z.; Nguyen, T.A.; Slimani, Y.; Nazir, M.T.; et al. Enhancing oxygen reduction reaction performance via CNTs/graphene supported iron protoporphyrin IX: A hybrid nanoarchitecture electrocatalyst. Diam. Relat. Mater. 2021, 113, 108272. [Google Scholar] [CrossRef]
- Costa, R.; Camacho, J.; Guimarães Júnior, S.; Salerno, C.H. The polymer electrolyte membrane fuel cell as electric energy source, steady state and dynamic behavior. Renew. Energy Power Qual. J. 2006, 4, 44–51. [Google Scholar] [CrossRef]
- Peighambardoust, S.J.; Rowshanzamir, S.; Amjadi, M. Review of the proton exchange membranes for fuel cell applications. Int. J. Hydrogen Energy 2010, 35, 9349–9384. [Google Scholar] [CrossRef]
- Ahuja, D.; Kalpna, V.; Varshney, P.K. Metal air battery: A sustainable and low cost material for energy storage. J. Phys. Conf. Ser. 2021, 1913, 012065. [Google Scholar] [CrossRef]
- Wang, Z.-L.; Xu, D.; Xu, J.-J.; Zhang, X.-B. Oxygen electrocatalysts in metal–air batteries: From aqueous to nonaqueous electrolytes. Chem. Soc. Rev. 2014, 43, 7746–7786. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-S.; Tai Kim, S.; Cao, R.; Choi, N.-S.; Liu, M.; Lee, K.T.; Cho, J. Metal–Air Batteries with High Energy Density: Li–Air versus Zn–Air. Adv. Energy Mater. 2011, 1, 34–50. [Google Scholar] [CrossRef]
- Nyaaba, A.A.; Wei, Z.; Liu, Y.; Kang, Z.; Naz, H.; Zhou, H.; Zhu, G. Iridium-based electrocatalysts for alkaline hydrogen oxidation reaction. J. Electroanal. Chem. 2024, 962, 118291. [Google Scholar] [CrossRef]
- Santoro, C.; Bollella, P.; Erable, B.; Atanassov, P.; Pant, D. Oxygen reduction reaction electrocatalysis in neutral media for bioelectrochemical systems. Nat. Catal. 2022, 5, 473–484. [Google Scholar] [CrossRef]
- Wang, X.X.; Prabhakaran, V.; He, Y.; Shao, Y.; Wu, G. Iron-Free Cathode Catalysts for Proton-Exchange-Membrane Fuel Cells: Cobalt Catalysts and the Peroxide Mitigation Approach. Adv. Mater. 2019, 31, 1805126. [Google Scholar] [CrossRef] [PubMed]
- Debe, M.K. Electrocatalyst approaches and challenges for automotive fuel cells. Nature 2012, 486, 43–51. [Google Scholar] [CrossRef]
- Yang, Y.; Peltier, C.R.; Zeng, R.; Schimmenti, R.; Li, Q.; Huang, X.; Yan, Z.; Potsi, G.; Selhorst, R.; Lu, X.; et al. Electrocatalysis in Alkaline Media and Alkaline Membrane-Based Energy Technologies. Chem. Rev. 2022, 122, 6117–6321. [Google Scholar] [CrossRef]
- Kiani, M.; Tian, X.Q.; Zhang, W. Single atom based electrocatalysts for oxygen reduction reaction in polymer electrolyte membrane fuel cell: Recent advances, challenges and future perspectives. J. Phys. Chem. Solids 2021, 153, 109989. [Google Scholar] [CrossRef]
- Liu, D.; Tao, L.; Yan, D.; Zou, Y.; Wang, S. Recent Advances on Non-precious Metal Porous Carbon-based Electrocatalysts for Oxygen Reduction Reaction. ChemElectroChem 2018, 5, 1775–1785. [Google Scholar] [CrossRef]
- Qin, D.-D.; Tang, Y.; Ma, G.; Qin, L.; Tao, C.-L.; Zhang, X.; Tang, Z. Molecular metal nanoclusters for ORR, HER and OER: Achievements, opportunities and challenges. Int. J. Hydrogen Energy 2021, 46, 25771–25781. [Google Scholar] [CrossRef]
- Zhao, Y.-M.; Yu, G.-Q.; Wang, F.-F.; Wei, P.-J.; Liu, J.-G. Bioinspired Transition-Metal Complexes as Electrocatalysts for the Oxygen Reduction Reaction. Chem. A Eur. J. 2019, 25, 3726–3739. [Google Scholar] [CrossRef] [PubMed]
- Pegis, M.L.; Wise, C.F.; Martin, D.J.; Mayer, J.M. Oxygen Reduction by Homogeneous Molecular Catalysts and Electrocatalysts. Chem. Rev. 2018, 118, 2340–2391. [Google Scholar] [CrossRef]
- Kumar, A.; Ubaidullah, M.; Pandit, B.; Yasin, G.; Gupta, R.; Zhang, G.-X. Fe-phthalocyanine derived highly conjugated 2D covalent organic framework as superior electrocatalyst for oxygen reduction reaction. Discov. Nano 2023, 18, 109. [Google Scholar] [CrossRef]
- Yuan, R.; George, S.L.; Chen, J.; Wu, Q.; Qiu, X.; Zhao, L. Meso-substituted Metalloporphyrin-based Composites for Electrocatalytic Oxygen Reduction Reactions. ChemNanoMat 2023, 9, e202300027. [Google Scholar] [CrossRef]
- Ma, R.; Lin, G.; Zhou, Y.; Liu, Q.; Zhang, T.; Shan, G.; Yang, M.; Wang, J. A review of oxygen reduction mechanisms for metal-free carbon-based electrocatalysts. NPJ Comput. Mater. 2019, 5, 78. [Google Scholar] [CrossRef]
- Alabi, A.; Popoola, P.; Popoola, O.; Mathe, N.; Abdulwahab, M. Materials for electrocatalysts in proton exchange membrane fuel cell: A brief review. Front. Energy Res. 2023, 11, 1091105. [Google Scholar] [CrossRef]
- Osmieri, L.; Park, J.; Cullen, D.A.; Zelenay, P.; Myers, D.J.; Neyerlin, K.C. Status and challenges for the application of platinum group metal-free catalysts in proton-exchange membrane fuel cells. Curr. Opin. Electrochem. 2021, 25, 100627. [Google Scholar] [CrossRef]
- Ali, A.; Khandelwal, S.; Panja, S.; Majumder, P.; Dutta, A. Chapter 1—Oxygen reduction reaction in nature and its importance in life. In Oxygen Reduction Reaction; Sengupta, K., Chatterjee, S., Dutta, K., Eds.; Elsevier: Amsterdam, The Netherlands, 2022; pp. 1–43. [Google Scholar]
- Liang, J.; Zheng, Y.; Chen, J.; Liu, J.; Hulicova-Jurcakova, D.; Jaroniec, M.; Qiao, S.Z. Facile Oxygen Reduction on a Three-Dimensionally Ordered Macroporous Graphitic C3N4/Carbon Composite Electrocatalyst. Angew. Chem. Int. Ed. 2012, 51, 3892–3896. [Google Scholar] [CrossRef] [PubMed]
- Jaouen, F.; Lefèvre, M.; Proietti, E.; Dodelet, J.-P. Iron-Based Catalysts with Improved Oxygen Reduction Activity in Polymer Electrolyte Fuel Cells. Science 2009, 324, 71–74. [Google Scholar] [CrossRef]
- Auffarth, S.; Dafinger, W.; Mehler, J.; Ardizzon, V.; Preuster, P.; Wasserscheid, P.; Thiele, S.; Kerres, J. Cross-linked proton-exchange membranes with strongly reduced fuel crossover and increased chemical stability for direct-isopropanol fuel cells. J. Mater. Chem. A 2022, 10, 17208–17216. [Google Scholar] [CrossRef]
- Tzelepis, S.; Kavadias, K. Progress on Platinum-Based Catalyst Layer Materials for H2-PEMFC. Energy Eng. 2022, 119, 1745–1769. [Google Scholar] [CrossRef]
- He, Q.; Mugadza, T.; Kang, X.; Zhu, X.; Chen, S.; Kerr, J.; Nyokong, T. Molecular catalysis of the oxygen reduction reaction by iron porphyrin catalysts tethered into Nafion layers: An electrochemical study in solution and a membrane-electrode-assembly study in fuel cells. J. Power Sources 2012, 216, 67–75. [Google Scholar] [CrossRef]
- Abbas, M.A.; Bang, J.H. Rising Again: Opportunities and Challenges for Platinum-Free Electrocatalysts. Chem. Mater. 2015, 27, 7218–7235. [Google Scholar] [CrossRef]
- Liang, Z.; Wang, H.-Y.; Zheng, H.; Zhang, W.; Cao, R. Porphyrin-based frameworks for oxygen electrocatalysis and catalytic reduction of carbon dioxide. Chem. Soc. Rev. 2021, 50, 2540–2581. [Google Scholar] [CrossRef]
- Tian, X.; Lu, X.F.; Xia, B.Y.; Lou, X.W. Advanced Electrocatalysts for the Oxygen Reduction Reaction in Energy Conversion Technologies. Joule 2020, 4, 45–68. [Google Scholar] [CrossRef]
- Zhang, W.; Lai, W.; Cao, R. Energy-Related Small Molecule Activation Reactions: Oxygen Reduction and Hydrogen and Oxygen Evolution Reactions Catalyzed by Porphyrin- and Corrole-Based Systems. Chem. Rev. 2017, 117, 3717–3797. [Google Scholar] [CrossRef]
- Amanullah, S.; Saha, P.; Dey, A. Recent developments in the synthesis of bio-inspired iron porphyrins for small molecule activation. Chem. Commun. 2022, 58, 5808–5828. [Google Scholar] [CrossRef]
- Li, X.; Lei, H.; Xie, L.; Wang, N.; Zhang, W.; Cao, R. Metalloporphyrins as Catalytic Models for Studying Hydrogen and Oxygen Evolution and Oxygen Reduction Reactions. Acc. Chem. Res. 2022, 55, 878–892. [Google Scholar] [CrossRef]
- Cook, L.P.; Brewer, G.; Wong-Ng, W. Structural Aspects of Porphyrins for Functional Materials Applications. Crystals 2017, 7, 223. [Google Scholar] [CrossRef]
- Li, Q.; Xu, Y.; Pedersen, A.; Wang, M.; Zhang, M.; Feng, J.; Luo, H.; Titirici, M.-M.; Jones, C.R. Investigating the Role of Fe-Pyrrolic N4 Configuration in the Oxygen Reduction Reaction via Covalently Bound Porphyrin Functionalized Carbon Nanotubes. Adv. Funct. Mater. 2024, 34, 2311086. [Google Scholar] [CrossRef]
- Poulos, T.L. Heme Enzyme Structure and Function. Chem. Rev. 2014, 114, 3919–3962. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Groves, J.T. Oxygen Activation and Radical Transformations in Heme Proteins and Metalloporphyrins. Chem. Rev. 2018, 118, 2491–2553. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.Y.; Xu, C.; Guo, P.P.; Sun, W.L.; Wei, P.J.; Liu, J.G. Axial Ligand Coordination Tuning of the Electrocatalytic Activity of Iron Porphyrin Electrografted onto Carbon Nanotubes for the Oxygen Reduction Reaction. Chemistry 2021, 27, 9898–9904. [Google Scholar] [CrossRef]
- de Visser, S.P.; Valentine, J.S.; Nam, W. A Biomimetic Ferric Hydroperoxo Porphyrin Intermediate. Angew. Chem. Int. Ed. 2010, 49, 2099–2101. [Google Scholar] [CrossRef]
- Layer, G.; Reichelt, J.; Jahn, D.; Heinz, D.W. Structure and function of enzymes in heme biosynthesis. Protein Sci. 2010, 19, 1137–1161. [Google Scholar] [CrossRef]
- Wikström, M.; Krab, K.; Sharma, V. Oxygen Activation and Energy Conservation by Cytochrome c Oxidase. Chem. Rev. 2018, 118, 2469–2490. [Google Scholar] [CrossRef]
- Collman, J.P.; Ghosh, S.; Dey, A.; Decréau, R.A.; Yang, Y. Catalytic Reduction of O2 by Cytochrome c Using a Synthetic Model of Cytochrome c Oxidase. J. Am. Chem. Soc. 2009, 131, 5034–5035. [Google Scholar] [CrossRef]
- Li, Y.; Wang, N.; Lei, H.; Li, X.; Zheng, H.; Wang, H.; Zhang, W.; Cao, R. Bioinspired N4-metallomacrocycles for electrocatalytic oxygen reduction reaction. Coord. Chem. Rev. 2021, 442, 213996. [Google Scholar] [CrossRef]
- Wu, G.; Zelenay, P. Nanostructured Nonprecious Metal Catalysts for Oxygen Reduction Reaction. Acc. Chem. Res. 2013, 46, 1878–1889. [Google Scholar] [CrossRef] [PubMed]
- Cheng, N.; Kemna, C.; Goubert-Renaudin, S.; Wieckowski, A. Reduction Reaction by Porphyrin-Based Catalysts for Fuel Cells. Electrocatalysis 2012, 3, 238–251. [Google Scholar] [CrossRef]
- Hua, Q.; Madsen, K.E.; Esposito, A.M.; Chen, X.; Woods, T.J.; Haasch, R.T.; Xiang, S.; Frenkel, A.I.; Fister, T.T.; Gewirth, A.A. Effect of Support on Oxygen Reduction Reaction Activity of Supported Iron Porphyrins. ACS Catal. 2022, 12, 1139–1149. [Google Scholar] [CrossRef]
- Chatterjee, S.; Sengupta, K.; Mondal, B.; Dey, S.; Dey, A. Factors Determining the Rate and Selectivity of 4e–/4H+ Electrocatalytic Reduction of Dioxygen by Iron Porphyrin Complexes. Acc. Chem. Res. 2017, 50, 1744–1753. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Zhang, X.-P.; Zhao, B.; Li, P.; Qi, J.; Guo, X.; Wang, B.; Lei, H.; Zhang, W.; Apfel, U.-P.; et al. Enzyme-Inspired Iron Porphyrins for Improved Electrocatalytic Oxygen Reduction and Evolution Reactions. Angew. Chem. Int. Ed. 2021, 60, 7576–7581. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.; Rao, R.R.; Peng, J.; Huang, B.; Stephens, I.E.L.; Risch, M.; Xu, Z.J.; Shao-Horn, Y. Recommended Practices and Benchmark Activity for Hydrogen and Oxygen Electrocatalysis in Water Splitting and Fuel Cells. Adv. Mater. 2019, 31, 1806296. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Zhao, L.; Yuan, R.; Xue, Z.; Mack, J.; Chiyumba, C.; Nyokong, T.; Zhang, J. Promotion of Catalytic Oxygen Reduction Reactions: The Utility of Proton Management Substituents on Cobalt Porphyrins. Inorg. Chem. 2022, 61, 13085–13095. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Qiao, J.; Yang, L.; Zhang, J. A Review of Graphene-Based Nanostructural Materials for Both Catalyst Supports and Metal-Free Catalysts in PEM Fuel Cell Oxygen Reduction Reactions. Adv. Energy Mater. 2014, 4, 1301523. [Google Scholar] [CrossRef]
- Das, S.; Ghosh, S.; Kuila, T.; Murmu, N.C.; Kundu, A. Biomass-Derived Advanced Carbon-Based Electrocatalysts for Oxygen Reduction Reaction. Biomass 2022, 2, 155–177. [Google Scholar] [CrossRef]
- Wu, Z.; Yang, G.; Zhang, Q.; Liu, Z.; Peng, F. Deciphering the high overpotential of the oxygen reduction reaction via comprehensively elucidating the open circuit potential. Energy Environ. Sci. 2024, 17, 3338–3346. [Google Scholar] [CrossRef]
- Zhou, R.; Zheng, Y.; Jaroniec, M.; Qiao, S.-Z. Determination of the Electron Transfer Number for the Oxygen Reduction Reaction: From Theory to Experiment. ACS Catal. 2016, 6, 4720–4728. [Google Scholar] [CrossRef]
- Yin, F.; Liu, Y.; Wang, C.; Liu, H. Assessing the electron transfer and oxygen mass transfer of the oxygen reduction reaction using a new electrode kinetic equation. Phys. Chem. Chem. Phys. 2018, 20, 16159–16166. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Kim, Y.; Higgins, D.; Yusuf, M.; Jaramillo, T.F.; Prinz, F.B. Building upon the Koutecky-Levich Equation for Evaluation of Next-Generation Oxygen Reduction Reaction Catalysts. Electrochim. Acta 2017, 255, 99–108. [Google Scholar] [CrossRef]
- Silva, G.; Perez, J. Oxygen Reduction Reaction on Pt-Y/C Catalysts: Activity and Long-Term Stability Study. J. Braz. Chem. Soc. 2023, 34, 549–559. [Google Scholar] [CrossRef]
- Santhanagopalan, S.; White, R.E. Estimating parameters from rotating ring disc electrode measurements. Russ. J. Electrochem. 2017, 53, 1087–1099. [Google Scholar] [CrossRef]
- Xu, Q.; Zhao, L.; Ma, Y.; Yuan, R.; Liu, M.; Xue, Z.; Li, H.; Zhang, J.; Qiu, X. Substituents and the induced partial charge effects on cobalt porphyrins catalytic oxygen reduction reactions in acidic medium. J. Colloid Interface Sci. 2021, 597, 269–277. [Google Scholar] [CrossRef]
- Zhou, Y.; Xing, Y.-F.; Wen, J.; Ma, H.-B.; Wang, F.-B.; Xia, X.-H. Axial ligands tailoring the ORR activity of cobalt porphyrin. Sci. Bull. 2019, 64, 1158–1166. [Google Scholar] [CrossRef]
- Jiao, Y.; Zheng, Y.; Jaroniec, M.; Qiao, S.Z. Origin of the Electrocatalytic Oxygen Reduction Activity of Graphene-Based Catalysts: A Roadmap to Achieve the Best Performance. J. Am. Chem. Soc. 2014, 136, 4394–4403. [Google Scholar] [CrossRef]
- Ge, X.; Sumboja, A.; Wuu, D.; An, T.; Li, B.; Goh, F.W.T.; Hor, T.S.A.; Zong, Y.; Liu, Z. Oxygen Reduction in Alkaline Media: From Mechanisms to Recent Advances of Catalysts. ACS Catal. 2015, 5, 4643–4667. [Google Scholar] [CrossRef]
- Kim, J.; Bard, A.J. Application of the Koutecký-Levich Method to the Analysis of Steady State Voltammograms with Ultramicroelectrodes. Anal. Chem. 2016, 88, 1742–1747. [Google Scholar] [CrossRef] [PubMed]
- Osmieri, L.; Monteverde Videla, A.H.A.; Ocón, P.; Specchia, S. Kinetics of Oxygen Electroreduction on Me–N–C (Me = Fe, Co, Cu) Catalysts in Acidic Medium: Insights on the Effect of the Transition Metal. J. Phys. Chem. C 2017, 121, 17796–17817. [Google Scholar] [CrossRef]
- Jaouen, F. O2 Reduction Mechanism on Non-Noble Metal Catalysts for PEM Fuel Cells. Part II: A Porous-Electrode Model To Predict the Quantity of H2O2 Detected by Rotating Ring-Disk Electrode. J. Phys. Chem. C 2009, 113, 15433–15443. [Google Scholar] [CrossRef]
- Xu, Q.; Zhao, L.; Yuan, R.; Chen, Y.; Xue, Z.; Zhang, J.; Qiu, X.; Qu, J. Interfacial charge transfer mechanism of oxygen reduction reaction in alkali media: Effects of molecular charge states and triphenylamine substituent on cobalt porphyrin electrocatalysts. Colloids Surf. A Physicochem. Eng. Asp. 2021, 629, 127435. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, L.; Yuan, R.; Xue, Z.; Wang, A.; Xu, H.; Cheng, M.; Xu, H.; Zhang, J. Donor-π-acceptor cobalt porphyrins for electrocatalytic oxygen reduction reaction in acidic medium. J. Alloys Compd. 2022, 902, 163790. [Google Scholar] [CrossRef]
- Wan, K.; Chu, T.; Li, B.; Ming, P.; Zhang, C. Rational Design of Atomically Dispersed Metal Site Electrocatalysts for Oxygen Reduction Reaction. Adv. Sci. 2023, 10, 2203391. [Google Scholar] [CrossRef]
- Yasin, G.; Ibrahim, S.; Ajmal, S.; Ibraheem, S.; Ali, S.; Nadda, A.K.; Zhang, G.; Kaur, J.; Maiyalagan, T.; Gupta, R.K.; et al. Tailoring of electrocatalyst interactions at interfacial level to benchmark the oxygen reduction reaction. Coord. Chem. Rev. 2022, 469, 214669. [Google Scholar] [CrossRef]
- Hong, Y.; Li, L.; Huang, B.; Tang, X.; Zhai, W.; Hu, T.; Yuan, K.; Chen, Y. Molecular Control of Carbon-Based Oxygen Reduction Electrocatalysts through Metal Macrocyclic Complexes Functionalization. Adv. Energy Mater. 2021, 11, 2100866. [Google Scholar] [CrossRef]
- Kulkarni, A.; Siahrostami, S.; Patel, A.; Nørskov, J.K. Understanding Catalytic Activity Trends in the Oxygen Reduction Reaction. Chem. Rev. 2018, 118, 2302–2312. [Google Scholar] [CrossRef]
- Pegis, M.L.; Martin, D.J.; Wise, C.F.; Brezny, A.C.; Johnson, S.I.; Johnson, L.E.; Kumar, N.; Raugei, S.; Mayer, J.M. Mechanism of Catalytic O2 Reduction by Iron Tetraphenylporphyrin. J. Am. Chem. Soc. 2019, 141, 8315–8326. [Google Scholar] [CrossRef]
- Surendran, A.K.; Pereverzev, A.Y.; Roithová, J. Intricacies of Mass Transport during Electrocatalysis: A Journey through Iron Porphyrin-Catalyzed Oxygen Reduction. J. Am. Chem. Soc. 2024, 146, 15619–15626. [Google Scholar] [CrossRef] [PubMed]
- Chilukuri, B.; Mazur, U.; Hipps, K.W. Structure, Properties, and Reactivity of Porphyrins on Surfaces and Nanostructures with Periodic DFT Calculations. Appl. Sci. 2020, 10, 740. [Google Scholar] [CrossRef]
- Rao, P.D.; Dhanalekshmi, S.; Littler, B.J.; Lindsey, J.S. Rational Syntheses of Porphyrins Bearing up to Four Different Meso Substituents. J. Org. Chem. 2000, 65, 7323–7344. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Xu, Q.; Shao, Z.; Chen, Y.; Xue, Z.; Li, H.; Zhang, J. Enhanced Oxygen Reduction Reaction Performance Using Intermolecular Forces Coupled with More Exposed Molecular Orbitals of Triphenylamine in Co-porphyrin Electrocatalysts. ACS Appl. Mater. Interfaces 2020, 12, 45976–45986. [Google Scholar] [CrossRef]
- Wei, Y.; Liang, Y.; Wu, Q.; Xue, Z.; Feng, L.; Zhang, J.; Zhao, L. Effects of tuning the structural symmetry of cobalt porphyrin on electrocatalytic oxygen reduction reactions. Dalton Trans. 2023, 52, 14573–14582. [Google Scholar] [CrossRef]
- Oveisi, A.R.; Karimi, P.; Delarami, H.S.; Daliran, S.; Khorramabadi-Zad, A.; Khajeh, M.; Sanchooli, E.; Ghaffari-Moghaddam, M. New porphyrins: Synthesis, characterization, and computational studies. Mol. Divers. 2020, 24, 335–344. [Google Scholar] [CrossRef]
- Rebelo, S.L.H.; Silva, A.M.N.; Medforth, C.J.; Freire, C. Iron(III) Fluorinated Porphyrins: Greener Chemistry from Synthesis to Oxidative Catalysis Reactions. Molecules 2016, 21, 481. [Google Scholar] [CrossRef]
- Sun, Z.-C.; She, Y.-B.; Zhou, Y.; Song, X.-F.; Li, K. Synthesis, Characterization and Spectral Properties of Substituted Tetraphenylporphyrin Iron Chloride Complexes. Molecules 2011, 16, 2960–2970. [Google Scholar] [CrossRef]
- Yuan, R.; Wei, Y.; Xue, Z.; Wang, A.; Zhang, J.; Xu, H.; Zhao, L. Effects of support material and electrolyte on a triphenylamine substituted cobalt porphyrin catalytic oxygen reduction reaction. Colloids Surf. A Physicochem. Eng. Asp. 2023, 665, 131214. [Google Scholar] [CrossRef]
- Gjuroski, I.; Furrer, J.; Vermathen, M. Probing the Interactions of Porphyrins with Macromolecules Using NMR Spectroscopy Techniques. Molecules 2021, 26, 1942. [Google Scholar] [CrossRef]
- Tachibana, Y.; Haque, S.A.; Mercer, I.P.; Durrant, J.R.; Klug, D.R. Electron Injection and Recombination in Dye Sensitized Nanocrystalline Titanium Dioxide Films: A Comparison of Ruthenium Bipyridyl and Porphyrin Sensitizer Dyes. J. Phys. Chem. B 2000, 104, 1198–1205. [Google Scholar] [CrossRef]
- Giovannetti, R. The Use of Spectrophotometry UV-Vis for the Study of Porphyrins. In Macro To Nano Spectroscopy; Intech: Rijeka, Croatia, 2012. [Google Scholar] [CrossRef]
- Bhunia, S.; Ghatak, A.; Rana, A.; Dey, A. Amine Groups in the Second Sphere of Iron Porphyrins Allow for Higher and Selective 4e–/4H+ Oxygen Reduction Rates at Lower Overpotentials. J. Am. Chem. Soc. 2023, 145, 3812–3825. [Google Scholar] [CrossRef] [PubMed]
- Chlistunoff, J.; Sansiñena, J.-M. Effects of Axial Coordination of the Metal Center on the Activity of Iron Tetraphenylporphyrin as a Nonprecious Catalyst for Oxygen Reduction. J. Phys. Chem. C 2014, 118, 19139–19149. [Google Scholar] [CrossRef]
- Ohta, T.; Nagaraju, P.; Liu, J.-G.; Ogura, T.; Naruta, Y. The secondary coordination sphere and axial ligand effects on oxygen reduction reaction by iron porphyrins: A DFT computational study. JBIC J. Biol. Inorg. Chem. 2016, 21, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Amanullah, S.; Singha, A.; Dey, A. Tailor made iron porphyrins for investigating axial ligand and distal environment contributions to electronic structure and reactivity. Coord. Chem. Rev. 2019, 386, 183–208. [Google Scholar] [CrossRef]
- Samanta, S.; Das, P.K.; Chatterjee, S.; Sengupta, K.; Mondal, B.; Dey, A. O2 Reduction Reaction by Biologically Relevant Anionic Ligand Bound Iron Porphyrin Complexes. Inorg. Chem. 2013, 52, 12963–12971. [Google Scholar] [CrossRef]
- Mittra, K.; Chatterjee, S.; Samanta, S.; Dey, A. Selective 4e−/4H+ O2 Reduction by an Iron(tetraferrocenyl)Porphyrin Complex: From Proton Transfer Followed by Electron Transfer in Organic Solvent to Proton Coupled Electron Transfer in Aqueous Medium. Inorg. Chem. 2013, 52, 14317–14325. [Google Scholar] [CrossRef]
- Ghatak, A.; Bhakta, S.; Bhunia, S.; Dey, A. Influence of the distal guanidine group on the rate and selectivity of O2 reduction by iron porphyrin. Chem. Sci. 2019, 10, 9692–9698. [Google Scholar] [CrossRef]
- Matson, B.D.; Carver, C.T.; Von Ruden, A.; Yang, J.Y.; Raugei, S.; Mayer, J.M. Distant protonated pyridine groups in water-soluble iron porphyrin electrocatalysts promote selective oxygen reduction to water. Chem. Commun. 2012, 48, 11100–11102. [Google Scholar] [CrossRef]
- Singha, A.; Mondal, A.; Nayek, A.; Dey, S.G.; Dey, A. Oxygen Reduction by Iron Porphyrins with Covalently Attached Pendent Phenol and Quinol. J. Am. Chem. Soc. 2020, 142, 21810–21828. [Google Scholar] [CrossRef]
- Brüller, S.; Liang, H.-W.; Kramm, U.I.; Krumpfer, J.W.; Feng, X.; Müllen, K. Bimetallic porous porphyrin polymer-derived non-precious metal electrocatalysts for oxygen reduction reactions. J. Mater. Chem. A 2015, 3, 23799–23808. [Google Scholar] [CrossRef]
- Suntivich, J.; Gasteiger, H.A.; Yabuuchi, N.; Nakanishi, H.; Goodenough, J.B.; Shao-Horn, Y. Design principles for oxygen-reduction activity on perovskite oxide catalysts for fuel cells and metal–air batteries. Nat. Chem. 2011, 3, 546–550. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Xin, Z.; Luo, Y.; Pan, J.; Liao, G.; Li, Q.; Sun, Y.; Feng, Z.; Tan, R. Recent advances in the development of single atom catalysts for oxygen evolution reaction. Int. J. Hydrogen Energy 2024, 82, 1081–1100. [Google Scholar] [CrossRef]
- He, H.; Qiu, Z.-Y.; Yin, Z.; Kong, J.; Dang, J.-S.; Lei, H.; Zhang, W.; Cao, R. The meso-substituent electronic effect of Fe porphyrins on the electrocatalytic CO2 reduction reaction. Chem. Commun. 2024, 60, 5916–5919. [Google Scholar] [CrossRef] [PubMed]
- Gujarathi, P.B. Review on synthetic advances in porphyrins and metalloporphyrins. Int. J. Chem. Stud. 2020, 8, 23–32. [Google Scholar] [CrossRef]
- Ma, Z.; Hada, M. Effect of meso-Substitution on the Selectivity of the Propene Reaction by Fe(IV)OCl–Porphyrin: A Density Functional Theory Mechanistic Study. J. Comput. Chem. Jpn.-Int. Ed. 2021, 7, 2020-0011. [Google Scholar] [CrossRef]
- Millheim, A.C.; Ponzano, E.; Moyano, A. Substituent Effects in the Photophysical and Electrochemical Properties of Meso-Tetraphenylporphyrin Derivatives. Molecules 2024, 29, 3689. [Google Scholar] [CrossRef]
- Lewtak, J.P.; Gryko, D.T. Synthesis of π-extended porphyrins via intramolecular oxidative coupling. Chem. Commun. 2012, 48, 10069–10086. [Google Scholar] [CrossRef] [PubMed]
- Qu, C.; Gong, X.; Sun, Y.; Gao, H.; Cai, F.; Zhao, Y.; Wu, F.; Shen, Z. Synergistic meso-β regulation of porphyrins: Squeezing the band gap into the near-infrared I/II region. Chem. Sci. 2024, 15, 10491–10498. [Google Scholar] [CrossRef]
- Shi, Z.; Zhang, J. Density Functional Theory Study of Transitional Metal Macrocyclic Complexes’ Dioxygen-Binding Abilities and Their Catalytic Activities toward Oxygen Reduction Reaction. J. Phys. Chem. C 2007, 111, 7084–7090. [Google Scholar] [CrossRef]
- Sun, B.; Ou, Z.; Yang, S.; Meng, D.; Lu, G.; Fang, Y.; Kadish, K.M. Synthesis and electrochemistry of β-pyrrole nitro-substituted cobalt(ii) porphyrins. The effect of the NO2 group on redox potentials, the electron transfer mechanism and catalytic reduction of molecular oxygen in acidic media. Dalton Trans. 2014, 43, 10809–10815. [Google Scholar] [CrossRef] [PubMed]
- Lü, A.; Fang, Y.; Zhu, M.; Huang, S.; Ou, Z.; Kadish, K.M. Dioxygen reduction catalyzed by substituted iron tetraphenylporphyrins in acidic media. J. Porphyr. Phthalocyanines 2012, 16, 310–315. [Google Scholar] [CrossRef]
- Jaouen, F.; Proietti, E.; Lefèvre, M.; Chenitz, R.; Dodelet, J.-P.; Wu, G.; Chung, H.T.; Johnston, C.M.; Zelenay, P. Recent advances in non-precious metal catalysis for oxygen-reduction reaction in polymer electrolyte fuel cells. Energy Environ. Sci. 2011, 4, 114–130. [Google Scholar] [CrossRef]
- Percástegui, E.G.; Jancik, V. Coordination-driven assemblies based on meso-substituted porphyrins: Metal-organic cages and a new type of meso-metallaporphyrin macrocycles. Coord. Chem. Rev. 2020, 407, 213165. [Google Scholar] [CrossRef]
- Araki, K.; Toma, H.E. Supramolecular Porphyrins as Electrocatalysts. In N4-Macrocyclic Metal Complexes; Zagal, J.H., Bedioui, F., Dodelet, J.-P., Eds.; Springer: New York, NY, USA, 2006; pp. 255–314. [Google Scholar]
- Torabi, M.; Tomapatanaget, B.; Karimi Shervedani, R. Bifunctional electrocatalyst for oxygen reduction/evolution reactions based on controlled morphology of nickel–cobalt nanoprickly particles composited by graphene and carbon nanotube. J. Mater. Sci. 2024, 59, 7218–7234. [Google Scholar] [CrossRef]
- Santra, A.; Das, A.; Kaur, S.; Jain, P.; Ingole, P.P.; Paria, S. Catalytic reduction of oxygen to water by non-heme iron complexes: Exploring the effect of the secondary coordination sphere proton exchanging site. Chem. Sci. 2024, 15, 4095–4105. [Google Scholar] [CrossRef]
- Rosenthal, J.; Nocera, D.G. Role of Proton-Coupled Electron Transfer in O–O Bond Activation. Acc. Chem. Res. 2007, 40, 543–553. [Google Scholar] [CrossRef]
- Savéant, J.-M. Proton Relays in Molecular Catalysis of Electrochemical Reactions: Origin and Limitations of the Boosting Effect. Angew. Chem. Int. Ed. 2019, 58, 2125–2128. [Google Scholar] [CrossRef]
- Han, J.; Wang, N.; Li, X.; Zhang, W.; Cao, R. Improving Electrocatalytic Oxygen Reduction Activity and Selectivity with a Cobalt Corrole Appended with Multiple Positively Charged Proton Relay Sites. J. Phys. Chem. C 2021, 125, 24805–24813. [Google Scholar] [CrossRef]
- Dung, T.P.; Chihaia, V.; Son, D.N. Effects of functional groups in iron porphyrin on the mechanism and activity of oxygen reduction reaction. RSC Adv. 2023, 13, 8523–8534. [Google Scholar] [CrossRef] [PubMed]
- Carver, C.T.; Matson, B.D.; Mayer, J.M. Electrocatalytic Oxygen Reduction by Iron Tetra-arylporphyrins Bearing Pendant Proton Relays. J. Am. Chem. Soc. 2012, 134, 5444–5447. [Google Scholar] [CrossRef] [PubMed]
- Hu, Z.; Shi, J.; Yang, H.; Huang, J.; Sheng, F. How Organic Substances Promote the Chemical Oxidative Degradation of Pollutants: A Mini Review. Sustainability 2021, 13, 10993. [Google Scholar] [CrossRef]
- Mazzucato, M.; Durante, C. Insights on Oxygen Reduction Reaction to H2O2: The role of functional groups and textural properties on the activity and selectivity of doped carbon electrocatalysts. Curr. Opin. Electrochem. 2022, 35, 101051. [Google Scholar] [CrossRef]
- Brezny, A.C.; Nedzbala, H.S.; Mayer, J.M. Multiple selectivity-determining mechanisms of H2O2 formation in iron porphyrin-catalysed oxygen reduction. Chem. Commun. 2021, 57, 1202–1205. [Google Scholar] [CrossRef]
- Wang, Z.-D.; Liang, S.; Bai, C.-K.; Guo, Z.-F.; Lu, G.-L.; Sun, H.; Liu, Z.-N.; Zang, H.-Y. Synergistically enhanced iron and zinc bimetallic sites as an advanced ORR electrocatalyst for flow liquid rechargeable Zn–air batteries. J. Mater. Chem. A 2022, 10, 3169–3177. [Google Scholar] [CrossRef]
- Arima, H.; Wada, M.; Nakazono, T.; Wada, T. Tuning Oxygen Reduction Catalysis of Dinuclear Cobalt Polypyridyl Complexes by the Bridging Structure. Inorg. Chem. 2021, 60, 9402–9415. [Google Scholar] [CrossRef]
- Collman, J.P.; Fu, L.; Herrmann, P.C.; Zhang, X. A Functional Model Related to Cytochrome c Oxidase and Its Electrocatalytic Four-Electron Reduction of O2. Science 1997, 275, 949–951. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, G.; Zhang, Z.; Lei, H.; Yao, Z.; Li, J.; Lin, J.; Cao, R. Significantly improved electrocatalytic oxygen reduction by an asymmetrical Pacman dinuclear cobalt(ii) porphyrin-porphyrin dyad. Chem. Sci. 2020, 11, 87–96. [Google Scholar] [CrossRef]
- Kodali, M.; Santoro, C.; Herrera, S.; Serov, A.; Atanassov, P. Bimetallic platinum group metal-free catalysts for high power generating microbial fuel cells. J. Power Sources 2017, 366, 18–26. [Google Scholar] [CrossRef]
- Li, W.; Ye, Q.; Chen, W.; Ouyang, Y.; Liu, R.; Yu, J.; Zhuo, H.; Zhang, Y.; Chen, Y. Synergistic effects of Co/Fe bimetallic polymer catalysts for enhanced ORR/OER: Insights from thermodynamic and in-situ kinetic study. Mol. Catal. 2024, 568, 114509. [Google Scholar] [CrossRef]
- Maruyama, J.; Baier, C.; Holger, W.; Bele, P.; Stimming, U. Enhancement of oxygen reduction at Fe tetrapyridyl porphyrin by pyridyl-N coordination to transition metal ions. Electrochim. Acta 2012, 63, 16–21. [Google Scholar] [CrossRef]
- Collman, J.P.; Ghosh, S.; Dey, A.; Decréau, R.A. Using a Functional Enzyme Model to Understand the Chemistry behind Hydrogen Sulfide Induced Hibernation. Proc. Natl. Acad. Sci. USA 2009, 106, 22090–22095. [Google Scholar] [CrossRef] [PubMed]
- Collman, J.P.; Devaraj, N.K.; Decréau, R.A.; Yang, Y.; Yan, Y.-L.; Ebina, W.; Eberspacher, T.A.; Chidsey, C.E.D. A Cytochrome c Oxidase Model Catalyzes Oxygen to Water Reduction Under Rate-Limiting Electron Flux. Science 2007, 315, 1565–1568. [Google Scholar] [CrossRef] [PubMed]
- Kuzmin, S.M.; Chulovskaya, S.A.; Filimonova, Y.A.; Parfenyuk, V.I. Synergistic effect of two metal porphyrins in a polymer catalyst for oxygen electroreduction. J. Electroanal. Chem. 2023, 947, 117798. [Google Scholar] [CrossRef]
- Feng, Y.-C.; Wang, X.; Wang, D. Metal porphyrins and metal phthalocyanines as designable molecular model electrocatalysts. Mater. Chem. Front. 2024, 8, 228–247. [Google Scholar] [CrossRef]
- Zhu, Y.; Han, C.; Chen, Z. Insights into the origin of Co-based bimetallic catalysts with a para-structure exhibiting ORR and OER bifunctional activities. Inorg. Chem. Front. 2024, 11, 4590–4602. [Google Scholar] [CrossRef]
- Tang, C.; Li, X.; Hu, Y.; Du, X.; Wang, S.; Chen, B.; Wang, S. Porphyrin-Based Metal-Organic Framework Materials: Design, Construction, and Application in the Field of Photocatalysis. Molecules 2024, 29, 467. [Google Scholar] [CrossRef]
- Jin, S. How to Effectively Utilize MOFs for Electrocatalysis. ACS Energy Lett. 2019, 4, 1443–1445. [Google Scholar] [CrossRef]
- Zion, N.; Friedman, A.; Levy, N.; Elbaz, L. Bioinspired Electrocatalysis of Oxygen Reduction Reaction in Fuel Cells Using Molecular Catalysts. Adv. Mater. 2018, 30, 1800406. [Google Scholar] [CrossRef]
- Liang, Z.; Guo, H.; Zhou, G.; Guo, K.; Wang, B.; Lei, H.; Zhang, W.; Zheng, H.; Apfel, U.-P.; Cao, R. Metal–Organic-Framework-Supported Molecular Electrocatalysis for the Oxygen Reduction Reaction. Angew. Chem. Int. Ed. 2021, 60, 8472–8476. [Google Scholar] [CrossRef]
- Li, C.; Zhang, H.; Liu, M.; Lang, F.-F.; Pang, J.; Bu, X.-H. Recent progress in metal–organic frameworks (MOFs) for electrocatalysis. Ind. Chem. Mater. 2023, 1, 9–38. [Google Scholar] [CrossRef]
- Liberman, I.; Shimoni, R.; Ifraemov, R.; Rozenberg, I.; Singh, C.; Hod, I. Active-Site Modulation in an Fe-Porphyrin-Based Metal–Organic Framework through Ligand Axial Coordination: Accelerating Electrocatalysis and Charge-Transport Kinetics. J. Am. Chem. Soc. 2020, 142, 1933–1940. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.-S.; Huang, Z.-Y.; Qi, X.-W.; Si, L.-P.; Zhang, H.; Liu, H.-Y. The optimization of iron porphyrin@MOF-5 derived FeNC electrocatalysts for oxygen reduction reaction in zinc-air batteries. J. Electroanal. Chem. 2023, 936, 117381. [Google Scholar] [CrossRef]
- Jahan, M.; Bao, Q.; Loh, K. Electrocatalytically Active Graphene−Porphyrin MOF Composite for Oxygen Reduction Reaction. J. Am. Chem. Soc. 2012, 134, 6707–6713. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, H.; Bae, Y.E.; Park, K.C.; Ji, H.; Jeong, N.C.; Lee, M.H.; Kwon, O.J.; Lee, C.Y. Dual-Functional Electrocatalyst Derived from Iron-Porphyrin-Encapsulated Metal–Organic Frameworks. ACS Appl. Mater. Interfaces 2017, 9, 28758–28765. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Zhang, P.; Sun, Y.; Chen, J.; Hou, C.; Wang, Z. Design and construction of porphyrin box-based metal–organic frameworks with hierarchical superstructures for efficient energy transfer and photooxidation. J. Mater. Chem. A 2024, 12, 30685–30691. [Google Scholar] [CrossRef]
- Saini, R.; Naaz, F.; Bashal, A.H.; Pandit, A.H.; Farooq, U. Recent advances in nitrogen-doped graphene-based heterostructures and composites: Mechanism and active sites for electrochemical ORR and HER. Green Chem. 2024, 26, 57–102. [Google Scholar] [CrossRef]
- Yuan, H.; Liu, P.; Ren, J.; Jiang, Z.; Wang, X.; Zhao, H. Carbon dot hybrid porous carbon nanofibers as efficient electrocatalysts for the oxygen reduction reaction. Mater. Chem. Front. 2024, 8, 1643–1650. [Google Scholar] [CrossRef]
- Tan, H.; Tang, J.; Kim, J.; Kaneti, Y.V.; Kang, Y.-M.; Sugahara, Y.; Yamauchi, Y. Rational design and construction of nanoporous iron- and nitrogen-doped carbon electrocatalysts for oxygen reduction reaction. J. Mater. Chem. A 2019, 7, 1380–1393. [Google Scholar] [CrossRef]
- Li, J.; Song, Y.; Zhang, G.; Liu, H.; Wang, Y.; Sun, S.; Guo, X. Pyrolysis of Self-Assembled Iron Porphyrin on Carbon Black as Core/Shell Structured Electrocatalysts for Highly Efficient Oxygen Reduction in Both Alkaline and Acidic Medium. Adv. Funct. Mater. 2017, 27, 1604356. [Google Scholar] [CrossRef]
- Choi, C.H.; Park, S.H.; Woo, S.I. Phosphorus–nitrogen dual doped carbon as an effective catalyst for oxygen reduction reaction in acidic media: Effects of the amount of P-doping on the physical and electrochemical properties of carbon. J. Mater. Chem. 2012, 22, 12107–12115. [Google Scholar] [CrossRef]
- Wang, H.; Qiu, X.; Wang, W.; Jiang, L.; Liu, H. Iron Sulfide Nanoparticles Embedded Into a Nitrogen and Sulfur Co-doped Carbon Sphere as a Highly Active Oxygen Reduction Electrocatalyst. Front. Chem. 2019, 7, 855. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Chu, L.; Wang, X.; Yao, Z.; Liu, F.; Ai, Y.; Zhuang, X.; Han, S. Boron, nitrogen, and phosphorous ternary doped graphene aerogel with hierarchically porous structures as highly efficient electrocatalysts for oxygen reduction reaction. New J. Chem. 2016, 40, 6022–6029. [Google Scholar] [CrossRef]
- Zhang, J.; Byeon, A.; Lee, J.W. Boron-doped carbon–iron nanocomposites as efficient oxygen reduction electrocatalysts derived from carbon dioxide. Chem. Commun. 2014, 50, 6349–6352. [Google Scholar] [CrossRef] [PubMed]
- Geng, H.; Wei, Z.; Li, Y.; Deng, Z.; Li, L.; Wang, Y.; Zhai, H.; Li, S.; Chen, W. Controlling the electron spin states ofsingle-atom catalysts for enhanced electrocatalytic performances. JES 2024, 1, 3630. [Google Scholar] [CrossRef]
- Gutru, R.; Turtayeva, Z.; Xu, F.; Maranzana, G.; Thimmappa, R.; Mamlouk, M.; Desforges, A.; Vigolo, B. Recent progress in heteroatom doped carbon based electrocatalysts for oxygen reduction reaction in anion exchange membrane fuel cells. Int. J. Hydrogen Energy 2023, 48, 3593–3631. [Google Scholar] [CrossRef]
- Winston Duo, W.; Dan, X. Active Sites Derived from Heteroatom Doping in Carbon Materials for Oxygen Reduction Reaction. In Electrocatalysts for Fuel Cells and Hydrogen Evolution; Abhijit, R., Indrajit, M., Ranjan, K.P., Eds.; IntechOpen: Rijeka, Croatia, 2018; Chapter 3; pp. 51–67. [Google Scholar]
- Zhao, H.; Chen, L.; Xu, Y.; Wang, H.; Li, J.-Y.; Xie, Y.; Wang, L. A Nitrogen and Sulfur Co-doped Iron-based Electrocatalyst Derived from Iron and Biomass Ligand towards the Oxygen Reduction Reaction in Alkaline Media. Dalton Trans. 2021, 50, 13943–13950. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, J.; He, F.; Chen, Y.; Zhu, J.; Wang, D.; Mu, S.; Yang, H.Y. Defect and Doping Co-Engineered Non-Metal Nanocarbon ORR Electrocatalyst. Nanomicro Lett. 2021, 13, 65. [Google Scholar] [CrossRef]
- Sanad, M.F.; Puente Santiago, A.R.; Tolba, S.A.; Ahsan, M.A.; Fernandez-Delgado, O.; Shawky Adly, M.; Hashem, E.M.; Mahrous Abodouh, M.; El-Shall, M.S.; Sreenivasan, S.T.; et al. Co–Cu Bimetallic Metal Organic Framework Catalyst Outperforms the Pt/C Benchmark for Oxygen Reduction. J. Am. Chem. Soc. 2021, 143, 4064–4073. [Google Scholar] [CrossRef]
- Wu, J.; Yang, H. Platinum-Based Oxygen Reduction Electrocatalysts. Acc. Chem. Res. 2013, 46, 1848–1857. [Google Scholar] [CrossRef]
- Hussain, S.; Erikson, H.; Kongi, N.; Sarapuu, A.; Solla-Gullón, J.; Maia, G.; Kannan, A.M.; Alonso-Vante, N.; Tammeveski, K. Oxygen reduction reaction on nanostructured Pt-based electrocatalysts: A review. Int. J. Hydrogen Energy 2020, 45, 31775–31797. [Google Scholar] [CrossRef]
- Lu, B.-A.; Tian, N.; Sun, S.-G. Surface structure effects of platinum-based catalysts for oxygen reduction reaction. Curr. Opin. Electrochem. 2017, 4, 76–82. [Google Scholar] [CrossRef]
- Xu, G.; Yang, L.; Li, J.; Liu, C.; Xing, W.; Zhu, J. Strategies for improving stability of Pt-based catalysts for oxygen reduction reaction. Adv. Sens. Energy Mater. 2023, 2, 100058. [Google Scholar] [CrossRef]
- Hötger, D.; Etzkorn, M.; Morchutt, C.; Wurster, B.; Dreiser, J.; Stepanow, S.; Grumelli, D.; Gutzler, R.; Kern, K. Stability of metallo-porphyrin networks under oxygen reduction and evolution conditions in alkaline media. Phys. Chem. Chem. Phys. 2019, 21, 2587–2594. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; He, X.; Ding, Y.; Shi, Z.; Wu, B. Supply and demand of platinum group metals and strategies for sustainable management. Renew. Sustain. Energy Rev. 2024, 204, 114821. [Google Scholar] [CrossRef]
- Li, S.; Ho, S.-H.; Hua, T.; Zhou, Q.; Li, F.; Tang, J. Sustainable biochar as an electrocatalysts for the oxygen reduction reaction in microbial fuel cells. Green Energy Environ. 2021, 6, 644–659. [Google Scholar] [CrossRef]
- Georgakilas, V.; Perman, J.A.; Tucek, J.; Zboril, R. Broad Family of Carbon Nanoallotropes: Classification, Chemistry, and Applications of Fullerenes, Carbon Dots, Nanotubes, Graphene, Nanodiamonds, and Combined Superstructures. Chem. Rev. 2015, 115, 4744–4822. [Google Scholar] [CrossRef]
- Machulek, A.; Quina, F.; Gozzi, F.; Silva, V.; Friedrich, L.; Moraes, J. Fundamental Mechanistic Studies of the Photo-Fenton Reaction for the Degradation of Organic Pollutants. In Organic Pollutants Ten Years After the Stockholm Convention-Environmental and Analytical Update; Intech: Rijeka, Croatia, 2012. [Google Scholar] [CrossRef]
- Sahoo, M. Degradation and mineralization of organic contaminants by Fenton and photo-Fenton processes: Review of mechanisms and effects of organic and inorganic additives. Res. J. Chem. Environ. 2011, 15, 96–112. [Google Scholar]
- Khan, I.A.A.; Meda, U.S.; Aman, A.; Suresh, R.; Mudbidre, R. Alternatives to Conventional Platinum-Based Catalysts in Polymer Electrolyte Membrane Fuel Cells. ECS Trans. 2022, 107, 5487. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst System | Electrolyte | Support Material | Eonset (mV) | E1/2 (mV) | Electron Transfer Number (n) | Ref. |
|---|---|---|---|---|---|---|
| FeIITPP-CNT | 0.1 M KOH | CNT | 790 vs. RHE | 640 vs. RHE | ≈3.8 | [44] |
| Complex 1 | 0.1 M KPF6 | EPG electrode | −200 vs. Ag/AgCl | - | - | [98] |
| Complex 2 | 0.1 M KPF6 | EPG Electrode | −300 vs. Ag/AgCl | - | - | [98] |
| Complex 3 | 0.1 M KOH | MWCNTs | 920 vs. RHE | 770 vs. RHE | ≈3.8 | [47] |
| Complex 4 | 0.1 M KOH | MWCNTs | 910 vs. RHE | 750 vs. RHE | ≈3.6 | [47] |
| Complex 5 | 0.1 M KOH | MWCNTs | 930 vs. RHE | 810 vs. RHE | ≈3.8 | [47] |
| Complex 6 | 0.1 M KOH | MWCNTs | 1040 vs. RHE | 870 vs. RHE | ≈4.0 | [47] |
| Complex 7 | 0.1 M KOH | CNT | 930 vs. RHE | 840 vs. RHE | 3.97 | [57] |
| Complex 8 | 0.1 M KOH | CNT | 850 vs. RHE | 680 vs. RHE | 3.84 | [57] |
| Complex 9 | 0.1 M KPF6 | Graphite disk | - | - | 3.7 (pH 7) | [99] |
| Complex 10 | Phosphate buffer solution | EPG electrode | −240 vs. Ag/AgCl | −255 vs. Ag/AgCl | - | [100] |
| Complex 13 | 0.5 M Triflic acid (HOTf) | - | 400 vs. NHE | - | - | [101] |
| Complex 14 | 0.5 M HOTf | - | 500 vs. NHE | - | - | [101] |
| Complex 17 | 0.1 M KPF6 | EPG electrode | −250 vs. Ag/AgCl | - | - | [102] |
| Complex 18 | 0.1 M KPF6 | EPG electrode | −310 vs. Ag/AgCl | - | - | [102] |
| Complex 19 | 0.1 M KPF6 | EPG electrode | −300 vs. Ag/AgCl | - | - | [102] |
| Complex 27 | 0.5 M H2SO4 | - | 880 vs. RHE | 780 vs. RHE | ≈4.0 | [103] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
George, S.L.; Zhao, L.; Wang, Z.; Xue, Z.; Zhao, L. Iron Porphyrin-Based Composites for Electrocatalytic Oxygen Reduction Reactions. Molecules 2024, 29, 5655. https://doi.org/10.3390/molecules29235655
George SL, Zhao L, Wang Z, Xue Z, Zhao L. Iron Porphyrin-Based Composites for Electrocatalytic Oxygen Reduction Reactions. Molecules. 2024; 29(23):5655. https://doi.org/10.3390/molecules29235655
Chicago/Turabian StyleGeorge, Stennard Leetroy, Linkai Zhao, Ziyi Wang, Zhaoli Xue, and Long Zhao. 2024. "Iron Porphyrin-Based Composites for Electrocatalytic Oxygen Reduction Reactions" Molecules 29, no. 23: 5655. https://doi.org/10.3390/molecules29235655
APA StyleGeorge, S. L., Zhao, L., Wang, Z., Xue, Z., & Zhao, L. (2024). Iron Porphyrin-Based Composites for Electrocatalytic Oxygen Reduction Reactions. Molecules, 29(23), 5655. https://doi.org/10.3390/molecules29235655