
Mechanism-Based Allylic Carbasugar Chlorides That Form Covalent Intermediates with α- and β-Galactosidases


Abstract

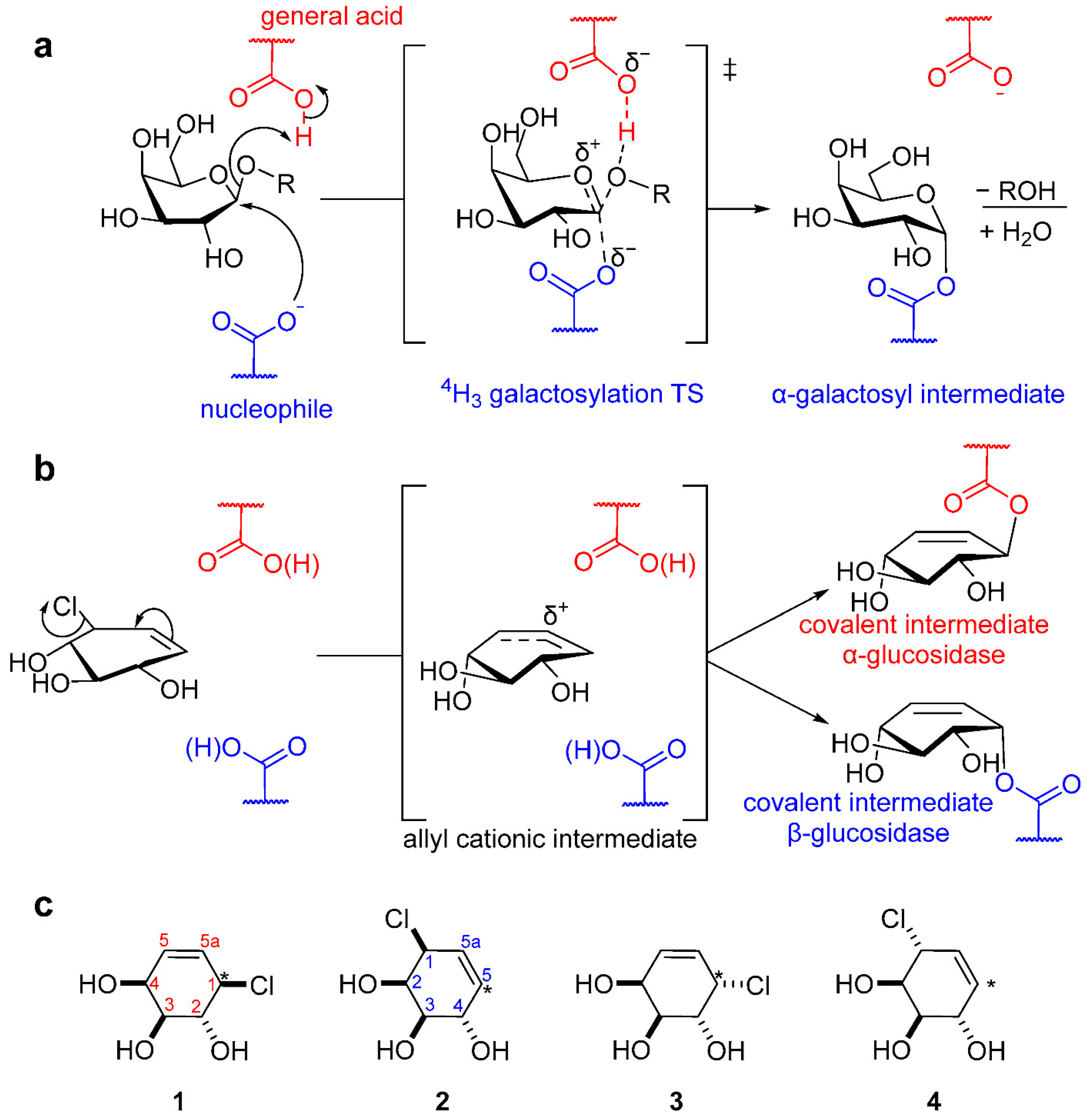
1. Introduction
2. Results and Discussion
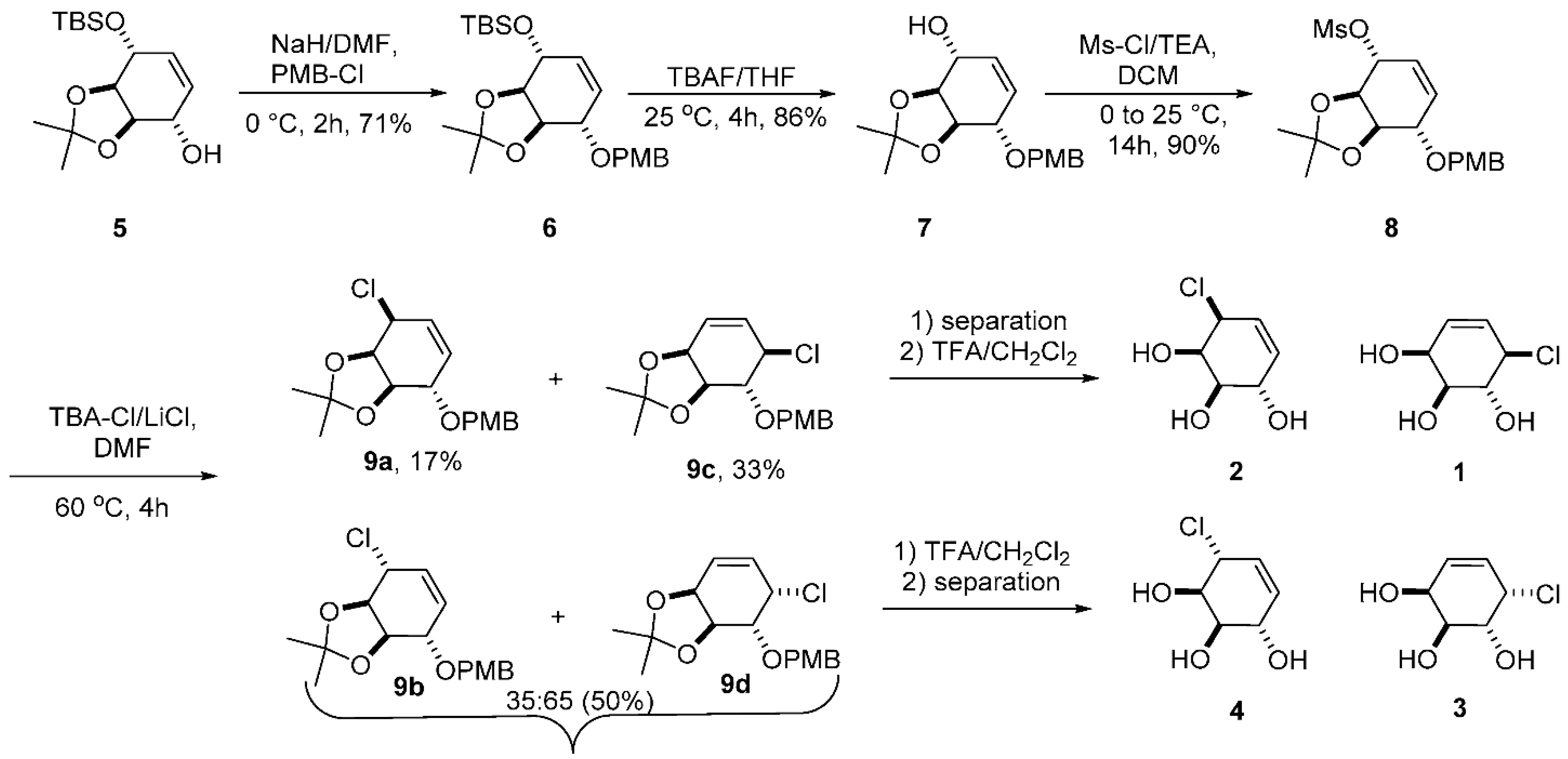
2.1. Synthesis of Galacto-Configured Allylic Carbasugars
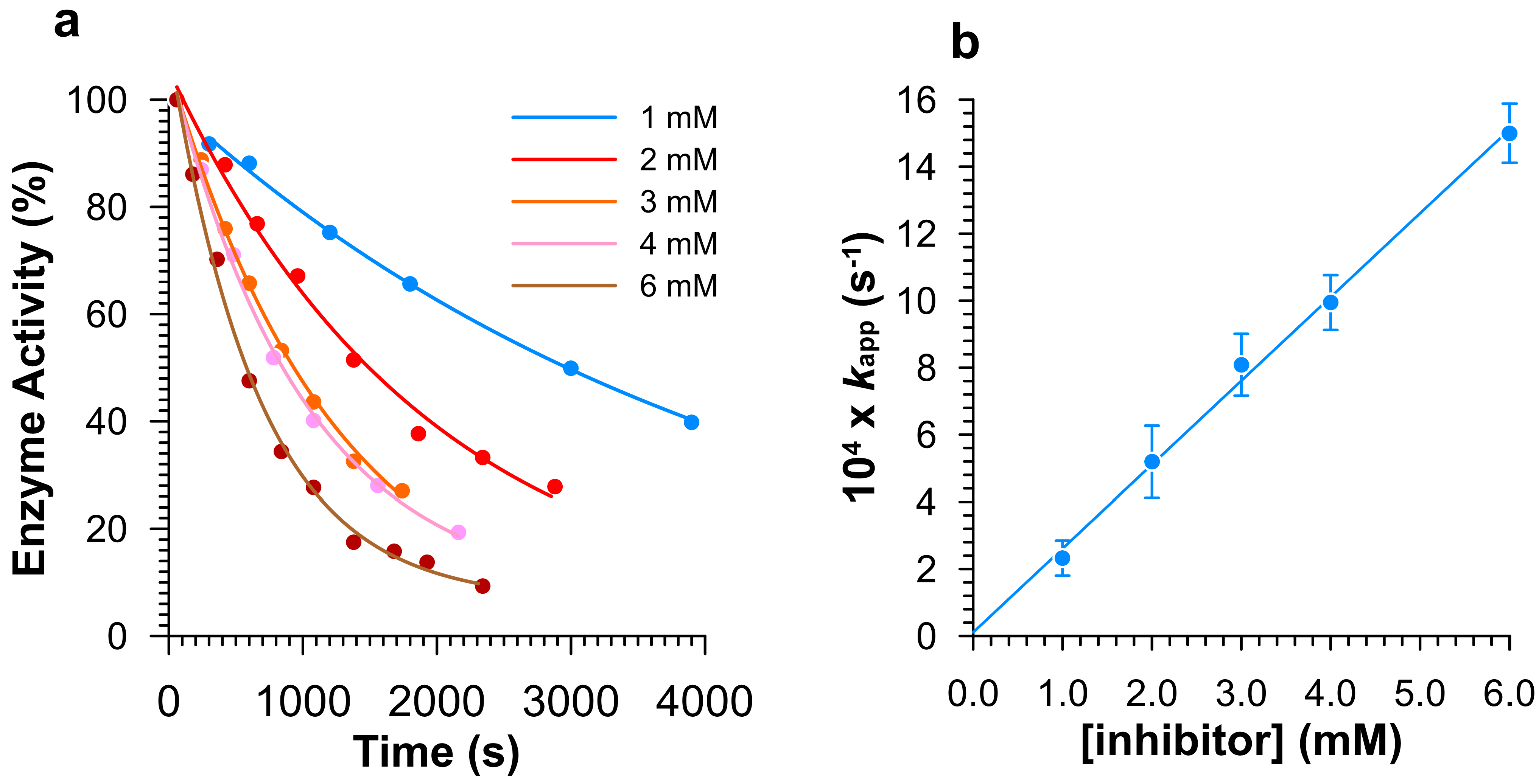
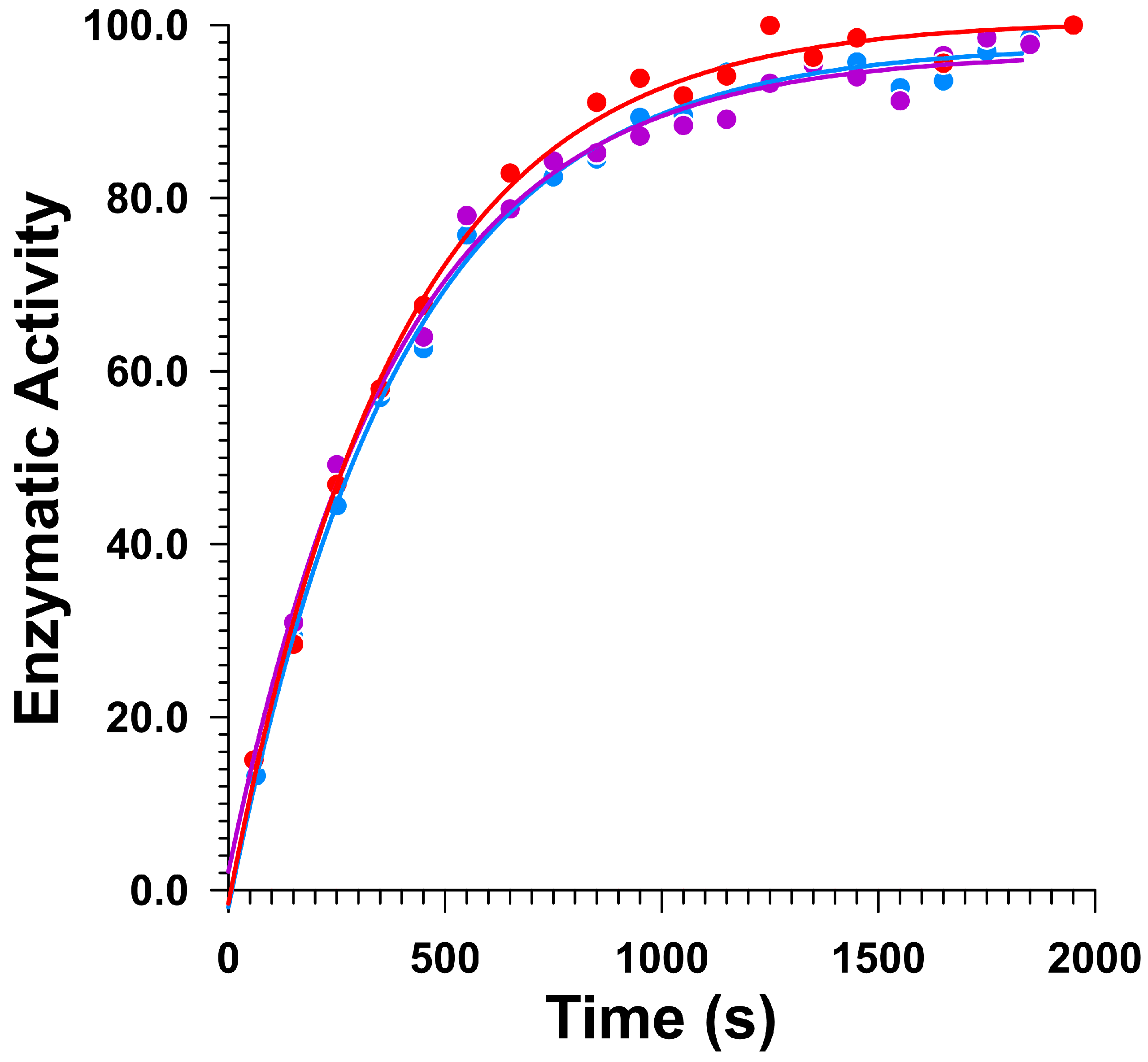
2.2. Kinetics of Galactosidase Covalent Inhibition and Reactivation
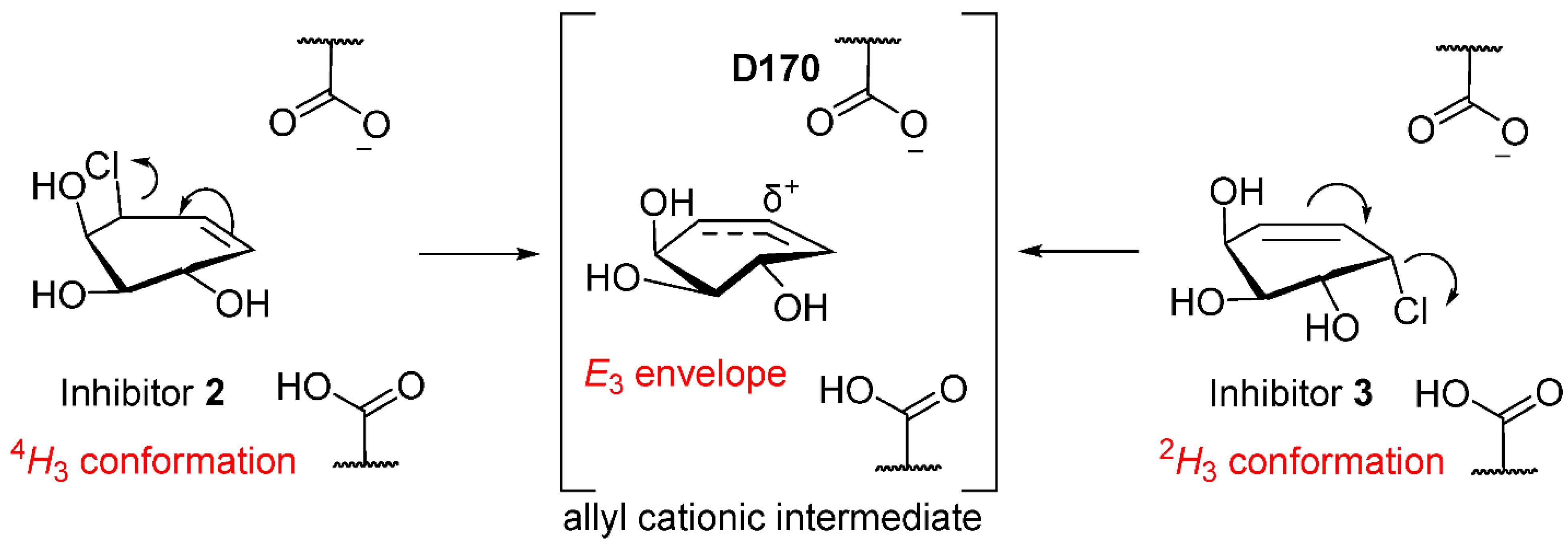
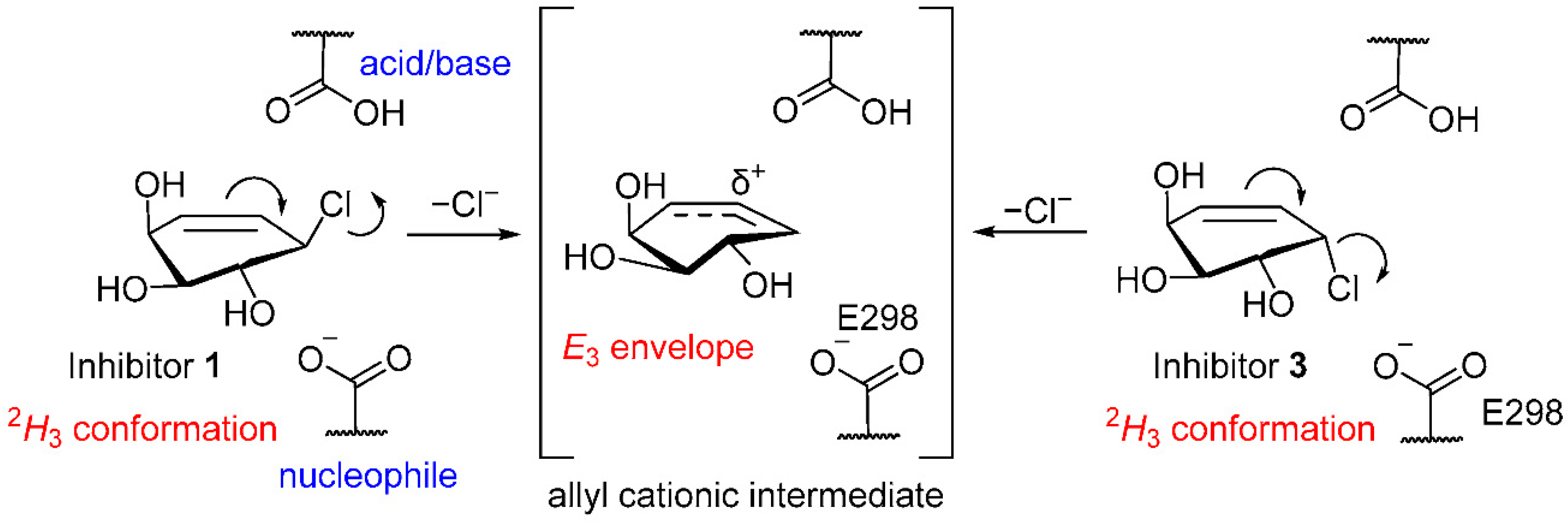
2.3. Inhibitory Mechanism Discussion
2.3.1. Covalent Inhibition and Reactivation of Human α-Galactosidase
2.3.2. Covalent Inhibition and Reactivation of A. Oryzae β-Galactosidase (LacA)
2.3.3. Conformational Itinerary of Carbasugar Inhibitors during Covalent Inhibition and Reactivation of Human α-Galactosidase (GalA) and A. Oryzae β-Galactosidase (LacA)
3. Materials and Methods
3.1. Chemistry
3.2. Measurement of Rate Constants for Galactosidase Covalent Inhibition
3.3. Measurement of Rate Constants for Galactosidase Reactivation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cantarel, B.L.; Coutinho, P.M.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active enZymes database (CAZy): An expert resource for glycogenomics. Nucleic Acids Res. 2009, 37, D233–D238. [Google Scholar] [CrossRef] [PubMed]
- Lairson, L.L.; Henrissat, B.; Davies, G.J.; Withers, S.G. Glycosyltransferases: Structures, functions, and mechanisms. Annu. Rev. Biochem. 2008, 77, 521–555. [Google Scholar] [CrossRef] [PubMed]
- Sinnott, M. Carbohydrate Chemistry and Biochemistry: Structure and Mechanism, 2nd ed.; Royal Society of Chemistry: London, UK, 2013. [Google Scholar]
- Sinnott, M.L. Catalytic mechanisms of enzymic glycosyl transfer. Chem. Rev. 1990, 90, 1171–1202. [Google Scholar] [CrossRef]
- Speciale, G.; Thompson, A.J.; Davies, G.J.; Williams, S.J. Dissecting conformational contributions to glycosidase catalysis and inhibition. Curr. Opin. Struct. Biol. 2014, 28, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Vocadlo, D.J.; Davies, G.J. Mechanistic insights into glycosidase chemistry. Curr. Opin. Chem. Biol. 2008, 12, 539–555. [Google Scholar] [CrossRef]
- Henrissat, B. A classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem. J. 1991, 280, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Drula, E.; Garron, M.L.; Dogan, S.; Lombard, V.; Henrissat, B.; Terrapon, N. The carbohydrate-active enzyme database: Functions and literature. Nucleic Acids Res. 2022, 50, D571–D577. [Google Scholar] [CrossRef]
- Boike, L.; Henning, N.J.; Nomura, D.K. Advances in covalent drug discovery. Nat. Rev. Drug Discov. 2022, 21, 881–898. [Google Scholar] [CrossRef]
- Patel, D.; Huma, Z.E.; Duncan, D. Reversible Covalent Inhibition-Desired Covalent Adduct Formation by Mass Action. ACS Chem. Biol. 2024, 19, 824–838. [Google Scholar] [CrossRef]
- Beenakker, T.J.M.; Wander, D.P.A.; Offen, W.A.; Artola, M.; Raich, L.; Ferraz, M.J.; Li, K.Y.; Houben, J.H.P.M.; van Rijssel, E.R.; Hansen, T.; et al. Carba-cyclophellitols are neutral retaining-glucosidase inhibitors. J. Am. Chem. Soc. 2017, 139, 6534–6537. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Armstrong, Z.; Schroder, S.P.; de Boer, C.; Artola, M.; Aerts, J.; Overkleeft, H.S.; Davies, G.J. An overview of activity-based probes for glycosidases. Curr. Opin. Chem. Biol. 2019, 53, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Rempel, B.P.; Withers, S.G. Covalent inhibitors of glycosidases and their applications in biochemistry and biology. Glycobiology 2008, 18, 570–586. [Google Scholar] [CrossRef] [PubMed]
- Chakladar, S.; Wang, Y.; Clark, T.; Cheng, L.; Ko, S.; Vocadlo, D.J.; Bennet, A.J. A mechanism-based inactivator of glycoside hydrolases involving formation of a transient non-classical carbocation. Nat. Commun. 2014, 5, 5590. [Google Scholar] [CrossRef] [PubMed]
- Adamson, C.; Pengelly, R.; Shamsi Kazem Abadi, S.; Chakladar, S.; Draper, J.; Britton, R.; Gloster, T.; Bennet, A.J. Structural snapshots for mechanism-based inactivation of a glycoside hydrolase by cyclopropyl-carbasugars. Angew. Chem. Int. Ed. 2016, 55, 14978–14982. [Google Scholar] [CrossRef]
- Adabala, P.J.P.; Shamsi Kazem Abadi, S.; Akintola, O.; Bhosale, S.; Bennet, A.J. Conformationally controlled reactivity of carbasugars uncovers the choreography of glycoside hydrolase catalysis. J. Org. Chem. 2020, 85, 3336–3348. [Google Scholar] [CrossRef]
- Shamsi Kazem Abadi, S.; Tran, M.; Yadav, A.K.; Adabala, P.J.P.; Chakladar, S.; Bennet, A.J. New class of glycoside hydrolase mechanism-based covalent inhibitors: Glycosylation transition state conformations. J. Am. Chem. Soc. 2017, 139, 10625–10628. [Google Scholar] [CrossRef] [PubMed]
- Danby, P.M.; Withers, S.G. Glycosyl cations versus allylic cations in spontaneous and enzymatic hydrolysis. J. Am. Chem. Soc. 2017, 139, 10629–10632. [Google Scholar] [CrossRef] [PubMed]
- Herrchen, M.; Legler, G. Identification of an essential carboxylate group at the active site of lacZ beta-galactosidase for Escherichia coli. Eur. J. Biochem. 1984, 138, 527–531. [Google Scholar] [CrossRef]
- Renaut, P.; Millet, J.; Sepulchre, C.; Theveniaux, J.; Barberousse, V. 5a-Carba-b-D-, 5a-carba-b-L- and 5-thio-beta-L-xylopyranosides as new orally active venous antithrombotic agents. Helv. Chim. Acta 1998, 2043–2052. [Google Scholar] [CrossRef]
- Jiang, J.B.; Beenakker, T.J.M.; Kallemeijn, W.W.; van der Marel, G.A.; van den Elst, H.; Codee, J.D.C.; Aerts, J.M.F.G.; Overkleeft, H.S. Comparing cyclophellitol N-alkyl and N-acyl cyclophellitol aziridines as activity-based glycosidase probes. Chem. Eur. J. 2015, 21, 10861–10869. [Google Scholar] [CrossRef]
- McGregor, N.G.S.; Kuo, C.L.; Beenakker, T.J.M.; Wong, C.S.; Offen, W.A.; Armstrong, Z.; Florea, B.I.; Codee, J.D.C.; Overkleeft, H.S.; Aerts, J.; et al. Synthesis of broad-specificity activity-based probes for exo-β-mannosidases. Org. Biomol. Chem. 2022, 20, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Bhosale, S.; Kandalkar, S.; Gilormini, P.A.; Akintola, O.; Rowland, R.; Adabala, J.P.J.; King, D.; Deen, M.C.; Chen, X.; Davies, G.J.; et al. Development of tunable mechanism-based carbasugar ligands that stabilize glycoside hydrolases through the formation of transient covalent intermediates. ACS Catal. 2024, 14, 14769–14779. [Google Scholar] [CrossRef]
- Katoh, T.; Izuhara, T.; Yokota, W.; Inoue, M.; Watanabe, K.; Nobeyama, A.; Suzuki, T. Enantiocontrolled synthesis of the epoxycyclohexenone moieties of scyphostatin, a potent and specific inhibitor of neutral sphingomyelinase. Tetrahedron 2006, 62, 1590–1608. [Google Scholar] [CrossRef]
- Wang, Z.X.; Miller, S.M.; Anderson, O.P.; Shi, Y. A class of C-2 and pseudo C-2 symmetric ketone catalysts for asymmetric epoxidation. Conformational effect on catalysis. J. Org. Chem. 1999, 64, 6443–6458. [Google Scholar] [CrossRef]
- Guce, A.I.; Clark, N.E.; Salgado, E.N.; Ivanen, D.R.; Kulminskaya, A.A.; Brumer, H.; Garman, S.C. Catalytic mechanism of human a-galactosidase. J. Biol. Chem. 2010, 285, 3625–3632. [Google Scholar] [CrossRef]
- Maksimainen, M.M.; Lampio, A.; Mertanen, M.; Turunen, O.; Rouvinen, J. The crystal structure of acidic β-galactosidase from Aspergillus oryzae. Int. J. Biol. Macromol. 2013, 60, 109–115. [Google Scholar] [CrossRef]
- Rico-Díaz, A.; Ramírez-Escudero, M.; Vizoso-Vázquez, A.; Cerdán, M.E.; Becerra, M.; Sanz-Aparicio, J. Structural features of Aspergillus niger β-galactosidase define its activity against glycoside linkages. FEBS J. 2017, 284, 1815–1829. [Google Scholar] [CrossRef]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR chemical shifts of trace impurities: Common laboratory solvents, organics, and gases in deuterated solvents relevant to the organometallic chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef]
- McWhirter, C. Kinetic mechanisms of covalent inhibition. In Design of Covalent-Based Inhibitors; Ward, R.A., Grimster, N.P., Eds.; Academic Press: Cambridge, MA, USA, 2021; Volume 56, pp. 1–31. [Google Scholar]
- Baici, A.; Schenker, P.; Wachter, M.; Ruedi, P. 3-Fluoro-2,4-dioxa-3-phosphadecalins as inhibitors of acetylcholinesterase. A reappraisal of kinetic mechanisms and diagnostic methods. Chem. Biodivers. 2009, 6, 261–282. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | kinact/Ki (M−1 s−1) | kreact (s−1) | t1/2 (min) |
|---|---|---|---|
| 1 | NIO a at 5 mM | – | |
| 2 | 0.25 ± 0.004 | (5.1 ± 0.2) × 10−4 | 22.5 ± 0.7 |
| 3 | 0.18 ± 0.01 | (4.9 ± 0.2) × 10−4 | 23.4 ± 0.9 |
| 4 | NIO a at 5 mM | – |
| Compound | kinact/Ki (M−1 s−1) | kreact (s−1) | t1/2 (min) |
|---|---|---|---|
| 1 | 17.0 ± 0.5 | (2.5 ± 0.1) × 10−3 | 4.6 ± 0.2 |
| 2 | 47 ± 2 | (2.6 ± 0.2) × 10−3 | 4.5 ± 0.3 |
| 3 | 2.7 ± 0.04 | (2.6 ± 0.2) × 10−3 | 4.5 ± 0.3 |
| 4 | NIO a at 1 mM | – | – |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Akintola, O.; Bhosale, S.; Bennet, A.J. Mechanism-Based Allylic Carbasugar Chlorides That Form Covalent Intermediates with α- and β-Galactosidases. Molecules 2024, 29, 4870. https://doi.org/10.3390/molecules29204870
Akintola O, Bhosale S, Bennet AJ. Mechanism-Based Allylic Carbasugar Chlorides That Form Covalent Intermediates with α- and β-Galactosidases. Molecules. 2024; 29(20):4870. https://doi.org/10.3390/molecules29204870
Chicago/Turabian StyleAkintola, Oluwafemi, Sandeep Bhosale, and Andrew J. Bennet. 2024. "Mechanism-Based Allylic Carbasugar Chlorides That Form Covalent Intermediates with α- and β-Galactosidases" Molecules 29, no. 20: 4870. https://doi.org/10.3390/molecules29204870
APA StyleAkintola, O., Bhosale, S., & Bennet, A. J. (2024). Mechanism-Based Allylic Carbasugar Chlorides That Form Covalent Intermediates with α- and β-Galactosidases. Molecules, 29(20), 4870. https://doi.org/10.3390/molecules29204870