
Thermal and Sono—Aqueous Reforming of Alcohols for Sustainable Hydrogen Production


Abstract

1. Introduction
2. Discussion—Thermal Aqueous-Phase Reforming of Alcohols
2.1. Al2O3 as Support
2.1.1. Al2O3 Supported Catalysts

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Catalyst | Reaction Condition | H2 Prod. Rate (mmol/g-cat/h) | Stability |
|---|---|---|---|---|
| 1 [a] | Pt/γ-Al2O3 [b] (3 wt.% Pt) | Fixed-bed, 29 bar, 4.5 g catalyst, 0.06 mL/min of 10 wt.% methanol, WHSV = 0.8 h−1 | 40 (at 225 °C) | Stable on stream for at least a week |
| 2 [c] | Pt/Al2O3 [d] (0.94 wt.% Pt) | Fixed bed, 29 bar, 1 g catalyst, 0.05 mL/min of 10 wt.% methanol, WHSV = 3 h−1 | 6 (at 210 °C) | Stable for at least 20 h on stream |
| 3 [e] | Pt/Al2O3 (2% wt.% Pt) | Batch, 20 bar, 0.1 g catalyst, 50 mL of 64 wt.% methanol, Time N.A. | 110 (at 240 °C) | N/A |
| 4 [f] | Pt/Al2O3 (20 nm) [g] (0.89% wt.%) | Batch, 20 bar, 0.1 g catalyst, 15 mL of 37 wt.% methanol, 1 h reaction | 86 (at 220° C) | N/A |
| 5 [h] | Pt/Al2O3 [i] (0.2 wt.% Pt) | Batch, 20 bar, 0.1 g catalyst, 15 g of 37 wt.% methanol in water and 0.3 g NaOH, 1 h reaction | 2.3 (at 220 °C) | N/A |
2.1.2. Catalysts Supported on Cobalt Aluminate and Hydrotalcite Related
2.1.3. Nickel Aluminate
2.1.4. SiO2-Al2O3
2.2. CeO2 as Support
2.2.1. CeO2-Supported Catalysts
2.2.2. CeO2-Supported Catalysts for Low-Temperature Aqueous Reforming (<150 °C)
| Entry | Catalyst | Reaction Condition | H2 Prod. Rate (mmol/g-cat/h) | Stability |
|---|---|---|---|---|
| 1 [a] | Pt/PN-CeO2 [b] (0.36 wt.%Pt) | Batch, 40 bar, 0.05 g catalyst, 58 mL of 63.8 wt.% methanol in water 1 h reaction | 20.4 (165 °C) 3.7 (at 135 °C) | 20% loss (to 16) after 10 cycles of one hour each at 165 °C |
| 2 [c] | Pt/PN-CeO2 [d] (1 wt.% Pt) | Batch, 1 atm 0.005 g catalyst, 5 mL of 56.4 wt.% methanol in 8M KOH (aq) 1 h reaction | 2.8 (at 60 °C) 73.4 (at 90 °C) | 22% loss (to 2.1) after 10 cycles of one hour each at 60 °C |
2.3. ZrO2 as Support
2.3.1. ZrO2-Supported Catalysts
| Entry | Catalyst | Reaction Condition | H2 prod. Rate (mmol/g-cat/h) | Stability |
|---|---|---|---|---|
| 1 [a] | Ni/ZrO2 [b] (9.4 wt.% Ni) | Fixed bed, 32 bar, 1.5 g catalyst, 2 mL/min of 5 wt.% methanol in water. WHSV = 80 h−1 | 60 (at 230 °C) | 18% loss in hydrogen production rate (to 49) after 12 h TOS. CO/CH4 selectivity changes to 7.8%/1.1% from 4.7%/1.5% |
| 2 [c] | β -Mo 2 C/m-ZrO2 [d] (10 wt.% Mo) | Batch, 6 bar, 0.04 g catalyst, 15 mL of 0.4M ethanol 1.5 h reaction | 20 (at 250 °C) [e] | 41% loss (to 12) after 4 cycles of 1.5 h each. |
2.3.2. Mixed Oxides of ZrO2
| Entry | Catalyst | Reaction Condition | H2 Prod. Rate (mmol/g-cat/h) | Stability |
|---|---|---|---|---|
| 1 [a] | Pt0.05Ce0.475Zr0.475O2 [b] | Fixed-bed, 50 bar, 0.25 g catalyst, 0.61 mL/min of 10 wt.% glycerol WHSV = 2.45 h−1 | 91 (at 250 °C) | Virtually no loss after 50 h on stream |
| 2 [c] | Ni/25Ce-ZrO2 [d] (9.3 wt.% Ni) | Fixed bed, 32 bar, 1.5 g catalyst, 2 mL/min of 5 wt.% methanol in water. WHSV = 80 h−1 | 151 (at 230 °C) | 30% loss in hydrogen production rate (to 106) after 12 h TOS. CO/CH4 selectivity changes to 7.8%/1.1% from 4.7%/1.5% |
| 3 [c] | Ni/17Ce-5La-ZrO2 [e] (10.1 wt.% Ni) | 128 (at 230 °C) | 15% loss in hydrogen production rate (to 109) after 12 h TOS. CO/CH4 selectivity changes to 5.9%/3.3%% from 4.6%/2.1% | |
| 4 [c] | Ni/10La-ZrO2 [f] (9.0 wt.% Ni) | 129 (at 230 °C) | 8% loss in hydrogen production rate (to 118) after 12 h TOS. CO/CH4 selectivity changes to 5.2%/2.6% from 4.2%/3.0% |
2.4. Other Metal-Supported Catalysts
2.4.1. TiO2-Related
2.4.2. MgO Hydrotalcite and Related Support
2.5. Carbon-Supported Catalysts
2.5.1. Activated Carbon
2.5.2. Ordered Mesoporous Carbon Support
| Entry | Catalyst | Reaction Condition | H2 Prod. Rate (mmol/g-cat/h) | Stability |
|---|---|---|---|---|
| 1 [a] | 7 wt.% Pt/CMK-3 [b] | Fixed bed, 45 atm, 0.3 g catalyst, 10 wt.% ethylene glycol in water, 0.1 mL/min, WHSV = 2 h−1 | 91 [c] (at 250 °C) | No deactivation was observed after 25 h TOS |
| 2 [d] | 7 wt.% Pt/CMK-9 [e] | 152 (at 250 °C) | ||
| 3 [f] | 3 wt.% Pt/CMK-9 | 79 (at 250 °C) | No deactivation was observed after 90 h TOS | |
| 4 [f] | 3 wt.% Pt-Fe/CMK-9 | 114 (at 250 °C) | ||
| 5 [f] | 7 wt.% Pt/3D-BMC-12 [g] | Fixed bed, 45 bar, 0.3 g catalyst, 10 vol. % ethylene glycol in water, 0.1 mL/min, WHSV = 2 h−1 | 161 [c] (at 250 °C) | No deactivation was observed after 25 h TOS |
2.5.3. Biomass-Derived Carbon Support
| Entry | Catalyst | Reaction Condition | H2 Prod. Rate (mmol/g-cat/h) | Stability |
|---|---|---|---|---|
| 1 [a] | Ni/HC-N1-S1 [b] (2.2 wt.% Ni) | Batch, 5 bar, 0.2 g catalyst, 40 mL of 10 wt.% methanol 1.5 h | 355 (at 250 °C) | Estimated 16% loss (to 298) after ten cycles of 1.5 h each. 5.5% loss in conversion 9.9% loss in H2 selectivity |
| 2 [c] | Cu@NC-200 [d] (44.9 wt.% Cu) | Fixed bed, 40 bar, 0.1 g catalyst, 64 wt.% methanol in water, 0.03 mL/min, WHSV = 15.8 h−1 | 34 (at 210 °C) | No significant loss after 200 h TOS. CO selectivity ≈ 0.03% |
| 3 [e] | Cu@CS19-G1-300 [f] (35 wt/% Cu) | Batch, 20 bar, 0.03 g catalyst, 10 mL of 37 wt.% methanol in water, 1.25 h | 139 (at 210 °C) | No significant loss are five cycles of 1.25 h each. H2 prod. rate fluctuated between 131 and 136. |
| 4 [g] | Ni@NC [h] (40 wt.% Ni) | Batch, 20 bar, 0.025 g catalyst, 10 mL of 25 mol.% methanol in water or 0.86 M KOH, 1 h | 152 (at 220 °C in water) 973 (at 220 °C, 0.86M KOH) | 4.2% loss (to 933) after nine cycles of 1 h each. |
| 5 [i] | Cu@Ca-Val-300 [j] | Fixed bed, 20 bar, 1 g catalyst, 64 wt.% methanol in water, 0.06 mL/min, WHSV = 3.22 h−1 | 3 (at 180 °C) | Stable for 110 h TOS |
2.5.4. Carbon-Encapsulated Metal Oxide Support
2.5.5. Carbon Nanotubes/Fibers
| Entry | Catalyst | Reaction Condition | H2 Prod. Rate (mmol/g-cat/h) | Stability |
|---|---|---|---|---|
| 1 [a] | Pt/CNF [b] (5 wt.% Pt) | Fixed bed, 29 bar, 0.1 g catalyst, 10 wt.% ethylene glycol (EG) in water, 0.05 mL/min WHSV = 3g-EG/g-cat/h | 39.6 ± 0.5 (at 230 °C) | No observable loss in activity after 50 h TOS |
| 2 [a] | Ni/CNF [c] (12.5 wt.% Ni) | with 0.5M KOH: 25.5 ± 0.8 (at 230 °C) | With 0.5M KOH: No observable loss in activity after 50 h TOS | |
| No KOH: 12 (at 230 °C at 2h TOS) | No KOH: 92% loss (to 1) in activity after 50 h TOS | |||
| 3 [d] | 12Ni/MWNT [e] (12 wt.% Ni) | Fixed bed, 40 bar, 0.15 g catalyst, 1 wt.% glycerol in water, 0.05 mL/min WHSV = 20 h−1 | 6.2 ± 0.4 (at 240 °C) [f] | 100% activity loss by 75 h TOS |
| 4 [d] | 1Cu-12Ni/MWNT [g] (1 wt.% Cu, 12 wt.% Ni) | 9.5 ± 0.4 (at 240 °C) | Stable for 110 h TOS | |
| 5 [h] | 5Pt-1.5Ni/MWNT [i] (4.7 wt.% Pt, 1.4 wt.% Ni) | Batch, 30 bar, 0.1 g catalyst, 15 mL of 10 wt.% glycerol in water, 4 h reaction | no CaO: 9.1 (at 230 °C) | N/A |
| With CaO: 18 (at 230 °C) | With CaO: 26% loss in activity [j] after five cycles of 4 h each. |
2.6. Molybdenum Carbide or Sulfide
3. Discussion—Sonolysis of Aqueous Alcohol for Hydrogen Production
3.1. Basic Theoretical Background
3.2. Sonolysis of Aqueous Alcohols
3.3. Insights into the Mechanism of Sonolysis
3.4. Sonocatalyst or Piezocatalyst
| Entry | Frequency (kHz) | Power (W) | Catalyst | Methanol in Water (Wt.%) | Optimal Temp. (°C) | Liquid Vol. (mL) | H2 Prod. Rate (µmol/mL-liquid/h) [a] |
|---|---|---|---|---|---|---|---|
| 1 | 40 | 50 | 0 | 21–25 | 150 | 0.0027 | |
| 2 | 40 | 50 | Au/TiO2 [b] | 0 | 21–25 | 150 | 0.144 |
| 3 | 40 | 50 | 4 | 21–25 | 150 | 0.023 | |
| 4 | 40 | 50 | Au/TiO2 [b] | 4 | 21–25 | 150 | 1.9 |
| 5 | 40 | 60 | BaTiO3 [c] | 8.1 | 35 | 100 | 0.6 |
| 6 | 200 | 6 W/cm2 | 100 | 5 | 10 | 1.3 | |
| 7 | 724 | 45 | 0 | 38–50 | 40 | 0.7 | |
| 8 | 724 | 45 | 3.2 | 29–47 | 40 | 12 | |
| 9 | 724 | 45 | 100 | −7 | 40 | 0.4 | |
| 10 | 1000 | 2 W/cm2 | 0 | N/A | 40 | 1.4 | |
| 11 | 1000 | 2 W/cm2 | 7 | N/A | 40 | 9.2 |
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Suleman, F.; Dincer, I.; Agelin-Chaab, M. Environmental impact assessment and comparison of some hydrogen production options. Int. J. Hydrogen Energy 2015, 40, 6976–6987. [Google Scholar] [CrossRef]
- Palo, D.R.; Dagle, R.A.; Holladay, J.D. Methanol Steam Reforming for Hydrogen Production. Chem. Rev. 2007, 107, 3992–4021. [Google Scholar] [CrossRef] [PubMed]
- Anil, S.; Indraja, S.; Singh, R.; Appari, S.; Roy, B. A review on ethanol steam reforming for hydrogen production over Ni/Al2O3 and Ni/CeO2 based catalyst powders. Int. J. Hydrogen Energy 2022, 47, 8177–8213. [Google Scholar] [CrossRef]
- Bepari, S.; Kuila, D. Steam reforming of methanol, ethanol and glycerol over nickel-based catalysts-A review. Int. J. Hydrogen Energy 2020, 45, 18090–18113. [Google Scholar] [CrossRef]
- Sharma, Y.C.; Kumar, A.; Prasad, R.; Upadhyay, S.N. Ethanol steam reforming for hydrogen production: Latest and effective catalyst modification strategies to minimize carbonaceous deactivation. Renew. Sustain. Energy Rev. 2017, 74, 89–103. [Google Scholar] [CrossRef]
- Achomo, M.A.; Kumar, A.; Peela, N.R.; Muthukumar, P. Hydrogen production from steam reforming of methanol: A comprehensive review on thermodynamics, catalysts, reactors, and kinetic studies. Int. J. Hydrogen Energy 2024, 58, 1640–1672. [Google Scholar] [CrossRef]
- Yang, W.-W.; Ma, X.; Tang, X.-Y.; Dou, P.-Y.; Yang, Y.-J.; He, Y.-L. Review on developments of catalytic system for methanol steam reforming from the perspective of energy-mass conversion. Fuel 2023, 345, 128234. [Google Scholar] [CrossRef]
- Fermoso, J.; He, L.; Chen, D. Sorption enhanced steam reforming (SESR): A direct route towards efficient hydrogen production from biomass-derived compounds. J. Chem. Technol. Biotechnol. 2012, 87, 1367–1374. [Google Scholar] [CrossRef]
- Dou, B.; Song, Y.; Wang, C.; Chen, H.; Xu, Y. Hydrogen production from catalytic steam reforming of biodiesel byproduct glycerol: Issues and challenges. Renew. Sustain. Energy Rev. 2014, 30, 950–960. [Google Scholar] [CrossRef]
- Shokrollahi Yancheshmeh, M.; Radfarnia, H.R.; Iliuta, M.C. High temperature CO2 sorbents and their application for hydrogen production by sorption enhanced steam reforming process. Chem. Eng. J. 2016, 283, 420–444. [Google Scholar] [CrossRef]
- Kothandaraman, J.; Kar, S.; Goeppert, A.; Sen, R.; Prakash, G.K.S. Advances in Homogeneous Catalysis for Low Temperature Methanol Reforming in the Context of the Methanol Economy. Top. Catal. 2018, 61, 542–559. [Google Scholar] [CrossRef]
- Kumar, A.; Daw, P.; Milstein, D. Homogeneous Catalysis for Sustainable Energy: Hydrogen and Methanol Economies, Fuels from Biomass, and Related Topics. Chem. Rev. 2022, 122, 385–441. [Google Scholar] [CrossRef] [PubMed]
- Sordakis, K.; Tang, C.; Vogt, L.K.; Junge, H.; Dyson, P.J.; Beller, M.; Laurenczy, G. Homogeneous Catalysis for Sustainable Hydrogen Storage in Formic Acid and Alcohols. Chem. Rev. 2018, 118, 372–433. [Google Scholar] [CrossRef]
- Davda, R.R.; Shabaker, J.W.; Huber, G.W.; Cortright, R.D.; Dumesic, J.A. A review of catalytic issues and process conditions for renewable hydrogen and alkanes by aqueous-phase reforming of oxygenated hydrocarbons over supported metal catalysts. Appl. Catal. B 2005, 56, 171–186. [Google Scholar] [CrossRef]
- Joshi, A.N.; Vaidya, P.D. Recent studies on aqueous-phase reforming: Catalysts, reactors, hybrid processes and techno-economic analysis. Int. J. Hydrogen Energy 2024, 49, 117–137. [Google Scholar] [CrossRef]
- Medeiros, D. DWSIM: Open-source process simulator, 8.8.0. 2024. Available online: https://sourceforge.net/projects/dwsim/files/DWSIM/DWSIM%208.8/8.8.0/ (accessed on 8 September 2024).
- Stryjek, R.; Vera, J.H. PRSV2: A cubic equation of state for accurate vapor—Liquid equilibria calculations. Can. J. Chem. Eng. 1986, 64, 820–826. [Google Scholar] [CrossRef]
- Lange, J.-P. Performance metrics for sustainable catalysis in industry. Nat. Catal. 2021, 4, 186–192. [Google Scholar] [CrossRef]
- Elliott, D.C.; Sealock, L.J., Jr.; Baker, E.G. Chemical processing in high-pressure aqueous environments. 2. Development of catalysts for gasification. Ind. Eng. Chem. Res. 1993, 32, 1542–1548. [Google Scholar] [CrossRef]
- Van Cleve, T.; Underhill, D.; Veiga Rodrigues, M.; Sievers, C.; Medlin, J.W. Enhanced Hydrothermal Stability of γ-Al2O3 Catalyst Supports with Alkyl Phosphonate Coatings. Langmuir 2018, 34, 3619–3625. [Google Scholar] [CrossRef]
- Huo, J.; Tessonnier, J.-P.; Shanks, B.H. Improving Hydrothermal Stability of Supported Metal Catalysts for Biomass Conversions: A Review. ACS Catal. 2021, 11, 5248–5270. [Google Scholar] [CrossRef]
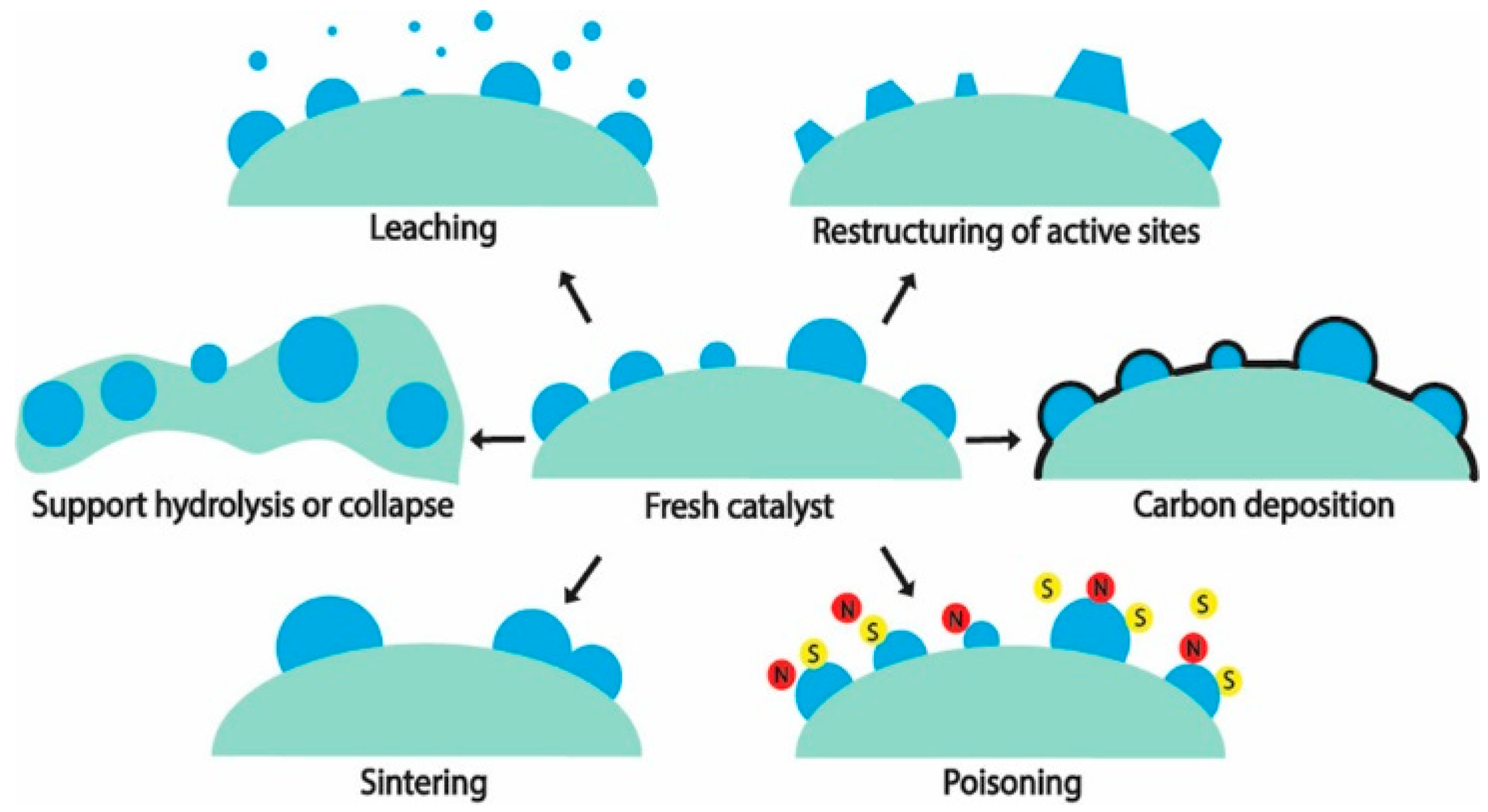
- Lin, F.; Xu, M.; Ramasamy, K.K.; Li, Z.; Klinger, J.L.; Schaidle, J.A.; Wang, H. Catalyst Deactivation and Its Mitigation during Catalytic Conversions of Biomass. ACS Catal. 2022, 12, 13555–13599. [Google Scholar] [CrossRef]
- Chen, W.-H.; Chen, C.-Y. Water gas shift reaction for hydrogen production and carbon dioxide capture: A review. Appl. Energy 2020, 258, 114078. [Google Scholar] [CrossRef]
- Mohanty, P.; Pant, K.K.; Mittal, R. Hydrogen generation from biomass materials: Challenges and opportunities. WIREs Energy Environ. 2015, 4, 139–155. [Google Scholar] [CrossRef]
- Davda, R.R.; Shabaker, J.W.; Huber, G.W.; Cortright, R.D.; Dumesic, J.A. Aqueous-phase reforming of ethylene glycol on silica-supported metal catalysts. Appl. Catal. B 2003, 43, 13–26. [Google Scholar] [CrossRef]
- Chen, G.-Y.; Li, W.-Q.; Chen, H.; Yan, B.-B. Progress in the aqueous-phase reforming of different biomass-derived alcohols for hydrogen production. J. Zhejiang Univ. Sci. A 2015, 16, 491–506. [Google Scholar] [CrossRef]
- Coronado, I.; Stekrova, M.; Reinikainen, M.; Simell, P.; Lefferts, L.; Lehtonen, J. A review of catalytic aqueous-phase reforming of oxygenated hydrocarbons derived from biorefinery water fractions. Int. J. Hydrogen Energy 2016, 41, 11003–11032. [Google Scholar] [CrossRef]
- Vaidya, P.D.; Lopez-Sanchez, J.A. Review of Hydrogen Production by Catalytic Aqueous-Phase Reforming. ChemistrySelect 2017, 2, 6563–6576. [Google Scholar] [CrossRef]
- Pipitone, G.; Zoppi, G.; Pirone, R.; Bensaid, S. A critical review on catalyst design for aqueous phase reforming. Int. J. Hydrogen Energy 2022, 47, 151–180. [Google Scholar] [CrossRef]
- Azizan, M.T.; Aqsha, A.; Ameen, M.; Syuhada, A.; Klaus, H.; Abidin, S.Z.; Sher, F. Catalytic reforming of oxygenated hydrocarbons for the hydrogen production: An outlook. Biomass Convers. Biorefinery 2023, 13, 8441–8464. [Google Scholar] [CrossRef]
- Tian, Z.; Lu, Y.; Wang, J.; Shu, R.; Wang, C.; Chen, Y. Advances in hydrogen production by aqueous phase reforming of biomass oxygenated derivatives. Fuel 2024, 357, 129691. [Google Scholar] [CrossRef]
- Xiong, H.; Pham, H.N.; Datye, A.K. Hydrothermally stable heterogeneous catalysts for conversion of biorenewables. Green Chem. 2014, 16, 4627–4643. [Google Scholar] [CrossRef]
- Shabaker, J.W.; Huber, G.W.; Davda, R.R.; Cortright, R.D.; Dumesic, J.A. Aqueous-Phase Reforming of Ethylene Glycol over Supported Platinum Catalysts. Catal. Lett. 2003, 88, 1–8. [Google Scholar] [CrossRef]
- Ravenelle, R.M.; Copeland, J.R.; Kim, W.-G.; Crittenden, J.C.; Sievers, C. Structural Changes of γ-Al2O3-Supported Catalysts in Hot Liquid Water. ACS Catal. 2011, 1, 552–561. [Google Scholar] [CrossRef]
- Pham, H.N.; Anderson, A.E.; Johnson, R.L.; Schmidt-Rohr, K.; Datye, A.K. Improved Hydrothermal Stability of Mesoporous Oxides for Reactions in the Aqueous Phase. Angew. Chem. Int. Ed. 2012, 51, 13163–13167. [Google Scholar] [CrossRef] [PubMed]
- Cortright, R.D.; Davda, R.R.; Dumesic, J.A. Hydrogen from catalytic reforming of biomass-derived hydrocarbons in liquid water. Nature 2002, 418, 964–967. [Google Scholar] [CrossRef]
- Liu, Y.; Li, S.; Zhu, S. Novel Pt/MoS2 nanosheet catalyst for hydrogen production via aqueous-phase reforming of methanol. React. Kinet. Mech. Catal. 2022, 135, 2579–2589. [Google Scholar] [CrossRef]
- Zhang, J.; Klasky, M.; Letellier, B.C. The aluminum chemistry and corrosion in alkaline solutions. J. Nucl. Mater. 2009, 384, 175–189. [Google Scholar] [CrossRef]
- Li, D.; Li, Y.; Liu, X.; Guo, Y.; Pao, C.-W.; Chen, J.-L.; Hu, Y.; Wang, Y. NiAl2O4 Spinel Supported Pt Catalyst: High Performance and Origin in Aqueous-Phase Reforming of Methanol. ACS Catal. 2019, 9, 9671–9682. [Google Scholar] [CrossRef]
- Lin, L.; Yu, Q.; Peng, M.; Li, A.; Yao, S.; Tian, S.; Liu, X.; Li, A.; Jiang, Z.; Gao, R.; et al. Atomically Dispersed Ni/α-MoC Catalyst for Hydrogen Production from Methanol/Water. J. Am. Chem. Soc. 2021, 143, 309–317. [Google Scholar] [CrossRef]
- Lv, Z.; Zhu, S.; Wang, S.; Dong, M.; Qin, Z.; Wang, J.; Fan, W. Aqueous-phase reforming of methanol to hydrogen over CoAl oxide-supported Pt catalyst. Appl. Catal. A 2023, 665, 119378. [Google Scholar] [CrossRef]
- Wen, G.; Xu, Y.; Ma, H.; Xu, Z.; Tian, Z. Production of hydrogen by aqueous-phase reforming of glycerol. Int. J. Hydrogen Energy 2008, 33, 6657–6666. [Google Scholar] [CrossRef]
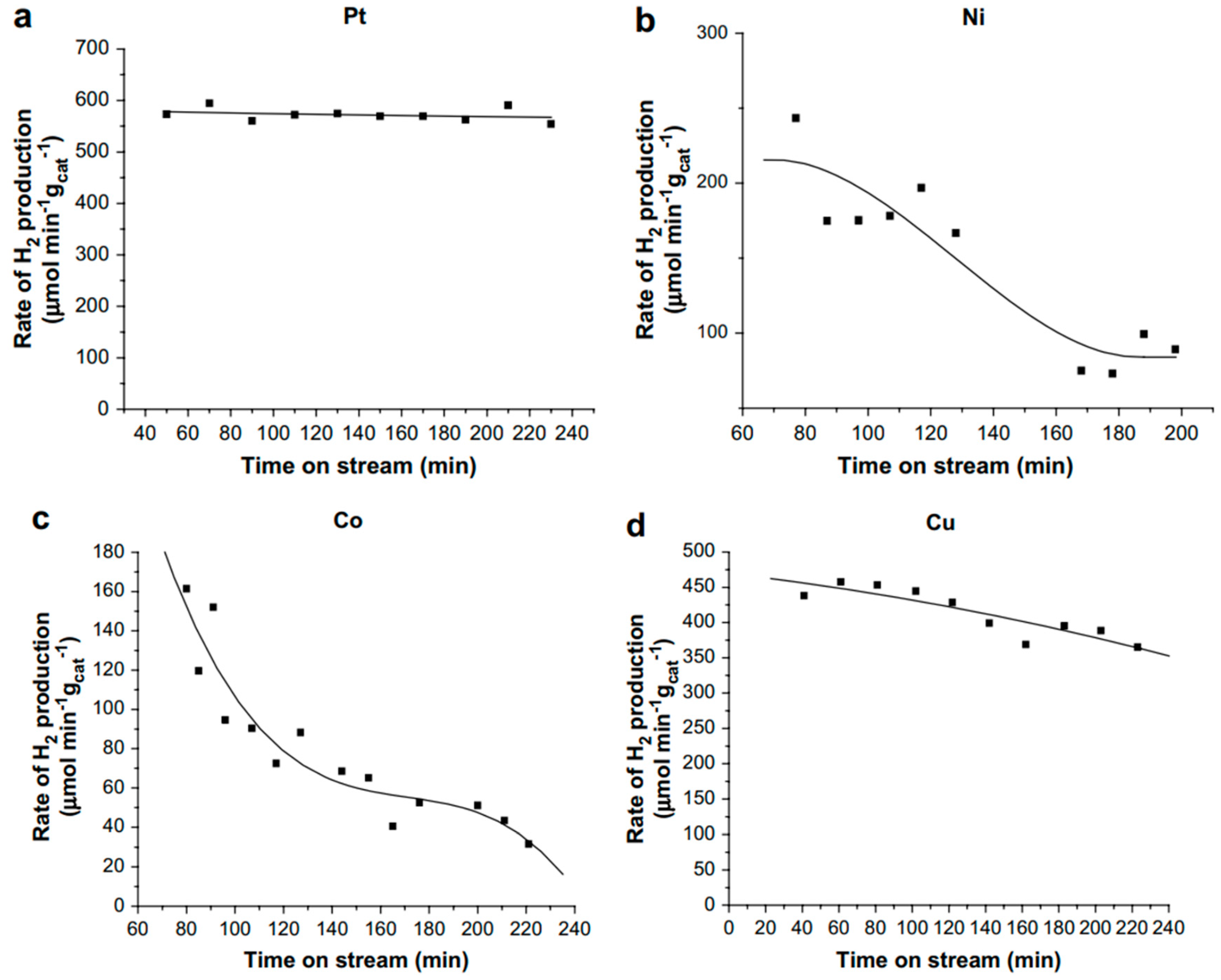
- El Doukkali, M.; Iriondo, A.; Cambra, J.F.; Gandarias, I.; Jalowiecki-Duhamel, L.; Dumeignil, F.; Arias, P.L. Deactivation study of the Pt and/or Ni-based γ-Al2O3 catalysts used in the aqueous phase reforming of glycerol for H2 production. Appl. Catal. A 2014, 472, 80–91. [Google Scholar] [CrossRef]
- El Doukkali, M.; Iriondo, A.; Arias, P.L.; Requies, J.; Gandarías, I.; Jalowiecki-Duhamel, L.; Dumeignil, F. A comparison of sol–gel and impregnated Pt or/and Ni based γ-alumina catalysts for bioglycerol aqueous phase reforming. Appl. Catal., B 2012, 125, 516–529. [Google Scholar] [CrossRef]
- Liu, Y.; Yu, S.; Wu, X.; Cao, X.; Geng, H.; Zhang, C.; Liu, S. Improving the hydrothermal stability and hydrogen selectivity of Ni-Cu based catalysts for the aqueous-phase reforming of methanol. Int. J. Hydrogen Energy 2023, 48, 12699–12711. [Google Scholar] [CrossRef]
- Kalekar, V.N.; Vaidya, P.D. Hydrogen production from aqueous-phase reforming of polyols over Ru/Al2O3 catalyst in a fixed-bed reactor. J. Indian Chem. Soc. 2024, 101, 101268. [Google Scholar] [CrossRef]
- Franck, J.P.; Freund, E.; Quéméré, E. Textural and structural changes in transition alumina supports. J. Chem. Soc. Chem. Commun. 1984, 10, 629–630. [Google Scholar] [CrossRef]
- Jun-Cheng, L.; Lan, X.; Feng, X.; Zhan-Wen, W.; Fei, W. Effect of hydrothermal treatment on the acidity distribution of γ-Al2O3 support. Appl. Surf. Sci. 2006, 253, 766–770. [Google Scholar] [CrossRef]
- Abi Aad, J.; Courty, P.; Decottignies, D.; Michau, M.; Diehl, F.; Carrier, X.; Marceau, E. Inhibition by Inorganic Dopants of γ-Alumina Chemical Weathering under Hydrothermal Conditions: Identification of Reactive Sites and their Influence in Fischer–Tropsch Synthesis. ChemCatChem 2017, 9, 2106–2117. [Google Scholar] [CrossRef]
- Liu, Y.; Jia, L.; Hou, B.; Sun, D.; Li, D. Cobalt aluminate-modified alumina as a carrier for cobalt in Fischer–Tropsch synthesis. Appl. Catal. A 2017, 530, 30–36. [Google Scholar] [CrossRef]
- Shen, J.; Hayes, R.E.; Wu, X.; Semagina, N. 100° Temperature Reduction of Wet Methane Combustion: Highly Active Pd–Ni/Al2O3 Catalyst versus Pd/NiAl2O4. ACS Catal. 2015, 5, 2916–2920. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, S.; Sun, T.; Gao, D.; Zhang, C.; Wang, S. Enhanced hydrothermal stability of high performance lean fuel combustion alumina-supported palladium catalyst modified by nickel. Appl. Catal. B 2012, 119–120, 321–328. [Google Scholar] [CrossRef]
- Reynoso, A.J.; Ayastuy, J.L.; Iriarte-Velasco, U.; Gutiérrez-Ortiz, M.A. Cobalt aluminate spinel-derived catalysts for glycerol aqueous phase reforming. Appl. Catal., B 2018, 239, 86–101. [Google Scholar] [CrossRef]
- Reynoso, A.J.; Iriarte-Velasco, U.; Gutiérrez-Ortiz, M.A.; Ayastuy, J.L. Highly stable Pt/CoAl2O4 catalysts in Aqueous-Phase Reforming of glycerol. Catal. Today 2021, 367, 278–289. [Google Scholar] [CrossRef]
- Reynoso, A.J.; Ayastuy, J.L.; Iriarte-Velasco, U.; Gutiérrez-Ortiz, M.A. Aqueous-phase reforming of glycerol over Pt-Co catalyst: Effect of process variables. J. Environ. Chem. Eng. 2022, 10, 107402. [Google Scholar] [CrossRef]
- Morales-Marín, A.; Ayastuy, J.L.; Iriarte-Velasco, U.; Gutiérrez-Ortiz, M.A. Nickel aluminate spinel-derived catalysts for the aqueous phase reforming of glycerol: Effect of reduction temperature. Appl. Catal. B 2019, 244, 931–945. [Google Scholar] [CrossRef]
- Leboda, R.; Mendyk, E.; Gierak, A.; Tertykh, V.A. Hydrothermal modification of silica gels (xerogels) 1. Effect of treatment temperature on their porous structure. Colloids Surf. A 1995, 105, 181–189. [Google Scholar] [CrossRef]
- Rohatgi, A. WebPlotDigitizer, 5.1. 2024. Available online: https://automeris.io/ (accessed on 13 October 2024).
- Seretis, A.; Tsiakaras, P. Crude bio-glycerol aqueous phase reforming and hydrogenolysis over commercial SiO2Al2O3 nickel catalyst. Renew. Energy 2016, 97, 373–379. [Google Scholar] [CrossRef]
- Ciftci, A.; Eren, S.; Ligthart, D.A.J.M.; Hensen, E.J.M. Platinum–Rhenium Synergy on Reducible Oxide Supports in Aqueous-Phase Glycerol Reforming. ChemCatChem 2014, 6, 1260–1269. [Google Scholar] [CrossRef]
- Wu, K.; Dou, B.; Zhang, H.; Liu, D.; Chen, H.; Xu, Y. Aqueous phase reforming of biodiesel byproduct glycerol over mesoporous Ni-Cu/CeO2 for renewable hydrogen production. Fuel 2022, 308, 122014. [Google Scholar] [CrossRef]
- Lu, Y.; Wang, C.; Luo, X.; Shu, R.; Lei, L.; Liu, J.; Tian, Z.; Liao, Y.; Chen, Y. Aqueous phase reforming of methanol for hydrogen production over highly-dispersed PtLa/CeO2 catalyst prepared by photochemical reduction method. Int. J. Hydrogen Energy 2024, 62, 1054–1066. [Google Scholar] [CrossRef]
- Zhang, G.; Qu, Z.; Tao, W.-Q.; Wang, X.; Wu, L.; Wu, S.; Xie, X.; Tongsh, C.; Huo, W.; Bao, Z.; et al. Porous Flow Field for Next-Generation Proton Exchange Membrane Fuel Cells: Materials, Characterization, Design, and Challenges. Chem. Rev. 2023, 123, 989–1039. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.; Alberico, E.; Baumann, W.; Drexler, H.-J.; Junge, H.; Gladiali, S.; Beller, M. Low-temperature aqueous-phase methanol dehydrogenation to hydrogen and carbon dioxide. Nature 2013, 495, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, Y.; Zhang, M.; Ma, Y.; Hu, J.; Qu, Y. Sustainable production of hydrogen with high purity from methanol and water at low temperatures. Nat. Commun. 2022, 13, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-N.; Hou, K.-P.; Liu, Y.-S.; Qi, Z.-Y.; Zheng, Q.; Lu, Y.-H.; Chen, J.-Y.; Chen, J.-L.; Pao, C.-W.; Wang, S.-B.; et al. Efficient Hydrogen Production from Methanol Using a Single-Site Pt1/CeO2 Catalyst. J. Am. Chem. Soc. 2019, 141, 17995–17999. [Google Scholar] [CrossRef]
- Mai, H.-X.; Sun, L.-D.; Zhang, Y.-W.; Si, R.; Feng, W.; Zhang, H.-P.; Liu, H.-C.; Yan, C.-H. Shape-Selective Synthesis and Oxygen Storage Behavior of Ceria Nanopolyhedra, Nanorods, and Nanocubes. J. Phys. Chem. B 2005, 109, 24380–24385. [Google Scholar] [CrossRef]
- Guo, Q.; Wang, Y.; Li, W.; Zou, Y.; Zhang, S. Oxygen vacancy of Pt/CeO2 enabled low-temperature hydrogen generation from methanol and water. J. Catal. 2024, 430, 115309. [Google Scholar] [CrossRef]
- Tanabe, K. Surface and catalytic properties of ZrO2. Mater. Chem. Phys. 1985, 13, 347–364. [Google Scholar] [CrossRef]
- Yamaguchi, T. Application of ZrO2 as a catalyst and a catalyst support. Catal. Today 1994, 20, 199–217. [Google Scholar] [CrossRef]
- Lin, Y.-S.; Chang, C.-H.; Gopalan, R. Improvement of Thermal Stability of Porous Nanostructured Ceramic Membranes. Ind. Eng. Chem. Res. 1994, 33, 860–870. [Google Scholar] [CrossRef]
- Duan, J.; Kim, Y.T.; Lou, H.; Huber, G.W. Hydrothermally stable regenerable catalytic supports for aqueous-phase conversion of biomass. Catal. Today 2014, 234, 66–74. [Google Scholar] [CrossRef]
- Bernard, P.; Stelmachowski, P.; Broś, P.; Makowski, W.; Kotarba, A. Demonstration of the Influence of Specific Surface Area on Reaction Rate in Heterogeneous Catalysis. J. Chem. Educ. 2021, 98, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Stekrova, M.; Rinta-Paavola, A.; Karinen, R. Hydrogen production via aqueous-phase reforming of methanol over nickel modified Ce, Zr and La oxide supports. Catal. Today 2018, 304, 143–152. [Google Scholar] [CrossRef]
- Pavesi Contreras, C.; Blanco, E.; Pazo, C.; Dongil, A.B.; Escalona, N. H2 production through aqueous phase reforming of ethanol over molybdenum carbide catalysts supported on zirconium oxide. Appl. Catal. A 2024, 670, 119535. [Google Scholar] [CrossRef]
- Karpenko, A.; Leppelt, R.; Cai, J.; Plzak, V.; Chuvilin, A.; Kaiser, U.; Behm, R.J. Deactivation of a Au/CeO2 catalyst during the low-temperature water–gas shift reaction and its reactivation: A combined TEM, XRD, XPS, DRIFTS, and activity study. J. Catal. 2007, 250, 139–150. [Google Scholar] [CrossRef]
- Larimi, A.S.; Kazemeini, M.; Khorasheh, F. Aqueous phase reforming of glycerol using highly active and stable Pt0.05CexZr0.95-xO2 ternary solid solution catalysts. Appl. Catal. A 2016, 523, 230–240. [Google Scholar] [CrossRef]
- Bastan, F.; Kazemeini, M.; Larimi, A.S. Aqueous-phase reforming of glycerol for production of alkanes over Ni/CexZr1-xO2 nano-catalyst: Effects of the support’s composition. Renew. Energy 2017, 108, 417–424. [Google Scholar] [CrossRef]
- Elliott, D.C.; Hart, T.R.; Neuenschwander, G.G. Chemical Processing in High-Pressure Aqueous Environments. 8. Improved Catalysts for Hydrothermal Gasification. Ind. Eng. Chem. Res. 2006, 45, 3776–3781. [Google Scholar] [CrossRef]
- Lin, L.; Zhou, W.; Gao, R.; Yao, S.; Zhang, X.; Xu, W.; Zheng, S.; Jiang, Z.; Yu, Q.; Li, Y.-W.; et al. Low-temperature hydrogen production from water and methanol using Pt/α-MoC catalysts. Nature 2017, 544, 80–83. [Google Scholar] [CrossRef] [PubMed]
- Nozawa, T.; Mizukoshi, Y.; Yoshida, A.; Naito, S. Aqueous phase reforming of ethanol and acetic acid over TiO2 supported Ru catalysts. Appl. Catal. B 2014, 146, 221–226. [Google Scholar] [CrossRef]
- Nozawa, T.; Yoshida, A.; Hikichi, S.; Naito, S. Effects of Re addition upon aqueous phase reforming of ethanol over TiO2 supported Rh and Ir catalysts. Int. J. Hydrogen Energy 2015, 40, 4129–4140. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, L.; Tan, Q.; Yang, F.; Faria, J.; Resasco, D. Synergistic bimetallic Ru–Pt catalysts for the low-temperature aqueous phase reforming of ethanol. AlChE J. 2019, 65, 151–160. [Google Scholar] [CrossRef]
- Valente, J.S.; Hernandez-Cortez, J.; Cantu, M.S.; Ferrat, G.; López-Salinas, E. Calcined layered double hydroxides Mg–Me–Al (Me: Cu, Fe, Ni, Zn) as bifunctional catalysts. Catal. Today 2010, 150, 340–345. [Google Scholar] [CrossRef]
- Dewoolkar, K.D.; Vaidya, P.D. Sorption-Enhanced Steam Reforming of Glycerol over Ni–hydrotalcite: Effect of Promotion with Pt. ChemCatChem 2016, 8, 3499–3509. [Google Scholar] [CrossRef]
- Ghungrud, S.A.; Vaidya, P.D. Improved Hydrogen Production from Sorption-Enhanced Steam Reforming of Ethanol (SESRE) Using Multifunctional Materials of Cobalt Catalyst and Mg-, Ce-, and Zr-Modified CaO Sorbents. Ind. Eng. Chem. Res. 2020, 59, 693–703. [Google Scholar] [CrossRef]
- Ghungrud, S.A.; Vaidya, P.D. Sorption-enhanced reaction process for glycerol-to-hydrogen conversion over cobalt catalyst supported on promoted hydrotalcites. Int. J. Hydrogen Energy 2020, 45, 9440–9450. [Google Scholar] [CrossRef]
- Huang, J.; Xie, L.; Luo, X.; Wang, C.; Shu, R.; Song, Q.; Liu, J.; Tian, Z.; Chen, Y. Hydrogen production by aqueous phase reforming over stable La-promoted Ni-based hydrotalcite catalysts. Int. J. Hydrogen Energy 2024, 50, 681–689. [Google Scholar] [CrossRef]
- Manfro, R.L.; Pires, T.P.M.D.; Ribeiro, N.F.P.; Souza, M.M.V.M. Aqueous-phase reforming of glycerol using Ni–Cu catalysts prepared from hydrotalcite-like precursors. Catal. Sci. Technol. 2013, 3, 1278. [Google Scholar] [CrossRef]
- Cruz, I.O.; Ribeiro, N.F.P.; Aranda, D.A.G.; Souza, M.M.V.M. Hydrogen production by aqueous-phase reforming of ethanol over nickel catalysts prepared from hydrotalcite precursors. Catal. Commun. 2008, 9, 2606–2611. [Google Scholar] [CrossRef]
- Huber, G.W.; Guymon, C.G.; Conrad, T.L.; Stephenson, B.C.; Bartholomew, C.H. Hydrothermal Stability of Co/SiO2 Fischer-Tropsch Synthesis Catalysts. In Studies in Surface Science and Catalysis; Spivey, J.J., Roberts, G.W., Davis, B.H., Eds.; Elsevier: Amsterdam, The Netherlands, 2001; Volume 139, pp. 423–430. [Google Scholar]
- Karim, A.M.; Howard, C.; Roberts, B.; Kovarik, L.; Zhang, L.; King, D.L.; Wang, Y. In Situ X-ray Absorption Fine Structure Studies on the Effect of pH on Pt Electronic Density during Aqueous Phase Reforming of Glycerol. ACS Catal. 2012, 2, 2387–2394. [Google Scholar] [CrossRef]
- King, D.L.; Zhang, L.; Xia, G.; Karim, A.M.; Heldebrant, D.J.; Wang, X.; Peterson, T.; Wang, Y. Aqueous phase reforming of glycerol for hydrogen production over Pt–Re supported on carbon. Appl. Catal. B 2010, 99, 206–213. [Google Scholar] [CrossRef]
- Wei, Z.; Karim, A.; Li, Y.; Wang, Y. Elucidation of the Roles of Re in Aqueous-Phase Reforming of Glycerol over Pt–Re/C Catalysts. ACS Catal. 2015, 5, 7312–7320. [Google Scholar] [CrossRef]
- Kim, T.-W.; Kim, H.-D.; Jeong, K.-E.; Chae, H.-J.; Jeong, S.-Y.; Lee, C.-H.; Kim, C.-U. Catalytic production of hydrogen through aqueous-phase reforming over platinum/ordered mesoporous carbon catalysts. Green Chem. 2011, 13, 1718. [Google Scholar] [CrossRef]
- Kim, M.-C.; Kim, T.-W.; Kim, H.J.; Kim, C.-U.; Bae, J.W. Aqueous phase reforming of polyols for hydrogen production using supported PtFe bimetallic catalysts. Renew. Energy 2016, 95, 396–403. [Google Scholar] [CrossRef]
- Wang, P.; Huang, Y.; Shu, R.; Wang, J.; Liu, J.; Wang, C.; Tian, Z.; Chen, Y. Efficient hydrogen production by methanol aqueous phase reforming over KMnO4 modified PtMnK/AC catalyst: Regulating the hydrophilicity of carbon support. Mol. Catal. 2024, 559, 114105. [Google Scholar] [CrossRef]
- Zheng, Z.; Fang, Y.; Ma, L.; Wu, X.; Meng, Q.; Wang, T. High-loaded sub-6 nm Cu catalyst with superior hydrothermal-stability and efficiency for aqueous phase reforming of methanol to hydrogen. Int. J. Hydrogen Energy 2022, 47, 22752–22762. [Google Scholar] [CrossRef]
- Kim, H.-D.; Kim, T.-W.; Park, H.J.; Jeong, K.-E.; Chae, H.-J.; Jeong, S.-Y.; Lee, C.-H.; Kim, C.-U. Hydrogen production via the aqueous phase reforming of ethylene glycol over platinum-supported ordered mesoporous carbon catalysts: Effect of structure and framework-configuration. Int. J. Hydrogen Energy 2012, 37, 12187–12197. [Google Scholar] [CrossRef]
- Park, H.J.; Kim, H.-D.; Kim, T.-W.; Jeong, K.-E.; Chae, H.-J.; Jeong, S.-Y.; Chung, Y.-M.; Park, Y.-K.; Kim, C.-U. Production of Biohydrogen by Aqueous Phase Reforming of Polyols over Platinum Catalysts Supported on Three-Dimensionally Bimodal Mesoporous Carbon. ChemSusChem 2012, 5, 629–633. [Google Scholar] [CrossRef]
- Gai, C.; Wang, X.; Liu, J.; Liu, Z.; Ok, Y.S.; Liu, W.; Yip, A.C.K. Ni/Hydrochar Nanostructures Derived from Biomass as Catalysts for H2 Production through Aqueous-Phase Reforming of Methanol. ACS Appl. Nano Mater. 2021, 4, 8958–8971. [Google Scholar] [CrossRef]
- Xiao, Z.; Meng, Q.; Qiu, C.; Qiu, S.; Wu, X.; Ma, L.; Wang, T. Promoting mechanism of alkali for aqueous phase reforming of bio-methanol towards highly efficient production of COx-free hydrogen. Fuel Process. Technol. 2022, 236, 107385. [Google Scholar] [CrossRef]
- Wu, X.; Zheng, Z.; Ma, L.; Hu, C.; Pi, Y.; Wang, T. Engineering of the Cu+/Cu0 interface by chitosan-glucose complex for aqueous phase reforming of methanol into hydrogen. Int. J. Hydrogen Energy 2023, 48, 33948–33959. [Google Scholar] [CrossRef]
- Li, J.; Lu, M.; Ge, Y.; Lu, W.; Liu, Z.; Xu, H.; Zhang, Y.; Li, Z.; Zheng, Z.; Gao, P.; et al. Efficient and sustainable H2 production from aqueous-phase reforming of methanol over Cu@CA-Val catalyst at low temperatures. Int. J. Hydrogen Energy 2024, 71, 775–784. [Google Scholar] [CrossRef]
- Chen, B.; Zheng, Z.; Hu, C.; Zengcai, Z.; Liu, Z.; Lu, M.; Meng, Q.; Wang, T. High-content graphitized N-doped carbon encapsulated Cu catalyst in aqueous phase reforming of methanol for efficient hydrogen production. Fuel 2024, 371, 131888. [Google Scholar] [CrossRef]
- Gao, C.; Lyu, F.; Yin, Y. Encapsulated Metal Nanoparticles for Catalysis. Chem. Rev. 2021, 121, 834–881. [Google Scholar] [CrossRef]
- Zheng, Z.; Fang, Y.; Yang, J.; Ma, L.; Meng, Q.; Lin, X.; Liu, Y.; Zhang, Q.; Wang, T. A highly active and hydrothermal-resistant Cu/ZnO@NC catalyst for aqueous phase reforming of methanol to hydrogen. Int. J. Hydrogen Energy 2022, 47, 950–961. [Google Scholar] [CrossRef]
- Lu, M.; Liu, S.; Zhu, H.; Huang, H.; Lin, C.; Li, J.; Zhang, B.; Zheng, Z.; Hu, C.; Wu, X.; et al. Highly efficient releasing of hydrogen from aqueous-phase reforming of methanol over Cu-SP/Al2O3–ZnO catalyst by carbon layer encapsulated hierarchical porous microsphere strategy. Int. J. Hydrogen Energy 2024, 52, 125–139. [Google Scholar] [CrossRef]
- De Volder, M.F.L.; Tawfick, S.H.; Baughman, R.H.; Hart, A.J. Carbon Nanotubes: Present and Future Commercial Applications. Science 2013, 339, 535–539. [Google Scholar] [CrossRef]
- Yan, Y.; Miao, J.; Yang, Z.; Xiao, F.-X.; Yang, H.B.; Liu, B.; Yang, Y. Carbon nanotube catalysts: Recent advances in synthesis, characterization and applications. Chem. Soc. Rev. 2015, 44, 3295–3346. [Google Scholar] [CrossRef] [PubMed]
- van Haasterecht, T.; Ludding, C.C.I.; de Jong, K.P.; Bitter, J.H. Toward stable nickel catalysts for aqueous phase reforming of biomass-derived feedstock under reducing and alkaline conditions. J. Catal. 2014, 319, 27–35. [Google Scholar] [CrossRef]
- van Haasterecht, T.; Ludding, C.C.I.; de Jong, K.P.; Bitter, J.H. Stability and activity of carbon nanofiber-supported catalysts in the aqueous phase reforming of ethylene glycol. J. Energy Chem. 2013, 22, 257–269. [Google Scholar] [CrossRef]
- Wang, X.; Li, N.; Pfefferle, L.D.; Haller, G.L. Pt–Co bimetallic catalyst supported on single walled carbon nanotube: XAS and aqueous phase reforming activity studies. Catal. Today 2009, 146, 160–165. [Google Scholar] [CrossRef]
- Wang, X.; Li, N.; Pfefferle, L.D.; Haller, G.L. Pt−Co Bimetallic Catalyst Supported on Single-Walled Carbon Nanotubes: Effect of Alloy Formation and Oxygen Containing Groups. J. Phys. Chem. C 2010, 114, 16996–17002. [Google Scholar] [CrossRef]
- Wang, X.; Li, N.; Webb, J.A.; Pfefferle, L.D.; Haller, G.L. Effect of surface oxygen containing groups on the catalytic activity of multi-walled carbon nanotube supported Pt catalyst. Appl. Catal. B 2010, 101, 21–30. [Google Scholar] [CrossRef]
- Wang, X.; Li, N.; Zhang, Z.; Wang, C.; Pfefferle, L.D.; Haller, G.L. High-Yield Hydrogen Production from Aqueous Phase Reforming over Single-Walled Carbon Nanotube Supported Catalysts. ACS Catal. 2012, 2, 1480–1486. [Google Scholar] [CrossRef]
- Rahman, M.M. H2 production from aqueous-phase reforming of glycerol over Cu–Ni bimetallic catalysts supported on carbon nanotubes. Int. J. Hydrogen Energy 2015, 40, 14833–14844. [Google Scholar] [CrossRef]
- He, C.; Zheng, J.; Wang, K.; Lin, H.; Wang, J.-Y.; Yang, Y. Sorption enhanced aqueous phase reforming of glycerol for hydrogen production over Pt-Ni supported on multi-walled carbon nanotubes. Appl. Catal. B 2015, 162, 401–411. [Google Scholar] [CrossRef]
- Tang, W.; Chen, Z.; Millan, M.; Zuo, X.; Yuan, G.; Cui, Z.; Dong, Z.; Cong, Y.; Li, X. Facile fabrication of porous carbon nanofibers encapsulated with nanoscale exposed Ni for producing high-purity hydrogen from cheap glycerol. Int. J. Hydrogen Energy 2023, 48, 38172–38187. [Google Scholar] [CrossRef]
- Dehane, A.; Nemdili, L.; Merouani, S.; Ashokkumar, M. Critical Analysis of Hydrogen Production by Aqueous Methanol Sonolysis. Top. Curr. Chem. 2023, 381, 9. [Google Scholar] [CrossRef]
- Yasui, K.; Tuziuti, T.; Kozuka, T.; Towata, A.; Iida, Y. Relationship between the bubble temperature and main oxidant created inside an air bubble under ultrasound. J. Chem. Phys. 2007, 127, 154502. [Google Scholar] [CrossRef] [PubMed]
- Merouani, S.; Ferkous, H.; Hamdaoui, O.; Rezgui, Y.; Guemini, M. New interpretation of the effects of argon-saturating gas toward sonochemical reactions. Ultrason. Sonochem. 2015, 23, 37–45. [Google Scholar] [CrossRef]
- Meroni, D.; Djellabi, R.; Ashokkumar, M.; Bianchi, C.L.; Boffito, D.C. Sonoprocessing: From Concepts to Large-Scale Reactors. Chem. Rev. 2022, 122, 3219–3258. [Google Scholar] [CrossRef]
- Merabet, N.; Kerboua, K. Sonolytic and ultrasound-assisted techniques for hydrogen production: A review based on the role of ultrasound. Int. J. Hydrogen Energy 2022, 47, 17879–17893. [Google Scholar] [CrossRef]
- Islam, M.H.; Burheim, O.S.; Pollet, B.G. Sonochemical and sonoelectrochemical production of hydrogen. Ultrason. Sonochem. 2019, 51, 533–555. [Google Scholar] [CrossRef] [PubMed]
- Kiss, A.A.; Geertman, R.; Wierschem, M.; Skiborowski, M.; Gielen, B.; Jordens, J.; John, J.J.; Van Gerven, T. Ultrasound-assisted emerging technologies for chemical processes. J. Chem. Technol. Biotechnol. 2018, 93, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Wood, R.J.; Lee, J.; Bussemaker, M.J. A parametric review of sonochemistry: Control and augmentation of sonochemical activity in aqueous solutions. Ultrason. Sonochem. 2017, 38, 351–370. [Google Scholar] [CrossRef] [PubMed]
- Suslick, K.S. Sonochemistry. Science 1990, 247, 1439–1445. [Google Scholar] [CrossRef]
- Didenko, Y.T.; Suslick, K.S. The energy efficiency of formation of photons, radicals and ions during single-bubble cavitation. Nature 2002, 418, 394–397. [Google Scholar] [CrossRef]
- Suslick, K.S.; Flannigan, D.J. Inside a Collapsing Bubble: Sonoluminescence and the Conditions during Cavitation. Annu. Rev. Phys. Chem. 2008, 59, 659–683. [Google Scholar] [CrossRef]
- Xu, H.; Zeiger, B.W.; Suslick, K.S. Sonochemical synthesis of nanomaterials. Chem. Soc. Rev. 2013, 42, 2555–2567. [Google Scholar] [CrossRef]
- Zhang, Y.; Khanbareh, H.; Dunn, S.; Bowen, C.R.; Gong, H.; Duy, N.P.H.; Phuong, P.T.T. High Efficiency Water Splitting using Ultrasound Coupled to a BaTiO3 Nanofluid. Adv. Sci. 2022, 9, 2105248. [Google Scholar] [CrossRef]
- Buettner, J.; Gutierrez, M.; Henglein, A. Sonolysis of water-methanol mixtures. J. Phys. Chem. 1991, 95, 1528–1530. [Google Scholar] [CrossRef]
- Rassokhin, D.N.; Kovalev, G.V.; Bugaenko, L.T. Temperature Effect on the Sonolysis of Methanol/Water Mixtures. J. Am. Chem. Soc. 1995, 117, 344–347. [Google Scholar] [CrossRef]
- Mizukoshi, Y.; Nakamura, H.; Bandow, H.; Maeda, Y.; Nagata, Y. Sonolysis of organic liquid: Effect of vapour pressure and evaporation rate. Ultrason. Sonochem. 1999, 6, 203–209. [Google Scholar] [CrossRef]
- Suslick, K.S.; Gawienowski, J.J.; Schubert, P.F.; Wang, H.H. Alkane sonochemistry. J. Phys. Chem. 1983, 87, 2299–2301. [Google Scholar] [CrossRef]
- Suslick, K.S.; Gawienowski, J.J.; Schubert, P.F.; Wang, H.H. Sonochemistry in non-aqueous liquids. Ultrasonics 1984, 22, 33–36. [Google Scholar] [CrossRef]
- Krishna, C.M.; Lion, Y.; Kondo, T.; Riesz, P. Thermal decomposition of methanol in the sonolysis of methanol-water mixtures. Spin-trapping evidence for isotope exchange reactions. J. Phys. Chem. 1987, 91, 5847–5850. [Google Scholar] [CrossRef]
- Rae, J.; Ashokkumar, M.; Eulaerts, O.; von Sonntag, C.; Reisse, J.; Grieser, F. Estimation of ultrasound induced cavitation bubble temperatures in aqueous solutions. Ultrason. Sonochem. 2005, 12, 325–329. [Google Scholar] [CrossRef]
- Kerboua, K.; Hamdaoui, O. Oxygen-argon acoustic cavitation bubble in a water-methanol mixture: Effects of medium composition on sonochemical activity. Ultrason. Sonochem. 2020, 61, 104811. [Google Scholar] [CrossRef]
- Thomas, R.G.; Jonnalagadda, U.S.; Kwan, J.J. Biomedical Applications for Gas-Stabilizing Solid Cavitation Agents. Langmuir 2019, 35, 10106–10115. [Google Scholar] [CrossRef] [PubMed]
- Jonnalagadda, U.S.; Fan, Q.; Su, X.; Liu, W.; Kwan, J.J. Nanostructured Sonophotocatalysts for spatially controlled inertial cavitation towards energy-efficient sonochemistry. ChemCatChem 2022, 14, e202200732. [Google Scholar] [CrossRef]
- Wang, Y.; Zhao, D.; Ji, H.; Liu, G.; Chen, C.; Ma, W.; Zhu, H.; Zhao, J. Sonochemical Hydrogen Production Efficiently Catalyzed by Au/TiO2. J. Phys. Chem. C 2010, 114, 17728–17733. [Google Scholar] [CrossRef]


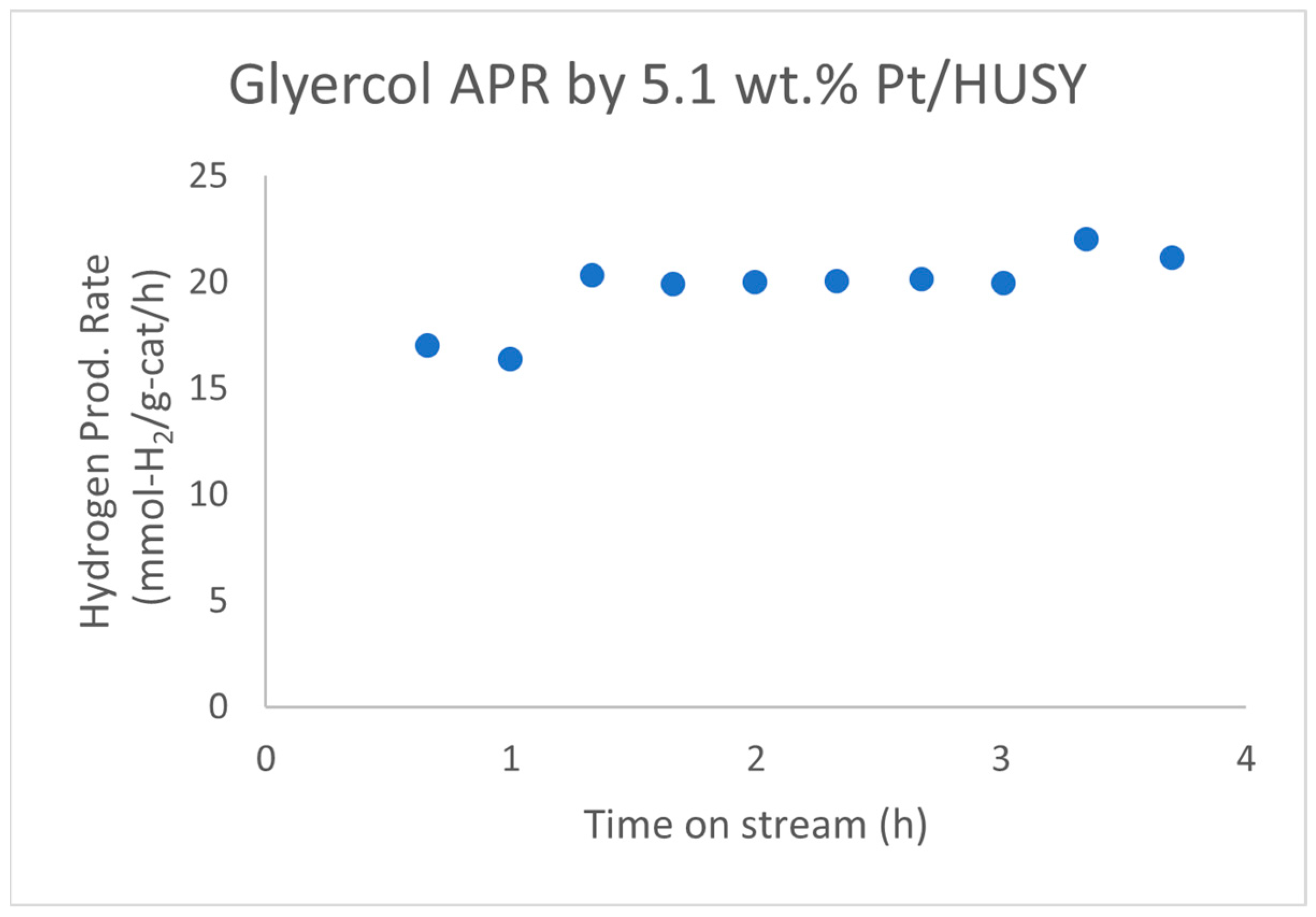

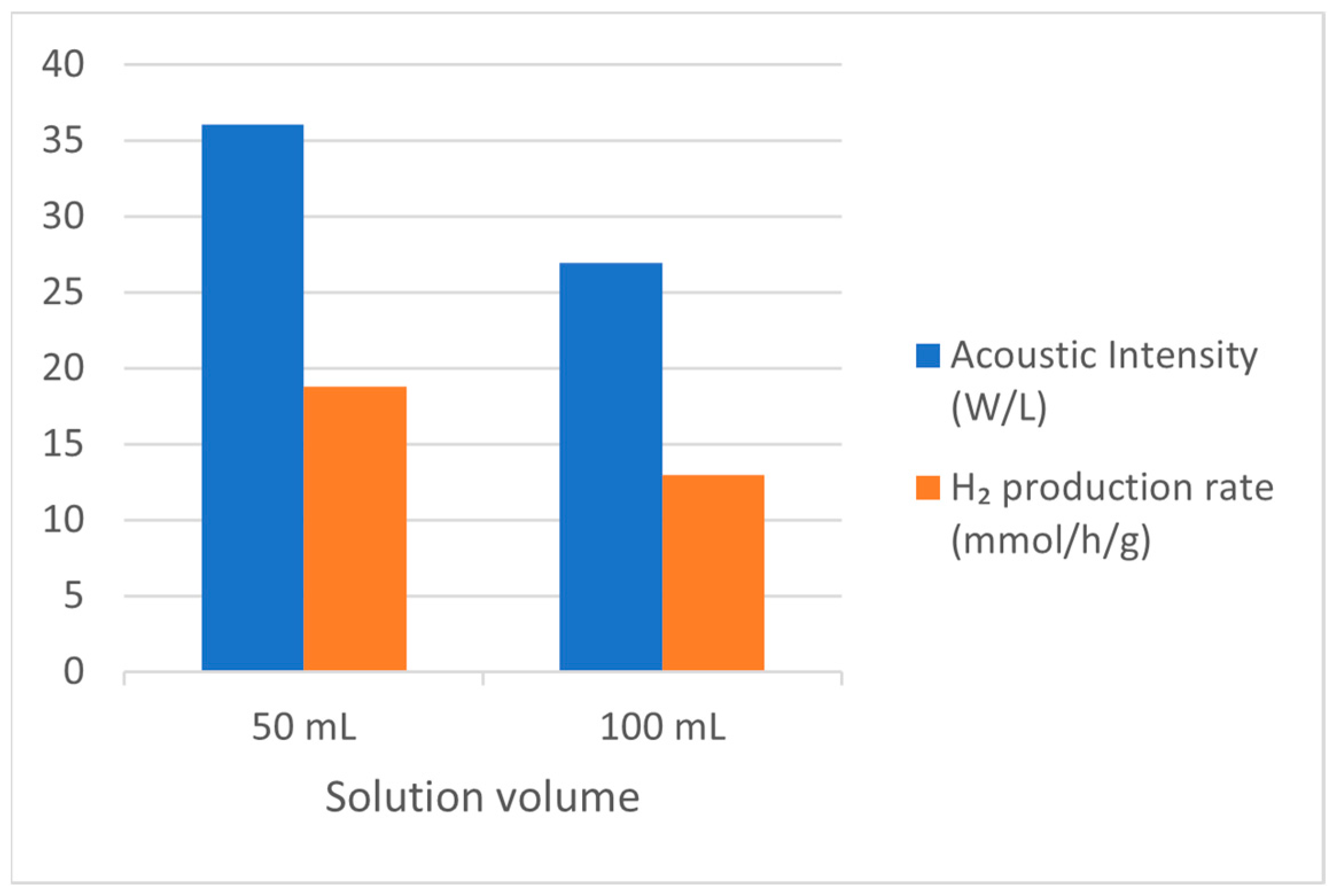
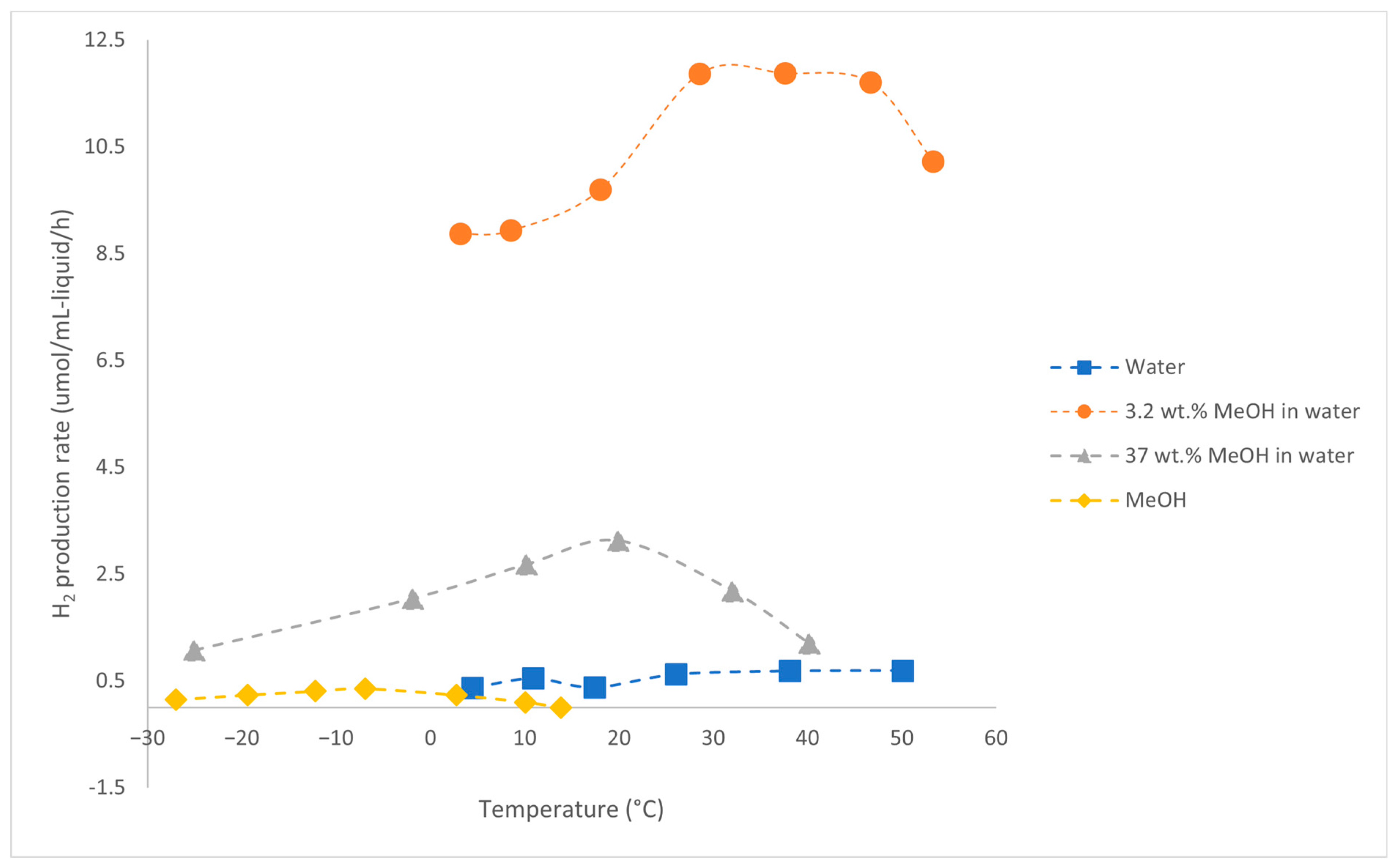
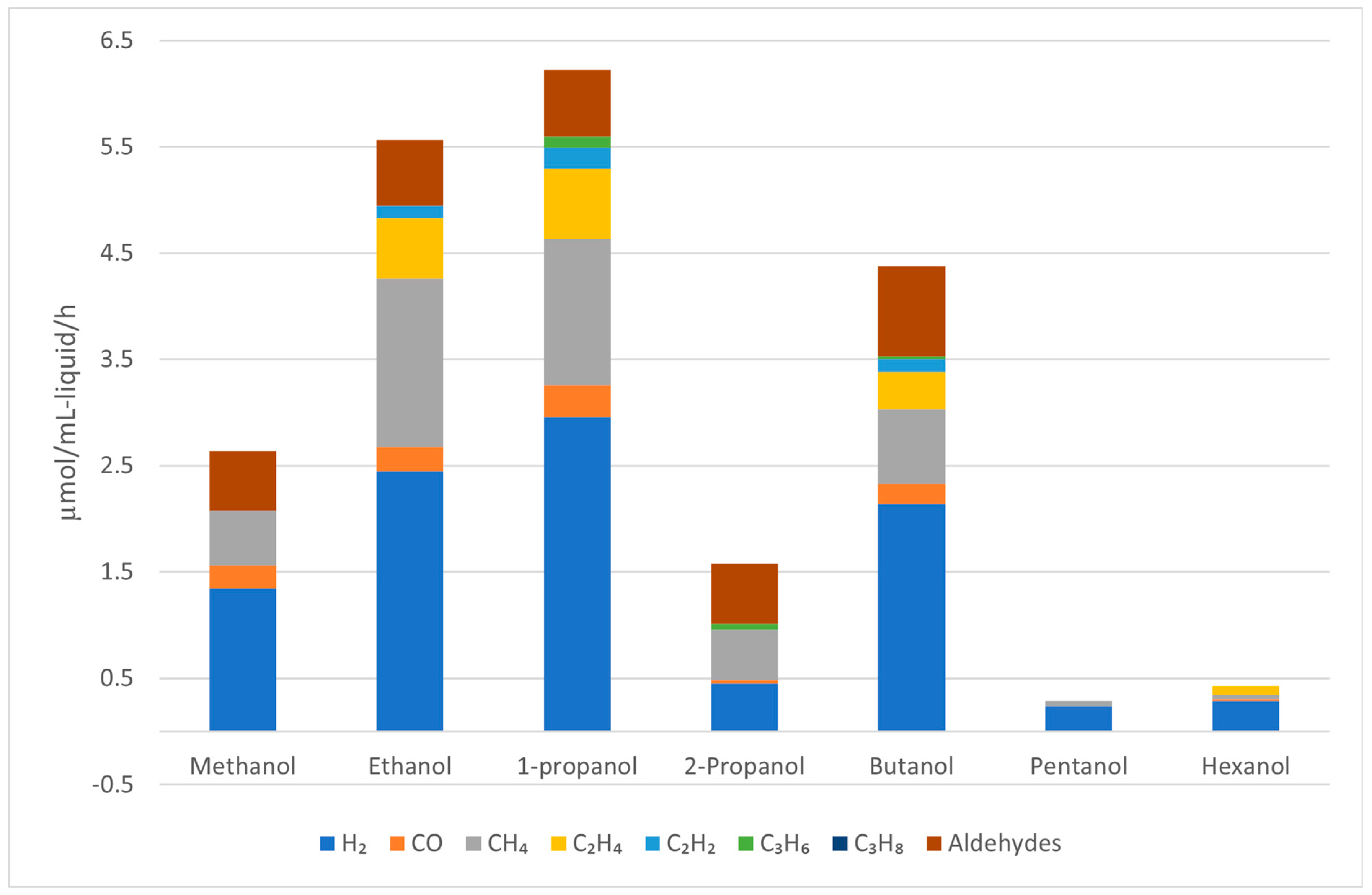
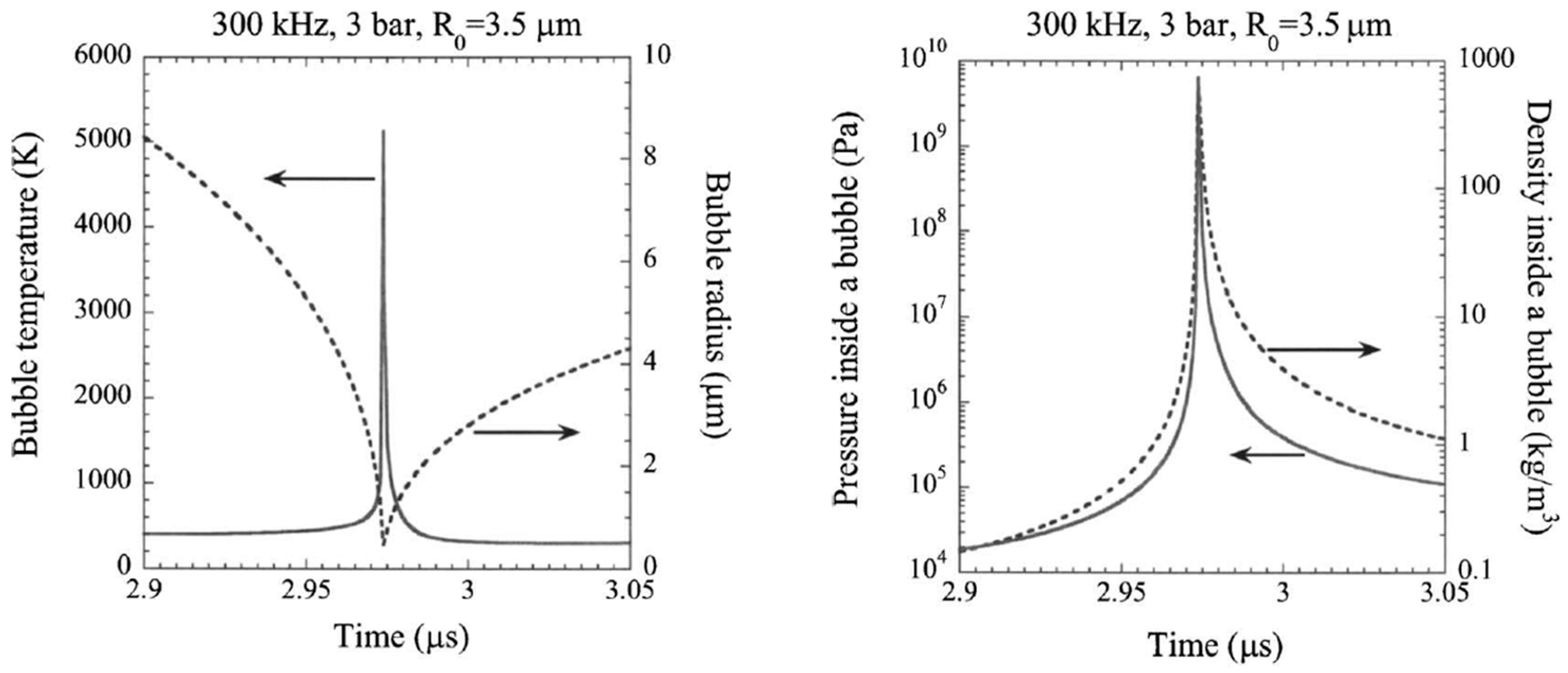
| Alcohols | wt.% (mol.%) in Water | Temperature (°C) | ||
|---|---|---|---|---|
| 200 | 225 | 250 | ||
| Methanol | 10 (5.9) | 18.5 | 29.9 | 46.2 |
| 50 (36) | 27.4 | 43.5 | 66.3 | |
| Ethanol | 10 (4.2) | 18.3 | 29.5 | 45.4 |
| 50 (28) | 25.5 | 40.6 | 61.5 | |
| Ethylene Glycol | 1 (0.3) | 15.5 | 25.4 | 39.7 |
| 10 (3.1) | 15.2 | 24.9 | 38.8 | |
| Glycerol | 1 (0.2) | 15.5 | 25.5 | 39.7 |
| 10 (2.1) | 15.2 | 24.9 | 38.8 | |
| Entry | Catalyst | Reaction Condition | H2 Prod. Rate (mmol/g-cat/h) | Stability |
|---|---|---|---|---|
| 1 [a] | ZnO/Ni-8Cu/Al2O3 [b] (10 wt.% Ni, 8 wt.% Cu) | Batch, 5 bar, 1 g catalyst, 100 mL of 10 wt.% methanol, 4 h reaction | 21.8 (at 250 °C) | 35% loss after 72 h |
| 2 [a] | Ni-8Cu/Al2O3 [c] (10 wt.% Ni, 8 wt.% Cu) | 21.3 (at 250 °C) | 75% loss after 72 h | |
| 3 [a] | Ni/γ-Al2O3 (10 wt.% Ni) | 10.8 (at 250 °C) | N/A | |
| 4 [a] | Cu/γ-Al2O3 (10 wt.% Cu) | 8.5 (at 250 °C) | N/A | |
| 5 [d] | Ru/γ-Al2O3 [e] (3.7 wt.% Ru) | Fixed bed, 24 bar, 4 g catalyst, 3.6 mL/h of 10 wt.% polyols, WHSV = 0.3 h−1 | Glycerol: 3.5 (at 225 °C) | 18% loss after 28 h TOS |
| Sorbitol: 3.3 (at 225 °C) | 24% loss after 28 h TOS | |||
| Xylitol: 2.5 (at 225 °C) | 22% loss after 28 h TOS |
| Entry | Catalyst | Reaction Condition | H2 Prod. Rate (mmol/g-cat/h) | Stability |
|---|---|---|---|---|
| 1 [a] | 0.625CoAl-600 [b] | Fixed bed, 50 bar, 0.5 g catalyst, 10 wt.% glycerol in water WHSV = 24.5 h−1 | 14 (at 260 °C) | 48% loss in H2 production rate after 30 h TOS (8 mmol-H2/g-cat/h) |
| 2 [c] | Pt/0.625CoAl [d] (0.3 wt.% Pt) | Fixed bed, 50 bar, 1.8 g catalyst, 10 wt.% glycerol in water 0.02 mL/min WHSV = 0.68 h−1 | 3.4 (at 260 °C, TOS: 10 h) 4.1 (at 260 °C, TOS: 100 h) | No decrease in glycerol conversion after 100 h TOS. H2 selectivity decreases from 53% to 49%. |
| 3 [e] | 0.5 mL/min WHSV = 17 h−1 | 19 (at 260 °C, TOS: 3 h) | No data | |
| 4 [f] | Pt/Co2Al-c700 [g] (0.98 wt.% Pt) | Batch, 20 bar, 0.1 g catalyst, 15 mL of 37 wt.% methanol in water, 1 h reaction | 202 (at 220 °C) | 9% loss in H2 production rate after 10 cycles of one hour each |
| Entry | Catalyst | Reaction Condition | H2 Prod. Rate (mmol/g-cat/h) | Stability |
|---|---|---|---|---|
| 1 [a] | NiAl2O4 [b] (33 wt.% Ni) | Fixed bed, 35 bar, 0.5 g catalyst, 10 wt.% glycerol in water 0.2 mL/min, WHSV = 24.5 h−1 | 26.2 (at 235 °C) | 12% loss in H2 production rate (to 23) after 50 h TOS. |
| 2 [c] | Pt/NiAl2O4 [d] (0.97 wt.% Pt) | Fixed bed, 29 bar, 1 g catalyst, 0.05 mL/min of 10 wt.% methanol in water. WHSV = 2.94 h−1 | 26.4 (at 210 °C) | 10% loss in conversion to gases after 600 h on stream |
| Entry | Catalyst | Reaction Condition | H2 Prod. Rate (mmol/g-cat/h) | Gas Selectivity | Stability |
|---|---|---|---|---|---|
| 1 [a] | Ni/mp-CeO2 [b] | Batch, 6 bar, 0.4 g catalyst 150 mL of 30 wt.% glycerol from biodiesel byproduct in water 2 h reaction | 7.5 (at 225 °C) | 75.65% H2 16.87% CO2 1.43% CH4 6.05% CO | N/A |
| 2 [a] | 1Ni-2Cu/CeO2 [c] (12.2 wt.%Ni, 23.3 wt.% Cu) | 10 (at 225 °C) | 82.72% H2 14.41% CO2 0.12% CH4 2.74% CO | 14% loss in H2 production rate (to 8), 10% increase in CO2, and 6% in CO content after 50 cycles of 2 h each | |
| 3 [a] | 1Ni-2Cu/CeO2 + 0.2 g CaO | 18 (at 225 °C) | 85.08% H2 14.25% CO2 0.06% CH4 0.61% CO | N/A |
| Entry | Catalyst | Reaction Condition | H2 Prod. Rate (mmol/g-cat/h) | Stability | Wt.% Pt in Fresh Catalyst/Spent Catalyst |
|---|---|---|---|---|---|
| 1 [a] | PtLa/CeO2 [b] (1.92 wt.% Pt, 1.29 wt.% La) | Batch, Autogenous pressure, 0.2 g catalyst, 20 mL of 10 wt.% methanol in water 6 h reaction | 30 (at 250 °C) | 17% loss in hydrogen production rate (25) after 5 cycles of six hours each at 250 °C | 1.92/1.27 |
| 2 [a] | Pt/CeO2-HT [c] (1.86 wt.% Pt) | 25 (at 250 °C) | 87% loss in hydrogen production rate (3.1) after 5 cycles of six hours each at 250 °C | 1.86/0.39 |
| Entry | Catalyst | Reaction Condition | H2 Prod. Rate (mmol/g-cat/h) | Stability |
|---|---|---|---|---|
| 1 [a] | 5La-NiMgAl [b] (38 wt.% Ni, 5.4 wt.% La) | Batch, Autogenous pressure, 0.2 g catalyst, 20 mL of 10 wt.% methanol in water, 6 h reaction | 41 (at 250 °C) | 20% loss in H2 production rate (to 33) after 5 cycles of 6 h each |
| 2 [a] | 0La-NiMgAl [c] (39 wt.% Ni) | 36 (at 250 °C) | 72% loss in H2 production rate (to 10) after 5 cycles of 6 h each | |
| 3 [d] | NiMg [e] (23 wt.% NiO, 51.5 wt.% MgO) | Fixed bed, 35 atm, 1.25 g catalyst., 10 vol. % glycerol in water, 0.102 mL/min, WHSV = 5 mL g−1 h−1 | 4.3 (at 250 °C) | N/A |
| 4 [d] | Ni5CuMg [f] (21.9 wt.% NiO, 5.9 wt.% CuO, 47.3 wt.% MgO) | 10 (at 250 °C) | No deactivation was observed after 6 h TOS |
| Entry | Catalyst | Reaction Condition | H2 Prod. Rate (mmol/g-cat/h) | Stability |
|---|---|---|---|---|
| 1 [a] | Pt/AC [b] (4.23 wt.% Pt) | Fixed bed, 32 bar, 5 mL catalyst, 10 wt.% glycerol in water, LHSV = 8.4 h−1 | 18 ± 1 (at 230 °C) | No significant deactivation over about 4 h TOS |
| 2 [c] | Pt/AC (7 wt.% Pt) | Fixed bed, 45 atm, 0.3 g cat., 10 wt.% ethylene glycol in water, 0.1 mL/min, WHSV = 2 h−1 | 38 ± 2 (at 250 °C) | No significant deactivation over about 25 h TOS |
| 3 [d] | Pt-Fe/AC [e] (3.11 wt.% Pt, 3.11 wt.% Fe) | 97 ± 3 (at 250 °C) | No significant deactivation over about 90 h TOS |
| Entry | Catalyst | Reaction Condition | H2 Prod. Rate (mmol/g-cat/h) | Stability |
|---|---|---|---|---|
| 1 [a] | 29 wt.% Cu/ZnO | Batch, 20 bar, 0.05 g catalyst, 23 mL of 37 wt.% methanol in 0.05M KOH (aq), 1.25 h | 94 (at 210 °C) | 81% loss (to 18) after four cycles of 1.25 h each. |
| 2 [a] | 27 wt.% Cu/ZnO@NC [b] | 320 (at 210 °C) | 17% loss (to 266) after four cycles of 1.25 h each. | |
| 3 [c] | Cu/ZnO/Al2O3 (53.1 wt.% Cu) | Batch, 20 bar, 0.05 g catalyst, 20 mL of 37 wt.% methanol, 1 h | 87 (at 210 °C) | 60% loss (to 35) after five cycles of 1.25 h each |
| 4 [c] | Cu-SP/Al2O3-ZnO [d] (52.8 wt.% Cu) | 221 (at 210 °C) | 44% loss (to 122) after five cycles of 1.25 h each |
| Entry | Catalyst | Reaction Condition | H2 Prod. Rate (mmol/g-cat/h) | Stability |
|---|---|---|---|---|
| 1 [a] | 2%Pt/α-Mo2C | Batch, 20 bar, 0.1 g catalyst, 50 mL of 37 wt.% methanol in water. 1.25 h reaction | 467 (at 190 °C) | 33% loss (to 313) after 11 cycles of 1.25 h. [b] |
| 2 [c] | Ni/α-Mo2C [d] (2.2 wt.% Ni) | Batch, 20 bar, 0.1 g catalyst, 50 mL of 64 wt.% methanol in water. | 626 (at 240 °C) | 30% loss (to 438) after 10 cycles. [e] |
| 3 [f] | Pt/MoS2 [g] (0.2 wt.% Pt) | Batch, 20 bar. 0.2 g catalyst, 15 g of 37 wt.% methanol in water and 0.3 g NaOH, 1 h reaction | 11.5 (@ 220 °C) | 24% loss after 4 cycles of 1 h. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kee, C.W.; Zheng, J.; Yap, W.J.; Ou Yong, R.; Liu, Y. Thermal and Sono—Aqueous Reforming of Alcohols for Sustainable Hydrogen Production. Molecules 2024, 29, 4867. https://doi.org/10.3390/molecules29204867
Kee CW, Zheng J, Yap WJ, Ou Yong R, Liu Y. Thermal and Sono—Aqueous Reforming of Alcohols for Sustainable Hydrogen Production. Molecules. 2024; 29(20):4867. https://doi.org/10.3390/molecules29204867
Chicago/Turabian StyleKee, Choon Wee, Jia’E Zheng, Wei Jie Yap, Roy Ou Yong, and Yan Liu. 2024. "Thermal and Sono—Aqueous Reforming of Alcohols for Sustainable Hydrogen Production" Molecules 29, no. 20: 4867. https://doi.org/10.3390/molecules29204867
APA StyleKee, C. W., Zheng, J., Yap, W. J., Ou Yong, R., & Liu, Y. (2024). Thermal and Sono—Aqueous Reforming of Alcohols for Sustainable Hydrogen Production. Molecules, 29(20), 4867. https://doi.org/10.3390/molecules29204867