Abstract
This paper describes the synthesis of two 6-azido-6-deoxy derivatives of phosphatidylinositol (PI), which contained different fatty acid chains. These syntheses, starting from methyl α-d-glucopyranoside, employed multiple regioselective transformations with Ferrier rearrangement as one of the key steps. The PI derivatives contained different fatty acid chains in the lipids and an azido group in the inositol residue to facilitate their further functionalization under bioorthogonal conditions. Therefore, they should be useful probes for the investigation of PI and related biology, such as PI phosphorylation, PI interaction with other molecules in cells, and the functions of lipid structures in these processes.
1. Introduction
Lipids are essential constituents of the cell membrane and play a critical role in many biological processes [1,2]. For example, phosphatidylinositol (PI) and its various phosphorylated forms, such as PI-3-phosphate (PI-3-P), PI-5-P and PI-3,4,5-P3 (Figure 1), are a class of lipids involved in not only structural modulations of the cell membrane but also the regulation of molecular transport and cellular signaling [3,4]. The unique and adaptable properties of PI have stimulated considerable enthusiasm for its structural investigation and modification to result in innovative lipid analogues with customized functionalities. These studies have broadened the range of lipid research and related biological applications [5]. Moreover, PI is also an important part in the core structure of glycosylphosphatidylinositol (GPI) anchors and GPI-anchored proteins (GPI-APs) [6,7].
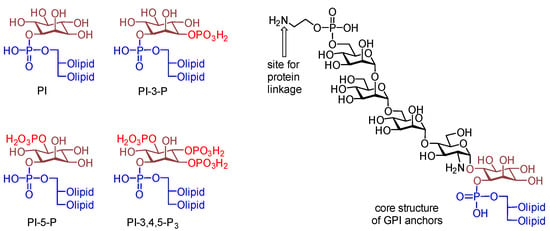
Figure 1.
Chemical structures of PI, PI-3-P, PI-5-P, PI-3,4,5-P3, and the conserved core structure of GPI anchors.
To study the functional roles and mechanisms of PIs in various biological processes, appropriate methods and molecular tools to enable specific labeling and examination of these biomolecules on cells are imperative. To this end, our group previously developed approaches to metabolically engineer cell-surface GPIs and GPI-APs to carry an azido tag using azide-modified myo-inositol and PI derivatives [8,9,10]. This enabled the functionalization of GPIs and GPI-APs with various molecular labels for their biological studies and applications. In the reported inositol and PI derivatives, the azide is linked to the 4-C-position of inositol, as it is the furthest site from the 1,2,6-C-positions involved in GPI biosynthesis—a design aimed to cause minimal interference to their metabolic incorporation by cells [8,11]. However, these inositol and PI derivatives are not suitable for the investigation of PIPs, as their 4-C-position that is critical for PIP biosynthesis [12] is modified. The current work aims to design new PI derivatives as probes for the study of PIPs and to develop efficient methods for the synthesis of these probes.
2. Results and Discussion
As mentioned above, the 3,4,5-C-positions of myo-inositol in PI are critical for its biosynthetic transformation into various PIPs (Figure 1). Therefore, PI derivatives useful for metabolic PIP engineering should have these positions undisrupted. Under this condition, the only sites left in the myo-inositol residue of PI for modifications are the 2- and 6-C-positions. However, the 2-C-position of myo-inositol is important for its distinction from other inositols, because only the hydroxyl group at this position is axial [13]. Accordingly, we designed PI derivatives 1 and 2 (Figure 2) having the inositol 6-OH group in PI replaced with an azido group as molecular tools for the metabolic engineering of PIPs. The azide in 1 and 2 will enable further modifications of these PI derivatives and their phosphorylated products for many biological applications. In the meantime, since the 6-C-position in 1 and 2 is blocked, but this position is required for GPI biosynthesis, these probes would not participate in GPI and GPI-AP metabolism [11], thereby showing selectivity for metabolic PIP engineering. Additionally, these probes are designed to contain different fatty acid chains (saturated vs. unsaturated) in the lipid, which can be used to investigate the impacts of lipid structures on the biosynthesis and biological functions of PIs and PIPs. In addition to the above properties, these new probes also have several other advantages over previously reported PI probes. For example, the azide group is relatively small, not much larger than a hydroxyl group, but definitely much smaller than any available fluorescent, radical, or affinity tags, which can minimize the steric hindrance during their interaction with other biomolecules, such as the enzymes involved in PI metabolism. In the meantime, the azide group is rather similar to the hydroxyl group in terms of polarity. Therefore, azides are widely utilized to replace hydroxyl groups in carbohydrates to mimic natural products for glycoengineering of cells and enzymatic synthesis of oligosaccharides.
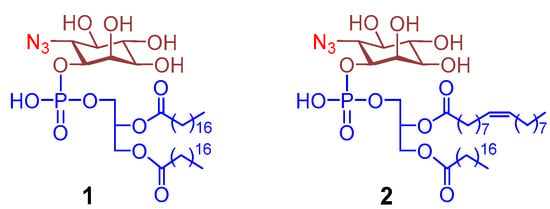
Figure 2.
Structures of the designed azido-PI analogues 1 and 2.
We commenced the synthesis of 1 and 2 with preparing the common key azido intermediate 8 (Scheme 1). First, methyl α-d-glucopyranoside 3 was smoothly converted into ketone 4 by a previously described procedure [14,15,16]. Here, the para-methoxybenzyl (PMB) group was utilized for global protection of hydroxyl groups, because PMB ethers can be easily deprotected under conditions that do not affect the azide, O-acyl groups, and unsaturated lipids in structures of the final products. The free hydroxyl group in 4 was then protected with a methoxymethyl (MOM) group to afford fully protected ketone 5. MOM was chosen since it can be introduced under conditions that would not interfere with other protecting groups in heavily substituted 4. Subsequent reduction of the keto group in 5 using NaBH4 was highly stereoselective, resulting in alcohol 6 as the single diastereomer in an excellent yield (90%) [15]. Azide was then introduced by a two-step protocol, including triflation of the hydroxyl group using triflic anhydride (Tf2O) and azide substitution of the triflate by SN2 mechanism, to afford 7. Finally, the O-acetyl group in 7 was chemoselectively removed with NaOMe to provide the key intermediate 6-azido-6-deoxy-inositol 8, which was utilized to synthesize the target molecules [14].
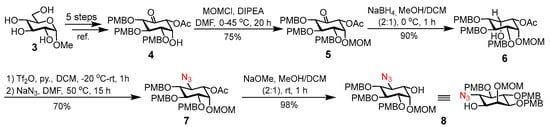
Scheme 1.
Synthesis of 6-azido-6-deoxy-inositol derivative 8.
Once the 6-azido-inositol derivative 8 was available, we turned our attention to the installation of phospholipids at its 1-O-position. We planned to accomplish this transformation by the well-established two-step one-pot protocol [17] using phosphoramidites 9 and 10 (Scheme 2) as the phosphorylating reagents. Both phosphoramidites were synthesized by a reported protocol [18] outlined in Scheme 2A,B. Condensation of alcohol 11 with oleic acid promoted by 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and 4-(dimethylamino)pyridine (DMAP) gave 12, which was followed by selective removal of the tert-butyldimethylsilyl (TBS) group with triethylamine trihydrofluoride (Et3N·3HF) to afford lipid 13. Finally, reactions of 13 and commercial 14 with 2-cyanoethyl N,N,N′,N′-tetraisopropylphosphorodiamidite 15 in the presence of diisopropylammonium tetrazolide provided 9 and 10, respectively.
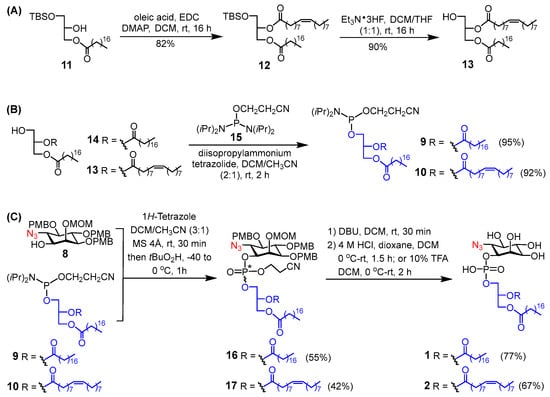
Scheme 2.
Synthesis of (A) lipid 13, (B) phosphoramidites 9 and 10, and (C) the target PI derivatives 1 and 2.
Phospholipidation of 8 was accomplished by a reaction with phosphoramidites 9 and 10 in the presence of 1H-tetrazole and then in situ oxidation of the resultant phosphites using t-BuO2H, to afford 16 and 17, respectively, as 1:1 inseparable diastereomeric mixtures due to the presence of a phosphorous stereogenic center in their structure. Interestingly, the 31P NMR spectrum of 17 showed two sets of four signals, like that observed with similar products in the literature [19]. We believe that this is due to the existence of rotamers resulting from the high steric hinderance caused by the protecting groups and rigid fatty acyl chains in 17, which was verified by the disappearance of the multiplet property of the 31P NMR signal after global deprotection to show a single peak for 2. We believe that this may cause their NMR spectra to be rather complex. It should be also pointed out that as phosphoramidites 9 and 10 were highly reactive and can decompose quickly, they were used in excess in the reaction [11]. Finally, global deprotection of 16 and 17 was carried out in two steps, including removal of the cyanoethyl group protecting the phosphate moiety with 1,8-diazabicyclo [5.4.0]undec-7-ene (DBU) and deprotection of the MOM and PMB ethers with HCl in dioxane or with 10% trifluoroacetic acid (TFA) [17,20], provided the desired PI analogues 1 and 2, whose purities were >96%, as determined by high-performance liquid chromatography (HPLC) (Figure S50, SI). The final products and all new synthetic intermediates were fully characterized with NMR and HR-MS data.
3. Materials and Methods
General methods: Commercial chemicals and materials were used without further purification unless specified otherwise. Flash chromatography used silica gel 60 (230–400 mesh). Thin-layer chromatography (TLC) was performed on silica gel 60G F254 25 glass plates detected with a UV lamp, with charring using 10% (v/v) H2SO4 in ethanol, and staining using p-anisaldehyde. 1H, 13C, COSY, and HSQC NMR spectra were obtained with Bruker (Billerica, MA, USA) 400 and 600 MHz NMR spectrometers. Coupling constants (J) were reported in hertz (Hz). Chemical shifts (δ) were reported in ppm referenced to CDCl3 (1H NMR: δ 7.26 ppm; 13C NMR: δ 77.00 ppm) or CD3OD (1H NMR: δ 3.31 ppm; 13C NMR: δ 49.0 ppm). The chemical shifts of 31P NMR signals were calculated using a lock signal in reference to 85% phosphoric acid in water. Abbreviations that describe peak splitting patterns are as follows: s, singlet; d, doublet; t, triplet; dd, double doublet; m, multiplet; br, broad. High-resolution electrospray ionization mass spectra (HR ESI MS) were obtained with an Agilent (Santa Clara, CA, USA) 6230 time-of-flight (TOF) machine. Aluminum heating blocks were used to heat the reaction mixtures.
(2R,3R,4S,5R,6S)-2-Acetoxy-4,5,6-tris[(4-methoxybenzyl)oxy]-3-methoxymethoxy-cyclohexan-1-one (5). To a solution of 4 [16] (100 mg, 0.172 mmol) in anhydrous DMF (0.6 mL), DIPEA (207 µL, 1.19 mmol) was added at 0 °C, which was followed by the addition of MOMCl (65 µL, 0.861 mmol). The reaction mixture was heated to 45 °C and stirred at this temperature for 20 h. The reaction mixture was diluted with water and extracted with DCM (10 mL × 3). The organic layers were combined, dried with Na2SO4, filtered, and condensed under reduced pressure. The crude product was purified by silica gel column chromatography to afford 5 (81 mg, 75%) as a colorless syrup. Rf = 0.40 (40% EtOAc in hexane); 1H NMR (600 MHz, CDCl3): δ 7.32 (d, J = 8.6 Hz, 2H), 7.27 (d, J = 9.3 Hz, 2H), 7.24 (d, J = 8.6 Hz, 2H), 6.89–6.83 (m, 6H), 5.15 (d, J = 2.4 Hz, 1H), 4.85 (d, J = 10.9 Hz, 1H), 4.80–4.77 (m, 2H), 4.73 (d, J = 10.3 Hz, 1H), 4.72–4.68 (m, 2H), 4.64 (d, J = 11.2 Hz, 1H), 4.48 (d, J = 10.9 Hz, 1H), 4.34 (t, J = 2.3 Hz, 1H), 4.08–4.04 (m, 2H), 3.82–3.79 (m, 10H), 3.39 (s, 3H), 2.21 (s, 3H); 13C NMR (151 MHz, CDCl3): δ 197.8, 169.8, 159.4, 159.4, 159.2, 130.6, 129.9, 129.7, 129.6, 129.5, 113.9, 113.8, 113.7, 96.8, 83.5, 81.9, 79.0, 75.7, 75.2, 73.4, 72.8, 72.6, 55.7, 55.3, 55.24, 55.23, 20.6; HR-ESI-TOF MS m/z: Calcd for C34H40NaO11 [M + Na]+ 647.2468; Found 647.2464.
3-O-Acetyl-1,5,6-tri-O-(4-methoxybenzyl)-2-O-methoxymethyl-epi-inositol (6). A mixture of 5 (310 mg, 0.496 mmol) and NaBH4 (28 mg, 0.744 mmol) in MeOH and DCM (2:1, 6 mL) was stirred at 0 °C for 1 h, and then saturated aqueous NH4Cl solution was added. The reaction mixture was diluted with DCM, dried over Na2SO4, filtered, and then concentrated in vacuo. The residue was purified by silica gel column chromatography (EtOAc/hexane, 2:1) to provide 6 (280 mg, 90%) as a white solid. Rf = 0.30 (50% EtOAc in hexane); 1H NMR (600 MHz, CDCl3): δ 7.34–7.24 (m, 7H), 6.90–6.85 (m, 6H), 4.88–4.83 (m, 2H), 4.79 (d, J = 10.2 Hz, 1H), 4.74 (d, J = 6.9 Hz, 1H), 4.71 (d, J = 11.4 Hz, 1H), 4.67–4.62 (m, 3H), 4.61 (t, J = 2.9 Hz, 1H), 4.33 (d, J = 1.8 Hz, 1H), 4.21 (d, J = 9.4 Hz, 1H), 4.11 (t, J = 9.8 Hz, 1H), 3.83 (s, 3H), 3.82 (s, 3H), 3.82 (s, 3H), 3.46 (s, 3H), 3.42 (dd, J = 9.8, 2.7 Hz, 1H), 3.38 (dd, J = 9.8, 3.3 Hz, 1H), 2.18 (s, 3H); 13C NMR (151 MHz, CDCl3): δ 170.2, 159.3, 159.2, 159.15, 131.1, 130.1, 130.0, 129.7, 129.5, 129.4, 113.8, 113.79, 113.7, 97.8, 79.9, 79.5, 78.4, 76.1, 75.8, 72.9, 71.9, 70.2, 68.9, 56.0, 55.3, 55.2, 21.0; HR-ESI-TOF MS m/z: Calcd for C34H46NO11 [M + NH4]+ 644.3065; Found 644.3063.
1-O-Acetyl-6-azido-6-deoxy-3,4,5-tri-O-(p-methoxybenzyl)-2-O-methoxymethyl-myo-inositol (7). To a solution of 6 (330 mg, 0.526 mmol) and pyridine (85 µL, 1.05 mmol) in DCM (5 mL), Tf2O (104 µL, 0.631 mmol) was added at −20 °C. The solution was allowed to warm at room temperature (rt) and was stirred for 1 h. After the disappearance of the starting material as shown by TLC, the reaction was quenched with saturated aqueous NaHCO3. The mixture was extracted with DCM, and the organic layers were combined, washed with brine, and dried over Na2SO4. The solvent was evaporated to provide crude triflate, which was dissolved in DMF (2 mL) and then treated with NaN3 (135 mg, 2.08 mmol, 4 equiv) at 40 °C overnight. The reaction mixture was diluted with ethyl acetate and washed with water and brine. The organic layer was dried over Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography to afford 7 as a colorless syrup (240 mg, 70%). Rf = 0.50 (40% EtOAc in Hexane); 1H NMR (600 MHz, CDCl3): δ 7.30–7.21 (m, 6H), 6.89–6.83 (m, 6H), 4.86–4.77 (m, 3H), 4.76–4.71 (m, 2H), 4.66 (d, J = 6.8 Hz, 1H), 4.62 (d, J = 11.0 Hz, 1H), 4.56 (dd, J = 10.9, 2.4 Hz, 1H), 4.54 (d, J = 11.1 Hz, 1H), 4.23 (t, J = 2.2 Hz, 1H), 3.96 (d, J = 10.6 Hz, 1H), 3.93 (d, J = 9.8 Hz, 1H), 3.81 (s, 3H), 3.80 (s, 6H), 3.41 (dd, J = 9.9, 2.2 Hz, 1H), 3.39 (s, 3H), 3.29 (t, J = 9.5 Hz, 1H), 2.16 (s, 3H); 13C NMR (151 MHz, CDCl3): δ 170.1, 159.3, 159.3, 159.2, 130.8, 130.1, 129.8, 129.8, 129.5, 129.4, 113.84, 113.8, 113.77, 97.5, 81.2, 81.1, 79.6, 75.5, 75.49, 72.5, 72.4, 71.7, 63.4, 55.7, 55.3, 20.9; HR-ESI-TOF MS m/z: Calcd for C34H45N4O10 [M + NH4]+ 669.3130; Found 669.3134.
6-Azido-6-deoxy-3,4,5-tri-O-(p-methoxybenzyl)-2-O-(methoxymethyl)-myo-inositol (8). To a solution of 7 (26 mg, 0.040 mmol) in MeOH and DCM (2:1, 1 mL), NaOMe (5 M in MeOH, 10 µL) was added at rt. After stirring the solution at rt for 2 h, Amberlite IR 120 H+ resin was added, followed by filtration and condensation under reduced pressure. The crude product was purified by silica gel column chromatography to afford 8 (25 mg, 98%) as a white solid. Rf = 0.35 (40% EtOAc in hexane); 1H NMR (600 MHz, CDCl3): δ 7.31–7.22 (m, 6H), 6.91–6.82 (m, 6H), 4.84 (d, J = 10.4 Hz, 1H), 4.83 (d, J = 6.8 Hz, 1H), 4.80 (d, J = 10.1 Hz, 1H), 4.74 (d, J = 10.2 Hz, 2H), 4.66 (d, J = 6.7 Hz, 1H), 4.64 (d, J = 11.4 Hz, 1H), 4.59 (d, J = 11.4 Hz, 1H), 3.92 (t, J = 2.4 Hz, 1H), 3.90 (t, J = 9.5 Hz, 1H), 3.81 (s, 6H), 3.80 (s, 3H), 3.62 (t, J = 10.1 Hz, 1H), 3.55 (d, J = 8.5 Hz, 1H), 3.45 (s, 3H), 3.37 (dd, J = 9.9, 2.6 Hz, 1H), 3.29–3.20 (m, 2H); 13C NMR (151 MHz, CDCl3): δ 159.4, 159.3, 159.2, 130.8, 130.2, 129.9, 129.8, 129.5, 129.47, 113.9, 113.8, 113.79, 98.6, 81.4, 81.3, 79.4, 79.2, 75.5, 75.4, 72.7, 70.3, 66.9, 56.1, 55.3; HR-ESI-TOF MS m/z: Calcd for C32H43N4O9 [M + NH4]+ 627.3025; Found 627.3032.
(R)-1-[(tert-butyldimethylsilyl)oxy]-3-(stearoyloxy)propan-2-yl (E)-octadec-9-enoate (12). To a solution of 11 [18,21] (0.20 g, 0.42 mmol) and oleic acid (0.3 mL, 0.97 mmol) in DCM (5.0 mL), EDC (0.17 g, 1.14 mmol) and DMAP (15.5 mg, 0.13 mmol) were added under a N2 atmosphere. The solution was stirred in the dark at rt for 16 h. The reaction mixture was diluted with water and extracted with DCM three times. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated under reduced pressure. The crude product was purified by column chromatography with 4% EtOAc in hexane as the eluent to give 12 (0.25 g, 82%) as colorless syrup. Rf = 0.45 (8% EtOAc in Hex); 1H NMR (400 MHz, CDCl3): δ 5.39–5.30 (m, 2H), 5.10–5.05 (m, 1H), 4.34 (dd, J = 11.8, 3.7 Hz, 1H), 4.16 (dd, J = 11.9, 6.3 Hz, 1H), 3.75–3.67 (m, 2H), 2.32–2.28 (m, 4H), 2.03–1.98 (m, 4H), 1.63–1.59 (m, 4H), 1.35−1.25 (m, 48H), 0.90–0.86 (m, 15H), 0.05 (s, 6H, SiMe2). The 1H NMR data match with those reported in the literature [22].
(S)-3-Hydroxy-1-(stearoyloxy)propan-2-yl (E)-octadec-9-enoate (13). Et3N·3HF (0.68 mL, 4.17 mmol) was slowly added to a solution of 12 (0.26 g, 0.35 mmol) in CH3CN and THF (1:1, 6.0 mL). After the solution was stirred at rt for 16 h, the reaction was quenched by dropwise addition of saturated aqueous NaHCO3 at 0 °C. The aqueous layer was extracted with DCM three times. The combined organic layers were washed with brine, dried over Na2SO4, and concentrated under vacuum. The crude product was purified by column chromatography with 12% EtOAc in hexane as the eluent to afford 13 (0.20 g, 90%) as colorless oil. Rf = 0.38 (20% EtOAc in Hex); 1H NMR (400 MHz, CDCl3): δ 5.35−5.31 (m, 2H), 5.08 (p, J = 5.0 Hz, 1H), 4.32 (dd, J = 11.9, 4.6 Hz, 1H), 4.24 (dd, J = 11.9, 5.6 Hz, 1H), 3.73 (t, J = 5.3 Hz, 2H), 2.37–2.31 (m, 4H), 2.06–1.98 (m, 4H), 1.62–1.60 (m, 4H), 1.34−1.25 (m, 48H), 0.87 (t, J = 8.0 Hz, 6H, CH3). The 1H NMR data match with those reported in the literature [23].
(2-Cyanoethoxyl)-(diisopropylamino)-[(R)-2-O-oleoyl-3-O-stearoyl-sn-glycerol)]-phosphine (10). To a solution of 13 (0.20 g, 0.31 mmol) and commercial 16 (0.30 mL, 0.94 mmol) in dry DCM and CH3CN (2:1, 3.0 mL), diisopropylammonium tetrazolide (161 mg, 0.94 mmol) was added at rt under a nitrogen atmosphere. After stirring for 2 h, the reaction mixture was diluted with DCM and poured into a saturated aqueous NaHCO3 solution. The mixture was extracted with DCM three times, and the combined organic layers were dried with Na2SO4 and concentrated under reduced pressure. The crude product was purified by column chromatography using triethylamine-neutralized silica gel with 10% EtOAc in hexane containing ~1% triethylamine as the eluent to provide 10 (0.24 g, 92%) as a colorless syrup. Rf = 0.33 (20% EtOAc in Hex); 1H NMR (400 MHz, CDCl3): 5.32–5.20 (m, 2H), 5.15–5.09 (m, 1H), 4.31–4.22 (m, 1H), 4.13–4.05 (m, 1H), 3.82–3.68 (m, 3H), 3.65–3.59 (m, 1H), 3.54–3.48 (m, 2H), 2.57 (t, J = 6.4 Hz, 2H), 2.28–2.22 (m, 4H), 1.96–1.91 (m, 4H), 1.57–1.52 (m, 4H), 1.26−1.18 (m, 54H), 1.11 (dd, J = 6.9, 5.0 Hz, 12H), 0.81 (t, J = 6.7 Hz, 7H); 31P NMR (CDCl3, 162 MHz): δ 149.51, 149.36. The 1H NMR data match with those reported in the literature [23].
(2-Cyanoethoxyl)-(diisopropylamino)-[(R)-2,3-di-O-stearoyl-sn-glycerol)]-phosphine (9). It was synthesized from commercial 14 in a 95% yield by the same method utilized to synthesize 10, and its 1H NMR data match those reported in the literature [11].
6-Azido-1-O-{(2-cyanoethoxy)-[(R)-2,3-di-O-stearoyl-sn-glycerol]-phosphono}-6-deoxy-3,4,5-tri-O-(4-methoxybenzyl)-2-O-(methoxymethyl)-myo-inositol (16). To a solution of 8 (15 mg, 0.024 mmol) in DCM and CH3CN (4:1, 1.5 mL), activated MS 4Å (50 mg) and 1H-tetrazole (0.45 M in acetonitrile, 0.53 mL, 0.236 mmol) were added at rt. This was followed by dropwise addition of 9 (72 mg, 0.086 mmol) dissolved in DCM (0.5 mL). After 30 min of stirring, the mixture was cooled to 0 °C, and then t-BuO2H (105 µL, 0.577 mmol) was added. After 1 h of stirring, the mixture was diluted with DCM and washed with saturated aqueous NaHCO3. The aqueous layer was extracted with DCM three times. The organic layers were combined, washed with brine, dried with Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by silica gel column chromatography to afford 16 (15 mg, 55%) as a white solid. Rf = 0.30 (40% EtOAc in hexane); 1H NMR (400 MHz, CDCl3): δ 7.31–7.20 (m, 8H), 6.90–6.82 (m, 4H), 5.35–5.25 (m, 1H), 4.90–4.80 (m, 3H), 4.78–4.62 (m, 4H), 4.54 (dd, J = 10.9, 1.8 Hz, 1H), 4.41–4.12 (m, 6H), 4.11–4.00 (m, 1H), 3.93 (td, J = 9.9, 2.6 Hz, 2H), 3.81 (m, 9H), 3.41–3.34 (m, 3H), 3.29 (td, J = 9.4, 2.9 Hz, 1H), 2.87–2.73 (m, 2H), 2.46–2.21 (m, 4H), 1.67–1.53 (m, 8H), 1.45–1.10 (m, 56H), 0.91–0.83 (m, 6H); 13C NMR (151 MHz, CDCl3): δ 173.2, 172.9, 159.3, 159.3, 159.2, 130.7, 129.9, 129.9, 129.8, 129.5, 116.3, 116.25, 113.8, 113.78, 113.77, 97.6, 97.5, 81.0, 80.9, 79.33, 79.3, 75.6, 75.5, 73.5, 73.3, 72.5, 72.4, 69.2, 64.3, 61.5, 55.94, 55.9, 55.3, 34.1, 34.0, 31.9, 29.7, 29.65, 29.5, 29.49, 29.4, 29.3, 29.29, 29.13, 29.12, 24.8, 22.7, 14.1; 31P NMR (162 MHz, CDCl3): δ −2.06, −2.12; HR-ESI-TOF MS m/z: Calcd for C74H121N5O16P [M + NH4]+ 1366.8541; Found 1366.8567.
6-Azido-1-O-[(2-cyanoethoxyl)-[(R)-2-O-oleoyl-3-O-stearoyl-sn-glycerol)]-phosphono-6-deoxy-3,4,5-tri-O-4-methoxybenzyl-2-O-(methoxymethyl)-myo-inositol (17). To a solution of 8 (35 mg, 0.057 mmol) in dry DCM and CH3CN (3:1, 6.4 mL), MS 4Å and 1H-tetrazole (0.45 M in acetonitrile, 1.26 mL, 0.57 mmol) were added, followed by dropwise addition of a fresh solution of 10 (0.24 g, 0.28 mmol) in DCM under nitrogen at rt. After the mixture was stirred at rt for 30 min, it was cooled to −40 °C and treated with t-BuO2H (5.5 M in decane, 0.21 mL, 1.14 mmol). After the solution was stirred at −40 °C for 1 h, Me2S (0.17 mL, 2.28 mmol) was added, with stirring for 1 h. While the reaction mixture was slowly warmed to rt, it was diluted with DCM and then filtered through a Celite pad. The mixture was poured into saturated aqueous NaHCO3 solution and extracted with DCM three times. The organic layers were combined, dried with Na2SO4, filtered, and condensed under reduced pressure. The crude product was purified by silica gel column chromatography with 12% EtOAc in toluene as the eluent to give 17 (33 mg, 42%) as a colorless syrup. Rf = 0.28 (40% EtOAc in Hex); 1H NMR (600 MHz, CDCl3): δ 7.29–7.24 (m, 6H), 6.89–6.86 (m, 6H), 5.39–5.34 (m, 2H), 5.33–5.31 (m, 1H), 4.87–4.83 (m, 3H), 4.78–4.66 (m, 4H), 4.56 (dd, J = 10.9, 2.6 Hz, 1H), 4.40–4.25 (m, 7H), 4.24–4.17 (m, 2H), 4.11–4.07 (m, 1H), 3.83 (d, J = 1.7 Hz, 9H), 3.41–3.39 (m, 4H), 3.32 (dd, J = 11.2, 7.9 Hz, 1H), 2.83–2.80 (m, 2H), 2.39–2.32 (m, 4H), 2.04–2.01 (m, 4H), 1.65–1.61 (m, 4H), 1.36–1.25 (m, 48H), 0.90 (t, J = 6.9 Hz, 6H); 13C NMR (151 MHz, CDCl3): δ 173.3, 172.9, 172.8, 159.4, 159.3, 159.2, 130.7, 130.0, 129.97, 129.92, 129.8 (2C), 129.72, 129.7, 129.5 (3C), 113.9 (3C), 113.82, 113.8 (3C), 97.6, 97.57, 97.55, 81.0, 80.9, 79.4, 79.3, 76.0, 75.6, 75.5, 73.5, 73.4, 73.35, 72.5, 72.48, 72.42, 72.4, 69.3, 69.2, 66.2, 65.9, 64.4, 64.3, 62.4, 62.3, 62.2, 62.1, 61.6, 61.5, 61.49, 56.0, 55.9, 55.3 (3C), 55.27, 34.2, 34.1 (2C), 34.0 (2C), 33.98, 31.94 (2C), 31.9 (2C), 29.8 (2C), 29.74, 29.72 (multiple C), 29.70, 29.67 (3C), 29.66 (2C), 29.54 (2C), 29.5 (2C), 29.4 (2C), 29.34 (2C), 29.3 (2C), 29.24, 29.2, 29.16, 29.14 (2C), 29.1, 27.24 (2C), 27.2 (2C), 24.9, 24.8, 22.7 (2C), 19.7, 19.65, 19.6, 14.1 (2C). 31P NMR (CDCl3, 243 MHz) δ −1.93, −2.06, −2.12, −2.16. HR-ESI-TOF MS m/z: Calcd for C74H119N4O16P [M + NH4]+ 1364.8383; Found 1364.8367.
6-Azido-6-deoxy-1-O-{[(R)-2,3-di-O-stearoyl-sn-glycerol]-phosphono}-myo-inositol (1). To a solution of 16 (15 mg, 0.011 mmol) in anhydrous DCM (3 mL), DBU (2 µL, 0.013 mmol) was added. After stirring at rt for 1 h, the solvent was evaporated under reduced pressure, and the product was briefly purified by silica gel column chromatography and then dissolved in DCM (3 mL). After the solution was cooled to 0 °C, a 20% TFA solution in DCM (3 mL) was added. The solution was warmed to rt, stirred for 2 h, diluted with toluene, and condensed under reduced pressure. The residue was co-evaporated with toluene five times, and the product was washed with diethyl ether three times to afford 1 as a white solid (7.5 mg, 77%). Rf = 0.1 (15% methanol in DCM); 1H NMR (600 MHz, MeOD:CDCl3 = 3:2): δ 5.26 (s, 1H), 4.41 (d, J = 11.9 Hz, 1H), 4.21–3.98 (m, 3H), 3.86 (br, 1H), 3.77–3.54 (m, 3H), 3.40–3.34 (m, 1H), 3.23–3.13 (m, 1H), 2.43–2.17 (m, 4H), 1.67–1.54 (m, 4H), 1.42–1.06 (m, 56H), 0.87 (t, J = 6.9 Hz, 6H); 13C NMR (151 MHz, MeOD:CDCl3 3:2): δ 174.0, 73.7, 73.3, 72.8, 71.1, 71.0, 70.3, 64.9, 63.9, 62.5, 54.5, 34.1, 34.0, 31.8, 29.6, 29.5, 29.4, 29.24, 29.2, 29.04, 29.0, 24.82, 24.8, 22.5, 13.8; 31P NMR (162 MHz, MeOD:CDCl3 = 3:2): δ −2.44; HR-ESI-TOF MS m/z: Calcd for C45H90N4O12P [M + NH4]+ 909.6287; Found 909.6282.
6-Azido-6-deoxy-1-O-{[(R)-2-O-oleoyl-3-O-stearoyl-sn-glycerol]-phosphono}-myo-inositol (2). DBU (7.2 µL, 0.048 mmol) was added to a solution of 17 (33 mg, 0.024 mmol) in dry DCM (5 mL) at rt. After stirring the solution for 1 h, glacial acetic acid (14 µL, 0.24 mmol) was added. The mixture was stirred for another 5 min and condensed under reduced pressure. The product was briefly purified by silica gel chromatography and then dissolved in dry DCM (6 mL), followed by dropwise addition of HCl in dioxane (4 M, 2.5 mL) at 0 °C. After the mixture was stirred for 10 min, it was warmed to rt with stirring for 1.5 h, diluted with toluene, and condensed under reduced pressure. The residue was co-evaporated with toluene six times, and the product was purified by silica gel column chromatography using 12% MeOH in DCM as the eluent to afford 2 as a white solid (14.5 mg, 67%). Rf = 0.18 (30% MeOH in CHCl3); 1H NMR (600 MHz, MeOD:CDCl3 = 3:2): δ 5.29–5.22 (m, 2H), 5.19–5.18 (m, 1H), 4.36–4.32 (m, 1H), 4.19 (br, 1H), 4.12–4.08 (m, 1H), 4.02–3.97 (m, 2H), 3.79 (br, 1H), 3.63–3.56 (m, 2H), 3.27–3.26 (m, 1H), 3.14–3.08 (m, 1H), 2.27–2.22 (m, 4H), 1.95–1.91 (m, 4H), 1.56–1.49 (m, 4H), 1.26–1.17 (m, 48H), 0.81–0.78 (m, 6H); 13C NMR: (151 MHz, MeOD:CDCl3 = 3:2): δ 173.9, 173.5, 129.8, 129.5, 73.7, 72.7, 71.1 (2C), 70.3, 65.1, 63.5, 62.4, 52.6, 34.0, 33.9, 31.8 (2C), 29.6 (2C), 29.5 (multiple C), 29.47 (3C), 29.3, 29.2, 29.15 (2C), 29.1, 29.0, 28.94, 28.9, 27.0, 26.96, 24.8, 24.7, 22.5 (3), 13.5 (2C). 31P NMR (243 MHz, MeOD:CDCl3 = 3:2): δ −4.74. HR-ESI-TOF MS m/z: Calcd for C45H88N3O12P [M + NH4]+ 907.6130; Found 907.6117.
4. Conclusions
We have designed and synthesized two azide-functionalized PI derivatives 1 and 2. The presence of an azido group within 1 and 2 would facilitate their further functionalization to install various molecular tags/labels, while the small size of the azide group would guarantee a minimal impact on the properties of 1 and 2 as PI analogues. These properties make 1 and 2 useful tools for biological investigations and applications [24,25]. Although this paper describes only the synthesis of these probes, they are currently under biological studies in our lab. For example, 1 and 2 are utilized as biosynthetic precursors of PIPs for metabolic PIP engineering to examine PI phosphorylation and related signaling processes [12,26]. On the other hand, attaching azide to the inositol 6-C-position within 1 and 2 can block them from participating in GPI biosynthesis, thereby enhancing their selectivity for metabolic PIP engineering. The synthesis of 1 and 2 is highlighted by the usage of and efficient access to a common intermediate 8 having hydroxyl groups protected with the MOM and PMB groups, which are easily removable under mildly acidic conditions compatible with unsaturated lipids, azide, and various other functional groups. Therefore, this synthetic intermediate and thus-based synthetic strategy are expected to be widely applicable to differently functionalized PI derivatives.
Supplementary Materials
The following supporting information, i.e., the NMR and MS spectra of all new compounds and the HPLC results of the final synthetic targets, can be downloaded at: https://www.mdpi.com/article/10.3390/molecules29214981/s1.
Author Contributions
M.R.M., P.G. and R.R. performed the design, synthesis, and data acquisition and analysis. Z.G. perceived the conception, supervised the whole project, and helped with designs and data analysis. All authors were involved in preparing the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This work is supported in part by NIH/NIGMS (1R35 GM131686). The MS instrument was funded by NIH (S10 OD021758 and S10 OD030250). ZG is grateful to Steven and Rebecca Scott for endowing our research.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
All the data underlying this research are available in the published article and its Supporting Information.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Brown, H.A.; Marnett, L.J. Introduction to lipid biochemistry, metabolism, and signaling. Chem. Rev. 2011, 111, 5817–5820. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Chen, Y.; Borgard, H.; Jijiwa, M.; Nasu, M.; He, M.; Deng, Y. The function and mechanism of lipid molecules and their roles in the diagnosis and prognosis of breast cancer. Molecules 2020, 25, 4864. [Google Scholar] [CrossRef] [PubMed]
- Dickson, E.J. Recent advances in understanding phosphoinositide signaling in the nervous system. F1000Res 2019, 8, 278. [Google Scholar] [CrossRef] [PubMed]
- Batrouni, A.G.; Baskin, J. The chemistry and biology of phosphatidylinositol 4-phosphate at the plasma membrane. Bioorg. Med. Chem. 2021, 40, 116190. [Google Scholar] [CrossRef] [PubMed]
- Routt, S.M.; Bankaitis, V.A. Biological functions of phosphatidylinositol transfer proteins. Biochem. Cell Biol. 2004, 82, 254–262. [Google Scholar] [CrossRef]
- Thomas, J.R.; Dwek, R.A.; Rademacher, T.W. Structure; biosynthesis, and function of glycosylphosphatidylinositols. Biochemistry 1990, 29, 5413–5422. [Google Scholar] [CrossRef]
- Ferguson, M.A.J. The structure, biosynthesis and functions of glycosylphosphatidylinositol anchors, and the contributions of trypanosome research. J. Cell Sci. 1999, 112, 2799–2809. [Google Scholar] [CrossRef]
- Lu, L.; Gao, J.; Guo, Z. Labeling cell surface GPIs and GPI-anchored proteins through cell metabolic engineering with artificial inositol derivatives. Angew. Chem. Int. Ed. 2015, 54, 9679–9682. [Google Scholar] [CrossRef]
- Jaiswal, M.; Zhu, S.; Jiang, W.; Guo, Z. Synthesis and evaluation of Nα,Nε-diacetyl-L-lysine-inositol conjugates for selective metabolic engineering of GPIs and GPI-anchored proteins on cancer cells. Org. Biomol. Chem. 2020, 18, 2938–2948. [Google Scholar] [CrossRef]
- Kundu, S.; Jaiswal, M.; Craig, K.C.; Guo, J.; Guo, Z. Labeling cell surface glycosylphosphatidylinositol-anchored proteins through metabolic engineering using an azide-modified phosphatidylinositol. Biochem. Biophys. Res. Commun. 2023, 645, 103–109. [Google Scholar] [CrossRef]
- Craig, K.C.; Guo, Z. Design and synthesis of 4-azido-phosphatidylinositol as a potential probe for metabolic engineering of glycosylphosphatidylinositol on cells. J. Carbohydr. Chem. 2022, 41, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Tolias, K.F.; Cantley, L.C. Pathways for phosphoinositide synthesis. Chem. Phys. Lipids 1999, 98, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Shashidhar, M.S.; Patil, N.T. Recent developments in the synthesis of biologically relevant inositol derivatives. In Carbohydrates in Drug Discovery and Development; Tiwari, V.K., Ed.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 283–329. [Google Scholar]
- Ausmus, A.P.; Hogue, M.; Snyder, J.L.; Rundell, S.R.; Bednarz, K.M.; Banahene, N.; Swarts, B.M. Ferrier carbocyclization-mediated synthesis of enantiopure azido inositol analogues. J. Org. Chem. 2020, 85, 3182–3191. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; Kittaka, H.; Ikegami, S. Novel synthesis of enantiomerically pure natural inositols and their diastereoisomers. J. Org. Chem. 2001, 66, 2705–2716. [Google Scholar] [CrossRef]
- Estevez, V.A.; Prestwich, G.D. Synthesis of enantiomerically pure, P-1-tethered inositol tetrakis (phosphate) affinity labels via a Ferrier rearrangement. J. Am. Chem. Soc. 1991, 113, 9885–9887. [Google Scholar] [CrossRef]
- Swarts, B.M.; Guo, Z. Synthesis of a glycosylphosphatidylinositol (GPI) anchor carrying unsaturated lipid chains. J. Am. Chem. Soc. 2010, 132, 6648–6650. [Google Scholar] [CrossRef]
- Burgula, S.; Swarts, B.M.; Guo, Z. Total synthesis of a glycosylphosphatidylinositol anchor of the human lymphoctye CD52 antigen. Chem. Eur. J. 2012, 18, 1194–1201. [Google Scholar] [CrossRef][Green Version]
- Anderson, R.J.; Osborne, S.L.; Meunier, F.A.; Painter, G.F. Regioselective approach to phosphatidylinositol 3,5-bisphosphates: Syntheses of the native phospholipid and biotinylated short-chain derivative. J. Org. Chem. 2010, 75, 3541–3551. [Google Scholar] [CrossRef]
- Hosoya, T.; Takashiro, E.; Matsumoto, T.; Suzuki, K. Total synthesis of the gilvocarcins. J. Am. Chem. Soc. 1994, 116, 1004–1015. [Google Scholar] [CrossRef]
- Ortuno, V.E.; Pulletikurti, S.; Veena, K.S.; Krishnamurthy, R. Synthesis and hydrolytic stability of cyclic phosphatidic acids: Implications for synthetic- and proto-cell studies. Chem. Commun. 2022, 58, 6231–6234. [Google Scholar] [CrossRef]
- Ardila-Fierro, K.J.; Pich, A.; Spehr, M.; Hernández, J.G.; Bolm, C. Synthesis of acylglycerol derivatives by mechanochemistry. Beilstein J. Org. Chem. 2019, 15, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.H.; Bang, S.; Park, S.M.; Ma, X.; Cassilly, C.; Graham, D.; Xavier, R.; Clardy, J. Revisiting Coley′s toxins: Immunogenic cardiolipins from Streptococcus pyogenes. J. Am. Chem. Soc. 2023, 145, 21183–21188. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Sletten, E.M.; Bertozzi, C.R. Bioorthogonal chemistry: Fishing for selectivity in a sea of functionality. Angew. Chem. Int. Ed. 2009, 48, 6974–6998. [Google Scholar] [CrossRef]
- Downes, C.P.; Macphee, C.H. myo-Inositol metabolites as cellular signals. Eur. J. Biochem. 1990, 193, 1–18. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).