
New Sulfenate Sources for Double Pallado-Catalyzed Cross-Coupling Reaction: Application in Symmetrical Biarylsulfoxide Synthesis, and Evidence of TADF Properties


Abstract
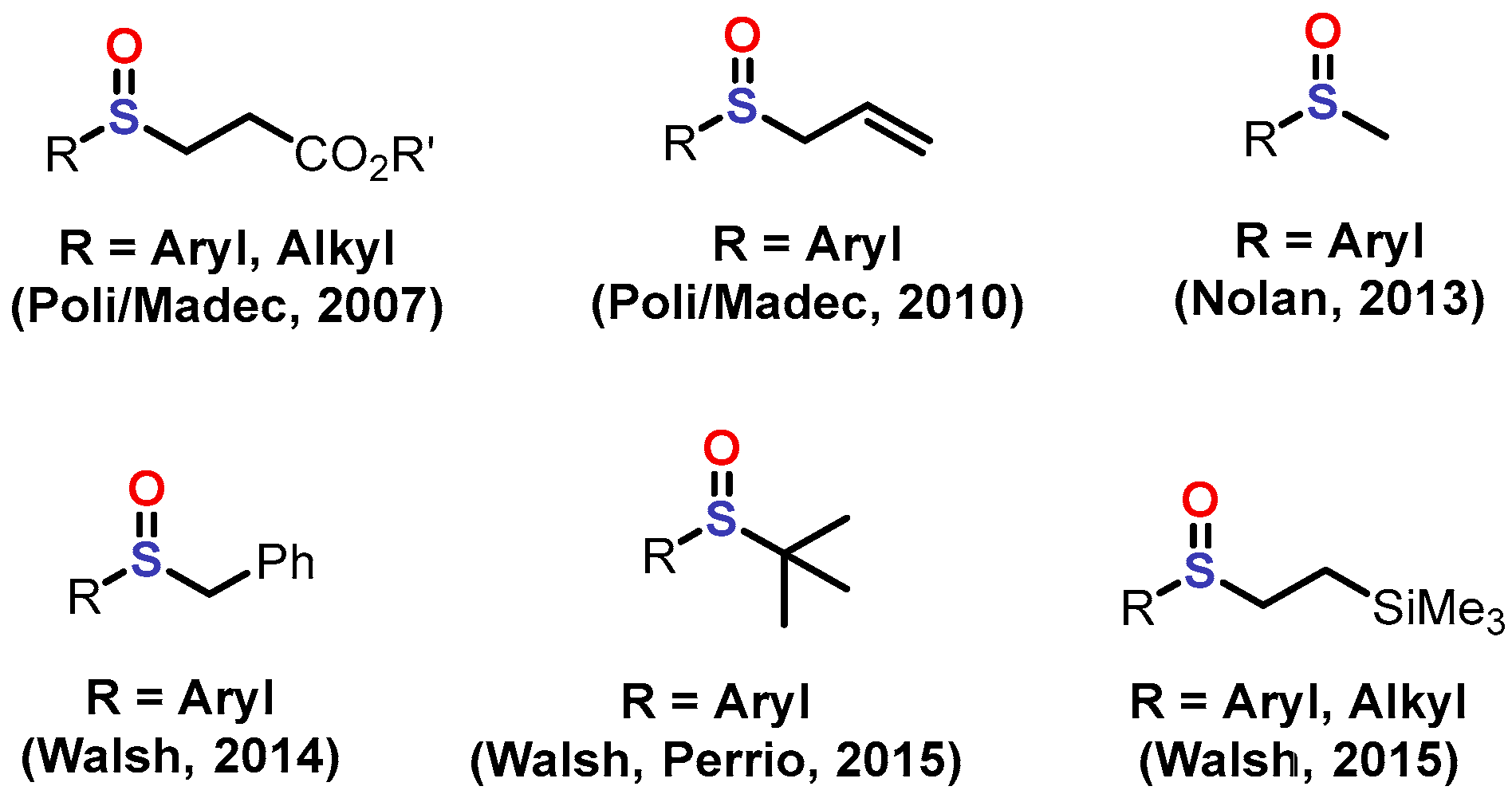
1. Introduction
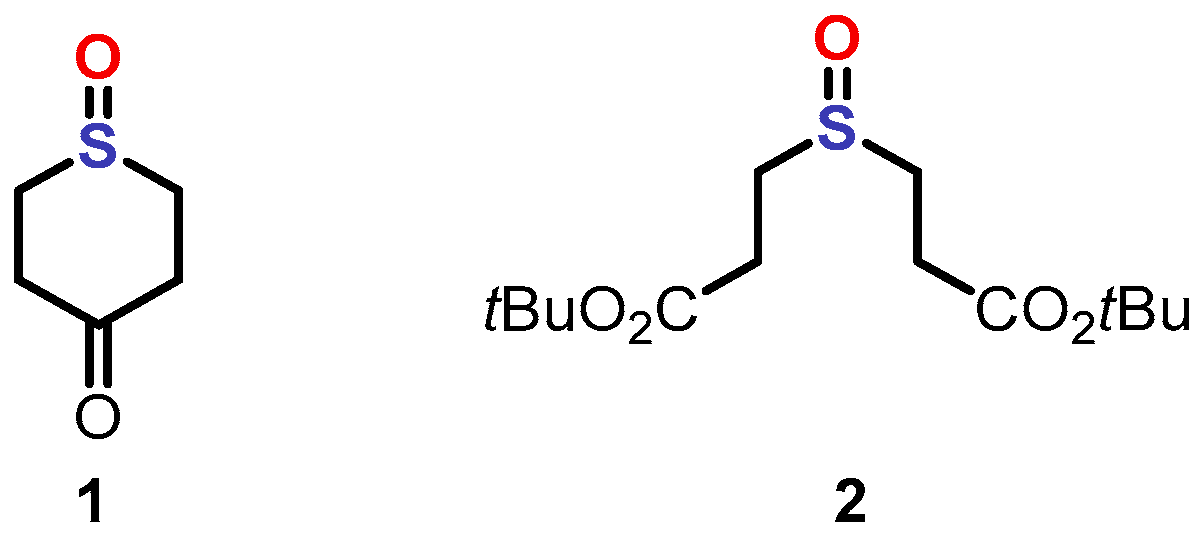
2. Results and Discussion
3. Materials and Methods
3.1. Reagents and Solvents
3.2. Analysis and Characterization
3.3. Synthesis of the Sulfenate Sources
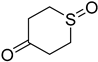
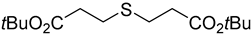
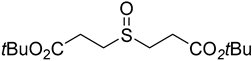
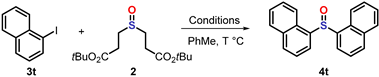
3.4. General Procedures for the Pallado-Catalyzed Cross Coupling Reactions
3.5. Biarylsulfoxide Characterization
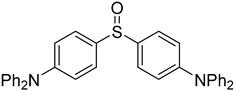
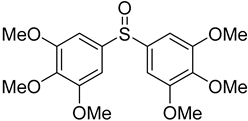
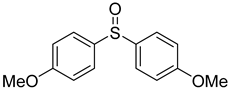
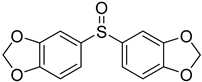
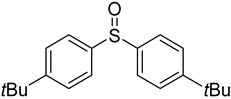
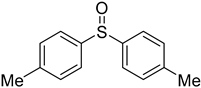
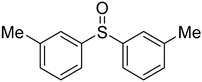
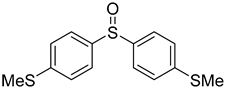
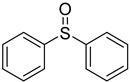
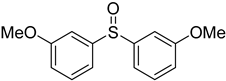
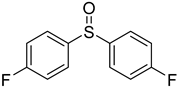
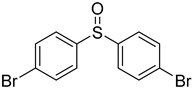
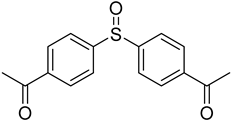

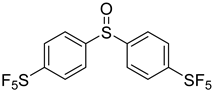
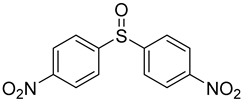

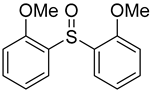
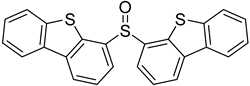
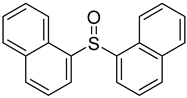

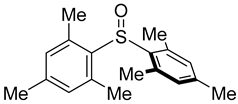
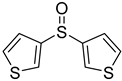
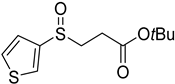
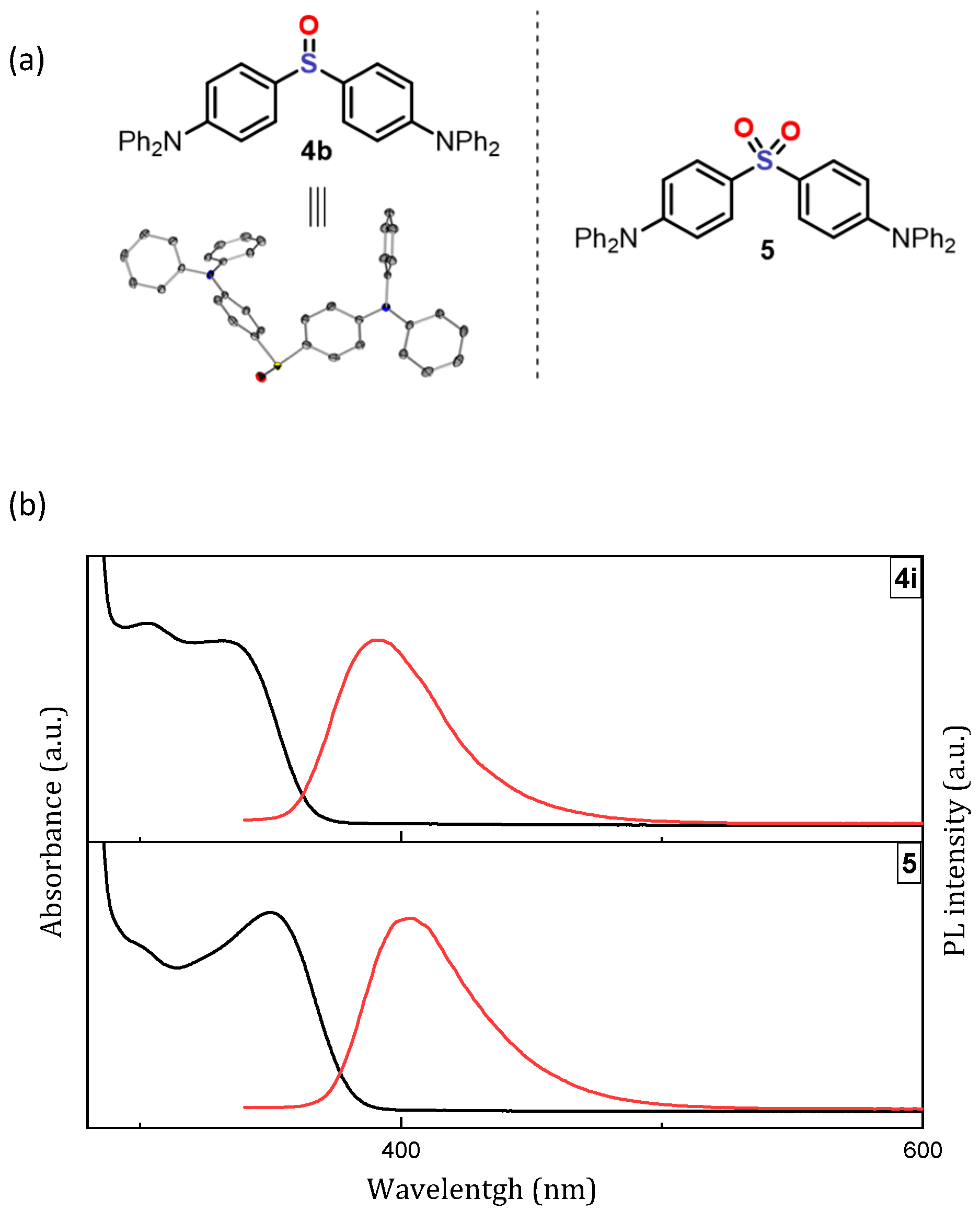
3.6. Photophysical Properties of 4,4′-Sulfinylbis(N,N-Diphenylaniline) 4b and 4,4′-Sulfonylbis(N,N-Diphenylaniline) 5
3.7. X-ray Data
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- David, N.A. The Pharmacology of Dimethyl Sulfoxide. Annu. Rev. Pharmacol. 1972, 12, 353–374. [Google Scholar] [CrossRef] [PubMed]
- Rubin, L.F. Toxicity of dimethyl sulfoxide, alone and in combination. Ann. N. Y. Acad. Sci. 1975, 243, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Brayton, C.F. Dimethyl sulfoxide (DMSO), a review. Cornell Vet. 1986, 76, 61–90. [Google Scholar] [PubMed]
- Aronson, J.K. (Ed.) Meyler’s Side Effects of Drugs, 16th ed.; Elsevier: Amsterdam, The Netherlands, 2016; pp. 992–993. [Google Scholar]
- Feng, M.; Tang, B.; Liang, S.H.; Jiang, X. Sulfur Containing Scaffolds in Drugs: Synthesis and Application in Medicinal Chemistry. Curr. Top. Med. Chem. 2016, 16, 1200–1216. [Google Scholar] [CrossRef]
- Bentley, R. Role of sulfur chirality in the chemical processes of biology. Chem. Soc. Rev. 2005, 34, 609–624. [Google Scholar] [CrossRef]
- Calligaris, M. Structure and bonding in metal sulfoxide complexes. Coord. Chem. Rev. 1996, 153, 83–154. [Google Scholar] [CrossRef]
- Calligaris, M. Structure and bonding in metal sulfoxide complexes: An update. Coord. Chem. Rev. 2004, 248, 351–375. [Google Scholar] [CrossRef]
- Mellah, M.; Voituriez, A.; Schulz, E. Chiral Sulfur Ligands for Asymmetric Catalysis. Chem. Rev. 2007, 107, 5133–5209. [Google Scholar] [CrossRef]
- Sipos, G.; Drinkel, E.E.; Dorta, R. The emergence of sulfoxides as efficient ligands in transition metal catalysis. Chem. Soc. Rev. 2015, 44, 3834–3860. [Google Scholar] [CrossRef]
- Lentz, N.; Mallet-Ladeira, S.; Baceiredo, A.; Kato, T.; Madec, D. Germylene–sulfoxide as a potential hemilabile ligand: Application in coordination chemistry. Dalton Trans. 2018, 47, 15751–15756. [Google Scholar] [CrossRef]
- Deak, N.; du Boullay, O.T.; Moraru, I.-T.; Mallet-Ladeira, S.; Madec, D.; Nemes, G. A non-symmetric sulfur-basedO,C,O-chelating pincer ligand leading to chiral germylene and stannylene. Dalton Trans. 2019, 48, 2399–2406. [Google Scholar] [CrossRef] [PubMed]
- Deak, N.; du Boullay, O.T.; Mallet-Ladeira, S.; Moraru, I.; Madec, D.; Nemes, G. Synthesis and Characterization of a Novel Bis-Sulfoxide and Its Evaluation as a Ligand in p-Block Chemistry. Eur. J. Inorg. Chem. 2020, 2020, 3729–3737. [Google Scholar] [CrossRef]
- Lentz, N.; Cuevas-Chavez, C.; Mallet-Ladeira, S.; Sotiropoulos, J.-M.; Baceiredo, A.; Kato, T.; Madec, D. Germylene-β-sulfoxide Hemilabile Ligand in Coordination Chemistry. Inorg. Chem. 2020, 60, 423–430. [Google Scholar] [CrossRef]
- Authesserre, U.; Hameury, S.; Dajnak, A.; Saffon-Merceron, N.; Baceiredo, A.; Madec, D.; Maerten, E. Complexes of Dichlorogermylene with Phosphine/Sulfoxide-Supported Carbone as Ligand. Molecules 2021, 26, 2005. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Takayama, T.; Nishio, H.; Matsushima, K.; Tanaka, Y.; Saito, S.; Sun, Y.; Oyaizu, K. Synthesis of colorless and high-refractive-index sulfoxide-containing polymers by the oxidation of poly(phenylene sulfide) derivatives. Polym. Chem. 2022, 13, 1705–1711. [Google Scholar] [CrossRef]
- Li, M.; Berritt, S.; Wang, C.; Yang, X.; Liu, Y.; Sha, S.-C.; Wang, B.; Wang, R.; Gao, X.; Li, Z.; et al. Sulfenate anions as organocatalysts for benzylic chloromethyl coupling polymerization via C=C bond formation. Nat. Commun. 2018, 9, 1754. [Google Scholar] [CrossRef]
- Kaiser, D.; Klose, I.; Oost, R.; Neuhaus, J.; Maulide, N. Bond-Forming and -Breaking Reactions at Sulfur(IV): Sulfoxides, Sulfonium Salts, Sulfur Ylides, and Sulfinate Salts. Chem. Rev. 2019, 119, 8701–8780. [Google Scholar] [CrossRef]
- Maitro, G.; Prestat, G.; Madec, D.; Poli, G. An escapade in the world of sulfenate anions: Generation, reactivity and applications in domino processes. Tetrahedron Asymmetry 2010, 21, 1075–1084. [Google Scholar] [CrossRef]
- Yang, L.; Wang, B.; Yin, X.; Zeng, Q. Advances of Sulfenate Anions in Catalytic Asymmetric Synthesis of Sulfoxides. Chem. Rec. 2021, 22, e202100242. [Google Scholar] [CrossRef]
- Riddell, A.B.; Smith, M.R.A.; Schwan, A.L. The generation and reactions of sulfenate anions. An update. J. Sulfur Chem. 2022, 43, 540–592. [Google Scholar] [CrossRef]
- Yin, X.; Zhang, Q.; Zeng, Q. Advance in the Synthesis of Sulfoxides and Sulfinamides from β-Sulfinyl Esters. Organics 2023, 4, 173–185. [Google Scholar] [CrossRef]
- Saito, F. Recent Developments on the Synthesis of Sulfoxides via Sulfenate Anions. Synthesis 2023, 56, 220–228. [Google Scholar] [CrossRef]
- Maitro, G.; Vogel, S.; Prestat, G.; Madec, D.; Poli, G. Aryl Sulfoxides via Palladium-Catalyzed Arylation of Sulfenate Anions. Org. Lett. 2006, 8, 5951–5954. [Google Scholar] [CrossRef]
- Maitro, G.; Vogel, S.; Sadaoui, M.; Prestat, G.; Madec, D.; Poli, G. Enantioselective Synthesis of Aryl Sulfoxides via Palladium-Catalyzed Arylation of Sulfenate Anions. Org. Lett. 2007, 9, 5493–5496. [Google Scholar] [CrossRef]
- Bernoud, E.; Le Duc, G.; Bantreil, X.; Prestat, G.; Madec, D.; Poli, G. Aryl Sulfoxides from Allyl Sulfoxides via [2,3]-Sigmatropic Rearrangement and Domino Pd-Catalyzed Generation/Arylation of Sulfenate Anions. Org. Lett. 2009, 12, 320–323. [Google Scholar] [CrossRef]
- Izquierdo, F.; Chartoire, A.; Nolan, S.P. Direct S-Arylation of Unactivated Arylsulfoxides Using [Pd(IPr*)(cin)Cl]. ACS Catal. 2013, 3, 2190–2193. [Google Scholar] [CrossRef]
- Gelat, F.; Lohier, J.; Gaumont, A.; Perrio, S. tert-Butyl Sulfoxides: Key Precursors for Palladium-Catalyzed Arylation of Sulfenate Salts. Adv. Synth. Catal. 2015, 357, 2011–2016. [Google Scholar] [CrossRef]
- Zhang, M.; Jia, T.; Sagamanova, I.K.; Pericás, M.A.; Walsh, P.J. tert-Butyl Phenyl Sulfoxide: A Traceless Sulfenate Anion Precatalyst. Org. Lett. 2015, 17, 1164–1167. [Google Scholar] [CrossRef] [PubMed]
- Jia, T.; Zhang, M.; Jiang, H.; Wang, C.Y.; Walsh, P.J. Palladium-Catalyzed Arylation of Alkyl Sulfenate Anions. J. Am. Chem. Soc. 2015, 137, 13887–13893. [Google Scholar] [CrossRef]
- Jiang, H.; Jia, T.; Zhang, M.; Walsh, P.J. Palladium-Catalyzed Arylation of Aryl Sulfenate Anions with Aryl Bromides under Mild Conditions: Synthesis of Diaryl Sulfoxides. Org. Lett. 2016, 18, 972–975. [Google Scholar] [CrossRef]
- Jia, T.; Bellomo, A.; Montel, S.; Zhang, M.; EL Baina, K.; Zheng, B.; Walsh, P.J. Diaryl Sulfoxides from Aryl Benzyl Sulfoxides: A Single Palladium-Catalyzed Triple Relay Process. Angew. Chem. Int. Ed. 2013, 53, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Jia, T.; Zhang, M.; Sagamanova, I.K.; Wang, C.Y.; Walsh, P.J. Palladium Catalyzed Diaryl Sulfoxide Generation from Aryl Benzyl Sulfoxides and Aryl Chlorides. Org. Lett. 2015, 17, 1168–1171. [Google Scholar] [CrossRef]
- Jia, T.; Zhang, M.; McCollom, S.P.; Bellomo, A.; Montel, S.; Mao, J.; Dreher, S.D.; Welch, C.J.; Regalado, E.L.; Williamson, R.T.; et al. Palladium-Catalyzed Enantioselective Arylation of Aryl Sulfenate Anions: A Combined Experimental and Computational Study. J. Am. Chem. Soc. 2017, 139, 8337–8345. [Google Scholar] [CrossRef]
- Wu, C.; Berritt, S.; Liang, X.; Walsh, P.J. Palladium-Catalyzed Enantioselective Alkenylation of Sulfenate Anions. Org. Lett. 2019, 21, 960–964. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Chen, M.; Zhang, P.; Li, W.; Zhang, J. Palladium/PC-Phos-Catalyzed Enantioselective Arylation of General Sulfenate Anions: Scope and Synthetic Applications. J. Am. Chem. Soc. 2018, 140, 3467–3473. [Google Scholar] [CrossRef]
- Zhang, J.; Suzuki, K.; Ohmori, K. Total Syntheses of Sparsomycin and Sparoxomycins A1 and A2 via Sulfenate-Anion-Mediated Iterative C–S Bond Formation. Org. Lett. 2023, 25, 9036–9040. [Google Scholar] [CrossRef]
- Christensen, P.R.; Patrick, B.O.; Caron, É.; Wolf, M.O. Oxidation-State-Dependent Photochemistry of Sulfur-Bridged Anthracenes. Angew. Chem. Int. Ed. 2013, 52, 12946–12950. [Google Scholar] [CrossRef] [PubMed]
- Magné, V.; Lenk, R.; Mallet-Ladeira, S.; Maerten, E.; Madec, D. Pentafluorophenyl Copper–Biarylsulfoxide Complexes: Synthesis and Photoreactivity. Molecules 2024, 29, 3332. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Li, J.; Shizu, K.; Huang, S.; Hirata, S.; Miyazaki, H.; Adachi, C. Design of Efficient Thermally Activated Delayed Fluorescence Materials for Pure Blue Organic Light Emitting Diodes. J. Am. Chem. Soc. 2012, 134, 14706–14709. [Google Scholar] [CrossRef]
- Wong, M.Y.; La-Placa, M.-G.; Pertegas, A.; Bolink, H.J.; Zysman-Colman, E. Deep-blue thermally activated delayed fluorescence (TADF) emitters for light-emitting electrochemical cells (LEECs). J. Mater. Chem. C 2017, 5, 1699–1705. [Google Scholar] [CrossRef]
- Hepguler, A.; Ulukan, P.; Catak, S. The photophysical properties of sulfone-based TADF emitters in relation to their structural properties. Phys. Chem. Chem. Phys. 2023, 25, 31457–31470. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Greenhalgh, R.P. Selective Oxidation of Phenyl Sulphides to Sulphoxides or Sulphones Using Oxone® and Wet Alumina. Synlett 1992, 1992, 235–236. [Google Scholar] [CrossRef]
- Johnson, J.A.; Zhang, X.; Reeson, T.C.; Chen, Y.-S.; Zhang, J. Facile Control of the Charge Density and Photocatalytic Activity of an Anionic Indium Porphyrin Framework via in Situ Metalation. J. Am. Chem. Soc. 2014, 136, 15881–15884. [Google Scholar] [CrossRef]
- Wright, S.W.; Hageman, D.L.; Wright, A.S.; McClure, L.D. Convenient preparations of t-butyl esters and ethers from t-butanol. Tetrahedron Lett. 1997, 38, 7345–7348. [Google Scholar] [CrossRef]
- Li, W.-X.; Yang, B.-W.; Ying, X.; Zhang, Z.-W.; Chu, X.-Q.; Zhou, X.; Ma, M.; Shen, Z.-L. Nickel-Catalyzed Direct Cross-Coupling of Diaryl Sulfoxide with Aryl Bromide. J. Org. Chem. 2022, 87, 11899–11908. [Google Scholar] [CrossRef]
- Zhu, Y.; Li, Y.; Zhang, B.; Zhang, F.; Yang, Y.; Wang, X. Palladium-Catalyzed Enantioselective C−H Olefination of Diaryl Sulfoxides through Parallel Kinetic Resolution and Desymmetrization. Angew. Chem. Int. Ed. 2018, 57, 5129–5133. [Google Scholar] [CrossRef]
- Chun, J.-H.; Morse, C.L.; Chin, F.T.; Pike, V.W. No-carrier-added [18F]fluoroarenes from the radiofluorination of diaryl sulfoxides. Chem. Commun. 2013, 49, 2151–2153. [Google Scholar] [CrossRef]
- Jeon, H.B.; Kim, K.T.; Kim, S.H. Selective oxidation of sulfides to sulfoxides with cyanuric chloride and urea–hydrogen peroxide adduct. Tetrahedron Lett. 2014, 55, 3905–3908. [Google Scholar] [CrossRef]
- Huang, M.; Wu, Z.; Krebs, J.; Friedrich, A.; Luo, X.; Westcott, S.A.; Radius, U.; Marder, T.B. Ni-Catalyzed Borylation of Aryl Sulfoxides. Chem.—A Eur. J. 2021, 27, 8149–8158. [Google Scholar] [CrossRef] [PubMed]
- Hampel, T.; Ruppenthal, S.; Sälinger, D.; Brückner, R. Desymmetrization of Prochiral Diaryl Sulfoxides by an Asymmetric Sulfoxide–Magnesium Exchange. Chem.—A Eur. J. 2012, 18, 3136–3140. [Google Scholar] [CrossRef] [PubMed]
- Bandgar, B.P.; Makone, S.S. Lithium/Sodium Perchlorate Catalyzed Synthesis of Symmetrical Diaryl Sulfoxides. Synth. Commun. 2004, 34, 743–750. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| Entry | Ligand | Base | Yield (%) b |
| 1 | Xantphos (12%) | Cs2CO3 | 48 (10) c |
| 2 | Xantphos (12%) | Cs2CO3 | n.r. d |
| 3 | SPhos (10%) | Cs2CO3 | n.r. c |
| 4 | XPhos (10%) | Cs2CO3 | n.r. c |
| 5 | DPEPhos (5%) | Cs2CO3 | n.r. c |
| 6 | CX21 (10%) e | Cs2CO3 | n.r. |
| 7 | Xantphos (5%) | Cs2CO3 | 33 (7) |
| 8 | Xantphos (2%) f | Cs2CO3 | 16 (3) |
| 9 | Xantphos (5%) | K3PO4 | 12 (4) |
| 10 | Xantphos (5%) | DIPEA | n.r. |
| 11 | Xantphos (5%) | DBU | 61 (2) |
| 12 | Xantphos (5%) | DBU | 62 (1) g |
 | ||
|---|---|---|
| Entry | Conditions | Yield (%) b |
| 1 | Pd(dba)2 (5%), Xantphos (5%), DBU, 80 °C | n.r. |
| 2 | Pd(dba)2 (5%), Xantphos (5%), Cs2CO3, 80 °C | 80 (6) |
| 3 | XantphosPdG3 (2%), Cs2CO3, 80 °C | 95 (3) |
| Compounds | λmax (nm) | Φ (%) | τ (ns/μs) |
|---|---|---|---|
| 4b | 391 | 25.7 | 0.93/94 |
| 5 | 401 | 69.1 | 2.43/111 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magné, V.; Cretoiu, I.; Mallet-Ladeira, S.; Maerten, E.; Madec, D. New Sulfenate Sources for Double Pallado-Catalyzed Cross-Coupling Reaction: Application in Symmetrical Biarylsulfoxide Synthesis, and Evidence of TADF Properties. Molecules 2024, 29, 4809. https://doi.org/10.3390/molecules29204809
Magné V, Cretoiu I, Mallet-Ladeira S, Maerten E, Madec D. New Sulfenate Sources for Double Pallado-Catalyzed Cross-Coupling Reaction: Application in Symmetrical Biarylsulfoxide Synthesis, and Evidence of TADF Properties. Molecules. 2024; 29(20):4809. https://doi.org/10.3390/molecules29204809
Chicago/Turabian StyleMagné, Valentin, Iulia Cretoiu, Sonia Mallet-Ladeira, Eddy Maerten, and David Madec. 2024. "New Sulfenate Sources for Double Pallado-Catalyzed Cross-Coupling Reaction: Application in Symmetrical Biarylsulfoxide Synthesis, and Evidence of TADF Properties" Molecules 29, no. 20: 4809. https://doi.org/10.3390/molecules29204809
APA StyleMagné, V., Cretoiu, I., Mallet-Ladeira, S., Maerten, E., & Madec, D. (2024). New Sulfenate Sources for Double Pallado-Catalyzed Cross-Coupling Reaction: Application in Symmetrical Biarylsulfoxide Synthesis, and Evidence of TADF Properties. Molecules, 29(20), 4809. https://doi.org/10.3390/molecules29204809