3. Materials and Methods
General Information.
All reactions were performed in a sealed tube with magnetic stirring. Unless otherwise stated, all commercially available reagents (innochem, Beijing, China) were used without further purification. Reactions were monitored using thin-layer chromatography (TLC), GC/MS, or LC/MS. NMR spectra were recorded on Bruker DRX-300 instruments (Bruker, Rheinstetten, Germany) and were calibrated using residual undeuterated solvent (CHCl3 at 7.26 ppm for 1H NMR and 77.16 ppm for 13C NMR). Data were reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, dd = doublet of doublets, td = triplet of doublets, qd = quartet of doublets, m = multiplet), coupling constants (Hz) and integration. High-resolution mass spectra (HRMS) were recorded on an Agilent LC/Xevo G2-XS QTOF mass spectrometer (Agilent, Palo Alto, CA, USA) using electrospray ionization time of flight reflectron experiments.
Experimental Procedure.
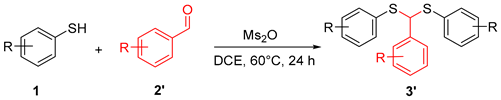
A 10 mL sealed tube was charged with substituted various thiols (0.6 mmol), methanesulfonic anhydride (Ms2O, 0.6 mmol), acetone (1 mL), and DCE (5 mL). The resulting solution was stirred at 60 °C and monitored using TLC until the reaction was complete. Saturated aqueous Na2CO3 solution (10 mL) and EtOAc (10 mL) was added to the mixture. The layers were separated, and the aqueous layer was washed with EtOAc (2 × 10 mL). Combined organic layers were washed with brine (10 mL), dried with anhydrous Na2SO4, filtered, and concentrated. The residue was purified using column chromatography on silica gel, with hexane/ethyl acetate = 20:1 as the eluent to provide the desired products 3.
A 10 mL sealed tube was charged with thiols (0.6 mmol), methanesulfonic anhydride (Ms2O, 0.6 mmol), 2′ (0.9 mmol), and DCE (5 mL). The resulting solution was stirred at 60 °C and monitored by TLC until the reaction was complete. Saturated aqueous Na2CO3 solution (10 mL) and EtOAc (10 mL) were added to the resulting mixture. The layers were separated, and the aqueous layer was washed with EtOAc (2 × 10 mL). The combined organic layers were washed with brine (10 mL), dried with anhydrous Na2SO4, filtered, and concentrated. The residue was purified using column chromatography on silica gel, with hexane/ethyl acetate = 50:1 as the eluent to provide the desired products 3′.
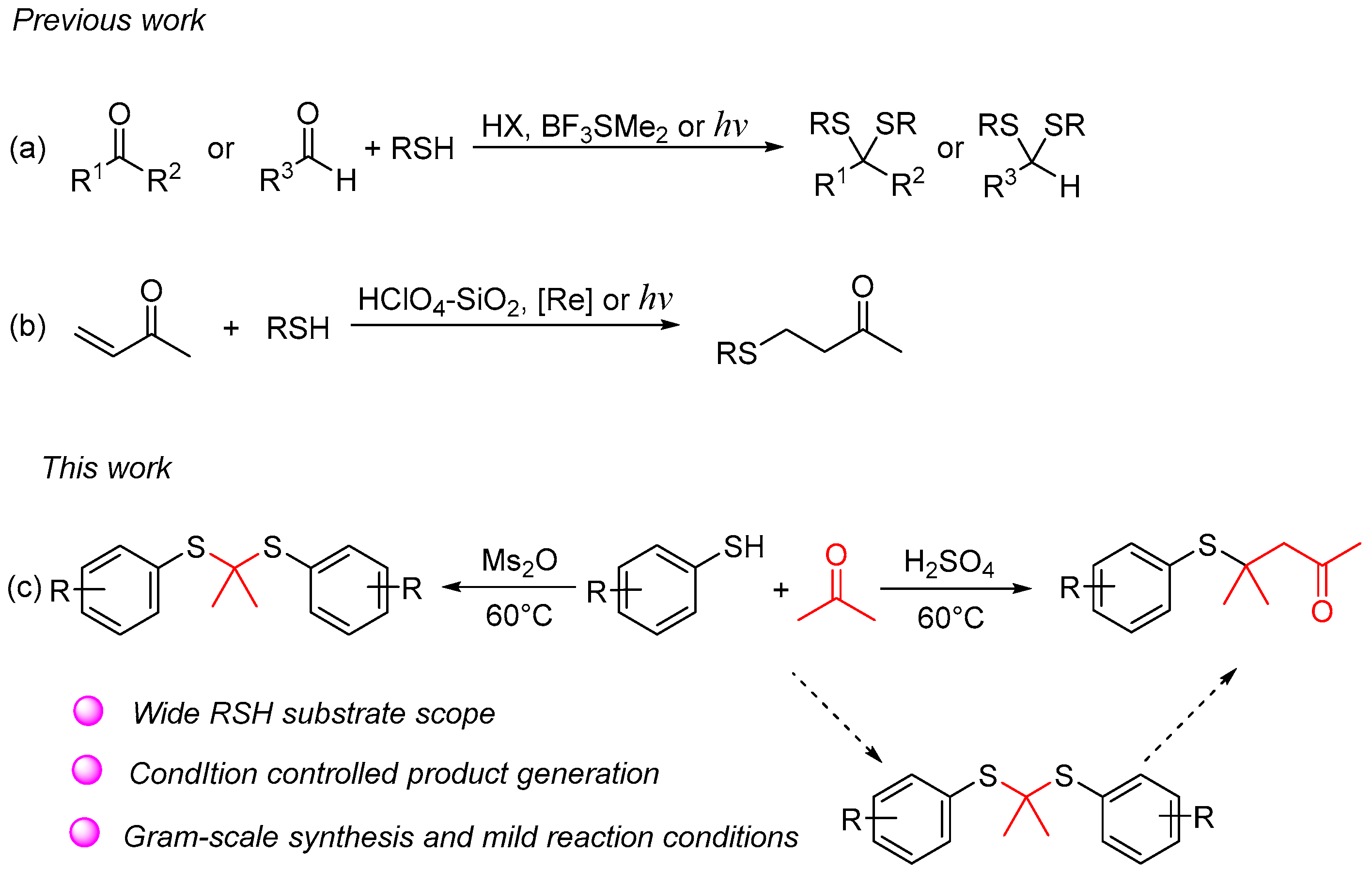
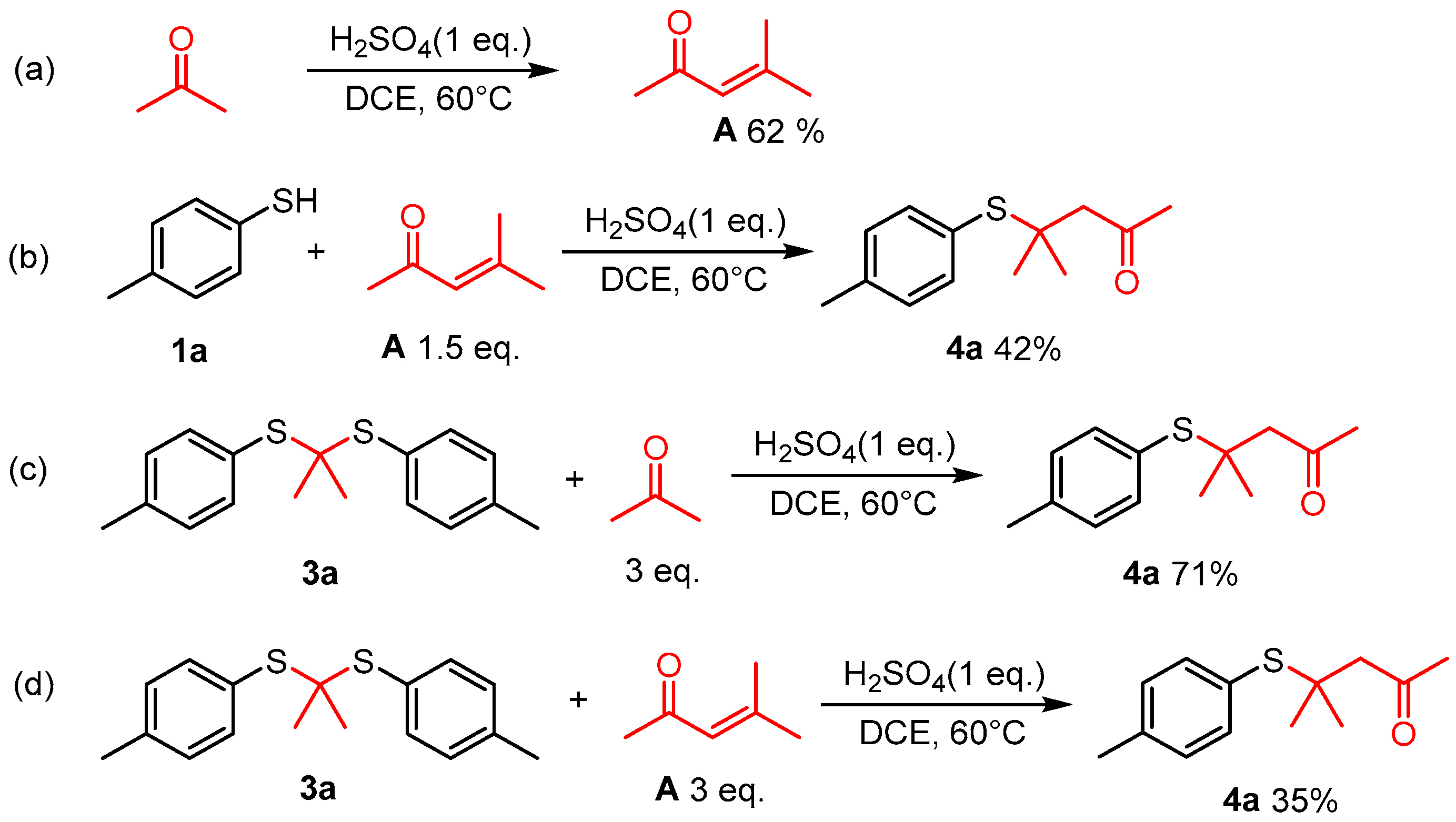
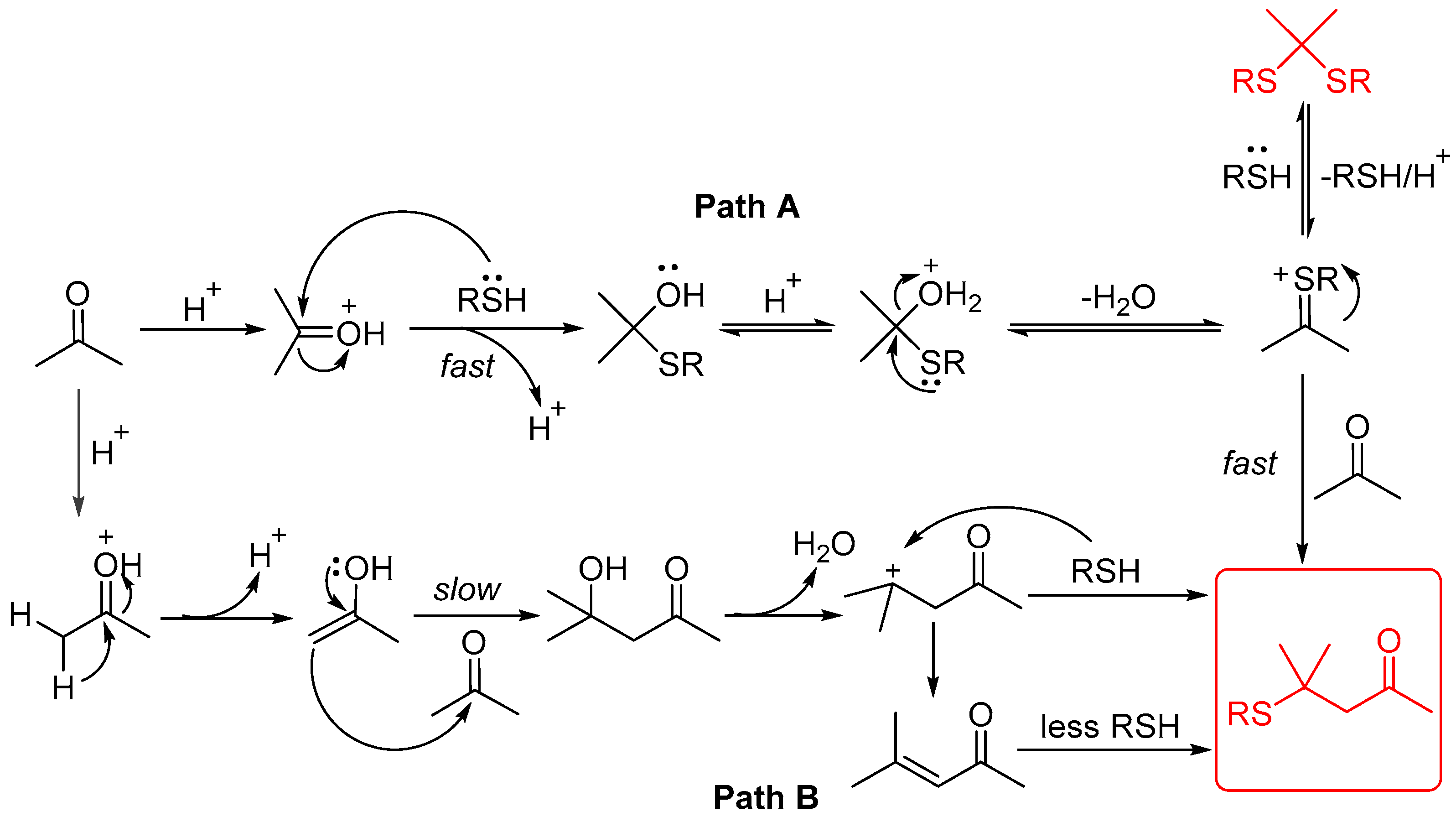
A 10 mL sealed tube was charged with thiols (0.6 mmol), H2SO4 (0.6 mmol), acetone (1 mL), and DCE (5 mL). The resulting solution was stirred at 60 °C and monitored using TLC until the reaction was complete. Saturated aqueous Na2CO3 solution (10 mL) and EtOAc (10 mL) were added to the resulting mixture. The layers were separated, and the aqueous layer was washed with EtOAc (2 × 10 mL). The combined organic layers were washed with brine (10 mL), dried with anhydrous Na2SO4, filtered, and concentrated. The residue was purified using column chromatography on silica gel, with hexane/ethyl acetate = 10:1 as the eluent to provide the desired products 4.
The gram-scale experiment procedure. A 250 mL round-bottomed flask was charged with 4-methylbenzenethiol (
1a) (10 mmol,1.2432 g), H
2SO
4 (10 mmol), acetone (16 mL), and DCE (84 mL). The bottle was sealed with a rubber stopper, and the resulting solution was stirred at 60 °C for 24 h. Saturated aqueous Na
2CO
3 solution (100 mL) and EtOAc (100 mL) were added to the resulting mixture. The layers were separated, and the aqueous layer was washed with EtOAc (2 × 100 mL). The combined organic layers were washed with brine (100 mL), dried with anhydrous Na
2SO
4, filtered, and concentrated. The residue was purified using column chromatography on silica gel, with hexane/ethyl acetate = 10:1 as the eluent to provide the desired product
4a. Characterization data for the products can be found in the
Supplementary Materials, along with references of previous reports which provide support for the identities of the products [
20,
26,
27,
28,
29,
30,
31].
Propane-2,2-diylbis(
p-tolylsulfane) (
3a) [
26]. White solid, 76% yield.
1H NMR (300 MHz, CDCl
3) δ 7.53 (d,
J = 8.0 Hz, 4H), 7.16 (d,
J = 7.9 Hz, 4H), 2.37 (s, 6H), 1.49 (s, 6H).
13C NMR (75 MHz, CDCl
3) δ 139.37, 137.16, 129.48, 128.92, 59.24, 30.77, 21.43.
Propane-2,2-diylbis(m-tolylsulfane) (3b). White solid, 61% yield. 1H NMR (300 MHz, Chloroform-d) δ 7.45 (d, J = 6.5 Hz, 4H), 7.20 (dd, J = 12.1, 7.7 Hz, 4H), 2.35 (s, 6H), 1.51 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 138.35, 137.66, 134.08, 132.14, 129.98, 128.44, 59.34, 30.99, 21.42.
Propane-2,2-diylbis((4-methoxyphenyl)sulfane) (3c). White solid, 67% yield. 1H NMR (300 MHz, CDCl3) δ 7.54 (d, J = 8.6 Hz, 4H), 6.88 (d, J = 8.7 Hz, 4H), 3.82 (s, 6H), 1.45 (s, 6H). 13C NMR (75 MHz, CDCl3) δ = 160.62, 138.82, 123.28, 114.14, 59.33, 55.41, 30.52.
Propane-2,2-diylbis((4-isopropylphenyl)sulfane) (3d). White solid, 45% yield. 1H NMR (300 MHz, Chloroform-d) δ 7.56 (d, J = 8.1 Hz, 4H), 7.21 (d, J = 8.0 Hz, 4H), 2.93 (hept, J = 6.8 Hz, 2H), 1.50 (s, 6H), 1.26 (d, J = 6.9 Hz, 12H). 13C NMR (75 MHz, CDCl3) δ 150.14, 137.19, 129.31, 126.83, 59.51, 34.02, 30.88, 24.01.
Propane-2,2-diylbis(benzylsulfane) (3e). White solid, 66% yield. 1H NMR (300 MHz, CDCl3) δ 7.34 (dt, J = 14.4, 7.5 Hz, 10H), 3.93 (s, 4H), 1.67 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 137.87, 129.23, 128.62, 127.03, 57.30, 35.19, 30.85.
Propane-2,2-diylbis((4-methylbenzyl)sulfane) (3f). White solid, 63% yield. 1H NMR (300 MHz, CDCl3) δ 7.45 (d, J = 7.8 Hz, 4H), 7.33 (d, J = 7.8 Hz, 4H), 4.07 (s, 4H), 2.54 (s, 6H), 1.83 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 136.62, 134.72, 129.31, 129.12, 57.18, 34.86, 30.84, 21.22.
Propane-2,2-diylbis((4-methoxybenzyl)sulfane) (3g). White solid, 67% yield. 1H NMR (300 MHz, CDCl3) δ 7.37 (d, J = 8.5 Hz, 4H), 6.95 (d, J = 8.5 Hz, 4H), 3.95 (s, 4H), 3.89 (s, 6H), 1.72 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 158.66, 130.28, 129.71, 114.03, 57.04, 55.36, 34.52, 30.85.
(Phenylmethylene)bis(
p-tolylsulfane) (
3′a) [
27]. White solid, 79% yield.
1H NMR (300 MHz, CDCl
3) δ 7.30 (dd,
J = 7.6, 1.7 Hz, 2H), 7.25–7.20 (m, 7H), 7.02 (d,
J = 7.9 Hz, 4H), 5.29 (s, 1H), 2.27 (s, 6H).
13C NMR (75 MHz, CDCl
3) δ 140.08, 138.13, 133.25, 130.95, 129.69, 128.48, 128.00, 61.39, 21.31.
(p-Tolylmethylene)bis(p-tolylsulfane) (3′b). White solid, 75% yield. 1H NMR (300 MHz, CDCl3) δ 7.35 (d, J = 8.0 Hz, 6H), 7.16 (t, J = 7.9 Hz, 6H), 5.43 (s, 1H), 2.42 (s, 3H), 2.40 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 137.91, 137.69, 137.14, 133.03, 131.29, 129.64, 129.17, 127.85, 61.18, 21.25.
(m-Tolylmethylene)bis(p-tolylsulfane) (3′c). White solid, 74% yield. 1H NMR (300 MHz, CDCl3) δ 7.29 (d, J = 8.1 Hz, 4H), 7.22–7.15 (m, 3H), 7.09 (d, J = 7.9 Hz, 5H), 5.34 (s, 1H), 2.34 (s,9H). 13C NMR (75 MHz, CDCl3) δ 139.91, 138.13, 138.00, 133.12, 131.10, 129.63, 128.78, 128.56, 128.31, 125.00, 61.44, 21.47, 21.26.
((3, 5-Dimethylphenyl)methylene)bis(p-tolylsulfane) (3′d). White solid, 77% yield. 1H NMR (300 MHz, CDCl3) δ 7.29 (d, J = 8.1 Hz, 4H), 7.09 (d, J = 7.9 Hz, 4H), 7.00 (s, 2H), 6.91 (s, 1H), 5.31 (s, 1H), 2.34 (s, 6H), 2.30 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 139.82, 137.98, 137.92, 133.05, 131.27, 129.73, 129.61, 125.64, 61.54, 21.35, 21.26.
((4-Ethylphenyl)methylene)bis(p-tolylsulfane) (3′e). White solid, 76% yield. 1H NMR (300 MHz, CDCl3) δ 7.31 (t, J = 8.5 Hz, 6H), 7.12 (dd, J = 16.9, 8.0 Hz, 6H), 5.38 (s, 1H), 2.66 (q, J = 7.6 Hz, 2H), 2.34 (s, 6H), 1.26 (t, J = 7.6 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 144.03, 137.89, 137.24, 132.99, 131.21, 129.62, 127.96, 127.84, 61.17, 28.61, 21.25, 15.52.
((4-Methoxyphenyl)methylene)bis(p-tolylsulfane) (3′f). White solid, 74% yield. 1H NMR (300 MHz, CDCl3) δ 7.24–7.20 (m, 6H), 7.03 (d, J = 7.9 Hz, 4H), 6.77 (d, J = 8.7 Hz, 2H), 5.29 (s, 1H), 3.77 (s, 3H), 2.29 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 159.27, 138.02, 133.12, 132.14, 131.15, 129.68, 129.22, 113.84, 60.70, 55.40, 21.31.
((4-Fluorophenyl)methylene)bis(p-tolylsulfane) (3′g). White solid, 87% yield. 1H NMR (300 MHz, CDCl3) δ 7.39–7.27 (m, 6H), 7.10 (d, J = 8.1 Hz, 4H), 6.97 (t, J = 8.6 Hz, 2H), 5.38 (s, 1H), 2.35 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 162.20 (d, J = 246.9 Hz), 138.27, 135.85 (d, J = 3.2 Hz), 133.35, 130.53, 129.72, 129.61, 115.28 (d, J = 21.7 Hz), 60.42, 21.25. 19F NMR (282 MHz, CDCl3) δ -113.83 (tt, J = 8.6, 5.3 Hz).
((4-Chlorophenyl)methylene)bis(p-tolylsulfane) (3′h). White solid, 88% yield. 1H NMR (300 MHz, CDCl3) δ 7.36–7.27 (m, 8H), 7.12 (d, J = 7.9 Hz, 4H), 5.37 (s, 1H), 2.37 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 138.62, 138.32, 133.51, 133.36, 130.38, 129.74, 129.31, 128.53, 60.51, 21.25.
((4-Bromophenyl)methylene)bis(p-tolylsulfane) (3′i). White solid, 87% yield. 1H NMR (300 MHz, CDCl3) δ 7.41 (d, J = 8.5 Hz, 2H), 7.26 (m, 6H), 7.10 (d, J = 8.0 Hz, 4H), 5.33 (s, 1H), 2.35 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 139.14, 138.34, 133.36, 131.49, 130.34, 129.75, 129.63, 121.72, 60.58, 21.28.
4-(Bis(p-tolylthio)methyl)benzonitrile (3′j). White solid, 86% yield. 1H NMR (300 MHz, CDCl3) δ 7.44 (d, J = 8.2 Hz, 2H), 7.31 (d, J = 8.2 Hz, 2H), 7.18 (d, J = 8.0 Hz, 4H), 7.01 (d, J = 8.0 Hz, 4H), 5.27 (s, 1H), 2.25 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 145.29, 138.64, 133.54, 132.02, 129.74, 129.50, 128.56, 118.57, 111.30, 60.55, 21.15.
4-(Bis(p-tolylthio)methyl)benzaldehyde (3′k). White solid, 86% yield. 1H NMR (300 MHz, CDCl3) δ 9.70 (s, 1H), 7.51 (d, J = 8.1 Hz, 2H), 7.21 (d, J = 8.1 Hz, 2H), 7.01 (d, J = 8.0 Hz, 4H), 6.81 (d, J = 7.9 Hz, 4H), 5.12 (s, 1H), 2.06 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 191.66, 146.82, 138.53, 135.66, 133.51, 129.87, 129.74, 128.54, 60.90, 21.19.
((4-Chlorophenyl)methylene)bis((4-methoxyphenyl)sulfane) (3′l). White solid, 72% yield. 1H NMR (300 MHz, CDCl3) δ 7.73–7.66 (m, 4H), 7.65–7.53 (m, 4H), 7.23–7.17 (m, 4H), 5.54 (s, 1H), 4.19 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 160.16, 138.83, 136.15, 133.45, 129.34, 128.50, 124.33, 114.50, 62.11, 55.39.
Dimethyl 4,4’-((phenylmethylene)bis(sulfanediyl))dibenzoate (3′m). White solid, 75% yield. 1H NMR (300 MHz, CDCl3) δ 7.89 (d, J = 8.5 Hz, 4H), 7.46 (dd, J = 7.7, 1.5 Hz, 2H), 7.35 (d, J = 8.5 Hz, 4H), 7.29 (d, J = 7.4 Hz, 2H), 5.69 (s, 1H), 3.87 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 166.49, 140.94, 138.13, 130.01, 129.97, 128.85, 128.71, 128.66, 127.93, 57.59, 52.18.
4-(Bis((4-bromophenyl)thio)methyl)benzonitrile (3′n). White solid, 75% yield. 1H NMR (300 MHz, CDCl3) δ 7.55 (d, J = 8.3 Hz, 2H), 7.38 (d, J = 8.4 Hz, 6H), 7.17 (dd, J = 8.8, 2.1 Hz, 4H), 5.33 (s, 1H). 13C NMR (75 MHz, CDCl3) δ 144.25, 134.79, 132.43, 132.30, 131.97, 128.59, 123.20, 118.39, 112.07, 60.03.
(p-tolylmethylene)bis((4-nitrophenyl)sulfane) (3′o). White solid, 70% yield. 1H NMR (300 MHz, CDCl3) δ 8.08 (d, J = 8.8 Hz, 4H), 7.42 (dd, J = 8.3, 6.5 Hz, 6H), 7.17 (d, J = 7.9 Hz, 2H), 5.80 (s, 1H), 2.34 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 146.34, 144.20, 139.42, 133.67, 129.97, 129.36, 127.88, 124.05, 56.18, 21.29.
(p-tolylmethylene)bis(naphthalen-2-ylsulfane) (3′p). White solid, 71% yield. 1H NMR (300 MHz, CDCl3) δ 7.86 (s, 2H), 7.78 (dd, J = 5.8, 3.5 Hz, 2H), 7.74–7.65 (m, 4H), 7.52–7.34 (m, 8H), 7.12 (d, J = 7.9 Hz, 2H), 5.69 (s, 1H), 2.34 (s, 3H). 13C NMR (75 MHz, CDCl3) δ 138.11, 136.69, 133.61, 132.62, 132.25, 131.51, 129.57, 129.42, 128.44, 127.90, 127.77, 127.69, 126.54, 126.43, 60.34, 21.31, 1.16.
(Phenylmethylene)bis(benzylsulfane) (
3′q) [
28]. White solid, 73% yield.
1H NMR (300 MHz, CDCl
3) δ 7.32–7.10 (m, 11H), 7.09–7.00 (m, 4H), 4.38 (s, 1H), 3.68 (d,
J = 13.4 Hz, 2H), 3.45 (d,
J = 13.4 Hz, 2H).
13C NMR (75 MHz, CDCl
3) δ 139.64, 137.81, 129.05, 128.68, 128.57, 128.09, 128.03, 127.07, 50.96, 36.61.
4-Methyl-4-(
p-tolylthio)pentan-2-one (
4a) [
20]. Oil liquid, 86% yield.
1H NMR (300 MHz, CDCl
3) δ 7.39 (d,
J = 8.0 Hz, 2H), 7.14 (d,
J = 7.9 Hz, 2H), 2.64 (s, 2H), 2.34 (s, 3H), 2.13 (s, 3H), 1.36 (s, 6H).
13C NMR (75 MHz, CDCl
3) δ 206.75, 139.31, 137.62, 129.54, 127.92, 54.41, 46.91, 32.26, 28.13, 21.30. HRMS (ESI-MS) [M + Na]
+: found 245.0985; calculated for C
13H
18NaOS
+: 245.0971.
4-Methyl-4-(
m-tolylthio)pentan-2-one (
4b) [
29]. Oil liquid, 84% yield.
1H NMR (300 MHz, CDCl
3) δ 7.38–7.26 (m, 2H), 7.20 (m,
J = 17.9, 7.3 Hz, 2H), 2.65 (s, 2H), 2.34 (s, 3H), 2.13 (s, 3H), 1.38 (s, 6H).
13C NMR (75 MHz, CDCl
3) δ 206.66, 138.39, 138.22, 134.61, 131.12, 129.91, 128.47, 54.43, 46.94, 32.21, 28.23, 21.27. HRMS (ESI-MS) [M + Na]
+: found 245.0982; calculated for C
13H
18NaOS
+: 245.0971.
4-Methyl-4-(
o-tolylthio)pentan-2-one (
4c) [
29]. Oil liquid, 78% yield.
1H NMR (300 MHz, CDCl
3) δ 7.54 (d,
J = 7.6 Hz, 1H), 7.34–7.25 (m, 2H), 7.23–7.13 (m, 1H), 2.75 (s, 2H), 2.54 (s, 3H), 2.17 (s, 3H), 1.42 (s, 6H).
13C NMR (75 MHz, CDCl3) δ 206.66, 144.03, 138.99, 130.99, 130.66, 129.32, 125.97, 54.63, 48.56, 32.27, 28.22, 21.87. HRMS (ESI-MS) [M + Na]
+: found 245.0984; calculated for C
13H
18NaOS
+: 245.0971.
4-((4-Isopropylphenyl)thio)-4-methylpentan-2-one (4d). Oil liquid, 77% yield. 1H NMR (300 MHz, CDCl3) δ 7.42 (d, J = 8.1 Hz, 2H), 7.18 (d, J = 8.1 Hz, 2H), 2.89 (dt, J = 13.8, 6.9 Hz, 1H), 2.65 (s, 2H), 2.13 (s, 3H), 1.36 (s, 6H), 1.23 (d, J = 6.9 Hz, 6H). 13C NMR (75 MHz, CDCl3) δ 206.81, 150.07, 137.69, 128.23, 126.87, 54.45, 46.93, 33.86, 32.26, 28.15, 23.90. HRMS (ESI-MS) [M + Na]+: found 273.1290; calculated for C15H22NaOS+: 273.1284.
4-((2,4-Dimethylphenyl)thio)-4-methylpentan-2-one (4e). Oil liquid, 89% yield. 1H NMR (300 MHz, CDCl3) δ 7.39 (d, J = 7.8 Hz, 1H), 7.11 (s, 1H), 6.97 (d, J = 7.7 Hz, 1H), 2.71 (s, 2H), 2.47 (s, 3H), 2.32 (s, 3H), 2.15 (s, 3H), 1.37 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 206.94, 143.90, 139.50, 139.06, 131.58, 127.59, 126.92, 54.73, 48.46, 32.37, 28.21, 21.84, 21.25. HRMS (ESI-MS) [M + Na]+: found 259.1136; calculated for C14H20NaOS+: 259.1127.
4-((4-Methoxyphenyl)thio)-4-methylpentan-2-one (
4f) [
20]. Oil liquid, 87% yield.
1H NMR (300 MHz, CDCl
3) δ 7.38 (d,
J = 8.6 Hz, 2H), 6.82 (d,
J = 8.6 Hz, 2H), 3.75 (s, 3H), 2.59 (s, 2H), 2.09 (s,3H), 1.31 (s, 6H).
13C NMR (75 MHz, CDCl
3) δ 206.71, 160.47, 139.01, 122.19, 114.15, 55.24, 54.22, 46.74, 32.16, 27.94. HRMS (ESI-MS) [M + Na]
+: found 261.0920; calculated for C
13H
18NaO
2S
+: 261.0920.
4-((4-Fluorophenyl)thio)-4-methylpentan-2-one (
4g) [
30]. Oil liquid, 76% yield.
1H NMR (300 MHz, CDCl
3) δ 7.67–7.33 (m, 2H), 7.01 (t,
J = 8.5 Hz, 2H), 2.62 (s, 2H), 2.12 (s, 3H), 1.34 (s, 6H).
13C NMR (75 MHz, CDCl
3) δ 206.53 (s), 163.68 (d,
J = 249.7 Hz), 139.63 (d,
J = 8.4 Hz), 126.94 (d,
J = 3.5 Hz), 115.93 (d,
J = 21.6 Hz), 54.39, 47.20 (d,
J = 1.3 Hz), 32.28, 28.18.
19F NMR (282 MHz, Chloroform-
d) δ -111.91 (tt,
J = 8.6, 5.5 Hz). HRMS (ESI-MS) [M + Na]
+: found 249.0726;calculated for C
12H
15FNaOS
+: 249.0720.
4-((4-Chlorophenyl)thio)-4-methylpentan-2-one (
4h) [
20]. Oil liquid, 85% yield.
1H NMR (300 MHz, CDCl
3) δ 7.43 (d,
J = 8.4 Hz, 2H), 7.30 (d,
J = 8.4 Hz, 2H), 2.63 (s, 2H), 2.12 (s, 3H), 1.36 (s, 6H).
13C NMR (75 MHz, CDCl
3) δ 206.35, 138.89, 135.74, 130.09, 128.99, 54.39, 47.45, 32.23, 28.24. HRMS (ESI-MS) [M + Na]
+: found 265.0429; calculated for C
12H
15ClNaOS
+: 265.0424.
4-((4-Bromophenyl)thio)-4-methylpentan-2-one (
4i) [
20]. Oil liquid, 79% yield.
1H NMR (300 MHz, CDCl
3) δ 7.45 (d,
J = 8.4 Hz, 2H), 7.35 (d,
J = 8.4 Hz, 2H), 2.62 (s, 2H), 2.11 (s, 3H), 1.35 (s, 6H).
13C NMR (75 MHz, CDCl
3) δ 206.32, 139.13, 131.93, 130.63, 124.02, 54.32, 47.40, 32.21, 28.21. HRMS (ESI-MS) [M + Na]
+: found 308.9923; calculated for C
12H
15BrNaOS
+: 308.9919.
4-((4-Iodophenyl)thio)-4-methylpentan-2-one (4j). Oil liquid, 76% yield. 1H NMR (300 MHz, CDCl3) δ 7.64 (d, J = 8.2 Hz, 2H), 7.21 (d, J = 8.2 Hz, 2H), 2.62 (s, 2H), 2.11 (s, 3H), 1.34 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 206.26, 139.25, 137.85, 131.32, 95.94, 54.27, 47.38, 32.20, 28.18. HRMS (ESI-MS) [M + Na]+: found 356.9786; calculated for C12H15INaOS+: 356.9780.
4-Methyl-4-((4-(trifluoromethyl)phenyl)thio)pentan-2-one (4k). Oil liquid, 63% yield. 1H NMR (300 MHz, CDCl3) δ 7.62 (q, J = 8.4 Hz, 4H), 2.68 (s, 2H), 2.15 (s, 3H), 1.40 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 206.28, 137.77, 136.40, 131.19 (q, J = 32.7 Hz), 125.59 (q, J = 3.6 Hz). 124.05 (q, J = 270.75 Hz), 54.46, 48.00, 32.25, 28.42. 19F NMR (282 MHz, CDCl3) δ = −62.74. HRMS (ESI-MS) [M + Na]+: found 299.0678; calculated for C13H15F3NaOS+: 299.0688.
4-Methyl-4-((4-nitrophenyl)thio)pentan-2-one (
4l) [
20]. Oil liquid, 68% yield.
1H NMR (300 MHz, CDCl
3) δ 8.18 (d,
J = 8.7 Hz, 2H), 7.68 (d,
J = 8.7 Hz, 2H), 2.71 (s,2H), 2.15 (s, 3H), 1.44 (s, 6H).
13C NMR (75 MHz, CDCl
3) δ 205.88, 148.19, 140.87, 137.59, 123.61, 54.47, 48.80, 32.21, 28.61. HRMS (ESI-MS) [M + Na]
+: found 276.0668; calculated for C
12H
15NNaO
3S
+: 276.0665.
Methyl 4-((2-methyl-4-oxopentan-2-yl)thio)benzoate (4m). Oil liquid, 69% yield. 1H NMR (300 MHz, CDCl3) δ 7.92 (d, J = 8.1 Hz, 2H), 7.52 (d, J = 8.1 Hz, 2H), 3.84 (s, 3H), 2.61 (s, 2H), 2.06 (s, 3H), 1.33 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 206.10, 166.44, 137.51, 137.08, 130.45, 129.56, 54.27, 52.22, 47.84, 32.06, 28.29. HRMS (ESI-MS) [M + Na]+: found 289.0872; calculated for C14H18NaO3S+: 289.0869.
4-(Benzylthio)-4-methylpentan-2-one (4n). Oil liquid, 85% yield. 1H NMR (300 MHz, CDCl3) δ 7.46–7.27 (m, 5H), 3.84 (s, 2H), 2.74 (s, 2H), 2.19 (s, 3H), 1.52 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 206.76, 137.99, 129.03, 128.57, 127.00, 54.56, 44.32, 33.32, 32.28, 28.49. HRMS (ESI-MS) [M + Na]+: found 245.0978; calculated for C13H18NaOS+: 245.0971.
4-Methyl-4-((4-methylbenzyl)thio)pentan-2-one (4o). Oil liquid, 79% yield. 1H NMR (300 MHz, CDCl3) δ 7.22 (d, J = 7.9 Hz, 2H), 7.10 (d, J = 7.8 Hz, 2H), 3.75 (s, 2H), 2.69 (s, 2H), 2.31 (s, 3H), 2.14 (s, 3H), 1.46 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 206.67, 136.47, 134.72, 129.17, 128.83, 54.47, 44.16, 32.89, 32.20, 28.42, 21.04. HRMS (ESI-MS) [M + Na]+: found 259.1130; calculated for C14H20NaOS+: 259.1127.
4-((4-Methoxybenzyl)thio)-4-methylpentan-2-one (
4p) [
31]. Oil liquid, 52% yield.
1H NMR (300 MHz, CDCl
3) δ 7.33 (d,
J = 8.6 Hz, 2H), 6.91 (d,
J = 8.6 Hz, 2H), 3.86 (s, 3H), 3.82 (s, 2H), 2.77 (s, 2H), 2.24 (s, 3H), 1.53 (s, 6H).
13C NMR (75 MHz, CDCl
3) δ 206.95, 158.66, 130.14, 129.79, 114.04, 55.35, 54.68, 44.30, 32.70, 32.40, 28.56. HRMS (ESI-MS) [M + Na]
+: found 275.0924; calculated for C
14H
20NaO
2S
+: 275.1076.
4-((4-Chlorobenzyl)thio)-4-methylpentan-2-one (
4q) [
31]. Oil liquid, 85% yield.
1H NMR (300 MHz, CDCl
3) δ 7.29 (s,4H), 3.77 (s, 2H), 2.72 (s, 2H), 2.18 (s, 3H), 1.47 (s, 6H).
13C NMR (75 MHz, CDCl
3) δ 206.52, 136.56, 132.65, 130.35, 128.63, 54.54, 44.43, 32.58, 32.24, 28.48. HRMS (ESI-MS) [M + Na]
+: found 279.0586; calculated for C
13H
17ClNaOS
+: 279.0581.
4-(Butylthio)-4-methylpentan-2-one (4r). Oil liquid, 36% yield. 1H NMR (300 MHz, CDCl3) δ 2.66 (s, 2H), 2.51 (t, J = 7.3 Hz, 2H), 2.16 (s, 3H), 1.59–1.45 (m, 2H), 1.45–1.29 (m, 8H), 0.89 (t, J = 7.2 Hz, 3H). 13C NMR (75 MHz, CDCl3) δ 207.10, 54.73, 43.35, 32.46, 31.64, 28.57, 27.86, 22.43, 13.81. HRMS (ESI-MS) [M + Na]+: found 211.1126; calculated for C10H20NaOS+: 211.1127.
4-(Cyclohexylthio)-4-methylpentan-2-one (4s). Oil liquid, 41% yield. 1H NMR (300 MHz, CDCl3) δ 2.67 (s, 2H), 2.66–2.57 (m, 1H), 2.15 (s, 3H), 1.91 (d, J = 9.0 Hz, 2H), 1.73–1.64 (m, 2H), 1.56–1.49 (m, 1H), 1.39 (s, 6H), 1.35–1.09 (m, 5H).13C NMR (75 MHz, CDCl3) δ 207.05, 55.50, 44.70, 41.22, 36.24, 32.54, 29.07, 26.44, 25.52. HRMS (ESI-MS) [M + Na]+: found 237.1288; calculated for C12H22NaOS+: 237.1284.
4-Methyl-4-(naphthalen-2-ylthio)pentan-2-one (
4t) [
29]. Oil liquid, 85% yield.
1H NMR (300 MHz, CDCl
3) δ 8.08 (s, 1H), 7.89–7.73 (m, 3H), 7.59 (dd,
J = 8.4, 1.4 Hz, 1H), 7.51 (dd,
J = 6.2, 3.3 Hz, 2H), 2.72 (s, 2H), 2.14 (s, 3H), 1.46 (s, 6H).
13C NMR (75 MHz, CDCl
3) δ 206.52, 137.45, 134.17, 133.32, 133.16, 128.85, 128.05, 127.91, 127.61, 126.94, 126.43, 54.38, 47.51, 32.16, 28.30. HRMS (ESI-MS) [M + Na]
+: found 281.0980; calculated for C
16H
18NaOS
+: 281.0971.
4-Methyl-4-(thiophen-2-ylthio)pentan-2-one (4u). Oil liquid, 21% yield. 1H NMR (300 MHz, CDCl3) δ 7.45 (dd, J = 5.4, 0.9 Hz, 1H), 7.17 (dd, J = 3.4, 0.9 Hz, 1H), 7.06 (dd, J = 5.3, 3.6 Hz, 1H), 2.70 (s, 2H), 2.16 (s, 3H), 1.41 (s, 6H). 13C NMR (75 MHz, CDCl3) δ 206.68, 137.63, 131.52, 130.63, 127.87, 54.04, 48.05, 32.30, 27.8 HRMS (ESI-MS) [M + Na]+: found 237.0383; calculated for C10H14NaOS2+: 237.0378.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}