
A Computational Study of Heteroatom Analogues of Selenoxide and Selenone syn Eliminations


Abstract

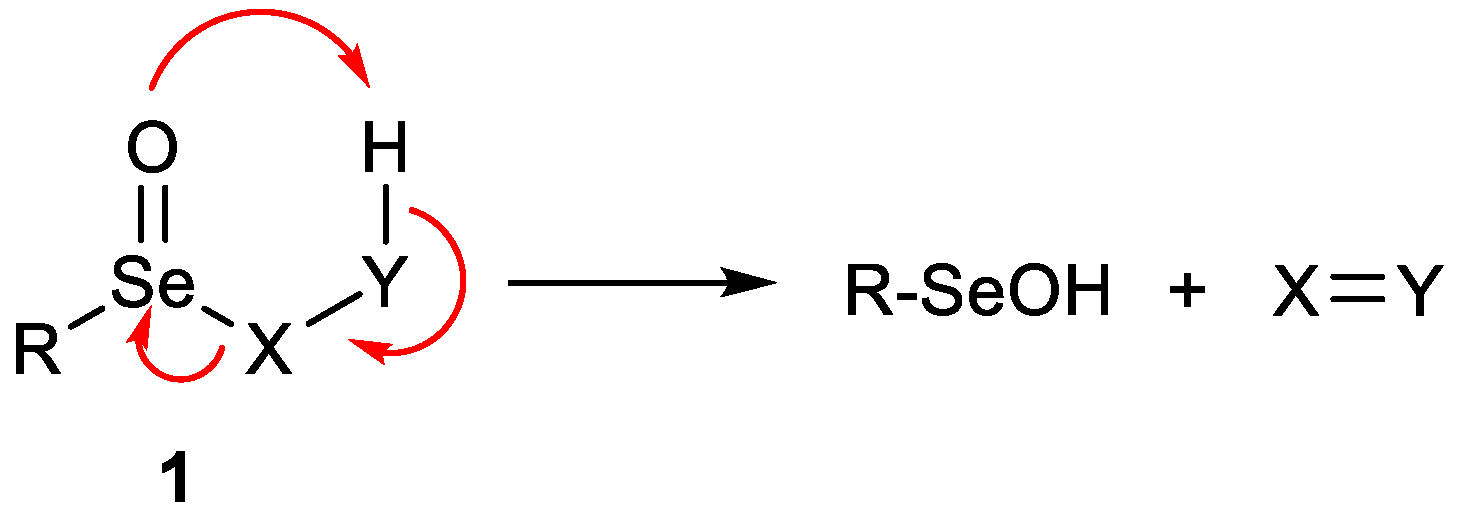
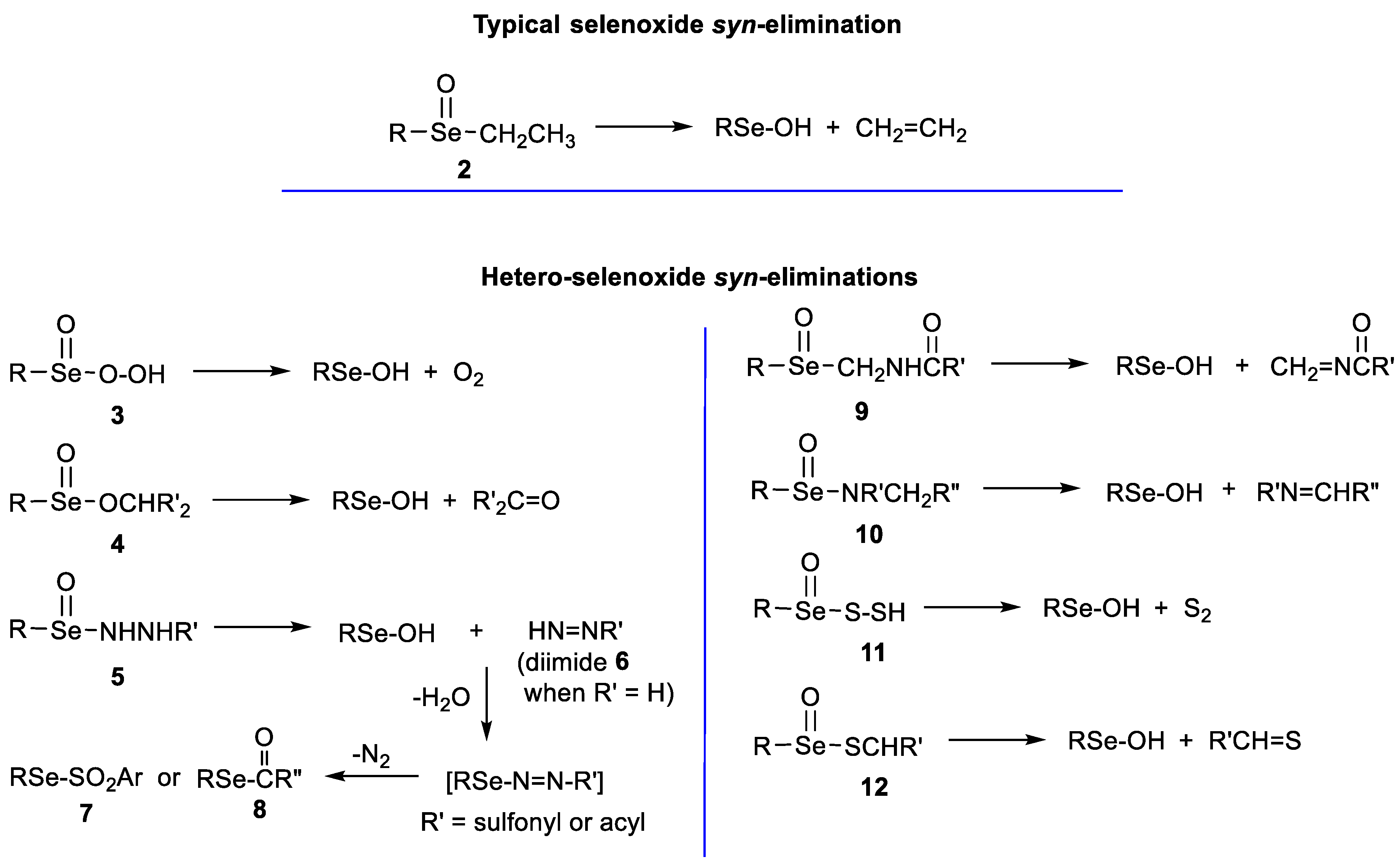
1. Introduction
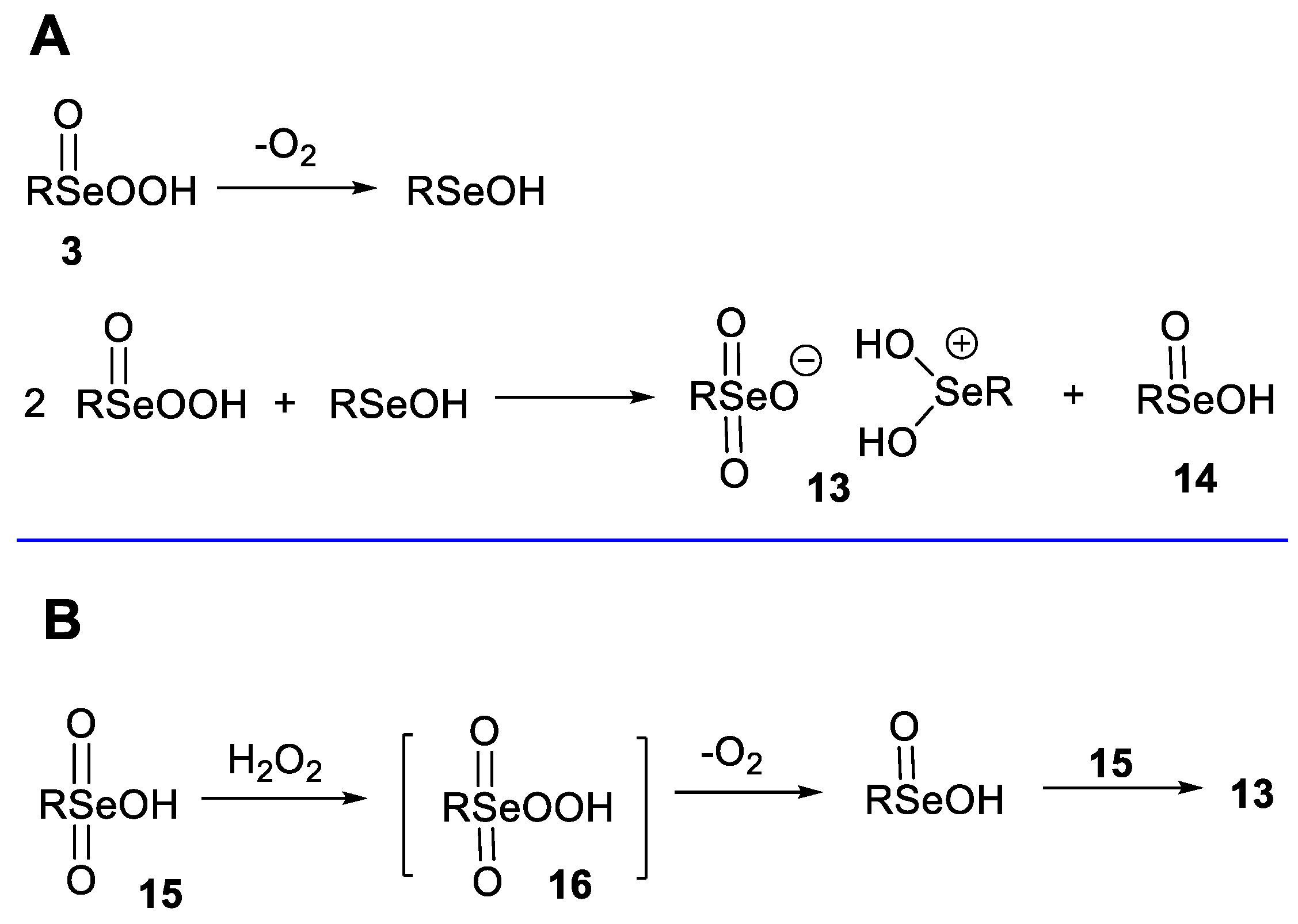
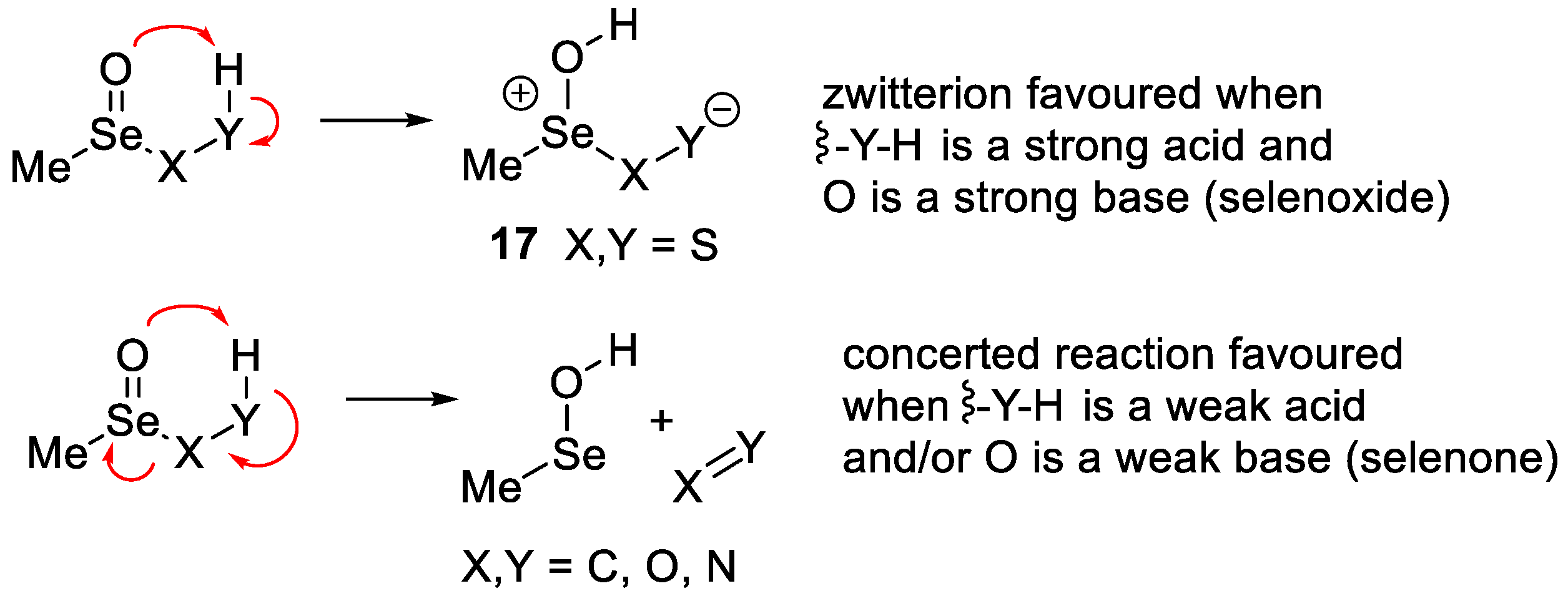
2. Results
3. Methods
4. Conclusions and Summary
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References and Notes
- In principle, concerted reactions may be synchronous or asynchronous. Both types proceed without the formation of intermediates, but bonding changes in asynchronous processes do not occur simultaneously. For example, see Tantillo, D.J. Recent Excursions to the Lands between Concerted and Stepwise: From Natural Products Biosynthesis to Reaction Design. J. Phys. Org. Chem. 2008, 21, 561–570. [Google Scholar] [CrossRef] To avoid these distinctions regarding the timing of events, some authors prefer to designate such reactions as “one-step” instead of “concerted”.
- Reich, H.J. Functional Group Manipulation Using Organoselenium Reagents. Acc. Chem. Res. 1979, 12, 22. [Google Scholar] [CrossRef]
- Reich, H.J.; Wollowitz, S. Preparation of α,β-Unsaturated Carbonyl Compounds and Nitriles by Selenoxide Elimination. Org. React. 1993, 44, 1–296. [Google Scholar]
- Back, T.G. Selenoxide Eliminations. In Organoselenium Chemistry—A Practical Approach; Chapter 2; Back, T.G., Ed.; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- Nishibayashi, Y.; Uemura, S. Selenoxide Elimination and [2,3] Sigmatropic Rearrangement. In Organoselenium Chemistry—Synthesis and Reactions; Chapter 7; Wirth, T., Ed.; Wiley—VCH: Weinheim, Germany, 2012. [Google Scholar]
- Walter, R.; Roy, J. Selenomethionine, a potential catalytic antioxidant in biological systems. J. Org. Chem. 1971, 36, 2561–2563. [Google Scholar] [CrossRef]
- Jones, D.N.; Mundy, D.; Whitehouse, R.D. Steroidal Selenoxides Diastereoisomeric at Selenium; Syn-Elimination, Absolute Configuration, and Optical Rotatory Dispersion Characteristics. J. Chem. Soc. D Chem. Commun. 1970, 2, 86–87. [Google Scholar] [CrossRef]
- Sharpless, K.B.; Lauer, R.F.; Teranishi, A.Y. Electrophilic and Nucleophilic Organoselenium Reagents. New Routes to α,β-Unsaturated Carbonyl Compounds. J. Am. Chem. Soc. 1973, 95, 6137–6139. [Google Scholar] [CrossRef]
- Reich, H.J.; Reich, I.L.; Renga, J.M. Organoselenium Chemistry. Alpha.-Phenylseleno Carbonyl Compounds as Precursors for α,β-Unsaturated Ketones and Esters. J. Am. Chem. Soc. 1973, 95, 5813–5815. [Google Scholar] [CrossRef]
- Clive, D.L.J. Fragmentation of selenoxides. New method for dehydrogenation of ketones. J. Chem. Soc. Chem. Commun. 1973, 18, 695–696. [Google Scholar] [CrossRef]
- Kwart, L.D.; Horgan, A.G.; Kwart, H. Structure of the Reaction Barrier in the Selenoxide-Mediated Formation of Olefins. J. Am. Chem. Soc. 1981, 103, 1232–1234. [Google Scholar] [CrossRef]
- Kondo, N.; Fueno, H.; Fujimoto, H.; Makino, M.; Nakaoka, H.; Aoki, I.; Uemura, S. Theoretical and Experimental Studies of Regioselectivity in Selenoxide Eliminations. J. Org. Chem. 1994, 59, 5254–5263. [Google Scholar] [CrossRef]
- Macdougall, P.E.; Smith, N.A.; Schiesser, C.H. Substituent Effects in Selenoxide Elimination Chemistry. Tetrahedron 2008, 64, 2824–2831. [Google Scholar] [CrossRef]
- Bayse, C.A.; Allison, B.D. Activation Energies of Selenoxide Elimination from Se-Substituted Selenocysteine. Mol. Model. 2007, 13, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Madabeni, A.; Zucchelli, S.; Nogara, P.A.; Rocha, J.B.T.; Orian, L. In the Chalcogenoxide Elimination Panorama: Systematic Insight into a Key Reaction. J. Org. Chem. 2022, 87, 11766–11775. [Google Scholar] [CrossRef] [PubMed]
- Sands, K.N.; Mendoza Rengifo, E.; George, G.N.; Pickering, I.J.; Gelfand, B.S.; Back, T.G. The Unexpected Role of Se(VI) Species in Epoxidations with Benzeneseleninic Acid and Hydrogen Peroxide. Angew. Chem. Int. Ed. 2020, 59, 4283–4287. [Google Scholar] [CrossRef] [PubMed]
- Sands, K.N.; Gelfand, B.S.; Back, T.G. One-Pot Synthesis of Aryl Selenonic Acids and Some Unexpected Byproducts. J. Org. Chem. 2021, 86, 9938–9944. [Google Scholar] [CrossRef]
- Syper, L.; Młochowski, J. Benzeneperoxyseleninic Acids—Synthesis and Properties. Tetrahedron 1987, 43, 207–213. [Google Scholar] [CrossRef]
- Barton, D.H.R.; Brewster, A.G.; Hui, R.A.H.F.; Lester, D.J.; Ley, S.V.; Back, T.G. Oxidation of Alcohols using Benzeneseleninic Anhydride. J. Chem. Soc. Chem. Commun. 1978, 21, 952–954. [Google Scholar] [CrossRef]
- Back, T.G. Oxidation of Azasteroid Lactams and Alcohols with Benzeneseleninic Anhydride. J. Org. Chem. 1981, 46, 1442–1446. [Google Scholar] [CrossRef]
- Kuwajima, I.; Shimizu, M.; Urabe, H. Oxidation of Alcohols with tert-Butyl Hydroperoxide and Diaryl Diselenide. J. Org. Chem. 1982, 47, 837–842. [Google Scholar] [CrossRef]
- Back, T.G. Dehydrogenation of Hydrazines and of 4-Azacholestan-3-one with Benzeneseleninic Acid and Benzeneseleninic Anhydride. J. Chem. Soc. Chem. Commun. 1978, 6, 278–279. [Google Scholar] [CrossRef]
- Back, T.G.; Collins, S.; Krishna, M.V. Reactions of Sulfonhydrazides with Benzeneseleninic Acid, Selenium Halides and Sulfur Halides. A Convenient Preparation of Selenosulfonates and Thiosulfonates. Can. J. Chem. 1987, 65, 38–42. [Google Scholar] [CrossRef]
- Back, T.G.; Collins, S.; Kerr, R.G. Oxidation of Hydrazines with Benzeneseleninic Acid and Anhydride. J. Org. Chem. 1981, 46, 1564–1570. [Google Scholar] [CrossRef]
- Branchaud, B.P.; Tsai, P. Relative Ease of Transient Imine Formation via Selenoxide, Sulfoxide and Sulfone β N-H Elimination. A Feasibility Study on the Preparation of Novel Peptide Analogues. J. Org. Chem. 1987, 52, 5475–5478. [Google Scholar] [CrossRef]
- Czarny, M.R. Oxidation of amines with diphenylseleninic anhydride. J. Chem. Soc. Chem. Commun. 1976, 3, 81. [Google Scholar] [CrossRef]
- Czarny, M.R. Oxidation of amines with benzeneseleninyl chloride. Synth. Commun. 1976, 6, 285–293. [Google Scholar] [CrossRef]
- Barton, D.H.R.; Lusinchi, X.; Milliet, P. Studies on the reaction of primary and secondary amines with phenylseleninic anhydride and with phenylseleninic acid. Tetrahedron 1985, 41, 4727–4738. [Google Scholar] [CrossRef]
- Steliou, K.; Gareau, Y.; Harpp, D.N. Molecular Sulfur (S2): Generation and Synthetic Application. J. Am. Chem. Soc. 1984, 106, 799–801. [Google Scholar] [CrossRef]
- Reich, H.J.; Jasperse, C.P. Organoselenium Chemistry. Redox Chemistry of Selenocysteine Model Systems. J. Am. Chem. Soc. 1987, 109, 5549–5551. [Google Scholar] [CrossRef]
- Glass, R.S.; Farooqui, F.; Sabahi, M.; Ehler, K.W. Formation of Thiocarbonyl Compounds in the Reaction of Ebselen Oxide with Thiols. J. Org. Chem. 1989, 54, 1092–1097. [Google Scholar] [CrossRef]
- Sands, K.N.; Burman, A.L.; Ansah-Asamoah, E.; Back, T.G. Chemistry Related to the Catalytic Cycle of the Antioxidant Ebselen. Molecules 2023, 28, 3732. [Google Scholar] [CrossRef]
- These results are based on the assumption that triplet oxygen is formed; singlet oxygen is reported to be 23 kcal mol−1 higher in energy, based on spectroscopic data. Lowry, T.H.; Schueller Richardson, K. Mechanism and Theory on Organic Chemistry, 2nd ed.; Harper and Row: New York, NY, USA, 1981; p. 960. [Google Scholar]
- The sum of the calculated energies of MeSeOH and S2 would be 1.2 kcal mol−1 higher than that of the transition state, if the triplet form 3S2 was produced, while formation of the singlet 1S2 would further increase the energy of products by another 13 kcal mol−1
- Steliou et al. indicated that the singlet sulfur species 1S2 is 13 kcal. mol−1 higher in energy than the triplet ground state, but the singlet might be formed by their method because of spin conservation rules. Steliou, K.; Gareau, Y.; Harpp, D.N. Molecular Sulfur (S2): Generation and Synthetic Application. J. Am. Chem. Soc. 1984, 106, 799–801. [Google Scholar] [CrossRef]
- Bailey, T.S.; Zakharov, L.N.; Pluth, M.D. Understanding Hydrogen Sulfide Storage: Probing Conditions for Sulfide Release from Hydrodisulfides. J. Am. Chem. Soc. 2014, 136, 10573–10576. [Google Scholar] [CrossRef] [PubMed]
- Paetzold, R.; Bochmann, G. Selenium compounds. XLVI. Aliphatic selenium oxides and selenones. Z. Anorg. Allg. Chem. 1968, 360, 293–299. [Google Scholar]
- Agenäs, L.-B. Organic Selenium Compounds: Their Chemistry and Biology; Klayman, D.L., Günther, W.H.H., Eds.; Wiley: New York, NY, USA, 1973; Chapter 5; p. 210. [Google Scholar]
- For a lead reference on electron density transfer, see: Domingo, L.R.; Ríos-Gutiérez, M.; Pérez, P. How does the global electron density transfer diminish activation energies in polar cycloaddition reactions? A Molecular Electron Density Theory study. Tetrahedron 2017, 73, 1718–1724. [Google Scholar] [CrossRef]
- Gaussian 16, Revision B.01, Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; et al. Gaussian, Inc.: Wallingford, CT, USA, 2016.
- Kendall, R.A.; Dunning, T.H., Jr.; Harrison, R.J. Electron Affinities of the First-Row Atoms Revisited. Systematic Basis Sets and Wave Functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- The forward IRC computation for the hydroperoxide in entry 3 did not proceed to the fragmentation products, but instead indicated formation of the corresponding zwitterion. Nevertheless, fragmentation appears to be more likely since the optimized zwitterion energy was 29.4 kcal mol−1 higher than that of the sum of the fragmentation products (triplet O2 and MeSeOH). This discrepancy, along with the need to freeze the methyl group in the transition state calculation to avoid additional imaginary frequencies, suggest that these results should be regarded with caution.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
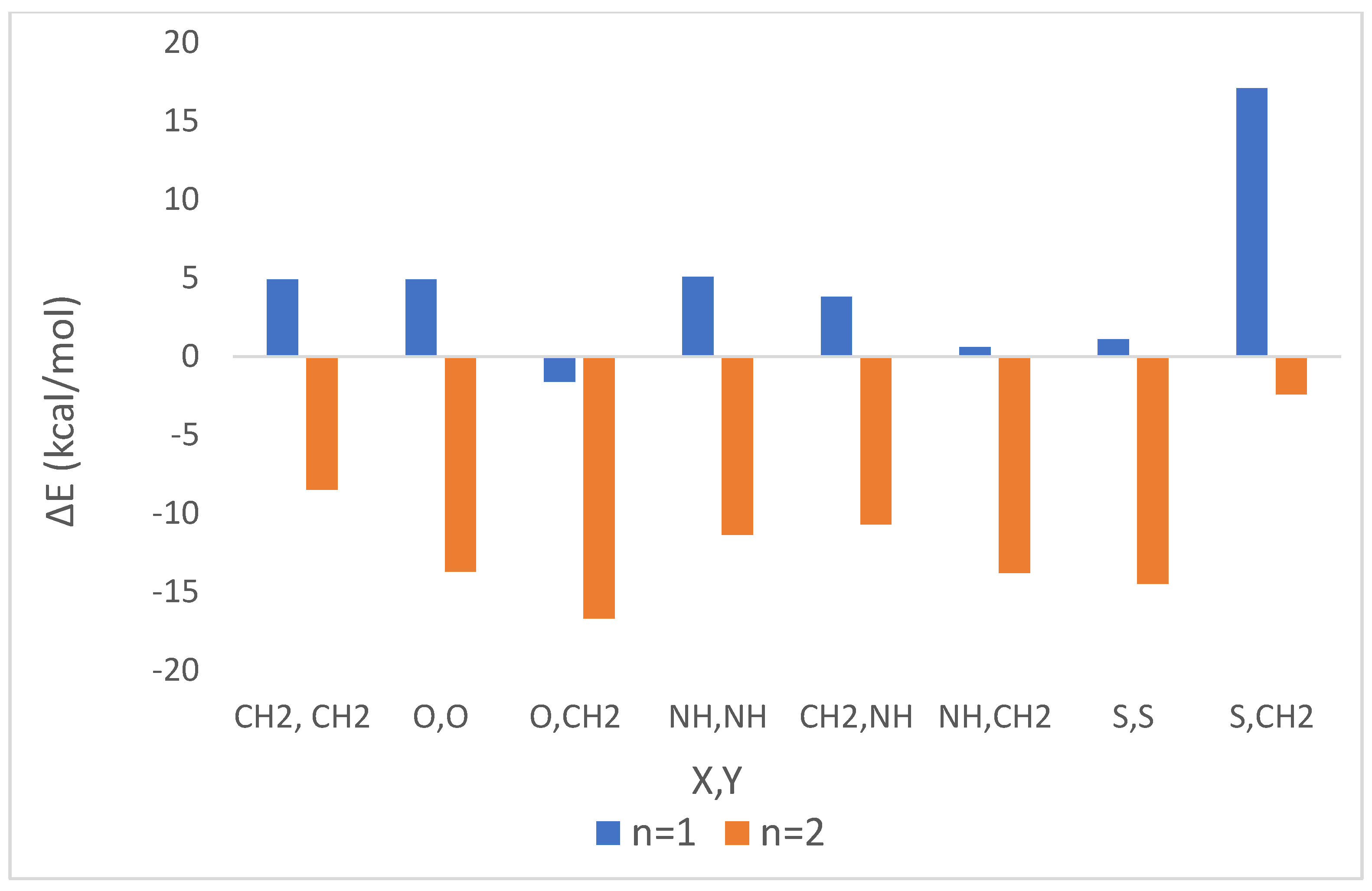
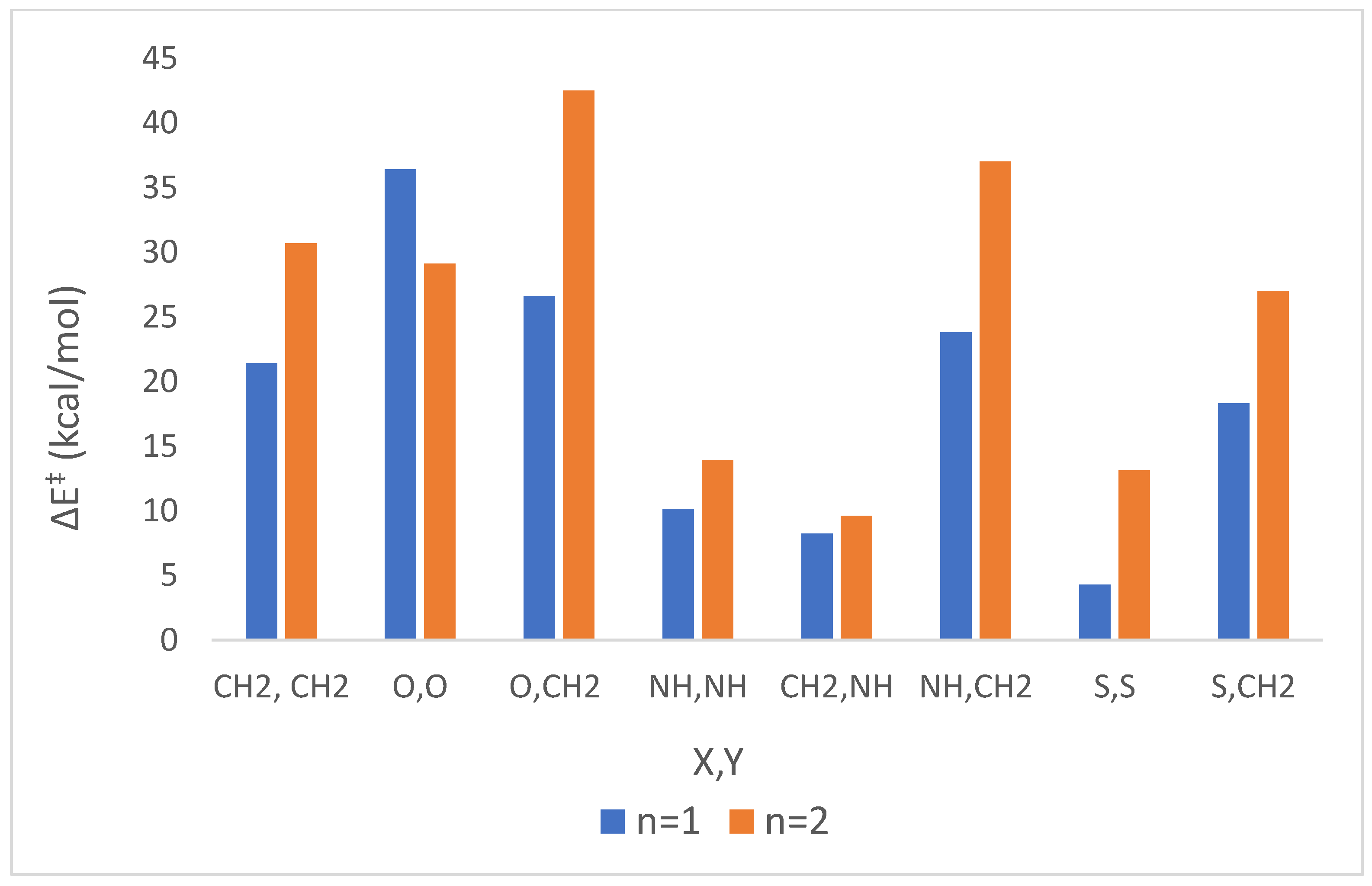
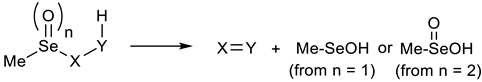 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | n | X | Y | ΔE‡ Gas Phase | ΔEGas Phase | ΔE‡ H2O | ΔE H2O | ΔE‡ CH2Cl2 | ΔE CH2Cl2 | ΔE‡ MeOH | ΔE MeOH |
| 1 | 1 | CH2 | CH2 | 21.4 | 4.9 | 24.3 | 7.4 | 23.8 | 7.0 | 24.2 | 7.3 |
| 2 | 2 | 30.7 | −8.5 | 31.4 | −6.0 | 31.3 | −6.4 | 31.4 | −6.1 | ||
| 3 c,d | 1 | O | O | 36.4 | 4.9 | 32.5 | 7.5 | 33.2 | 7.1 | 32.6 | 7.4 |
| 4 c | 2 | 29.1 | −13.7 | 28.7 | −12.9 | 28.8 | −12.9 | 28.7 | −12.9 | ||
| 5 | 1 | O | CH2 | 26.6 | −1.6 | 28.1 | −2.5 | 27.9 | −2.3 | 28.1 | −2.4 |
| 6 | 2 | 42.5 | −16.7 | 42.7 | −19.2 | 42.7 | −18.7 | 42.7 | −19.1 | ||
| 7 e | 1 | NH | NH | 10.1 | 5.1 | 12.3 | 6.0 | 11.9 | 5.8 | 12.2 | 6.0 |
| 8 e | 2 | 13.9 | −11.4 | 9.9 | −11.9 | 11.5 | −11.8 | 10.1 | −11.8 | ||
| 9 | 1 | CH2 | NH | 8.2 | 3.8 | 10.1 | 5.0 | 9.8 | 4.8 | 10.1 | 5.0 |
| 10 | 2 | 9.6 | −10.7 | 5.0 | −9.2 | 6.2 | −9.4 | 5.3 | −9.2 | ||
| 11 | 1 | NH | CH2 | 23.8 | 0.6 | 26.4 | 0.6 | 26.0 | 0.6 | 26.3 | 0.6 |
| 12 | 2 | 37.0 | −13.8 | 37.8 | −15.3 | 37.7 | −15.0 | 37.8 | −15.2 | ||
| 13 f | 1 | S | S | 4.3 | 1.1 | 3.7 | −1.6 | 3.8 | −1.1 | 3.7 | −1.5 |
| 14 | 2 | 13.1 | −14.5 | 12.3 | −13.9 | 12.5 | −14.0 | 12.4 | −13.9 | ||
| 15 | 1 | S | CH2 | 18.3 | 17.1 | 19.7 | 17.0 | 19.4 | 17.0 | 19.6 | 17.0 |
| 16 | 2 | 27.0 | −2.4 | 28.3 | −3.2 | 28.1 | −3.1 | 28.3 | −3.2 | ||
 | 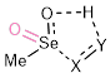 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Starting Material | Transition State | ||||||||||
| Entry | n, X, Y | X-Se | X-Y | H-OSe | H-Y | Se=O | X-Se | X-Y | H-OSe | H-Y | Se=O |
| 1 | 1, C, C | 2.005 | 1.521 | 2.492 | 1.093 | 1.651 | 2.481 | 1.415 | 1.278 | 1.334 | 1.707 |
| 2 | 2, C, C | 1.966 | 1.524 | 2.995 b | 1.091 | 1.618, 1.618 | 2.513 | 1.415 | 1.384 | 1.278 | 1.689, 1.629 |
| 3 | 1, O, O | 1.858 | 1.443 | 2.332 | 0.975 | 1.630 | 1.824 | 1.426 | 1.180 | 1.298 | 1.999 |
| 4 | 2, O, O | 1.831 | 1.446 | 2.744 | 0.971 | 1.605, 1.600 | 2.055 | 1.337 | 1.103 | 1.376 | 1.707, 1.615 |
| 5 | 1, O, CH2 | 1.818 | 1.435 | 2.420 | 1.093 | 1.629 | 2.283 | 1.308 | 1.310 | 1.300 | 1.685 |
| 6 | 2, O, CH2 | 1.775 | 1.446 | 2.950 b | 1.092 | 1.607, 1.607 | 2.237 | 1.317 | 1.370 | 1.278 | 1.621, 1.682 |
| 7 | 1, NH, NH | 1.940 | 1.418 | 2.190 | 1.027 | 1.647 | 2.499 | 1.314 | 1.334 | 1.180 | 1.708 |
| 8 | 2, NH, NH | 1.883 | 1.414 | 2.425 c | 1.014 | 1.613, 1.617 | 2.581 | 1.292 | 1.700 | 1.059 | 1.677, 1.633 |
| 9 | 1, CH2, NH | 2.074 | 1.421 | 2.214 | 1.023 | 1.660 | 2.530 | 1.335 | 1.341 | 1.181 | 1.715 |
| 10 | 2, CH2, NH | 2.029 | 1.420 | 3.006 | 1.014 | 1.622, 1.622 | 2.641 | 1.320 | 1.632 | 1.072 | 1.682, 1.634 |
| 11 | 1, NH, CH2 | 1.885 | 1.475 | 2.554 | 1.096 | 1.642 | 2.370 | 1.370 | 1.310 | 1.303 | 1.693 |
| 12 | 2, NH, CH2 | 1.840 | 1.474 | 2.870 d | 1.093 | 1.613, 1.612 | 2.370 | 1.371 | 1.372 | 1.281 | 1.684, 1.628 |
| 13 | 1, S, S | 2.371 | 2.086 | 3.142 | 1.352 | 1.636 | 2.338 | 2.094 | 1.328 | 1.555 | 1.684 |
| 14 | 2, S, S | 2.324 | 2.086 | 3.262 | 1.352 | 1.612, 1.612 | 2.290 | 2.105 | 1.243 | 1.634 | 1.677, 1.607 |
| 15 | 1, S, CH2 | 2.330 | 1.830 | 2.190 | 1.096 | 1.642 | 2.631 | 1.699 | 1.182 | 1.417 | 1.710 |
| 16 | 2, S, CH2 | 2.279 | 1.837 | 2.356 | 1.090 | 1.617, 1.610 | 2.576 | 1.705 | 1.247 | 1.375 | 1.697, 1.620 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Doig, A.I.; Stadel, J.T.; Back, T.G. A Computational Study of Heteroatom Analogues of Selenoxide and Selenone syn Eliminations. Molecules 2024, 29, 4915. https://doi.org/10.3390/molecules29204915
Doig AI, Stadel JT, Back TG. A Computational Study of Heteroatom Analogues of Selenoxide and Selenone syn Eliminations. Molecules. 2024; 29(20):4915. https://doi.org/10.3390/molecules29204915
Chicago/Turabian StyleDoig, Adrian I., Jessica T. Stadel, and Thomas G. Back. 2024. "A Computational Study of Heteroatom Analogues of Selenoxide and Selenone syn Eliminations" Molecules 29, no. 20: 4915. https://doi.org/10.3390/molecules29204915
APA StyleDoig, A. I., Stadel, J. T., & Back, T. G. (2024). A Computational Study of Heteroatom Analogues of Selenoxide and Selenone syn Eliminations. Molecules, 29(20), 4915. https://doi.org/10.3390/molecules29204915