

Reactivity and Stability of (Hetero)Benzylic Alkenes via the Wittig Olefination Reaction




Abstract

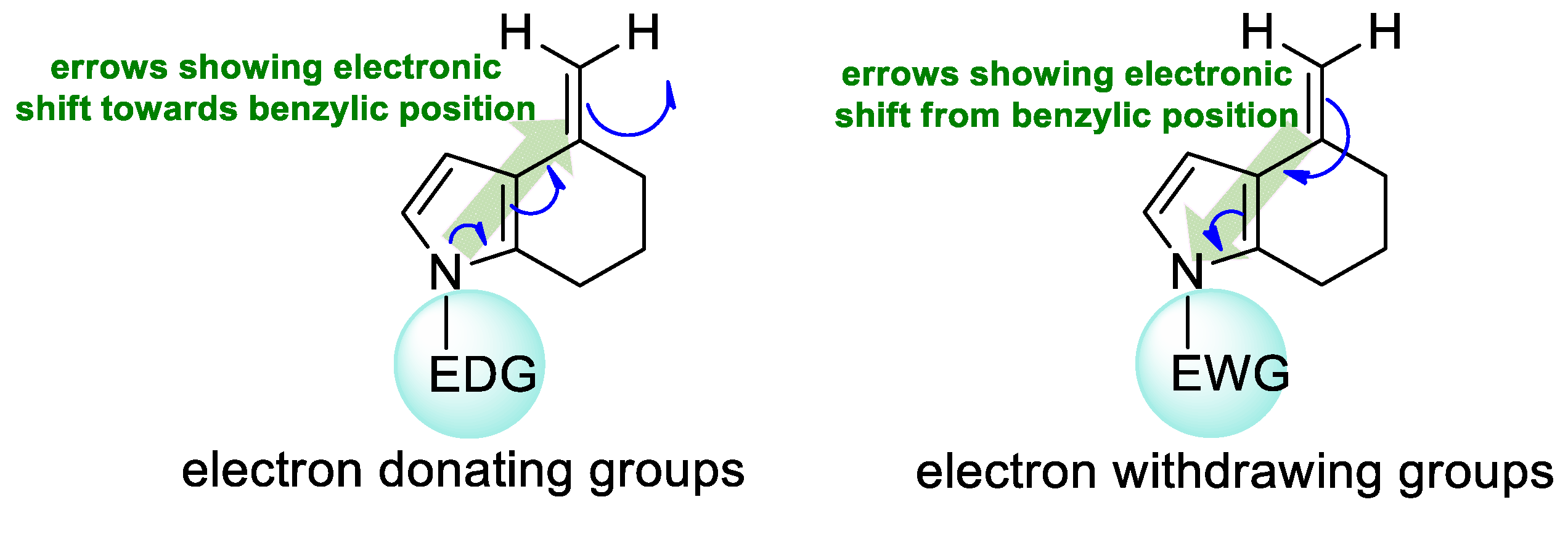
1. Introduction
2. Results and Discussion
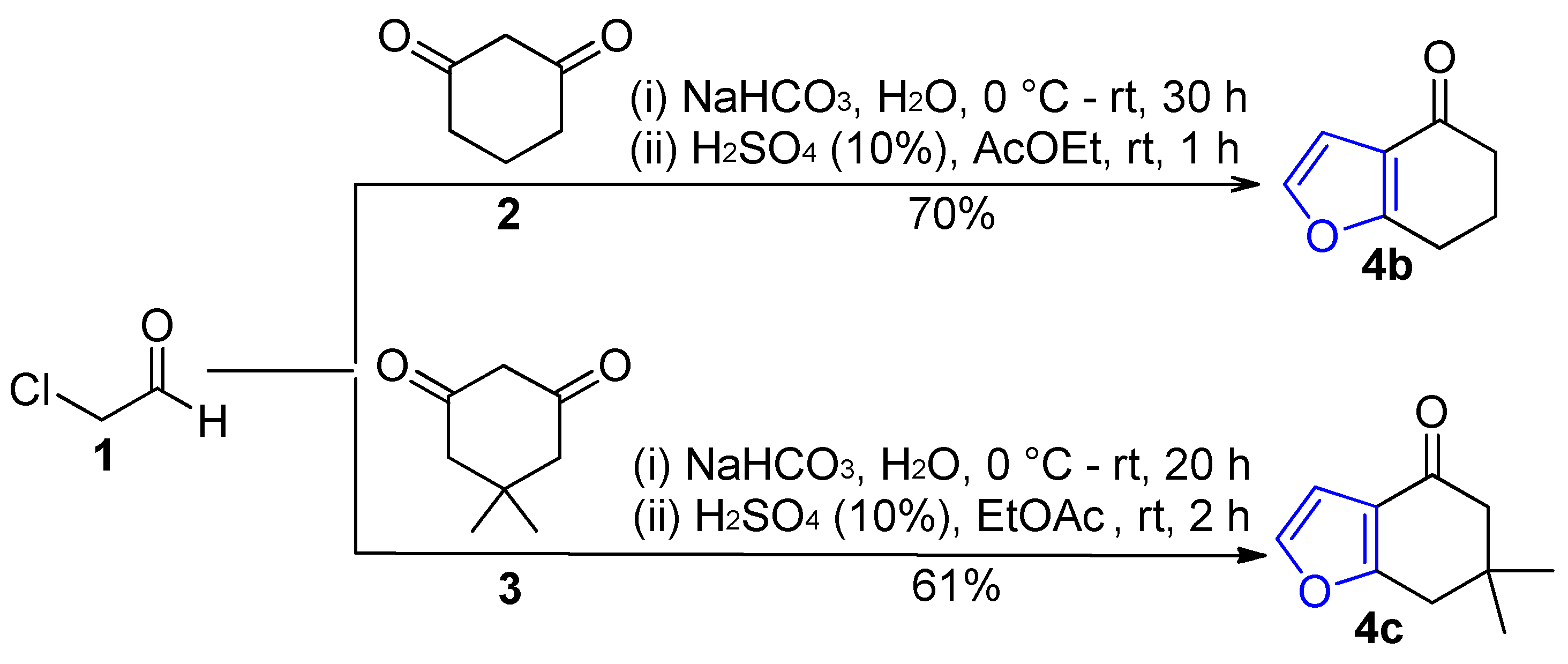
2.1. Synthesis of Furans
2.2. Synthesis of Indoles
2.3. Synthesis of Fused Pyrroles
2.4. Synthesis of Tetrahydroisoquinoline
2.5. Protection of Indole and Pyrrole Derivatives
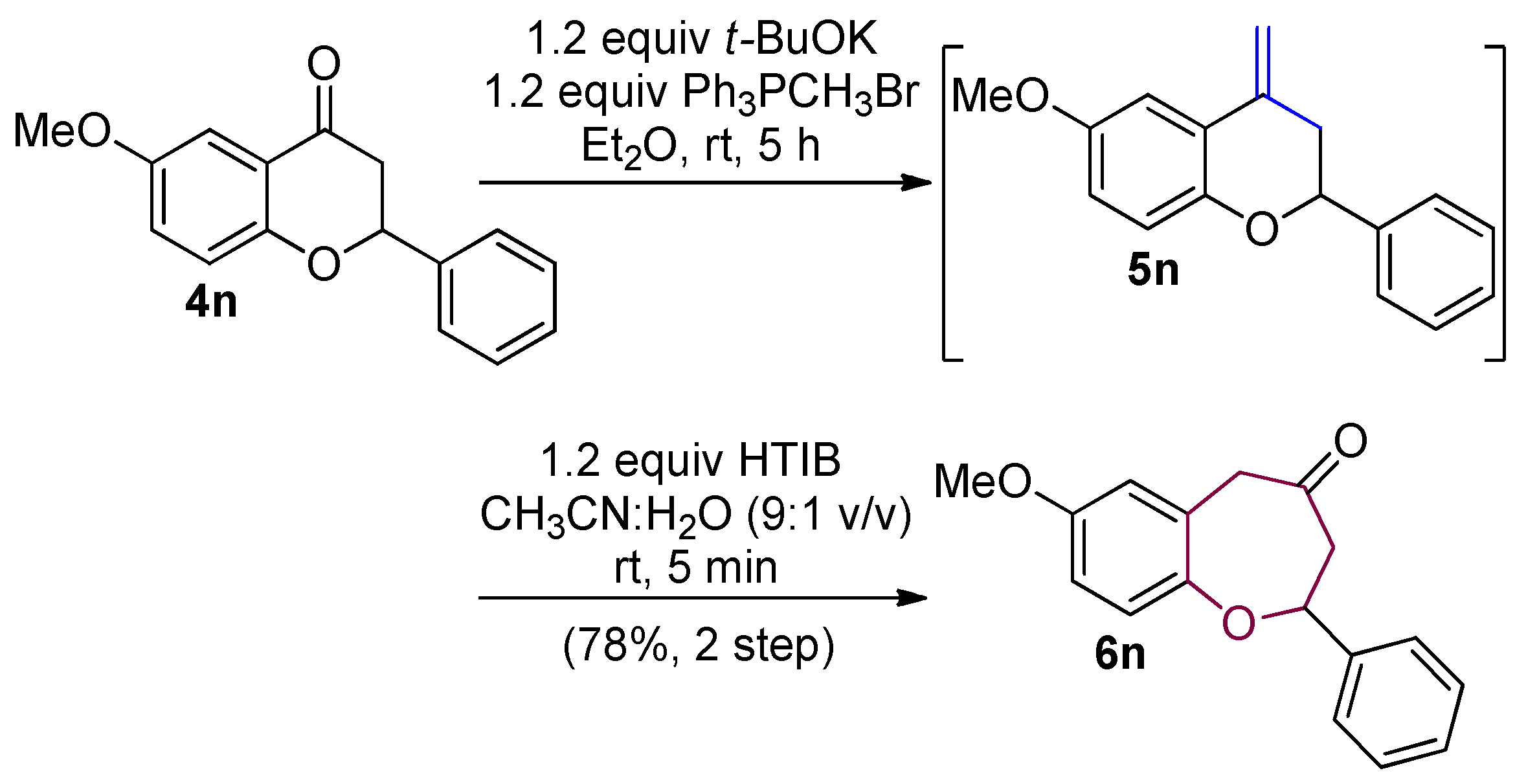
2.6. Heterocyclic Wittig Olefination
3. Experimental Procedures
3.1. General Information
- Buchi B-545 apparatus for melting point (Merck, Darmstadt, Germany);
- Perkin Elmer 1750-FT equipment for IR analysis (Perkin Elmer, Waltham, MA, USA);
- Bruker Daltonics microTOF electrospray for HRMS (Bruker, Billerica, MA, USA);
- Perkin Elmer 2400 Series II for elemental analysis (Perkin Elmer, Waltham, MA, USA);
- INOVA 300 MHz, (1H and 13C NMR analysis) (INOVA, Purcellville, VA, USA);
- Bruker AIII 300 MHz (1H and 13C NMR analysis) (Bruker, Billerica, MA, USA); and
- Bruker AIII 500 MHz (1H and 13C NMR analysis) (Bruker, Billerica, MA, USA).
3.2. Synthesis of Isoquinolin Derivatives
3.3. Conversion of Furan Derivatives to Pyrrole Derivatives
3.4. General Protocol for the Protection of Heterocyclic Derivatives
3.5. General Protocol for Ring Expansion
3.6. General Procedure for Wittig Olefination
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Al-Mulla, A. A Review: Biological Importance of Heterocyclic Compounds. Der Pharma Chem. 2017, 9, 141–147. [Google Scholar]
- Arora, P.; Arora, V.; Lamba, H.S.; Wadhwa, D. Importance of Heterocyclic Chemistry: A Review. Int. J. Pharm. Sci. Res. 2012, 3, 2947. [Google Scholar]
- Alvarez-Builla, J.; Barluenga, J. Heterocyclic Compounds: An Introduction; Wiley: Hoboken, NJ, USA, 2011; pp. 1–9. [Google Scholar]
- Kabir, E.; Uzzaman, M. A Review on Biological and Medicinal Impact of Heterocyclic Compounds. Results Chem. 2022, 4, 100606. [Google Scholar] [CrossRef]
- Chinchilla, R.; Nájera, C.; Yus, M. Metalated Heterocycles and Their Applications in Synthetic Organic Chemistry. Chem. Rev. 2004, 104, 2667–2722. [Google Scholar] [CrossRef] [PubMed]
- Karanam, P.; Reddy, G.M.; Lin, W. Strategic Exploitation of the Wittig Reaction: Facile Synthesis of Heteroaromatics and Multifunctional Olefins. Synlett 2018, 29, 2608–2622. [Google Scholar]
- Das, U.; Tsai, Y.-L.; Lin, W. Preparation of Functionalized Heteroaromatics Using an Intramolecular Wittig Reaction. Org. Biomol. Chem. 2014, 12, 4044–4050. [Google Scholar] [CrossRef]
- Rocha, D.H.A.; Pinto, D.C.G.A.; Silva, A.M.S. Applications of the Wittig Reaction on the Synthesis of Natural and Natural-Analogue Heterocyclic Compounds. Eur. J. Org. Chem. 2018, 2018, 2443–2457. [Google Scholar] [CrossRef]
- Heravi, M.M.; Zadsirjan, V.; Hamidi, H.; Daraie, M.; Momeni, T. Recent Applications of the Wittig Reaction in Alkaloid Synthesis. Alkaloids Chem. Biol. 2020, 84, 201–334. [Google Scholar]
- Hoffmann, R.W. Wittig and His Accomplishments: Still Relevant beyond His 100th Birthday. Angew. Chem. Int. Ed. 2001, 40, 1411–1416. [Google Scholar] [CrossRef]
- Byrne, P.A.; Gilheany, D.G. The Modern Interpretation of the Wittig Reaction Mechanism. Chem. Soc. Rev. 2013, 42, 6670–6696. [Google Scholar] [CrossRef]
- Maryanoff, B.E.; Reitz, A.B. The Wittig Olefination Reaction and Modifications Involving Phosphoryl-Stabilized Carbanions. Stereochemistry, Mechanism, and Selected Synthetic Aspects. Chem. Rev. 1989, 89, 863–927. [Google Scholar] [CrossRef]
- Edmonds, M.; Abell, A. The Wittig Reaction. In Modern Carbonyl Olefination; Wiley: Hoboken, NJ, USA, 2004; pp. 1–17. [Google Scholar]
- Nicolaou, K.C.; Härter, M.W.; Gunzner, J.L.; Nadin, A. The Wittig and Related Reactions in Natural Product Synthesis. Liebigs Ann. 1997, 1997, 1283–1301. [Google Scholar] [CrossRef]
- Takeda, T. Modern Carbonyl Olefination: Methods and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2006; ISBN 352760538X. [Google Scholar]
- Heravi, M.M.; Ghanbarian, M.; Zadsirjan, V.; Alimadadi Jani, B. Recent Advances in the Applications of Wittig Reaction in the Total Synthesis of Natural Products Containing Lactone, Pyrone, and Lactam as a Scaffold. Monatshefte Chem. Chem. Mon. 2019, 150, 1365–1407. [Google Scholar] [CrossRef]
- Khaskin, E.; Milstein, D. Catalytic, Oxidant-Free, Direct Olefination of Alcohols Using Wittig Reagents. Chem. Commun. 2015, 51, 9002–9005. [Google Scholar] [CrossRef] [PubMed]
- Tukhtaev, H.B.; Ivanov, K.L.; Bezzubov, S.I.; Cheshkov, D.A.; Melnikov, M.Y.; Budynina, E.M. Aza-Wittig Reaction with Nitriles: How Carbonyl Function Switches from Reacting to Activating. Org. Lett. 2019, 21, 1087–1092. [Google Scholar] [CrossRef] [PubMed]
- Fitjer, L.; Quabeck, U. The Wittig Reaction Using Potassium-Tert-Butoxide High Yield Methylenations of Sterically Hindered Ketones. Synth. Commun. 1985, 15, 855–864. [Google Scholar] [CrossRef]
- Rathke, M.W.; Nowak, M. The Horner-Wadsworth-Emmons Modification of the Wittig Reaction Using Triethylamine and Lithium or Magnesium Salts. J. Org. Chem. 1985, 50, 2624–2626. [Google Scholar] [CrossRef]
- Taylor, R.J.K.; Reid, M.; Foot, J.; Raw, S.A. Tandem Oxidation Processes Using Manganese Dioxide: Discovery, Applications, and Current Studies. Acc. Chem. Res. 2005, 38, 851–869. [Google Scholar] [CrossRef]
- Jeena, V.; Robinson, R.S. Recent Developments in One-Pot Tandem Oxidation Process Coupling Reactions. RSC Adv. 2014, 4, 40720–40739. [Google Scholar] [CrossRef]
- Khan, A.; Silva, L.F., Jr.; Rabnawaz, M. Iodine (III)-Promoted Ring Expansion Reactions: A Metal-Free Approach toward Seven-Membered Heterocyclic Rings. Asian J. Org. Chem. 2021, 10, 2549–2552. [Google Scholar] [CrossRef]
- Lussari, N.; Khan, A.; Pilli, R.A.; Dos Santos, A.A.; Silva, L.F.; Braga, A.A.C. A DFT Study on the Formation of Heterocycles via Iodine (Iii)-Promoted Ring Expansion Reactions. New J. Chem. 2022, 46, 20817–20827. [Google Scholar] [CrossRef]
- Plouvier, B.; Beatch, G.N.; Jung, G.L.; Zolotoy, A.; Sheng, T.; Clohs, L.; Barrett, T.D.; Fedida, D.; Wang, W.Q.; Zhu, J.J.; et al. Synthesis and Biological Studies of Novel 2-Aminoalkylethers as Potential Antiarrhythmic Agents for the Conversion of Atrial Fibrillation. J. Med. Chem. 2007, 50, 2818–2841. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.R.; Morehead, A.T. A New Route for the Synthesis of Furanoflavone and Furanochalcone Natural Products. Tetrahedron 1995, 51, 4909–4922. [Google Scholar] [CrossRef]
- Peng, Y.; Luo, J.; Feng, Q.; Tang, Q. Understanding the Scope of Feist–Bénary Furan Synthesis: Chemoselectivity and Diastereoselectivity of the Reaction Between α-Halo Ketones and β-Dicarbonyl Compounds. Eur. J. Org. Chem. 2016, 2016, 5169–5179. [Google Scholar] [CrossRef]
- Arai, M.; Miyauchi, Y.; Miyahara, T.; Ishikawa, T.; Saito, S. Synthesis of 4-Acetoxyindoles and Related Derivatives by Means of Air Oxidation of 4-Oxo-4,5,6,7-Tetrahydroindoles Obtained from Nitroalkenes and Cyclohexane-1,3-Diones. Synlett 2008, 2009, 122–126. [Google Scholar] [CrossRef]
- de Candia, M.; Zaetta, G.; Denora, N.; Tricarico, D.; Majellaro, M.; Cellamare, S.; Altomare, C.D. New Azepino[4,3-b]Indole Derivatives as Nanomolar Selective Inhibitors of Human Butyrylcholinesterase Showing Protective Effects against NMDA-Induced Neurotoxicity. Eur. J. Med. Chem. 2017, 125, 288–298. [Google Scholar] [CrossRef] [PubMed]
- Kadam, S.A.; Haav, K.; Toom, L.; Haljasorg, T.; Leito, I. NMR Method for Simultaneous Host-Guest Binding Constant Measurement. J. Org. Chem. 2014, 79, 2501–2513. [Google Scholar] [CrossRef]
- Azizi, N.; Khajeh-Amiri, A.; Ghafuri, H.; Bolourtchian, M.; Saidi, M. Iron-Catalyzed Inexpensive and Practical Synthesis of N-Substituted Pyrroles in Water. Synlett 2009, 2009, 2245–2248. [Google Scholar] [CrossRef]
- Ueda, K.; Amaike, K.; Maceiczyk, R.M.; Itami, K.; Yamaguchi, J. β-Selective C-H Arylation of Pyrroles Leading to Concise Syntheses of Lamellarins C and I. J. Am. Chem. Soc. 2014, 136, 13226–13232. [Google Scholar] [CrossRef]
- Lardenois, P.; Frost, J.; Dargazanli, G.; George, P. A Convenient Synthesis 7,8-Dihydroisoquinolin-5(6H)-One. Synth. Commun. 1996, 26, 2305–2308. [Google Scholar] [CrossRef]
- Khan, A.; Silva, L.F.; Rabnawaz, M. A Comparative Study of Thallium (Iii) and Iodine (Iii)-Mediated Ring Contraction Reactions for the Synthesis of Indane. New J. Chem. 2021, 45, 2078–2084. [Google Scholar] [CrossRef]
- Zuo, Y.; He, X.; Ning, Y.; Wu, Y.; Shang, Y. Rh(III)-Catalyzed C–H Activation/Intramolecular Cyclization: Access to N-Acyl-2,3-Dihydro-1 H-Carbazol-4(9H)-Ones from Cyclic 2-Diazo-1,3-Diketones and N-Arylamides. ACS Omega 2017, 2, 8507–8516. [Google Scholar] [CrossRef] [PubMed]
- Gabbutt, C.D.; Hepworth, J.D.; Heron, B.M. Reactions of Some 2H-Chromenes and 2H-Thiochromenes with Triazolinediones. Tetrahedron 1995, 51, 13277–13290. [Google Scholar] [CrossRef]
- Davies, H.M.L.; Manning, J.R. C-H Activation as a Strategic Reaction: Enantioselective Synthesis of 4-Substituted Indoles. J. Am. Chem. Soc. 2006, 128, 1060–1061. [Google Scholar] [CrossRef]
- Cho, H.; Iwama, Y.; Sugimoto, K.; Mori, S.; Tokuyama, H. Regioselective Synthesis of Heterocycles Containing Nitrogen Neighboring an Aromatic Ring by Reductive Ring Expansion Using Diisobutylaluminum Hydride and Studies. J. Org. Chem 2010, 75, 627–636. [Google Scholar] [CrossRef]
- Bardakos, V.; Sucrow, W. Enhydrazine, 21: Lactame Aus 1,2,3,9-Tetrahydro-4H-Carbazol-4-Onen. Chem. Ber. 1978, 111, 853–859. [Google Scholar] [CrossRef]
- Montalban, A.G.; Baum, S.M.; Cowell, J.; McKillop, A. Formation of N-Substituted 4-and 7-Oxo-4, 5, 6, 7-Tetrahydroindoles Revisited: A Mechanistic Interpretation and Conversion into 4-and 7-Oxoindoles. Tetrahedron Lett. 2012, 53, 4276–4279. [Google Scholar] [CrossRef]
- Laurila, M.L.; Magnus, N.A.; Staszak, M.A. Green N-Methylation of Electron Deficient Pyrroles with Dimethylcarbonate. Org. Process Res. Dev. 2009, 13, 1199–1201. [Google Scholar] [CrossRef]
- Lee, I.-S.H. Synthesis of N-Aryl-4,5,6,7-Tetrahydroindoles. Korean Chem. Soc. 2012, 33, 341–343. [Google Scholar] [CrossRef]
- McEachern, E.J.; Yang, W.; Chen, G.; Skerlj, R.T.; Bridger, G.J. Convenient Synthesis of 5, 6, 7, 8-Tetrahydroquinolin-8-Ylamine and 6, 7-Dihydro-5 H-Quinolin-8-One. Synth. Commun. 2003, 33, 3497–3502. [Google Scholar] [CrossRef]
- Gartshore, C.J.; Lupton, D.W. Studies on the Enantioselective Synthesis of Carbazolones as Intermediates in Aspidosperma and Kopsia Alkaloid Synthesis. Aust. J. Chem. 2013, 66, 882–890. [Google Scholar] [CrossRef]
- Piras, L.; Ghiron, C.; Minetto, G.; Taddei, M. Microwave-Assisted Synthesis of Tetrahydroindoles. Tetrahedron Lett. 2008, 49, 459–462. [Google Scholar] [CrossRef]
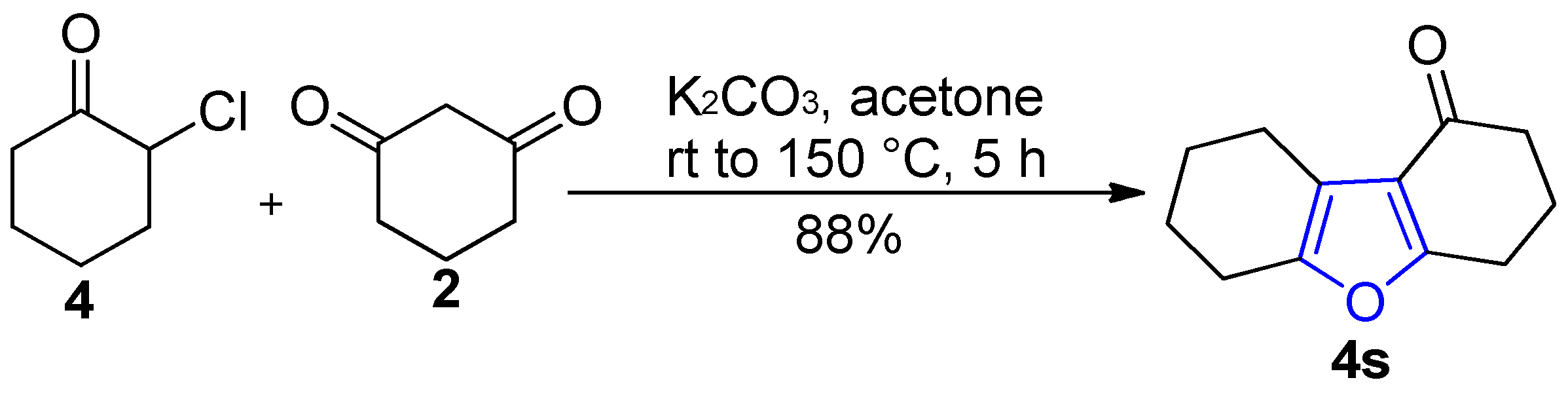

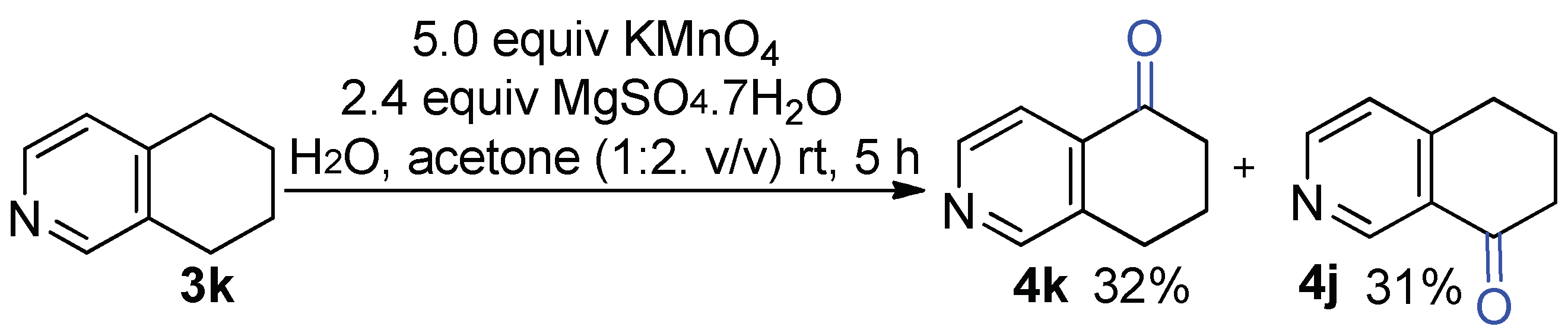



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Substrate | Conditions | Product | Yield % |
|---|---|---|---|---|
| 1 | 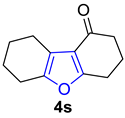 | CH3NH2, PTSA (few crystals) rt to 125 °C, 20 h | 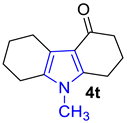 | 25% |
| 2 | 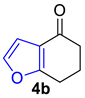 | CH3NH2, PTSA (few crystals) rt to 125 °C, 20 h | 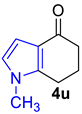 | 98% |
| 3 | 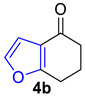 | 5.0 equiv m-chloroaniline xylene, PTSA rt to 160 °C, 24 h | 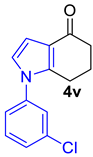 | 73% |
| 4 | 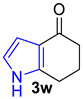 | 1.0 equiv n-BuLi, 1.0 equiv MeI THF, 0 °C to rt, 1 h | 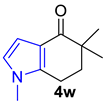 | 11% |
| Entry | Substrate | Conditions | Product | %Yield |
|---|---|---|---|---|
| 1 | 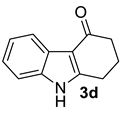 | (i) 1.5 equiv NaH, THF, 0 °C, 30 min (ii) 1.5 equiv AcCl, 0 °C to rt, 3 h | 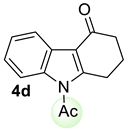 | 92 |
| 2 |  | (i) 1.5 equiv NaH, THF, 0 °C, 30 min (ii) 1.5 equiv TsCl, 0 °C to rt, 2 h | 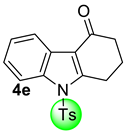 | 62 |
| 3 | 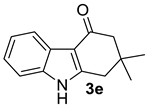 | (i) 1.5 equiv NaH, DMF, 0 °C, 30 min (ii) 1.5 equiv AcCl, 0 °C to rt, 3 h | 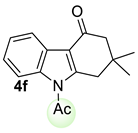 | 84 |
| 4 | 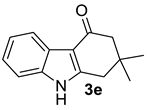 | (i) 1.5 equiv NaH, THF, 0 °C, 30 min (ii) 1.5 equiv TsCl, 0 °C to rt, 2 h |  | 72 |
| 5 | 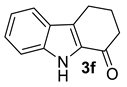 | (i) 3.0 equiv NaH, THF, 0 °C, 30 min (ii) 3.0 equiv Boc2O, 0 °C to rt, 6 h |  | 67 |
| 6 | 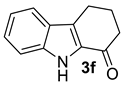 | (i) 3.0 equiv NaOH, DCE, 0 ºC, 30 min (ii) 3.0 equiv MsCl, 0 ºC to rt, 24 h |  | 64 |
| 7 | 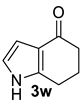 | (i) 3.0 equiv NaOH, DCE, 0 °C, 30 min (ii) 3.0 equiv MsCl, 0 °C to rt, 15 h |  | 81 |
| 8 | 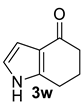 | (i) 1.5 equiv NaH, THF, 0 °C, 30 min (ii) 1.5 equiv Boc2O, 0 °C to rt, 3 h | 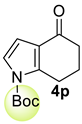 | 98 |
| 9 |  | (i) 1.5 equiv NaH, THF, 0 °C, 30 min (ii) 1.5 equiv TBS, 0 °C to rt, 20 h | 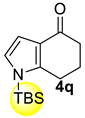 | 75 |
| 10 | 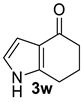 | (i) 4.0 equiv KOH, DMSO, 0 °C, 30 min (ii) 1.2 equiv PhCH2Br, 0 °C to rt, 12 h |  | 90 |
| Entry | Substrate | Conditions | Product | Yield |
|---|---|---|---|---|
| 1 | 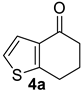 | 1.5 equiv t-BuOK 1.5 equiv Ph3PCH3Br Et2O, rt, 3 h | 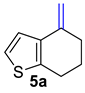 | 97% |
| 2 | 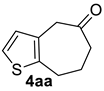 | 1.5 equiv t-BuOK 1.5 equiv Ph3PCH3Br Et2O, rt, 3 h | 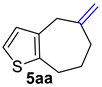 | 69% |
| 3 | 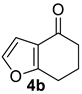 | 1.5 equiv t-BuOK 1.5 equiv Ph3PCH3Br Et2O, rt, 3 h | 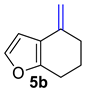 | 86% |
| 4 | 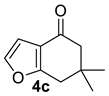 | 1.5 equiv t-BuOK 1.5 equiv Ph3PCH3Br Et2O, rt, 3 h | 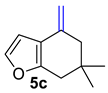 | 48% |
| 5 |  | 1.5 equiv t-BuOK 1.5 equiv Ph3PCH2Br Et2O, rt, 3 h | 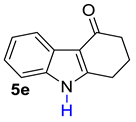 | 83% |
| 6 | 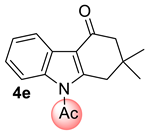 | 1.5 equiv t-BuOK 1.5 equiv Ph3PCH3Br Et2O, rt, 3 h |  | 66% |
| 7 |  | 1.8 equiv t-BuOK 1.8 equiv Ph3PCH3Br Et2O, rt, 10 h | 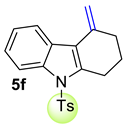 | 82% |
| 8 |  | 2.4 equiv t-BuOK 2.4 equiv Ph3PCH3Br Et2O, rt, 26 h | 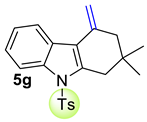 | 58% |
| 9 | 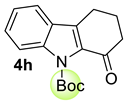 | 2.0 equiv t-BuOK 2.0 equiv Ph3PCH3Br Et2O, rt, 3 h |  | 91% |
| 10 | 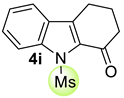 | 1.5 equiv t-BuOK 1.5 equiv Ph3PCH3Br Et2O:DCM (4:1), rt, 15 h | 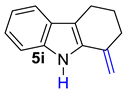 | 49% |
| 11 |  | 1.5 equiv t-BuOK 1.5 equiv Ph3PCH3Br Et2O, rt, 3 h | 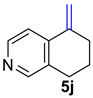 | 88% |
| 12 | 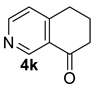 | 1.5 equiv t-BuOK 1.5 equiv Ph3PCH3Br Et2O, rt, 3 h | 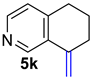 | 62% |
| 13 |  | 1.5 equiv t-BuOK 1.5 equiv Ph3PCH3Br Et2O, rt, 5 h | 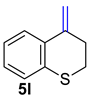 | 72% |
| 14 | 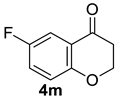 | 1.5 equiv t-BuOK 1.5 equiv Ph3PCH3Br Et2O, rt, 5 h | 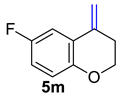 | 90% |
| 15 | 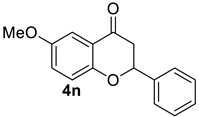 | 1.5 equiv t-BuOK 1.5 equiv Ph3PCH3Br Et2O, rt, 5 h | decomposed (complex mixture) |
| Entry | Substrate | Conditions | Product | |
|---|---|---|---|---|
| 1 | 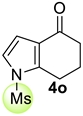 | 1.5 equiv t-BuOK 1.5 equiv Ph3PCH3Br Et2O, rt, 24 h | 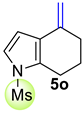 | 50% |
| 2 | 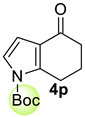 | 1.5 equiv t-BuOK 1.5 equiv Ph3PCH3Br Et2O, rt, 6 h | 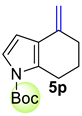 | 58% |
| 3 |  | 1.5 equiv t-BuOK 1.5 equiv Ph3PCH3Br Et2O, rt, 5 h | Decomposed (complex mixture) | |
| 4 | 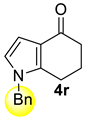 | 1.5 equiv t-BuOK 1.5 equiv Ph3PCH3Br Et2O, rt, 5 h | Decomposed (complex mixture) | |
| 5 | 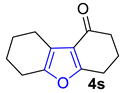 | 1.5 equiv t-BuOK 1.5 equiv Ph3PCH3Br Et2O, rt, 3 h | Decomposed (complex mixture) | |
| 6 |  | 1.5 equiv t-BuOK 1.5 equiv Ph3PCH3Br Et2O, rt, 10 h | Decomposed (complex mixture) | |
| 7 | 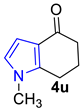 | 1.5 equiv t-BuOK 1.5 equiv Ph3PCH3Br Et2O, rt, 12 h | Decomposed (complex mixture) | |
| 8 |  | 1.5 equiv t-BuOK 1.5 equiv Ph3PCH3Br Et2O, rt, 10 h | Decomposed (complex mixture) | |
| 9 | 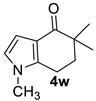 | 1.5 equiv t-BuOK 1.5 equiv Ph3PCH3Br Et2O, rt, 10 h | No reaction |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, A.; Sarwar, M.G.; Ali, S. Reactivity and Stability of (Hetero)Benzylic Alkenes via the Wittig Olefination Reaction. Molecules 2024, 29, 501. https://doi.org/10.3390/molecules29020501
Khan A, Sarwar MG, Ali S. Reactivity and Stability of (Hetero)Benzylic Alkenes via the Wittig Olefination Reaction. Molecules. 2024; 29(2):501. https://doi.org/10.3390/molecules29020501
Chicago/Turabian StyleKhan, Ajmir, Mohammed G. Sarwar, and Sher Ali. 2024. "Reactivity and Stability of (Hetero)Benzylic Alkenes via the Wittig Olefination Reaction" Molecules 29, no. 2: 501. https://doi.org/10.3390/molecules29020501
APA StyleKhan, A., Sarwar, M. G., & Ali, S. (2024). Reactivity and Stability of (Hetero)Benzylic Alkenes via the Wittig Olefination Reaction. Molecules, 29(2), 501. https://doi.org/10.3390/molecules29020501