Abstract
The interaction of a series of metal derivatives of 5, 10, 15, 20-tetrakis(4-sulfonato-phenyl)porphyrin (MTPPS4, M = Cu(II), Pt(II), Ni(II), Zn(II) and Co(II)), including the metal free porphyrin (TPPS4), with the aromatic amino acids L-tryptophan (L-Trp), L-and D-phenylalanine (L-and D-Phe) and L-histidine (L-His) have been investigated through UV/Vis spectroscopy. The amino acid L-serine (L-Ser) has been included as reference compound. The spectroscopic changes induced by adding the amino acids have been exploited to evaluate the extent of interaction between the molecular components in the supramolecular adducts. The binding constants have been estimated for most of the investigated systems, assuming a simple 1:1 equilibrium. The bathochromic shifts of the B-bands, the extent of hypochromicity and the binding constants have been analyzed through two chemical descriptors. All the data point to the important role played by the steric hindrance introduced by axial ligands coordinated to the metal ions and to the degree of hydrophobicity and size of the aromatic moiety in the amino acids.
1. Introduction
Chirality in supramolecular chemistry is a highly investigated topic [1,2,3]. From a fundamental point of view, it is important to understand the mechanism of chirality transfer or propagation from a chiral monomeric building block to a complex supramolecular architecture [1,4]. More intriguing is the case when chirality occurs upon supramolecular assembling processes starting from achiral building blocks, in the presence of a proper chiral bias [3,5]. Many reports in the literature have pointed out the possibility of controlling the handedness of the final assemblies through chiral chemical [1,6,7,8,9,10,11] or physical stimuli [12,13,14,15,16,17,18,19,20]. In such a context, porphyrins are good model systems to access a variety of supramolecular aggregates. These compounds possess a quite large aromatic region that can lead to stacking interactions among neighboring porphyrins. Moreover, they are easily tunable by peripheral chemical modifications, introducing further side groups with different molecular recognition ability [21,22,23,24,25]. In addition, due to the presence of four central nitrogen atoms, these macrocycles are able to act as ligands for a large number of metal ions, thus exhibiting a rich coordination chemistry [26,27,28]. The size and extent of electronic conjugation of the aromatic core are responsible for their characteristic absorption in the visible range of the UV/Vis spectrum, which is dominated by a very intense B-band in the higher-energy portion accompanied by a variable number of weaker and lower-energy Q-bands [26,27,29]. Because the electronic spectra of porphyrins are very sensitive to slight changes in the microenvironment, their aggregation state or degree of interaction with other species can be easily accessed through modification of the energy and intensity of these bands. In the plethora of aggregated porphyrins, J-aggregates represent an interesting class due to their peculiar electronic features. According to Kasha [30], a lateral geometric arrangement of the chromophores determines a shift of the bands to lower energy, leading to a characteristic red-shifted band. Furthermore, motional narrowing is responsible for the sharpening of the so called J-band [31]. Meso-phenyl substituted porphyrins bearing a variable number of sulphonato groups in para position on the ring are able to self-assemble to form these aggregated species [13,21,32,33]. In particular, the tetra-anionic 5, 10, 15, 20-tetrakis(4-sulfonatophenyl)porphyrin (TPPS4) has been extensively investigated [34,35,36,37,38,39]. Aggregation can be easily triggered by lowering the pH and increasing the ionic strength, or in the presence of a variety of cationic species [40,41,42,43]. The main species—due to the initial protonation of the nitrogen core and eventually of a couple of sulphonato groups—are able to self-organize through an interplay of non-covalent interactions, including electrostatic, hydrogen-bonding and π-stacking contacts. The resulting nanoaggregates exhibit a variety of architectures [13,32], among which nanotubes have been widely characterized [39]. Chirality can be expressed in these latter ones when they are prepared in the presence of chiral stimuli [40,44,45,46,47,48,49,50,51,52,53,54] or even by spontaneous symmetry breaking [55,56,57]. Kinetic investigations have pointed out that the mechanism of growth is based on an initial nucleation stage that leads to the rate-determining formation of a trimer or tetramer of porphyrin. These latter species are the starting seeds of the auto-catalytic pathway for the eventual J-aggregates [41]. Therefore, all the experimental evidence suggests that the factor influencing the rates, i.e., counter-anions [58] and chiral inducers, seems to be operative at this level. Anyway, many reports in the literature deal with the interaction of simple amino acids with porphyrin receptors. Induction of chirality has been observed in a series of properly functionalized porphyrins, when the contact with the guest amino acid is effective [52,59,60,61,62,63,64,65,66]. In this context, it is important to understand whether the TPPS4 porphyrin, under conditions disfavoring the formation of J-aggregates, is able at monomeric level to form any kind of supramolecular species with a simple amino acid. Considering that electrostatics, hydrogen bonding and solvophobic interactions largely contribute to the stabilization of such species, we expect that the presence of an aromatic moiety on the side chain of the amino acid could provide a further driving force to the overall binding. Here we report an investigation on the interaction of some aromatic amino acids with a series of divalent metal ion derivatives of TPPS4, including the metal free porphyrin. In particular, Cu(II), Pt(II), Ni(II), Zn(II) and Co(II) have been selected to study the progressive impact of the coordination geometry imposed by the metal ion. Besides hampering the formation of J-aggregates, we anticipate that the absence/presence of axial ligands bound to the metal and generating steric hindrance on the porphyrin plane has a deep effect on the degree of interaction, as measured by the modification of the electronic absorption properties and the binding constants. The results are in line with the extent of hydrophobicity and the size of the aromatic region of the amino acids.
2. Results
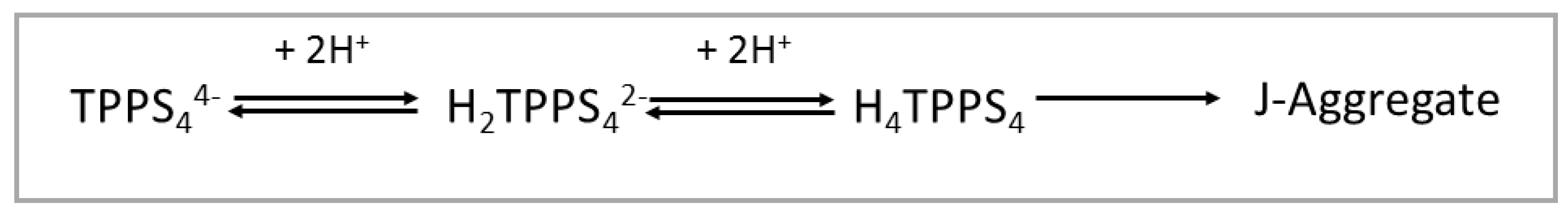
TPPS4 porphyrin leads to various species with different protonation degrees and overall charges depending on the acidity of the medium [35]. Under neutral pH conditions, this compound has an overall −4 charge (TPPS44−), since all the sulphonato groups are ionized. Since the pKa for the protonation of the nitrogen atoms of the macrocycle is 4.9 [67], below this value of pH the main prevailing species is a di-anion (H2TPPS42−). At pH lower than 3, a neutral zwitter-ion (H4TPPS4) forms due to protonation of two sulphonato groups [35]. This latter species can eventually self-organize into J-aggregates, under the proper conditions of pH, ionic strength and presence of cationic species (Scheme 1) [35].
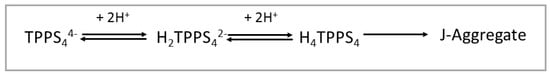
Scheme 1.
Protonation state for TPPS4 porphyrin and formation of J-aggregates as function of pH.
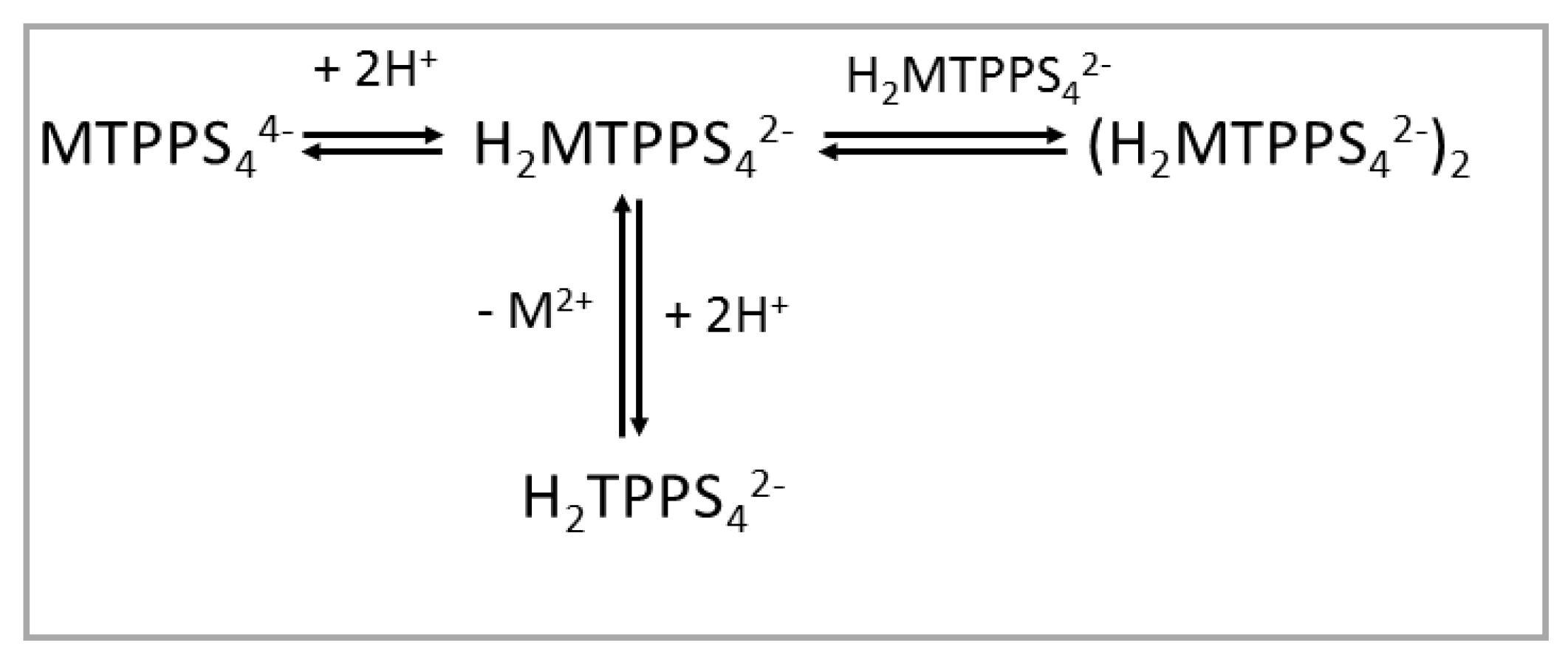
When the TPPS4 porphyrin is bound to divalent metal ions forming MTPPS44− coordination complexes, the protonation behavior is obviously different, as the porphyrin core is blocked by the presence of the metal ion and the nitrogen atoms are not available for binding hydrogen ions. The effect of increasing the proton concentration is (i) protonation of at least two of the sulphonato groups, thus leading to a H2MTPPS42− species, and (ii) that depending on the lability of the metal ion, demetallation can take place (e.g., for the labile ZnTPPS44−) [42], with formation of the diacid species H2TPPS42−. In the case of CuTPPS44−, under quite acidic conditions, demetallation is still slow, and dimerization can occur (Scheme 2) [68]. Considering the higher inertness of Ni(II), Co(II), Cu(II) and Pt(II) with respect to the acid solvolysis reaction, at rather low porphyrin concentration and mild acidic conditions, all the relative metal derivatives do not undergo demetallation.
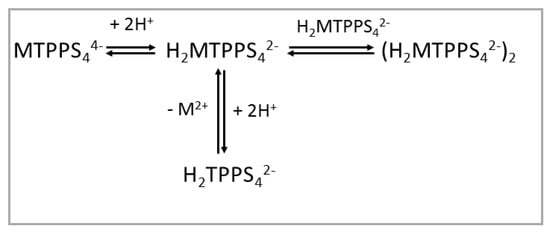
Scheme 2.
Protonation state for MTPPS44− porphyrins as a function of pH.
The conditions used in our experiments (pH = 4 in acetate buffer and porphyrin concentration 1.5 µM) ensure that all the porphyrin metal complexes are actually present as tetra-anions and that the metal free ligand is in its di-anionic H2TPPS42− form. As far as the amino acids are concerned, the values of their isoelectric points are higher than 4 (Trp, 5.89; Phe, 5.48; His, 7.59; Ser, 5.68). This condition implies that the species in solution is the mono-cation, histidine being the only exception due to the double protonation of the α-amino group and the imidazole nitrogen on the side chain.
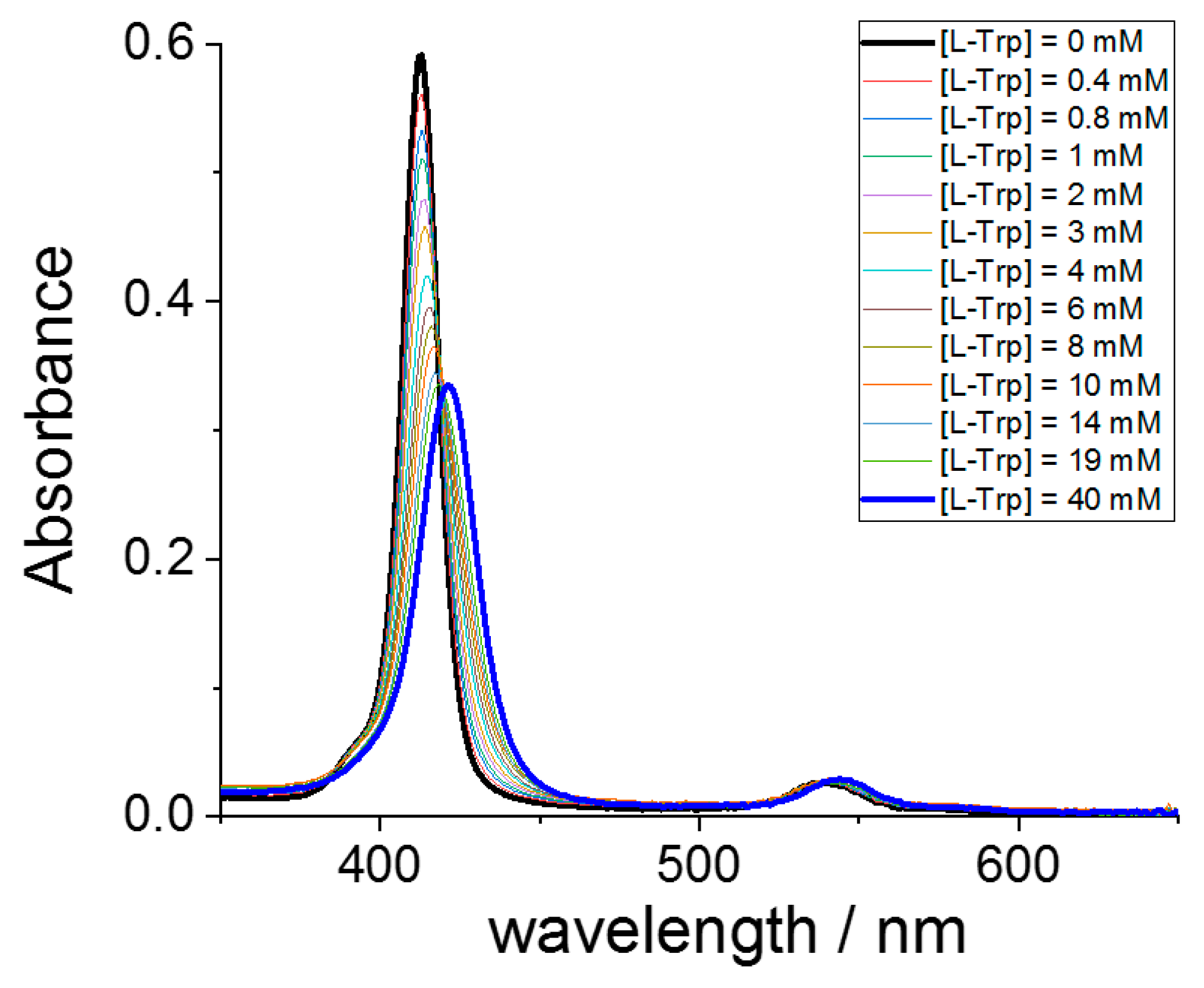
To study the interaction between the two components, we have performed a series of UV/Vis titrations by adding the various amino acids to the selected metal derivatives of TPPS4, including the metal free ligand. As an example, Figure 1 shows a typical spectral change observed for the interaction of CuTPPS44− and L-Trp. This porphyrin exhibits a strong B-band at 413 nm, accompanied by two weaker Q-bands at lower energy. The progressive addition of L-Trp leads to a bathochromic shift of the B-band that eventually moves to 421 nm (Δλ = +8 nm), accompanied by a consistent hypochromicity. Two distinct isosbestic points can be detected at 419 and 538 nm. Under these experimental conditions, J-aggregates do not form, as is possible to observe by the absence of the characteristic J-band at 490 nm.
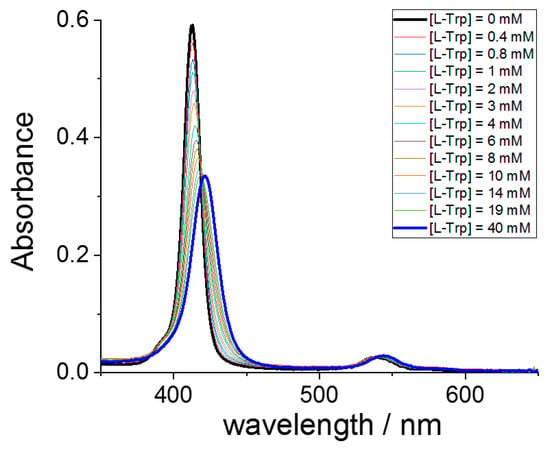
Figure 1.
UV/Vis absorbance spectral changes for the progressive titration of CuTPPS44− (black line) with L-Trp up to a concentration of 40 mM (blue line). Experimental conditions: [CuTPPS44−] = 1.5 μM; [L-Trp] = 0–40 mM; acetate buffer 100 mM pH = 4, T = 298 K, cell path length 1 cm.
The circular dichroism spectrum (CD) measured on the solution containing the higher L-Trp concentration shows no induced bands in the region of the porphyrin absorption, suggesting that tryptophan is not able to transfer the chiral information to the supramolecular adduct.
Analogous titration experiments have been performed for the other amino acids and porphyrins. In all these cases, the extent of the observed bathochromic shift and the hypochromicity are lower. In particular, in the case of CuTPPS44− with L-His and L-Ser, and CoTPPS44− with L-Trp, no shift of the B-band has been detected. The diacid porphyrin H2TPPS42− does not evidence any red shift, even if its Soret band undergoes a discrete reduction in intensity. To evaluate the extent of hypochromicity, we have applied the following relationship: H% = 100 × (Absmax − Absmin)/Absmax, where H% is the percentage of hypochromicity, Absmax is the maximum absorbance value for the initial concentration of porphyrin in the absence of amino acid at the B-band and Absmin is the final absorbance value for the adduct with the amino acid. The observation of bathochromic shifts in the B-bands, in association with hypochromicity, points to the electronic interaction between the porphyrin (P) and the amino acids (AA), thus leading to the supramolecular species P@AA, in agreement with similar findings in the literature. The amount of red shift is generally related to the stacking interaction of the aromatic portion of the amino acid and the porphyrin macrocycle. All the data relative to the bathochromic shifts and the hypochromicity (H%) are collected in Table 1. For all the final adducts, the CD spectra do not display any induced signal in the B-band region. This observation is different from what is normally observed for the aggregated form of the free ligand, since chirality is easily induced by amino acids and other chiral species in J-aggregates of the parent TPPS4 porphyrin [52]. The occurrence of a chiral arrangement of the porphyrin building blocks in the growing supramolecular structure at the nano- and mesoscopic levels is proved by the appearance of strong CD bands, whose sign is controlled by the handedness of the added chiral templating reagents. Our experimental evidence suggests that the chiral bias should play a role at the level of the rate-determining step of the J-aggregates formation. Kinetic investigations have already pointed out that, after a preliminary nucleation period, the auto-catalytic pathway leading to the final aggregated species is controlled by the formation of trimers or tetramers [35,52]. Consequently, the formation of a chiral assembly should involve such species and not the simple monomeric porphyrin.

Table 1.
Bathochromic shift of the B-band (Δλ), hypochromicity (H%), equilibrium binding constants (Keq) and molar extinction coefficients (εP@AA) for the P@AA supramolecular adducts a.
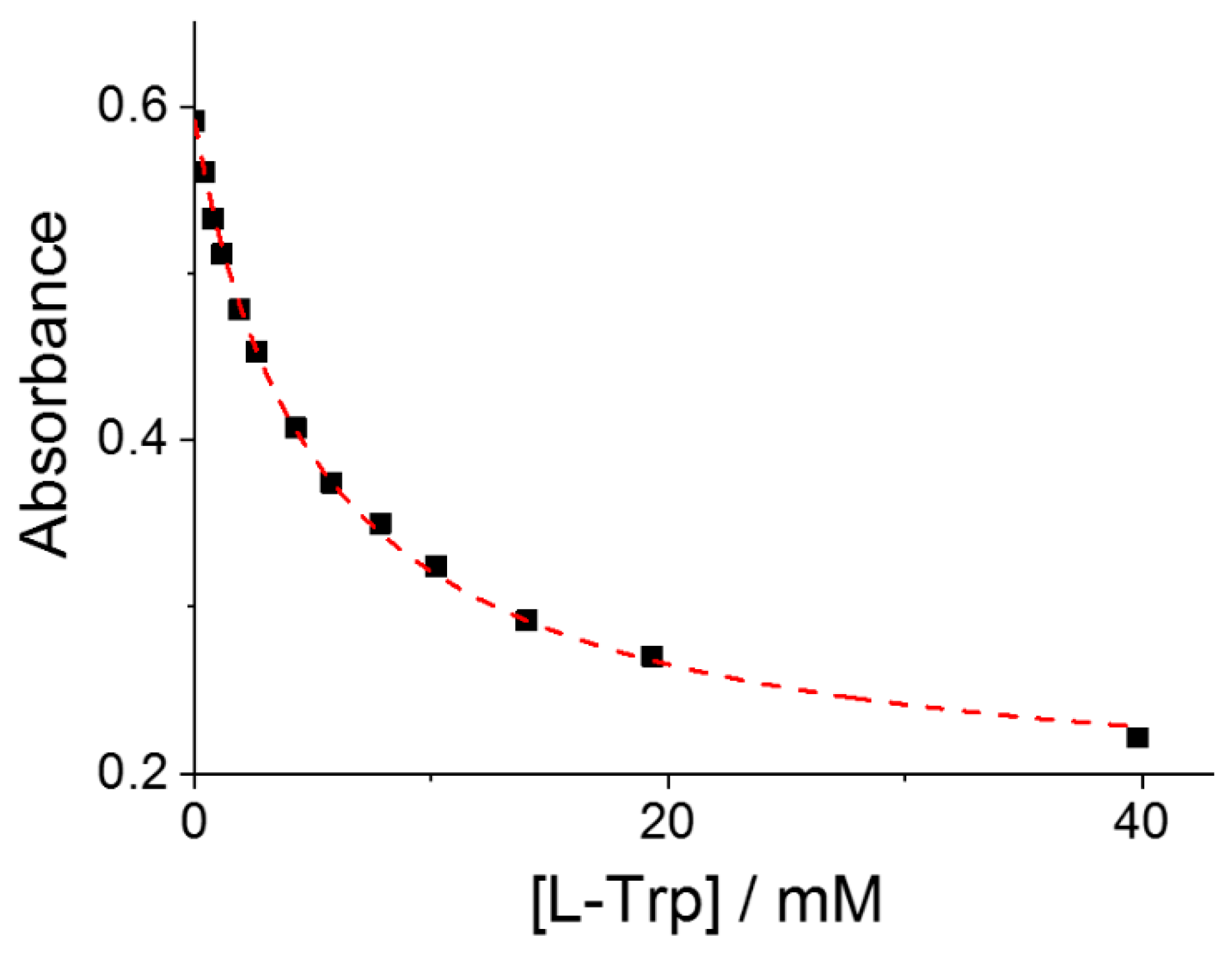
The binding constants (Keq) for the formation of the supramolecular adduct P@AA have been evaluated assuming a simple 1:1 equilibrium between the porphyrin and the selected amino acid. Consequently, the absorbance data collected at the wavelength corresponding to the maximum spectral change, usually at the B-band of the starting porphyrin, have been fitted to Equation (6) as a function of the total amino acid concentration (see Section 3). Figure 2 shows a typical curve fitting obtained for the titration of CuTPPS44− with L-Trp. This procedure allowed us to obtain the values of Keq and the molar extinction coefficient, εP@AA, for the various adducts P@AA, except for the cases where the spectral changes were negligible (CuTPPS44− with L-Ser and CoTPPS44− with L-Trp). All these data are collected in Table 1 (Figures S1–S11).
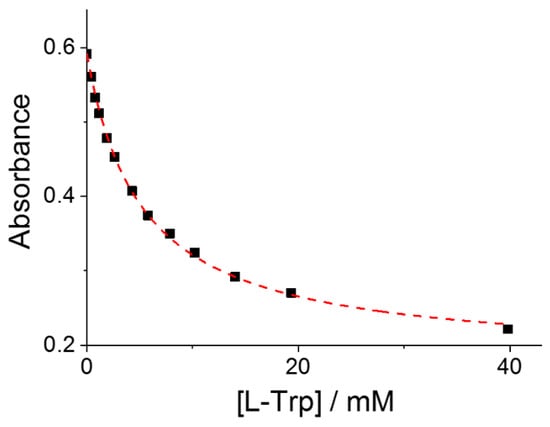
Figure 2.
Absorbance changes at the B-band of CuTPPS44− as a function of the total concentration of added L-Trp. The red curve is the best fitting of the experimental data to Equation (6) (see Section 3): Keq = 212 ± 5; εP@AA = (1.14 ± 0.03) × 105 M−1cm−1; R2 = 0.9989. Experimental conditions: [CuTPPS44−] = 1.5 μM; [L-Trp] = 0–40 mM; acetate buffer 100 mM, pH = 4; T = 298 K; cell path length 1 cm.
The experimental findings allow us to correlate the degree of interaction in the various supramolecular adducts and the structural features of both porphyrins and amino acids. As previously mentioned, the choice of the metal ions in MTPPS44− has been oriented to the possibility of modulating the steric properties of the macrocycle. Whereas Pt(II) and Cu(II) usually form tetra-coordinated complexes with a square planar geometry, Ni(II) has the ability to vary the coordination number spanning from 4 to 6 [69], Zn(II) is penta-coordinated and Co(II) in most cases binds its ligands, affording hexa-coordinated complexes. In our experimental pH conditions, the axial ligand should be water in all the metal complexes with a coordination number higher than 4. Therefore, we can organize the various metal complexes MTPPS44− in increasing order of steric hindrance, according to the following series: CuTPPS44− ~ PtTPPS44− ~ NiTPPS44− < ZnTPPS44− < CoTPPS44−. The metal free ligand H2TPPS42− used for the sake of comparison is not planar, but in a saddled conformation, i.e., the pyrrole rings are alternatively oriented up and down with respect to the mean plane of the macrocycle. Considering that tryptophan is the amino acid that binds more tightly in comparison with the others, we decided to select this compound as a reference point.
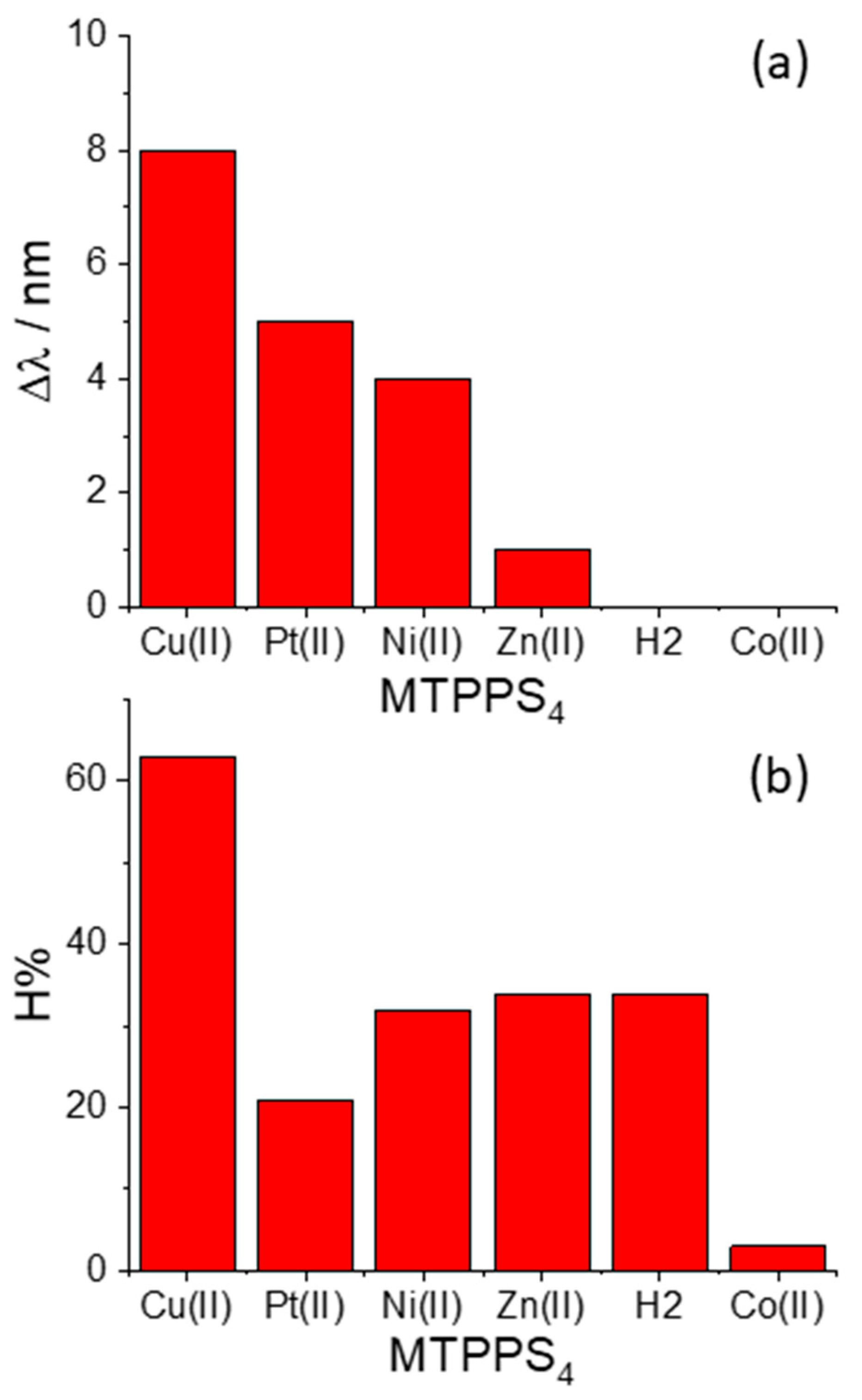
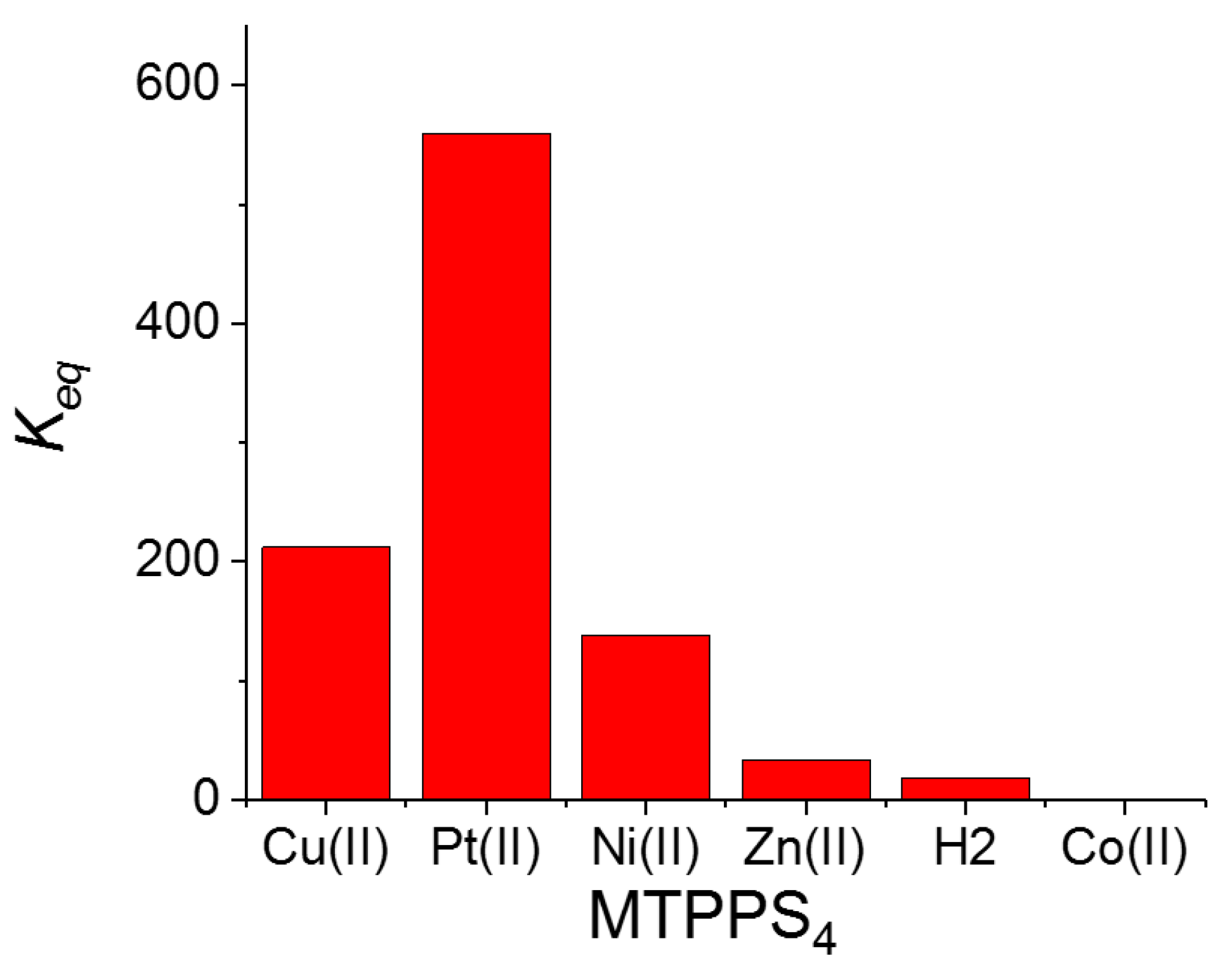
The pattern of behavior that the bathochromic shifts (Δλ) and the hypochromicity (H%) exhibit in the various species MTPPS44−@L-Trp is displayed in Figure 3a,b, respectively. An inspection of the data reveals that the Δλ values indicate the following sequence: CuTPPS44− > PtTPPS44− > NiTPPS44− >> ZnTPPS44− >> H2TPPS42− ~ CoTPPS44−. This result is in line with (i) the absence of axial ligands in the metal complexes containing Cu(II), Pt(II) and Ni(II), and (ii) an axial ligand in the case of the Zn(II) porphyrin and (iii) two axial ligands for Co(II) porphyrin. Therefore, the steric hindrance caused by two axial ligands coordinated above and below the porphyrin plane seems to play a role as important as the deformation of the macrocycle core induced by the protonation of the nitrogen atoms. When considering the hypochromicity, the data do not exhibit any relevant trend related to the number of coordinated solvent molecules, apart from the steep decrease observed for CoTPPS44− (Figure 3b). The values of the binding constants Keq are reported in Figure 4 and follow the trend: PtTPPS44− >> CuTPPS44− > NiTPPS44− >> ZnTPPS44− > H2TPPS42− > CoTPPS44−, which is in agreement with the gradual decrease of the binding force on increasing the number of the axial ligands on the porphyrin plane.
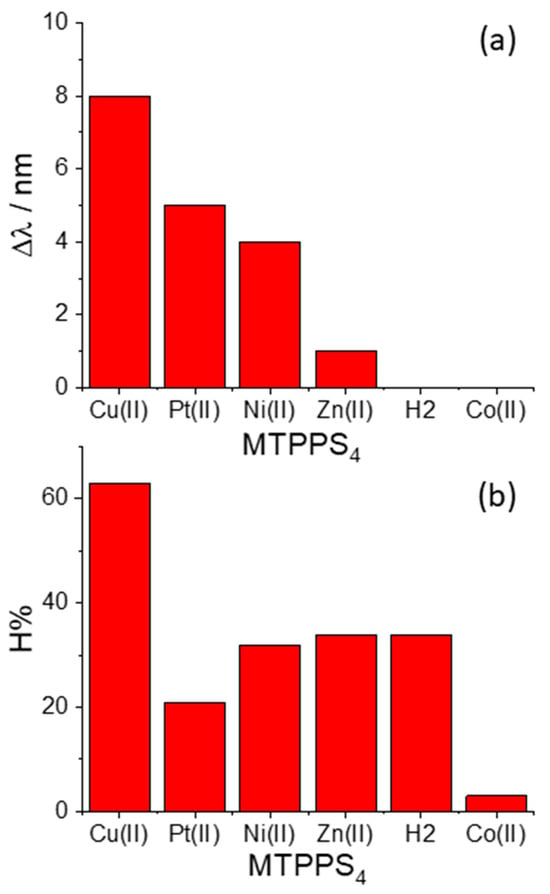
Figure 3.
Plot of the values for (a) the bathochromic shifts (Δλ) and (b) the hypochromicity degree (H%) for the various metal derivatives MTPPS44− with the amino acid L-Trp.
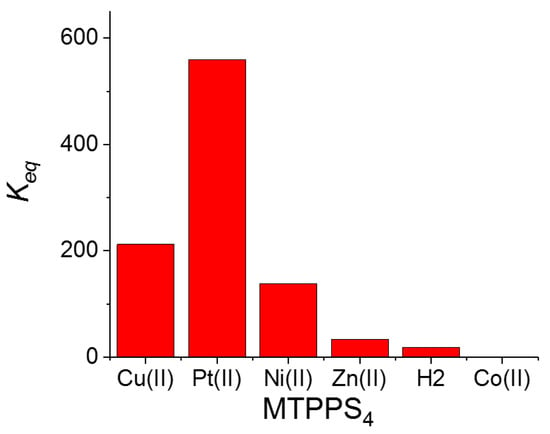
Figure 4.
Plot of the values for the equilibrium binding constants Keq for the various metal derivatives MTPPS44− with the amino acid L-Trp.
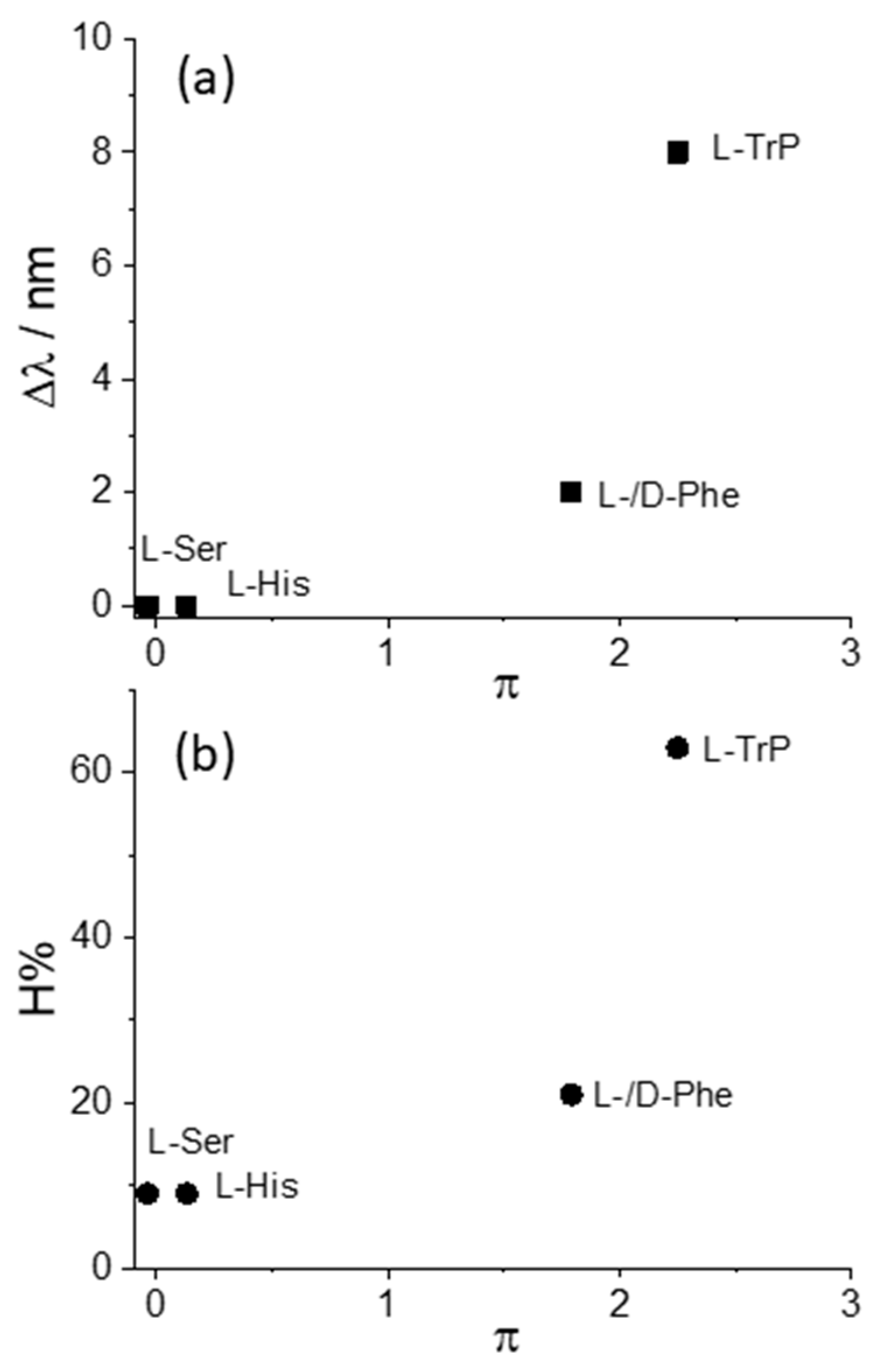
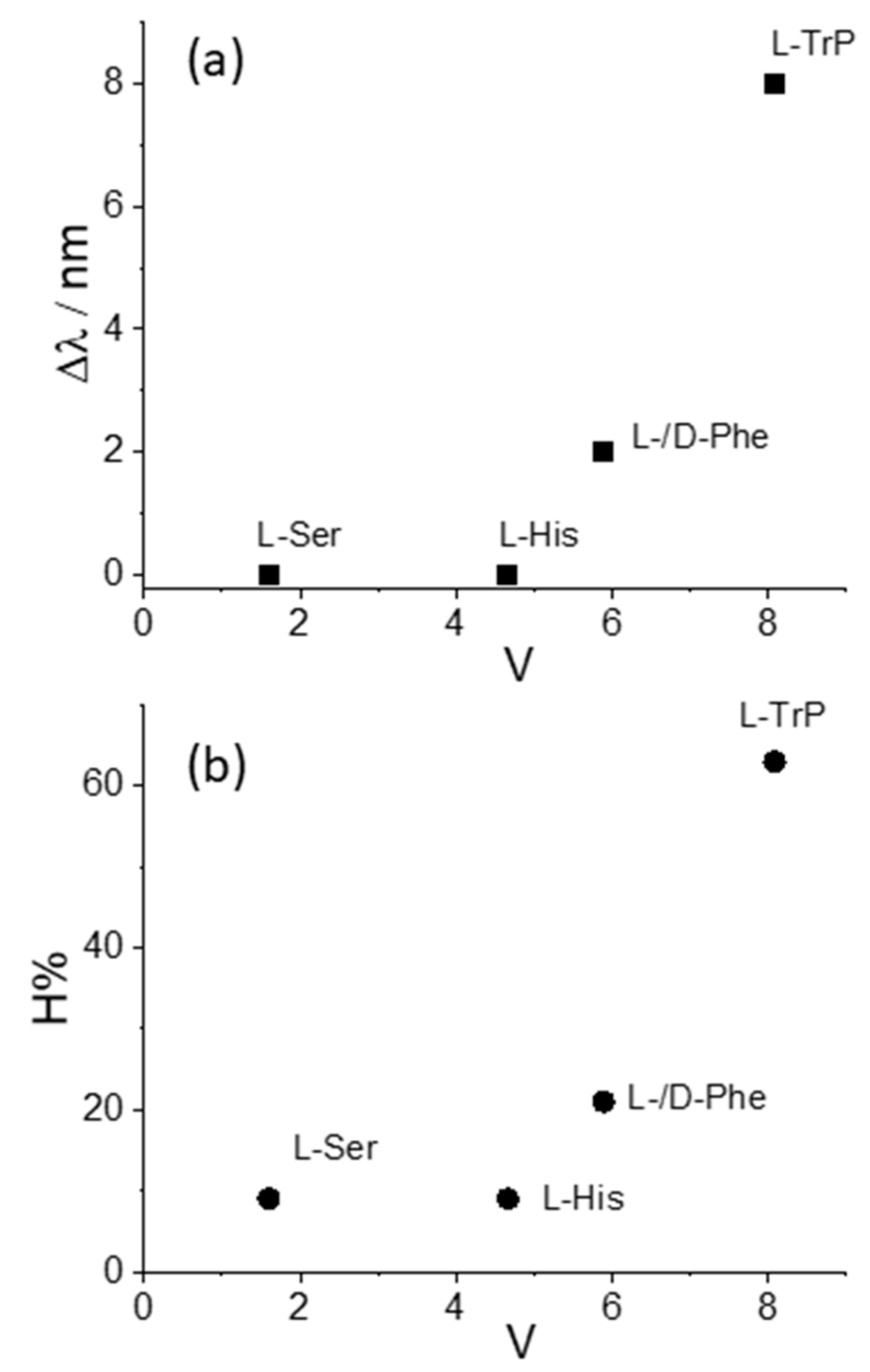
We decided to take advantage of some chemical descriptors, commonly used to analyze properties of the amino acids in proteins, to find a rationale in our data [70]. Taking into account the Cu derivative revealed to be the most sensible to interact with L-Trp, we adopted (i) hydrophobicity, π, defined as log P(amino acid) − log P(glycine), where log P is the partition coefficient of the specific amino acid in 1-octanol/water mixture, and (ii) the normalized van der Waals volume, V, of the side chain, calculated by the equation V = [V(side chain) − V(H)]/V(CH2) [70]. This parameter is 0 for glycine and 1 for alanine. The values of these two descriptors for the amino acids investigated in our study decrease in the order Trp > Phe > His > Ser, as expected based on the relative molecular size. Figure 5 and Figure 6 report the plots of Δλ and H% as functions of the hydrophobicity π and normalized van der Waals volume V, respectively, for the various amino acids. A monotonically increasing trend is evident in all these graphics, following the increase of the size of the aromatic side chain and the hydrophobicity of the amino acids.
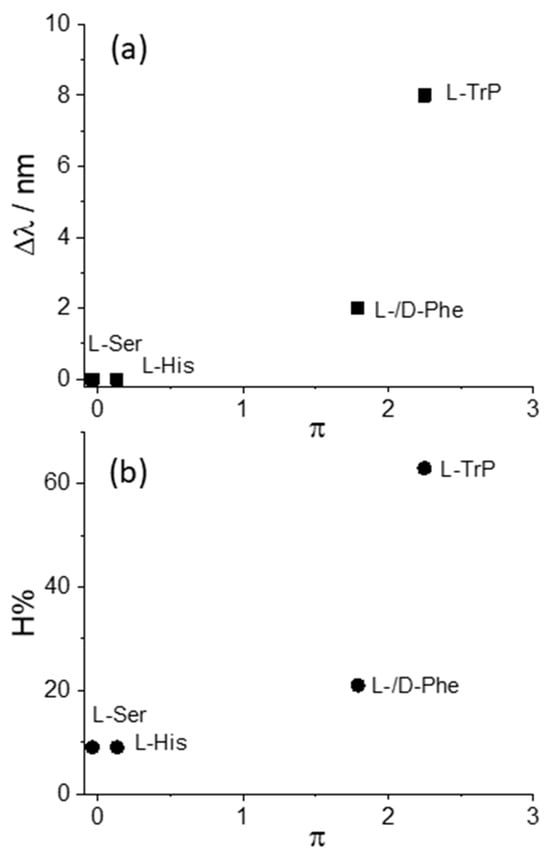
Figure 5.
Plot of the values for the (a) bathochromic shift values, Δλ, and (b) the hypochromicity extent, H%, as functions of the hydrophobicity parameter, π, for CuTPPS44− interacting with the various amino acids.
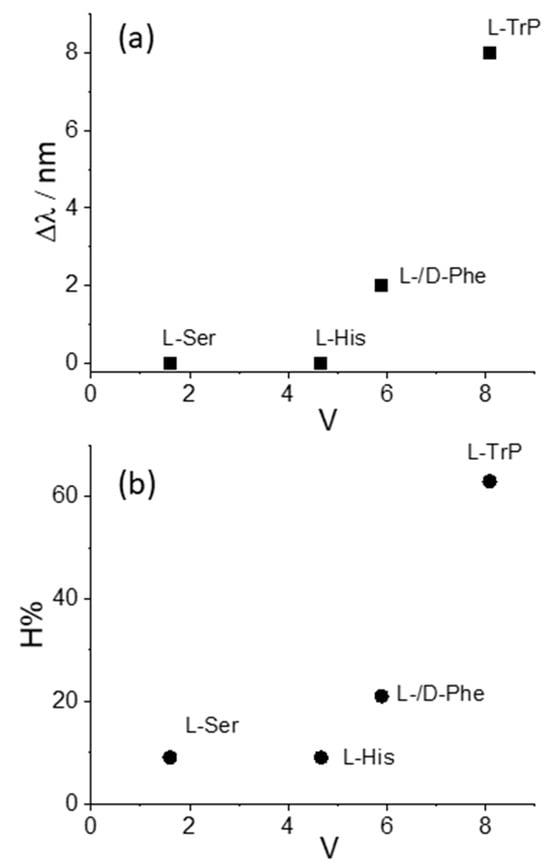
Figure 6.
Plot of the values for the (a) bathochromic shift values, Δλ, and (b) the hypochromicity extent, H%, as functions of the normalized van der Waals volume, V, for CuTPPS44− interacting with the various amino acids.
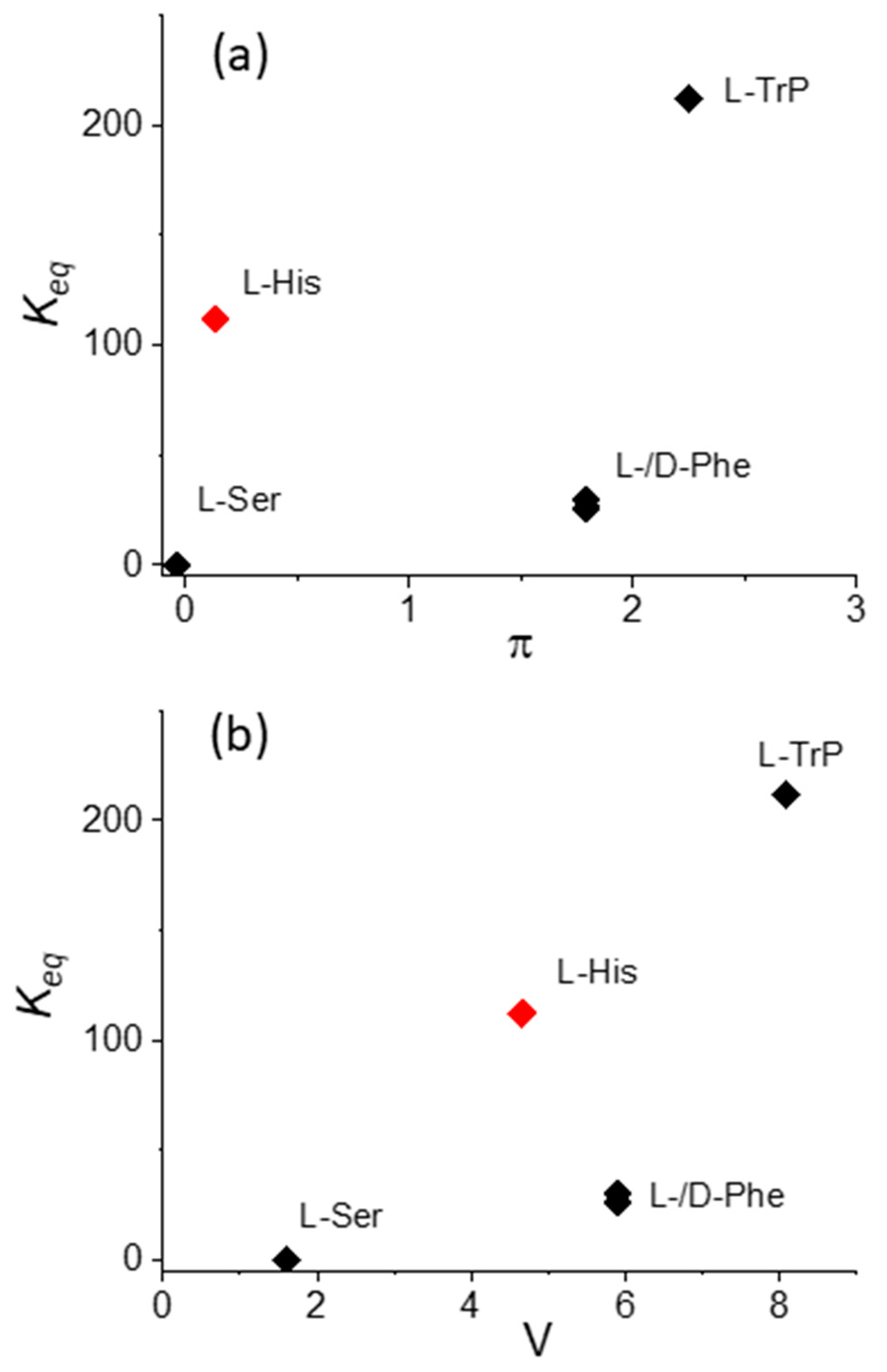
If the L-His data is excluded, Figure 7 shows that also in the case of the binding constants Keq, a monotonically increasing trend, with both π and V increasing, is evident. The anomalous position of the value for L-His should be explained by (i) the presence of the donor nitrogen atom on the imidazole residue, which is able to coordinate or interact with the Cu(II) metal center in the macrocycle, and (ii) the overall charge on this specific amino acid, which, as pointed out above, at pH 4 is +2, due to the protonation of both the α-amino group and the imidazole nitrogen on the side chain. It is interesting to observe that, as expected, an inspection of the data relative to the enantiomers L- and D-Phe does not evidence any difference in terms of electronic interactions and binding constant values.
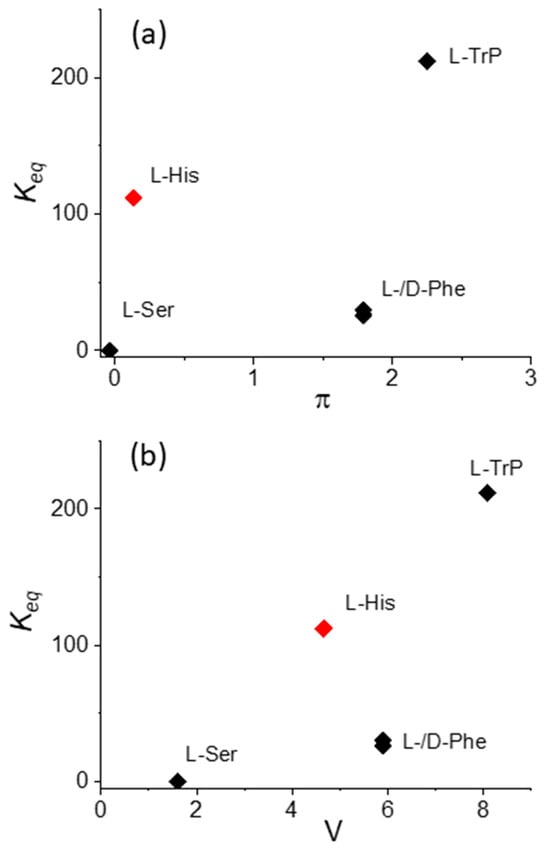
Figure 7.
Plot of the values for the equilibrium binding constants Keq versus (a) hydrophobicity, π, and (b) the normalized van der Waals volume, V, for CuTPPS44− interacting with the various amino acids.
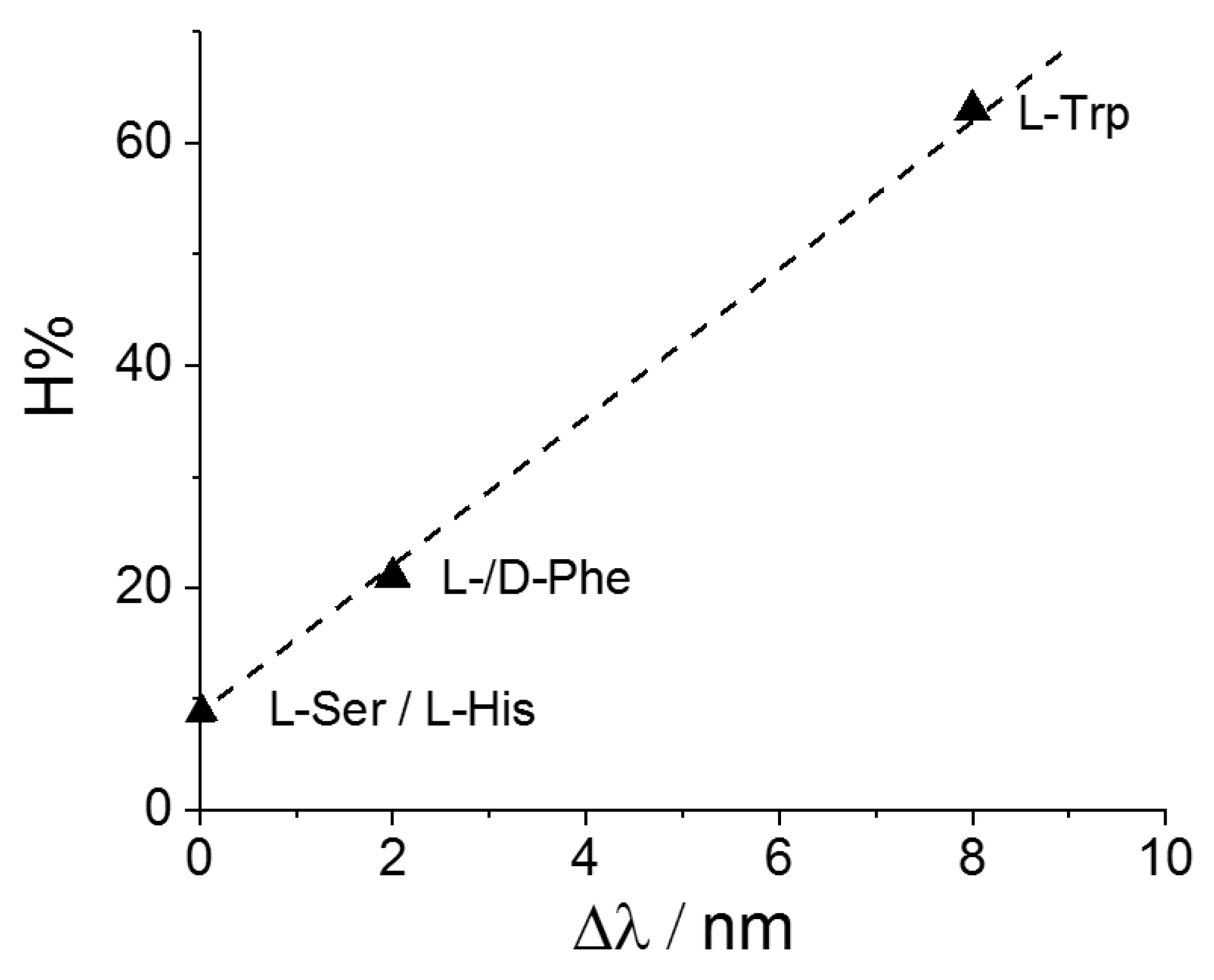
Figure 8 displays a plot of the hypochromicity extent H% versus the bathochromic shift Δλ for the adducts CuTPPS44−@AA. It is interesting to observe that these two spectroscopic parameters are linearly dependent, at least for this specific metal derivative. This evidence suggests that a common mechanism is operative and related to the extent of perturbation exerted by the aromatic moiety of the amino acid on the electronic distribution of the porphyrin core, reflected both in the hypochromicity and the bathochromic shift of the band. On the other hand, no clear correlation has been observed when the different MTPPS44−@L-Trp adducts are considered. All the spectroscopic data suggest that electrostatics and stacking interactions are the main driving forces in stabilizing these supramolecular species.
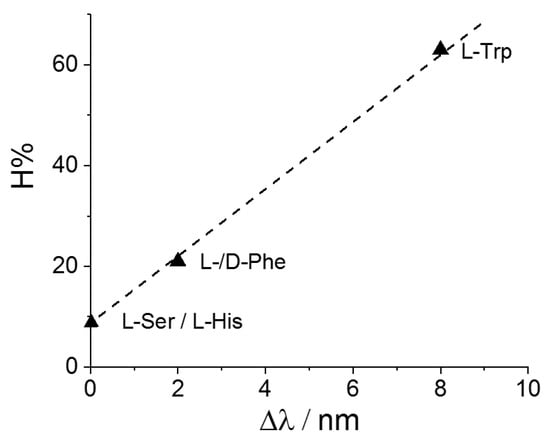
Figure 8.
Plot of the values for the hypochromicity extent, H%, as a function of the observed bathochromic shift values, Δλ, for CuTPPS44− interacting with the various amino acids.
3. Materials and Methods
3.1. Materials
5, 10, 15, 20-tetrakis(4-sulfonatophenyl)porphyrin (TPPS4), as sodium salt, was received from Aldrich (Milan, Italy). All the amino acids (L-tryptophan, L-phenylalanine, D-phenylalanine, L-histidine and L-serine), sodium acetate and acetic acid were of the highest commercial grade available and were used as received, without further purification, from Sigma-Aldrich (Milan, Italy). All the aqueous solutions were prepared in high-purity doubly distilled water (HPLC grade, Fluka, Milan, Italy). Stock solutions of the various porphyrins (100–200 μM) were freshly prepared and stored in the dark to avoid photo-damage. The concentration of the samples used in the experiments was calculated by UV/Vis absorption spectroscopy using the molar extinction coefficients at the B-band (TPPS4: 5.33 × 105 M−1 cm−1, λ = 414 nm; NiTPPS4: 2.7 × 105 M−1 cm−1, λ = 409 nm; ZnTPPS4: 6.8 × 105 M−1 cm−1, λ = 422 nm; CuTPPS4: 4.2 × 105 M−1 cm−1, λ = 413 nm; CoTPPS4: 2.7 × 105 M−1 cm−1, λ = 425 nm; PtTPPS4: 3.7 × 105 M−1 cm−1, λ = 397 nm). The amino acid solutions for the titration experiments have been prepared by solubilizing these compounds in 100 mM acetate buffer at pH = 4.
3.2. Methods
UV/Vis extinction spectra were collected on an Agilent 8453 diode array spectrophotometer. In order to prevent the light-induced degradation of the porphyrin solutions during the experiments, we used a UV filter (Hoya glass type UV-34, cut-off: 340 nm) set between the lamp and the samples. Temperature was controlled at 298 K by an external water-circulating bath. CD spectra were recorded on a Jasco J-710 spectropolarimeter, Jasco Europe.
Titration experiments were performed by collecting UV/Vis spectra on solutions contained in quartz Hellma cells placed in the thermostatic holder of the spectrophotometer. In a general procedure, small aliquots of a stock solution containing the selected AA (40 mM) and porphyrin (1.5 µM) were progressively added to 2 mL of a prediluted MTPPS44− 1.5 μM solution in a 1 cm pathlength cell. The absorbance values (Abs) have been collected at the wavelength of maximum change, generally at the B-band of the initial MTPPS44− species. In order to evaluate the equilibrium binding constants, we have considered a simple 1:1 equilibrium between porphyrin and AA ligand:
with an equilibrium binding constant
P + AA ⇄ P@AA
Keq = [P@AA]/([P] × [AA])
Considering the mass balance of porphyrin,
[P]T = [P] + [P@AA]
([P] and [P]T are the free and total porphyrin concentration, respectively, while [P@AA] is the supramolecular adduct concentration) and AA ligand,
[<=>AA]T = [AA] + [P@AA]
([AA] and [AA]T are the free and total AA concentration, respectively), the following equation can be derived:
from which the value of the free porphyrin concentration [P] can be obtained:
Keq[P]2 + (1 + Keq([AA]T − [P]T)) [P] − [P]T = 0
[P] = −(1 + Keq ([AA]T − [P]T)) + ((1 + Keq ([AA]T − [P]T))2 + 4 Keq [P]T)1/2}/(2 Keq)
The absorbance data have been analyzed through a non-linear best fitting procedure of the absorbance data at the B-band maximum as a function of the total AA concentration, according to the following equation:
where εP and εP@AA are the molar extinction coefficients of the metal derivatives and the supramolecular adducts with the AA, respectively.
Abs = εP [P] + εP@AA ([P]T − [P])
4. Conclusions
Amino acids bearing an aromatic residue on the side chain are able to interact with TPPS4 and its metal complexes MTPPS4 through a combination of weak interactions. Among these forces, electrostatics play a very important role as, under the experimental conditions adopted in our experiments, the AA are in cationic form and the MTPPS4 derivatives are tetra-anions, while TPPS4 is a di-anion. In addition, hydrogen bonding and hydrophobic interactions through π-stacking of the aromatic moieties are present on both the interacting species in the supramolecular adducts. Experimental evidence based on the electronic perturbation and the binding equilibrium constant values indicates that the absence of steric hindrance above and below the porphyrin plane favors the interaction with AA. This condition is ensured by transition metal ions exhibiting square planar coordination geometry (i.e., Cu(II), Pt(II) and Ni(II)). The introduction of axial ligands leads to a steep decrease in the extent of interaction. In the case of Co(II)TPPS4, the presence of two solvent molecules bound to the metal center reduces even further the possibility of π-stacking contacts. The role of the size of the aromatic region of the AA finds a rationale considering descriptor parameters, i.e., the hydrophobicity and the normalized van der Waals volume, that have been developed in the literature to model the behavior of these compounds in proteins and other biological systems.
The absence of any detectable induced CD spectra of the adducts between the metallo-porphyrins and the AA points to a role of these latter compounds in the aggregation pathway to chiral J-aggregates only at the level of the rate-determining step, when trimers or tetramers must be formed.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules29020472/s1, Figures S1–S8: UV/Vis absorption spectral changes and absorbance changes at the B-band of MTPPS44− as function of the total concentration of different added aminoacids. Figure S9: UV/Vis absorption spectral changes and absorbance changes at the B-band of H2TPPS44− as function of the total concentration of added L-Trp. Figure S10: UV/Vis spectra of CoTPPS44− with and without L-Trp. Figure S11: UV/Vis spectra of CuTPPS44− with and without L-Ser.
Author Contributions
R.Z. and M.A.C. equally contributed to this work; Conceptualization, L.M.S.; investigation, M.A.C. and R.Z.; formal analysis, R.Z. and M.A.C.; data curation, M.A.C., M.T. and A.R.; writing—original draft preparation, L.M.S.; writing—review and editing, all authors. All authors have read and agreed to the published version of the manuscript.
Funding
The Authors thank MUR-FFARB for financial support and Next Generation EU, PNRR Samothrace Project (ECS00000022).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data are available upon request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Jia, S.Z.; Tao, T.T.; Xie, Y.J.; Yu, L.Y.; Kang, X.; Zhang, Y.; Tang, W.W.; Gong, J.B. Chirality Supramolecular Systems: Helical Assemblies, Structure Designs, and Functions. Small 2023. [Google Scholar] [CrossRef]
- Ariga, K.; Mori, T.; Kitao, T.; Uemura, T. Supramolecular Chiral Nanoarchitectonics. Adv. Mat. 2020, 32, 1905657. [Google Scholar] [CrossRef]
- Mateos-Timoneda, M.A.; Crego-Calama, M.; Reinhoudt, D.N. Supramolecular chirality of self-assembled systems in solution. Chem. Soc. Rev. 2004, 33, 363–372. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, L.; Wang, T. Supramolecular Chirality in Self-Assembled Systems. Chem. Rev. 2015, 115, 7304–7397. [Google Scholar] [CrossRef] [PubMed]
- Travagliante, G.; Gaeta, M.; Purrello, R.; D′Urso, A. Supramolecular Chirality in Porphyrin Self-Assembly Systems in Aqueous Solution. Curr. Org. Chem. 2022, 26, 563–579. [Google Scholar] [CrossRef]
- Palmans, A.R.A.; Meijer, E.W. Amplification of Chirality in Dynamic Supramolecular Aggregates. Angew. Chem. Int. Ed. 2007, 46, 8948–8968. [Google Scholar] [CrossRef]
- Besenius, P.; Portale, G.; Bomans, P.H.H.; Janssen, H.M.; Palmans, A.R.A.; Meijer, E.W. Controlling the growth and shape of chiral supramolecular polymers in water. Proc. Natl. Acad. Sci. USA 2010, 107, 17888–17893. [Google Scholar] [CrossRef] [PubMed]
- Dou, X.Q.; Mehwish, N.; Zhao, C.L.; Liu, J.Y.; Xing, C.; Feng, C.L. Supramolecular Hydrogels with Tunable Chirality for Promising Biomedical Applications. Acc. Chem. Res. 2020, 53, 852–862. [Google Scholar] [CrossRef] [PubMed]
- Dorca, Y.; Greciano, E.E.; Valera, J.S.; Gómez, R.; Sánchez, L. Hierarchy of Asymmetry in Chiral Supramolecular Polymers: Toward Functional, Helical Supramolecular Structures. Chem. Eur. J. 2019, 25, 5848–5864. [Google Scholar] [CrossRef]
- Xing, P.Y.; Zhao, Y.L. Controlling Supramolecular Chirality in Multicomponent Self-Assembled Systems. Acc. Chem. Res. 2018, 51, 2324–2334. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.Y.; Chen, Z.H.; Shao, K.N.; Qing, G.Y.; Sun, T.L. Stimuli-Directed Helical Chirality Inversion and Bio-Applications. Polymers 2016, 8, 310. [Google Scholar] [CrossRef] [PubMed]
- Ribo, J.M.; Crusats, J.; Sagues, F.; Claret, J.; Rubires, R. Chiral Sign Induction by Vortices During the Formation of Mesophases in Stirred Solutions. Science 2001, 292, 2063–2066. [Google Scholar] [PubMed]
- Escudero, C.; Crusats, J.; Díez-Pérez, I.; El-Hachemi, Z.; Ribó, J.M. Folding and Hydrodynamic Forces in J-Aggregates of 5-Phenyl-10,15,20-tris(4-sulfophenyl)porphyrin. Angew. Chem. Int. Ed. 2006, 45, 8032–8035. [Google Scholar] [CrossRef]
- Arteaga, O.; Canillas, A.; Purrello, R.; Ribo, J.M. Evidence of induced chirality in stirred solutions of supramolecular nanofibers. Opt. Lett. 2009, 34, 2177–2179. [Google Scholar] [CrossRef] [PubMed]
- El-Hachemi, Z.; Balaban, T.S.; Campos, J.L.; Cespedes, S.; Crusats, J.; Escudero, C.; Kamma-Lorger, C.S.; Llorens, J.; Malfois, M.; Mitchell, G.R.; et al. Effect of Hydrodynamic Forces on meso-(4-Sulfonatophenyl)-Substituted Porphyrin J-Aggregate Nanoparticles: Elasticity, Plasticity and Breaking. Chem. Eur. J. 2016, 22, 9740–9749. [Google Scholar] [CrossRef] [PubMed]
- Crusats, J.; El-Hachemi, Z.; Ribo, J.M. Hydrodynamic effects on chiral induction. Chem. Soc. Rev. 2010, 39, 569. [Google Scholar] [PubMed]
- D′Urso, A.; Randazzo, R.; Lo Faro, L.; Purrello, R. Vortexes and Nanoscale Chirality. Angew. Chem. Int. Ed. 2010, 49, 108–112. [Google Scholar] [CrossRef]
- Sun, J.S.; Li, Y.K.; Yan, F.S.; Liu, C.; Sang, Y.T.; Tian, F.; Feng, Q.; Duan, P.F.; Zhang, L.; Shi, X.H.; et al. Control over the emerging chirality in supramolecular gels and solutions by chiral microvortices in milliseconds. Nat. Commun. 2018, 9, 2599. [Google Scholar] [CrossRef] [PubMed]
- Nicosia, A.; Vento, F.; Marletta, G.; Messina, G.M.L.; Satriano, C.; Villari, V.; Micali, N.; De Martino, M.T.; Schotman, M.J.G.; Mineo, P.G. Porphyrin-Based Supramolecular Flags in the Thermal Gradients′ Wind: What Breaks the Symmetry, How and Why. Nanomaterials 2021, 11, 1673. [Google Scholar] [CrossRef]
- Mineo, P.; Villari, V.; Scamporrino, E.; Micali, N. New Evidence about the Spontaneous Symmetry Breaking: Action of an Asymmetric Weak Heat Source. J. Phys. Chem. B 2015, 119, 12345–12353. [Google Scholar] [CrossRef]
- Pasternack, R.F.; Huber, P.R.; Boyd, P.; Engasser, G.; Francesconi, L.; Gibbs, E.; Fasella, P.; Cerio Venturo, G.; Hinds, L.d. Aggregation of meso-substituted water-soluble porphyrins. J. Am. Chem. Soc. 1972, 94, 4511–4517. [Google Scholar] [CrossRef]
- Kobayashi, T. J-Aggregates; World Scientific: Singapore, 1996. [Google Scholar]
- Monti, D.; Cantonetti, V.; Venanzi, M.; Ceccacci, F.; Bombelli, C.; Mancini, G. Interaction of a chirally functionalised porphyrin derivative with chiral micellar aggregates. Chem. Commun. 2004, 8, 972–973. [Google Scholar] [CrossRef] [PubMed]
- Balaban, T.S.; Bhise, A.D.; Fischer, M.; Linke-Schaetzel, M.; Roussel, C.; Vanthuyne, N. Controlling Chirality and Optical Properties of Artificial Antenna Systems with Self-Assembling Porphyrins. Angew. Chem. Int. Ed. 2003, 42, 2140–2144. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Ma, X.; Duan, P.; Liu, M. Chirality Amplification of Porphyrin Assemblies Exclusively Constructed from Achiral Porphyrin Derivatives. ChemPhysChem 2006, 7, 2419–2423. [Google Scholar] [CrossRef] [PubMed]
- Hambright, P. Chemistry of Water-Soluble Porphyrins. In The Porphyrin Handbook; Kadish, K.M., Smith, K.M., Guilard, R., Eds.; Academic Press: New York, NY, USA, 2000; Volume 3, pp. 129–210. [Google Scholar]
- Hambright, P. Coordination chemistry of metalloporphyrins. Coord. Chem. Rev. 1971, 6, 247–268. [Google Scholar] [CrossRef]
- Smykalla, L.; Mende, C.; Fronk, M.; Siles, P.F.; Hietschold, M.; Salvan, G.; Zahn, D.R.T.; Schmidt, O.G.; Rüffer, T.; Lang, H. (Metallo)porphyrins for potential materials science applications. Beilstein J. Nanotechnol. 2017, 8, 1786–1800. [Google Scholar] [CrossRef]
- Baker, H.; Hambrigh, P.; Wagner, L.; Ross, L. Metal-ion interactions with porphyrins exchange and substitution-reactions. Inorg. Chem. 1973, 12, 2200–2202. [Google Scholar] [CrossRef]
- Kasha, M.; Rawls, H.R.; Ashraf El-Bayoumi, M. The exciton model in molecular spectroscopy. Pure Appl. Chem. 1965, 11, 371–392. [Google Scholar] [CrossRef]
- Knapp, E.W. Lineshapes of molecular aggregates, exchange narrowing and intersite correlation. Chem. Phys. 1984, 85, 73–82. [Google Scholar] [CrossRef]
- Pleckaitis, M.; Habach, F.; Kontenis, L.; Steinbach, G.; Jarockyte, G.; Kalnaityte, A.; Domonkos, I.; Akhtar, P.; Alizadeh, M.; Bagdonas, S.; et al. Structure and principles of self-assembly of giant “sea urchin” type sulfonatophenyl porphine aggregates. Nano Res. 2022, 15, 5527–5537. [Google Scholar] [CrossRef]
- Elemans, J.; Van Hameren, R.; Nolte, R.J.M.; Rowan, A.E. Molecular materials by self-assembly of porphyrins, phthalocyanines, and perylenes. Adv. Mat. 2006, 18, 1251–1266. [Google Scholar] [CrossRef]
- Snitka, V.; Rackaitis, M.; Rodaite, R. Assemblies of TPPS4 porphyrin investigated by TEM, SPM and UV–vis spectroscopy. Sens. Actuators B Chem. 2005, 109, 159–166. [Google Scholar] [CrossRef]
- Zagami, R.; Castriciano, M.A.; Romeo, A.; Scolaro, L.M. J-aggregates of 5, 10, 15, 20-tetrakis(4-sulfonatophenyl)-porphyrin. An overview of the supramolecular self-assembling mechanism. J. Porphyr. Phthalocyanines 2023, 27, 463–470. [Google Scholar] [CrossRef]
- Ribo, J.M.; Crusats, J.; Farrera, J.A.; Valero, M.L. Aggregation in Water Solutions of Tetrasodium Diprotonated Meso-Tetrakis(4-Sulfonatophenyl)Porphyrin. J. Am. Chem. Soc. Chem. Commun. 1994, 6, 681–682. [Google Scholar] [CrossRef]
- Ribo, J.M.; Rubires, R.; El-Hachemi, Z.; Farrera, J.A.; Campos, L.; Pakhomov, G.L.; Vendrell, M. Self-assembly to ordered films of the homoassociate solutions of the tetrasodium salt of 5, 10, 15, 20-tetrakis(4-sulfonatophenyl) porphyrin dihydrochloride. Mater. Sci. Eng. C Biomim. Supramol. Syst. 2000, 11, 107–115. [Google Scholar]
- Sorrenti, A.; El-Hachemi, Z.; Crusats, J.; Ribo, J.M. Effects of flow-selectivity on self-assembly and auto-organization processes: An example. Chem. Commun. 2011, 47, 8551–8553. [Google Scholar] [CrossRef]
- Short, J.M.; Berriman, J.A.; Kübel, C.; El-Hachemi, Z.; Naubron, J.-V.; Balaban, T.S. Electron Cryo-Microscopy of TPPS4⋅2HCl Tubes Reveals a Helical Organisation Explaining the Origin of their Chirality. ChemPhysChem 2013, 14, 3209–3214. [Google Scholar] [CrossRef]
- El-Hachemi, Z.; Escudero, C.; Acosta-Reyes, F.; Casas, M.T.; Altoe, V.; Aloni, S.; Oncins, G.; Sorrenti, A.; Crusats, J.; Campos, J.L.; et al. Structure vs. properties—Chirality, optics and shapes—In amphiphilic porphyrin J-aggregates. J. Mater. Chem. C 2013, 1, 3337–3346. [Google Scholar] [CrossRef]
- Occhiuto, I.G.; Zagami, R.; Trapani, M.; Castriciano, M.A.; Romeo, A.; Scolaro, L.M. Kinetic Investigation on Tetrakis(4-Sulfonatophenyl)Porphyrin J-Aggregates Formation Catalyzed by Cationic Metallo-Porphyrins. Molecules 2020, 25, 5742. [Google Scholar] [CrossRef]
- Occhiuto, I.G.; Castriciano, M.A.; Trapani, M.; Zagami, R.; Romeo, A.; Pasternack, R.F.; Monsù Scolaro, L. Controlling J-Aggregates Formation and Chirality Induction through Demetallation of a Zinc(II) Water Soluble Porphyrin. Int. J. Mol. Sci. 2020, 21, 4001. [Google Scholar] [CrossRef]
- Toncelli, C.; Pino-Pinto, J.P.; Sano, N.; Picchioni, F.; Broekhuis, A.A.; Nishide, H.; Moreno-Villoslada, I. Controlling the aggregation of 5, 10, 15, 20-tetrakis-(4-sulfonatophenyl)-porphyrin by the use of polycations derived from polyketones bearing charged aromatic groups. Dye Pigment 2013, 98, 51–63. [Google Scholar] [CrossRef]
- Zhang, L.; Yuan, J.; Liu, M. Supramolecular Chirality of Achiral TPPS Complexed with Chiral Molecular Films. J. Phys. Chem. B 2003, 107, 12768–12773. [Google Scholar] [CrossRef]
- Jiang, S.; Liu, M. Aggregation and Induced Chirality of an Anionic meso-Tetraphenylsulfonato Porphyrin (TPPS) on a Layer-by-Layer Assembled DNA/PAH Matrix. J. Phys. Chem. B 2004, 108, 2880–2884. [Google Scholar]
- Zhang, L.; Liu, M.H. Supramolecular Chirality and Chiral Inversion of Tetraphenylsulfonato Porphyrin Assemblies on Optically Active Polylysine. J. Phys. Chem. B 2009, 113, 14015–14020. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Wang, X.; Li, Y.; Ma, R.; An, Y.; Shi, L. Chiral Micelles of Achiral TPPS and Diblock Copolymer Induced by Amino Acids. Macromolecules 2009, 42, 6253–6260. [Google Scholar] [CrossRef]
- Trapani, M.; Castriciano, M.A.; Romeo, A.; De Luca, G.; Machado, N.; Howes, B.D.; Smulevich, G.; Scolaro, L.M. Nanohybrid Assemblies of Porphyrin and Au-10 Cluster Nanoparticles. Nanomaterials 2019, 9, 1026. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, T.Y.; Jiang, J.Z.; Liu, M.H. Chiral porphyrin assemblies. Aggregate 2023, 4, e198. [Google Scholar] [CrossRef]
- Randazzo, R.; Gaeta, M.; Gangemi, C.M.A.; Fragalà, M.E.; Purrello, R.; D’Urso, A. Chiral Recognition of L- and D- Amino Acid by Porphyrin Supramolecular Aggregates. Molecules 2019, 24, 84. [Google Scholar]
- Oliveras-González, C.; Linares, M.; Amabilino, D.B.; Avarvari, N. Large Synthetic Molecule that either Folds or Aggregates through Weak Supramolecular Interactions Determined by Solvent. ACS Omega 2019, 4, 10108–10120. [Google Scholar] [CrossRef]
- Zagami, R.; Castriciano, M.A.; Romeo, A.; Scolaro, L.M. Kinetic Investigations on the Chiral Induction by Amino Acids in Porphyrin J-Aggregates. Int. J. Mol. Sci. 2023, 24, 1695. [Google Scholar] [CrossRef]
- Zhao, L.; Liu, M.; Li, S.; Li, A.; An, H.; Ye, H.; Zhang, Y. Aggregation and supramolecular chirality of 5, 10, 15, 20-tetrakis-(4-sulfonatophenyl)-porphyrin on an achiral poly(2-(dimethylamino)ethyl methylacrylate)-grafted ethylene-vinyl alcohol membrane. J. Mater. Chem. C 2015, 3, 3650–3658. [Google Scholar] [CrossRef]
- Lu, J.; Wu, L.; Jiang, J.; Zhang, X. Helical Nanostructures of an Optically Active Metal-Free Porphyrin with Four Optically Active Binaphthyl Moieties: Effect of Metal–Ligand Coordination on the Morphology. Eur. J. Inorg. Chem. 2010, 2010, 4000–4008. [Google Scholar] [CrossRef]
- Castriciano, M.A.; Cardillo, S.; Zagami, R.; Trapani, M.; Romeo, A.; Scolaro, L.M. Effects of the Mixing Protocol on the Self-Assembling Process of Water Soluble Porphyrins. Int. J. Mol. Sci. 2021, 22, 797. [Google Scholar] [CrossRef]
- Zagami, R.; Romeo, A.; Castriciano, M.A.; Monsù Scolaro, L. Inverse Kinetic and Equilibrium Isotope Effects on Self-Assembly and Supramolecular Chirality of Porphyrin J-Aggregates. Chem. Eur. J. 2017, 23, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Arlegui, A.; Soler, B.; Galindo, A.; Arteaga, O.; Canillas, A.; Ribó, J.M.; El-Hachemi, Z.; Crusats, J.; Moyano, A. Spontaneous mirror-symmetry breaking coupled to top-bottom chirality transfer: From porphyrin self-assembly to scalemic Diels-Alder adducts. Chem. Commun. 2019, 55, 12219–12222. [Google Scholar] [CrossRef] [PubMed]
- Occhiuto, I.G.; Zagami, R.; Trapani, M.; Bolzonello, L.; Romeo, A.; Castriciano, M.A.; Collini, E.; Monsu Scolaro, L. The role of counter-anions in the kinetics and chirality of porphyrin J-aggregates. Chem. Commun. 2016, 52, 11520–11523. [Google Scholar] [CrossRef]
- Monti, D.; Venanzi, M.; Stefanelli, M.; Sorrenti, A.; Mancini, G.; Di Natale, C.; Paolesse, R.J. Chiral amplification of chiral porphyrin derivatives by templated heteroaggregation. J. Am. Chem. Soc. 2007, 129, 6688–6689. [Google Scholar]
- Wang, Q.; Chen, Y.; Ma, P.; Lu, J.; Zhang, X.; Jiang, J.J. Morphology and chirality controlled self-assembled nanostructures of porphyrin–pentapeptide conjugate: Effect of the peptide secondary conformation. J. Mater. Chem. C 2011, 21, 8057–8065. [Google Scholar] [CrossRef]
- Wang, S.J.; Ruan, W.J.; Zhao, X.J.; Luo, D.B.; Zhu, Z.A.J. Molecular recognition of chiral zinc porphyrin with amino acid esters. Chin. J. Chem. 2005, 23, 44–49. [Google Scholar] [CrossRef]
- Charalambidis, G.; Georgilis, E.; Panda, M.K.; Anson, C.E.; Powell, A.K.; Doyle, S.; Moss, D.; Jochum, T.; Horton, P.N.; Coles, S.J.; et al. A switchable self-assembling and disassembling chiral system based on a porphyrin-substituted phenylalanine–phenylalanine motif. Nat. Commun. 2016, 7, 12657. [Google Scholar] [CrossRef]
- Jintoku, H.; Sagawa, T.; Takafuji, M.; Ihara, H. Chirally self-assembled porphyrin nanowires assisted by L-glutamide-derived lipid for excitation energy transfer. Org. Biomol. Chem. 2009, 7, 2430–2434. [Google Scholar] [CrossRef]
- Jintoku, H.; Shimoda, S.; Takafuji, M.; Sagawa, T.; Ihara, H. Tuning of molecular orientation of porphyrin assembly according to monitoring the chiroptical signals. J. Mol. Cryst. Liq. Cryst. 2011, 539, 63/[403]–67/[407]. [Google Scholar] [CrossRef]
- Rananaware, A.; La, D.D.; Al Kobaisi, M.; Bhosale, R.S.; Bhosale, S.V.; Bhosale, S.V.J. Controlled chiral supramolecular assemblies of water soluble achiral porphyrins induced by chiral counterions. Chem. Commun. 2016, 52, 10253–10256. [Google Scholar] [CrossRef] [PubMed]
- De Luca, G.; Romeo, A.; Scolaro, L.M.; Pasternack, R.F. Conformations of a model protein revealed by an aggregating CuII porphyrin: Sensing the difference. Chem. Commun. 2010, 46, 389–391. [Google Scholar] [CrossRef] [PubMed]
- Kalyanasundaram, K. Photochemistry of Polypyridine and Porphyrin Complexes; Academic Press: London, UK, 1992; p. 428. [Google Scholar]
- Trapani, M.; Occhiuto, I.G.; Zagami, R.; De Luca, G.; Castriciano, M.A.; Romeo, A.; Scolaro, L.M.; Pasternack, R.F. Mechanism for Copper(II)-Mediated Disaggregation of a Porphyrin J-Aggregate. ACS Omega 2018, 3, 18843–18848. [Google Scholar] [CrossRef]
- Pasternack, R.F.; Francesconi, L.; Raff, D.; Spiro, E. Aggregation of nickel(II), copper(II), and zinc(II) derivatives of water-soluble porphyrins. Inorg. Chem. 1973, 12, 2606–2611. [Google Scholar] [CrossRef]
- Meiler, J.; Müller, M.; Zeidler, A.; Schmäschke, F. Generation and evaluation of dimension-reduced amino acid parameter representations by artificial neural networks. J. Mol. Model 2001, 7, 360–369. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).