
CYP1-Activation and Anticancer Properties of Synthetic Methoxylated Resveratrol Analogues

 , , , , and
, , , , and
Abstract
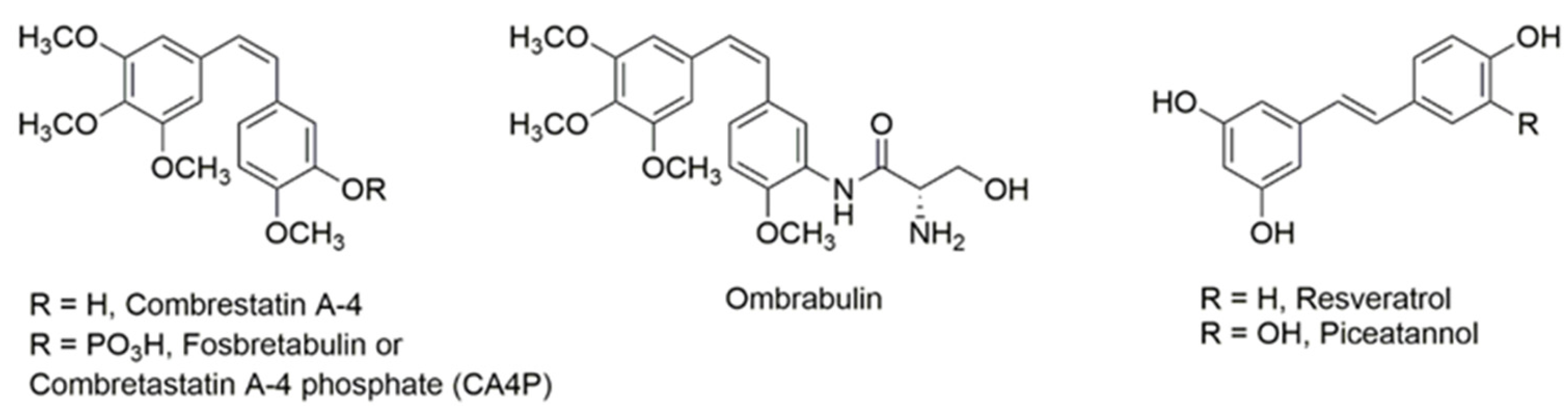
1. Introduction
2. Results
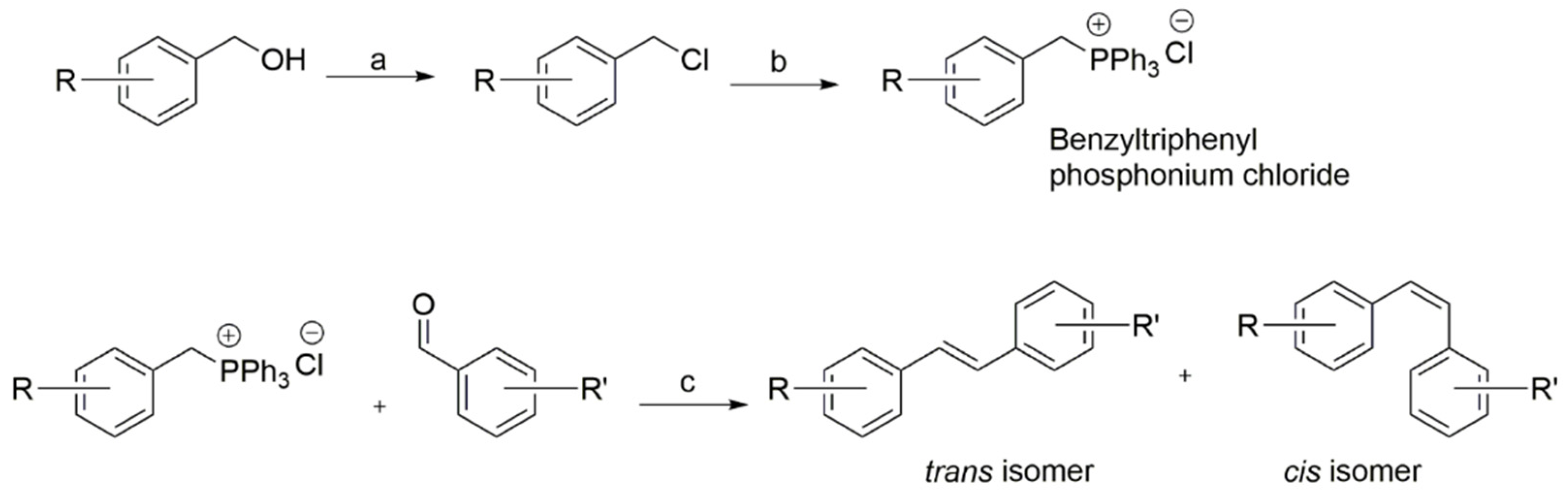
2.1. Synthesis of New Resveratrol Analogues
2.2. Determination of Antiproliferative Activity
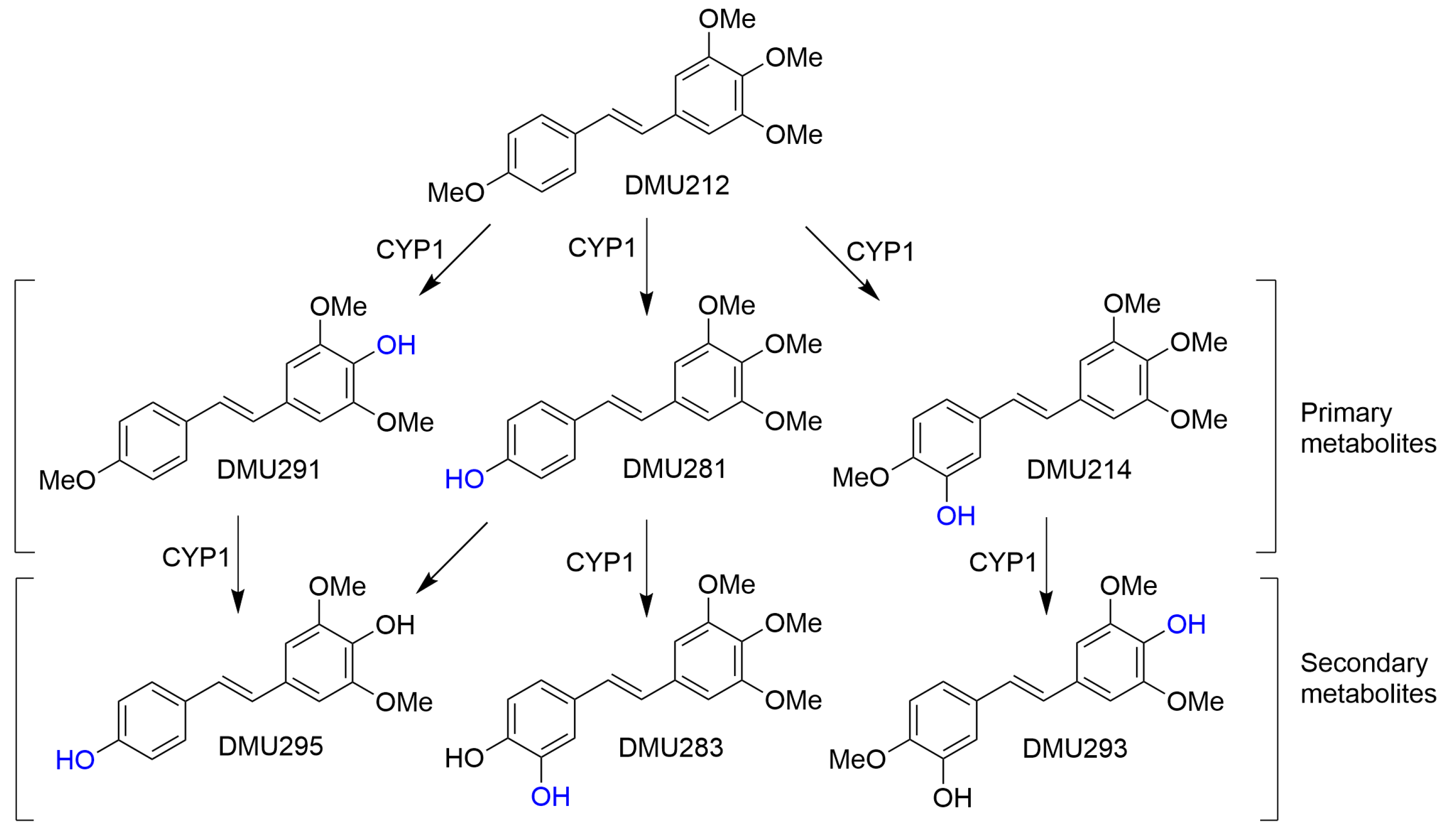
2.3. CYP1-Catalysed Bioconversion of DMU212
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. General Synthetic Procedures
4.3. General Procedure for the Preparation of Stilbenes
4.4. Biological Assays
4.4.1. MTT Cytotoxicity Assay
4.4.2. Enzyme Assays
4.4.3. HPLC Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M.; Snader, K.M. Natural products as sources of new drugs over the period 1981–2002. J. Nat. Prod. 2003, 66, 1022–1037. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmann, V.K.; Fürst, R. Plant-derived vascular disrupting agents: Compounds, actions, and clinical trials. Phytochem. Rev. 2014, 13, 191–206. [Google Scholar] [CrossRef]
- Pecyna, P.; Wargula, J.; Murias, M.; Kucinska, M. More Than Resveratrol: New Insights into Stilbene-Based Compounds. Biomolecules 2020, 10, 1111. [Google Scholar] [CrossRef] [PubMed]
- Eskens, F.A.; Tresca, P.; Tosi, D.; Van Doorn, L.; Fontaine, H.; Van der Gaast, A.; Veyrat-Follet, C.; Oprea, C.; Hospitel, M.; Dieras, V. A phase I pharmacokinetic study of the vascular disrupting agent ombrabulin (AVE8062) and docetaxel in advanced solid tumours. Br. J. Cancer 2014, 110, 2170–2177. [Google Scholar] [CrossRef] [PubMed]
- Birrer, M.J.; Bondarenko, I.; Tjulandin, S.; Vergote, I.; Cibula, D.; Ray-Coquard, I.; Colombo, N.; Allard, A.; Oprea, C.; Rey, A.A.; et al. Opsalin: A phase II placebo (Pbo)-controlled randomized study of ombrabulin in patients with platinum-sensitive recurrent ovarian cancer (OC) treated with carboplatin (Cb) and paclitaxel (P). J. Clin. Oncol. 2013, 31 (Suppl. S15), 5516. [Google Scholar] [CrossRef]
- Bhullar, K.S.; Hubbard, B.P. Lifespan and healthspan extension by resveratrol. Biochim. Biophys. Acta 2015, 1852, 1209–1218. [Google Scholar] [CrossRef]
- Kulkarni, S.S.; Cantó, C. The molecular targets of resveratrol. Biochim. Biophys. Acta 2015, 1852, 1114–1123. [Google Scholar] [CrossRef]
- Berman, A.Y.; Motechin, R.A.; Wiesenfeld, M.Y.; Holz, M.K. The therapeutic potential of resveratrol: A review of clinical trials. NPJ Precis. Oncol. 2017, 1, 35. [Google Scholar] [CrossRef]
- Behroozaghdam, M.; Dehghani, M.; Zabolian, A.; Kamali, D.; Javanshir, S.; Hasani Sadi, F.; Hashemi, M.; Tabari, T.; Rashidi, M.; Mirzaei, S.; et al. Resveratrol in breast cancer treatment: From cellular effects to molecular mechanisms of action. Cell Mol. Life Sci. 2022, 79, 539. [Google Scholar] [CrossRef]
- Şöhretoğlu, D.; Barut, B.; Sari, S.; Özel, A.; Kuruüzüm-Uz, A.; Arroo, R. In Vitro and In Silico Investigation of DNA Interaction, Topoisomerase I and II Inhibitory Properties of Polydatin. Chem. Biodivers. 2022, 19, e202200352. [Google Scholar] [CrossRef]
- Lalani, A.R.; Fakhari, F.; Radgoudarzi, S.; Rastegar-Pouyani, N.; Moloudi, K.; Khodamoradi, E.; Taeb, S.; Najafi, M. Immunoregulation by resveratrol; implications for normal tissue protection and tumour suppression. Clin. Exp. Pharmacol. Physiol. 2023, 50, 353–368. [Google Scholar] [CrossRef]
- Zhu, B.T.; Liehr, J.G. Inhibition of catechol O-methyltransferase-catalyzed O-methylation of 2- and 4-hydroxyestradiol by quercetin: Possible role in estradiol-induced tumorigenesis. J. Biol. Chem. 1996, 271, 1357–1363. [Google Scholar] [CrossRef] [PubMed]
- Takemura, H.; Itoh, T.; Yamamoto, K.; Sakakibara, H.; Shimoi, K. Selective inhibition of methoxyflavonoids on human CYP1B1 activity. Bioorganic Med. Chem. 2010, 18, 6310–6315. [Google Scholar] [CrossRef] [PubMed]
- D’Uva, G.; Baci, D.; Albini, A.; Noonan, D.M. Cancer chemoprevention revisited: Cytochrome P450 family 1B1 as a target in the tumor and the microenvironment. Cancer Treat Rev. 2018, 63, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cui, J.; Jia, J. The Activation of Procarcinogens by CYP1A1/1B1 and Related Chemo-Preventive Agents: A Review. Curr Cancer Drug Targets 2021, 21, 21–54. [Google Scholar] [CrossRef]
- Iacopetta, D.; Ceramella, J.; Catalano, A.; Scali, E.; Scumaci, D.; Pellegrino, M.; Aquaro, S.; Saturnino, C.; Sinicropi, M.S. Impact of Cytochrome P450 Enzymes on the Phase I Metabolism of Drugs. Appl. Sci. 2023, 13, 6045. [Google Scholar] [CrossRef]
- Foti, R.S. Cytochrome P450 and Other Drug-Metabolizing Enzymes as Therapeutic Targets. Drug Metab. Dispos. 2023, 51, 936–949. [Google Scholar] [CrossRef]
- Molina-Ortiz, D.; Camacho-Carranza, R.; González-Zamora, J.F.; Shalkow-Kalincovstein, J.; Cárdenas-Cardós, R.; Ností-Palacios, R.; Vences-Mejía, A. Differential expression of cytochrome P450 enzymes in normal and tumor tissues from childhood rhabdomyosarcoma. PLoS ONE 2014, 9, e93261. [Google Scholar] [CrossRef]
- Li, F.; Zhu, W.; Gonzalez, F.J. Potential role of CYP1B1 in the development and treatment of metabolic diseases. Pharmacol. Ther. 2017, 178, 18–30. [Google Scholar] [CrossRef]
- Androutsopoulos, V.P.; Arroo, R.R.J.; Hall, J.F.; Surichan, S.; Potter, G.A. Antiproliferative and cytostatic effects of the natural product eupatorin on MDA-MB-468 human breast cancer cells due to CYP1 mediated metabolism. Breast Cancer Res. 2008, 10, R39. [Google Scholar] [CrossRef]
- Androutsopoulos, V.; Wilsher, N.; Arroo, R.R.; Potter, G.A. Bioactivation of the phytoestrogen diosmetin by CYP1 cytochromes P450. Cancer Lett. 2009, 274, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Breinholt, V.M.; Offord, E.A.; Brouwer, C.; Nielsen, S.E.; Brosen, K.; Friedberg, T.H. In vitro investigation of cytochrome; P450-mediated metabolism of dietary flavonoids. Food Chem. Toxicol. 2002, 40, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Surichan, S.; Ruparelia, K.; Arroo, R.; Boarder, M.R. Tangeretin and its metabolite 4′-hydroxytetramethoxyflavone attenuate EGF-stimulated cell cycle progression in hepatocytes; role of inhibition at the level of mTOR/p70S6K. Br. J. Pharmacol. 2011, 162, 1781–1791. [Google Scholar] [CrossRef] [PubMed]
- Nagayoshi, H.; Murayama, N.; Tsujino, M.; Takenaka, S.; Katahira, J.; Kim, V.; Kim, D.; Komori, M.; Yamazaki, H.; Guengerich, F.P.; et al. Preference for O-demethylation reactions in the oxidation of 2′-, 3′-, and 4′-methoxyflavones by human cytochrome P450 enzymes. Xenobiotica 2020, 50, 1158–1169. [Google Scholar] [CrossRef] [PubMed]
- Wilsher, N.E.; Arroo, R.R.; Matsoukas, M.T.; Tsatsakis, A.M.; Spandidos, D.A.; Androutsopoulos, V.P. Cytochrome P450 CYP1 metabolism of hydroxylated flavones and flavonols: Selective bioactivation of luteolin in breast cancer cells. Food Chem. Toxicol. 2017, 110, 383–394. [Google Scholar] [CrossRef]
- Gazit, A.; Yaish, P.; Gilon, C.; Levitzki, A. Tyrphostins I: Synthesis and biological activity of protein tyrosine kinase inhibitors. J. Med. Chem. 1989, 32, 2344–2352. [Google Scholar] [CrossRef]
- Potter, G.A.; Patterson, L.H.; Wanogho, E.; Perry, P.J.; Butler, P.C.; Ijaz, T.; Ruparelia, K.C.; Lamb, J.H.; Farmer, P.B.; Stanley, L.A.; et al. The cancer preventative agent resveratrol is converted to the anticancer agent piceatannol by the cytochrome P450 enzyme CYP1B1. Br. J. Cancer 2002, 86, 774–778. [Google Scholar] [CrossRef]
- Chang, T.S. An updated review of tyrosinase inhibitors. Int. J. Mol. Sci. 2009, 10, 2440–2475. [Google Scholar] [CrossRef]
- Gandhi, H.; Mahant, S.; Sharma, A.K.; Kumar, D.; Dua, K.; Chellappan, D.K.; Singh, S.K.; Gupta, G.; Aljabali, A.A.A.; Tambuwala, M.M.; et al. Exploring the therapeutic potential of naturally occurring piceatannol in non-communicable diseases. Biofactors 2023. [Google Scholar] [CrossRef]
- Wenzel, E.; Somoza, V. Metabolism and bioavailability of trans-resveratrol. Mol. Nutr. Food Res. 2005, 49, 472–481. [Google Scholar] [CrossRef] [PubMed]
- Spink, D.C.; Spink, B.C.; Cao, J.Q.; DePasquale, J.A.; Pentecost, B.T.; Fasco, M.J.; Li, Y.; Sutter, T.R. Differential expression of CYP1A1 and CYP1B1 in human breast epithelial cells and breast tumor cells. Carcinogenesis 1998, 19, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Döhr, O.; Vogel, C.; Abel, J. Different response of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-sensitive genes in human breast cancer MCF-7 and MDA-MB 231 cells. Arch. Biochem. Biophys. 1995, 321, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Gibson, P.; Gill, J.H.; Khan, P.A.; Seargent, J.M.; Martin, S.W.; Batman, P.A.; Griffith, J.; Bradley, C.; Double, J.A.; Bibby, M.C.; et al. Cytochrome P450 1B1 (CYP1B1) is overexpressed in human colon adenocarcinomas relative to normal colon: Implications for drug development. Mol. Cancer Ther. 2003, 2, 527–534. [Google Scholar] [PubMed]
- Soule, H.D.; Maloney, T.M.; Wolman, S.R.; Peterson, W.D., Jr.; Brenz, R.; McGrath, C.M.; Russo, J.; Pauley, R.J.; Jones, R.F.; Brooks, S.C. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res. 1990, 50, 6075–6086. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Meth. 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Angus, W.G.; Larsen, M.C.; Jefcoate, C.R. Expression of CYP1A1 and CYP1B1 depends on cell-specific factors in human breast cancer cell lines: Role of estrogen receptor status. Carcinogenesis 1999, 20, 947–955. [Google Scholar] [CrossRef]
- Woods, J.A.; Hadfield, J.A.; Pettit, G.R.; Fox, B.W.; McGown, A.T. The interaction with tubulin of a series of stilbenes based on combretastatin A-4. Br. J. Cancer 1995, 71, 705–711. [Google Scholar] [CrossRef]
- Karatoprak, G.Ş.; Küpeli Akkol, E.; Genç, Y.; Bardakci, H.; Yücel, Ç.; Sobarzo-Sánchez, E. Combretastatins: An Overview of Structure, Probable Mechanisms of Action and Potential Applications. Molecules 2020, 25, 2560. [Google Scholar] [CrossRef]
- Surichan, S.; Androutsopoulos, V.P.; Sifakis, S.; Koutala, E.; Tsatsakis, A.; Arroo, R.R.; Boarder, M.R. Bioactivation of the citrus flavonoid nobiletin by CYP1 enzymes in MCF7 breast adenocarcinoma cells. Food Chem. Toxicol. 2012, 50, 3320–3328. [Google Scholar] [CrossRef]
- Yao, C.; Dai, S.; Wang, C.; Fu, K.; Wu, R.; Zhao, X.; Yao, Y.; Li, Y. Luteolin as a potential hepatoprotective drug: Molecular mechanisms and treatment strategies. Biomed Pharmacother. 2023, 167, 115464. [Google Scholar] [CrossRef] [PubMed]
- Sale, S.; Verschoyle, R.D.; Boocock, D.; Jones, D.J.; Wilsher, N.; Ruparelia, K.C.; Potter, G.A.; Farmer, P.B.; Steward, W.P.; Gescher, A.J. Pharmacokinetics in mice and growth-inhibitory properties of the putative cancer chemopreventive agent resveratrol and the synthetic analogue trans 3,4,5,4′-tetramethoxystilbene. Br. J. Cancer 2004, 90, 736–744. [Google Scholar] [CrossRef] [PubMed]
- Sale, S.; Tunstall, R.G.; Ruparelia, K.C.; Potter, G.A.; Steward, W.P.; Gescher, A.J. Comparison of the effects of the chemopreventive agent resveratrol and its synthetic analog trans 3,4,5,4′-tetramethoxystilbene (DMU-212) on adenoma development in the Apc(Min+) mouse and cyclooxygenase-2 in human-derived colon cancer cells. Int. J. Cancer 2005, 115, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Cushman, M.; Nagarathnam, D.; Gopal, D.; He, H.M.; Lin, C.M.; Hamel, E. Synthesis and evaluation of analogues of (Z)-1-(4-methoxyphenyl)-2-(3,4,5-trimethoxyphenyl)ethene as potential cytotoxic and antimitotic agents. J. Med. Chem. 1992, 35, 2293–2306. [Google Scholar] [CrossRef]
- Cavalieri, E.L.; Stack, D.E.; Devanesan, P.D.; Todorovic, R.; Dwivedy, I.; Higginbotham, S.; Johansson, S.L.; Patil, K.D.; Gross, M.L.; Gooden, J.K.; et al. Molecular origin of cancer: Catechol estrogen-3,4-quinones as endogenous tumor initiators. Proc. Natl. Acad. Sci. USA 1997, 94, 10937–10942. [Google Scholar] [CrossRef]
- Wang, W.L.; Porter, W.; Burghardt, R.; Safe, S.H. Mechanism of inhibition of MDA-MB-468 breast cancer cell growth by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Carcinogenesis 1997, 18, 925–933. [Google Scholar] [CrossRef][Green Version]
- Park, J.G.; Kramer, B.S.; Steinberg, S.M.; Carmichael, J.; Collins, J.M.; Minna, J.D.; Gazdar, A.F. Chemosensitivity testing of human colorectal carcinoma cell lines using a tetrazolium-based colorimetric assay. Cancer Res. 1987, 47, 5875–5879. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| DMU Code | R (A-Ring) | R’ (B-Ring) | IC50 (µM) a | AF | IC50 (µM) a | TS | ||
| MCF7 | MCF7 (TCDD) | MCF10A | MDA468 | |||||
| 205 | 4-OMe- | 4-OMe- | 28 | 28 | 1 | 100 | 100 | 1 |
| 241 | 4-OMe- | 3-OMe- | 27 | 24 | 1 | >100 | >100 | 1 |
| 216T | 4-OMe- | 3-Me- | 25 | 23 | 1 | >100 | >100 | 1 |
| 271 | 4-OMe- | 3,5-(OMe)2- | 14 | 14 | 1 | >100 | >100 | 1 |
| 507 | C6H5 | 3,4,5-(OMe)3 | 50 | 50 | 1 | 70 | 10 | 7 |
| 212 | 4-OMe | 3,4,5-(OMe)3 | 2.6 | 0.12 | 22 | 4.3 | 0.001 | 4300 |
| 547 | 4-OMe- | 2,3,4-(OMe)3 | 80 | 1 | 80 | 20 | 4.5 | 4 |
| 260 | 4-OEt- | 3,4,5-(OMe)3 | 5 | 5 | 1 | 3.7 | 1.8 | 2 |
| 555 | 4-Cl | 3,4,5-(OMe)3 | 100 | 100 | 1 | 8 | 12 | 0.7 |
| 557 | 4-Br | 3,4,5-(OMe)3 | 7 | 8 | 0.9 | 20 | 7 | 3 |
| 509 | 4-I | 3,4,5-(OMe)3 | 15 | 15 | 1 | 13 | 13 | 1 |
| 567 | 6-OMe-Naphthyl- | 3,4,5-(OMe)3 | 100 | 42 | 2.5 | 0.11 | 0.04 | 3 |
| 569 | 4-OMe-Naphthyl- | 3,4,5-(OMe)3 | 30 | 8.6 | 3.5 | 40 | 4.5 | 9 |
| 2417 | 3,4-(OMe)2- | 3,4,5-(OMe)3 | 14 | 14 | 1 | 18 | 9 | 2 |
| 219 | 2,4-(OMe)2- | 3,4,5-(OMe)3 | 1.7 | 1.4 | 1 | 1.4 | 0.8 | 2 |
| 220 | 3,4-OCH2O- | 3,4,5-(OMe)3 | 3.4 | 2.3 | 1.5 | 3.9 | 0.99 | 4 |
| Resveratrol | 4-OH- | 3,5-(OH)2 | 31 | 33 | 1 | 50 | 29 | 1.7 |
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| DMU Code | R (A-Ring) | R’ (B-Ring) | IC50 (µM) a | AF | IC50 (µM) a | TS | ||
| MCF7 | MCF7 (TCDD) | MCF10A | MDA468 | |||||
| 265 | 4-OMe- | 4-OMe | 25 | 20 | 1 | >100 | >100 | 1 |
| 241C | 4-OMe- | 3-OMe | 29 | 26 | 1 | >100 | >100 | 1 |
| 216C | 4-OMe- | 3-Me | 26 | 30 | 1 | >100 | >100 | 1 |
| 210 | 4-OMe- | 3,5-(OMe)2 | 26 | 24 | 1 | >100 | >100 | 1 |
| 508 | C6H5 | 3,4,5-(OMe)2 | 5 | 4 | 1 | 7 | 9 | 0.9 |
| 213 | 4-OMe | 3,4,5-(OMe)3 | 2.5 | 0.44 | 6 | 2 | 0.5 | 4 |
| 548 | 4-OMe | 3,4,5-(OMe)3 | 8 | 7.1 | 1 | 100 | 30 | 3 |
| 261 | 4-OEt | 3,4,5-(OMe)3 | 0.04 | 0.13 | 0.3 | 0.06 | 0.07 | 0.9 |
| 556 | 4-Cl | 3,4,5-(OME)3 | 0.8 | 0.8 | 1 | 35 | 7 | 5 |
| 558 | 4-Br | 3,4,5-(OMe)3 | 0.08 | 0.05 | 2 | 0.05 | 0.04 | 1 |
| 510 | 4-I | 3,4,5-(OMe)3 | 0.06 | 0.06 | 1 | 0.01 | 0.006 | 2 |
| 568 | 6-OMe-Naphthyl- | 3,4,5-(OMe)3 | 0.05 | 0.05 | 1 | 100 | 1 | 100 |
| 570 | 4-OMe-Naphthyl- | 3,4,5-(OMe)3 | 0.82 | 0.45 | 1.8 | 100 | 4 | 25 |
| 1024 | 3,4-(OMe)2-Ph- | 3,4,5-(OMe)3 | 9 | 9 | 1 | 10 | 6 | 2 |
| 222 | 2,4-(OMe)2-Ph- | 3,4,5-OMe)3 | 0.07 | 0.007 | 10 | 0.06 | 0.005 | 10 |
| 299 | 3,4-OCH2O | 3,4,5-(OMe)3 | 0.05 | 0.05 | 1 | 20 | 20 | 1 |
| CA4 | 4-OMe-3-OH | 3,4,5-(OMe)3 | 0.002 | 0.002 | 1 | 0.002 | 0.003 | 0.7 |
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| DMU Code | R (A-Ring) | R’ (B-Ring) | IC50 (µM) a | AF | IC50 (µM) a | TS | ||
| MCF7 | MCF7 (TCDD) | MCF10A | MDA468 | |||||
| 283 | 3,4-(OH)2 | 3,4,5-(OMe)3 | 0.02 | 0.14 | 0.1 | 0.008 | 0.05 | 0.16 |
| 214 | 3-OH, 4-OMe- | 3,4,5-(OMe)3 | 0.06 | 0.11 | 0.6 | 0.05 | 0.05 | 1 |
| 281 | 4-OH | 3,4,5-(OMe)3 | 30 | 28 | 1 | 23 | 3.6 | 6 |
| 293 | 3-OH, 4-OMe- | 4-OH, 3,5-(OMe)2 | 100 | 100 | 1 | 110 | 10 | 10 |
| 291 | 4-OMe | 4-OH, 3,5-(OMe)2 | 20 | 20 | 1 | 100 | 1 | 100 |
| 295 | 4-OH | 4-OH, 3,5-(OMe)2 | 50 | 4.2 | 12 | 51 | 5.1 | 10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruparelia, K.C.; Zeka, K.; Beresford, K.J.M.; Wilsher, N.E.; Potter, G.A.; Androutsopoulos, V.P.; Brucoli, F.; Arroo, R.R.J. CYP1-Activation and Anticancer Properties of Synthetic Methoxylated Resveratrol Analogues. Molecules 2024, 29, 423. https://doi.org/10.3390/molecules29020423
Ruparelia KC, Zeka K, Beresford KJM, Wilsher NE, Potter GA, Androutsopoulos VP, Brucoli F, Arroo RRJ. CYP1-Activation and Anticancer Properties of Synthetic Methoxylated Resveratrol Analogues. Molecules. 2024; 29(2):423. https://doi.org/10.3390/molecules29020423
Chicago/Turabian StyleRuparelia, Ketan C., Keti Zeka, Kenneth J. M. Beresford, Nicola E. Wilsher, Gerry A. Potter, Vasilis P. Androutsopoulos, Federico Brucoli, and Randolph R. J. Arroo. 2024. "CYP1-Activation and Anticancer Properties of Synthetic Methoxylated Resveratrol Analogues" Molecules 29, no. 2: 423. https://doi.org/10.3390/molecules29020423
APA StyleRuparelia, K. C., Zeka, K., Beresford, K. J. M., Wilsher, N. E., Potter, G. A., Androutsopoulos, V. P., Brucoli, F., & Arroo, R. R. J. (2024). CYP1-Activation and Anticancer Properties of Synthetic Methoxylated Resveratrol Analogues. Molecules, 29(2), 423. https://doi.org/10.3390/molecules29020423