Abstract
Amino acids with unusual types of chirality and their derivatives have recently attracted attention as precursors in the synthesis of chiral catalysts and peptide analogues with unique properties. In this study, we have synthesized a new nido-carborane-based planar-chiral amino acid, in the molecule of which the amino group is directly bonded to the B(3) atom, and the carboxyl group is attached to the B(9) atom through the CH2S+(Me) fragment. 3-Amino-9-dimethylsulfonio-nido-carborane, prepared in three steps from 3-amino-closo-carborane in a high yield, was a key intermediate in the synthesis of the target planar-chiral amino acid. The carboxymethyl group at the sulfur atom was introduced by the demethylation reaction of the dimethylsulfonio derivative, followed by S-alkylation. The structure of new 3,9-disubstituted nido-carboranes was studied for the first time using NMR spectroscopy. The resonances of all boron atoms in the 11B NMR spectrum of 3-amino-9-dimethylsulfonio-nido-carborane were assigned based on the 2D NMR correlation experiments. The nido-carborane-based planar-chiral amino acid and related compounds are of interest as a basis for peptide-like compounds and chiral ligands.
1. Introduction
Chirality is a unique property of matter that underlies molecular recognition and life itself [1,2,3,4]. To date, significant progress has been achieved in the preparation of organic and organoelement compounds with different types of chirality (central, axial, planar, etc.) [5,6,7,8,9,10].
Amino acids are one of the most important groups of chiral compounds. As a rule, natural amino acids contain an asymmetric carbon atom at position 2. Derivatives of amino acids with the property of central chirality are widely used in the design of medicinal agents, functional materials, and chiral catalysts [11,12,13,14,15,16].
Recently, the preparation and application of compounds (including amino acid derivatives) possessing unusual types of chirality have attracted more and more attention (Figure 1). Synthetic approaches to racemic and enantiomerically pure derivatives and analogs of amino acids possessing axial (biphenyl and binaphthyl derivatives [17,18,19,20,21,22,23,24,25,26]), planar (ferrocene derivatives [27,28,29,30], [2.2]paracyclophane [31,32,33,34,35,36] and macrocyclic cyclophanes [37,38,39]), and helical chirality [40,41,42,43,44] have been proposed. Amino acid derivatives containing adamantane [45,46,47], calix[4]arene [48,49], and fullerene [50,51,52] residues as a chirality element, as well as catenane and rotaxane motifs possessing topological chirality [53,54,55], have been synthesized. It has been shown that the introduction of amino acid fragments allows obtaining stable conformational isomers of pillar[5]arenes [56,57,58,59]. Axial and planar-chiral derivatives of amino acids find application in chiral organocatalysis [19,20,21,23,34,60,61,62,63] and the design of peptides with original structures [28,64].
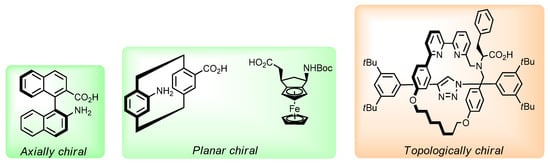
Figure 1.
Selected examples of amino acid derivatives possessing unusual forms of chirality.
A special group of unnatural amino acids is represented by derivatives of 1,2-dicarba-closo-dodecaborane and 7,8-dicarba-nido-undecaborane (carboranes). Molecules of ortho-closo- and nido-carboranes have an icosahedral structure and contain ten or nine boron atoms, respectively, and two carbon atoms located at adjacent vertices of the cluster [65,66]. Carborane derivatives are of interest primarily as potential agents for boron delivery in boron neutron capture therapy (BNCT) of tumors [67,68,69,70,71], as well as in combined approaches such as BNCT/GdNCT [72,73,74]. Carborane-based amino acids, in the molecules of which the carboxyl and amino groups are attached to different cluster vertices, are planar chiral [75]. We have developed methods for the preparation of a number of planar-chiral amino acid derivatives based on closo- [76,77,78] and nido-carboranes [79,80] (Figure 2), methods for analyzing their stereoisomeric composition and carried out the assignment of the absolute configuration. It has been shown that crystals of planar-chiral closo-carborane–based pseudo-peptides can have significant piezoelectric activity [77,81].
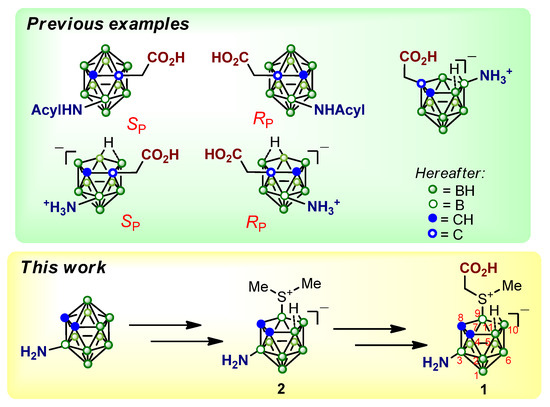
Figure 2.
Planar-chiral carborane-based amino acids.
Recently, we have synthesized a series of carborane-containing derivatives of natural amino acids in which the nido-carborane residue carries a negative charge, and the positive charge is localized on the nitrogen or sulfur atoms [82,83,84]. In these sulfur-containing α-amino acids, a nido-carborane residue is located in the side chain [83,84]. A number of primary amines and carboxylic acids were obtained earlier on the basis of 9-methylthio-nido-carborane [85]. In the present work, we synthesized for the first time a planar-chiral nido-carborane-based amino acid 1 (Figure 2), in which the amino group is attached to the B(3) atom, and the carboxyl group is linked to the B(9) position via the sulfur atom carrying a positive charge. Using the example of dimethylsulfonio derivative 2, the precursor of amino acid 1, we have, for the first time, carried out a detailed NMR spectroscopy study of the structure of a nido-carborane derivative containing amino and dialkysulfonio substituents at positions 3 and 9.
2. Results and Discussion
The 3-amino-nido-carborane derivative 2 is a key intermediate in the synthesis of planar-chiral amino acid 1. We studied the possibilities of obtaining compound 2 starting from 3-amino-closo-carborane (3a) and its N-formyl derivative 3b (Scheme 1).
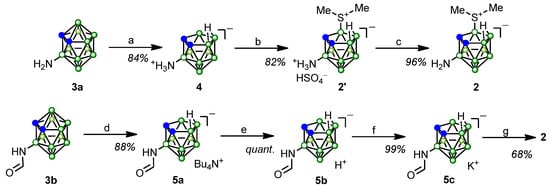
Scheme 1.
Synthesis of 3-amino-9-dimethylsulfonio-nido-carborane (2). (a) piperidine, benzene, rt, 48 h [86]; (b) DMSO, H2SO4, H2O, 80 °C, 4.5 h; (c) NaHCO3, H2O, rt, 0.5 h; (d) TBAF, THF/H2O, rt, 9 days; (e) Amberlite IR-120 (H form), EtOH, rt, 1 h; (f) 1. KOH, EtOH, 0.5 h; 2. CO2 solid (excess), rt, 0.5 h; (g) 1. DMSO, H2SO4, H2O, 80 °C, 4.5 h; 2. KOH, rt, 1 h.
Deboronation of compound 3a according to the known method [86] smoothly led to 3-ammonio-nido-carborane (4). Treatment of compound 4 with dimethyl sulfoxide (DMSO) in the presence of sulfuric acid resulted in the corresponding 9-dimethylsulfonio derivative in the form of a salt with sulfuric acid (2′) (Scheme 1). Previously, the introduction of the dimethylsulfonio group by an acid-promoted reaction with DMSO was carried out only at position 9 of unsubstituted nido-carborane [87,88,89] or nido-carborane with substituents at carbon atoms [90,91,92]. In our case, compound 4 with an amino group at position 3 of nido-carborane was highly reactive, and compound 2′ was isolated in high yield.
The structure of compound 2′ was confirmed by NMR spectroscopy and chromato-mass spectrometry. In the 1H NMR spectrum recorded in DMSO-d6 (Figure S1), we observed a set of signals characteristic of 9-substituted nido-carboranes: a broadened signal in the region δ −3.1…−3.0 ppm corresponding to the bridging hydrogen atom and singlets at δ 2.37 and 3.04 ppm, corresponding to nonequivalent CH protons of carborane. The protons of the S(Me)2 diastereotopic methyl groups resonated at δ 2.65 and 2.77 ppm, respectively. In the 11B NMR spectrum of compound 2′ (Figure S2), there were nine separate peaks, which indicated an asymmetric structure of the molecule and pointed to the presence of a substituent at position 9. The resonances of two nonequivalent carbon atoms of 9-substituted nido-carborane in the 13C NMR spectrum (Figure S3) were in the characteristic region (broadened peaks at δ 41 and 54 ppm, respectively).
Neutralization of salt 2′ with aqueous NaHCO3 solution afforded 3-amino derivative 2 in high yield.
An alternative method for the synthesis of compound 2 starting from 3-formamido-closo-carborane (3b) involved its deboronation followed by the introduction of a dimethylsulfonio group. The interaction of the closo-derivative 3b with tetrabutylammonium fluoride (TBAF) at room temperature, by analogy with the approach proposed by us earlier [93], proceeded regioselectively and led to the nido-derivative 5a in high yield (Scheme 1). Treatment of tetrabutylammonium salt 5a with a strongly acidic ion exchange resin afforded the protonated nido-carborane 5b, which was further converted to the corresponding potassium salt 5c. Potassium salt 5c was highly hygroscopic and prone to solvate with ethanol; even after drying in vacuo at 70 °C for 6 h, the ethanol content was 50 mol% (according to 1H NMR spectroscopy). The reaction of the formyl derivative 5c with DMSO in aqueous sulfuric acid resulted in the simultaneous introduction of a dimethylsulfonio group into position 9 of nido-carborane and hydrolysis of the formamido group, thus yielding compound 2′ as the only reaction product. After adjusting the reaction mixture with potassium hydroxide to pH ~ 8, subsequent extraction with EtOAc, and chromatographic purification, free 3-amino-9-dimethylsulfonio-nido-carborane (2) was isolated in 68% yield.
In the NMR spectra of compounds 5a–c (DMSO-d6), we observed double sets of signals corresponding to trans- and cis-isomers due to hindered rotation around the amide bond (Figures S4–S12). A rotamer ratio of approximately 85:15 is typical for secondary formamides [94,95,96].
The structure of 3-amino-9-dimethylsulfonio-nido-carborane (2) was studied using a combination of 2D NMR experiments: 11B–11B{1H} COSY, 1H–1H{11B} COSY, 1H–13C HSQC and 1H{11B}–11B HMQC (Figure 3). 1H NMR spectrum of compound 2 was recorded with broad-band 11B-decoupling. Analysis of the correlation spectra allowed, for the first time, a complete and unambiguous assignment of the 1H, 11B, and 13C resonances of the 3,9-disubstituted nido-carborane derivative. It is significant that in the 1H{11B}–11B HMQC spectrum, the bridging “extra” (endocyclic) proton H-ec, located above the open pentagonal plane C2B3, has the most intense cross-peak with the B-10 boron, which indicates its probable localization near this atom.
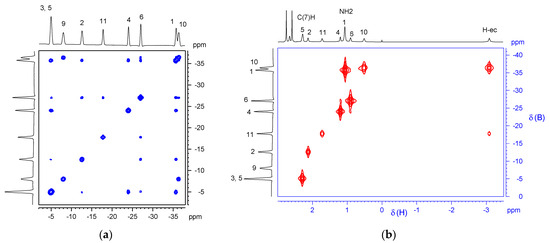
Figure 3.
2D NMR spectra of compound 2: (a) 11B–11B COSY spectrum (160 MHz, CDCl3); (b) 2D 1H{11B}–11B{1H} HMQC spectrum (500 MHz, CDCl3).
Demethylation of compound 2 with sodium naphthalene by analogy with the known approach [83,97] smoothly led to 3-ammonio-9-methylthio-nido-carborane (6) as an internal salt in good yield (Scheme 2). Compound 6 contains a chiral plane in the structure, so it is a mixture of two enantiomers (1:1) distinguishable by chiral HPLC (Figure S33). Zwitterion 6 is a valuable building block from which various charge-compensated planar-chiral derivatives of 3-amino-nido-carborane can be prepared.
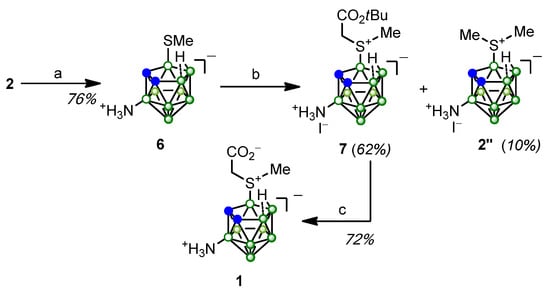
Scheme 2.
Synthesis of planar chiral nido-carborane-based amino acid 1: (a) naphthalene, Na, THF, 10 °C → rt, 7 days; (b) ICH2COOtBu, EtOH, Δ, 10 h; (c) CF3COOH, CH2Cl2, rt, 3 h.
Alkylation of thioether 6 with tert-butyl iodoacetate led to the 3-ammonio-nido-carboran-9-yl derivative of S-methylthioglycolic acid as an iodide (compound 7), in the molecule of which the nido-carborane cluster is linked to positively charged nitrogen and sulfur atoms. A by-product, 3-amino-9-dimethylsulfonio-nido-carborane as a hydroiodide (2″), was isolated in 10% yield. Cleavage of tert-butyl ester group in compound 7 under the action of trifluoroacetic acid followed by crystallization and chromatographic purification successively led to compound 1 containing positively charged nitrogen and sulfur atoms along with carboxylate group and nido-carborane cage bearing negative charges.
From the literature, it is known that sulfonium derivatives can be highly reactive alkylating and arylating agents [98,99,100]. We have previously noted the instability of dialkylsulfonio nido-carborane derivatives under basic conditions [83]. Examples of the formation of dimethylsulfonio-nido-carborane as a by-product in the alkylation reactions of the corresponding methylthio derivative were reported [101], but the mechanism of this process has not been studied. It has been shown that dialkylsulfonio dodecaborane derivatives exhibit alkylating ability, including when interacting with methylthio-dodecaborane [102,103]. In our case, the formation of compound 2″ apparently occurs as a result of the transfer of a methyl group from the molecule of the main reaction product 7 to the molecule of the starting thioether 6.
The alkylating ability of compound 7 was illustrated in a model reaction with the cesium salt of 9-methylthio-7,8-dicarba-nido-undecaborane (8) (Scheme 3). Keeping an equimolar mixture of compounds 7 and 8 in refluxing ethanol for 28 h resulted in the formation of a complex mixture of transalkylation products. We managed to isolate the predominant product, 9-dimethylsulfonio-7,8-dicarba-nido-undecaborane (9), in pure form from the reaction mixture. The formation of compound 9 provides clear evidence that compound 7 is prone to demethylation in the presence of suitable nucleophiles.
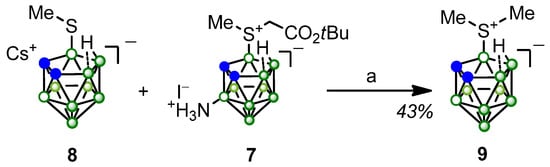
Scheme 3.
Interaction of compound 7 with 9-methylthio-nido-carborane cesium salt (8): (a) EtOH, Δ, 28 h.
The negative charge of the nido-carborane cluster in compounds 1, 2′, 2″, and 7 is compensated by the positive charge localized on the sulfur atom. The amino group at position 3 of nido-carborane is prone to protonation; the counterion, in this case is hydrosulfate or iodide anions. Examples of stable nido-carborane derivatives containing two substituents bearing a positive charge at different vertices of the cluster are very rare [91,104]. The derivatives we obtained were stable when stored in solid form for more than 2 months but were prone to decomposition during chromatographic purification on silica gel and storage in DMSO solutions.
The structure of the obtained compounds was proved by 1H, 11B, and 13C NMR spectroscopy and high-resolution mass spectrometry. Compounds 1 and 7, containing a chiral plane and an asymmetric sulfur atom in their structure, are mixtures of four diastereomers with similar physicochemical properties. We failed to select the conditions for their HPLC separation in various normal and reversed-phase modes.
The ratio of the integral intensities of the proton signals of the diastereomers of compounds 1 and 7 in the 1H NMR spectra was 7:3 and 6:4, respectively, which is consistent with the previously observed formation of diastereomers of 9-dialkylsulfonio nido-carboranes in nonequivalent amounts [84,101,105]. Apparently, this phenomenon is due to the fact that the lone pair of electrons of the sulfur atom in thioether 6 is more accessible for interaction with the alkylating agent in the case when the methyl group is turned “upwards” with respect to the “open pentagonal face” of nido-carborane, and lone pair of electrons of sulfur atom is not shielded by a high electronic density near the open face of nido-carborane (Scheme 4). This results in the preferential formation of isomer 7, in which the tert-butoxycarbonylmethyl substituent occupies a “down-directed position” relative to the “open face” of the carborane, and the methyl substituent is in an “up-directed position”. The formation of diastereomers of compound 7 in nonequivalent amounts may also be due to the fact that one of them is more thermodynamically stable than the other.
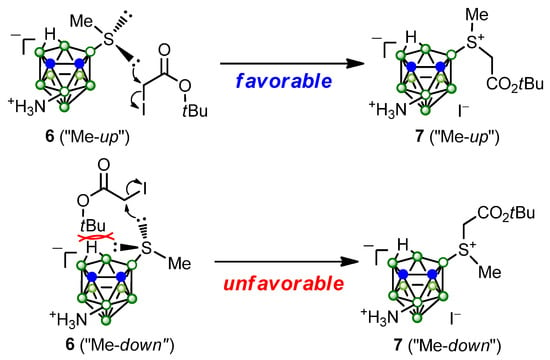
Scheme 4.
Possible orientations of SMe group in compound 6 leading to the formation of different diastereomers of compound 7 (only compounds of (RP)-configuration are shown).
Accordingly, in the 1H NMR spectra, the methylene protons of the predominant diastereomers of compounds 1 and 7 are down-field shifted as compared to the resonances corresponding to the minor diastereomers; the peaks of the methyl group protons of the predominant diastereomers are up-field shifted (Figure 4). This is due to the presence of an induced diatropic ring current in the nido-carborane cage, which causes an up-field shift of the endocyclic proton and apical boron-1 [106].
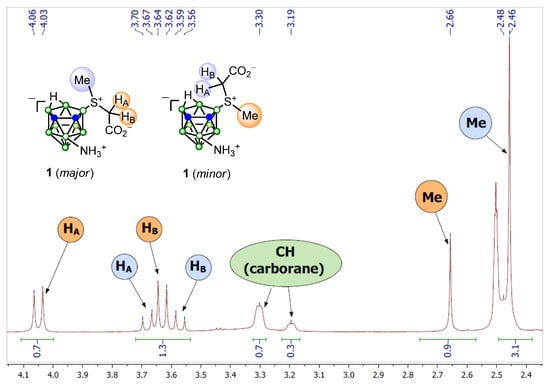
Figure 4.
Fragment of 1H NMR spectrum of compound 1.
3. Materials and Methods
3-Ammonio-7,8-dicarba-nido-undecaborane (4) [86], 3-formamido-1,2-dicarba-closo-dodecaborane (3b) [107], cesium salt of 9-methylthio-7,8-dicarba-nido-undecaborane (8) [83] and tert-butyl iodoacetate [108] were obtained according to known procedures. Other reagents were commercially available and were purchased from Alfa Aesar (Heysham, UK). Solvents were purified according to traditional methods [109] and used freshly distilled. Melting points were obtained on a SMP3 apparatus (Barloworld Scientific, Staffordshire, UK). The 1H, 11B, and 13C NMR spectra were recorded on Bruker DRX-400 (operating frequencies of 400 (1H) and 128 (11B) MHz), Bruker AVANCE 500 (operating frequencies of 500 (1H), 160 (11B), and 126 (13C) MHz) or Bruker AVANCE NEO 600 (operating frequencies of 193 (11B) and 151 (13C) MHz) instruments (Bruker, Karlsruhe, Germany) at ambient temperature using TMS as an internal standard and BF3·Et2O as an external standard. 1H NMR spectrum of compound 2 was recorded with broad-band 11B-decoupling. For compound 2, signals in 1H, 11B and 13C spectra were assigned based on 2D 1H–1H{11B} and 11B–11B{1H} COSY, 1H–13C HSQC and 1H{11B}–11B HMQC experiments. NMR spectra of the compounds obtained, see the Supplementary Materials, Figures S1–S32. Analytical TLC was performed using Sorbfil plates (Imid, Krasnodar, Russia); visualization under UV light (254 nm) and by treatment with a 0.75% KMnO4 solution in 0.016 M NaOH solution containing 5% K2CO3. Flash column chromatography was performed using Silica gel 60 (230–400 mesh) (Alfa Aesar, Heysham, UK). The high-resolution mass spectra were obtained on a Bruker maXis Impact HD mass spectrometer (Bruker, Karlsruhe, Germany), electrospray ionization or chemical ionization at atmospheric pressure in positive or negative mode with direct sample inlet (4 L/min flow rate). Analytical HPLC of compound 6 was carried out on an AZURA instrument (KNAUER, Berlin, Germany) using a Chiralpak AS-H column (250 × 4.6 mm, 5 µm) (Daicel Corp., Osaka, Japan); flow rate 1.0 mL/min, detection at 220 nm, n-hexane–iPrOH 5:1 as an eluent. HPLC of compound 6, see the Supplementary Materials, Figure S33.
3-Ammonio-9-dimethylsulfonio-7,8-dicarba-nido-undecaborane Hydrosulfate (2′). Water (0.48 mL) and H2SO4 conc. (0.60 mL) were slowly added to a solution of compound 4 (0.063 g, 0.42 mmol) in DMSO (0.13 mL) under argon atmosphere under stirring at room temperature. The reaction mixture was stirred at 80 °C for 4.5 h, then cooled to room temperature. Water (5 mL) and solid KOH were added to the reaction mixture to pH ~ 7, and then EtOH (7 mL) was added. The precipitate was filtered off and washed with EtOH (3 mL). The filtrate and washings were evaporated to dryness under reduced pressure. The residue was purified by flash chromatography on silica gel (eluent CHCl3–EtOH from 8:2 to 4:6). Yield 0.11 g (82%). Light beige powder, m.p. 217–222 °C (decomp.). TLC (CHCl3–EtOH–AcOH 5:5:0.1): Rf 0.17. 1H NMR (DMSO-d6, 400 MHz) δ (ppm): −3.08 (br. s, 1H, BHB), 0.0–2.4 (br. m, 7H, 7 × BH), 2.37 (s, 1H, CH-carborane), 2.65 (s, 3H, Me), 2.77 (s, 3H, Me), 3.04 (s, 1H, CH-carborane), 7.05 (br. s, 3H, NH3+). 11B NMR (DMSO-d6, 193 MHz) δ (ppm): −37.4 (d, J = 138.5 Hz, 1B), −34.4 (br. s, 1B), −26.8 (d, J = 145.5 Hz, 1B), −23.9 (d, J = 132.6 Hz, 1B), −18.2 (1B), −16.7 (br. s, 1B), −13.8 (br. s, 1B), −10.7 (s, 1B), −5.5 (br. m, 2B). 13C NMR (DMSO-d6, 151 MHz) δ (ppm): 25.3 (Me), 27.1 (Me), 41.4 (CH-carborane), 54.4 (CH-carborane). HRMS (ESI) (m/z) [M − HSO4]+: calcd. for [C4H1911B9NS]+: 212.2079, found: 212.2081.
3-Formamido-7,8-dicarba-nido-undecaborane Tetrabutylammonium Salt (5a). A mixture of compound 3b (0.59 g, 3.15 mmol), 75% aqueous TBAF solution (5.75 mL, 15.75 mmol), and THF (16 mL) was kept at room temperature for 9 days, then EtOAc (50 mL) was added, and the mixture was washed successively with water (3 × 25 mL) and saturated NaCl solution (20 mL). The organic layer was dried over Na2SO4 and evaporated to dryness under reduced pressure. The residue was purified by flash chromatography on silica gel (eluent benzene—EtOAc from 95:5 to 9:1). Yield 1.16 g (88%). Colorless powder, m.p. 84.5–86.7 °C. TLC (CHCl3–EtOH 95:5): Rf 0.52. 1H NMR (DMSO-d6, 400 MHz) δ (ppm) (rotamers I and II, 85:15): −2.85 (br. s, 1H, BHB), −0.92–2.20 (br. m, 8H, 8 × BH), 0.94 (t, 12H, Bu4N+), 1.31 (sex, J = 7.4 Hz, 8H, Bu4N+), 1.53–1.61 (m, 8H, Bu4N+), 1.86 (s, 1.7H, 2 × CH-carborane, I), 2.23 (s, 0.3H, 2 × CH-carborane, II), 3.00–3.06 (m, 1.2H, Bu4N+, II), 3.14–3.18 (m, 6.8H, Bu4N+, I), 7.02 (br. s, 0.85H, NH, I), 7.18 (br. s, 0.15H, NH, II), 7.91 (s, 0.15H, CHO, II), 8.11 (d, J = 12.8 Hz, 0.85H, CHO, I). 11B NMR (DMSO-d6, 193 MHz) δ (ppm) (major rotamer): −37.8 (d, J = 132.4 Hz, 2B), −22.7 (d, J = 147.6 Hz, 2B), −17.3 (d, J = 132.2 Hz, 2B), −11.4 (d, J = 132.2 Hz, 2B), −8.97 (s, 1B). 13C NMR (DMSO-d6, 151 MHz) δ (ppm) (rotamers I and II): 14.0 (4C, Bu4N+), 19.7 (4C, Bu4N+), 23.5 (4C, Bu4N+), 45.3 (br. s, 2C, CH-carborane, II), 46.4 (br. s, 2C, CH-carborane, I), 58.0 (4C, Bu4N+), 164.5 (CHO, II), 166.7 (CHO, I). HRMS (ESI) (m/z) [M − Bu4N]−: calcd. for [C3H1311B9NO]−: 178.1846, found: 178.1845.
3-Formamido-7,8-dicarba-nido-undecaborane Protonated Form (5b). Amberlite IR-120 ion exchange resin (H form) (4 g) was added to a solution of compound 5a (0.90 g, 2.15 mmol) in EtOH (70 mL). The mixture was stirred at room temperature for 1 h and filtered. A fresh portion of Amberlite IR-120 ion exchange resin (H form) (4 g) was added to the filtrate, and the mixture was stirred for 1 h and filtered. The filtrate was evaporated to dryness. Yield 0.38 g (100%). Yellowish hygroscopic foam. 1H NMR (DMSO-d6, 400 MHz) δ (ppm) (rotamers I and II, 85:15): −2.82 (br. s, 1H, BHB), −0.83–2.38 (br. m, 8H, 8 × BH), 1.86 (s, 1.7H, 2 × CH-carborane, I), 2.23 (s, 0.3H, 2 × CH-carborane, II), 4.63 (br. s, 1H, H+, overlapped by H2O signal), 7.04 (br. s, 0.85H, NH, I), 7.23 (br. s, 0.15H, NH, II), 7.91 (s, 0.15H, CHO, II), 8.11 (d, J = 12.5 Hz, 0.85H, CHO, I). 11B NMR (DMSO-d6, 193 MHz) δ (ppm) (major rotamer): −37.8 (d, J = 132.6 Hz, 2B), −22.8 (d, J = 147.2 Hz, 2B), −17.3 (d, J = 130.2 Hz, 2B), −11.4 (d, J = 135.9 Hz, 2B), −9.0 (s, 1B). 13C NMR (DMSO-d6, 151 MHz) δ (ppm) (rotamers I and II): 45.2 (br. s, 2C, CH-carborane, II), 46.4 (br. s, 2C, CH-carborane, I), 164.9 (CHO, II), 166.9 (CHO, I). HRMS (ESI) (m/z) [M − H]−: calcd. for [C3H1311B9NO]−: 178.1846, found: 178.1844.
3-Formamido-7,8-dicarba-nido-undecaborane Potassium Salt, Solvate with EtOH (5c × 0.5EtOH). Compound 5b (0.38 g, 2.14 mmol) was added to a solution of KOH (0.18 g, 3.21 mmol) in EtOH (20 mL). The reaction mixture was stirred at room temperature for 30 min, then solid CO2 (excess) was added, and the precipitate was filtered off. The filtrate was evaporated to dryness under reduced pressure. Yield 0.51 g (100%). Colorless hygroscopic foam. 1H NMR (DMSO-d6, 400 MHz) δ (ppm) (rotamers I and II, 85:15): −2.87 (br. s, 1H, BHB), −0.80–2.45 (br. m, 8H, 8 × BH), 1.86 (s, 1.7H, 2 × CH-carborane, I), 2.24 (s, 0.3H, 2 × CH-carborane, II), 7.03 (br. s, 0.85H, NH, I), 7.19 (br. s, 0.15H, NH, II), 7.91 (s, 0.15H, CHO, II), 8.11 (d, J = 12.5 Hz, 0.85H, CHO, I). 11B NMR (DMSO-d6, 193 MHz) δ (ppm) (major rotamer): −37.8 (d, J = 133.8 Hz, 2B), −22.8 (d, J = 148.1 Hz, 2B), −17.3 (d, J = 131.2 Hz, 2B), −11.4 (d, J = 132.5 Hz, 2B), −9.0 (s, 1B). 13C NMR (DMSO-d6, 151 MHz) δ (ppm) (rotamers I and II): 45.3 (br. s, 2C, CH-carborane, II), 46.5 (br. s, 2C, CH-carborane, I), 164.6 (CHO, II), 166.8 (CHO, I). HRMS (ESI) (m/z) [M − K]−: calcd. for [C3H1311B9NO]−: 178.1846, found: 178.1846.
3-Amino-9-dimethylsulfonio-7,8-dicarba-nido-undecaborane (2).
Method A. Compound 2′ (0.10 g, 0.33 mmol) was added to 5% NaHCO3 solution (7 mL) with stirring at room temperature, then EtOAc (5 mL) was added. The aqueous layer was separated and washed with EtOAc (3 × 5 mL). The combined organic layers were washed with water (7 mL) and saturated NaCl solution (7 mL), dried with Na2SO4, and evaporated to dryness under reduced pressure. Yield 0.07 g (96%). Colorless powder, m.p. 110–114 °C. TLC (CHCl3–EtOH 4:6): Rf 0.24. 1H NMR (CDCl3, 500 MHz) δ (ppm): −3.09 (br. s, 1H, BHB), 0.1–2.4 (br. m, 7H, 7 × BH), 1.07 (s, 2H, NH2), 2.27 (s, 1H, CH-carborane), 2.59 (s, 3H, Me), 2.66 (s, 1H, CH-carborane), 2.75 (s, 3H, Me). 1H{11B} NMR (CDCl3, 500 MHz) δ (ppm): −3.09 (s, 1H, H-ec), 0.51 (s, 1H, H-10), 0.90 (s, 1H, H-6),1.07 (s, 3H, H-1, NH2), 1.20 (s, 1H, H-4), 2.12 (s, 1H, H-2), 2.28 (br. s, 2H, H-5, H-7), 2.58 (s, 3H, Me), 2.65 (s, 1H, H-8), 2.75 (s, 3H. Me). 11B NMR (CDCl3, 160 MHz) δ (ppm): −36.4 (d, J =139.5 Hz, 1B, B-10), −35.8 (d, J =140.5 Hz, 1B, B-1), −27.1 (d, J = 144.7 Hz, 1B, B-6), −24.1 (d, J = 150.9 Hz, 1B, B-4), −17.8 (d, J = 140.6 Hz, 1B, B-11), −12.6 (d, J = 153.9 Hz, 1B, B-2), −8.0 (s, 1B, B-9), −5.2 (d, J = 111.1 Hz, 1B, B-5), −4.9 (s, 1B, B-3). 13C NMR (CDCl3, 126 MHz) δ (ppm): 25.5 (Me), 28.6 (Me), 45.8 (br. s, C-7), 58.4 (br. s, C-8). HRMS (ESI) (m/z) [M + H]+: calcd. for [C4H1911B9NS]+: 212.2079, found: 212.2080.
Method B. Water (2 mL) and H2SO4 conc. (2.53 mL) were slowly added to a solution of compound 5c × 0.5EtOH (0.42 g, 1.76 mmol) in DMSO (0.55 mL) under argon atmosphere under stirring at room temperature. The reaction mixture was stirred at 80 °C for 4.5 h, then cooled to room temperature. Solid KOH was added to the reaction mixture to pH ~ 8, then the reaction mixture was extracted with EtOAc (4 × 15 mL). The combined organic layers were washed with water (10 mL), saturated NaCl solution (10 mL), dried with Na2SO4, and evaporated to dryness under reduced pressure. The residue was purified by flash chromatography on silica gel (eluent CHCl3–EtOH from 8:2 to 2:8). Yield 0.25 g (68%). Colorless amorphous solid. TLC (CHCl3–EtOH 4:6): Rf 0.24. 1H, 11B, and 13C NMR spectra were identical to those of compound 2 obtained according to method A. HRMS (ESI) (m/z) [M + H]+: calcd. for [C4H1911B9NS]+: 212.2079, found: 212.2081.
3-Ammonio-9-methylthio-7,8-dicarba-nido-undecaborane (6). Sodium (0.50 g, 21.92 mmol) was added to a solution of naphthalene (3.09 g, 24.11 mmol) in THF (26 mL) under stirring at 10 °C. The reaction mixture was stirred at 10 °C for 15 min, then a solution of compound 2 (1.70 g, 8.12 mmol) in THF (6 mL) was added dropwise under stirring. The reaction mixture was stirred at room temperature for 7 days and cooled to 10 °C. MeOH (14 mL) was added to the reaction mixture. The reaction mixture was evaporated to dryness under reduced pressure. The residue was dissolved in water (30 mL), and solid NaOH was added to pH = 14. The reaction mixture was extracted with n-hexane (3 × 15 mL). The combined organic layers were washed with 2N NaOH (2 × 5 mL). The combined aqueous layers were acidified with HCl conc. to pH ~ 2 and extracted with EtOAc (4 × 15 mL). The combined organic layers were washed with saturated aqueous NaCl solution (2 × 10 mL), dried over Na2SO4, and evaporated to dryness under reduced pressure. The residue was purified by flash chromatography on silica gel (eluent CHCl3–EtOH from 95:5 to 8:2). Yield 1.21 g (76%). Beige powder, m.p. 271–277 °C (decomp.). TLC (CHCl3–EtOH 9:1): Rf 0.30. HPLC (Chiralpak AS-H, n-hexane–iPrOH 5:1, 1.0 mL/min, detection at 220 nm): τ1 8.68 min, τ2 12.75 min. 1H NMR (DMSO-d6, 500 MHz) δ (ppm): −2.50 (br. s, 1H, BHB), −0.4–2.5 (br. m, 7H, 7×BH), 1.93 (s, 1H, CH-carborane), 1.95 (s, 3H, Me), 2.34 (s, 1H, CH-carborane), 7.23 (br. s, 3H, NH3+). 11B{1H} NMR (DMSO-d6, 128 MHz) δ (ppm): −38.6 (1B), −36.1 (1B), −23.3 (3B), −11.9 (3B), 1.8 (1B). 13C NMR (DMSO-d6, 126 MHz) δ (ppm): 15.6 (Me), 37.9 (CH-carborane), 48.7 (br. s, CH-carborane). HRMS (ESI) (m/z) [M − H]−: calcd. for [C3H1511B9NS]−: 196.1776, found: 196.1776.
Alkylation of Compound 6 with tert-Butyl Iodoacetate. A solution of compound 6 (0.26 g, 1.32 mmol) and tert-butyl iodoacetate (0.32 g, 1.32 mmol) in EtOH (15 mL) was stirred under reflux for 10 h, then cooled to room temperature and evaporated to dryness under reduced pressure. The residue was separated by flash chromatography on silica gel (CHCl3 to CHCl3–EtOH 7:3) to afford amino ester 7 (in the form of hydroiodide) and compound 2” as fast and slow eluting components, respectively.
tert-Butyl 2-[S-(3-Ammonio-7,8-dicarba-nido-undecaboran-9-yl)-S-methylsulfonio]ethanoate Iodide (7). Yield 0.32 g (55%). Yellowish powder, m.p. 77–79 °C. TLC (CHCl3–EtOH 9: 1): Rf 0.48. 1H NMR (DMSO-d6, 500 MHz) δ (ppm) (diastereomers I and II, appr. 6:4): −3.11 (br. s, 1H, BHB (I and II)), −0.2–2.5 (br. m, 7H, 7 × BH), 1.46 (s, 3.6H, tBu (II)), 1.49 (s, 5.4H, tBu (I)), 2.38–2.45 (br. s, 1H, CH-carborane (I and II)), 2.70 (s, 1.8H, SMe (I)), 2.84 (s, 1.2H, SMe (II)), 3.07–3.12 (br. s, 1H, CH-carborane (I and II)), 4.04 (d, J = 15.8 Hz, 0.4H, HB (II)), 4.20 (d, J = 15.6 Hz, 0.6H, HB (I)), 4.37 (d, J = 15.8 Hz, 0.4H, HA (II)), 4.50 (d, J = 15.6 Hz, 0.6H, HA (I)), 7.49 (br. s, 3H, NH3+). 11B{1H} NMR (DMSO-d6, 160 MHz) δ (ppm): −37.6 (br. s, 1B), −33.2 (br. s, 1B), −26.7 (1B), −24.1 (1B), −12.4 (2B), −5.1 (br. s, 3B). 13C NMR (DMSO-d6, 126 MHz) δ (ppm) (diastereomers I and II): 22.8 (SMe (I)), 25.2 (SMe (II)), 27.49 (tBu (II)), 27.52 (tBu (I)), 37.9 (br. s, carborane (I and II)), 44.4 (CH2 (II)), 46.7 (CH2 (I)), 52.6 (br. s, CH-carborane (II)), 53.6 (br. s, CH-carborane (I)), 83.6 (tBu (II)), 83.8 (tBu (I)), 164.0 (CO (II)), 164.4 (CO (I)). HRMS (ESI) (m/z) [M − I]+: calcd. for [C9H2711B9NO2S]+: 312.2611, found: 312.2609. HRMS (APCI) (m/z) [I]− calcd.: 126.9050, found 126.9050.
3-Ammonio-9-dimethylsulfonio-7,8-dicarba-nido-undecaborane Iodide (2″). Yield 0.045 g (10%). Beige amorphous powder. TLC (CHCl3–EtOH 9:1): Rf 0.19. 1H NMR (DMSO-d6, 500 MHz) δ (ppm): −3.09 (br. s, 1H, BHB), 0.1–2.5 (br. m, 7H, 7 × BH), 2.42 (s, 1H, CH-carborane), 2.67 (s, 3H, Me), 2.81 (s, 3H, Me), 3.10 (s, 1H, CH-carborane), 7.40 (br. s, 3H, NH3). 11B NMR (DMSO-d6, 160 MHz) δ (ppm): −37.7 (d, J = 140.2 Hz, 1B), −33.9 (br. s, 1B), −26.8 (d, J = 141.2 Hz, 1B), −24.1 (d, J = 136.8 Hz, 1B), −12.1 (br. s, 2B), −5.2 (br. s, 3B). 13C NMR (DMSO-d6, 126 MHz) δ (ppm): 24.7 (Me), 26.75 (Me), 39.5 (CH-carborane, overlapped by DMSO-d6 signal), 53.0 (br. s, CH-carborane). HRMS (ESI) (m/z) [M − I]+: calcd. for [C4H1911B9NS]+: 212.2079, found: 212.2079.
2-[S-(3-Amino-7,8-dicarba-nido-undecaboran-9-yl)-S-methylsulfonio]ethanoic Acid (1). Trifluoroacetic acid (1.9 mL) was added to a solution of compound 7 (0.19 g, 0.44 mmol) in CH2Cl2 (2.6 mL). The reaction mixture was stirred at room temperature for 3 h, then evaporated to dryness under reduced pressure. The residue was treated with Et2O (15 mL). The precipitate was filtered off and then purified by flash chromatography on silica gel (CHCl3–EtOH from 7:3 to 1:1). Yield 0.08 g (72%). Colorless powder, m.p. 252–255 °C. TLC (CHCl3–EtOH 1:1): Rf 0.39. 1H NMR (DMSO-d6, 500 MHz) δ (ppm) (diastereomers I and II, appr. 7:3): −3.14 (br. s, 1H, BHB (I and II)), −0.1–2.5 (br. m, 7H, 7×BH), 2.46 (s, 2.1H, SMe (I)), 2.48 (br. s, 1H, CH-carborane (I and II)), 2.66 (s, 0.9H, SMe (II)), 3.20 (br. s, 0.3H, CH-carborane (II)), 3.31 (br. s, 0.7H, CH-carborane (I)), 3.57 (d, J = 15.0 Hz, 0.3H, H-2B (II)), 3.63 (d, J = 14.5 Hz, 0.7H, H-2B (I)), 3.68 (d, J = 15.0 Hz, 0.3H, H-2A (II)), 4.05 (d, J = 14.5 Hz, 0.7H, H-2A (I)), 7.93 (br. s, 3H, NH3+). 11B{1H} NMR (DMSO-d6, 160 MHz) δ (ppm): −37.7 (1B), −34.2 (br. s, 1B), −27.0 (1B), −24.0 (br. s, 1B), −14.5 (br. s, 2B), −12.0 (1B), −4.9 (br. s, 2B). 13C NMR (DMSO-d6, 126 MHz) δ (ppm) (diastereomers I and II): 21.7 (SMe (I)), 24.2 (SMe (II)), 39.5 (CH-carborane (I and II), overlapped by DMSO-d6 signal), 47.6 (CH2 (II)), 51.4 (CH2 (I)), 52.7 (br. s, CH-carborane (II)), 54.4 (br. s, CH-carborane (I)), 165.2 (CO2H (II)), 166.2 (CO2H (I)). HRMS (ESI) (m/z) [M + H]+: calcd. for [C5H1911B9NO2S]+: 256.1980, found: 256.1978.
9-Dimethylsulfonio-7,8-dicarba-nido-undecaborane (9). A solution of compound 7 (17.8 mg, 0.041 mmol) and compound 8 (12.7 mg, 0.041 mmol) in EtOH (1 mL) was stirred under reflux for 28 h, then evaporated to dryness under reduced pressure. The residue was purified by flash chromatography (n-hexane–EtOAc from 8:2 to 4:6). Yield 3.4 mg (43%). Beige powder, m.p. 138–141 °C (lit. m.p. 147–148 °C [110]). TLC (n-hexane–EtOAc 6:4): Rf 0.34. HRMS (ESI) (m/z) [M + Na]+: calcd. for [C4H1711B9NaS]+: 219.1790, found: 219.1788. 1H NMR spectrum was identical to that described in [87].
4. Conclusions
Thus, we have developed a method for the preparation of a new nido-carborane-based bifunctional building block, namely 3-ammonio-9-methylthio-7,8-dicarba-nido-undecaborane. Its alkylation with tert-butyl iodoacetate yielded a charge-compensated nido-carborane-based amino ester as a mixture of diastereomers in a ratio of approximately 6:4. Removal of the ester protecting group led to a new planar-chiral amino acid with free functional groups. In its molecule, the amino group is directly attached to the B(3) atom, and the carboxyl group is linked to the B(9) atom via the CH2S+(Me) fragment. For the first time, the assignment of boron atom signals in the 11B NMR spectrum of the 3,9-disubstituted nido-carborane derivative was carried out. The obtained nido-carborane-based planar-chiral amino acid, containing a positively charged sulfur atom, and related compounds are of interest as a basis for peptide-like compounds and chiral ligands.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules29184487/s1, Figures S1–S32: 1H, 11B, and 13C NMR spectra of compounds 2′, 5a–c, 2, 6, 7, 2″ and 1; Figure S33: HPLC chromatogram of compound 6; Figures S34–S43: HRMS data for compounds 2′, 5a–c, 2, 6, 7, 2″, 1 and 9.
Author Contributions
Conceptualization and methodology, D.A.G. and A.A.T.; investigation, A.A.T.; NMR. analysis, M.A.E. and M.I.K.; writing—original draft preparation, D.A.G. and A.A.T.; writing—review and editing, G.L.L. and V.P.K.; project administration, D.A.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was financially supported by the Ministry of Higher Education and Science of the Russian Federation (themes no. 124020200038-6 and no. 124020500023-9).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
Acknowledgments
Equipment of the Centre for Joint Use “Spectroscopy and Analysis of Organic Compounds” at the Postovsky Institute of Organic Synthesis was used. The authors express their gratitude to Iliya N. Ganebnykh for recording high-resolution mass spectra, and Oleg S. Yeltsov for assistance in recording the NMR spectra.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Eliel, E.L.; Wilen, S.H.; Mander, L.N. Stereochemistry of Organic Compounds; John Wiley & Sons, Inc.: New York, NY, USA, 1994; 1267p. [Google Scholar]
- Wang, Y.; Xu, J.; Wang, Y.; Chen, H. Emerging chirality in nanoscience. Chem. Soc. Rev. 2013, 42, 2930–2962. [Google Scholar] [CrossRef] [PubMed]
- Tverdislov, V.A.; Malyshko, E.V. Chiral Dualism as an Instrument of Hierarchical Structure Formation in Molecular Biology. Symmetry 2020, 12, 587. [Google Scholar] [CrossRef]
- Peluso, P.; Chankvetadze, B. Recognition in the Domain of Molecular Chirality: From Noncovalent Interactions to Separation of Enantiomers. Chem. Rev. 2022, 122, 13235–13400. [Google Scholar] [CrossRef] [PubMed]
- Hassan, Z.; Spuling, E.; Knoll, D.M.; Lahann, J.; Bräse, S. Planar chiral [2.2]paracyclophanes: From synthetic curiosity to applications in asymmetric synthesis and materials. Chem. Soc. Rev. 2018, 47, 6947–6963. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Peng, C.; Wang, Y.-T.; Zhan, G.; Han, B. Recent progress on the construction of axial chirality through transition-metal-catalyzed benzannulation. Org. Chem. Front. 2021, 8, 2772–2785. [Google Scholar] [CrossRef]
- López, R.; Palomo, C. Planar Chirality: A Mine for Catalysis and Structure Discovery. Angew. Chem. Int. Ed. 2022, 61, e202113504. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, Z.-G.; Shi, F. Advances in catalytic enantioselective synthesis of chiral helicenes and helicenoids. Chem Catal. 2022, 2, 3077–3111. [Google Scholar] [CrossRef]
- Wang, Z.; Meng, L.; Liu, X.; Zhang, L.; Yu, Z.; Wu, G. Recent progress toward developing axial chirality bioactive compounds. Eur. J. Med. Chem. 2022, 243, 114700. [Google Scholar] [CrossRef]
- Laws, D., III; Poff, C.D.; Heyboer, E.M.; Blakey, S.B. Synthesis, stereochemical assignment, and enantioselective catalytic activity of late transition metal planar chiral complexes. Chem. Soc. Rev. 2023, 52, 6003–6030. [Google Scholar] [CrossRef]
- Blaskovich, M.A.T. Unusual Amino Acids in Medicinal Chemistry. J. Med. Chem. 2016, 59, 10807–10836. [Google Scholar] [CrossRef]
- Agirre, M.; Arrieta, A.; Arrastia, I.; Cossío, F.P. Organocatalysts Derived from Unnatural α-Amino Acids: Scope and Applications. Chem.—Asian J. 2019, 14, 44–66. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Deng, H.; Li, X.; Quan, Z.-S. Application of Amino Acids in the Structural Modification of Natural Products: A Review. Front. Chem. 2021, 9, 650569. [Google Scholar] [CrossRef] [PubMed]
- Zahradníčková, H.; Opekar, S.; Řimnáčová, L.; Šimek, P.; Moos, M. Chiral secondary amino acids, their importance, and methods of analysis. Amino Acids 2022, 54, 687–719. [Google Scholar] [CrossRef] [PubMed]
- Bongioanni, A.; Bueno, M.S.; Mezzano, B.A.; Longhi, M.R.; Garnero, C. Amino acids and its pharmaceutical applications: A mini-review. Int. J. Pharm. 2022, 613, 121375. [Google Scholar] [CrossRef] [PubMed]
- Kamanna, K. Organocatalysts based on natural and modified amino acids for asymmetric reactions. Phys. Sci. Rev. 2022, 7, 429–467. [Google Scholar] [CrossRef]
- Tichý, M.; Holanová, J.; Závada, J. ω-Amino acids and lactams with chiral biaryl axis. Tetrahedron Asymmetry 1998, 9, 3497–3504. [Google Scholar] [CrossRef]
- Ridvan, L.; Buděšínský, M.; Tichý, M.; Maloň, P.; Závada, J.; Podlaha, J.; Císařová, I. Axially Chiral Bis(α-Amino Acid)s and Dioxopiperazines. Synthesis and Configurational Assignment. Tetrahedron 1999, 55, 12331–12348. [Google Scholar] [CrossRef]
- Kano, T.; Takai, J.; Tokuda, O.; Maruoka, K. Design of an Axially Chiral Amino Acid with a Binaphthyl Backbone as an Organocatalyst for a Direct Asymmetric Aldol Reaction. Angew. Chem. Int. Ed. 2005, 44, 3055–3057. [Google Scholar] [CrossRef]
- Kano, T.; Tokuda, O.; Takai, J.; Maruoka, K. Design of a Binaphthyl-Based Axially Chiral Amino Acid as an Organocatalyst for Direct Asymmetric Aldol Reactions. Chem.—Asian J. 2006, 1–2, 210–215. [Google Scholar] [CrossRef]
- Furuta, T.; Yamamoto, J.; Kitamura, Y.; Hashimoto, A.; Masu, H.; Azumaya, I.; Kan, T.; Kawabata, T. Synthesis of Axially Chiral Amino Acid and Amino Alcohols via Additive–Ligand-free Pd-Catalyzed Domino Coupling Reaction and Subsequent Transformations of the Product Amidoaza [5]helicene. J. Org. Chem. 2010, 75, 7010–7013. [Google Scholar] [CrossRef]
- Furuta, T. Biaryl Amino Acids and Their Surrogates: A Unique Class of Unnatural Amino Acid. In Designed Molecular Space in Material Science and Catalysis; Shirakawa, S., Ed.; Springer Nature: Singapore, 2018; pp. 123–145. [Google Scholar] [CrossRef]
- Bulman Page, P.C.; Kinsey, F.S.; Chan, Y.; Strutt, I.R.; Slawin, A.M.Z.; Jones, G.A. Novel binaphthyl and biphenyl α- and β-amino acids and esters: Organocatalysis of asymmetric Diels–Alder reactions. A combined synthetic and computational study. Org. Biomol. Chem. 2018, 16, 7400–7416. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Shi, Q.; Hu, W.; Chen, T.; Guo, Y.; Hu, Z.; Gong, M.; Guo, J.; Wei, D.; Fu, Z.; et al. Organocatalytic asymmetric N-sulfonyl amide C–N bond activation to access axially chiral biaryl amino acids. Nat. Commun. 2020, 11, 946. [Google Scholar] [CrossRef] [PubMed]
- Cen, S.; Li, S.-S.; Zhao, Y.; Zhao, M.-X.; Zhang, Z. Catalytic Asymmetric Synthesis of Unnatural Axially Chiral Biaryl δ-Amino Acid Derivatives via a Chiral Phenanthroline-Potassium Catalyst-Enabled Dynamic Kinetic Resolution. Angew. Chem. Int. Ed. 2024, 63, e202407920. [Google Scholar] [CrossRef] [PubMed]
- Ansari, S.; Knipe, P.C. Atropisomeric Foldamers. ChemPlusChem 2024, 89, e202400218. [Google Scholar] [CrossRef] [PubMed]
- Salter, R.; Pickett, T.E.; Richards, C.J. 2-Nitroferrocenyloxazolines: Precursors of nitrofulvalenes and derivatives of (pS)- and (pR)-2-aminoferrocenecarboxylic acids. Tetrahedron Asymmetry 1998, 9, 4239–4247. [Google Scholar] [CrossRef]
- Hunold, A.; Neundorf, I.; James, P.; Neudörfl, J.; Schmalz, H.-G. Stereoselective Synthesis of New Ferrocene-Derived Amino Acid Building Blocks. Eur. J. Org. Chem. 2009, 2009, 4429–4440. [Google Scholar] [CrossRef]
- Werner, G.; Butenschön, H. Improved Syntheses of 1,2-Disubstituted Ferrocenes. Eur. J. Inorg. Chem. 2017, 2017, 378–387. [Google Scholar] [CrossRef]
- Kadari, L.; Erb, W.; Halauko, Y.S.; Ivashkevich, O.A.; Matulis, V.E.; Lyakhov, D.; Roisnel, T.; Krishna, P.R.; Mongin, F. On the N-Arylation of Acetamide using 2-, 3- and 1′-Substituted Iodoferrocenes. Eur. J. Inorg. Chem. 2021, 2021, 377–391. [Google Scholar] [CrossRef]
- Pelter, A.; Crump, R.A.N.C.; Kidwell, H. Chiral [2:2]Paracyclophanes. 1. Synthesis and Characterization of Unique Homochiral Amino-Acids derived from [2:2]Paracyclophane. Tetrahedron Lett. 1996, 37, 1273–1276. [Google Scholar] [CrossRef]
- Pelter, A.; Crump, R.A.N.C.; Kidwell, H. Chiral [2.2]paracyclophanes—III. The preparation of unique homochiral amino-acids derived from [2.2]paracyclophane. Tetrahedron Asymmetry 1997, 8, 3873–3880. [Google Scholar] [CrossRef]
- Marchand, A.; Maxwell, A.; Mootoo, B.; Pelter, A.; Reid, A. Oxazoline Mediated Routes to a Unique Amino-Acid, 4-Amino-13-carboxy[2.2]paracyclophane, of Planar Chirality. Tetrahedron 2000, 56, 7331–7338. [Google Scholar] [CrossRef]
- Jayasundera, K.P.; Kusmus, D.N.M.; Deuilhé, L.; Etheridge, L.; Farrow, Z.; Lun, D.J.; Kaur, G.; Rowlands, G.J. The synthesis of substituted amino[2.2]paracyclophanes. Org. Biomol. Chem. 2016, 14, 10848–10860. [Google Scholar] [CrossRef] [PubMed]
- Felder, S.; Micouin, L.; Benedetti, E. Para-Functionalization of N-Substituted 4-Amino[2.2]paracyclophanes by Regioselective Formylation. Eur. J. Org. Chem. 2021, 2021, 4015–4018. [Google Scholar] [CrossRef]
- Yu, S.; Bao, H.; Zhang, D.; Yang, X. Kinetic resolution of substituted amido[2.2]paracyclophanes via asymmetric electrophilic amination. Nat. Commun. 2023, 14, 5239. [Google Scholar] [CrossRef] [PubMed]
- Gibson, S.E.; Jones, J.O.; Kalindjian, S.B.; Knight, J.D.; Mainolfi, N.; Rudd, M.; Steed, J.W.; Tozer, M.J.; Wright, P.T. Synthesis of meta- and paracyclophanes containing unsaturated amino acid residues. Tetrahedron 2004, 60, 6945–6958. [Google Scholar] [CrossRef]
- Kotha, S.; Halder, S.; Damodharan, L.; Pattabhi, V. First and unexpected synthesis of macrocyclic cyclophane-based unusual α-amino acid derivatives by phosphazene base without high dilution conditions. Bioorg. Med. Chem. Lett. 2002, 12, 1113–1115. [Google Scholar] [CrossRef]
- Kotha, S.; Halder, S. Synthesis of macrocyclic cyclophane-based unusual α-amino acid derivatives. Arkivoc 2005, iii, 56–66. [Google Scholar] [CrossRef]
- Tanaka, M.; Anan, K.; Demizu, Y.; Kurihara, M.; Doi, M.; Suemune, H. Side-Chain Chiral Centers of Amino Acid and Helical-Screw Handedness of Its Peptides. J. Am. Chem. Soc. 2005, 127, 11570–11571. [Google Scholar] [CrossRef]
- Barišić, L.; Čakić, M.; Mahmoud, K.A.; Liu, Y.; Kraatz, H.-B.; Pritzkow, H.; Kirin, S.I.; Metzler-Nolte, N.; Rapić, V. Helically Chiral Ferrocene Peptides Containing 1′-Aminoferrocene-1-Carboxylic Acid Subunits as Turn Inducers. Chem.—Eur. J. 2006, 12, 4965–4980. [Google Scholar] [CrossRef]
- Liu, Y.; Gao, X.; Lu, F.; Hu, M.; Shi, L.; Zheng, L. Reversible helical chirality of perylene bisimide aggregates: Amino acid-directed chiral transfer and chiral inversion. Soft Matter 2017, 13, 3072–3075. [Google Scholar] [CrossRef]
- Ueda, A.; Makura, Y.; Kakazu, S.; Kato, T.; Umeno, T.; Hirayama, K.; Doi, M.; Oba, M.; Tanaka, M. E-Selective Ring-Closing Metathesis in α-Helical Stapled Peptides Using Carbocyclic α,α-Disubstituted α-Amino Acids. Org. Lett. 2022, 24, 1049–1054. [Google Scholar] [CrossRef] [PubMed]
- Ousaka, N.; MacLachlan, M.J.; Akine, S. Stapling strategy for slowing helicity interconversion of α-helical peptides and isolating chiral auxiliary-free one-handed forms. Nat. Commun. 2023, 14, 6834. [Google Scholar] [CrossRef] [PubMed]
- Wanka, L.; Cabrele, C.; Vanejews, M.; Schreiner, P.R. γ-Aminoadamantanecarboxylic Acids through Direct C–H Bond Amidations. Eur. J. Org. Chem. 2007, 2007, 1474–1490. [Google Scholar] [CrossRef]
- Aoyama, M.; Hara, S. Synthesis of optically active fluoroadamantane derivative having different substituents on its tert-carbons and its use as non-racemizable source for new optically active adamantine derivatives. Tetrahedron 2013, 69, 10357–10360. [Google Scholar] [CrossRef][Green Version]
- Yasue, R.; Yoshida, K. Enantioselective Desymmetrization of 1,3-Disubstituted Adamantane Derivatives via Rhodium-Catalyzed C–H Bond Amination: Access to Optically Active Amino Acids Containing Adamantane Core. Adv. Synth. Catal. 2021, 363, 1662–1671. [Google Scholar] [CrossRef]
- Shirakawa, S.; Tanaka, Y.; Kobari, T.; Shimizu, S. Synthesis and optical resolution of an inherently chiral calix[4]arene amino acid. New J. Chem. 2008, 32, 1835–1838. [Google Scholar] [CrossRef]
- Shirakawa, S.; Shimizu, S. Synthesis of an Inherently Chiral Calix[4]arene Amino Acid and Its Derivatives: Their Application to Asymmetric Reactions as Organocatalysts. Eur. J. Org. Chem. 2009, 2009, 1916–1924. [Google Scholar] [CrossRef]
- Bianco, A.; Maggini, M.; Scorrano, G.; Toniolo, C.; Marconi, G.; Villani, C.; Prato, M. Synthesis, Chiroptical Properties, and Configurational Assignment of Fulleroproline Derivatives and Peptides. J. Am. Chem. Soc. 1996, 118, 4072–4080. [Google Scholar] [CrossRef]
- Ioutsi, V.A.; Zadorin, A.A.; Khavrel, P.A.; Belov, N.M.; Ovchinnikova, N.S.; Goryunkov, A.A.; Kharybin, O.N.; Nikolaev, E.N.; Yurovskaya, M.A.; Sidorov, L.N. Diastereoselective lithium salt-assisted 1,3-dipolar cycloaddition of azomethine ylides to the fullerene C60. Tetrahedron 2010, 66, 3037–3041. [Google Scholar] [CrossRef]
- Levitskiy, O.A.; Grishin, Y.K.; Semivrazhskaya, O.O.; Ambartsumyan, A.A.; Kochetkov, K.A.; Magdesieva, T.V. Individual (f,tA)- and (f,tC)-Fullerene-Based Nickel(II) Glycinates: Protected Chiral Amino Acids Directly Linked to a Chiral π-Electron System. Angew. Chem. Int. Ed. 2017, 56, 2704–2708. [Google Scholar] [CrossRef]
- Rodríguez-Rubio, A.; Savoini, A.; Modicom, F.; Butler, P.; Goldup, S.M. A Co-conformationally “Topologically” Chiral Catenane. J. Am. Chem. Soc. 2022, 144, 11927–11932. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Rodríguez-Rubio, A.; Saady, A.; Tizzard, G.J.; Goldup, S.M. A chiral macrocycle for the stereoselective synthesis of mechanically planar chiral rotaxanes and catenanes. Chem 2023, 9, 1195–1207. [Google Scholar] [CrossRef]
- Bao, S.-J.; Zhang, H.-N.; Jin, G.-X. Stereoselective Self-Assembly of Chiral Metalla[2]Catenanes and Lemniscular Macrocycles. CCS Chem. 2024, 6, 2000–2010. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, Z.; Lv, S.; Zeng, X.; Sun, Y.; Li, H.; Zhang, R. The chiral interfaces fabricated by D/L-alanine-pillar[5]arenes for selectively absorbing ctDNA. Chem. Commun. 2019, 55, 778–781. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Fu, L.; Sun, B.; Qian, C.; Wang, R.; Jiang, J.; Lin, C.; Ma, J.; Wang, L. Competitve Selection of Conformation Chirality of Water-Soluble Pillar[5]arene Induced by Amino Acid Derivatives. Org. Lett. 2020, 22, 2266–2270. [Google Scholar] [CrossRef] [PubMed]
- Nazarova, A.; Padnya, P.; Kharlamova, A.; Petrov, K.; Yusupov, G.; Zelenikhin, P.; Bukharov, M.; Hua, B.; Huang, F.; Stoikov, I. Peptidomimetics based on ammonium decasubstituted pillar[5]arenes: Influence of the alpha-amino acid residue nature on cholinesterase inhibition. Bioorg. Chem. 2023, 141, 106927. [Google Scholar] [CrossRef]
- Sultanaev, V.; Yakimova, L.; Nazarova, A.; Mostovaya, O.; Sedov, I.; Davletshin, D.; Gilyazova, E.; Bulatov, E.; Li, Z.-T.; Zhang, D.-W.; et al. Decasubstituted Pillar[5]arene Derivatives Containing L-Tryptophan and L-Phenylalanine Residues: Non-Covalent Binding and Release of Fluorescein from Nanoparticles. Int. J. Mol. Sci. 2023, 24, 7700. [Google Scholar] [CrossRef]
- Shibatomi, K.; Kitahara, K.; Okimi, T.; Abe, Y.; Iwasa, S. Enantioselective fluorination of α-branched aldehydes and subsequent conversion to α-hydroxyacetals via stereospecific C–F bond cleavage. Chem. Sci. 2016, 7, 1388–1392. [Google Scholar] [CrossRef]
- Shibatomi, K.; Kitahara, K.; Sasaki, N.; Kawasaki, Y.; Fujisawa, I.; Iwasa, S. Enantioselective decarboxylative chlorination of β-ketocarboxylic acids. Nat. Commun. 2017, 8, 15600. [Google Scholar] [CrossRef]
- Lu, W.; Pei, X.; Murai, T.; Sasamori, T.; Tokitoh, N.; Kawabata, T.; Furuta, T. Asymmetric Intramolecular C–H Insertion Promoted by Dirhodium(II) Carboxylate Catalyst Bearing Axially Chiral Amino Acid Derivatives. Synlett 2017, 28, 679–683. [Google Scholar] [CrossRef]
- Mizutani, H.; Kawanishi, R.; Shibatomi, K. Enantioselective decarboxylative protonation and deuteration of β-ketocarboxylic acids. Chem. Commun. 2021, 57, 6676–6679. [Google Scholar] [CrossRef] [PubMed]
- Thoß, M.; Seidel, R.W.; Feigel, M. The artificial binaphthyl amino acid 6-amino-6′-carboxyethyl-2-methoxy-2′-hydroxy-1,1′-binaphthyl (Bna): Synthesis and assembly of Bna peptides. Tetrahedron 2010, 66, 8503–8511. [Google Scholar] [CrossRef]
- Grimes, R.N. Carboranes, 3rd ed.; Academic Press: Cambridge, MA, USA, 2016; 1041p. [Google Scholar]
- Sivaev, I.B. Polyhedral Boranes and Carboranes. In Comprehensive Organometallic Chemistry IV; Aldridge, S., Ed.; Elsevier: Amsterdam, The Netherlands, 2022; Volume 9, pp. 196–262. [Google Scholar] [CrossRef]
- Takagaki, M.; Kazuko, U.; Hosmane, N.S. Chapter 5: An Overview of Clinical and Biological Aspects of Current Boron Neutron Capture Therapy (BNCT) for Cancer Treatment. In Handbook of Boron Science with Applications in Organometallics, Catalysis, Materials and Medicine; Hosmane, N.S., Egaling, R., Eds.; World Scientific: Hackensack, NJ, USA, 2018; Volume 4, pp. 101–143. [Google Scholar] [CrossRef]
- Marfavi, A.; Kavianpoor, P.; Rendina, L.M. Carboranes in drug discovery, chemical biology and molecular imaging. Nat. Rev. Chem. 2022, 6, 486–504. [Google Scholar] [CrossRef] [PubMed]
- Marforio, T.D.; Carboni, A.; Calvaresi, M. In Vivo Application of Carboranes for Boron Neutron Capture Therapy (BNCT): Structure, Formulation and Analytical Methods for Detection. Cancers 2023, 15, 4944. [Google Scholar] [CrossRef]
- Xu, H.; Liu, J.; Li, R.; Lin, J.; Gui, L.; Wang, Y.; Jin, Z.; Xia, W.; Liu, Y.; Cheng, S.; et al. Novel promising boron agents for boron neutron capture therapy: Current status and outlook on the future. Coord. Chem. Rev. 2024, 511, 215795. [Google Scholar] [CrossRef]
- Couto, M.; Cerecetto, H. Advancements in the Synthesis and Biological Properties of Carboranes and High-Boron Related Compounds: A Comprehensive Exploration with Emphasis on BNCT Applications. J. Braz. Chem. Soc. 2024, 35, e-20240109. [Google Scholar] [CrossRef]
- Lanfranco, A.; Alberti, D.; Parisotto, S.; Renzi, P.; Lecomte, V.; Geninatti Crich, S.; Deagostino, A. Biotinylation of a MRI/Gd BNCT theranostic agent to access a novel tumour-targeted delivery system. Org. Biomol. Chem. 2022, 20, 5342–5354. [Google Scholar] [CrossRef]
- Lee, W.; Kim, K.W.; Lim, J.E.; Sarkar, S.; Kim, J.Y.; Chang, Y.; Yoo, J. In vivo evaluation of the effects of combined boron and gadolinium neutron capture therapy in mouse models. Sci. Rep. 2022, 12, 13360. [Google Scholar] [CrossRef]
- Shanmugam, M.; Kuthala, N.; Kong, X.; Chiang, C.-S.; Hwang, K.C. Combined Gadolinium and Boron Neutron Capture Therapies for Eradication of Head-and-Neck Tumor Using Gd10B6 Nanoparticles under MRI/CT Image Guidance. JACS Au 2023, 3, 2192–2205. [Google Scholar] [CrossRef]
- Gruzdev, D.A.; Levit, G.L.; Krasnov, V.P.; Charushin, V.N. Carborane-containing amino acids and peptides: Synthesis, properties, and applications. Coord. Chem. Rev. 2021, 433, 213753. [Google Scholar] [CrossRef]
- Levit, G.L.; Krasnov, V.P.; Gruzdev, D.A.; Demin, A.M.; Bazhov, I.V.; Sadretdinova, L.S.; Olshevskaya, V.A.; Kalinin, V.N.; Cheong, C.S.; Chupakhin, O.N.; et al. Synthesis of N-[(3-amino-1,2-dicarba-closo-dodecaboran-1-yl)acetyl] derivatives of α-amino acids. Collect. Czech. Chem. Commun. 2007, 72, 1697–1706. [Google Scholar] [CrossRef]
- Gruzdev, D.A.; Nuraeva, A.S.; Slepukhin, P.A.; Levit, G.L.; Zelenovskiy, P.S.; Shur, V.Y.; Krasnov, V.P. Piezoactive amino acid derivatives containing fragments of planar-chiral ortho-carboranes. J. Mater. Chem. C 2018, 6, 8638–8645. [Google Scholar] [CrossRef]
- Gruzdev, D.A.; Ustinova, V.O.; Chulakov, E.N.; Ol’shevskaya, V.A.; Slepukhin, P.A.; Levit, G.L.; Krasnov, V.P.; Charushin, V.N. Preparation of enantiomerically pure derivatives of (3-amino-1,2-dicarba-closo-dodecaboran-1-yl)acetic acid. J. Organomet. Chem. 2018, 876, 50–55. [Google Scholar] [CrossRef]
- Telegina, A.A.; Gruzdev, D.A.; Levit, G.L.; Krasnov, V.P. Synthesis of a novel planar-chiral nido-carborane amino acid. Russ. Chem. Bull. 2021, 70, 539–544. [Google Scholar] [CrossRef]
- Gruzdev, D.A.; Telegina, A.A.; Chulakov, E.N.; Levit, G.L.; Krasnov, V.P. (7,8-Dicarba-nido-undecaboran-7-yl)acetic acid: Synthesis of individual enantiomers and the first example of the determination of the absolute configuration of chiral monosubstituted nido-carborane. New J. Chem. 2022, 46, 17338–17347. [Google Scholar] [CrossRef]
- Nuraeva, A.S.; Vasileva, D.S.; Vasilev, S.G.; Zelenovskiy, P.S.; Gruzdev, D.A.; Krasnov, V.P.; Ol’shevskaya, V.A.; Kalinin, V.N.; Shur, V.Y. Piezoelectric and ferroelectric properties of organic single crystals and films derived from chiral 2-methoxy and 2-amino acids. Ferroelectrics 2016, 496, 1–9. [Google Scholar] [CrossRef]
- Gruzdev, D.A.; Telegina, A.A.; Levit, G.L.; Krasnov, V.P. N-Aminoacyl-3-amino-nido-carboranes as a Group of Boron-Containing Derivatives of Natural Amino Acids. J. Org. Chem. 2022, 87, 5437–5441. [Google Scholar] [CrossRef]
- Gruzdev, D.A.; Telegina, A.A.; Levit, G.L.; Ezhikova, M.A.; Kodess, M.I.; Krasnov, V.P. Synthesis of Charge-Compensated nido-Carboranyl Derivatives of Sulfur-Containing Amino Acids and Biotin. J. Org. Chem. 2023, 88, 14022–14032. [Google Scholar] [CrossRef]
- Telegina, A.A.; Gruzdev, D.A.; Ezhikova, M.A.; Kodess, M.I.; Ol’shevskaya, V.A.; Levit, G.L.; Krasnov, V.P. Synthesis of novel zwitterionic nido-carborane-containing derivatives of cysteine and methionine. Russ. Chem. Bull. 2024, 73, 1716–1724. [Google Scholar] [CrossRef]
- Anufriev, S.A.; Zakharova, M.V.; Sivaev, I.B.; Bregadze, V.I. New carborane-containing acids and amines. Russ. Chem. Bull. 2017, 66, 1643–1649. [Google Scholar] [CrossRef]
- Zakharkin, L.I.; Ol’shevskaya, V.A.; Sulaimankulova, D.D.; Antonovich, V.A. Cleavage of 3-amino-o-carborane and its N-derivatives by bases into 3-amino-7,8-dicarbaundecaborate anion and its N-derivatives. Russ. Chem. Bull. 1991, 40, 1026–1032. [Google Scholar] [CrossRef]
- Plešek, J.; Janoušek, Z.; Heřmánek, S. Four new (CH3)2S.C2B9H11 isomers. Collect. Czech. Chem. Commun. 1978, 43, 2862–2868. [Google Scholar] [CrossRef]
- Plešek, J.; Janoušek, Z.; Heřmánek, S. (9-Dimethyl sulfide)-7,8-dicarba-nido-undecaborane(11), 9-[(CH3)2S]-7,8-C2B9H11. Inorg. Synth. 1983, 22, 239–241. [Google Scholar] [CrossRef]
- Yan, Y.-K.; Mingos, D.M.P.; Müller, T.E.; Williams, D.J.; Kurmoo, M. Synthesis and Structure of a Charge-compensated Ferracarborane, commo-[3,3′-Fe{4-(Me2S)-l,2-C2B9H10}2], and Its Charge-transfer Salt with 2,3-Dichloro-5,6-dicyano-p-benzoquinone. J. Chem. Soc. Dalton Trans. 1994, 1735–1741. [Google Scholar] [CrossRef]
- Rosair, G.M.; Welch, A.J.; Weller, A.S.; Zahn, S.K. Sterically encumbered charge-compensated carbaboranes: Synthesis and reactivity. Molecular structure of 7-Ph-11-SMe2-7,8-nido-C2B9H10 and 1-Ph-3,3-(CO)2-7-SMe2-3,1,2-closo-RhC2B9H8. J. Organomet. Chem. 1997, 536–537, 299–308. [Google Scholar] [CrossRef]
- Meshcheryakov, V.I.; Kitaev, P.S.; Lyssenko, K.A.; Starikova, Z.A.; Petrovskii, P.V.; Janoušek, Z.; Corsini, M.; Laschi, F.; Zanello, P.; Kudinov, A.R. (Tetramethylcyclobutadiene)cobalt complexes with monoanionic carborane ligands [9-L-7,8-C2B9H10]− (L = SMe2, NMe3 and py). J. Organomet. Chem. 2005, 690, 4745–4754. [Google Scholar] [CrossRef]
- Vinogradov, M.M.; Nelyubina, Y.V.; Aliyeu, T.M. New aspects of acid-assisted nucleophilic substitution reactions of 11-vertex nido-carboranes. Polyhedron 2022, 214, 115654. [Google Scholar] [CrossRef]
- Telegina, A.A.; Gruzdev, D.A.; Levit, G.L.; Krasnov, V.P. Synthesis of new nido-carborane-containing 6-thiopurine derivatives. Russ. Chem. Bull. 2023, 72, 2860–2866. [Google Scholar] [CrossRef]
- Stewart, W.E.; Siddall, T.H., III. Nuclear Magnetic Resonance Studies of Amides. Chem. Rev. 1970, 70, 517–551. [Google Scholar] [CrossRef]
- Li, Y.-S.; Escobar, L.; Zhu, Y.-J.; Cohen, Y.; Ballester, P.; Rebek, J., Jr.; Yu, Y. Relative hydrophilicities of cis and trans formamides. Proc. Natl. Acad. Sci. USA 2019, 116, 19815–19820. [Google Scholar] [CrossRef]
- Chouhan, K.K.; Chowdhury, D.; Mukherjee, A. Transamidation of Aromatic Amines with Formamides Using Cyclic Dihydrogen Tetrametaphosphate. Org. Biomol. Chem. 2022, 20, 7929–7935. [Google Scholar] [CrossRef] [PubMed]
- Plešek, J.; Grüner, B.; Maloń, P. Synthesis and properties of (±)- and (+)-4-MeS-3-C2H5-1,2,3-C2CoB9H10. Collect. Czech. Chem. Commun. 1993, 58, 1087–1092. [Google Scholar] [CrossRef]
- Tian, Z.-Y.; Hu, Y.-T.; Teng, H.-B.; Zhang, C.-P. Application of arylsulfonium salts as arylation reagents. Tetrahedron Lett. 2018, 59, 299–309. [Google Scholar] [CrossRef]
- Tian, Z.-Y.; Ma, Y.; Zhang, C.-P. Alkylation Reactions with Alkylsulfonium Salts. Synthesis 2022, 54, 1478–1502. [Google Scholar] [CrossRef]
- Chen, J.; Liu, S.; Su, S.; Fan, R.; Zhang, R.; Meng, W.; Tan, J. Sulfonium-based precise alkyl transposition reactions. Sci. Adv. 2023, 9, eadi1370. [Google Scholar] [CrossRef]
- Anufriev, S.A.; Sivaev, I.B.; Suponitsky, K.Y.; Bregadze, V.I. Practical synthesis of 9-methylthio-7,8-nido-carborane [9-MeS-nido-7,8-C2B9H11]−. Some evidences of BH···X hydride-halogen bonds in 9-XCH2(Me)S-7,8-C2B9H11 (X = Cl, Br, I). J. Organomet. Chem. 2017, 849–850, 315–323. [Google Scholar] [CrossRef]
- Kultyshev, R.G.; Liu, J.; Meyers, E.A.; Shore, S.G. Synthesis and Characterization of Sulfide, Sulfide–Sulfonium, and Bissulfide Derivatives of [B12H12]2−. Additivity of Me2S and MeS− Substituent Effects in 11B NMR Spectra of Disubstituted Icosahedral Boron Clusters. Inorg. Chem. 2000, 39, 3333–3341. [Google Scholar] [CrossRef]
- Kultyshev, R.G.; Liu, J.; Liu, S.; Tjarks, W.; Soloway, A.H.; Shore, S.G. S-Alkylation and S-Amination of Methyl Thioethers–Derivatives of closo-[B12H12]2−. Synthesis of a Boronated Phosphonate, gem-Bisphosphonates, and Dodecaborane-ortho-carborane Oligomers. J. Am. Chem. Soc. 2002, 124, 2614–2624. [Google Scholar] [CrossRef]
- Kabytaev, K.Z.; Safronov, A.V.; Sevryugina, Y.V.; Barnes, C.L.; Jalisatgi, S.S.; Hawthorne, M.F. Novel Synthetic Approach to Charge-Compensated Phosphonio-nido-Carboranes. Synthesis and Structural Characterization of Neutral Mono and Bis(Phosphonio) nido-ortho-Carboranes. Inorg. Chem. 2015, 54, 4143–4150. [Google Scholar] [CrossRef]
- Zakharova, M.V.; Sivaev, I.B.; Anufriev, S.A.; Timofeev, S.V.; Suponitsky, K.Y.; Godovikov, I.A.; Bregadze, V.I. A new approach to the synthesis of functional derivatives of nido-carborane: Alkylation of [9-MeS-nido-7,8-C2B9H11]−. Dalton Trans. 2014, 43, 5044–5053. [Google Scholar] [CrossRef]
- Poater, J.; Viñas, C.; Bennour, I.; Escayola, S.; Solà, M.; Teixidor, F. Too Persistent to Give Up: Aromaticity in Boron Clusters Survives Radical Structural Changes. J. Am. Chem. Soc. 2020, 142, 9396–9407. [Google Scholar] [CrossRef] [PubMed]
- Zakharkin, L.I.; Kalinin, V.N.; Gedymin, V.V. Synthesis of 1,2-Dicarba-closo-dodecaboran(12)-3-yl-carboxylic Acids (3-O-Carboranecarboxylic Acids). Synth. Inorg. Met.-Org. Chem. 1971, 1, 45–50. [Google Scholar] [CrossRef]
- Meguellati, K.; Koripelly, G.; Ladame, S. DNA-Templated Synthesis of Trimethine Cyanine Dyes: A Versatile Fluorogenic Reaction for Sensing G-Quadruplex Formation. Angew. Chem. Int. Ed. 2010, 49, 2738–2742. [Google Scholar] [CrossRef] [PubMed]
- Armarego, W.L.F.; Chai, C.L.L. Purification of Laboratory Chemicals, 6th ed.; Butterworth Heinemann: Burlington, MA, USA, 2009; 743p. [Google Scholar]
- Janoušek, Z.; Heřmánek, S.; Plešek, J.; Štíbr, B. Tetracarba-dinido-docosaborane (C4B18H22), a new type of carborane, its chemistry and structure. Collect. Czech. Chem. Commun. 1974, 39, 2363–2373. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).