
Design, Synthesis, and Anticancer Activity of Novel Enmein-Type Diterpenoid Derivatives Targeting the PI3K/Akt/mTOR Signaling Pathway

Abstract
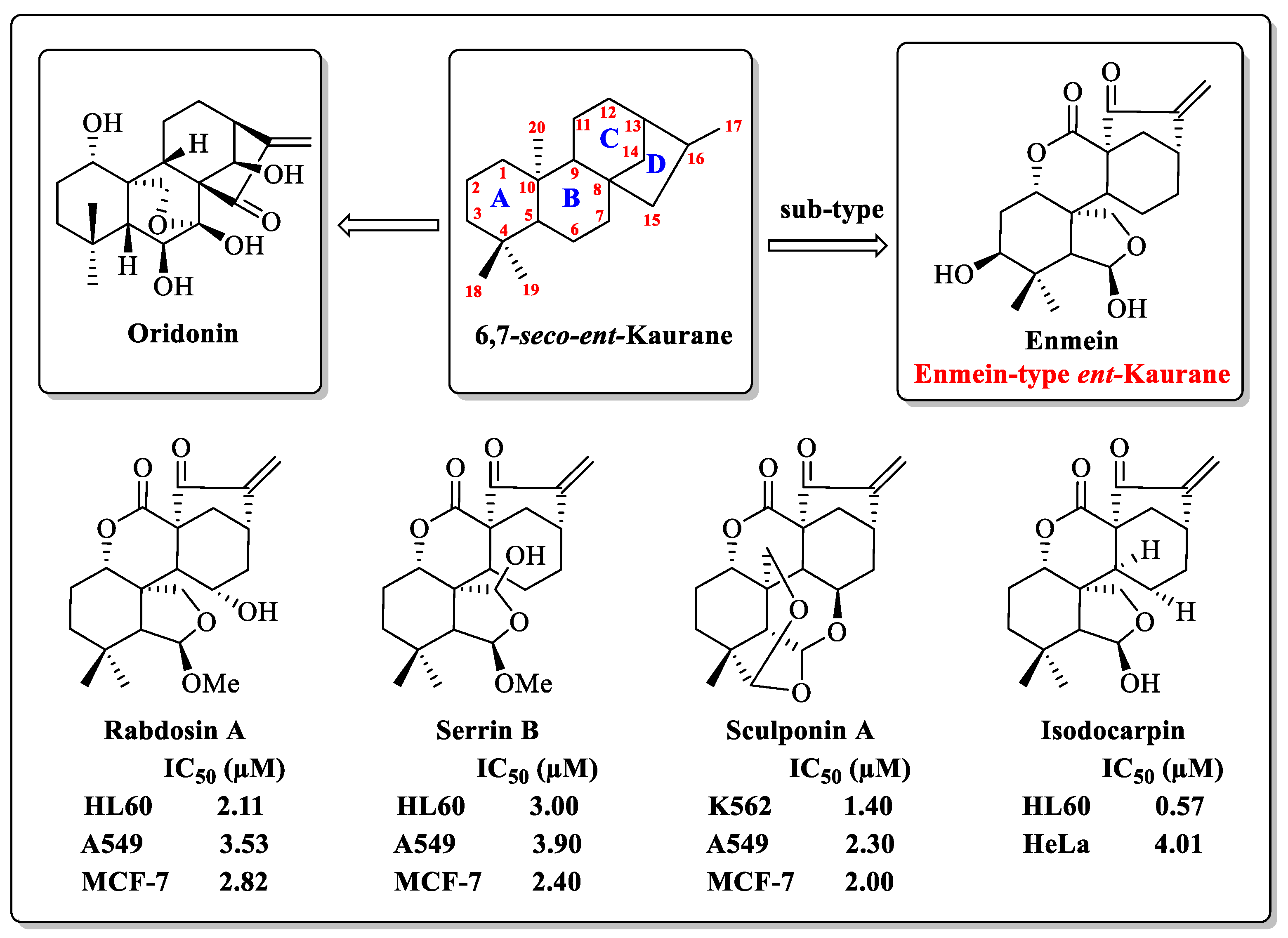
1. Introduction
2. Results and Discussion
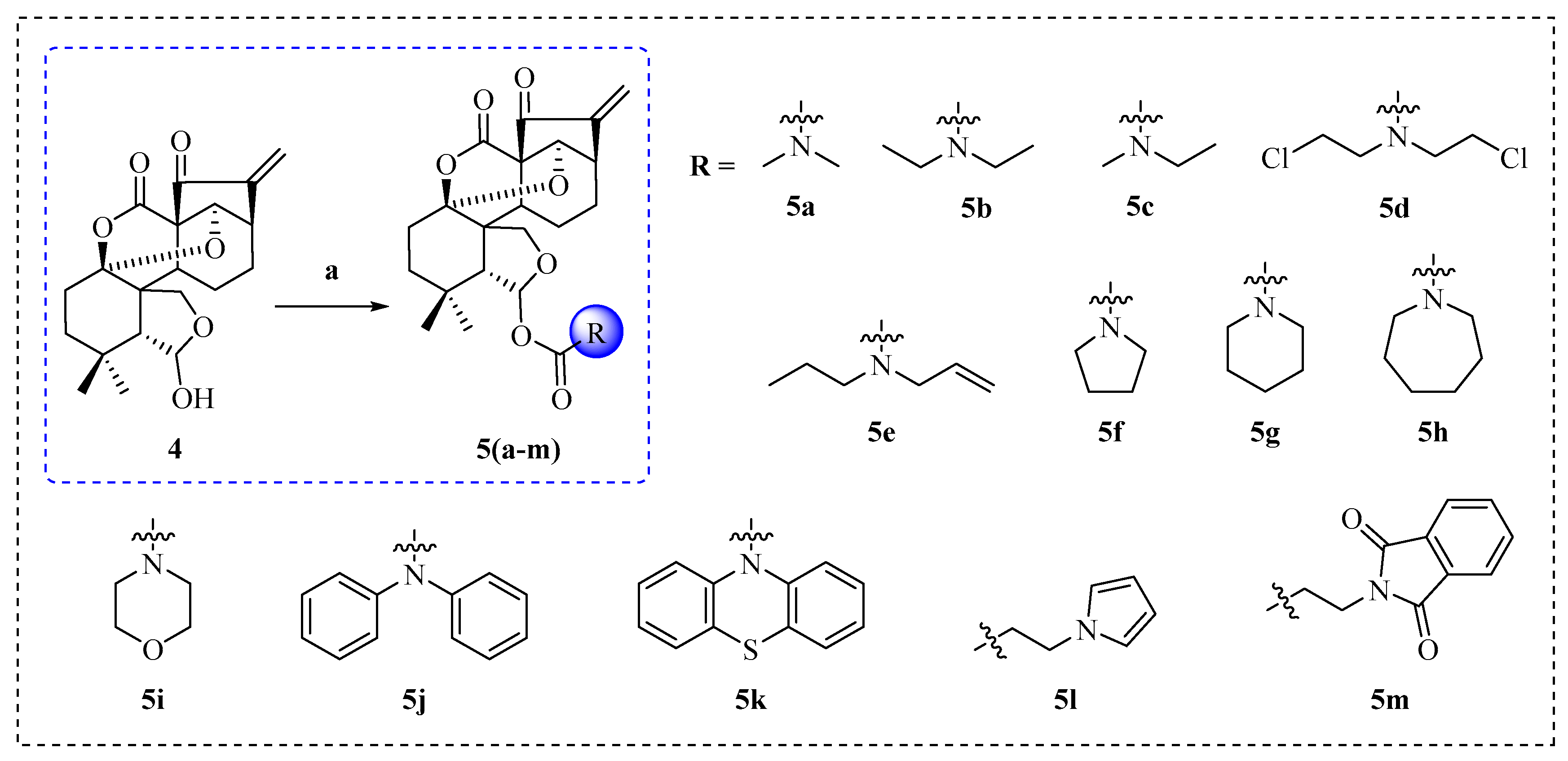
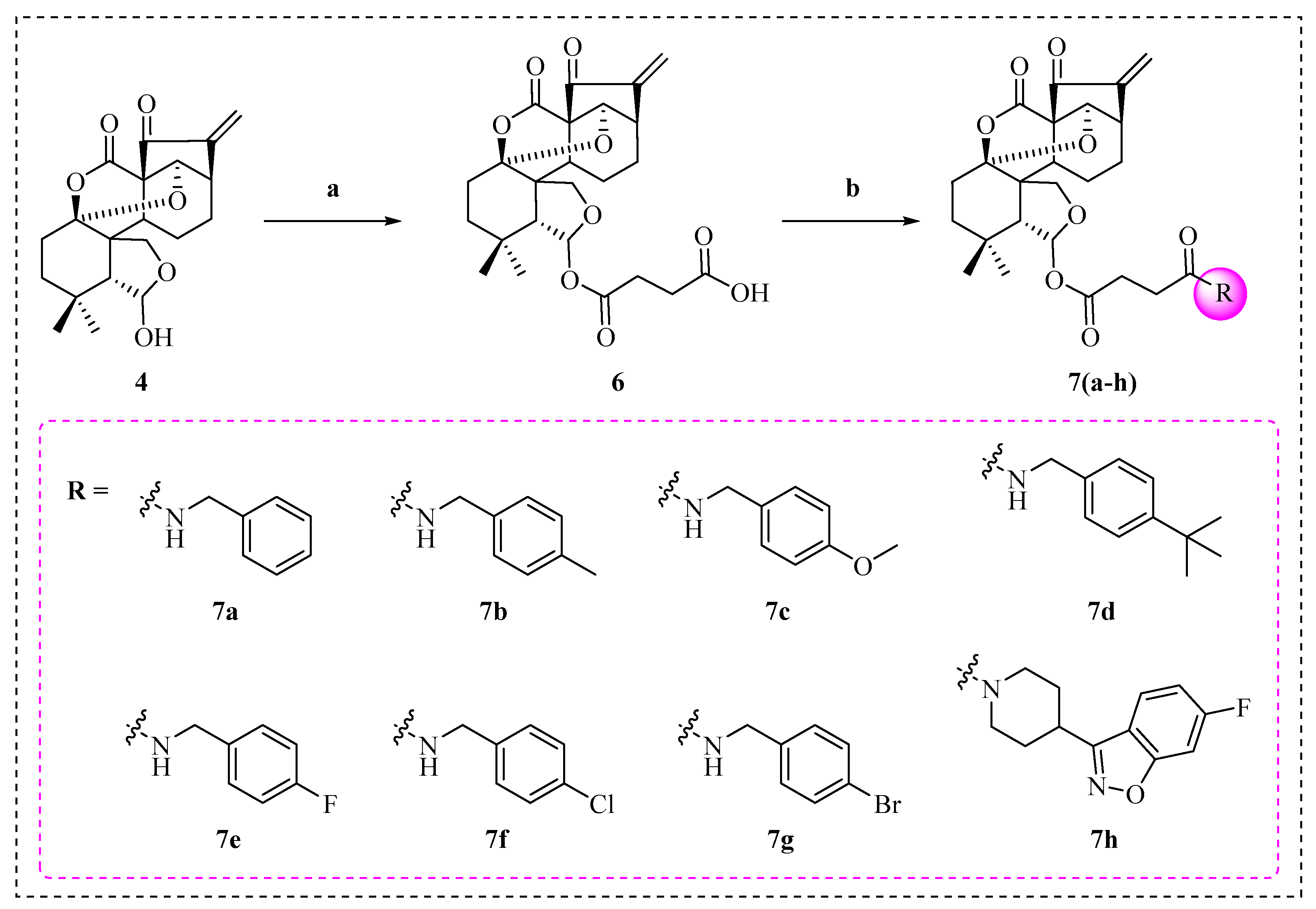
2.1. Synthesis of Enmein-Type Diterpenoid Derivatives
2.2. In Vitro Anti-Proliferative Activity and SAR Analysis
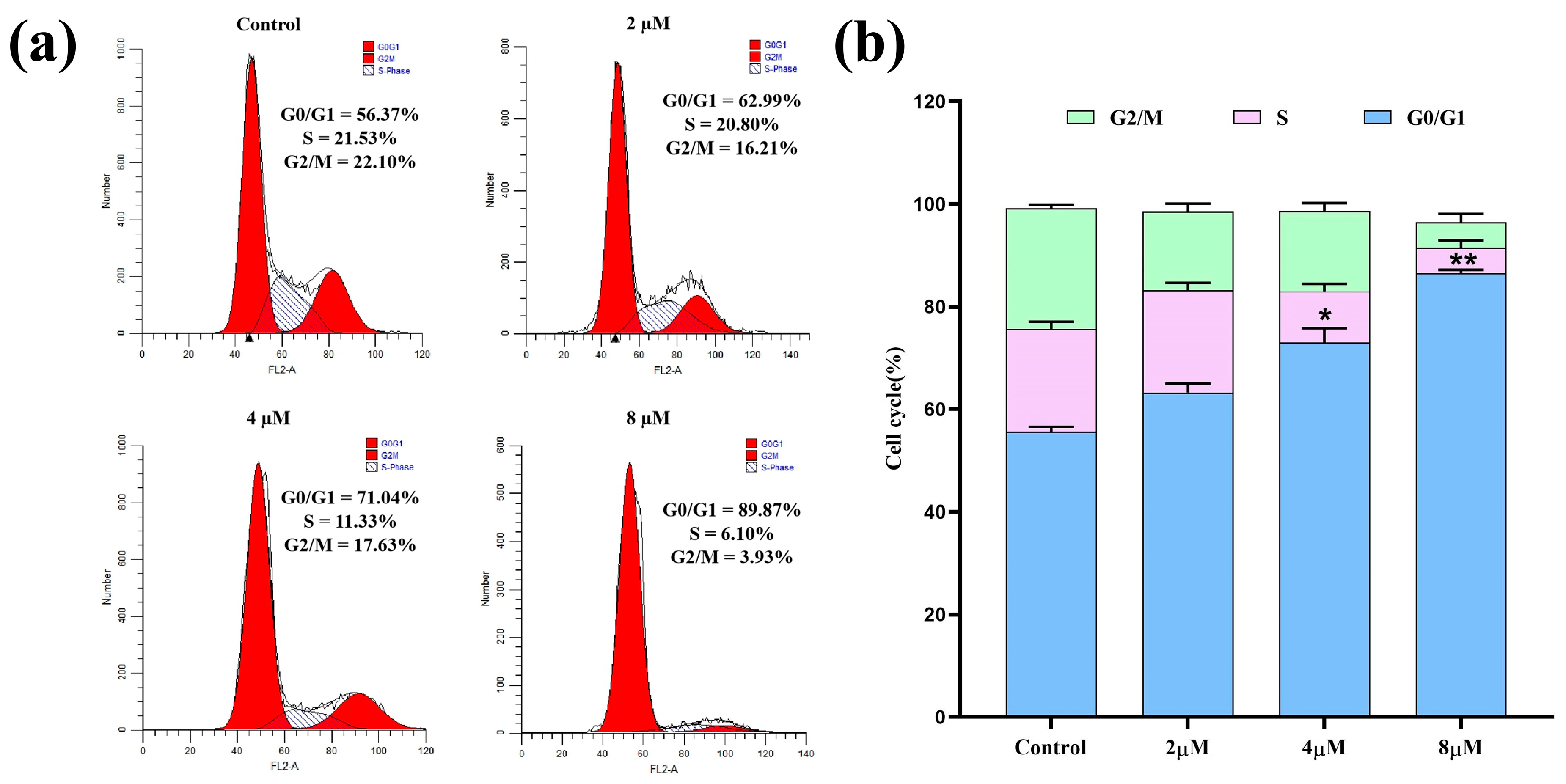
2.3. Cell Cycle Analysis
2.4. Apoptosis Analysis
2.5. Reactive Oxygen Species (ROS) Analysis
2.6. Mitochondrial Membrane Potential (MMP) Analysis
2.7. The Predicted Potential Targets and Pathways of 7h
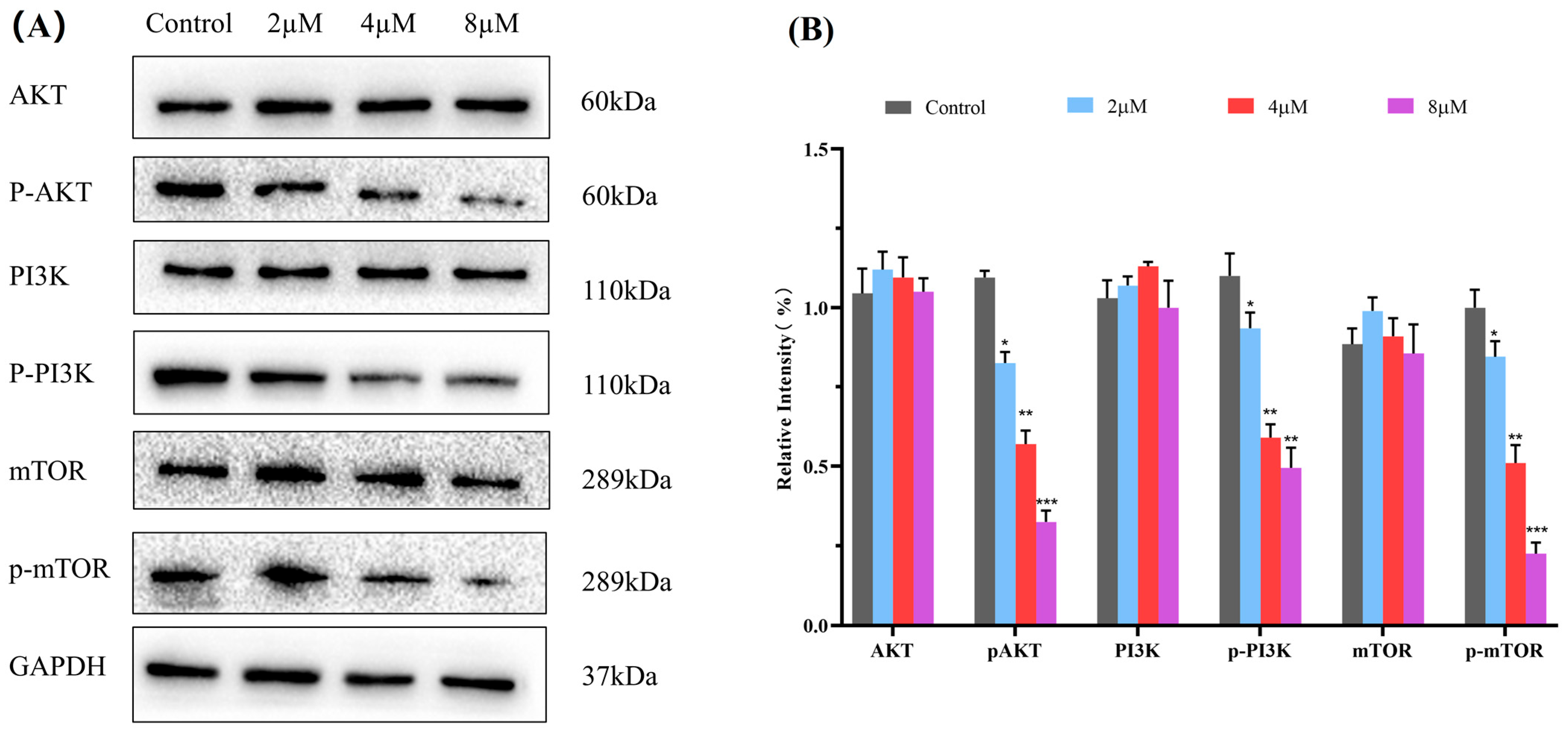
2.8. Western Blot Analysis
2.9. Molecular Docking Study
3. Conclusions
4. Materials and Methods
4.1. Chemistry
4.1.1. Reagents and Apparatus
4.1.2. Synthesis of Ent-6β,7β,14β-trihydroxy-1,15-dioxo-7,20-epoxy-16-kaurene (2)
4.1.3. Synthesis of Ent-6β,14α-Dihydroxy-(1,7–1,14)-epoxy-7,15-dioxo-6,20-hemiketal-6,7-seco-16-kaurene (4)
4.1.4. General Procedure for the Synthesis of Target Derivatives 5a–m
4.1.5. Synthesis of 4-(Ent-6β,14α-dihydroxy-(1,7–1,14)-epoxy-7,15-dioxo-6,20-hemi-ketal-6,7-seco-16-kaurene-6β-yloxy)-4-oxobutanoic Acid (6)
4.1.6. General Procedure for the Synthesis of Target Derivatives 7a–h
4.2. Biological Testing
4.2.1. Cell Culture
4.2.2. In Vitro Anti-Proliferative Activity
4.2.3. Experimental Method for Cell Cycle Analysis
4.2.4. Experimental Methods for Apoptosis Analysis
Cell Apoptosis Morphological Detection
Cell Apoptosis Rate Detection
4.2.5. Experimental Methods for ROS Measurement
Cell Morphological Detection
ROS Content Determination
4.2.6. Experimental Method for MMP Measurement
Cell Morphological Detection
Cell Morphological Detection
4.2.7. Experimental Methods for GO and KEGG Analysis
4.2.8. Experimental Method of Western Blot Analysis
4.2.9. Experimental Method of Molecular Docking Study
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Gaidai, O.; Yan, P.; Xing, Y.H. Future world cancer death rate prediction. Sci. Rep. 2023, 13, 303. [Google Scholar] [CrossRef]
- Herrero Álvarez, N.; Bauer, D.; Hernández-Gil, J.; Lewis, J.S. Recent Advances in Radiometals for Combined Imaging and Therapy in Cancer. ChemMedChem 2021, 16, 2909–2941. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Liu, X. Radionanomedicine: Advanced Strategy for Precision Theranostics of Breast Cancer. J. Biomed. Nanotechnol. 2022, 18, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Bian, X.; Li, P.; Huang, Y.; Li, C. Carrier-Free Nanomedicine for Cancer Immunotherapy. J. Biomed. Nanotechnol. 2022, 18, 939–956. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.C.; Ni, J.J.; Cui, W.Y.; Wang, B.Y.; Zhuo, W. Emerging roles of lncRNA in cancer and therapeutic opportunities. Am. J. Cancer Res. 2019, 9, 1354–1366. [Google Scholar] [PubMed]
- Fehrenbacher, J.C. Chemotherapy-induced peripheral neuropathy. Prog. Mol. Biol. Transl. Sci. 2015, 131, 471–508. [Google Scholar] [PubMed]
- Zhang, J.; Chai, H. Recent Advances in Drug Discovery and Cancer Diagnoses. Curr. Top. Med. Chem. 2020, 20, 1855–1857. [Google Scholar] [CrossRef]
- Barot, K.P.; Nikolova, S.; Ivanov, I.; Ghate, M.D. Novel anticancer agents and targets: Recent advances and future perspectives. Mini Rev. Med. Chem. 2013, 13, 1239–1255. [Google Scholar] [CrossRef]
- Zhang, L.; Song, J.; Kong, L.; Yuan, T.; Li, W.; Zhang, W.; Hou, B.; Lu, Y.; Du, G. The strategies and techniques of drug discovery from natural products. Pharmacol. Ther. 2020, 216, 107686. [Google Scholar] [CrossRef]
- Chan, W.J.; Adiwidjaja, J.; McLachlan, A.J.; Boddy, A.V.; Harnett, J.E. Interactions between natural products and cancer treatments: Underlying mechanisms and clinical importance. Cancer Chemother. Pharmacol. 2023, 91, 103–119. [Google Scholar] [CrossRef]
- Hashem, S.; Ali, T.A.; Akhtar, S.; Nisar, S.; Sageena, G.; Ali, S.; Al-Mannai, S.; Therachiyil, L.; Mir, R.; Elfaki, I.; et al. Targeting cancer signaling pathways by natural products: Exploring promising anticancer agents. Biomed. Pharmacother. 2022, 150, 113054. [Google Scholar] [CrossRef] [PubMed]
- Shaik, B.B.; Katari, N.K.; Jonnalagadda, S.B. Role of Natural Products in Developing Novel Anticancer Agents: A Perspective. Chem. Biodivers. 2022, 19, e202200535. [Google Scholar] [CrossRef] [PubMed]
- Najmi, A.; Javed, S.A.; Al Bratty, M.; Alhazmi, H.A. Modern Approaches in the Discovery and Development of Plant-Based Natural Products and Their Analogues as Potential Therapeutic Agents. Molecules 2022, 27, 349. [Google Scholar] [CrossRef] [PubMed]
- Cabral, C.; Efferth, T.; Pires, I.M.; Severino, P.; Lemos, M.F.L. Natural Products as a Source for New Leads in Cancer Research and Treatment. Evid.-Based Complement. Altern. Med. 2018, 2018, 8243680. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural Products as Sources of New Drugs over the Nearly Four Decades from 01/1981 to 09/2019. J. Nat. Prod. 2020, 83, 770–803. [Google Scholar] [CrossRef] [PubMed]
- Naeem, A.; Hu, P.; Yang, M.; Zhang, J.; Liu, Y.; Zhu, W.; Zheng, Q. Natural Products as Anticancer Agents: Current Status and Future Perspectives. Molecules 2022, 27, 8367. [Google Scholar] [CrossRef] [PubMed]
- Varghese, R.; Dalvi, Y.B. Natural Products as Anticancer Agents. Curr. Drug Targets 2021, 22, 1272–1287. [Google Scholar] [CrossRef]
- Ibrahim, T.S.; Khongorzul, P.; Muyaba, M.; Alolga, R.N. Ent-kaurane diterpenoids from the Annonaceae family: A review of research progress and call for further research. Front. Pharmacol. 2023, 14, 1227574. [Google Scholar] [CrossRef]
- Sarwar, M.S.; Xia, Y.X.; Liang, Z.M.; Tsang, S.W.; Zhang, H.J. Mechanistic Pathways and Molecular Targets of Plant-Derived Anticancer ent-Kaurane Diterpenes. Biomolecules 2020, 10, 144. [Google Scholar] [CrossRef]
- Luo, H.; Vong, C.T.; Chen, H.; Gao, Y.; Lyu, P.; Qiu, L.; Zhao, M.; Liu, Q.; Cheng, Z.; Zou, J.; et al. Naturally occurring anticancer compounds: Shining from Chinese herbal medicine. Chin. Med. 2019, 14, 48. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, C.T.; Ma, W.; Xie, X.; Huang, Q. Oridonin: A Review of Its Pharmacology, Pharmacokinetics and Toxicity. Front. Pharmacol. 2021, 12, 645824. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Chen, X.; Qu, S.; Yao, B.; Xu, Y.; Wu, J.; Jin, Y.; Ma, C. Oridonin induces G2/M cell cycle arrest and apoptosis via the PI3K/Akt signaling pathway in hormone-independent prostate cancer cells. Oncol. Lett. 2017, 13, 2838–2846. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.H.; Pi, J.; Jin, H.; Cai, J.Y. Oridonin-induced mitochondria-dependent apoptosis in esophageal cancer cells by inhibiting PI3K/AKT/mTOR and Ras/Raf pathways. J. Cell. Biochem. 2019, 120, 3736–3746. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.X.; He, Z.A.; Chen, Z.Y.; Wang, Y.X.; Bai, S.P.; Sun, H.D. Cytotoxic ent-kaurane diterpenoids from Isodon macrophyllus. J. Asian Nat. Prod. Res. 2009, 11, 693–697. [Google Scholar] [CrossRef]
- Yang, J.; Wang, W.G.; Wu, H.Y.; Du, X.; Li, X.N.; Li, Y.; Pu, J.X.; Sun, H.D. Bioactive Enmein-Type ent-Kaurane Diterpenoids from Isodon phyllostachys. J. Nat. Prod. 2016, 79, 132–140. [Google Scholar] [CrossRef]
- Li, L.M.; Li, G.Y.; Ding, L.S.; Lei, C.; Yang, L.B.; Zhao, Y.; Weng, Z.Y.; Li, S.H.; Huang, S.X.; Xiao, W.L.; et al. Sculponins A-C, three new 6,7-seco-ent-kauranoids from Isodon sculponeatus. Tetrahedron Lett. 2007, 48, 9100–9103. [Google Scholar] [CrossRef]
- Yan, F.L.; Guo, L.Q.; Wang, C.M.; Zhang, J.X. Chemical constituents of Isodon nervosus and their cytotoxicity. J. Asian Nat. Prod. Res. 2009, 11, 326–331. [Google Scholar] [CrossRef]
- Wang, L.; Li, D.; Xu, S.; Cai, H.; Yao, H.; Zhang, Y.; Jiang, J.; Xu, J. The conversion of oridonin to spirolactone-type or enmein-type diterpenoid: Synthesis and biological evaluation of ent-6,7-seco-oridonin derivatives as novel potential anticancer agents. Eur. J. Med. Chem. 2012, 52, 242–250. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Brindisi, M. Organic carbamates in drug design and medicinal chemistry. J. Med. Chem. 2015, 58, 2895–2940. [Google Scholar] [CrossRef] [PubMed]
- Bu, M.; Cao, T.; Li, H.; Guo, M.; Yang, B.B.; Zeng, C.; Hu, L. Synthesis of 5α,8α-Ergosterol Peroxide 3-Carbamate Derivatives and a Fluorescent Mitochondria-Targeting Conjugate for Enhanced Anticancer Activities. ChemMedChem 2017, 12, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.X.; Chen, C.; Liu, X.Q.; Li, Y.; Lin, Y.L.; Wu, X.T.; Kong, L.Y.; Luo, J.G. Discovery and optimization of withangulatin A derivatives as novel glutaminase 1 inhibitors for the treatment of triple-negative breast cancer. Eur. J. Med. Chem. 2021, 210, 112980. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.J.; Liu, M.C.; Xiang, H.M.; Zhao, Q.; Xue, W.; Yang, S. Synthesis and in vitro antitumor evaluation of betulin acid ester derivatives as novel apoptosis inducers. Eur. J. Med. Chem. 2015, 102, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Hameed, R.H.; Mohamed, M.S.; Awad, S.M.; Hassan, B.B.; Khodair, M.A.E.; Mansour, Y.E. Novel benzo chromene derivatives: Design, synthesis, molecular docking, cell cycle arrest, and apoptosis induction in human acute myeloid leukemia HL-60 cells. J. Enzym. Inhib. Med. Chem. 2023, 38, 405–422. [Google Scholar] [CrossRef] [PubMed]
- Wei, T.; Zheng, Z.; Wei, X.; Liu, Y.; Li, W.; Fang, B.; Yun, D.; Dong, Z.; Yi, B.; Li, W.; et al. Rational design, synthesis, and pharmacological characterisation of dicarbonyl curcuminoid analogues with improved stability against lung cancer via ROS and ER stress mediated cell apoptosis and pyroptosis. J. Enzym. Inhib. Med. Chem. 2022, 37, 2357–2369. [Google Scholar] [CrossRef]
- Wang, X.; Pan, C.X.; Gong, J.Y.; Liu, X.F.; Li, H.L. Enhancing the Enrichment of Pharmacophore-Based Target Prediction for the Polypharmacological Profiles of Drugs. J. Chem. Inf. Model. 2016, 56, 1175–1183. [Google Scholar] [CrossRef]
- Wang, X.; Shen, Y.; Wang, S.; Li, S.; Zhang, W.; Liu, X.; Lai, L.; Pei, J.; Li, H. PharmMapper 2017 update: A web server for potential drug target identification with a comprehensive target pharmacophore database. Nucleic Acids Res. 2017, 45, W356–W360. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, Y.H.; Wang, S.; Zhang, Y.; Huang, T.; Cai, Y.D. Prediction and analysis of essential genes using the enrichments of gene ontology and KEGG pathways. PLoS ONE 2017, 12, e0184129. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, Y.H.; Lu, G.; Huang, T.; Cai, Y.D. Analysis of cancer-related lncRNAs using gene ontology and KEGG pathways. Artif. Intell. Med. 2017, 76, 27–36. [Google Scholar] [CrossRef]
- Yu, L.; Wei, J.; Liu, P. Attacking the PI3K/Akt/mTOR signaling pathway for targeted therapeutic treatment in human cancer. Semin. Cancer Biol. 2022, 85, 69–94. [Google Scholar] [CrossRef] [PubMed]
- Tewari, D.; Patni, P.; Bishayee, A.; Sah, A.N.; Bishayee, A. Natural products targeting the PI3K-Akt-mTOR signaling pathway in cancer: A novel therapeutic strategy. Semin. Cancer Biol. 2022, 80, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.C. Targeting the PI3K/Akt/mTOR pathway in non-small cell lung cancer (NSCLC). Thorac. Cancer 2020, 11, 511–518. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.; Yang, J.; Yu, W.; Li, H.; Cai, M.; Xu, J.L.; Xu, H.D.; Shi, Y.F.; Guan, X.; Cheng, X.D.; et al. Discovery of benzochalcone derivative as a potential antigastric cancer agent targeting signal transducer and activator of transcription 3 (STAT3). J. Enzym. Inhib. Med. Chem. 2022, 37, 2004–2016. [Google Scholar] [CrossRef]
- Ahmed, R.I.; Osman, E.E.; Awadallah, F.M.; El-Moghazy, S.M. Design, synthesis and molecular docking of novel diarylcyclohexenone and diarylindazole derivatives as tubulin polymerization inhibitors. J. Enzym. Inhib. Med. Chem. 2017, 32, 176–188. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | IC50 (µM) a | |||
|---|---|---|---|---|
| A549 | HepG2 | MCF-7 | L-02 | |
| 5a | 14.72 ± 1.54 | 17.12 ± 1.39 | 30.72 ± 1.16 | >50.00 |
| 5b | 15.84 ± 1.50 | 15.22 ± 1.58 | 17.57 ± 2.40 | >50.00 |
| 5c | 20.25 ± 1.46 | 22.54 ± 3.42 | 21.87 ± 2.72 | >50.00 |
| 5d | 8.76 ± 1.56 | 11.04 ± 1.45 | 15.56 ± 1.61 | 30.36 ± 3.44 |
| 5e | 10.71 ± 1.81 | 11.57 ± 1.96 | 14.11 ± 2.35 | 36.68 ± 2.43 |
| 5f | 13.43 ± 2.19 | 18.30 ± 1.36 | 20.71 ± 1.20 | 45.88 ± 2.30 |
| 5g | 15.26 ± 2.40 | 21.10 ± 2.50 | 22.81 ± 2.13 | >50.00 |
| 5h | 18.37 ± 1.13 | 20.18 ± 1.33 | 24.04 ± 2.55 | >50.00 |
| 5i | 14.39 ± 1.42 | 23.65 ± 3.40 | 25.12 ± 3.34 | 38.15 ± 2.82 |
| 5j | 23.30 ± 2.25 | 34.07 ± 3.52 | 32.84 ± 2.95 | >50.00 |
| 5k | 17.16 ± 1.43 | 25.68 ± 2.16 | 26.27 ± 2.30 | >50.00 |
| 5l | 8.30 ± 2.55 | 11.15 ± 1.82 | 15.87 ± 1.90 | >50.00 |
| 5m | 6.85 ± 1.68 | 10.87 ± 1.25 | 12.34 ± 1.11 | 32.51 ± 1.62 |
| 7a | 10.21 ± 1.10 | 13.77 ± 2.19 | 10.80 ± 1.65 | >50.00 |
| 7b | 7.48 ± 1.31 | 10.53 ± 1.68 | 12.76 ± 1.64 | >50.00 |
| 7c | 8.37 ± 1.53 | 12.83 ± 1.73 | 8.35 ± 1.96 | 34.42 ± 4.37 |
| 7d | 6.41 ± 1.90 | 11.54 ± 1.58 | 7.64 ± 2.52 | 35.74 ± 2.66 |
| 7e | 7.12 ± 1.52 | 11.60 ± 1.34 | 9.85 ± 1.53 | >50.00 |
| 7f | 7.38 ± 0.96 | 8.61 ± 1.14 | 8.20 ± 1.21 | >50.00 |
| 7g | 8.60 ± 2.65 | 8.78 ± 1.46 | 13.00 ± 1.52 | >50.00 |
| 7h | 2.16 ± 1.40 | 6.10 ± 1.24 | 6.74 ± 1.94 | >50.00 |
| 4 | 23.83 ± 1.86 | 27.35 ± 0.83 | 25.75 ± 1.14 | 30.25 ± 1.58 |
| Oridonin | 21.10 ± 1.58 | 22.62 ± 1.20 | 18.46 ± 1.37 | 23.62 ± 1.65 |
| Taxol | 4.21 ± 0.73 | 6.60 ± 1.61 | 5.15 ± 0.80 | 8.31 ± 1.82 |
| Compound | Receptor Protein | Binding Energy (kcal/mol) a |
|---|---|---|
| 7h | PI3Kα | −11.6 |
| Akt1 | −11.3 | |
| mTOR | −11.4 | |
| 4 | PI3Kα | −10.1 |
| Akt1 | −9.0 | |
| mTOR | −10.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Wang, L.; Zhang, Y.; Pan, S.; Lin, Y.; Wu, J.; Bu, M. Design, Synthesis, and Anticancer Activity of Novel Enmein-Type Diterpenoid Derivatives Targeting the PI3K/Akt/mTOR Signaling Pathway. Molecules 2024, 29, 4066. https://doi.org/10.3390/molecules29174066
Wang J, Wang L, Zhang Y, Pan S, Lin Y, Wu J, Bu M. Design, Synthesis, and Anticancer Activity of Novel Enmein-Type Diterpenoid Derivatives Targeting the PI3K/Akt/mTOR Signaling Pathway. Molecules. 2024; 29(17):4066. https://doi.org/10.3390/molecules29174066
Chicago/Turabian StyleWang, Jiafeng, Lu Wang, Yingbo Zhang, Siwen Pan, Yu Lin, Jiale Wu, and Ming Bu. 2024. "Design, Synthesis, and Anticancer Activity of Novel Enmein-Type Diterpenoid Derivatives Targeting the PI3K/Akt/mTOR Signaling Pathway" Molecules 29, no. 17: 4066. https://doi.org/10.3390/molecules29174066
APA StyleWang, J., Wang, L., Zhang, Y., Pan, S., Lin, Y., Wu, J., & Bu, M. (2024). Design, Synthesis, and Anticancer Activity of Novel Enmein-Type Diterpenoid Derivatives Targeting the PI3K/Akt/mTOR Signaling Pathway. Molecules, 29(17), 4066. https://doi.org/10.3390/molecules29174066