
Identify Regioselective Residues of Ginsenoside Hydrolases by Graph-Based Active Learning from Molecular Dynamics


Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Data Collection and Structure Acquisition
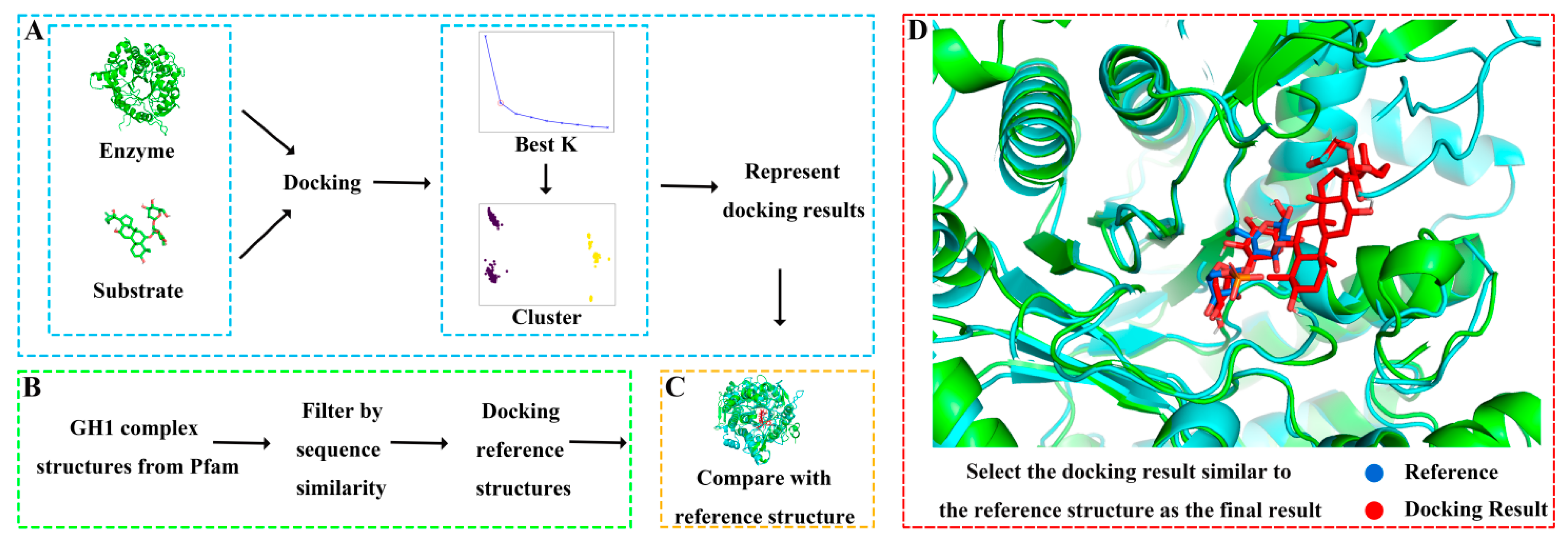
2.2. Molecular Docking and Validation
2.3. Molecular Dynamics Simulations
2.4. Trajectory Analysis Methods
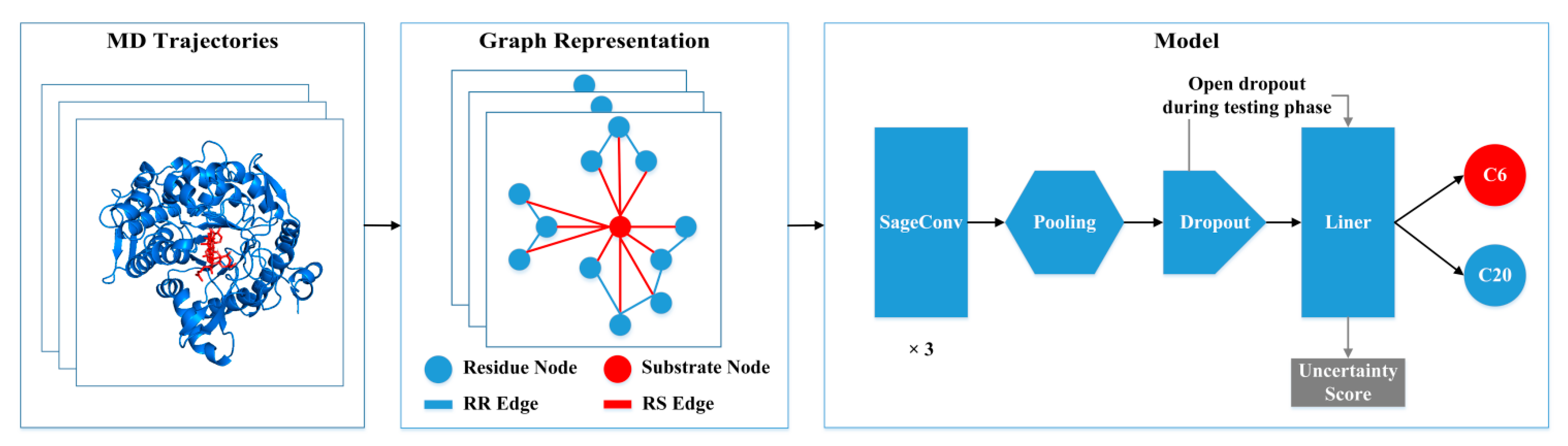
2.5. Graph Neural Network
2.6. Uncertainty Calculation
2.7. Evaluation Metrics
2.8. Interpretability Algorithm
3. Results and Discussion
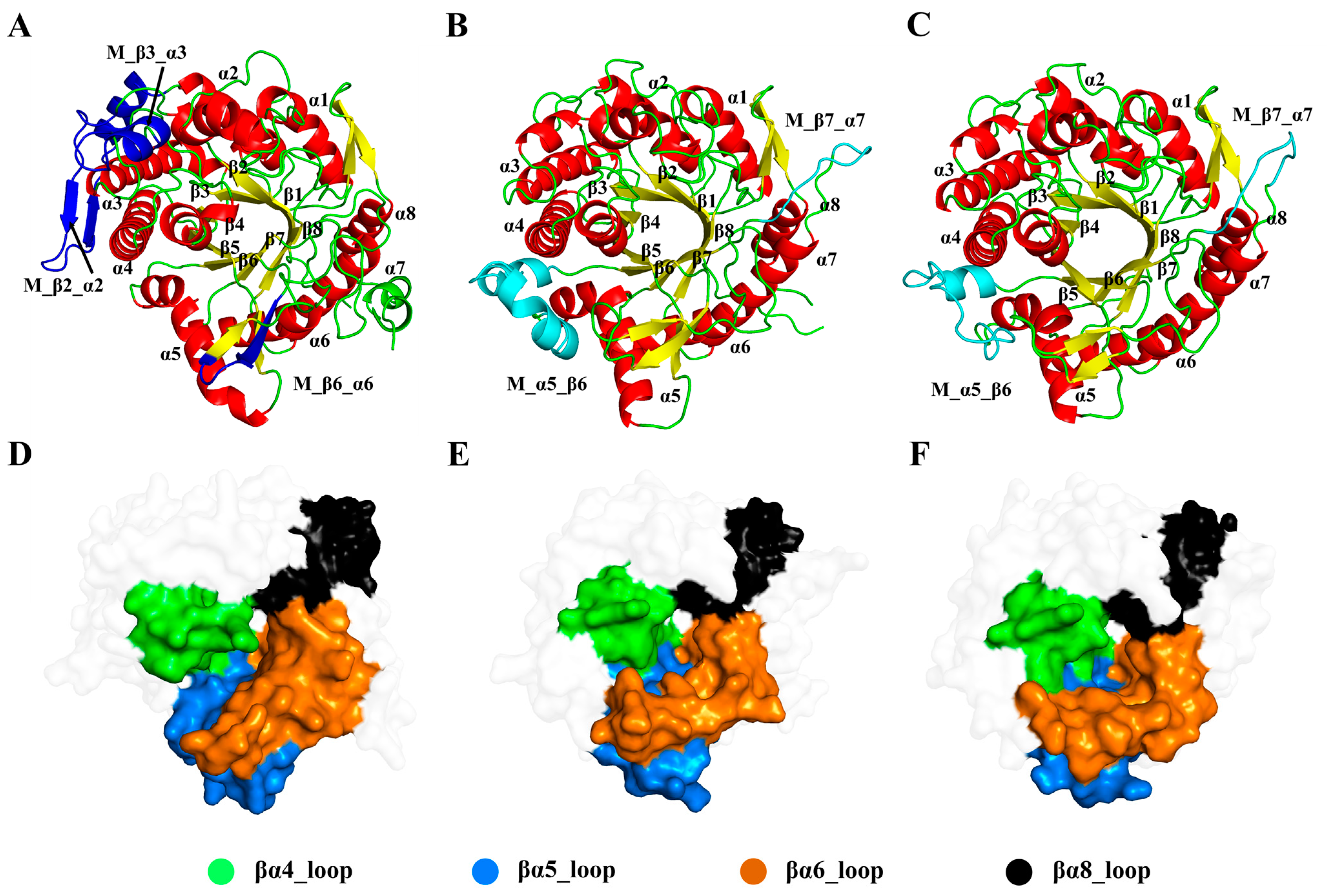
3.1. Sequence and Structure Alignments of Ginsenoside Hydrolase
3.2. Cluster Analysis of Substrate Docking Poses
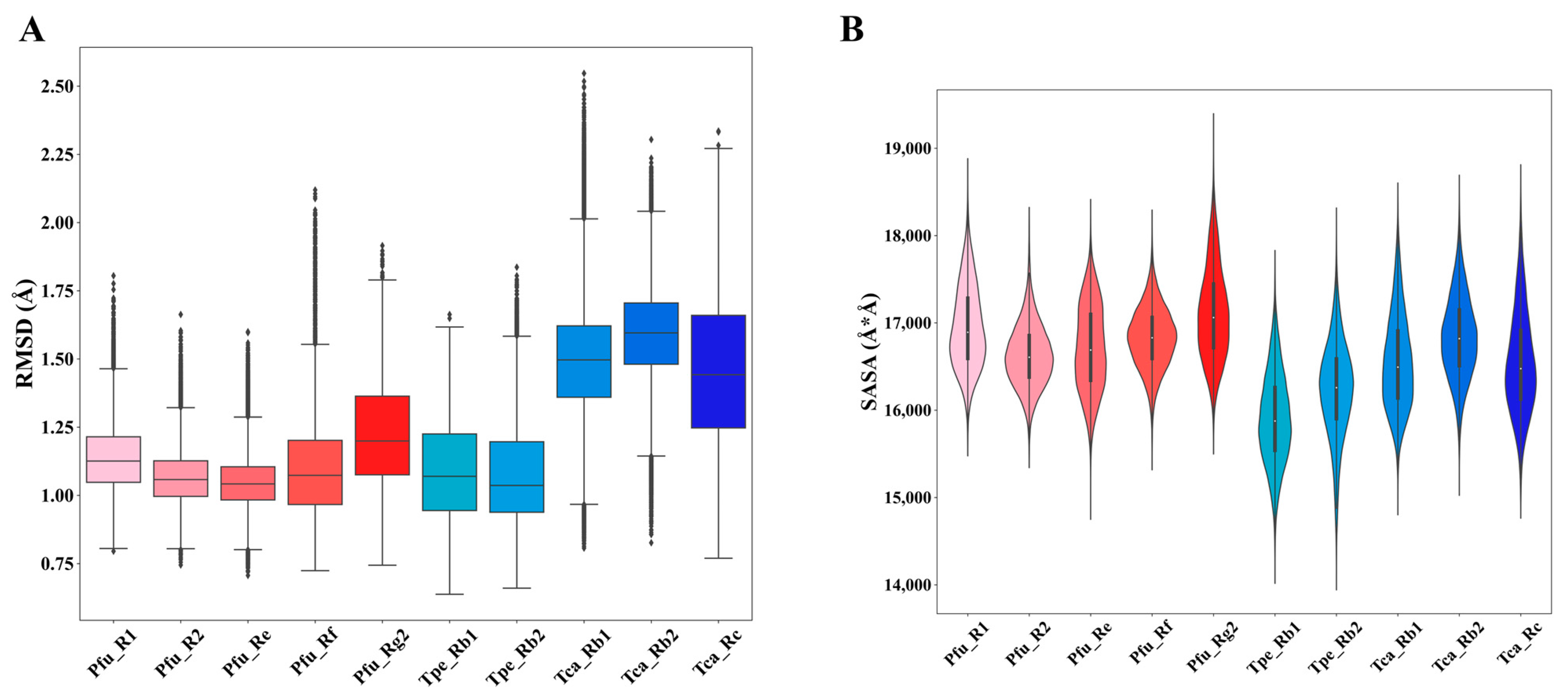
3.3. Different Systems Exhibit Similar Dynamic Macroscopic Properties
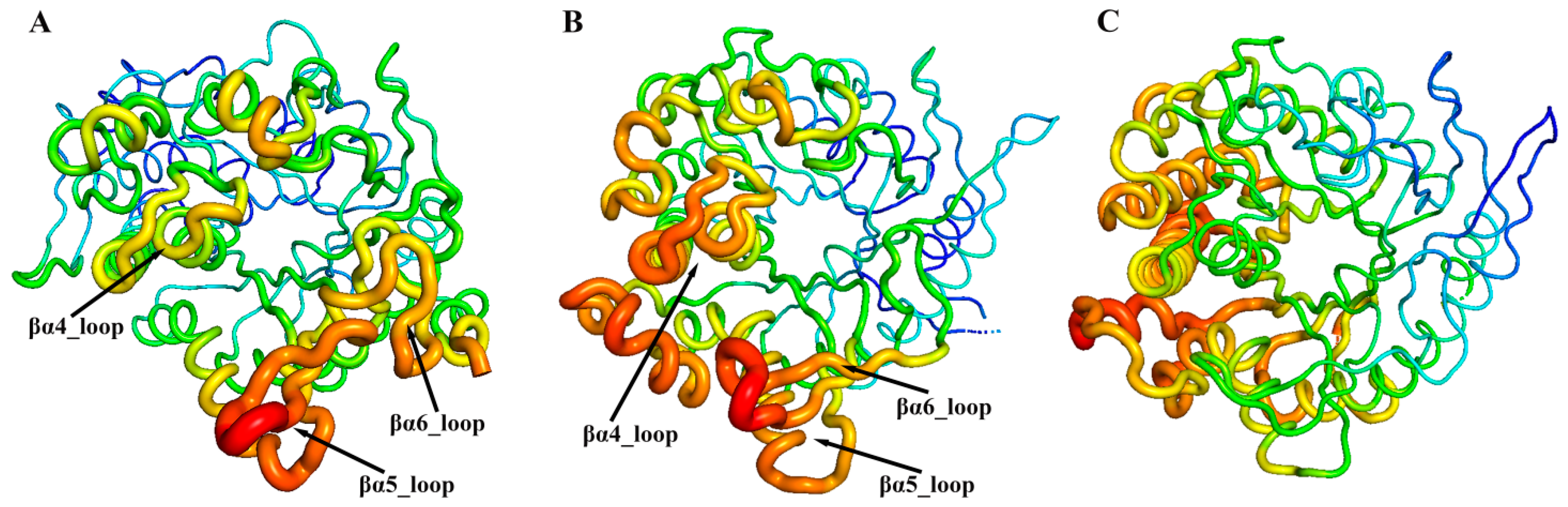
3.4. Residue Dynamics Exacerbate the Challenge of Distinguishing Regioselectivity
3.5. Classification Performance of Graph Neural Network
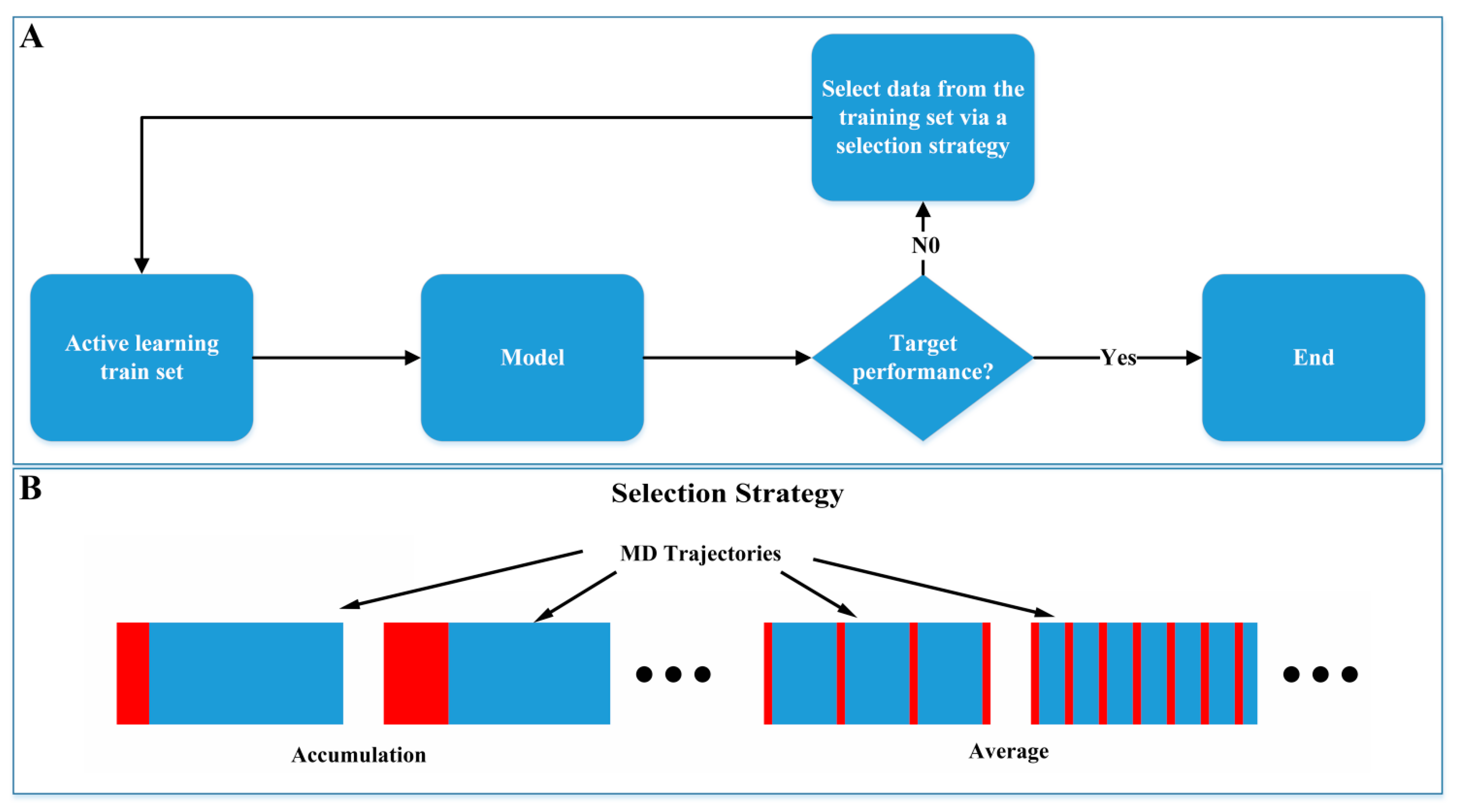
3.6. Active Learning Weakens Reliance on Molecular Dynamics
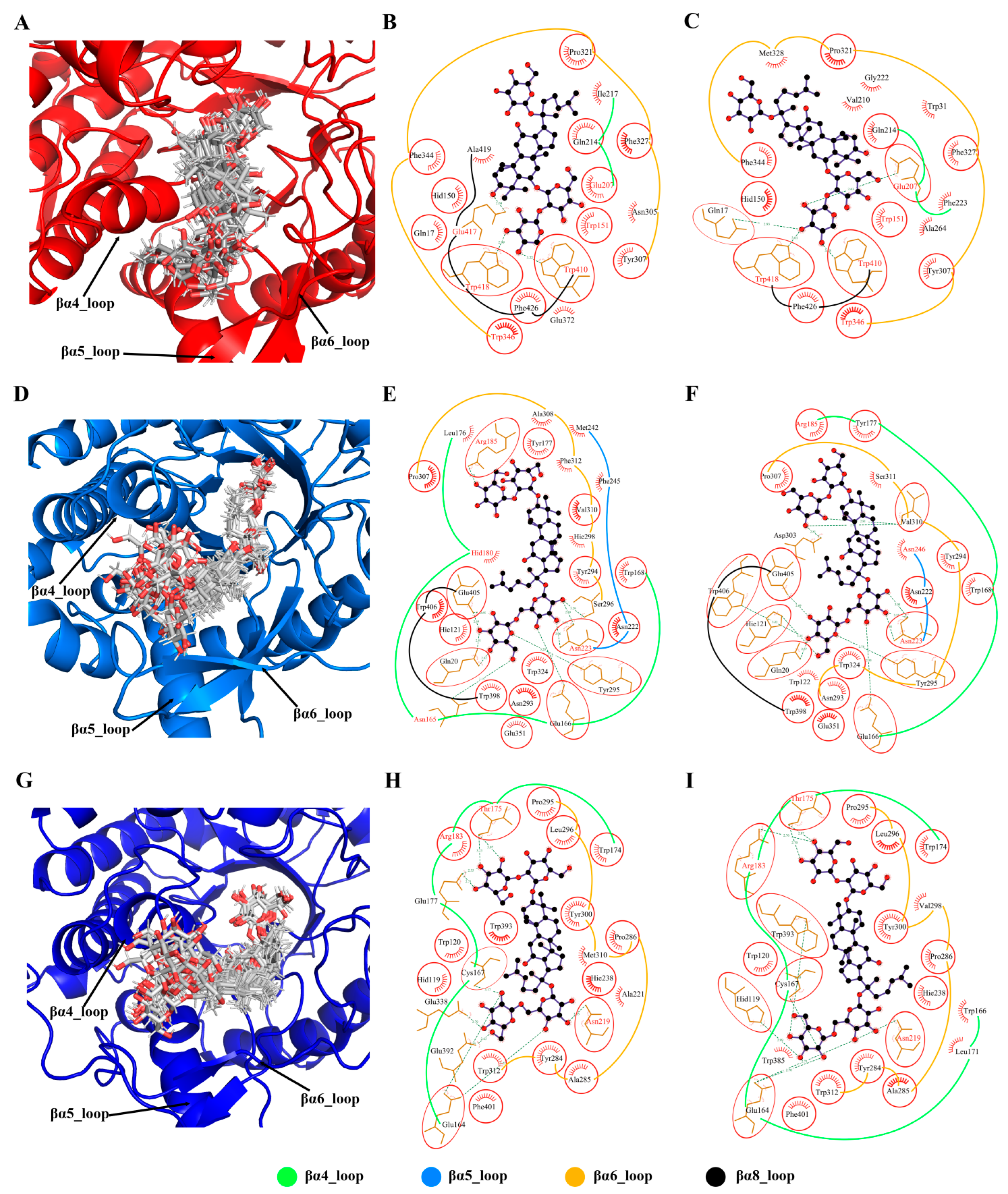
3.7. Model Interpretability and Its Important Elements
3.8. Regioselective Residues Extracted from Model Interpretability
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Liu, L.; Xu, F.-R.; Wang, Y.-Z. Traditional uses, chemical diversity and biological activities of Panax L. (Araliaceae): A review. J. Ethnopharmacol. 2020, 263, 112792. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.L.; Huy, N.Q.; Tung, N.H. Microorganisms for Ginsenosides Biosynthesis: Recent Progress, Challenges, and Perspectives. Molecules 2023, 28, 1437. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-J.; Yamabe, N.; Choi, P.; Lee, J.W.; Ham, J.; Kang, K.S. Efficient Thermal Deglycosylation of Ginsenoside Rd and Its Contribution to the Improved Anticancer Activity of Ginseng. J. Agric. Food Chem. 2013, 61, 9185–9191. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Wang, L.; Fan, D. Insights into Recent Studies on Biotransformation and Pharmacological Activities of Ginsenoside Rd. Biomolecules 2022, 12, 512. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-N.; Fan, D.-D. Biocatalytic strategies for the production of ginsenosides using glycosidase: Current state and perspectives. Appl. Microbiol. Biotechnol. 2020, 104, 3807–3823. [Google Scholar] [CrossRef] [PubMed]
- Son, J.W.; Kim, H.J.; Oh, D.K. Ginsenoside Rd production from the major ginsenoside Rb(1) by beta-glucosidase from Thermus caldophilus. Biotechnol. Lett. 2008, 30, 713–716. [Google Scholar] [CrossRef]
- Oh, H.J.; Shin, K.C.; Oh, D.K. Production of ginsenosides Rg1 and Rh1 by hydrolyzing the outer glycoside at the C-6 position in protopanaxatriol-type ginsenosides using beta-glucosidase from Pyrococcus furiosus. Biotechnol. Lett. 2014, 36, 113–119. [Google Scholar] [CrossRef]
- Zhang, S.; Luo, J.; Xie, J.; Wang, Z.; Xiao, W.; Zhao, L. Cooperated biotransformation of ginsenoside extracts into ginsenoside 20(S)-Rg3 by three thermostable glycosidases. J. Appl. Microbiol. 2020, 128, 721–734. [Google Scholar] [CrossRef]
- Li, Q.; Wu, T.; Qi, Z.; Zhao, L.; Pei, J.; Tang, F. Characterization of a novel thermostable and xylose-tolerant GH 39 beta-xylosidase from Dictyoglomus thermophilum. BMC Biotechnol. 2018, 18, 29. [Google Scholar] [CrossRef]
- Xie, J.; Zhao, D.; Zhao, L.; Pei, J.; Xiao, W.; Ding, G.; Wang, Z.; Xu, J. Characterization of a novel arabinose-tolerant alpha-L-arabinofuranosidase with high ginsenoside Rc to ginsenoside Rd bioconversion productivity. J. Appl. Microbiol. 2016, 120, 647–660. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, H.; Luo, X.; Deng, Y.; Zhang, W.; Li, S.; Liang, J.; Pang, Z. Enzymatic biotransformation of Rb3 from the leaves of Panax notoginseng to ginsenoside rd by a recombinant beta-xylosidase from Thermoascus aurantiacus. World J. Microbiol. Biotechnol. 2022, 39, 21. [Google Scholar] [CrossRef]
- Park, C.-S.; Yoo, M.-H.; Noh, K.-H.; Oh, D.-K. Biotransformation of ginsenosides by hydrolyzing the sugar moieties of ginsenosides using microbial glycosidases. Appl. Microbiol. Biotechnol. 2010, 87, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Quan, L.-H.; Min, J.-W.; Jin, Y.; Wang, C.; Kim, Y.-J.; Yang, D.-C. Enzymatic Biotransformation of Ginsenoside Rb1 to Compound K by Recombinant β-Glucosidase from Microbacterium esteraromaticum. J. Agric. Food Chem. 2012, 60, 3776–3781. [Google Scholar] [CrossRef] [PubMed]
- Bai, Q.; Liu, S.; Tian, Y.; Xu, T.; Banegas-Luna, A.J.; Pérez-Sánchez, H.; Huang, J.; Liu, H.; Yao, X. Application advances of deep learning methods for de novo drug design and molecular dynamics simulation. WIREs Comput. Mol. Sci. 2022, 12, e1581. [Google Scholar] [CrossRef]
- Plante, A.; Shore, D.M.; Morra, G.; Khelashvili, G.; Weinstein, H.J.M. A machine learning approach for the discovery of ligand-specific functional mechanisms of GPCRs. Molecules 2019, 24, 2097. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, M.; Moroni, E.; Ippoliti, E.; Rinaldi, S.; Sanchez-Martin, C.; Rasola, A.; Pavarino, L.F.; Colombo, G. Machine learning of allosteric effects: The analysis of ligand-induced dynamics to predict functional effects in TRAP1. J. Phys. Chem. B 2020, 125, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Liu, J.; Chen, J.; Yuan, Y.; Yu, J.; Gou, Q.; Guo, Y.; Pu, X. Modeling. An interpretable convolutional neural network framework for analyzing molecular dynamics trajectories: A case study on functional states for g-protein-coupled receptors. J. Chem. Inf. Model. 2022, 62, 1399–1410. [Google Scholar] [CrossRef]
- UniProt, C. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar] [CrossRef]
- Varadi, M.; Bertoni, D.; Magana, P.; Paramval, U.; Pidruchna, I.; Radhakrishnan, M.; Tsenkov, M.; Nair, S.; Mirdita, M.; Yeo, J.; et al. AlphaFold Protein Structure Database in 2024: Providing structure coverage for over 214 million protein sequences. Nucleic Acids Res. 2023, 52, D368–D375. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2023 update. Nucleic Acids Res. 2022, 51, D1373–D1380. [Google Scholar] [CrossRef]
- Madeira, F.; Pearce, M.; Tivey, A.R.N.; Basutkar, P.; Lee, J.; Edbali, O.; Madhusoodanan, N.; Kolesnikov, A.; Lopez, R. Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic Acids Res. 2022, 50, W276–W279. [Google Scholar] [CrossRef]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Case, D.; Aktulga, H.M.; Belfon, K.; Ben-Shalom, I.; Berryman, J.; Brozell, S.; Cerutti, D.; Cheatham, T.; Cisneros, G.A.; Cruzeiro, V.; et al. Amber 2022; University of California: San Francisco, CA, USA, 2022. [Google Scholar] [CrossRef]
- Price, D.J.; Brooks, C.L., III. A modified TIP3P water potential for simulation with Ewald summation. J. Chem. Phys. 2004, 121, 10096–10103. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- He, X.; Man, V.H.; Yang, W.; Lee, T.-S.; Wang, J. A fast and high-quality charge model for the next generation general AMBER force field. J. Chem. Phys. 2020, 153, 114502. [Google Scholar] [CrossRef]
- Elber, R.; Ruymgaart, A.P.; Hess, B. SHAKE parallelization. Eur. Phys. J. Spec. Top. 2011, 200, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Zhang, S.; Krieger, J.M.; Zhang, Y.; Kaya, C.; Kaynak, B.; Mikulska-Ruminska, K.; Doruker, P.; Li, H.; Bahar, I. ProDy 2.0: Increased scale and scope after 10 years of protein dynamics modelling with Python. Bioinformatics 2021, 37, 3657–3659. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple Ligand–Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Fey, M.; Lenssen, J.E. Fast graph representation learning with PyTorch Geometric. arXiv 2019, arXiv:1903.02428. [Google Scholar]
- Paszke, A.; Gross, S.; Massa, F.; Lerer, A.; Bradbury, J.; Chanan, G.; Killeen, T.; Lin, Z.; Gimelshein, N.; Antiga, L. Pytorch: An imperative style, high-performance deep learning library. arXiv 2019, arXiv:1912.01703. [Google Scholar]
- Hamilton, W.; Ying, Z.; Leskovec, J. Inductive representation learning on large graphs. arXiv 2017, arXiv:1706.02216. [Google Scholar]
- Senge, R.; Bösner, S.; Dembczyński, K.; Haasenritter, J.; Hirsch, O.; Donner-Banzhoff, N.; Hüllermeier, E. Reliable classification: Learning classifiers that distinguish aleatoric and epistemic uncertainty. Inf. Sci. 2014, 255, 16–29. [Google Scholar] [CrossRef]
- Amara, K.; Ying, R.; Zhang, Z.; Han, Z.; Shan, Y.; Brandes, U.; Schemm, S.; Zhang, C. Graphframex: Towards systematic evaluation of explainability methods for graph neural networks. arXiv 2022, arXiv:2206.09677. [Google Scholar]
- Kokhlikyan, N.; Miglani, V.; Martin, M.; Wang, E.; Alsallakh, B.; Reynolds, J.; Melnikov, A.; Kliushkina, N.; Araya, C.; Yan, S.J.; et al. Captum: A unified and generic model interpretability library for pytorch. arXiv 2020, arXiv:2009.07896. [Google Scholar]
- Wierenga, R.K. The TIM-barrel fold: A versatile framework for efficient enzymes. FEBS Lett. 2001, 492, 193–198. [Google Scholar] [CrossRef]
- McIntosh, L.P.; Hand, G.; Johnson, P.E.; Joshi, M.D.; Körner, M.; Plesniak, L.A.; Ziser, L.; Wakarchuk, W.W.; Withers, S.G. The pKa of the General Acid/Base Carboxyl Group of a Glycosidase Cycles during Catalysis: A 13C-NMR Study of Bacillus circulans Xylanase. Biochemistry 1996, 35, 9958–9966. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
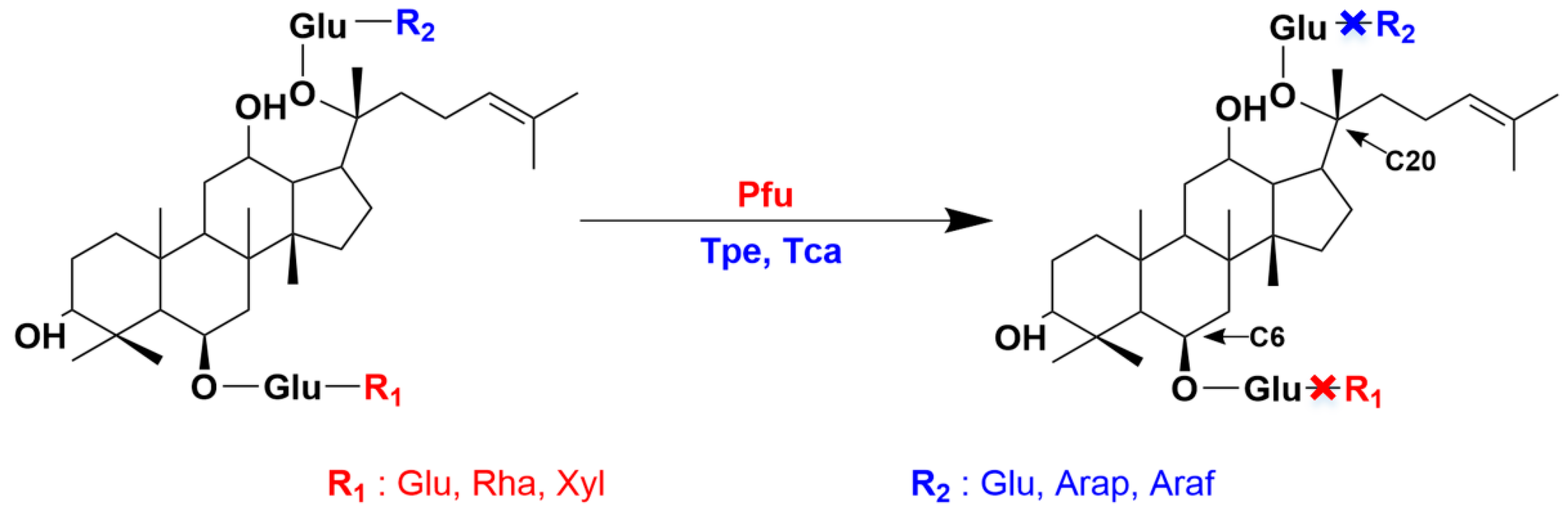
| Regioselectivity | Ginsenoside Hydrolase | Substrate |
|---|---|---|
| C-6 | Pfubgl1 (pfu) | R1, R2, Re, Rf, Rg2 |
| C-20 | Tpebgl1 (Tpe) | Rb1, Rb2 |
| Tcabgl1 (Tca) | Rb1, Rb2, Rc |
| Dataset | Precision | Recall | Acc | Mcc | Uncertainty |
|---|---|---|---|---|---|
| Glycan test set | 96.6 ± 3.8 | 100.0 ± 0.0 | 98.1 ± 2.1 | 96.4 ± 4.0 | 0.0214 ± 0.0187 |
| Replica test set | 98.5 ± 1.4 | 100.0 ± 0.0 | 99.2 ± 0.8 | 98.5 ± 1.5 | 0.0075 ± 0.0069 |
| Cumulative Data | Accumulation | Average | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Precision | Recall | Acc | Mcc | Uncertainty | Precision | Recall | Acc | Mcc | Uncertainty | |
| 3000 (5 ns) | 84.2 ± 8.7 | 100.0 ± 0.0 | 90.0 ± 6.4 | 82.0 ± 10.8 | 0.0395 ± 0.0033 | 91.6 ± 6.6 | 100.0 ± 0.0 | 95.1 ± 4.0 | 90.9 ± 7.3 | 0.0335 ± 0.0180 |
| 6000 (10 ns) | 86.6 ± 9.2 | 100.0 ± 0.0 | 91.6 ± 6.2 | 84.9 ± 10.8 | 0.0357 ± 0.0132 | 92.8 ± 4.0 | 100.0 ± 0.0 | 95.9 ± 2.3 | 92.3 ± 4.3 | 0.0111 ± 0.0058 |
| 12,000 (20 ns) | 92.1 ± 8.4 | 100.0 ± 0.0 | 95.2 ± 5.4 | 91.2 ± 9.7 | 0.0233 ± 0.0104 | 96.6 ± 4.2 | 100.0 ± 0.0 | 98.1 ± 2.3 | 96.4 ± 4.4 | 0.0165 ± 0.0160 |
| Ginsenoside Hydrolases | Feature Importance | Edge Importance | Fidelity Score | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Bond | Angle | Dih | Vdw14 | Elec14 | Vdw | Elec | RR Edge | RS Edge | Fid+ Score (↑) | Fid− Score (↓) | |
| Pfu | 0% | 0% | 0% | 0% | 96% | 0% | 4% | 54% | 46% | 0.95 | 0.05 |
| Tpe | 0% | 1% | 0% | 0% | 69% | 0% | 30% | 0% | 100% | 1.00 | 0 |
| Tca | 0% | 0% | 0% | 0% | 62% | 0% | 38% | 1% | 99% | 0.99 | 0.01 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Peng, H.-Q.; Wen, M.-L.; Yang, L.-Q. Identify Regioselective Residues of Ginsenoside Hydrolases by Graph-Based Active Learning from Molecular Dynamics. Molecules 2024, 29, 3614. https://doi.org/10.3390/molecules29153614
Li Y, Peng H-Q, Wen M-L, Yang L-Q. Identify Regioselective Residues of Ginsenoside Hydrolases by Graph-Based Active Learning from Molecular Dynamics. Molecules. 2024; 29(15):3614. https://doi.org/10.3390/molecules29153614
Chicago/Turabian StyleLi, Yi, Hong-Qian Peng, Meng-Liang Wen, and Li-Quan Yang. 2024. "Identify Regioselective Residues of Ginsenoside Hydrolases by Graph-Based Active Learning from Molecular Dynamics" Molecules 29, no. 15: 3614. https://doi.org/10.3390/molecules29153614
APA StyleLi, Y., Peng, H.-Q., Wen, M.-L., & Yang, L.-Q. (2024). Identify Regioselective Residues of Ginsenoside Hydrolases by Graph-Based Active Learning from Molecular Dynamics. Molecules, 29(15), 3614. https://doi.org/10.3390/molecules29153614