
Enzymatic Fructosylation of Phenolic Compounds: A New Alternative for the Development of Antidiabetic Drugs


 ,
,  and
and
Abstract

1. Introduction
2. Results
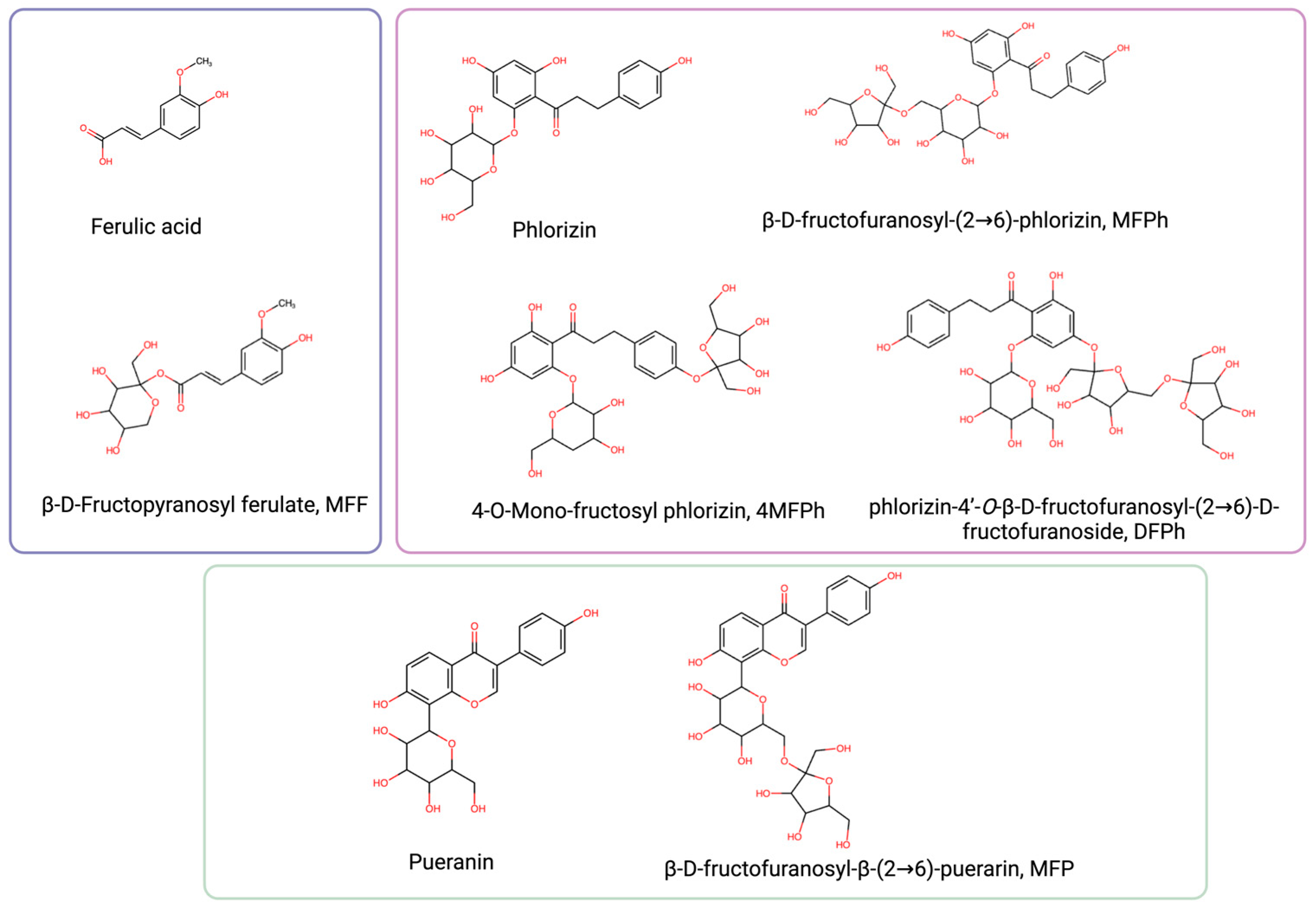
2.1. Synthesis and Physicochemical Properties of Phenolic Fructosides
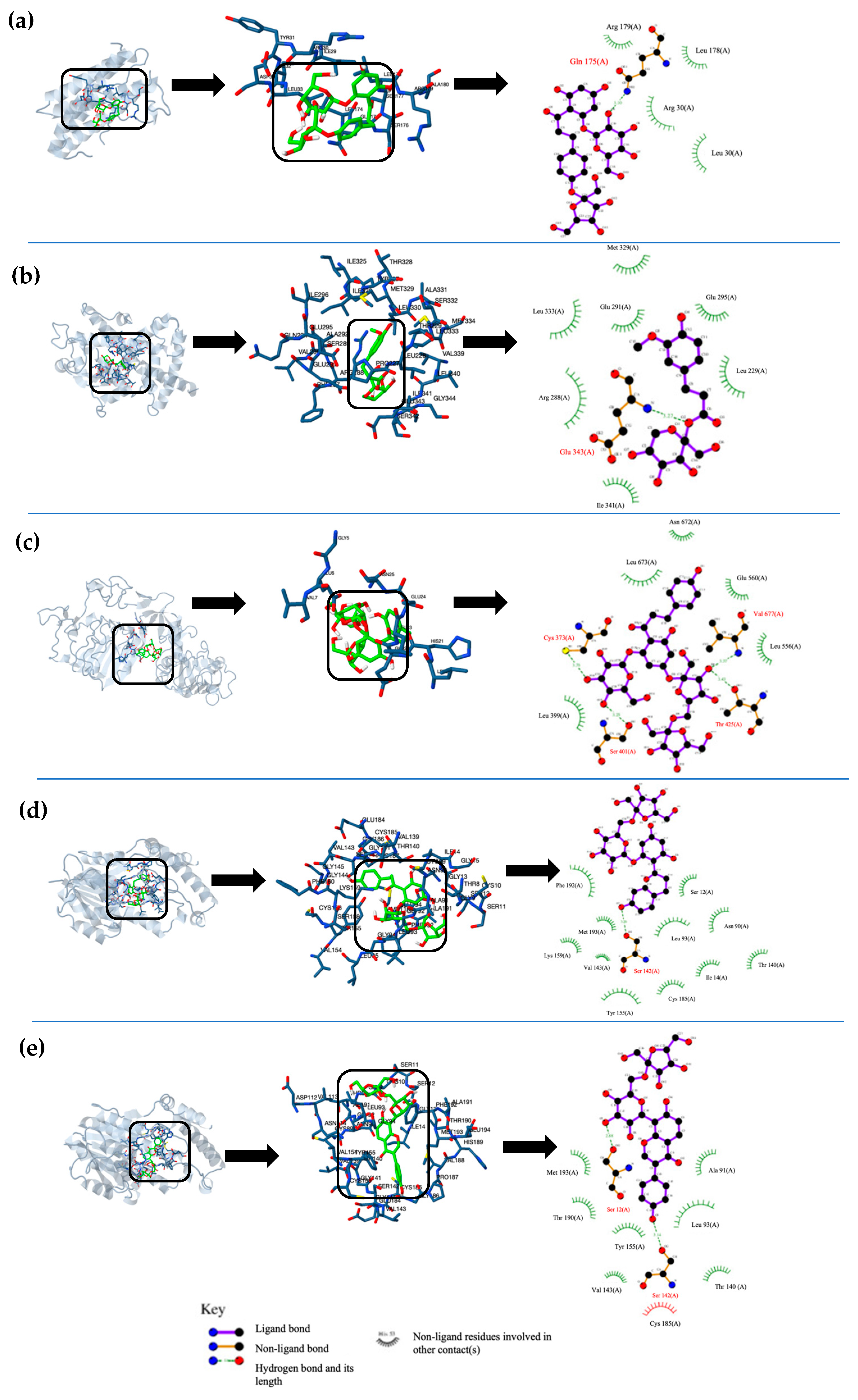
2.2. In Silico Analysis of Phenolic Fructosides
2.3. ADMET Properties of Phenolic Fructosides
3. Discussion
4. Materials and Methods
4.1. Synthesis and Physicochemical Properties of Phenolic Fructosides
4.2. Molecular Docking Analysis
4.2.1. T2DM Key Protein Selection and Preparation
4.2.2. Molecular Docking Simulations
4.3. Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) Interactions
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lizárraga-Velázquez, C.E.; Leyva-López, N.; Hernández, C.; Gutiérrez-Grijalva, E.P.; Salazar-Leyva, J.A.; Osuna-Ruíz, I.; Martínez-Montaño, E.; Arrizon, J.; Guerrero, A.; Benitez-Hernández, A.; et al. Antioxidant molecules from plant waste: Extraction techniques and biological properties. Processes 2020, 8, 1566. [Google Scholar] [CrossRef]
- Zhou, Y.-X.; Zhang, H.; Peng, C. Puerarin: A review of pharmacological effects. Phytother. Res. 2014, 28, 961–975. [Google Scholar] [CrossRef]
- Meng, F.; Guo, B.; Ma, Y.-Q.; Li, K.-W.; Niu, F.-J. Puerarin: A review of its mechanisms of action and clinical studies in ophthalmology. Phytomedicine 2022, 107, 154465. [Google Scholar] [CrossRef]
- Tian, L.; Cao, J.; Zhao, T.; Liu, Y.; Khan, A.; Cheng, G. The Bioavailability, Extraction, Biosynthesis and Distribution of Natural Dihydrochalcone: Phloridzin. Int. J. Mol. Sci. 2021, 22, 962. [Google Scholar] [CrossRef] [PubMed]
- Ehrenkranz, J.R.; Lewis, N.G.; Kahn, C.R.; Roth, J. Phlorizin: A review. Diabetes Metab. Res. Rev. 2005, 21, 31–38. [Google Scholar] [CrossRef]
- Kalra, S. Sodium Glucose Co-Transporter-2 (SGLT2) Inhibitors: A Review of Their Basic and Clinical Pharmacology. Diabetes Ther. 2014, 5, 355–366. [Google Scholar] [CrossRef]
- Li, D.; Rui, Y.-X.; Guo, S.-D.; Luan, F.; Liu, R.; Zeng, N. Ferulic acid: A review of its pharmacology, pharmacokinetics and derivatives. Life Sci. 2021, 284, 119921. [Google Scholar] [CrossRef] [PubMed]
- Deep-Raj, N.; Singh, D. A critical appraisal on ferulic acid: Biological profile, biopharmaceutical challenges and nano formulations. Health Sci. Rev. 2022, 5, 100063. [Google Scholar] [CrossRef]
- Herrera-González, A.; Núñez-López, G.; Morel, S.; Amaya-Delgado, A.; Sandoval, G.; Gschaedler, A.; Remaud-Simeon, M.; Arrizon, J. Functionalization of natural compounds by enzymatic fructosylation. Appl. Microbiol. Biotechnol. 2017, 101, 5223–5234. [Google Scholar] [CrossRef]
- Núñez-López, G.; Herrera-González, A.; Hernández, L.; Amaya-Delgado, L.; Sandoval, G.; Gschaedler, A.; Arrizon, J.; Remaud-Simeon, M.; Morel, S. Fructosylation of phenolic compounds by Gluconacetobacter diazatrophicus. Enzym. Microb. Technol. 2019, 122, 19–25. [Google Scholar] [CrossRef]
- Herrera-González, A.; Núñez-López, G.; Núñez-Dallos, N.; Amaya-Delgado, L.; Sandoval, G.; Remaud-Simeon, M.; Morel, S.; Arrizon, J.; Hernández, L. Enzymatic synthesis of phlorizin fructosides. Enzym. Microb. Technol. 2021, 147, 109783. [Google Scholar] [CrossRef] [PubMed]
- Adelusi, T.I.; Oyedele, A.Q.K.; Boyenle, I.D.; Ogunlana, A.T.; Adeyemi, R.O.; Ukachi, C.D.; Idris, M.O.; Olaoba, O.T.; Adedotun, I.O.; Kolawole, O.E.; et al. Molecular modeling in drug discovery. Inform. Med. Unlocked 2022, 29, 100880. [Google Scholar] [CrossRef]
- Sargsyan, K.; Grauffel, C.; Lim, C. How Molecular Size Impacts RMSD Applications in Molecular Dynamics Simulations. J. Chem. Theory Comput. 2017, 13, 1518–1524. [Google Scholar] [CrossRef]
- Tri, M.D.; Phat, N.T.; Minh, P.N.; Chi, M.T.; Hao, B.X.; Minh An, T.N.; Alam, M.; Van Kieu, N.; Dang, V.S.; Mai, T.T.N.; et al. In vitro anti-inflammatory, in silico molecular docking and molecular dynamics simulation of oleanane-type triterpenes from aerial parts of Mussaenda recurvata. RSC Adv. 2023, 13, 5324–5336. [Google Scholar] [CrossRef]
- Chigurupati, S.; Al-Murikhy, A.; Almahmoud, S.A.; Almoshari, Y.; Saber-Ahmed, A.; Vijayabalan, S.; Ghazi-Felemban, S.; Raj-Palanimuthu, V. Molecular docking of phenolic compounds and screening of antioxidant and antidiabetic potential of Moringa oleifera ethanolic leaves extract from Qassim region, Saudi Arabia. Saudi J. Biol. Sci. 2022, 29, 854–859. [Google Scholar] [CrossRef] [PubMed]
- Damian-Medina, K.; Salinas-Moreno, Y.; Milenkovic, D.; Figueroa-Yáñez, L.; Marino-Marmolejo, E.; Higuera-Ciapara, I.; Vallejo-Cardona, A.; Lugo-Cervantes, E. In silico análisis of antidiabetic potential of phenolic compounds from blue corn (Zea mays L.) and black bean (Phaseolus vulgaris L). Helyion 2020, 6, e03632. [Google Scholar] [CrossRef]
- Nguyen, N.; Le, L. Targeted proteins for diabetes drug design. Adv. Nat. Sci. Nanosci. Nanotechnol. 2012, 3, 013001. [Google Scholar] [CrossRef]
- Rathore, P.; Arathy, V.; Attimarad, V.; Kumar, P.; Roy, S. In-silico analysis of gymnemagenin from Gymnema sylvestre (Retz.) R. Br. with targets related to diabetes. J Theor. Biol. 2016, 21, 95–101. [Google Scholar] [CrossRef]
- Defronzo, R. Banting Lecture. From the triumvirate to the ominous octet: A new paradigm for the treatment of type 2 diabetes mellitus. Diabetes 2009, 58, 773–795. [Google Scholar] [CrossRef] [PubMed]
- Jelén, M.; Ying, P.T.C.; Hao, Y.J.; Balachandran, A.; Anamalay, K.; Morak-Młodawska, B.; Gaurav, A.; Lavilla, C.A.; Uy, M.M.; Billacura, M.P.; et al. In vitro study of antioxidant, antigylycation, sugar hydrolysis enzyme inhibitory effect and molecular in silico docking study of regulatory condensed diquinothiazines. J. Mol. Struct. 2024, 1296, 136856. [Google Scholar] [CrossRef]
- Jagadeesan, S.; Karpagam, S.; Noor, A.; Basu, R. Indole 3-heterocyclic derivative: A potential antioxidant, antibiabetic agent and their docking study on alpha amylase. J. Mol. Struct. 2023, 1291, 136027. [Google Scholar] [CrossRef]
- Guzmán-Flores, J.M.; Pérez-Vázquez, P.; Martínez-Esquivias, F.; Isiordia-Espinoza, M.A.; Viveros-Paredes, J.M. Molecular docking integrated with network pharmacology explores the therapeutic mechanism of Cannabis sativa against type 2 Diabetes. Curr. Issues Mol. Biol. 2023, 45, 7228–7241. [Google Scholar] [CrossRef]
- Ortiz, C.L.D.; Completo, G.C.; Nacario, R.C.; Nellas, R.B. Potential inhibitors of galactofuranosyltransferase 2 (GlfT2): Molecular docking, 3D-QSAR, and in silico ADMETox studies. Sci. Rep. 2019, 9, 17096. [Google Scholar] [CrossRef]
- Satoh, T.; Fujisawa, H.; Nakamura, A.; Takahashi, N.; Watanabe, K. Inhibitory effects of eight green tea catechins on cytochrome P450 1A2, 2C9, 2D6, and 3A4 activities. J. Pharm. Pharm. Sci. 2016, 19, 188–197. [Google Scholar] [CrossRef]
- Mattio, L.M.; Marengo, M.; Parravicini, C.; Eberini, I.; Dallavalle, S.; Bonomi, F.; Lametti, S.; Pinto, A. Inhibition of Pancreatic α-amylase by Resveratrol derivatives: Biological activity and molecular modelling evidence for cooperativity between viniferin enantiomers. Molecules 2019, 24, 3225. [Google Scholar] [CrossRef] [PubMed]
- Jakimiuk, K.; Sari, S.; Milewski, R.; Supuran, C.T.; Şöhretoğlu, D.; Tomczyk, M. Flavonoids as tyrosinase inhibitors in in silico and in vitro models: Basic framework of SAR using a statistical modelling approach. J. Enzym. Inhib. Med. Chem. 2022, 37, 427–436. [Google Scholar] [CrossRef]
- Zou, H.; Ben, T.; Wu, P.; Waterhouse, G.I.N.; Chen, Y. Effective anti-inflammatory phenolic compounds from dandelion: Identification and mechanistic insights using UHPLC-ESI-MS/MS, fluorescence quenching and anisotropy, molecular docking and dynamics simulation. Food Sci. Hum. Wellness 2023, 12, 2184–21941. [Google Scholar] [CrossRef]
- Srinivasan, V.; Sandhya, N.; Sampathkumar, R.; Farooq, S.; Mohan, V.; Balasubramanyam, M. Glutamine fructose-6-phosphate amidotransferase (GFAT) gene expression and activity in patients with type 2 diabetes: Inter-relationships with hyperglycaemia and oxidative stress. Clin. Biochem. 2007, 40, 952–957. [Google Scholar] [CrossRef]
- Akbari, M.; Hassan-Zadeh, V. IL-6 signalling pathways and the development of type 2 diabetes. Inflammopharmacology 2018, 26, 685–698. [Google Scholar] [CrossRef]
- Ambhore, J.P.; Laddha, P.R.; Nandedkar, A.; Ajmire, P.V.; Chumbhale, D.S.; Navghare, A.B.; Kuchake, V.G.; Chaudhari, P.J.; Adhao, V.S. Medicinal chemistry of non-peptidomimetic dipeptidyl peptidase IV (DPP IV) inhibitors for treatment of Type-2 diabetes mellitus: Insights on recent development. J. Mol. Struct. 2023, 1284, 135249. [Google Scholar] [CrossRef]
- Sarhangi, N.; Sharifi, F.; Hashemian, L.; Doabsari, M.H.; Heshmatzad, K.; Rahbaran, M.; Jamaldini, S.H.; Meybodi, H.R.A.; Hasanzad, M. PPARG (Pro12Ala) genetic variant and risk of T2DM: A systematic review and meta-analysis. Sci. Rep. 2020, 10, 12764. [Google Scholar] [CrossRef] [PubMed]
- Entezari, M.; Hashemi, D.; Taheriazam, A.; Zabolian, A.; Mohammadi, S.; Fakhri, F.; Hashemi, M.; Hushmandi, K.; Ashrafizadeh, M.; Zarrabi, A.; et al. AMPK signaling in diabetes mellitus, insulin resistance and diabetic complications: A pre-clinical and clinical investigation. Biomed. Pharmacother. 2022, 146, 112563. [Google Scholar] [CrossRef] [PubMed]
- Batista, T.M.; Haider, N.; Ronald Kahn, C.R. Defining the underlying defect in insulin action in type 2 diabetes. Diabetologia 2021, 64, 994–1006. [Google Scholar] [CrossRef] [PubMed]
- Bešlo, D.; Golubić, N.; Rastija, V.; Dejan Agić, D.; Karnaš, M.; Šubarić, D.; Lučić, B. Antioxidant Activity, Metabolism, and Bioavailability of Polyphenols in the Diet of Animals. Antioxidants 2023, 12, 1141. [Google Scholar] [CrossRef] [PubMed]
- Lagorce, D.; Douguet, D.; Miteva, M.A.; Villoutreix, B.O. Computational analysis of calculated physicochemical and ADMET properties of protein-protein interaction inhibitors. Sci. Rep. 2017, 7, 46277. [Google Scholar] [CrossRef]
- Fang, Y.; Cao, W.; Xia, M.; Pan, S.; Xu, X. Study of Structure and Permeability Relationship of Flavonoids in Caco-2 Cells. Nutrients 2017, 9, 1301. [Google Scholar] [CrossRef] [PubMed]
- Bitew, M.; Desalegn, T.; Demissie, T.B.; Belayneh, A.; Endale, M.; Eswaramoorthy, R. Pharmacokinetics and drug-likeness of antidiabetic flavonoids: Molecular docking and DFT study. PLoS ONE 2021, 16, e0260853. [Google Scholar] [CrossRef] [PubMed]
- López-Yerena, A.; Perez, M.; Vallverdú-Queralt, A.; Escribano-Ferrer, E. Insights into the Binding of Dietary Phenolic Compounds to Human Serum Albumin and Food-Drug Interactions. Pharmaceutics 2020, 12, 1123. [Google Scholar] [CrossRef]
- Bhatt, S.; Dhiman, S.; Kumar, V.; Gour, A.; Manhas, D.; Sharma, K.; Ojha, P.K.; Nandi, U. Assessment of the CYP1A2 inhibition-mediated drug interaction potential for pinocembrin using in silico, in vitro, and in vivo approaches. ACS Omega 2022, 7, 20321–20331. [Google Scholar] [CrossRef]
- Kiani, H.S.; Ahmad, W.; Nawaz, S.; Farah, M.A.; Akhtar Ali, A. Optimized Extraction of Polyphenols from Unconventional Edible Plants: LC-MS/MS Profiling of Polyphenols, Biological Functions, Molecular Docking, and Pharmacokinetics Study. Molecules 2023, 28, 6703. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Halgren, T.A. Characterization of MMFF94, MMFF94s, and other widely available force fields for conformational energies and for intermolecular-interaction energies and geometries. Abstr. Pap. Am. Chem. Soc. 1998, 216, U702. [Google Scholar] [CrossRef]
- Bikadi, Z.; Hazai, E. Application of the PM6 semi-empirical method to modeling proteins enhances docking accuracy of AutoDock. J. Cheminformatics 2009, 1, 15. [Google Scholar] [CrossRef] [PubMed]
- Solis, F.J.; Wets, R.J.B. Minimization by random search techniques. Math. Oper. Res. 1981, 6, 19–30. Available online: http://www.jstor.org/stable/3689263 (accessed on 20 December 2023). [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Compound | Solubility g L−1 | DPPH % Free Radical Scavenging Activity | ABTS % Free Radical Scavenging Activity | NO % Free Radical Scavenging Activity |
|---|---|---|---|---|
| Ferulic acid (FA) | 0.354 ± 0.02 | 70.04 ± 2.3 | 90.69 ± 0.14 | 22.21 ± 1.39 |
| β-D-Fructopyranosyl-β-(2→6)- ferulate (MFF) | 8.69 ± 0.45 | 17.16 ± 0.81 | 90.95 ± 0.18 | 3.27 ± 1.02 |
| Puerarin (PU) | 0.7 ± 0.09 | 33.3% | NR | NR |
| β-D-Fructofuranosyl-β-(2→6)-puerarin (MFP) | 16.2 ± 1.7 | 26.2 ± 1.3% | NR | NR |
| Phlorizin (PH) | 1.93 ± 0.03 | 62.13 ± 1.8% | NR | NR |
| D-β-D-Fructofuranosyl-(2→6)-phlorizin (MFPh) | 30.57 ± 0.1 | 44.56 ± 8.4% | NR | NR |
| 4-O-Mono-fructosyl phlorizin (4MFPh) | NR | NR | NR | NR |
| Phlorizin-4′-O-β-D-fructofuranosyl-(2→6)-D-fructofuranoside (DFPh) | NR | NR | NR | NR |
| Protein | Abbreviation | PDB Code | Function |
|---|---|---|---|
| Glucokinase | GK | 1V4S | Catalyzes the transfer of phosphate from ATP to glucose to generate glucose 6-phosphate [17]. |
| AMP-activated protein kinase | AMPK | 2H6D | Involved in the stimulation of glucose transport and fatty acid oxidation [17]. |
| 11 β-hydroxysteroid dehydrogenase 1 | 11β-HS1 | 1BHS | Produces insulin resistance throuh the conversion of cortisone to cortisol [17,18]. |
| Insulin receptor substrate | IRS | 1K3A | Impairment of IRS-2 signaling in the β-cell produces β-cell loss in T2DM [18]. |
| Interleukin 1 beta | IL-1ß | 9ILB | Contributes to inflammation of beta cells in pancreas [19]. |
| Dipeptidyl peptidase IV | DPPIV | 1J2E | Inhibits the action of GIP and GLP-1, increasing glucose levels [17]. |
| C-reactive protein | CRP | 1GNH | Involved in chronic inflammation in adipose tissue and leads to insulin resistance [19]. |
| Glutamine fructose-6-phosphate amidotransferase | GFAT | 2ZJ3 | Increased the flux of glucose through the pathway where GFAT is a key catalyst that can lead to insulin resistance [17,18]. |
| Ligands | Proteins | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 11β-HS1 | CRP | DPPIV | IRS | PPAR-γ | GK | AMPK | IR | GFAT | IL-1ß | IL-6 | TNF-α | |
| 4-O-Mono-fructosyl phlorizin (4MFPh) | −11.88 | +864.05 | −10.01 | −6.62 | −5.79 | +13.38 | −3.49 | −11.37 | −11.79 | +606.71 | −12.11 | +141.26 |
| β-D-Fructopyranosyl-β-(2→6)- ferulate(MFF) | −8.90 | +110.52 | −6.45 | −7.36 | −9.71 | −7.69 | −9.10 | −6.76 | −8.14 | +27.95 | −6.95 | −2.21 |
| Phlorizin-4′-O-β-D-fructofuranosyl-(2→6)-D-fructofuranoside (DFPh) | −13.98 | +1.64 | −15.48 | +6.54 | +13.34 | +107.26 | +16.42 | −16.8 | −16.9 | +950.39 | +53.21 | +470.95 |
| Ferulic acid (FA) | −4.78 | +4.48 | −2.90 | −3.60 | −5.01 | −4.64 | −5.26 | −4.24 | −4.28 | −4.21 | −4.58 | −5.63 |
| ß-D-fructofuranosyl-ß-(2→6)-phlorizin (MFPh) | −13.99 | +915.68 | −10.77 | −10.49 | −5.21 | +22.38 | −7.66 | −10.92 | −12.55 | +407.72 | −6.80 | +113.82 |
| ß-D-fructofuranosyl-ß-(2→6)-puerarin (MFPu) | −12.02 | +775.58 | −9.90 | −7.85 | −9.17 | +23.3 | −6.47 | −10.22 | −10.26 | +326.27 | −10.29 | +184.51 |
| Phlorizin (PH) | −8.36 | +321.76 | −5.65 | −6.97 | −8.61 | +0.73 | −5.77 | −5.21 | −8.01 | +139.38 | −5.88 | +8.28 |
| Puerarin (PU) | −10.25 | +498.85 | −7.50 | −8.24 | −11.76 | +7.73 | −7.66 | −7.57 | −8.96 | +213.72 | −8.01 | +76.95 |
| Metformin | −3.18 | −3.87 | −4.84 | −4.97 | −6.06 | −5.79 | −4.00 | −4.83 | −4.37 | −3.80 | −3.43 | −2.62 |
| Sitagliptin | −8.90 | +133.74 | −7.47 | −8.30 | −12.73 | −7.60 | −8.58 | −7.30 | −10.39 | +28.82 | −7.28 | −7.41 |
| Ligand | Protein | Free Energy of Binding (kcal/mol) | Inhibition Constant (Ki) nM | Type of Interaction | Amino Acid Residue Interaction |
|---|---|---|---|---|---|
| 4-O-Mono-fructosyl phlorizin (4MFPh) | 11β-HS1 | −11.88 | 1.96 | HB | O14-SER11 [2.75]; O4-TYR155 [2.88]; H31-SER11 [2.70]; H31-SER12 [3.64] |
| GFAT | −11.79 | 2.28 | P | O2-SER401 [3.69]; H9-SER401 [3.76]; O15-SER422 [3.87]; O2-ASP427 [3.00]; H9-ASP427 [2.32]; O7-GLU560 [3.53]; H15-GLU560 [3.89] | |
| IL-6 | −12.11 | 1.33 | HB | O9-GLN175 [3.10] | |
| β-D-Fructofuranosyl-β -(2→6) phlorizin (MFPh) | 11β-HS1 | −13.91 | 55.28 | HB | O7-SER142 [3.26]; H20-SER142 [2.63]; H20-VAL143 [3.27] |
| GFAT | -12.55 | 627.45 | HB | O15-CYS373 [3.25]; O2-ALA674 [3.27]; H28-CYS373 [3.58]; H28-ASP427 [3.61] | |
| IR | −10.92 | 9.87 | P | O11-GLU24 [3.41]; O10-GLU24 [3.63]; H21-GLU24 [3.82]; O5-ASN25 [3.32]; O9-ASN25 [3.36]; H22-ASN25 [3.86]; H10-ASN25 [3.77] | |
| Phlorizin-4′-O-β-D-fructofuranosyl-(2→6)-D-fructofuranoside (DFPh) | DPPIV | −15.48 | 4.52 | P | O7-ASN85 [3.56]; O15-ASN85 [3.57]; O1-SER86 [3.35]; H9-SER86 [2.62]; H10-SER87 [3.64] |
| IR | −16.80 | 481.43 | P | H44-GLU6 [2.73]; O20-GLU6 [3.02]; O2-GLU22 [2.92]; H10-GLU22 [1.94]; O3-GLU24 [3.42] | |
| GFAT | −16.87 | 431.06 | HB | O18-CYS373 [3.75]; O17-SER401 [3.25]; O18-SER401 [3.23]; O6-THR425 [3.43]; O6-VAL677 [3.37]; H38-LEU399 [3.81]; H37-SER401 [3.68]; H38-SER401 [3.18]; H21-THR425 [3.46] | |
| β-D-Fructopyranosyl-β-(2→6) ferulate (MFF) | 11β-HS1 | −8.9 | 298.32 | HB | O9-TYR155 [3.00]; O8-TYR155 [2.87]; O4-LYS159 [3.47]; H13-TYR155 [3.88]; H22-TYR155 [3.36]; H21-TYR155 [3.20] |
| PPAR-γ | −9.71 | 76.66 | HB | O2-GLU343 [3.27] | |
| AMPK | −9.1 | 213.35 | HB | O7-LYS141 [3.02]; O4-ASP157 [3.26]; H13-LYS45 [3.59]; H20-LYS141 [3.53]; H20-ASN144 [3.32]; H13-ASP157 [3.51] | |
| β-D-Fructofuranosyl-β-(2→6)-puerarin (MFP) | 11β-HS1 | −12.02 | 1.55 | HB | O7-SER12 [2.88]; O3-SER142 [3.14]; H16-SER12 [2.91]; H8-SER142 [3.32]; H8-VAL143 [3.73] |
| GFAT | −10.26 | 30.14 | HB | O7-THR425 [3.24]; H16-THR425 [3.51] | |
| IL-6 | −10.29 | 28.47 | P | O13-ARG30 [3.52]; O4-ARG30 [3.27]; H9-ARG30 [3.75]; O14-ARG30 [3.48]; H30-ARG30 [3.33]; H27-ARG30 [3.85]; O11-ASP34 [3.01]; H23-ASP34 [2.06]; O12-ASP34 [3.01]; H24-ASP34 [3.72]; O8-GLN175 [3.11]; H17-GLN175 [3.66]; O2-ARG182 [3.51] |
| Ligands | Absorption | Distribution | Metabolism | Excretion | Toxicity | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C2P | R | HIA | R | PPB (%) | R | CYP1A2 Inhibitor | R | CL | R | T ½ | R | AMES | R | H-HT | R | |
| 4-O-Mono-fructosyl phlorizin (4MFPh) | −6.505 | + | 0.998 | + | 61.59 | + | 0.016 | + | 3.15 | − | 0.436 | − | 0.125 | − | 0.159 | + |
| Phlorizin-4-O-β-D-fructofuranosyl-(2→6)-D-fructofuranoside (DFPh) | −6.634 | + | 1.0 | + | 37.56 | + | 0.002 | + | 1.79 | − | 0.362 | − | 0.083 | − | 0.125 | + |
| β-D-Fructopyranosyl- β-(2→6) phlorizin (MFPh) | −6.492 | + | 0.997 | + | 57.84 | + | 0.013 | + | 4.33 | − | 0.54 | − | 0.161 | − | 0.823 | + |
| β-D-Fructopyranosyl- β-(2→6) ferulate (MFF) | −5.907 | + | 0.935 | + | 72.93 | + | 0.023 | + | 5.67 | − | 0.855 | − | 0.086 | − | 0.133 | + |
| β-D-Fructopyranosyl- β-(2→6) puerarin (MFP) | −6.350 | + | 0.989 | + | 78.88 | + | 0.009 | + | 2.58 | − | 0.352 | − | 0.353 | − | 0.084 | + |
| Phlorizin | −6.318 | + | 0.953 | + | 65.29 | + | 0.104 | + | 8.40 | − | 0.706 | − | 0.523 | − | 0.057 | + |
| Puerarin | −6.038 | + | 0.83 | − | 89.30 | + | 0.046 | + | 2.68 | − | 0.491 | − | 0.531 | − | 0.08 | + |
| Ferulic acid | −4.902 | − | 0.03 | − | 89.75 | + | 0.059 | + | 7.48 | − | 0.926 | − | 0.224 | − | 0.345 | + |
| Metformin | −8.670 | + | 1.0 | 55.34 | + | 0.365 | + | 1.25 | + | 0.856 | − | 0.496 | + | 0.245 | − | |
| Sitagliptin | 0.536 | + | 0.989 | 34.56 | + | 0.547 | + | 3.25 | + | 0.478 | - | 0.148 | + | 0.152 | + | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Damian-Medina, K.; Herrera-González, A.; Figueroa-Yáñez, L.J.; Arrizon, J. Enzymatic Fructosylation of Phenolic Compounds: A New Alternative for the Development of Antidiabetic Drugs. Molecules 2024, 29, 3072. https://doi.org/10.3390/molecules29133072
Damian-Medina K, Herrera-González A, Figueroa-Yáñez LJ, Arrizon J. Enzymatic Fructosylation of Phenolic Compounds: A New Alternative for the Development of Antidiabetic Drugs. Molecules. 2024; 29(13):3072. https://doi.org/10.3390/molecules29133072
Chicago/Turabian StyleDamian-Medina, Karla, Azucena Herrera-González, Luis J. Figueroa-Yáñez, and Javier Arrizon. 2024. "Enzymatic Fructosylation of Phenolic Compounds: A New Alternative for the Development of Antidiabetic Drugs" Molecules 29, no. 13: 3072. https://doi.org/10.3390/molecules29133072
APA StyleDamian-Medina, K., Herrera-González, A., Figueroa-Yáñez, L. J., & Arrizon, J. (2024). Enzymatic Fructosylation of Phenolic Compounds: A New Alternative for the Development of Antidiabetic Drugs. Molecules, 29(13), 3072. https://doi.org/10.3390/molecules29133072