

piRNA in Machine-Learning-Based Diagnostics of Colorectal Cancer


Abstract
1. Introduction
2. Results
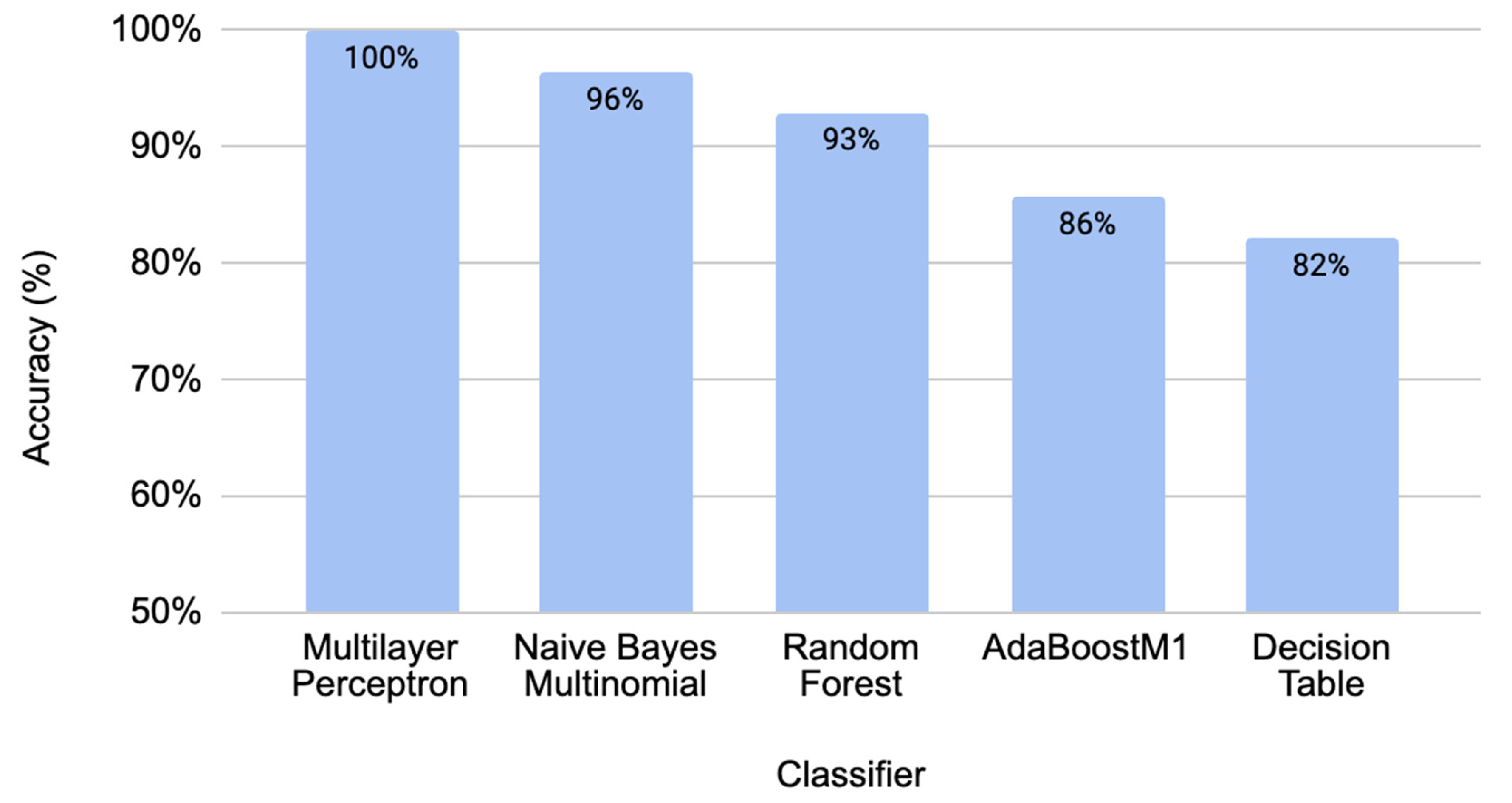
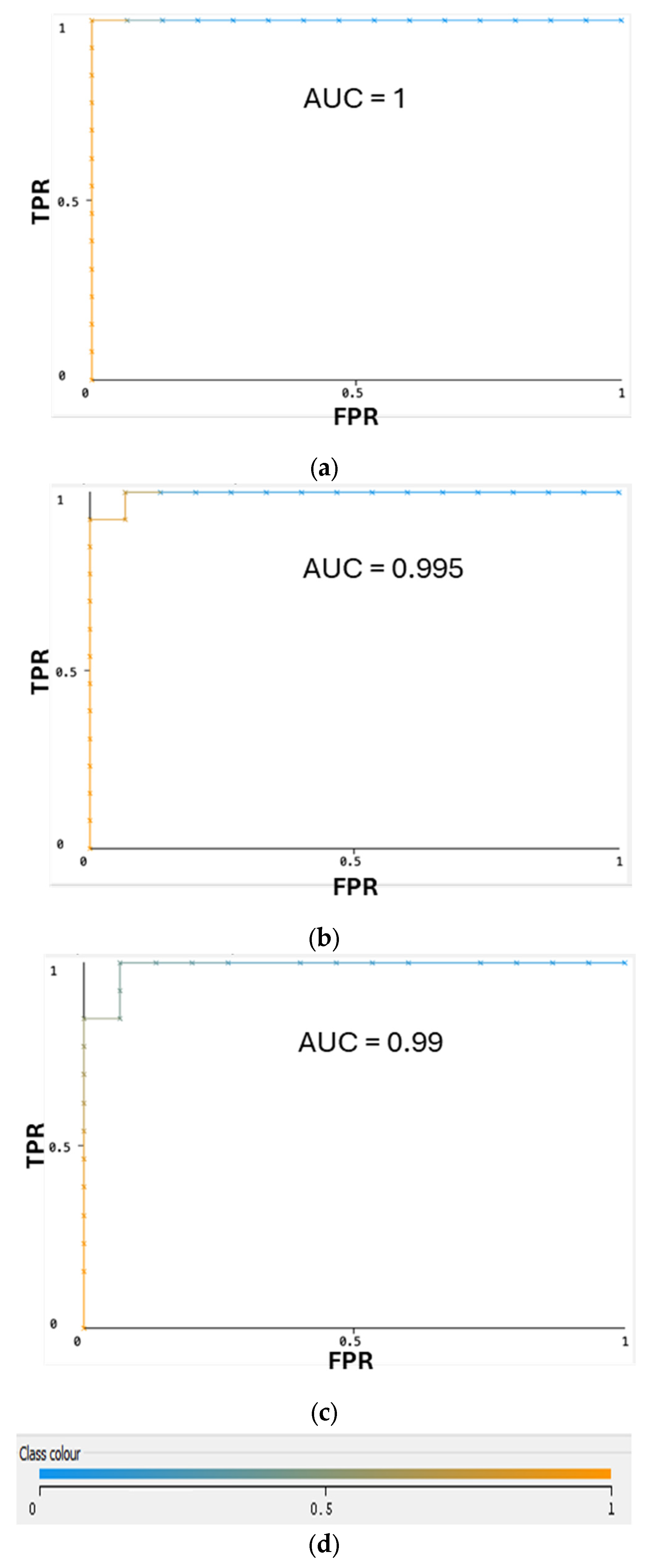
2.1. Performance Comparison for Different Classifiers through Cross-Validation
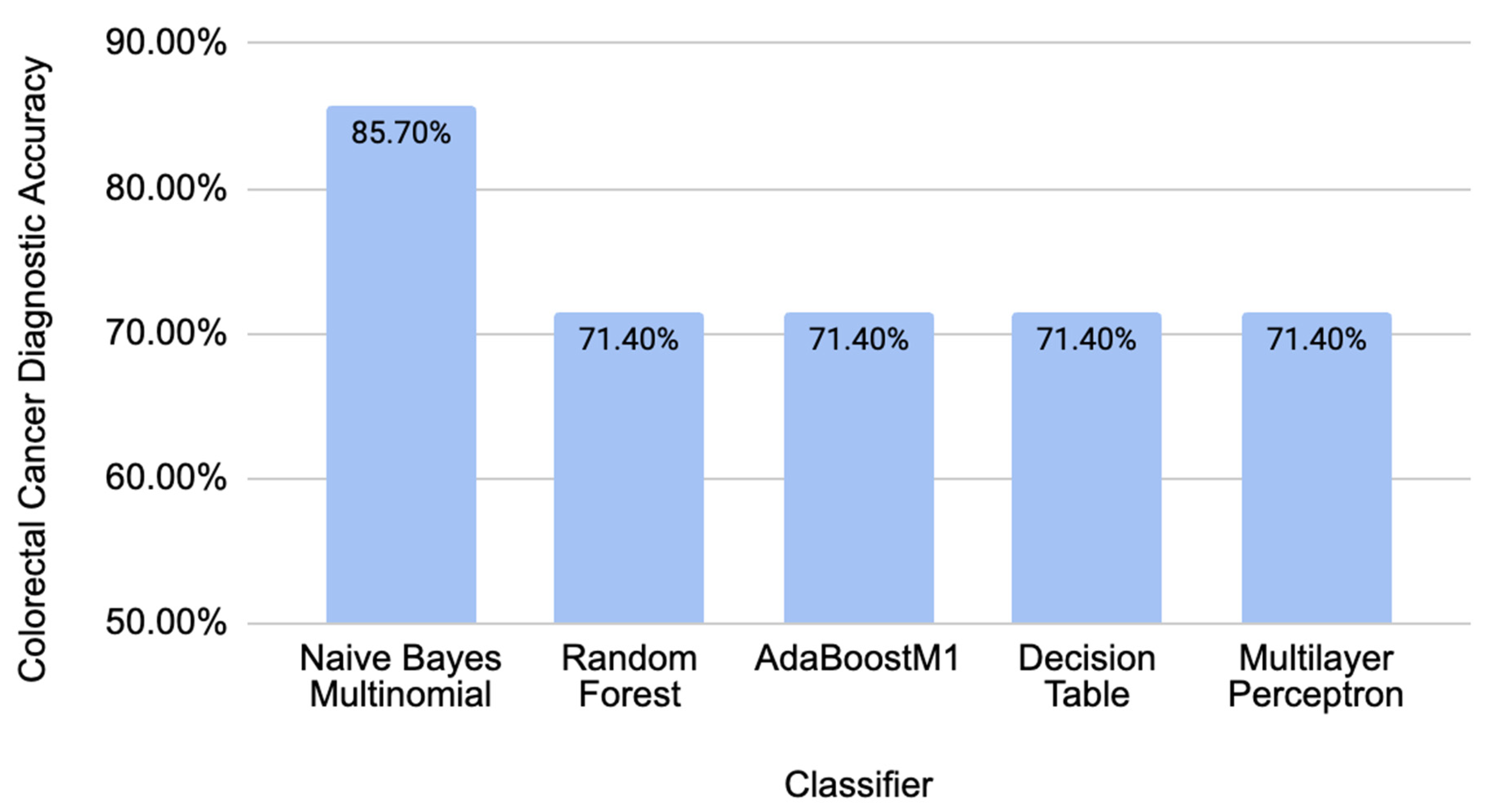
2.2. Comparison of Different Classifiers’ Performance on Independent CRC-Related Data
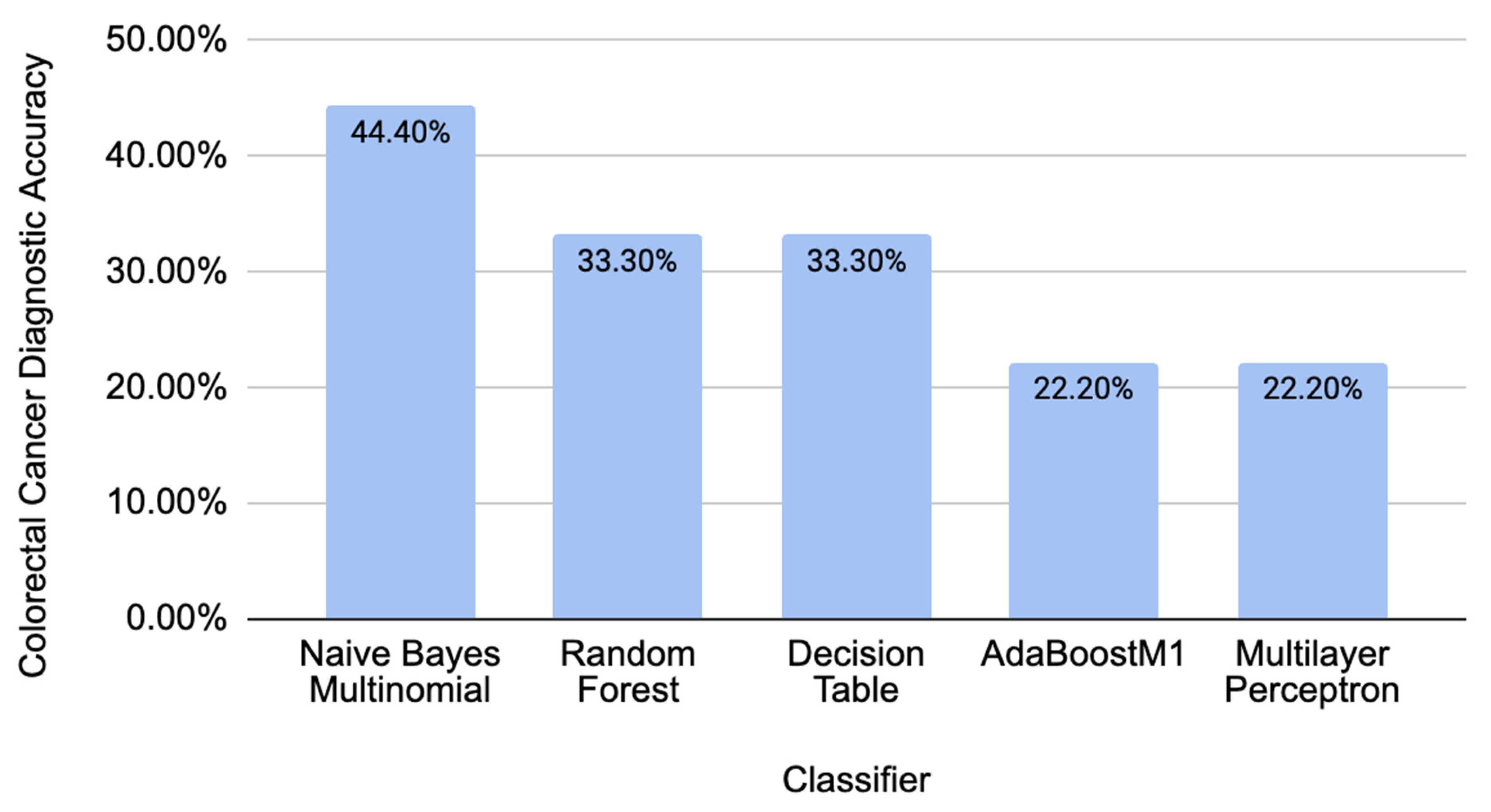
2.3. Comparison of Different Classifiers’ Performance on Independent CRC-Unrelated Data
3. Discussion
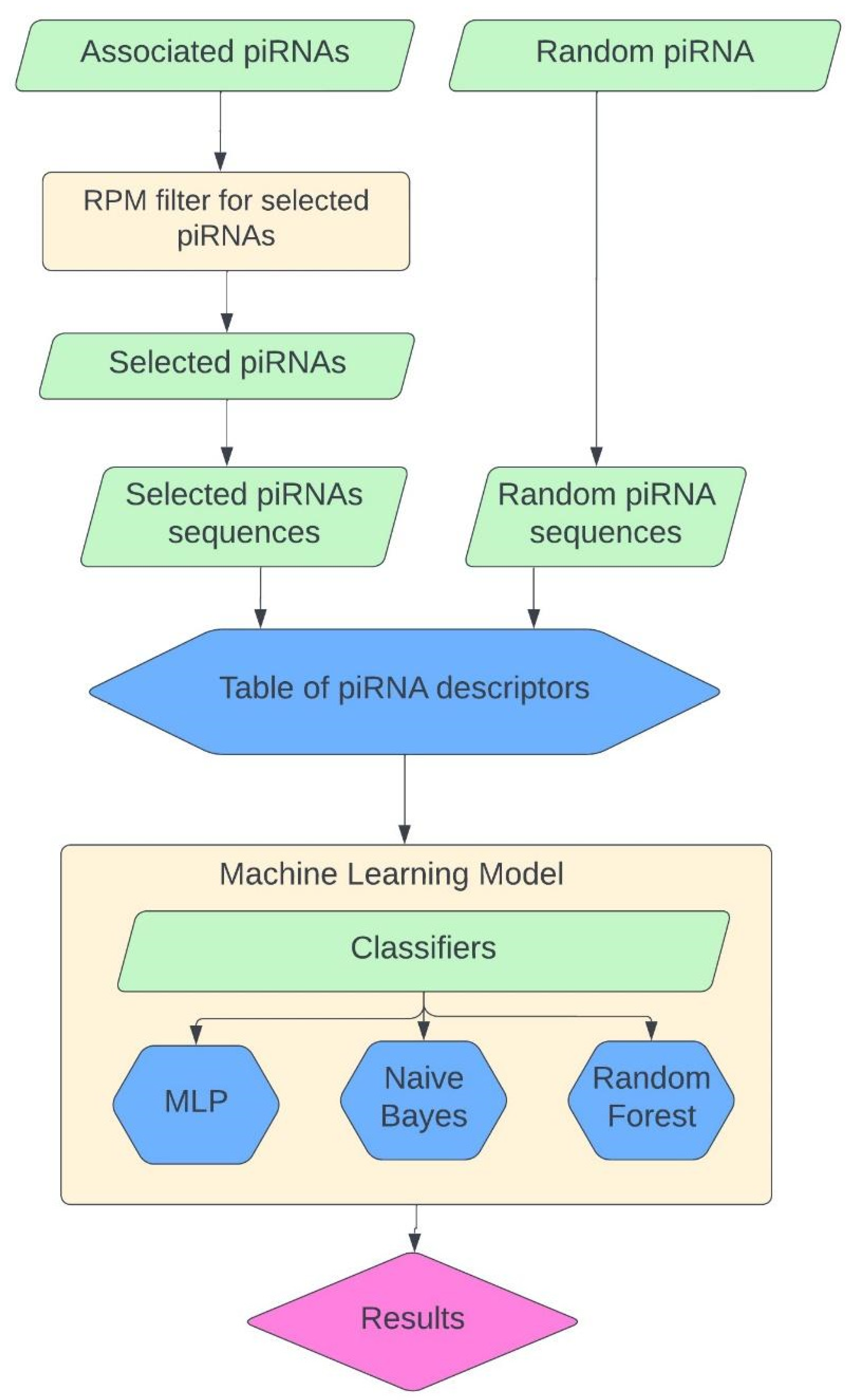
4. Materials and Methods
4.1. Classification Model
4.2. Sequence Descriptor System
4.3. Classifier Descriptions
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Liu, Y.; Dou, M.; Song, X.; Dong, Y.; Liu, S.; Liu, H.; Tao, J.; Li, W.; Yin, X.; Xu, W. The emerging role of the piRNA/piwi complex in cancer. Mol. Cancer 2019, 18, 123. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Mai, D.; Zhang, B.; Jiang, X.; Zhang, J.; Bai, R.; Ye, Y.; Li, M.; Pan, L.; Su, J.; et al. PIWI-interacting RNA-36712 restrains breast cancer progression and chemoresistance by interaction with SEPW1 pseudogene SEPW1P RNA. Mol. Cancer 2019, 18, 9. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Wagle, N.S.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2023. CA Cancer J. Clin. 2023, 73, 233–254. [Google Scholar] [CrossRef] [PubMed]
- Qu, A.; Wang, W.; Yang, Y.; Zhang, X.; Dong, Y.; Zheng, G.; Wu, Q.; Zou, M.; Du, L.; Wang, Y.; et al. A serum piRNA signature as promising non-invasive diagnostic and prognostic biomarkers for colorectal cancer. Cancer Manag. Res. 2019, 11, 3703–3720. [Google Scholar] [CrossRef] [PubMed]
- Weng, W.; Liu, N.; Toiyama, Y.; Kusunoki, M.; Nagasaka, T.; Fujiwara, T.; Wei, Q.; Qin, H.; Lin, H.; Ma, Y.; et al. Novel evidence for a PIWI-interacting RNA (piRNA) as an oncogenic mediator of disease progression, and a potential prognostic biomarker in colorectal cancer. Mol. Cancer 2018, 17, 16. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhu, Y.; Cao, X.; Tan, W.; Yu, J.; Lu, Y.; Kang, R.; Wang, X.; Li, E. The epigenetic regulatory mechanism of PIWI/piRNAs in human cancers. Mol. Cancer 2023, 22, 45. [Google Scholar] [CrossRef] [PubMed]
- Cai, A.; Hu, Y.; Zhou, Z.; Qi, Q.; Wu, Y.; Dong, P.; Chen, L.; Wang, F. PIWI-interacting RNAs (piRNAs): Promising applications as emerging biomarkers for digestive system cancer. Front. Mol. Biosci. 2022, 9, 848105. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.; Xie, H.; Zhang, L.; Zhao, Q.; Lü, J.; Yu, Z. Piwi-interacting RNAs: A new class of regulator in human breast cancer. Front. Oncol. 2021, 11, 695097. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Luo, Y.; Gao, Y.; Yang, Y.; Wang, Y.; Xu, Y.; Tan, S.; Zhang, Y.; Duan, J.; Yang, Y. piR-651 promotes tumor formation in non-small cell lung carcinoma through the upregulation of cyclin D1 and CDK4. Int. J. Mol. Med. 2016, 38, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Guo, J.-M.; Xiao, B.-X.; Miao, Y.; Jiang, Z.; Zhou, H.; Li, Q.-N. piRNA, the new non-coding RNA, is aberrantly expressed in human cancer cells. Clin. Chim. Acta 2011, 412, 1621–1625. [Google Scholar] [CrossRef] [PubMed]
- Kang, W.; Kouznetsova, V.L.; Tsigelny, I.F. miRNA in machine-learning-based diagnostics of cancers. Cancer Screen. Prev. 2022, 1, 32–38. [Google Scholar] [CrossRef]
- Xu, A.; Kouznetsova, V.L.; Tsigelny, I.F. Alzheimer’s disease diagnostics using miRNA biomarkers and machine learning. J. Alzheimer’s Dis. 2022, 86, 841–859. [Google Scholar] [CrossRef] [PubMed]
- Piuco, R.; Galante, P.A.F. piRNAdb: A piwi-interacting RNA database. bioRxiv 2021, 2021.09.21.461238. [Google Scholar] [CrossRef]
- Wang, J.; Shi, Y.; Zhou, H.; Zhang, P.; Song, T.; Ying, Z.; Yu, H.; Li, Y.; Zhao, Y.; Zeng, X.; et al. piRBase: Integrating piRNA annotation in all aspects. Nucleic Acids Res. 2022, 50, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Ansa, B.E.; Coughlin, S.S.; Alema-Mensah, E.; Smith, S.A. Evaluation of colorectal cancer incidence trends in the United States (2000–2014). J. Clin. Med. 2018, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Kim, H.-S.; Park, H.J. Adverse events related to colonoscopy: Global trends and future challenges. World J. Gastroenterol. 2019, 25, 190–204. [Google Scholar] [CrossRef] [PubMed]
- Abdelsattar, Z.M.; Wong, S.L.; Regenbogen, S.E.; Jomaa, D.M.; Hardiman, K.M.; Hendren, S. Colorectal cancer outcomes and treatment patterns in patients too young for average-risk screening. Cancer 2016, 122, 929–934. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Bai, J.Y.; Ren, H.T. PiRNAs biogenesis and its functions. Bioorg. Khim. 2014, 40, 320–326. [Google Scholar] [PubMed]
- Hall, M.; Frank, E.; Holmes, G.; Pfahringer, B.; Reutemann, P.; Witten, I.H. The WEKA data mining software. ACM SIGKDD Explor. 2009, 11, 10–18. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Classifier | TPR | FPR | Precision | Recall | F-Measure | MCC | AUC | AUPRC |
|---|---|---|---|---|---|---|---|---|
| Multilayer Perceptron | 100% | 0% | 100% | 100% | 100% | 100% | 100% | 100% |
| Naïve Bayes Multinomial | 96.40% | 3.10% | 96.70% | 96.40% | 96.40% | 93.10% | 99.50% | 99.50% |
| Random Forest | 92.90% | 8.20% | 93.70% | 92.90% | 92.80% | 86.40% | 99.00% | 99.10% |
| AdaBoostM1 | 85.70% | 15.50% | 86.30% | 85.70% | 85.60% | 71.70% | 89.20% | 90.30% |
| Decision Table | 82.10% | 19.60% | 83.50% | 82.10% | 81.80% | 65.10% | 71.50% | 71.40% |
| Descriptor | Explanation |
|---|---|
| C | Number of C nucleotides |
| C/N | Frequency of C nucleotides |
| CU | Number of CU dinucleotides |
| UUC | Number of UUC trinucleotides |
| CGC | Number of CGC trinucleotides |
| 5sCAG | Number of CAG trinucleotides in the first 5 nucleotides of piRNA |
| 5sAAG | Number of AAG trinucleotides in the first 5 nucleotides of piRNA |
| 5sGGU | Number of GGU trinucleotides in the first 5 nucleotides of piRNA |
| 5sGGC | Number of GGC trinucleotides in the first 5 nucleotides of piRNA |
| 5eCA | Number of CA dinucleotides in the last 5 nucleotides of piRNA |
| 5eUGA | Number of UGA trinucleotides in the last 5 nucleotides of piRNA |
| 5eGGA | Number of GGA trinucleotides in the last 5 nucleotides of piRNA |
| 5eAGG | Number of AAG trinucleotides in the last 5 nucleotides of piRNA |
| AGGC | Number of AGGC four nucleotides’ motifs |
| AUCA | Number of AUCA four nucleotides’ motifs |
| GAAA | Number of GAAA four nucleotides’ motifs |
| GAGU | Number of GAGU four nucleotides’ motifs |
| GGCA | Number of GGCA four nucleotides’ motifs |
| GUAG | Number of GUAG four nucleotides’ motifs |
| GUGU | Number of GUGU four nucleotides’ motifs |
| CUUC | Number of GUUC four nucleotides’ motifs |
| UAAA | Number of UAAA four nucleotides’ motifs |
| UCCA | Number of UCCA four nucleotides’ motifs |
| UCCC | Number of UCCC four nucleotides’ motifs |
| UCUG | Number of UCUG four nucleotides’ motifs |
| UUGU | Number of UUGU four nucleotides’ motifss |
| piRNA | A | G | C | U | AA | GG | UU | CC | AAA | GGG | UUU | CCC | N | A/N | G/N | C/N | U/N | Mass/N |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| piR-001312 | 7 | 8 | 3 | 6 | 0 | 4 | 1 | 0 | 0 | 0 | 0 | 0 | 29 | 0.24 | 0.28 | 0.1 | 0.21 | 111.88 |
| piR-004150 | 7 | 5 | 9 | 2 | 2 | 1 | 0 | 3 | 0 | 0 | 1 | 0 | 30 | 0.23 | 0.17 | 0.3 | 0.07 | 98.44 |
| piR-004153 | 9 | 5 | 7 | 4 | 1 | 2 | 0 | 1 | 1 | 0 | 0 | 0 | 30 | 0.3 | 0.17 | 0.2 | 0.13 | 108.45 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, S.; Kouznetsova, V.L.; Kesari, S.; Tsigelny, I.F. piRNA in Machine-Learning-Based Diagnostics of Colorectal Cancer. Molecules 2024, 29, 4311. https://doi.org/10.3390/molecules29184311
Li S, Kouznetsova VL, Kesari S, Tsigelny IF. piRNA in Machine-Learning-Based Diagnostics of Colorectal Cancer. Molecules. 2024; 29(18):4311. https://doi.org/10.3390/molecules29184311
Chicago/Turabian StyleLi, Sienna, Valentina L. Kouznetsova, Santosh Kesari, and Igor F. Tsigelny. 2024. "piRNA in Machine-Learning-Based Diagnostics of Colorectal Cancer" Molecules 29, no. 18: 4311. https://doi.org/10.3390/molecules29184311
APA StyleLi, S., Kouznetsova, V. L., Kesari, S., & Tsigelny, I. F. (2024). piRNA in Machine-Learning-Based Diagnostics of Colorectal Cancer. Molecules, 29(18), 4311. https://doi.org/10.3390/molecules29184311