
Multiclass Analysis for the Determination of Pharmaceuticals and Their Main Metabolites in Leafy and Root Vegetables

 ,
,  , , and
, , and 
Abstract
1. Introduction
2. Results and Discussion
2.1. Method Optimisation
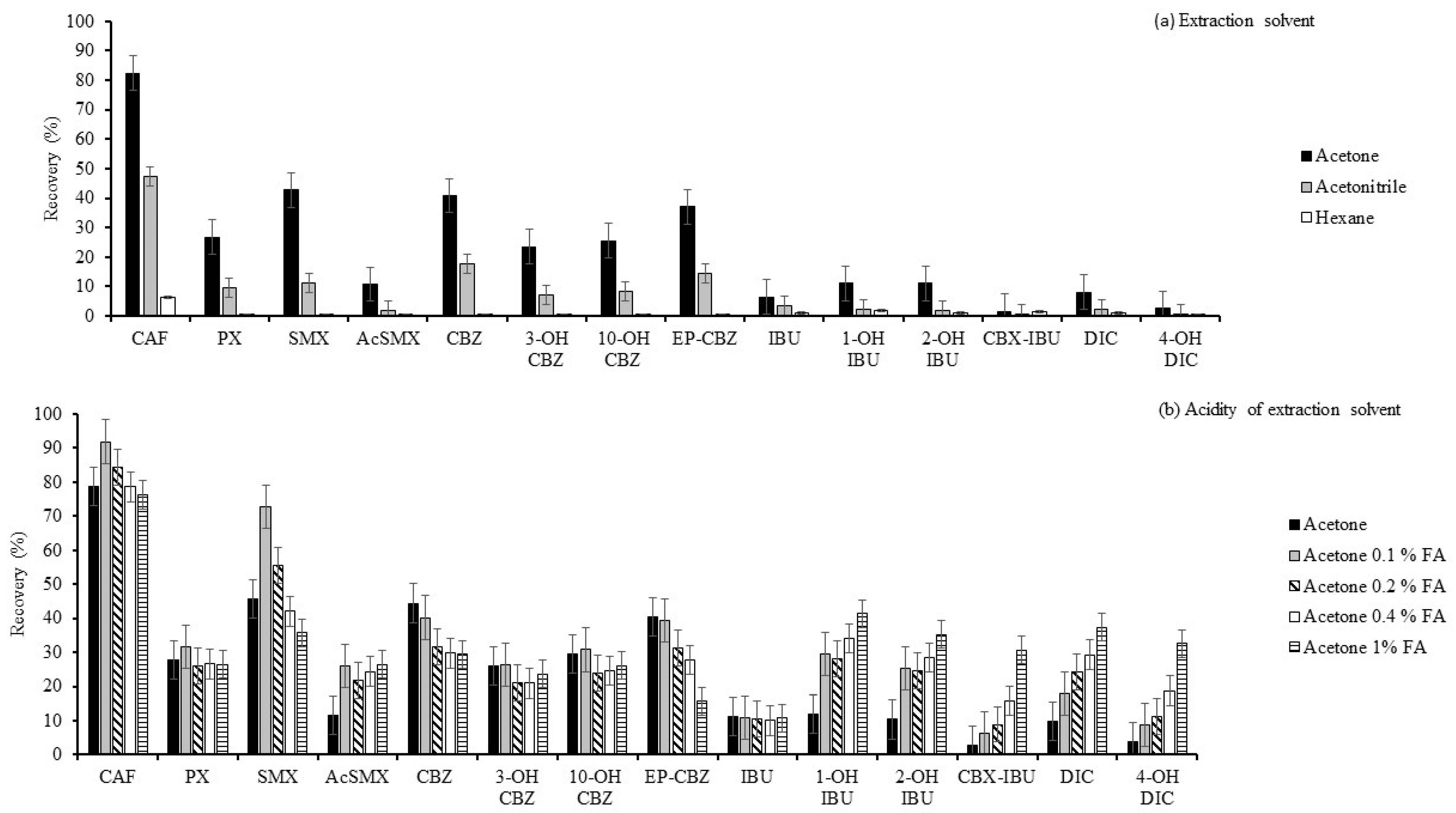
2.1.1. Optimisation of the Extraction Solvent
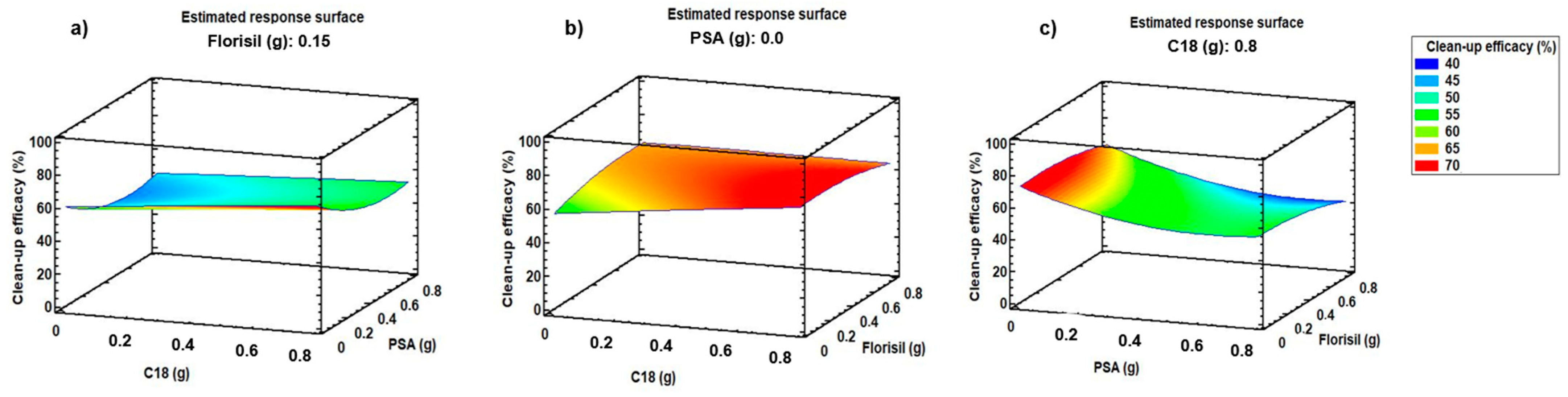
2.1.2. Optimisation of d-SPE Sorbents and Their Amount
2.1.3. Optimisation of Extraction Solvent Volume, UAE Time, and Number of Extraction Cycles
2.2. Method Validation
2.3. Method Comparison
2.4. Method Application
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Sample Collection and Treatment
3.3. Liquid Chromatography–Tandem Mass Spectrometry
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, X.; Dodgen, L.K.; Conkle, J.L.; Gan, J. Plant uptake of pharmaceutical and personal care products from recycled water and biosolids: A review. Sci. Total Environ. 2015, 536, 655–666. [Google Scholar] [CrossRef]
- Nkoh-Nkoh, J.; Shang, C.; Okeke, E.S.; Ejeromedoghene, O.; Oderinde, O.; Etafo, N.O.; Mgbechidinma, C.L.; Bakare, O.C.; Meugang, E.F. Antibiotics soil-solution chemistry: A review of environmental behavior and uptake and transformation by plants. J. Environ. Manag. 2024, 354, 120312. [Google Scholar] [CrossRef]
- Miller, E.; Nason, S.L.; Karthikeyan, K.G.; Pedersen, J.A. Root uptake of pharmaceuticals and personal care products ingredients. Environ. Sci. Technol 2016, 50, 525–541. [Google Scholar] [CrossRef]
- Malvar, J.L.; Santos, J.L.; Martín, J.; Aparicio, I.; Alonso, E. Occurrence of the main metabolites of the most recurrent pharmaceuticals and personal care products in Mediterranean soils. J. Environ. Manag. 2020, 278, 111584. [Google Scholar] [CrossRef]
- Santos, J.L.; Martín, J.; Mejías, C.; Aparicio, I.; Alonso, E. Pharmaceuticals and their metabolites in sewage sludge and soils: Distribution and environmental risk assessment. Emerging pollutants in sewage sludge and soils. In Handbook of Environmental Chemistry; Núñez-Delgado, A., Arias-Estévez, M., Eds.; Springer: Cham, Switzerland, 2023; Volume 114, pp. 19–36. [Google Scholar] [CrossRef]
- Mejías, C.; Martín, J.; Santos, J.L.; Aparicio, I.; Alonso, E. Occurrence of pharmaceuticals and their metabolites in sewage sludge and soil: A review on their distribution and environmental risk assessment. Trends Environ. Anal. Chem. 2021, 30, e00125. [Google Scholar] [CrossRef]
- Ahmed, M.B.M.; Rajapaksha, A.U.; Lim, J.E.; Vu, N.T.; Kim, I.S.; Kang, H.M.; Lee, S.S.; Ok, Y.S. Distribution and accumulative pattern of tetracyclines and sulfonamides in edible vegetables of cucumber, tomato and lettuce. J. Agric. Food Chem 2015, 63, 398–405. [Google Scholar] [CrossRef]
- Azanu, D.; Mortey, C.; Darko, G.; Weisser, J.J.; Styrishave, B.; Abaidoo, R.C. Uptake of antibiotics from irrigation water by plants. Chemosphere 2016, 157, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Abril, C.; Santos, J.L.; Martín, J.; Aparicio, I.; Alonso, E. Uptake and translocation of multiresidue industrial and household contaminants in radish grown under controlled conditions. Chemosphere 2021, 268, 128823. [Google Scholar] [CrossRef]
- Herklotz, P.A.; Gurung, P.; Heuvel, B.V.; Kinney, C.A. Uptake of human pharmaceuticals by plants grown under hydroponic conditions. Chemosphere 2010, 78, 1416–1421. [Google Scholar] [CrossRef] [PubMed]
- Shargil, D.; Gerstl, Z.; Fine, P.; Nitsan, I.; Kurtzman, D. Impact of biosolids and wastewater effluent application to agricultural land on steroidal hormone content in lettuce plants. Sci. Total Environ 2015, 505, 357–366. [Google Scholar] [CrossRef]
- Riemenschneider, C.; Seiwert, B.; Moeder, M.; Schwarz, D.; Reemstsma, T. Extensive transformation of the pharmaceutical carbamazepine following uptake into intact tomato plants. Environ. Sci. Technol. 2017, 51, 6100–6109. [Google Scholar] [CrossRef]
- Malchi, T.; Maor, Y.; Tadmor, G.; Shenker, M.; Chefetz, B. Irrigation of root vegetables with treated wastewater: Evaluating uptake of pharmaceuticals and the associated human health risks. Environ. Sci. Technol. 2014, 48, 9325–9333. [Google Scholar] [CrossRef]
- Geng, J.; Liu, X.; Li, S. Accumulation and risk assessment of antibiotics in edible plants grown in contaminated farmlands: A review. Sci. Total Environ. 2022, 853, 158616. [Google Scholar] [CrossRef] [PubMed]
- Masiá, A.; Suárez-Varela, M.M.; Llopis-González, A.; Picó, Y. Determination of pesticides and veterinary drug residues in food by liquid chromatography-mass spectrometry: A review. Anal. Chim. Acta 2016, 936, 40–61. [Google Scholar] [CrossRef]
- Malvar, J.L.; Santos, J.L.; Martín, J.; Aparicio, I.; Alonso, E. Routine analytical method for monitoring the main metabolites for a recurrent group of parabens and pharmaceuticals in wastewater and tap water. Anal. Bioanal. Chem. 2019, 411, 6625–6635. [Google Scholar] [CrossRef]
- Merlo, F.; Centenaro, D.; Maraschi, F.; Profumo, A.; Speltini, A. Green and Efficient Determination of Fluoroquinolone Residues in Edible Green Fruits and Leafy Vegetables by Ultrasound-Assisted Extraction Followed by HPLC-MS/MS. Molecules 2022, 27, 6595. [Google Scholar] [CrossRef]
- Feng, Y.; Zhang, W.J.; Liu, Y.W.; Xue, J.M.; Zhang, S.Q.; Li, Z.J. A simple, sensitive, and reliable method for the simultaneous determination of multiple antibiotics in vegetables through SPE-HPLC-MS/MS. Molecules 2018, 23, 1953. [Google Scholar] [CrossRef] [PubMed]
- Calderón-Preciado, D.; Jiménez-Cartagena, C.; Peñuela, G.; Bayona, J.M. Development of an analytical procedure for the determination of emerging and priority organic pollutants in leafy vegetables by pressurized solvent extraction followed by GC-MS determination. Anal. Bioanal. Chem. 2009, 394, 1319–1327. [Google Scholar] [CrossRef]
- Chuang, Y.H.; Zhang, Y.; Zhang, W.; Boyd, S.A.; Li, H. Comparison of accelerated solvent extraction and quick, easy, cheap effective, rugged and safe method for the extraction and determination of pharmaceuticals in vegetables. J. Chromatog. A 2015, 1404, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Hoff, R.B.; Futigami, L.S.; Pierezan, M.D.; Deolindo, C.T.P.; de Melo, A.P.Z.; Molognoni, L.; Pimenta, R.; Burin, V.M.; de Francisco, A.; Daguer, H. Cassava-based materials for matrix solid phase dispersion: An alternative for sample preparation in food analysis. J. Chromatog. B 2022, 1201–1202, 123263. [Google Scholar] [CrossRef]
- Yu, X.; Liu, H.; Pu, C.; Chen, J.; Sun, Y.; Hu, L. Determination of multiple antibiotics in leafy vegetables using QuEChERS-UHPLC-MS/MS. J. Sep. Sci. 2017, 41, 713–722. [Google Scholar] [CrossRef]
- Hu, F.; Bian, K.; Liu, Y.; Su, Y.; Zhou, T.; Song, X.; He, L. Development of a modified QUick, Easy, CHeap, Effective, Ruffed and Safe method for the determination of multi-class antimicrobials in vegetables by liquid chromatography tandem mass spectrometry. J. Chromatog. A 2014, 1368, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Tadic, D.; Matamoros, V.; Bayona, J.M. Simultaneous determination of multiclass antibiotics and their metabolites in four types of field-grown vegetables. Anal. Bioanal. Chem. 2019, 411, 5209–5222. [Google Scholar] [CrossRef]
- Aparicio, I.; Martín, J.; Abril, C.; Santos, J.L.; Alonso, E. Determination of household and industrial chemicals, personal care products and hormones in leafy and root vegetables by liquid chromatography-tandem mass spectrometry. J. Chromatog. A 2018, 1533, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Albero, B.; Sánchez-Brunete, E.M.; Tadeo, J.L. Application of matrix solid-phase dispersion followed by GC-MS/MS to the analysis of emerging contaminants in vegetables. Food Chem 2017, 217, 660–667. [Google Scholar] [CrossRef] [PubMed]
- AOAC International. Method Validation Programs (OMA/PVM Department), Including Appendix D: Guidelines for Collaborative Study Procedures to Validate Characteristics of a Method of Analysis. 2000. Available online: http://www.aoac.org/vmeth/devmethno.htm (accessed on 18 July 2024).
- Tobiszewski, M.; Namieśnik, J. Greener organic solvents in analytical chemistry. Curr. Opin. Green Sustain. Chem. 2017, 5, 1–4. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Compound | Lettuce | Carrot | ||||
|---|---|---|---|---|---|---|
| MDL (ng g−1 dw) | MQL (ng g−1 dw) | R2 | MDL (ng g−1 dw) | MQL (ng g−1 dw) | R2 | |
| CAF | 0.011 | 0.027 | 0.998 | 0.022 | 0.217 | 0.995 |
| PX | 0.013 | 0.313 | 0.998 | 0.018 | 0.045 | 0.997 |
| CBZ | 0.003 | 0.008 | 0.998 | 0.017 | 0.050 | 0.995 |
| 3-OH CBZ | 0.010 | 0.052 | 0.998 | 0.027 | 0.068 | 0.995 |
| 10-OH CBZ | 0.004 | 0.012 | 0.997 | 0.018 | 0.053 | 0.995 |
| EP-CBZ | 0.003 | 0.010 | 0.995 | 0.020 | 0.059 | 0.995 |
| DIC | 0.009 | 0.086 | 0.999 | 0.016 | 0.040 | 0.996 |
| 4-OH DIC | 0.002 | 0.007 | 0.997 | 0.039 | 0.781 | 0.994 |
| IBU | 0.833 | 2.25 | 0.997 | 0.833 | 2.25 | 0.996 |
| 1-OH IBU | 0.164 | 0.820 | 0.998 | 0.179 | 0.446 | 0.996 |
| 2-OH IBU | 0.003 | 0.009 | 0.998 | 0.023 | 2.326 | 0.998 |
| CBX-IBU | 1.020 | 2.25 | 0.999 | 0.018 | 1.761 | 0.998 |
| SMX | 0.008 | 0.024 | 0.997 | 0.417 | 1.667 | 0.996 |
| AcSMX | 0.007 | 0.070 | 0.999 | 0.049 | 0.196 | 0.996 |
| Compound | Lettuce | Carrot | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2.5 (ng g−1 dw) | 40 (ng g−1 dw) | 100 (ng g−1 dw) | 2.5 (ng g−1 dw) | 40 (ng g−1 dw) | 100 (ng g−1 dw) | |||||||
| R (%) | RSD (%) | R (%) | RSD (%) | R (%) | RSD (%) | R (%) | RSD (%) | R (%) | RSD (%) | R (%) | RSD (%) | |
| CAF | 94 | 12 | 34 | 0 | 37 | 11 | 115 | 10 | 41 | 4 | 33 | 7 |
| PX | 80 | 21 | 49 | 5 | 28 | 6 | 55 | 15 | 41 | 6 | 38 | 9 |
| CBZ | 66 | 22 | 47 | 9 | 55 | 7 | 20 | 24 | 19 | 1 | 27 | 8 |
| 3-OH CBZ | 48 | 16 | 48 | 7 | 51 | 8 | 37 | 3 | 52 | 10 | 47 | 14 |
| 10-OH CBZ | 43 | 18 | 45 | 8 | 51 | 10 | 19 | 25 | 17 | 4 | 26 | 8 |
| EP-CBZ | 48 | 18 | 20 | 1 | 21 | 10 | 17 | 18 | 26 | 24 | 19 | 3 |
| DIC | 58 | 25 | 24 | 16 | 27 | 6 | 62 | 23 | 24 | 14 | 21 | 3 |
| 4-OH DIC | 67 | 23 | 61 | 2 | 38 | 4 | 64 | 12 | 34 | 18 | 31 | 7 |
| IBU | 13 | 27 | 23 | 1 | 31 | 7 | 20 | 30 | 48 | 21 | 60 | 28 |
| 1-OH IBU | 61 | 28 | 45 | 9 | 53 | 9 | 56 | 7 | 48 | 9 | 40 | 11 |
| 2-OH IBU | 55 | 14 | 44 | 5 | 41 | 7 | 43 | 18 | 42 | 11 | 37 | 9 |
| CBX-IBU | 98 | 16 | 43 | 5 | 52 | 9 | 142 | 19 | 48 | 16 | 42 | 13 |
| SMX | 21 | 30 | 21 | 3 | 25 | 8 | 16 | 22 | 26 | 17 | 27 | 2 |
| AcSMX | 71 | 30 | 51 | 6 | 45 | 9 | 51 | 11 | 44 | 14 | 42 | 13 |
| Compound | Lettuce | Carrot | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2.5 (ng g−1 dw) | 40 (ng g−1 dw) | 100 (ng g−1 dw) | 2.5 (ng g−1 dw) | 40 (ng g−1 dw) | 100 (ng g−1 dw) | |||||||
| A (%) | ME (%) | A (%) | ME (%) | A (%) | ME (%) | A (%) | ME (%) | A (%) | ME (%) | A (%) | ME (%) | |
| CAF | 87 | −27.6 | 95 | −34.8 | 102 | −0.34 | 81 | −62.4 | 99 | −37.6 | 107 | −35.0 |
| PX | 88 | −13.2 | 94 | −19.9 | 103 | −0.22 | 84 | −54.2 | 95 | −64.3 | 99 | −27.1 |
| CBZ | 86 | −7.07 | 92 | −1.77 | 101 | −0.01 | 82 | −28.8 | 96 | −0.81 | 98 | −6.33 |
| 3-OH CBZ | 89 | −15.9 | 93 | −11.9 | 100 | −0.10 | 83 | −44.8 | 98 | −24.4 | 100 | −10.6 |
| 10-OH CBZ | 81 | −7.41 | 96 | −5.00 | 99 | −0.03 | 85 | −2.73 | 97 | −7.37 | 102 | −2.93 |
| EP-CBZ | 84 | −30.8 | 91 | −12.1 | 97 | −0.10 | 87 | −11.2 | 100 | −19.8 | 104 | −8.05 |
| DIC | 82 | −15.3 | 91 | −39.0 | 98 | −0.31 | 88 | −53.5 | 92 | −55.2 | 107 | −15.8 |
| 4-OH DIC | 83 | −32.0 | 94 | −11.7 | 100 | −0.10 | 86 | −6.27 | 94 | −26.02 | 105 | −4.11 |
| IBU | 88 | −11.2 | 93 | −8.76 | 101 | −0.32 | 89 | −35.7 | 93 | −22.2 | 103 | −8.41 |
| 1-OH IBU | 87 | −9.76 | 95 | −4.92 | 104 | −0.02 | 91 | −28.7 | 91 | −0.08 | 106 | −0.42 |
| 2-OH IBU | 87 | −6.06 | 98 | −4.31 | 106 | −0.04 | 92 | −45.6 | 100 | −27.7 | 101 | −6.85 |
| CBX-IBU | 85 | −7.98 | 96 | −0.45 | 101 | −0.01 | 90 | −49.5 | 97 | −6.49 | 97 | −1.24 |
| SMX | 84 | −20.3 | 94 | −20.8 | 99 | −0.19 | 85 | −79.3 | 98 | −75.7 | 99 | −26.1 |
| AcSMX | 81 | −1.81 | 97 | 0.02 | 98 | −0.01 | 82 | −33.4 | 99 | −29.9 | 101 | −1.32 |
| Compounds | Sample Amount (g) | Extraction Technique | Time (min) | Solvent (mL) | Clean-Up Step | Analytical Determination | MQL (ng g−1 dw) | Reference |
|---|---|---|---|---|---|---|---|---|
| 1 CAF, 1 analgesic, 8 antibiotics, and 1 anticonvulsant | 0.5 | QuEChERS | 3.5 | ACN, MeOH 5 | - | LC-MS/MS | 0.7–8.0 (MDL) | [20] |
| 1 CAF, 1 analgesic, 8 antibiotics, and 1 anticonvulsant | 0.5 | PLE | 20 | ACN, MeOH 40 | SPE | LC-MS/MS | 1.9–15.8 (MDL) | [20] |
| 7 antibiotics | 0.25–0.5 | UAE | 6 | 18 (Alkaline solution with Mg2+) | SPE | LC-MS/MS | 2–10 | [17] |
| 1 anticonvulsant | 0.5 | PLE | 27 | n.d. (Acetone, hexane) | - | GC-MS | 7.6–61.7 | [19] |
| 11 antibiotics | 1.0 | UAE | 30 | 20 (ACN) | SPE | LC-MS/MS | 0.2–6.25 | [18] |
| 10 antibiotics and 6 of their metabolites | 1.0 | UAE | 30 | 20 (MeOH) | SPE | LC-MS/MS | 0.2–9.2 | [24] |
| 26 antibiotics | 10 | QuEChERS | 27 | 10 (ACN, MeOH) | - | LC-MS/MS | 0.02–1.5 | [23] |
| 20 antibiotics | 10 | QuEChERS | 35 | 10 (ACN, MeOH) | - | LC-MS/MS | 0.33–2.92 (MDL) | [22] |
| 1 CAF, 2 NSAIDs, 1 antibiotic, 1 anticonvulsant, and 9 of their main metabolites | 0.5 | UAE | 27 | 12 (Acetone) | d-SPE | LC-MS/MS | 0.007–2.25 | Proposed methodology |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mejías, C.; Arenas, M.; Martín, J.; Santos, J.L.; Aparicio, I.; Alonso, E. Multiclass Analysis for the Determination of Pharmaceuticals and Their Main Metabolites in Leafy and Root Vegetables. Molecules 2024, 29, 3471. https://doi.org/10.3390/molecules29153471
Mejías C, Arenas M, Martín J, Santos JL, Aparicio I, Alonso E. Multiclass Analysis for the Determination of Pharmaceuticals and Their Main Metabolites in Leafy and Root Vegetables. Molecules. 2024; 29(15):3471. https://doi.org/10.3390/molecules29153471
Chicago/Turabian StyleMejías, Carmen, Marina Arenas, Julia Martín, Juan Luis Santos, Irene Aparicio, and Esteban Alonso. 2024. "Multiclass Analysis for the Determination of Pharmaceuticals and Their Main Metabolites in Leafy and Root Vegetables" Molecules 29, no. 15: 3471. https://doi.org/10.3390/molecules29153471
APA StyleMejías, C., Arenas, M., Martín, J., Santos, J. L., Aparicio, I., & Alonso, E. (2024). Multiclass Analysis for the Determination of Pharmaceuticals and Their Main Metabolites in Leafy and Root Vegetables. Molecules, 29(15), 3471. https://doi.org/10.3390/molecules29153471