3.1.1. General Procedure 1 for the Synthesis of Compounds 7a–r
Ketone
4 (2.24 g, 0.01 mol, 1 equiv.) was dissolved in dimethylformamide dimethyl acetal (13 mL, 10 equiv.) and heated under reflux for 18 h. The volatiles were removed on a rotary evaporator, and the residue was co-evaporated again with toluene. The residue was dissolved in methanol (10 mL) and then was added dropwise at 0 °C to a solution of corresponding amidine hydrochloride (0.015 mol, 1.5 equiv) and sodium methoxide (0.9 g, 0.017 mol, 1.7 equiv.) in methanol (30 mL). The resulting mixture was heated at reflux for 8 h. The solvent was removed in vacuo and the residue was partitioned between 5% aqueous citric acid (50 mL) and dichloromethane (100 mL). The organic phase was separated, washed with water, dried over anhydrous Na
2SO
4, filtered, and concentrated in vacuo. The residue was purified by column chromatography on silica gel eluting with 50% → 100% ethyl acetate in hexane. Compounds
7a–
d were synthesized according to this procedure previously, and the spectra were reported [
24].
tert-butyl 2′-(4-fluorophenyl)-1H,7′H-spiro[azetidine-3,5′-furo[3,4-d]pyrimidine]-1-carboxylate (7e).
The compound was synthesized according to General Procedure 1. Yield 1.75 g (49%), white solid, m.p. 132-133 °C, 1H NMR (300 MHz, DMSO-d6) δ 9.12 (s, 1H), 8.45 (dd, J = 8.8, 5.7 Hz, 2H), 7.36 (t, J = 8.8 Hz, 2H), 5.09 (s, 2H), 4.29 (d, J = 10.0 Hz, 2H), 4.15 (d, J = 9.7 Hz, 2H), 1.43 (s, 9H); 13C NMR (75 MHz, DMSO-d6) δ 170.72, 163.20, 162.84, 156.01, 151.65, 133.69, 130.77, 130.65, 130.30, 116.31, 116.02, 81.28, 79.55, 71.31, 63.57, 28.46. HRMS (ESI) m/z calcd for C19H21FN3O3 (M+H+) 358.1566, found 358.1568.
tert-butyl 2′-(4-chlorophenyl)-1H,7′H-spiro[azetidine-3,5′-furo[3,4-d]pyrimidine]-1-carboxylate (7f).
The compound was synthesized according to General Procedure 1. Yield 2.06 g (55%), white solid, m.p. 138–139 °C. 1H NMR (300 MHz, DMSO-d6) δ 9.14 (s, 1H), 8.41 (d, J = 8.4 Hz, 2H), 7.60 (d, J = 8.6 Hz, 2H), 5.10 (s, 2H), 4.29 (d, J = 9.6 Hz, 2H), 4.15 (d, J = 9.8 Hz, 2H), 1.43 (s, 9H); 13C NMR (75 MHz, DMSO-d6) δ 170.78, 163.12, 156.01, 151.70, 136.40, 135.99, 130.65, 130.03, 129.32, 81.28, 79.56, 71.30, 28.46. HRMS (ESI) m/z calcd for C19H21ClN3O3 (M+H+) 374.1271, found 374.1274.
tert-butyl 2′-(3-chlorophenyl)-1H,7′H-spiro[azetidine-3,5′-furo[3,4-d]pyrimidine]-1-carboxylate (7g).
The compound was synthesized according to General Procedure 1. Yield 1.91 g (51%), white solid, m.p. 122-123 °C. 1H NMR (300 MHz, DMSO-d6) δ 9.17 (s, 1H), 8.40–8.31 (m, 2H), 7.68–7.53 (m, 2H), 5.11 (s, 2H), 4.30 (d, J = 9.7 Hz, 2H), 4.15 (d, J = 9.7 Hz, 2H), 1.43 (s, 9H); 13C NMR (75 MHz, DMSO-d6) δ 170.88, 162.67, 156.01, 151.75, 139.21, 134.11, 131.24, 131.03, 127.77, 126.82, 81.27, 79.57, 71.31, 63.54, 28.46. HRMS (ESI) m/z calcd for C19H21ClN3O3 (M+H+) 374.1271, found 374.1272.
tert-butyl 2′-(4-isopropylphenyl)-1H,7′H-spiro[azetidine-3,5′-furo[3,4-d]pyrimidine]-1- carboxylate (7h).
The compound was synthesized according to General Procedure 1. Yield 1.68 g (44%), white solid, m.p. 94–95 °C. 1H NMR (300 MHz, DMSO-d6) δ 9.11 (s, 1H), 8.33 (d, J = 7.8 Hz, 2H), 7.41 (d, J = 7.9 Hz, 2H), 5.08 (s, 2H), 4.29 (d, J = 9.6 Hz, 2H), 4.15 (d, J = 9.7 Hz, 2H), 2.97 (m, 1H), 1.43 (s, 9H), 1.25 (d, J = 7.0 Hz, 6H); 13C NMR (75 MHz, DMSO-d6) δ 170.53, 164.11, 156.00, 152.08, 151.61, 134.83, 130.04, 128.38, 127.18, 81.26, 79.55, 71.33, 33.76, 29.40, 28.44, 24.06. HRMS (ESI) m/z calcd for C22H28N3O3 (M+H+) 382.2130, found 382.2130.
tert-butyl 2′-[(4-methoxyphenoxy)methyl]-1H,7′H-spiro[azetidine-3,5′-furo[3,4-d]pyrimidine] -1-carboxylate (7i).
The compound was synthesized according to General Procedure 1. Yield 2.68 g (67%), white solid, m.p. 88–89 °C. 1H NMR (300 MHz, DMSO-d6) δ 9.07 (s, 1H), 6.92 (d, J = 9.2 Hz, 2H), 6.84 (d, J = 9.2 Hz, 2H), 5.22 (s, 2H), 5.04 (s, 2H), 4.26 (d, J = 9.6 Hz, 2H), 4.12 (d, J = 9.7 Hz, 2H), 3.68 (s, 3H), 1.42 (s, 9H). 13C NMR (75 MHz, DMSO-d6) δ 170.45, 166.19, 155.98, 153.91, 152.56, 151.47, 131.04, 115.85, 114.96, 81.14, 79.56, 71.18, 71.07, 55.70, 55.31, 28.42. HRMS (ESI) m/z calcd for C21H26N3O5 (M+H+) 400.1872, found 400.1872.
tert-butyl 2′-pyridin-3-yl-1H,7′H-spiro[azetidine-3,5′-furo[3,4-d]pyrimidine]-1-carboxylate (7j).
The compound was synthesized according to General Procedure 1. Yield 1.90 g (56%), white solid, m.p. 107–108 °C. 1H NMR (300 MHz, DMSO-d6) 9.51 (s, 1H), 9.19 (s, 1H), 8.77–8.63 (m, 2H), 7.58 (dd, J = 7.9, 4.8 Hz, 1H), 5.12 (s, 2H), 4.31 (d, J = 9.7 Hz, 2H), 4.16 (d, J = 9.7 Hz, 2H), 1.44 (s, 9H); 13C NMR (75 MHz, DMSO-d6) δ 170.88, 162.55, 156.01, 152.04, 151.81, 149.39, 135.62, 132.66, 131.06, 124.31, 81.28, 79.57, 71.32, 63.54, 28.46. HRMS (ESI) m/z calcd for C18H21N4O3 (M+H+) 341.1613, found 341.1617.
tert-butyl 2′-(2-methoxyethyl)-1H,7′H-spiro[azetidine-3,5′-furo[3,4-d]pyrimidine]-1-carboxylate (7k).
The compound was synthesized according to General Procedure 1. Yield 1.57 g (49%), white solid, m.p. 65–66 °C. 1H NMR (300 MHz, DMSO-d6) δ 8.96 (s, 1H), 4.99 (s, 2H), 4.23 (d, J = 9.8 Hz, 2H), 4.12 (d, J = 9.7 Hz, 2H), 3.81 (t, J = 6.5 Hz, 2H), 3.22 (s, 3H), 3.14 (t, J = 6.5 Hz, 2H), 1.42 (s, 9H); 13C NMR (75 MHz, DMSO-d6) δ 169.96, 168.79, 156.00, 151.06, 129.74, 81.19, 79.51, 71.16, 70.62, 63.59, 58.21, 39.14, 28.44. HRMS (ESI) m/z calcd for C16H24N3O4 (M+H+) 322.1766, found 322.1765.
tert-butyl 2′-(4-bromophenyl)-1H,7′H-spiro[azetidine-3,5′-furo[3,4-d]pyrimidine]-1-carboxylate (7l).
The compound was synthesized according to General Procedure 1. Yield 2.13 g (51%), white solid, m.p. 139-140 °C. 1H NMR (300 MHz, DMSO-d6) δ 9.14 (s, 1H), 8.33 (d, J = 8.4 Hz, 2H), 7.74 (d, J = 8.4 Hz, 2H), 5.09 (s, 2H), 4.29 (d, J = 9.6 Hz, 2H), 4.15 (d, J = 9.6 Hz, 2H), 1.43 (s, 9H); 13C NMR (75 MHz, DMSO-d6) δ 170.78, 163.14, 155.98, 151.78, 136.28, 132.29, 130.69, 130.23, 125.38, 81.25, 79.56, 71.30, 63.70, 28.44. HRMS (ESI) m/z calcd for C19H21BrN3O3 (M+H+) 418.0766, found 418.0768.
tert-butyl 2′-(3-bromophenyl)-1H,7′H-spiro[azetidine-3,5′-furo[3,4-d]pyrimidine]-1-carboxylate (7m).
The compound was synthesized according to General Procedure 1. Yield 0.54 g (13%), white solid, m.p. 128–129 °C. 1H NMR (300 MHz, DMSO-d6) δ 9.16 (s, 1H), 8.54–8.51 (m, 1H), 8.39 (d, J = 7.9, 1H), 7.75 (dd, J = 8.0 Hz, 1H), 7.52 (t, J = 7.9 Hz, 1H), 5.10 (s, 2H), 4.30 (d, J = 9.6 Hz, 2H), 4.16 (d, J = 9.6 Hz, 2H), 1.43 (s, 9H); 13C NMR (75 MHz, DMSO-d6) δ 170.88, 162.56, 156.00, 151.75, 139.40, 134.13, 131.50, 131.02, 130.70, 127.19, 122.58, 81.27, 79.56, 71.31, 63.55, 28.46. HRMS (ESI) m/z calcd for C19H21BrN3O3 (M+H+) 418.0766, found 418.0767.
tert-butyl 2′-pyridin-2-yl-1H,7′H-spiro[azetidine-3,5′-furo[3,4-d]pyrimidine]-1-carboxylate (7n).
The compound was synthesized according to General Procedure 1. Yield 2.07 g (61%), white solid, m.p. 107–108 °C. 1H NMR (300 MHz, DMSO-d6) δ 9.20 (s, 1H), 8.75 (d, J = 2.7 Hz, 1H), 8.38 (d, J = 7.9 Hz, 1H), 8.03–7.94 (m, 1H), 7.59–7.51 (m, 1H), 5.11 (s, 2H), 4.32 (d, J = 9.6 Hz, 2H), 4.16 (d, J = 9.7 Hz, 2H), 1.43 (s, 9H); 13C NMR (75 MHz, DMSO-d6) δ 170.79, 163.82, 156.03, 154.59, 151.57, 150.13, 137.55, 131.34, 125.59, 124.01, 81.24, 79.56, 71.33, 63.56, 28.45. HRMS (ESI) m/z calcd for C18H21N4O3 (M+H+) 341.1613, found 341.1613.
tert-butyl 2′-pyridin-4-yl-1H,7′H-spiro[azetidine-3,5′-furo[3,4-d]pyrimidine]-1-carboxylate (7o).
The compound was synthesized according to General Procedure 1. Yield 2.04 g (60%), white solid, m.p. 100–101 °C. 1H NMR (300 MHz, DMSO-d6) δ 9.24 (s, 1H), 8.78 (d, J = 5.5 Hz, 2H), 8.26 (d, J = 4.4 Hz, 2H), 5.13 (s, 2H), 4.31 (d, J = 9.7 Hz, 2H), 4.16 (d, J = 9.7 Hz, 2H), 1.43 (s, 9H); 13C NMR (75 MHz, DMSO-d6) δ 171.09, 162.28, 156.00, 151.90, 151.01, 144.18, 131.99, 122.01, 81.27, 79.58, 71.30, 63.52, 40.79, 40.51, 40.23, 39.96, 39.68, 39.40, 39.12, 28.45. HRMS (ESI) m/z calcd for C18H21N4O3 (M+H+) 341.1613, found 341.1615.
tert-butyl 2′-pyrazin-2-yl-1H,7′H-spiro[azetidine-3,5′-furo[3,4-d]pyrimidine]-1-carboxylate (7p).
The compound was synthesized according to General Procedure 1. Yield 1.80 g (53%), white solid, m.p. 114–115 °C. 1H NMR (300 MHz, DMSO-d6) δ 9.53 (s, 1H), 9.25 (s, 1H), 8.82 (d, J = 7.0 Hz, 2H), 5.14 (s, 2H), 4.33 (d, J = 9.6 Hz, 2H), 4.18 (d, J = 9.7 Hz, 2H), 1.44 (s, 9H); 13C NMR (75 MHz, DMSO-d6) δ 171.10, 162.17, 156.01, 151.85, 149.91, 146.34, 145.08, 132.05, 81.26, 79.59, 71.32, 63.52, 28.46. HRMS (ESI) m/z calcd for C17H20N5O3 (M+H+) 342.1566, found 342.1566.
tert-butyl 2′-ethyl-1H,7′H-spiro[azetidine-3,5′-furo[3,4-d]pyrimidine]-1-carboxylate (7q).
Prepared according to General Procedure 1. Yield 1.39 g (48%), white solid, m.p. 64–65 °C. 1H NMR (300 MHz, DMSO-d6) δ 8.95 (d, J = 1.7 Hz, 1H), 4.99 (s, 1H), 4.23 (d, J = 9.6 Hz, 2H), 4.12 (d, J = 9.6 Hz, 2H), 2.93 (q, J = 7.4 Hz, 1H), 1.42 (s, 9H), 1.28 (t, J = 7.6 Hz, 3H); 13C NMR (75 MHz, DMSO-d6) δ 171.89, 169.94, 155.99, 151.13, 129.45, 81.14, 79.52, 71.18, 63.58, 32.15, 28.42, 12.99. HRMS (ESI) m/z calcd for C15H22N3O3 (M+H+) 292.1661, found 292.1664.
tert-butyl 2′-isopropyl-1H,7′H-spiro[azetidine-3,5′-furo[3,4-d]pyrimidine]-1-carboxylate (7r).
The compound was synthesized according to General Procedure 1. Yield 2.22 g (73%), white solid, m.p. 75–76 °C. 1H NMR (300 MHz, DMSO-d6) δ 8.97 (d, J = 1.0 Hz, 1H), 5.00 (s, 1H), 4.23 (d, J = 9.6 Hz, 2H), 4.10 (d, J = 9.7 Hz, 2H), 3.18 (dt, J = 14.2, 6.9 Hz, 0H), 1.42 (s, 9H), 1.27 (dd, J = 6.9, 1.1 Hz, 6H); 13C NMR (75 MHz, DMSO-d6) δ 175.12, 169.93, 155.99, 151.15, 129.54, 81.17, 79.52, 71.23, 63.60, 37.28, 28.43, 22.13. HRMS (ESI) m/z calcd for C16H24N3O3 (M+H+) 306.1817, found 306.1818.
3.1.2. General Procedure 2 for the Synthesis of Compounds 3a–r
Solution A. To a solution of 5-nitrofuranoic acid (75 mg, 0.47 mmol) in DMF (3 mL), CDI (97 mg, 0.6 mmol) was added at 0 °C, and the mixture was stirred at 0 °C for 1 h.
Solution B. To a solution of compound 7 (0.6 mmol) in dichloromethane (5 mL), trifluoroacetic acid (1 mL) was added dropwise at 0 °C, and the resulting mixture was stirred for 1 h. The volatiles were removed in vacuo (bath temperature < 30 °C) and the residue was dissolved in DMF (3 mL). Triethylamine (0.19 g, 1.9 mmol) was added; the mixture was stirred for 30 min and added dropwise to solution A.
The reaction mixture was stirred at room temperature for 18 h, poured into water (25 mL), and the mixture was extracted with ethyl acetate (3 × 20 mL). The combined organic extracts were washed with brine, dried over anhydrous Na
2SO
4, filtered, and concentrated in vacuo. The residue was purified by column chromatography on silica gel eluting with 10:1 dichloromethane-methanol. Compounds
3a–
d were synthesized according to this procedure previously, and the spectra were reported [
24].
2′-(4-fluorophenyl)-1-(5-nitro-2-furoyl)-7′H-spiro[azetidine-3,5′-furo[3,4-d]pyrimidine] (3e).
Yield 44 mg (24%), white solid, m.p. 125-126 °C. 1H NMR (300 MHz, CDCl3) δ 8.90 (s, 1H), 8.55–8.42 (m, 2H), 7.43–7.32 (m, 2H), 7.19 (t, J = 8.7 Hz, 2H), 5.18 (s, 2H), 5.10 (d, J = 11.0 Hz, 1H), 4.99 (d, J = 11.0 Hz, 1H), 4.67 (d, J = 11.6 Hz, 1H), 4.56 (d, J = 11.6 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 170.06, 164.96 (d, J = 251.6 Hz), 164.83, 156.32, 150.02, 147.97, 132.96 (d, J = 3.2 Hz), 130.62 (d, J = 8.8 Hz), 129.06, 123.36, 117.26, 115.63 (d, J = 21.7 Hz), 111.66, 81.70, 72.01, 66.91, 63.35. HRMS (ESI) m/z calcd for C19H14FN4O5 (M+H+) 397.0948, found 397.0948.
2′-(4-chlorophenyl)-1-(5-nitro-2-furoyl)-7′H-spiro[azetidine-3,5′-furo[3,4-d]pyrimidine] (3f).
Yield 38 mg (20%), white solid, m.p. 129-128 °C. 1H NMR (300 MHz, CDCl3) δ 8.91 (s, 1H), 8.43 (d, J = 8.3 Hz, 2H), 7.49 (d, J = 8.3 Hz, 2H), 7.37 (d, J = 9.2 Hz, 2H), 5.19 (s, 2H), 5.13–4.97 (m, 2H), 4.72–4.51 (m, 2H); 13C NMR (75 MHz, CDCl3) δ 170.11, 164.80, 156.31, 150.04, 147.96, 137.57, 135.23, 129.76, 129.39, 128.88, 117.25, 111.65, 81.70, 72.00, 66.89, 63.33. HRMS (ESI) m/z calcd for C19H14ClN4O5 (M+H+) 413.0652, found 413.0654.
2′-(3-chlorophenyl)-1-(5-nitro-2-furoyl)-7′H-spiro[azetidine-3,5′-furo[3,4-d]pyrimidine] (3g).
Yield 66 mg (34%), white solid, m.p. 124–126 °C. 1H NMR (300 MHz, CDCl3) δ 8.91 (s, 1H), 8.47 (s, 1H), 8.35 (d, J = 7.5 Hz, 1H), 7.54–7.32 (m, 4H), 5.18 (s, 2H), 5.09 (d, J = 11.0 Hz, 1H), 4.99 (d, J = 11.0 Hz, 1H), 4.66 (d, J = 11.4 Hz, 1H), 4.55 (d, J = 11.6 Hz, 1H); 13C NMR (75 MHz, CDCl3) δ 170.18, 164.46, 156.31, 150.11, 147.93, 138.53, 134.84, 131.21, 129.93, 129.75, 128.53, 126.51, 117.34, 111.76, 81.68, 72.03, 66.95, 63.39. HRMS (ESI) m/z calcd for C19H14ClN4O5 (M+H+) 413.0652, found 413.0652.
2′-(4-isopropylphenyl)-1-(5-nitro-2-furoyl)-7′H-spiro[azetidine-3,5′-furo[3,4-d]pyrimidine] (3h).
Yield 117 mg (59%), white solid, m.p. 116-117 °C. 1H NMR (300 MHz, CDCl3) δ 8.90 (s, 1H), 8.38 (d, J = 8.6 Hz, 2H), 7.43 -7.34 (m, 4H), 5.19 (s, 2H), 5.10 (d, J = 11.0 Hz, 1H), 5.00 (d, J = 11.0 Hz, 1H), 4.67 (d, J = 11.5 Hz, 1H), 4.56 (d, J = 11.6 Hz, 1H), 3.01 (p, J = 7.0 Hz, 1H), 1.32 (d, J = 6.9 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 169.90, 165.91, 156.31, 152.51, 149.96, 148.02, 134.46, 128.74, 128.53, 126.77, 117.22, 111.66, 81.73, 72.06, 66.95, 63.38, 34.07, 23.71. HRMS (ESI) m/z calcd for C22H21N4O5 (M+H+) 421.1511, found 421.1512.
2′-[(4-methoxyphenoxy)methyl]-1-(5-nitro-2-furoyl)-7′H-spiro[azetidine-3,5′-furo[3,4-d]pyrimidine] (3i).
Yield 119 mg (58%), white solid, m.p. 118-119 °C. 1H NMR (300 MHz, CDCl3) δ 8.90 (s, 1H), 7.42–7.30 (m, 2H), 6.96 (d, J = 8.8 Hz, 2H), 6.83 (d, J = 8.9 Hz, 2H), 5.32 (s, 2H), 5.16 (s, 2H), 5.07 (d, J = 11.0 Hz, 1H), 4.96 (d, J = 11.0 Hz, 1H), 4.64 (d, J = 11.6 Hz, 1H), 4.53 (d, J = 11.6 Hz, 1H), 3.77 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 170.33, 167.43, 156.29, 154.30, 152.33, 150.13, 147.89, 130.46, 117.28, 115.93, 114.60, 111.66, 81.58, 71.96, 71.31, 66.83, 63.28, 55.59. HRMS (ESI) m/z calcd for C21H19N4O7 (M+H+) 439.1253, found 439.1253.
1-(5-nitro-2-furoyl)-2′-pyridin-3-yl-7′H-spiro[azetidine-3,5′-furo[3,4-d]pyrimidine] (3j).
Yield 61 mg (34%), white solid, m.p. 118-119 °C. 1H NMR (300 MHz, DMSO-d6) δ 9.50 (d, J = 2.0 Hz, 1H), 9.27 (s, 1H), 8.72 (dd, J = 4.8, 1.7 Hz, 1H), 8.67 (dt, J = 8.0, 2.0 Hz, 1H), 7.82–7.68 (m, 1H), 7.57 (dd, J = 8.0, 4.8 Hz, 1H), 7.49–7.35 (m, 1H), 5.16 (s, 2H), 5.01 (d, J = 10.6 Hz, 1H), 4.86 (d, J = 10.6 Hz, 1H), 4.59 (d, J = 11.4 Hz, 1H), 4.37 (d, J = 11.5 Hz, 1H); 13C NMR (75 MHz, DMSO-d6)170.92, 162.61, 156.84, 152.25, 151.97, 151.87, 149.33, 147.84, 135.73, 132.67, 130.64, 124.34, 123.07, 117.55, 113.52, 82.10, 71.58, 66.60, 63.21. HRMS (ESI) m/z calcd for C18H14N5O5 (M+H+) 380.0994, found 380.0996.
2′-(2-methoxyethyl)-1-(5-nitro-2-furoyl)-7′H-spiro[azetidine-3,5′-furo[3,4-d]pyrimidine] (3k).
Yield 66 mg (39%), white solid, m.p. 92-93 °C. 1H NMR (300 MHz, CDCl3) δ 8.81 (s, 1H), 7.37 (d, J = 3.8 Hz, 1H), 7.33 (d, J = 3.8 Hz, 1H), 5.11 (s, 2H), 5.04 (d, J = 10.9 Hz, 1H), 4.94 (d, J = 11.0 Hz, 1H), 4.62 (d, J = 11.6 Hz, 1H), 4.51 (d, J = 11.8 Hz, 1H), 3.92 (t, J = 6.4 Hz, 2H), 3.37 (s, 3H), 3.30 (t, J = 6.4 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 169.95, 156.27, 149.75, 147.93, 129.10, 117.28, 111.73, 109.53, 81.63, 72.00, 70.72, 66.91, 63.31, 58.70, 39.41. HRMS (ESI) m/z calcd for C16H17N4O6 (M+H+) 361.1148, found 361.1148.
2′-(4-bromophenyl)-1-(5-nitro-2-furoyl)-7′H-spiro[azetidine-3,5′-furo[3,4-d]pyrimidine] (3l).
Yield 110 mg (51%), white solid, m.p. 140-141 °C. 1H NMR (300 MHz, DMSO-d6) δ 9.25 (s, 1H), 8.35 (d, J = 8.6 Hz, 2H), 7.80 (d, J = 3.9 Hz, 1H), 7.75 (d, J = 8.6 Hz, 2H), 7.40 (d, J = 3.9 Hz, 1H), 5.16 (s, 2H), 5.01 (d, J = 10.6 Hz, 1H), 4.87 (d, J = 10.6 Hz, 1H), 4.60 (d, J = 11.5 Hz, 1H), 4.38 (d, J = 11.5 Hz, 1H); 13C NMR (75 MHz, DMSO-d6) δ 170.85, 163.32, 156.86, 152.16, 147.83, 136.34, 132.28, 130.29, 125.39, 117.55, 113.54, 82.11, 71.55, 63.22. HRMS (ESI) m/z calcd for C19H14BrN4O5 (M+H+) 457.0147, found 457.0149.
2′-(3-bromophenyl)-1-(5-nitro-2-furoyl)-7′H-spiro[azetidine-3,5′-furo[3,4-d]pyrimidine] (3m).
Yield 52 mg (24%), white solid, m.p. 136-137 °C. 1H NMR (300 MHz, DMSO-d6) δ 9.27 (s, 1H), 8.53 (s, 1H), 8.41 (d, J = 7.8 Hz, 1H), 7.84-7.72 (m, 2H), 7.52 (t, J = 8.0 Hz, 1H), 7.41 (s, 1H), 5.17 (s, 2H), 5.07-4.97 (m, 1H), 4.94–4.82 (m, 1H), 4.60 (d, J = 11.1 Hz, 1H), 4.39 (d, J = 11.4 Hz, 1H); 13C NMR (75 MHz, DMSO-d6) δ 170.93, 162.66, 156.86, 152.20, 147.84, 139.39, 134.17, 131.51, 130.73, 130.59, 127.23, 122.58, 117.55, 113.54, 82.10, 71.56, 66.63, 63.21. HRMS (ESI) m/z calcd for C19H14BrN4O5 (M+H+) 457.0147, found 457.0147
1-(5-nitro-2-furoyl)-2′-pyridin-2-yl-7′H-spiro[azetidine-3,5′-furo[3,4-d]pyrimidine] (3n).
Yield 91 mg (51%), white solid, m.p. 117-118 °C. 1H NMR (300 MHz, CDCl3) δ 9.01 (s, 1H), 8.86 (d, J = 4.1 Hz, 1H), 8.55 (d, J = 8.0 Hz, 1H), 7.94–7.84 (m, 1H), 7.49–7.41 (m, 1H), 7.34 (m, J = 13.3, 3.8 Hz, 2H), 5.23 (s, 2H), 5.04 (dd, J = 30.9, 10.7 Hz, 2H), 4.60 (dd, J = 32.7, 11.2 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 170.82, 164.13, 156.39, 153.51, 150.42, 149.87, 147.99, 137.67, 130.94, 125.56, 124.14, 117.35, 111.74, 81.71, 72.19, 66.91, 63.40. HRMS (ESI) m/z calcd for C18H14N5O5 (M+H+) 380.0994, found 380.0994.
1-(5-nitro-2-furoyl)-2′-pyridin-4-yl-7′H-spiro[azetidine-3,5′-furo[3,4-d]pyrimidine] (3o).
Yield 62 mg (35%), white solid, m.p. 115-116 °C. 1H NMR (300 MHz, CDCl3) δ 8.94 (s, 1H), 8.71 (d, J = 5.0 Hz, 2H), 8.24 (d, J = 5.1 Hz, 2H), 7.37–7.23 (m, 2H), 5.13 (s, 2H), 4.97 (d, J = 14.7 Hz, 2H), 4.53 (d, J = 15.6 Hz, 2H); 13C NMR (75 MHz, CDCl3) δ 170.44, 163.55, 150.35, 147.83, 144.04, 130.94, 123.76, 122.07, 117.31, 111.81, 81.63, 71.91, 66.80, 63.25. HRMS (ESI) m/z calcd for C18H14N5O5 (M+H+) 380.0994, found 380.0999.
1-(5-nitro-2-furoyl)-2′-pyrazin-2-yl-7′H-spiro[azetidine-3,5′-furo[3,4-d]pyrimidine] (3p).
Yield 107 mg (60%), white solid, m.p. 122–123 °C. 1H NMR (300 MHz, DMSO-d6) δ 9.55 (s, 1H), 9.36 (s, 1H), 8.88–8.78 (m, 2H), 7.80 (d, J = 3.8 Hz, 1H), 7.40 (d, J = 3.9 Hz, 1H), 5.20 (s, 2H), 5.04 (d, J = 10.5 Hz, 1H), 4.89 (d, J = 10.7 Hz, 1H), 4.62 (d, J = 11.3 Hz, 1H), 4.40 (d, J = 11.5 Hz, 1H); 13C NMR (75 MHz, DMSO-d6) δ 171.22, 162.33, 156.94, 152.38, 151.95, 149.98, 147.93, 146.45, 145.18, 145.16, 131.72, 117.62, 113.60, 82.17, 71.66, 66.67, 63.28. HRMS (ESI) m/z calcd for C17H13N5O5 (M+H+) 381.0947, found 381.0946.
2′-ethyl-1-(5-nitro-2-furoyl)-7′H-spiro[azetidine-3,5′-furo[3,4-d]pyrimidine] (3q).
Yield 62 mg (40%), white solid, m.p. 96-97 °C. 1H NMR (300 MHz, CDCl3) δ 8.80 (s, 1H), 7.39 (d, J = 3.8 Hz, 1H), 7.34 (d, J = 3.8 Hz, 1H), 5.12 (s, 2H), 5.06 (d, J = 10.9 Hz, 1H), 4.95 (d, J = 11.0 Hz, 1H), 4.63 (d, J = 11.6 Hz, 1H), 4.52 (d, J = 11.5 Hz, 1H), 3.06 (q, J = 7.6 Hz, 2H), 1.40 (t, J = 7.6 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 173.51, 169.68, 156.28, 149.76, 147.98, 128.67, 117.22, 111.65, 81.65, 71.97, 66.91, 32.51, 12.67. HRMS (ESI) m/z calcd for C15H15N4O5 (M+H+) 331.1042, found 331.1043.
2′-(4-isopropylphenyl)-1-(5-nitro-2-furoyl)-7′H-spiro[azetidine-3,5′-furo[3,4-d]pyrimidine] (3r).
Yield 90 mg (56%), white solid, m.p. 114–115 °C. 1H NMR (300 MHz, CDCl3) 1H NMR (300 MHz, CDCl3) δ 8.80 (s, 1H), 7.38 (d, J = 3.8 Hz, 1H), 7.34 (d, J = 3.8 Hz, 1H), 5.12 (s, 2H), 5.05 (d, J = 11.0 Hz, 1H), 4.95 (d, J = 10.9 Hz, 1H), 4.63 (d, J = 11.9 Hz, 1H), 4.52 (d, J = 11.6 Hz, 1H), 3.30 (hept, J = 6.9 Hz, 1H), 1.37 (d, J = 6.9 Hz, 6H); 13C NMR (75 MHz, CDCl3) δ 176.77, 169.63, 156.27, 149.68, 147.99, 128.68, 117.20, 111.65, 81.64, 72.00, 66.94, 63.30, 37.59, 21.66. HRMS (ESI) m/z calcd for C16H17N4O5 (M+H+) 345.1198, found 345.1198.
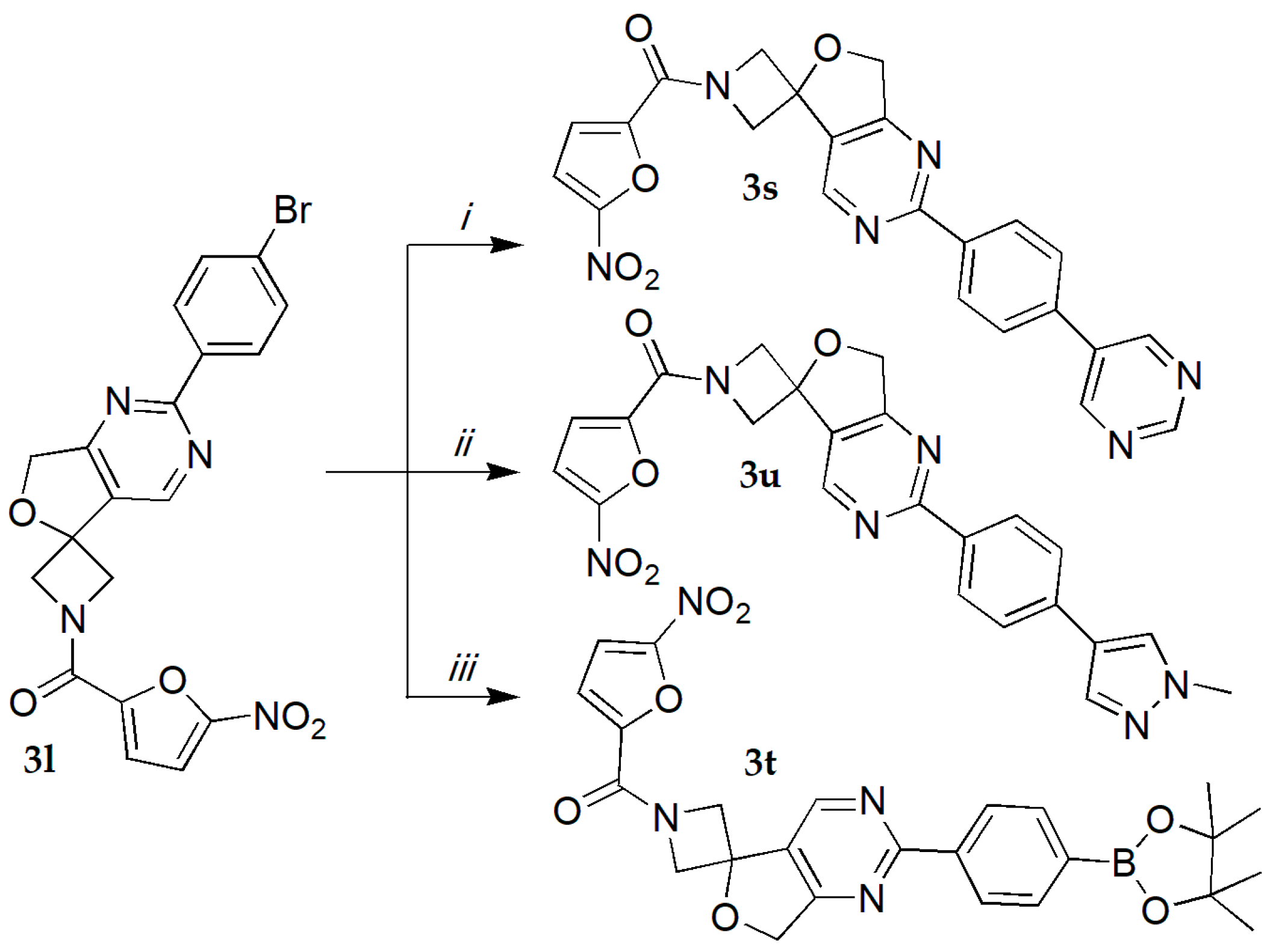
1-(5-nitro-2-furoyl)-2′-(4-pyrimidin-5-ylphenyl)-7′H-spiro[azetidine-3,5′-furo[3,4-d]pyrimidine] (3s).
Pyrimidine-5-boronic acid (0.034 g, 0.273 mmol), compound 3l (0.1 g, 0.219 mmol), [(C5H4P(C6H5)2)2)Fe]PdCl2·CH2Cl2 (0.016 g, 0.022 mmol) and Cs2CO3 (0.143 g, 0.438 mmol) were dissolved in a 100 mL screw cap vial under argon atmosphere in degassed dioxane/H2O 10:1 (10 mL). The mixture was heated for 6 h at 105 °C, H2O (50 mL) was added, and the mixture was extracted with EtOAc (2 × 50 mL). The combined organic layers were dried (Na2SO4), then evaporated and dried to constant mass on evaporating rotor. The residue was purified by column chromatography (silica gel, CH2Cl2 with 2.5% MeOH). Yield 50 mg (50%), white solid, m.p. 136–137 °C. 1H NMR (300 MHz, DMSO-d6) δ 9.28 (s, 1H), 9.24 (d, J = 3.2 Hz, 3H), 8.57 (d, J = 8.3 Hz, 2H), 8.02 (d, J = 8.4 Hz, 2H), 7.81 (d, J = 3.9 Hz, 1H), 7.41 (d, J = 3.9 Hz, 1H), 5.19 (s, 2H), 5.04 (d, J = 10.2 Hz, 1H), 4.89 (d, J = 11.1 Hz, 1H), 4.62 (d, J = 11.6 Hz, 1H), 4.40 (d, J = 11.2 Hz, 1H). 13C NMR (75 MHz, DMSO-d6) δ 170.85, 163.55, 158.04, 156.86, 155.25, 152.20, 151.89, 147.81, 137.44, 136.66, 132.88, 130.25, 129.09, 127.79, 117.58, 113.58, 82.12, 71.61, 66.65, 63.25. HRMS (ESI) m/z calcd for C23H17N6O5 (M+H+) 457.1260, found 457.1261.
1-(5-nitro-2-furoyl)-2′-[4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl]-7′H-spiro[azetidine-3,5′-furo[3,4-d]pyrimidine] (3t).
Bis(pinacolato)diboron (0.133 g, 0.525 mmol), compound 3l (0.2 g, 0.437 mmol), [(C5H4P(C6H5)2)2)Fe]PdCl2·CH2Cl2 (0.032 g, 0.044 mmol) and AcOK (0.129 g, 1.31 mmol) were dissolved in a 100 mL screw cap vial under argon atmosphere in degassed dioxane (10 mL). The mixture was heated for 6 h at 105 °C, H2O (50 mL) was added, and the mixture was extracted with EtOAc (2 × 50 mL). The combined organic layers were dried (Na2SO4) and concentrated in vacuo. Residue was purified by column chromatography (silica gel, CH2Cl2 with 2.5% MeOH). Yield 49 mg (45%), white solid, m.p. 119–120 °C. 1H NMR (300 MHz, CDCl3) δ 8.94 (s, 1H), 8.47 (d, J = 8.1 Hz, 2H), 7.96 (d, J = 8.1 Hz, 2H), 7.43–7.32 (m, 2H), 5.20 (s, 2H), 5.10 (d, J = 11.0 Hz, 1H), 5.00 (d, J = 11.0 Hz, 1H), 4.67 (d, J = 11.6 Hz, 1H), 4.57 (d, J = 11.6 Hz, 1H), 1.39 (s, 12H); 13C NMR (75 MHz, CDCl3) δ 170.03, 165.69, 156.31, 150.01, 147.98, 139.06, 135.01, 129.34, 127.53, 117.22, 111.65, 83.96, 81.71, 72.03, 66.90, 63.35, 24.83. HRMS (ESI) m/z calcd for C25H26BN4O7 (M+H+) 505.1894, found 505.1895.
2′-[4-(1-methyl-1H-pyrazol-4-yl)phenyl]-1-(5-nitro-2-furoyl)-7′H-spiro[azetidine-3,5′-furo[3,4-d]pyrimidine] (3u).
1-methyl-1H-pyrazole-4-boronic acid pinacol ester (0.057 g, 0.273 mmol), compound 3l (0.1 g, 0.219 mmol), [(C5H4P(C6H5)2)2)Fe]PdCl2·CH2Cl2 (0.016 g, 0.022 mmol), and Cs2CO3 (0.143 g, 0.438 mmol) were dissolved in a 100 mL screw cap vial under argon atmosphere in degassed dioxane/H2O 10:1 (10 mL). The mixture was heated for 6 h at 105 °C, H2O (50 mL) was added, and the mixture was extracted with EtOAc (2 × 50 mL). The combined organic layers were dried (Na2SO4) and concentrated in vacuo. Residue was purified by column chromatography (silica gel, CH2Cl2 with 2.5% MeOH). Yield 40 mg (40%), white solid, m.p. 138-139 °C. 1H NMR (300 MHz, DMSO-d6) δ 9.21 (s, 1H), 8.39 (d, J = 8.3 Hz, 3H), 8.25 (s, 1H), 7.96 (s, 1H), 7.80 (d, J = 3.9 Hz, 1H), 7.73 (d, J = 8.2 Hz, 3H), 7.40 (d, J = 3.9 Hz, 1H), 5.15 (s, 2H), 5.01 (d, J = 10.6 Hz, 1H), 4.87 (d, J = 10.7 Hz, 1H), 4.59 (d, J = 11.5 Hz, 1H), 4.38 (d, J = 11.5 Hz, 1H), 3.89 (s, 4H); 13C NMR (75 MHz, DMSO-d6) δ 170.60, 164.06, 156.87, 152.00, 151.89, 147.83, 136.78, 135.86, 134.56, 129.55, 128.93, 128.82, 125.49, 121.64, 117.55, 113.54, 82.12, 80.18, 71.58, 66.64, 64.38, 63.26. HRMS (ESI) m/z calcd for C23H19N6O5 (M+H+) 459.1416, found 459.1416.


 ,
, 


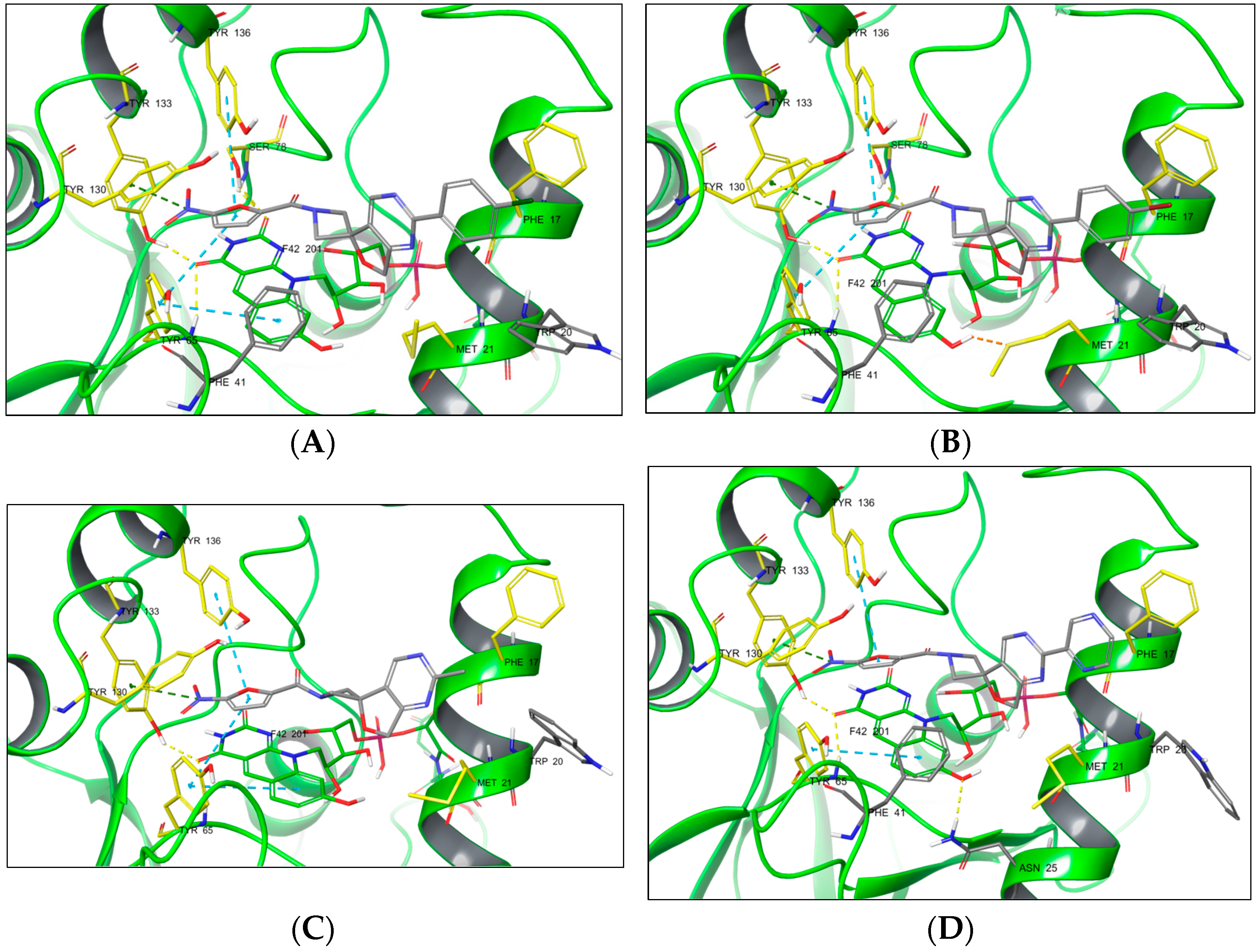

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}