
The Molecular Mechanisms of Cuproptosis and Small-Molecule Drug Design in Diabetes Mellitus

Abstract
1. Introduction
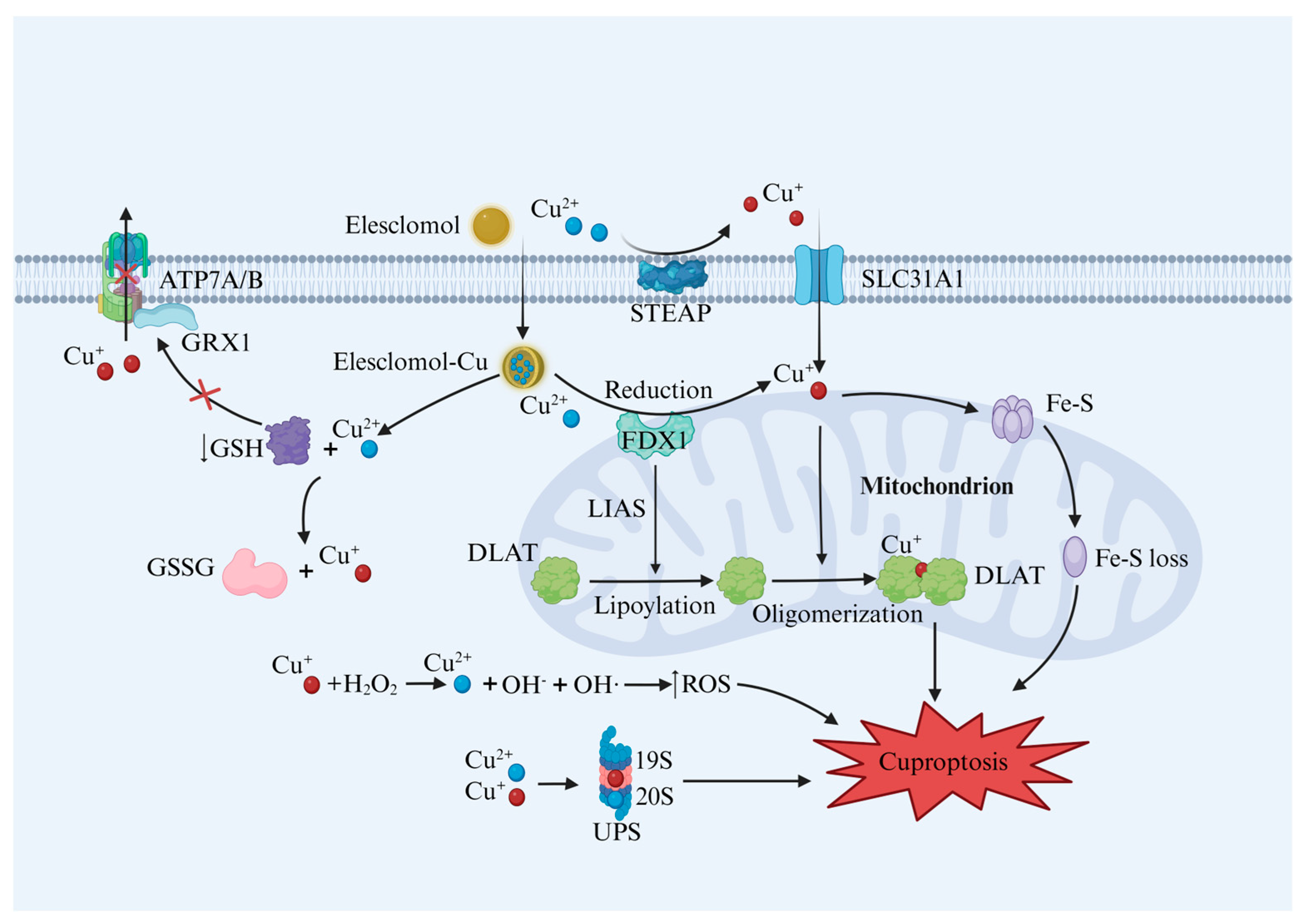
2. Cu Homeostasis and Cuproptosis Mechanism
2.1. Cu Metabolism
2.2. Cuproptosis Mechanism
2.2.1. Cu Homeostasis Imbalance and Cuproptosis
2.2.2. Protein Lipoylation and Cuproptosis
2.2.3. GSH Depletion and Cuproptosis
2.2.4. ROS and Cuproptosis
2.2.5. UPS and Cuproptosis
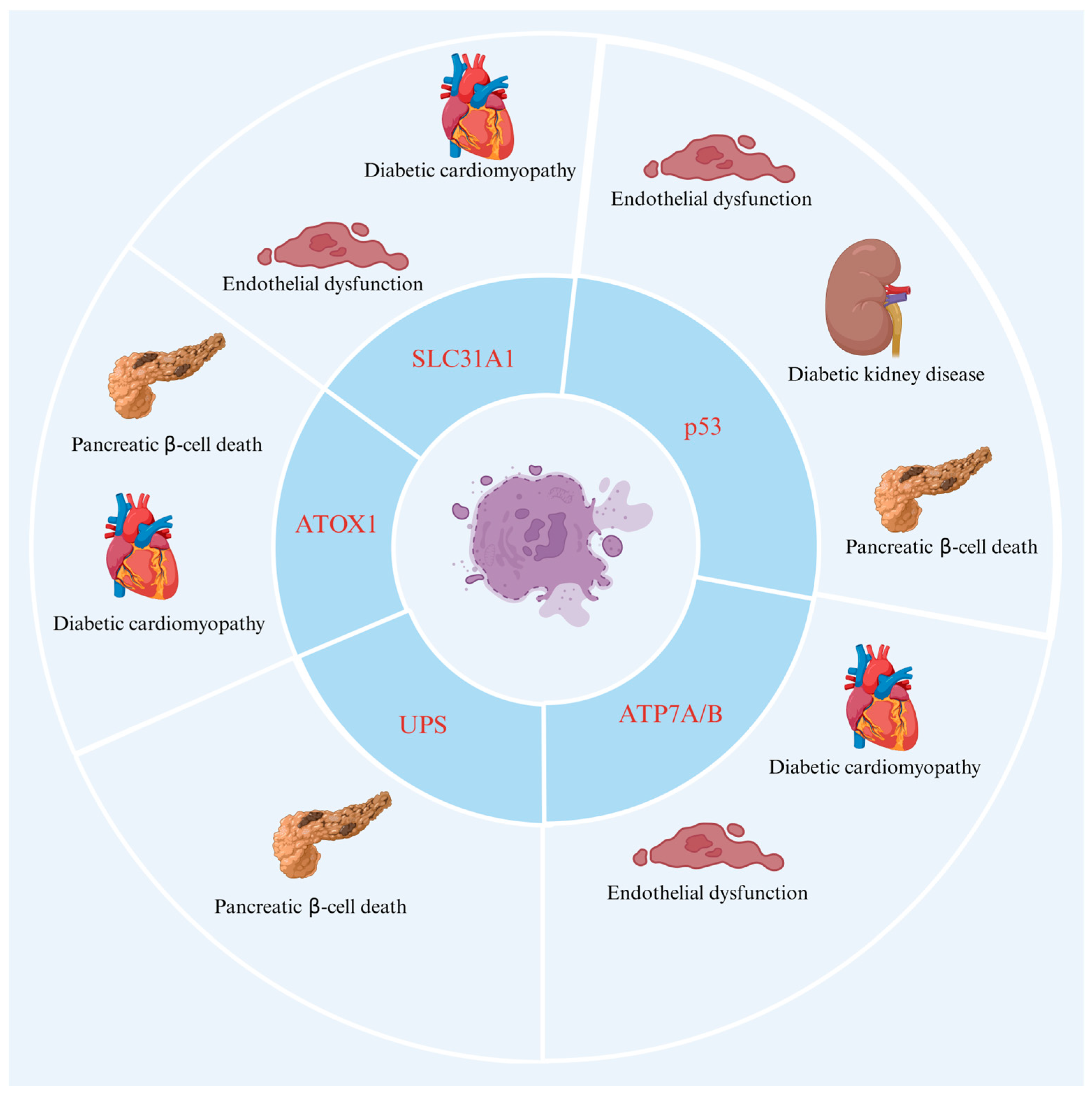
3. Cuproptosis in DM
3.1. The Role of Key Proteins of Cuproptosis in DM
3.1.1. ATOX1
3.1.2. FDX1
3.1.3. ATP7A and ATP7B
3.1.4. SLC31A1
3.2. The Role of Potential Proteins of Cuproptosis in DM
3.2.1. p53
3.2.2. UPS
4. Cuproptosis Drugs
Advances in Research into Small-Molecule Drugs Targeting Cuproptosis
5. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Warnes, G. Flow Cytometric Characterization of Accidental Cell Death Highlights Connections to Regulated Cell Death. J. Cell. Signal. 2020, 1, 1–3. [Google Scholar] [CrossRef]
- Chen, Y.; Hua, Y.; Li, X.; Arslan, I.M.; Zhang, W.; Meng, G. Distinct Types of Cell Death and the Implication in Diabetic Cardiomyopathy. Front. Pharmacol. 2020, 11, 42. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Coy, S.; Petrova, B.; Dreishpoon, M.; Verma, A.; Abdusamad, M.; Rossen, J.; Joesch-Cohen, L.; Humeidi, R.; Spangler, R.D.; et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 2022, 375, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Arredondo, M.; Núñez, M.T. Iron and copper metabolism. Mol. Asp. Med. 2005, 26, 313–327. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, L.; Zhou, F. Cuproptosis: A new form of programmed cell death. Cell. Mol. Immunol. 2022, 19, 867–868. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Min, J.; Wang, F. Copper homeostasis and cuproptosis in health and disease. Signal Transduct. Target. Ther. 2022, 7, 378. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, N.; Zhang, M.; Sun, M.; Bian, J.; Yang, B.; Li, Z.; Wang, J.; Li, F.; Shi, X.; et al. APEX2-based Proximity Labeling of Atox1 Identifies CRIP2 as a Nuclear Copper-binding Protein that Regulates Autophagy Activation. Angew. Chem. (Int. Ed. Engl.) 2021, 60, 25346–25355. [Google Scholar] [CrossRef] [PubMed]
- Braiterman, L.; Nyasae, L.; Leves, F.; Hubbard, A.L. Critical roles for the COOH terminus of the Cu-ATPase ATP7B in protein stability, trans-Golgi network retention, copper sensing, and retrograde trafficking. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G69–G81. [Google Scholar] [CrossRef]
- Yang, H.; Zhong, C.; Tan, X.; Chen, G.; He, Y.; Liu, S.; Luo, Z. Transcriptional Responses of Copper-Transport-Related Genes ctr1, ctr2 and atox1 and Their Roles in the Regulation of Cu Homeostasis in Yellow Catfish Pelteobagrus fulvidraco. Int. J. Mol. Sci. 2022, 23, 12243. [Google Scholar] [CrossRef]
- Clifford, R.J.; Maryon, E.B.; Kaplan, J.H. Dynamic internalization and recycling of a metal ion transporter: Cu homeostasis and CTR1, the human Cu+ uptake system. J. Cell Sci. 2016, 129, 1711–1721. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, H.; Amarsingh, G.V.; Cheung, C.C.H.; Hogl, S.; Narayanan, U.; Zhang, L.; McHarg, S.; Xu, J.; Gong, D.; et al. Diabetic cardiomyopathy is associated with defective myocellular copper regulation and both defects are rectified by divalent copper chelation. Cardiovasc. Diabetol. 2014, 13, 100. [Google Scholar] [CrossRef] [PubMed]
- Sudhahar, V.; Okur, M.N.; Bagi, Z.; O’bryan, J.P.; Hay, N.; Makino, A.; Patel, V.S.; Phillips, S.A.; Stepp, D.; Ushio-Fukai, M.; et al. Akt2 (Protein Kinase B Beta) Stabilizes ATP7A, a Copper Transporter for Extracellular Superoxide Dismutase, in Vascular Smooth Muscle: Novel Mechanism to Limit Endothelial Dysfunction in Type 2 Diabetes Mellitus. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Makova, M.; Alomar, S.Y.; Alwasel, S.H.; Nepovimova, E.; Kuca, K.; Rhodes, C.J.; Valko, M. Essential metals in health and disease. Chem.-Biol. Interact. 2022, 367, 110173. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Flores, S.R.; Ha, J.-H.; Doguer, C.; Woloshun, R.R.; Xiang, P.; Grosche, A.; Vidyasagar, S.; Collins, J.F. Intestinal DMT1 Is Essential for Optimal Assimilation of Dietary Copper in Male and Female Mice with Iron-Deficiency Anemia. J. Nutr. 2018, 148, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Rupp, J.C.; Locatelli, M.; Grieser, A.; Ramos, A.; Campbell, P.J.; Yi, H.; Steel, J.; Burkhead, J.L.; Bortz, E. Host Cell Copper Transporters CTR1 and ATP7A are important for Influenza A virus replication. Virol. J. 2017, 14, 11. [Google Scholar] [CrossRef] [PubMed]
- Maryon, E.B.; Molloy, S.A.; Kaplan, J.H. Cellular glutathione plays a key role in copper uptake mediated by human copper transporter 1. Am. J. Physiol.-Cell Physiol. 2013, 304, C768–C779. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y.; O’Halloran, T.V. Posttranslational Modifications in Cu,Zn-Superoxide Dismutase and Mutations Associated with Amyotrophic Lateral Sclerosis. Antioxid. Redox Signal. 2006, 8, 847–867. [Google Scholar] [CrossRef] [PubMed]
- Horng, Y.-C.; Cobine, P.A.; Maxfield, A.B.; Carr, H.S.; Winge, D.R. Specific copper transfer from the Cox17 metallochaperone to both Sco1 and Cox11 in the assembly of yeast cytochrome C oxidase. J. Biol. Chem. 2004, 279, 35334–35340. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zheng, L.; Ma, S.; Lin, R.; Li, J.; Yang, S. Cuproptosis: Emerging biomarkers and potential therapeutics in cancers. Front. Oncol. 2023, 13, 1288504. [Google Scholar] [CrossRef]
- Li, J.; Jiang, Y.; Xu, T.; Zhang, Y.; Xue, J.; Gao, X.; Yang, X.; Wang, X.; Jia, X.; Cheng, W.; et al. Wilson Disease With Novel Compound Heterozygote Mutations in the ATP7B Gene Presenting with Severe Diabetes. Diabetes Care 2020, 43, 1363–1365. [Google Scholar] [CrossRef]
- Tang, D.; Chen, X.; Kroemer, G. Cuproptosis: A copper-triggered modality of mitochondrial cell death. Cell Res. 2022, 32, 417–418. [Google Scholar] [CrossRef] [PubMed]
- Lillig, C.H.; Berndt, C.; Holmgren, A. Glutaredoxin systems. Biochim. Et Biophys. Acta (BBA)—Gen. Subj. 2008, 1780, 1304–1317. [Google Scholar] [CrossRef] [PubMed]
- Gallogly, M.M.; Starke, D.W.; Mieyal, J.J. Mechanistic and Kinetic Details of Catalysis of Thiol-Disulfide Exchange by Glutaredoxins and Potential Mechanisms of Regulation. Antioxid. Redox Signal. 2009, 11, 1059–1081. [Google Scholar] [CrossRef] [PubMed]
- Singleton, W.C.; McInnes, K.T.; Cater, M.A.; Winnall, W.R.; McKirdy, R.; Yu, Y.; Taylor, P.E.; Ke, B.-X.; Richardson, D.R.; Mercer, J.F.; et al. Role of Glutaredoxin1 and Glutathione in Regulating the Activity of the Copper-transporting P-type ATPases, ATP7A and ATP7B. J. Biol. Chem. 2010, 285, 27111–27121. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Qian, J.; Hou, G.; Wang, T.; Wang, J.; Wang, Y.; Yang, L.; Cui, X.; Suo, A. A Hollow Amorphous Bimetal Organic Framework for Synergistic Cuproptosis/Ferroptosis/Apoptosis Anticancer Therapy via Disrupting Intracellular Redox Homeostasis and Copper/Iron Metabolisms. Adv. Funct. Mater. 2022, 32, 2205013. [Google Scholar] [CrossRef]
- Graham, R.E.; Elliott, R.J.R.; Munro, A.F.; Carragher, N.O. A cautionary note on the use of N-acetylcysteine as a reactive oxygen species antagonist to assess copper mediated cell death. Biochemistry 2023, 18, e0294297. [Google Scholar] [CrossRef]
- Perillo, B.; Di Donato, M.; Pezone, A.; Di Zazzo, E.; Giovannelli, P.; Galasso, G.; Castoria, G.; Migliaccio, A. ROS in cancer therapy: The bright side of the moon. Exp. Mol. Med. 2020, 52, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Husain, N.; Mahmood, R. Copper(II) generates ROS and RNS, impairs antioxidant system and damages membrane and DNA in human blood cells. Environ. Sci. Pollut. Res. 2019, 26, 20654–20668. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Cui, Y.; Wei, X.; Zang, Y.; Chen, X.; Cheng, L.; Wang, X. CuCo2O4 Nanoflowers with Multiple Enzyme Activities for Treating Bacterium-Infected Wounds via Cuproptosis-like Death. ACS Nano 2024. [Google Scholar] [CrossRef]
- Wu, H.; Zhang, Z.; Cao, Y.; Hu, Y.; Li, Y.; Zhang, L.; Cao, X.; Wen, H.; Zhang, Y.; Lv, H.; et al. A Self-Amplifying ROS-Responsive Nanoplatform for Simultaneous Cuproptosis and Cancer Immunotherapy. Adv. Sci. 2024, 2401047. [Google Scholar] [CrossRef]
- Guo, B.; Yang, F.; Zhang, L.; Zhao, Q.; Wang, W.; Yin, L.; Chen, D.; Wang, M.; Han, S.; Xiao, H.; et al. Cuproptosis Induced by ROS Responsive Nanoparticles with Elesclomol and Copper Combined with αPD-L1 for Enhanced Cancer Immunotherapy. Adv. Mater. 2023, 35, 2212267. [Google Scholar] [CrossRef]
- Nalepa, G.; Rolfe, M.; Harper, J.W. Drug discovery in the ubiquitin–proteasome system. Nat. Rev. Drug Discov. 2006, 5, 596–613. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Bi, C.; Schmitt, S.M.; Fan, Y.; Dong, L.; Zuo, J.; Dou, Q.P. 1,10-Phenanthroline promotes copper complexes into tumor cells and induces apoptosis by inhibiting the proteasome activity. JBIC J. Biol. Inorg. Chem. 2012, 17, 1257–1267. [Google Scholar] [CrossRef]
- Opazo, C.M.; Lotan, A.; Xiao, Z.; Zhang, B.; Greenough, M.A.; Lim, C.M.; Trytell, H.; Ramírez, A.; Ukuwela, A.A.; Mawal, C.H.; et al. Nutrient copper signaling promotes protein turnover by allosteric activation of ubiquitin E2D conjugases. bioRxiv 2021. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, X.; Chen, J.; Yang, Q.; Yang, L.; Xu, D.; Zhang, P.; Wang, X.; Liu, J. Hinokitiol copper complex inhibits proteasomal deubiquitination and induces paraptosis-like cell death in human cancer cells. Eur. J. Pharmacol. 2017, 815, 147–155. [Google Scholar] [CrossRef]
- Zhang, P.; Zhou, C.; Ren, X.; Jing, Q.; Gao, Y.; Yang, C.; Shen, Y.; Zhou, Y.; Hu, W.; Jin, F.; et al. Inhibiting the compensatory elevation of xCT collaborates with disulfiram/copper-induced GSH consumption for cascade ferroptosis and cuproptosis. Redox Biol. 2024, 69, 103007. [Google Scholar] [CrossRef] [PubMed]
- Petersmann, A.; Müller-Wieland, D.; Müller, U.A.; Landgraf, R.; Nauck, M.; Freckmann, G.; Heinemann, L.; Schleicher, E. Definition, Classification and Diagnosis of Diabetes Mellitus. Exp. Clin. Endocrinol. Diabetes 2019, 127, S1–S7. [Google Scholar] [CrossRef]
- Ong, K.L.; Stafford, L.K.; McLaughlin, S.A.; Boyko, E.J.; Vollset, S.E.; Smith, A.E.; Dalton, B.E.; Duprey, J.; Cruz, J.A.; Hagins, H.; et al. Global, regional, and national burden of diabetes from 1990 to 2021, with projections of prevalence to 2050: A systematic analysis for the Global Burden of Disease Study 2021. Lancet 2023, 402, 203–234. [Google Scholar] [CrossRef] [PubMed]
- Mikłosz, A.; Chabowski, A. Efficacy of adipose-derived mesenchymal stem cell therapy in the treatment of chronic micro- and macrovascular complications of diabetes. Diabetes Obes. Metab. 2023, 26, 793–808. [Google Scholar] [CrossRef]
- Öhrvik, H.; Wittung-Stafshede, P. Identification of New Potential Interaction Partners for Human Cytoplasmic Copper Chaperone Atox1: Roles in Gene Regulation? Int. J. Mol. Sci. 2015, 16, 16728–16739. [Google Scholar] [CrossRef]
- Roberts, E.A.; Sarkar, B. Liver as a key organ in the supply, storage, and excretion of copper. Am. J. Clin. Nutr. 2008, 88, 851S–854S. [Google Scholar] [CrossRef] [PubMed]
- Ahn, C.; Choi, J.; Jeung, E. Organ-specific expression of the divalent ion channel proteins NCKX3, TRPV2, CTR1, ATP7A, IREG1 and HEPH in various canine organs. Mol. Med. Rep. 2018, 18, 1773–1781. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.-M.; Zhou, B.; Cosco, D.; Gitschier, J. The copper transporter CTR1 provides an essential function in mammalian embryonic development. Proc. Natl. Acad. Sci. USA 2001, 98, 6836–6841. [Google Scholar] [CrossRef] [PubMed]
- Flores, A.G.; Unger, V.M. Atox1 Contains Positive Residues that Mediate Membrane Association and Aid Subsequent Copper Loading. J. Membr. Biol. 2013, 246, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Hatori, Y.; Lutsenko, S. An Expanding Range of Functions for the Copper Chaperone/Antioxidant Protein Atox1. Antioxid. Redox Signal. 2013, 19, 945–957. [Google Scholar] [CrossRef] [PubMed]
- Hatori, Y.; Lutsenko, S. The role of copper chaperone Atox1 in coupling redox homeostasis to intracellular copper distribution. Antioxidants 2016, 5, 25. [Google Scholar] [CrossRef]
- Zou, M.; Zhang, W.; Zhu, Y.; Xu, Y. Identification of 6 cuproptosis-related genes for active ulcerative colitis with both diagnostic and therapeutic values. Medicine 2023, 102, e35503. [Google Scholar] [CrossRef]
- Liu, M.; Yu, W.; Jin, J.; Ma, M.; An, T.; Nie, Y.; Teng, C.-B. Copper promotes sheep pancreatic duct organoid growth by activation of an antioxidant protein 1-dependent MEK-ERK pathway. Am. J. Physiol.-Cell Physiol. 2020, 318, C806–C816. [Google Scholar] [CrossRef] [PubMed]
- Ahn, E.H.; Kim, D.W.; Shin, M.J.; Ryu, E.J.; Yong, J.I.; Chung, S.Y.; Cha, H.J.; Kim, S.J.; Choi, Y.J.; Kim, D.-S.; et al. Tat-ATOX1 inhibits streptozotocin-induced cell death in pancreatic RINm5F cells and attenuates diabetes in a mouse model. Int. J. Mol. Med. 2016, 38, 217–224. [Google Scholar] [CrossRef]
- Zulkifli, M.; Spelbring, A.N.; Zhang, Y.; Soma, S.; Chen, S.; Li, L.; Le, T.; Shanbhag, V.; Petris, M.J.; Chen, T.-Y.; et al. FDX1-dependent and independent mechanisms of elesclomol-mediated intracellular copper delivery. Proc. Natl. Acad. Sci. USA 2023, 120, e2216722120. [Google Scholar] [CrossRef]
- Lu, H.; Zhou, L.; Zhang, B.; Xie, Y.; Yang, H.; Wang, Z. Cuproptosis key gene FDX1 is a prognostic biomarker and associated with immune infiltration in glioma. Front. Med. 2022, 9, 939776. [Google Scholar] [CrossRef] [PubMed]
- Cai, C.; Zhou, K.; Jing, J.; Ren, Y.; Weng, G.; Cen, D.; Wang, X.; Huang, S. Confirmation of the predictive function of cuproptosis-related gene FDX1 in clear cell renal carcinoma using qRT-PCR and western blotting. Aging 2023, 15, 6117–6134. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Detappe, A.; Cai, K.; Keys, H.R.; Brune, Z.; Ying, W.; Thiru, P.; Reidy, M.; Kugener, G.; Rossen, J.; et al. Mitochondrial metabolism promotes adaptation to proteotoxic stress. Nat. Chem. Biol. 2019, 15, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Cai, K.; Tonelli, M.; Frederick, R.O.; Markley, J.L. Human Mitochondrial Ferredoxin 1 (FDX1) and Ferredoxin 2 (FDX2) Both Bind Cysteine Desulfurase and Donate Electrons for Iron–Sulfur Cluster Biosynthesis. Biochemistry 2017, 56, 487–499. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, M.C.F.; Anderson, C.P.; Neschen, S.; Zumbrennen-Bullough, K.B.; Romney, S.J.; Kahle-Stephan, M.; Rathkolb, B.; Gailus-Durner, V.; Fuchs, H.; Wolf, E.; et al. Irp2 regulates insulin production through iron-mediated Cdkal1-catalyzed tRNA modification. Nat. Commun. 2020, 11, 296. [Google Scholar] [CrossRef] [PubMed]
- Nokhoijav, E.; Guba, A.; Kumar, A.; Kunkli, B.; Kalló, G.; Káplár, M.; Somodi, S.; Garai, I.; Csutak, A.; Tóth, N.; et al. Metabolomic Analysis of Serum and Tear Samples from Patients with Obesity and Type 2 Diabetes Mellitus. Int. J. Mol. Sci. 2022, 23, 4534. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Ding, L.; Zhang, S.; Jiang, X.; Wang, Q.; Luo, Y.; Tian, X. Bioinformatics analysis of copper death gene in diabetic immune infiltration. Medicine 2023, 102, e35241. [Google Scholar] [CrossRef] [PubMed]
- Sudhahar, V.; Urao, N.; Oshikawa, J.; McKinney, R.D.; Llanos, R.M.; Mercer, J.F.; Ushio-Fukai, M.; Fukai, T. Copper transporter ATP7A protects against endothelial dysfunction in type 1 diabetic mice by regulating extracellular superoxide dismutase. Diabetes 2013, 62, 3839–3850. [Google Scholar] [CrossRef] [PubMed]
- Abdelsaid, K.; Sudhahar, V.; Harris, R.A.; Das, A.; Liu, Y.; McMenamin, M.; Hou, Y.; Fulton, D.; Hamrick, M.W.; Tang, Y.; et al. Exercise improves angiogenic function of circulating exosomes in type 2 diabetes: Role of exosomal SOD3. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2022, 36, e22177. [Google Scholar] [CrossRef]
- Arciello, M.; Longo, A.; Viscomi, C.; Capo, C.; Angeloni, A.; Rossi, L.; Balsano, C. Core domain mutant Y220C of p53 protein has a key role in copper homeostasis in case of free fatty acids overload. BioMetals 2015, 28, 1017–1029. [Google Scholar] [CrossRef]
- Schweigel-Röntgen, M. The Families of Zinc (SLC30 and SLC39) and Copper (SLC31) Transporters. Curr. Top. Membr. 2014, 73, 321–355. [Google Scholar] [CrossRef] [PubMed]
- Huo, S.; Wang, Q.; Shi, W.; Peng, L.; Jiang, Y.; Zhu, M.; Guo, J.; Peng, D.; Wang, M.; Men, L.; et al. ATF3/SPI1/SLC31A1 Signaling Promotes Cuproptosis Induced by Advanced Glycosylation End Products in Diabetic Myocardial Injury. Int. J. Mol. Sci. 2023, 24, 1667. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Dong, Y.-J.; Hong, C.; Li, Y.-H.; Xiao, C.-X.; Liu, X.-H.; Chang, J. ASH2L upregulation contributes to diabetic endothelial dysfunction in mice through STEAP4-mediated copper uptake. Acta Pharmacol. Sin. 2023, 45, 558–569. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zhang, C.; Zheng, D.; Guo, Q.; Maierhaba, M.; Xue, L.; Zeng, X.; Wu, Y.; Gao, W. An original cuproptosis-related genes signature effectively influences the prognosis and immune status of head and neck squamous cell carcinoma. Front. Genet. 2023, 13, 1084206. [Google Scholar] [CrossRef] [PubMed]
- Tsymbal, S.A.; Refeld, A.G.; Kuchur, O.A. The p53 Tumor Suppressor and Copper Metabolism: An Unrevealed but Important Link. Mol. Biol. 2022, 56, 979–992. [Google Scholar] [CrossRef]
- Tsymbal, S.; Refeld, A.; Zatsepin, V.; Kuchur, O. The p53 Protein is a Suppressor of Atox1 Copper Chaperon in Tumor Cells Under Genotoxic Effects. Cancer Biol. 2023, 18, e0295944. [Google Scholar] [CrossRef]
- Kung, C.-P.; Murphy, M.E. The role of the p53 tumor suppressor in metabolism and diabetes. J. Endocrinol. 2016, 231, R61–R75. [Google Scholar] [CrossRef]
- Si, R.; Zhang, Q.; Tsuji-Hosokawa, A.; Watanabe, M.; Willson, C.; Lai, N.; Wang, J.; Dai, A.; Scott, B.T.; Dillmann, W.H.; et al. Overexpression of p53 due to excess protein O-GlcNAcylation is associated with coronary microvascular disease in type 2 diabetes. Cardiovasc. Res. 2020, 116, 1186–1198. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Li, L.; Livingston, M.J.; Zhang, D.; Mi, Q.; Zhang, M.; Ding, H.-F.; Huo, Y.; Mei, C.; Dong, Z. p53/microRNA-214/ULK1 axis impairs renal tubular autophagy in diabetic kidney disease. J. Clin. Investig. 2020, 130, 5011–5026. [Google Scholar] [CrossRef]
- Gu, J.; Wang, S.; Guo, H.; Tan, Y.; Liang, Y.; Feng, A.; Liu, Q.; Damodaran, C.; Zhang, Z.; Keller, B.B.; et al. Inhibition of p53 prevents diabetic cardiomyopathy by preventing early-stage apoptosis and cell senescence, reduced glycolysis, and impaired angiogenesis. Cell Death Dis. 2018, 9, 82. [Google Scholar] [CrossRef]
- Ao, H.; Liu, B.; Li, H.; Lu, L. Egr1 mediates retinal vascular dysfunction in diabetes mellitus via promoting p53 transcription. J. Cell. Mol. Med. 2019, 23, 3345–3356. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, H.D.; Bergmann, A.; Gonen, H.; Ciechanover, A.; Steller, H. Regulation of Drosophila IAP1 degradation and apoptosis by reaper and ubcD1. Nat. Cell Biol. 2002, 4, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Saville, M.K.; Sparks, A.; Xirodimas, D.P.; Wardrop, J.; Stevenson, L.F.; Bourdon, J.-C.; Woods, Y.L.; Lane, D.P. Regulation of p53 by the ubiquitin-conjugating enzymes UbcH5B/C in vivo. J. Biol. Chem. 2004, 279, 42169–42181. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Xiong, Y.; Lv, X.; Xia, Y.; Zhang, S.; Chen, H.; Fan, J.; Wu, W.; Liu, F.; Wu, H.; et al. UbcD1 regulates Hedgehog signaling by directly modulating Ci ubiquitination and processing. EMBO Rep. 2017, 18, 1922–1934. [Google Scholar] [CrossRef] [PubMed]
- Opazo, C.M.; Lotan, A.; Xiao, Z.; Zhang, B.; Greenough, M.A.; Lim, C.M.; Trytell, H.; Ramirez, A.; Ukuwela, A.A.; Mawal, C.H.; et al. Copper Signaling Promotes Proteostasis and Animal Development Via Allosteric Activation of Ubiquitin E2 Conjugates. bioExiv 2021. [Google Scholar]
- Zhang, B. Regulation of Copper Homeostasis by the Ubiquitin Proteasome System. Ph.D. Thesis, Monash University, Melbourne, Australia, 2021. [Google Scholar]
- Chen, X.; Dou, Q.P.; Liu, J.; Tang, D. Targeting Ubiquitin–Proteasome System With Copper Complexes for Cancer Therapy. Front. Mol. Biosci. 2021, 8, 649151. [Google Scholar] [CrossRef]
- Kitiphongspattana, K.; Mathews, C.E.; Leiter, E.H.; Gaskins, H.R. Proteasome Inhibition Alters Glucose-stimulated (Pro)insulin Secretion and Turnover in Pancreatic β-Cells. J. Biol. Chem. 2005, 280, 15727–15734. [Google Scholar] [CrossRef]
- Zhou, Y.; Zheng, Z.; Wu, S.; Zhu, J. Ubiquitin-conjugating enzyme E2 for regulating autophagy in diabetic cardiomyopathy: A mini-review. J. Diabetes 2023, 16, e13511. [Google Scholar] [CrossRef] [PubMed]
- Wing, S.S. The UPS in diabetes and obesity. BMC Biochem. 2008, 9, S6. [Google Scholar] [CrossRef]
- Bugliani, M.; Liechti, R.; Cheon, H.; Suleiman, M.; Marselli, L.; Kirkpatrick, C.; Filipponi, F.; Boggi, U.; Xenarios, I.; Syed, F.; et al. Microarray analysis of isolated human islet transcriptome in type 2 diabetes and the role of the ubiquitin–proteasome system in pancreatic beta cell dysfunction. Mol. Cell. Endocrinol. 2013, 367, 1–10. [Google Scholar] [CrossRef]
- Costes, S.; Vandewalle, B.; Tourrel-Cuzin, C.; Broca, C.; Linck, N.; Bertrand, G.; Kerr-Conte, J.; Portha, B.; Pattou, F.; Bockaert, J.; et al. Degradation of cAMP-responsive element–binding protein by the ubiquitin-proteasome pathway contributes to glucotoxicity in β-cells and human pancreatic islets. Diabetes 2009, 58, 1105–1115. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, X.; Sun, R.; Ji, J.; Yang, F.; Tian, W.; Ji, W.; Huang, Q. A broad cuproptosis landscape in inflammatory bowel disease. Front. Immunol. 2022, 13, 1031539. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhu, N.; Shi, Y.; Liu, Q.; Gu, J.; Qin, L. Sensitivity of renal cell carcinoma to cuproptosis and cuproptosis related genes FDX1 combined with DLAT as an immunological and prognostic biomarker. Res. Sq. 2022. [Google Scholar] [CrossRef]
- Guo, Q.; Ma, M.; Yu, H.; Han, Y.; Zhang, D. Dexmedetomidine enables copper homeostasis in cerebral ischemia/reperfusion via ferredoxin 1. Ann. Med. 2023, 55, 2209735. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhang, X.; Shen, K.; Wang, Z.; Liu, G.; Huang, K.; He, Z.; Li, Y.; Hou, Z.; Lv, S.; et al. Cuproptosis-related genes signature and validation of differential expression and the potential targeting drugs in temporal lobe epilepsy. Front. Pharmacol. 2023, 14, 1033859. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, J.; Wang, F.; Zhang, B.; Tang, X. Comprehensive analysis of cuproptosis-related genes in immune infiltration and diagnosis in ulcerative colitis. Front. Immunol. 2022, 13, 1008146. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Drug Name | Drug Structure | Targeting Protein | Binding Affinity | Binding Sites | Combined Score | Mechanism of Action | Refs. |
|---|---|---|---|---|---|---|---|
| Methotrexate | 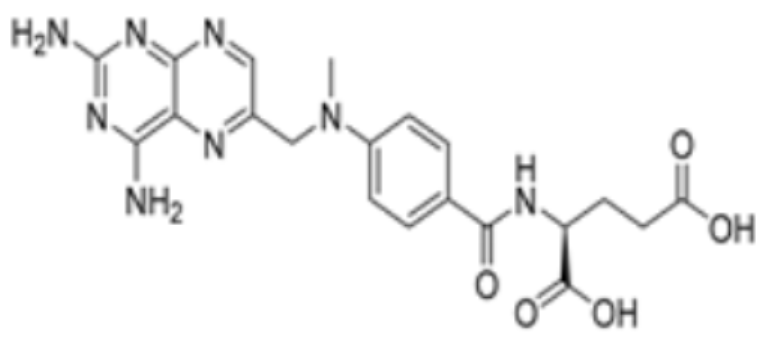 | FDX1 | −9.8 | Arg74, Thr114, His116, Tyr142, Glu169, Val 171 | / | Treats IBD, may have some effect in combating cuproptosis, due to its ability to tightly bind to FDX1. | [83] |
| Olsalazine | 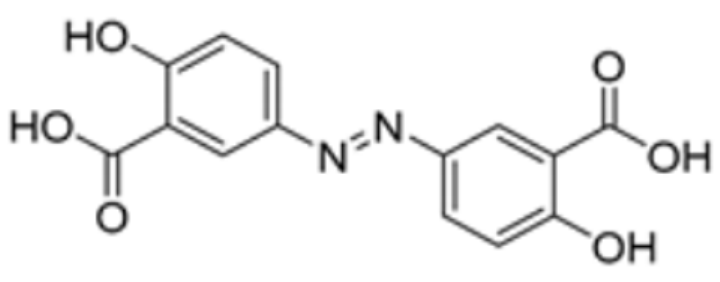 | FDX1 | −9.1 | Leu140, Tyr142, Val171, Arg74, Thr 114 | / | [83] | |
| Mitotane |  | FDX1 | −8.1 | Leu 140, Tyr 142, Val 171, Arg 74, Thr 114 | / | Found to bind to multiple amino acid sites of FDX1. | [84] |
| Nicotinamide adenine dinucleotide | 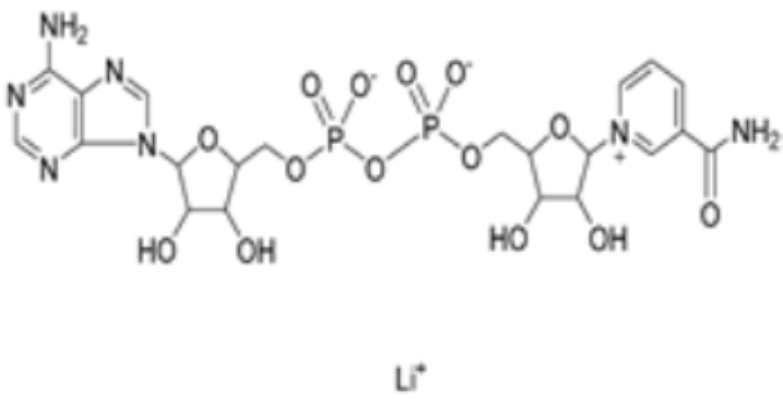 | DLAT | −8.1 | Phe48/35, His168, Leu27 | / | Found to bind to multiple amino acid sites of DLAT. | [84] |
| Radicicol |  | DLAT | −6.4 | Phe35/32,His168, Leu164, Gln167, Thr44, Lys173, Met47 | / | [84] | |
| Dihydrolipoic acid |  | DLAT | −5.3 | Phe35/48, Asn39, Gln-31 | / | [84] | |
| Dexmedetomidine |  | FDX1, SLC31A1 | / | / | / | Reduction in FDX1 and SLC31A1 levels prevents cuproptosis. | [85] |
| Chlorzoxazone |  | LIPT1, FDX1, DLD, PDHB | / | / | 301.8211 | Suppression of LIPT1, FDX1 expression and DLD to impede hippocampal neuron cuproptosis in TLE. | [86] |
| Piperlongumine | 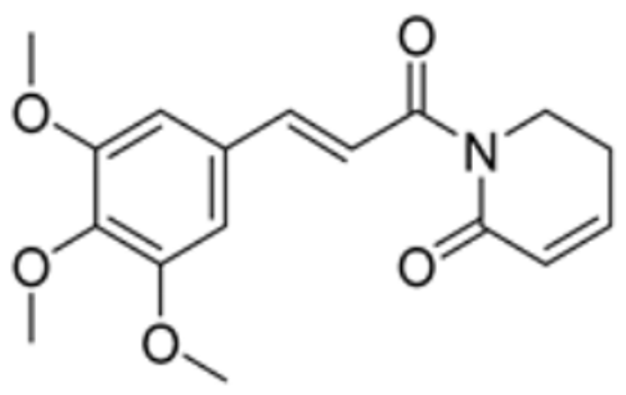 | LIPT1, PDHB | / | / | 309.921 | Suppression of LIPT1 and PDHB expression to impede hippocampal neuron cuproptosis in TLE. | [86] |
| Llatamoxef |  | PDHA1, FDX1, DBT, DLAT, LIAS | / | / | 173.328 | Potential cuproptosis-related gene-targeting drugs | [87] |
| Vitinoin | 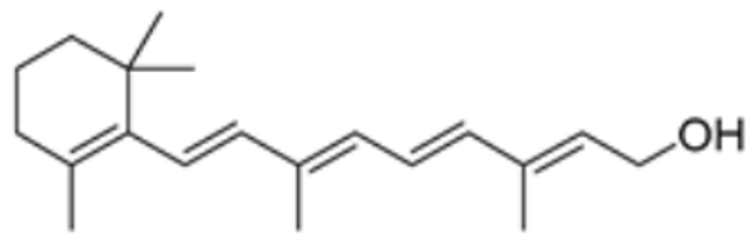 | PDHA1, DBT, PDHB, DLD | / | / | 219.439 | [87] | |
| Clomipramine | 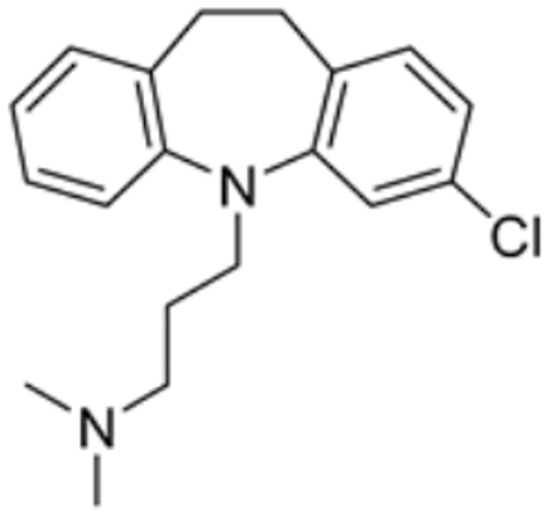 | DBT, LIAS | / | / | 446.560 | [87] | |
| Chlorzoxazone | 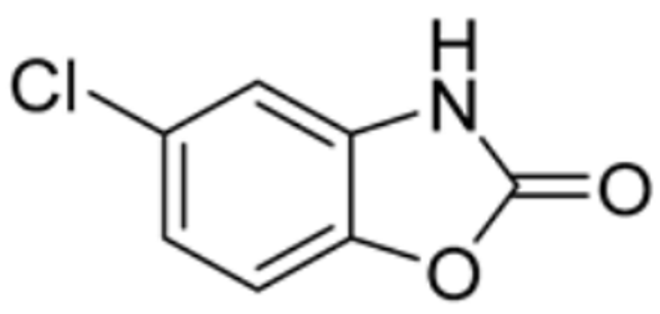 | PDHA1, FDX1, DLAT, DLD | / | / | 101.323 | [87] | |
| Glibenclamide | 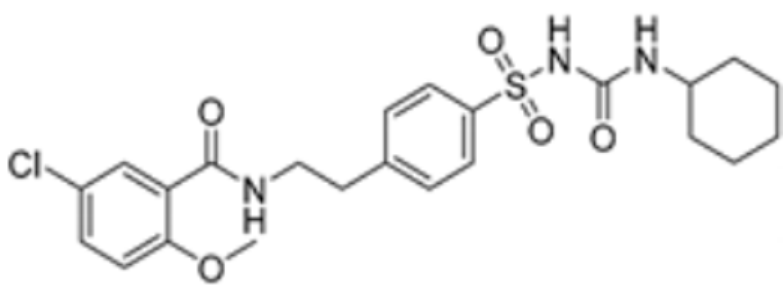 | PDHA1, FDX1, DLAT, DLD | / | / | 95.586 | [87] | |
| Pyruvic acid |  | LIAS, DLD | / | / | 270.716 | [87] | |
| Clindamycin |  | PDHA1, FDX1, DLAT, LIAS | / | / | 68.639 | [87] | |
| Medrysone |  | FDX1, DLAT, LIAS | / | / | 90.444 | [87] | |
| Flavin adenine dinucleotide | 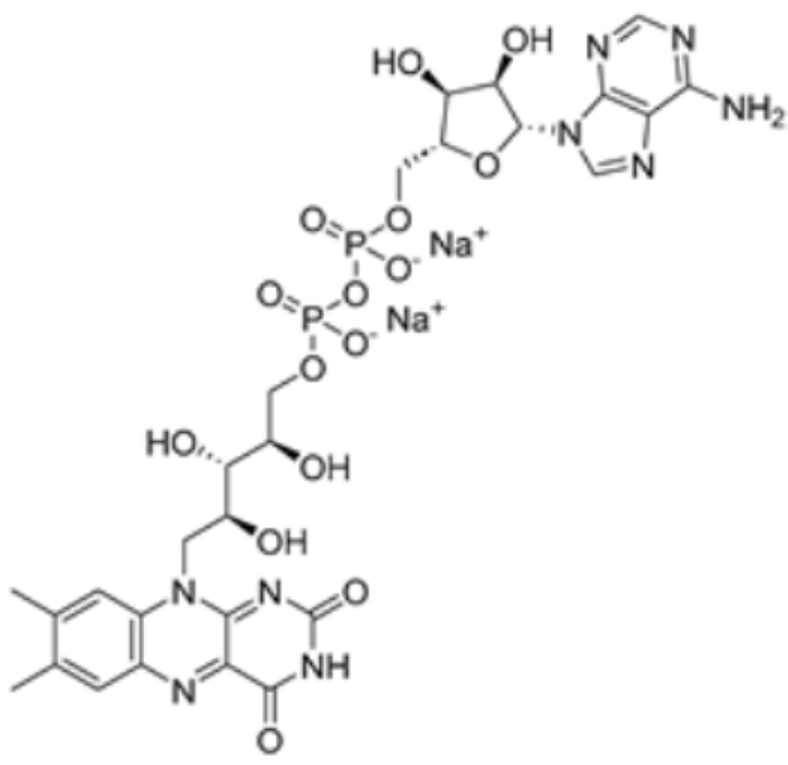 | DLD | / | / | 1549.437 | [87] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pan, Z.; Huang, L.; Gan, Y.; Xia, Y.; Yu, W. The Molecular Mechanisms of Cuproptosis and Small-Molecule Drug Design in Diabetes Mellitus. Molecules 2024, 29, 2852. https://doi.org/10.3390/molecules29122852
Pan Z, Huang L, Gan Y, Xia Y, Yu W. The Molecular Mechanisms of Cuproptosis and Small-Molecule Drug Design in Diabetes Mellitus. Molecules. 2024; 29(12):2852. https://doi.org/10.3390/molecules29122852
Chicago/Turabian StylePan, Zhaowen, Lan Huang, Yuanyuan Gan, Yan Xia, and Wei Yu. 2024. "The Molecular Mechanisms of Cuproptosis and Small-Molecule Drug Design in Diabetes Mellitus" Molecules 29, no. 12: 2852. https://doi.org/10.3390/molecules29122852
APA StylePan, Z., Huang, L., Gan, Y., Xia, Y., & Yu, W. (2024). The Molecular Mechanisms of Cuproptosis and Small-Molecule Drug Design in Diabetes Mellitus. Molecules, 29(12), 2852. https://doi.org/10.3390/molecules29122852