
Selective Oxidative Cleavage of Benzyl C–N Bond under Metal-Free Electrochemical Conditions


Abstract
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Materials and Instruments
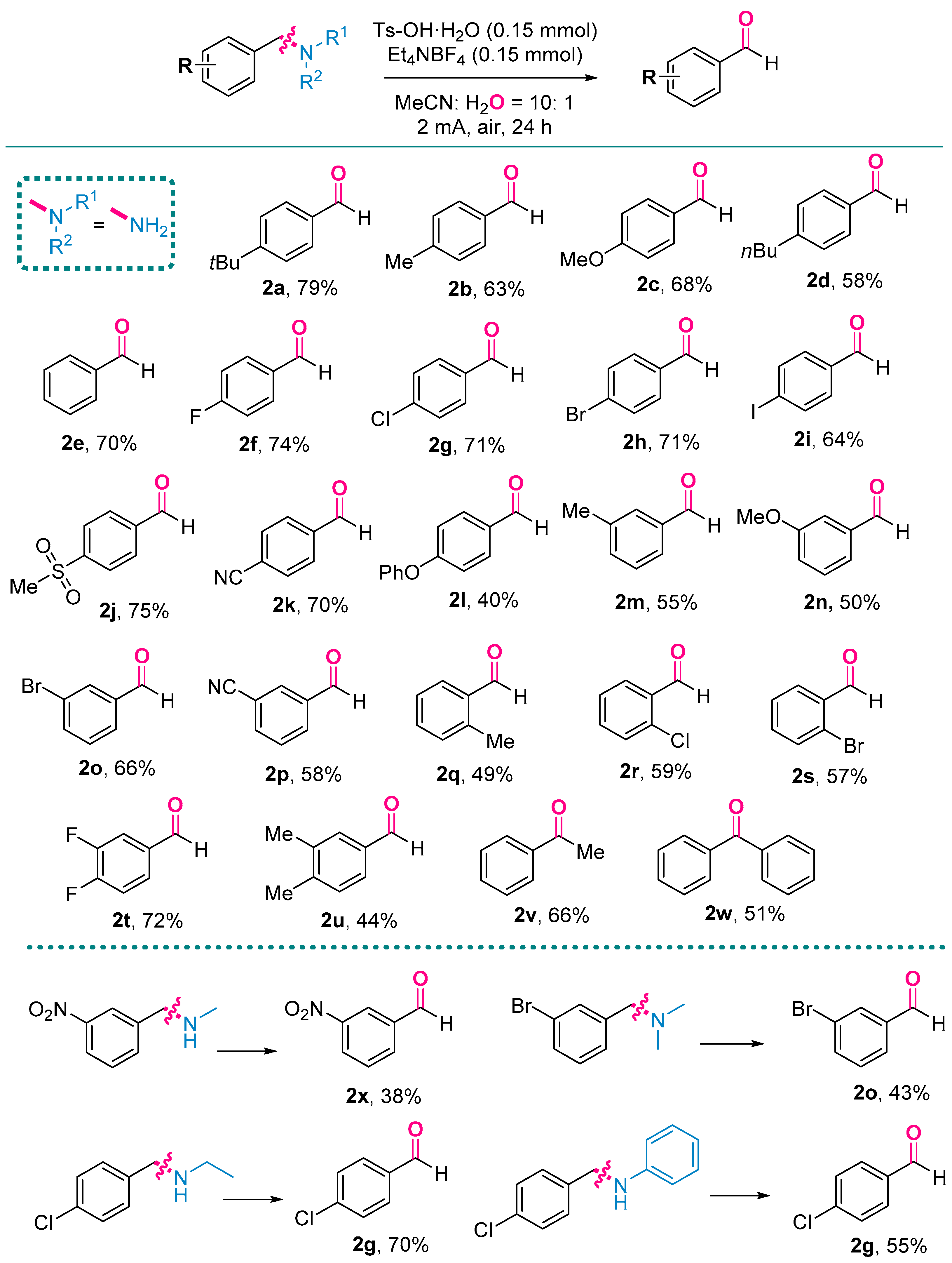
3.2. The General Procedure for Electrochemical Oxidation Cracking of C–N Bond
3.3. Characterization Data of Products
- 4-(Tert-butyl)benzaldehyde (2a) [27]. Following the general procedure with 4-(tert-butyl)benzaldehyde (48.9 mg, 0.3 mmol), 2a was obtained as a white solid (38.4 mg, 79%). This target product was purified by column chromatography on silica gel (PE:EA = 10:1). 1H NMR (400 MHz, CDCl3) δ 9.98 (s, 1H), 7.81 (d, J = 8.4 Hz, 2H), 7.55 (d, J = 8.4 Hz, 2H), 1.35 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 192.1, 158.4, 134.1, 129.7, 126.0, 35.3, 31.1.
- 4-Methylbenzaldehyde (2b) [27]. Following the general procedure with p-tolylmethanamine (36.3 mg, 0.3 mmol), 2b was obtained as a colorless oil (22.7 mg, 63%). This target product was purified by column chromatography on silica gel (PE:EA = 10:1). 1H NMR (400 MHz, CDCl3) δ 9.96 (s, 1H), 7.77 (d, J = 8.0 Hz, 2H), 7.33 (d, J = 8.0 Hz, 2H), 2.44 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 192.0, 145.5, 134.2, 129.8, 129.7, 21.9.
- 4-Methoxybenzaldehyde (2c) [27]. Following the general procedure with (4-methoxyphenyl)methanamine (41.1 mg, 0.3 mmol), 2c was obtained as a colorless oil (27.7 mg, 68%). This target product was purified by column chromatography on silica gel (PE:EA = 10:1). 1H NMR (400 MHz, CDCl3) δ 9.87 (s, 1H), 7.82 (d, J = 8.8 Hz, 2H), 6.99 (d, J = 8.8 Hz, 2H), 3.87 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 190.8, 164.6, 131.9, 129.9, 114.3, 55.5.
- 4-Nutylbenzaldehyde (2d) [27]. Following the general procedure with (4-butylphenyl)methanamine (48.9 mg, 0.3 mmol), 2d was obtained as a colorless oil (28.2 mg, 58%). This target product was purified by column chromatography on silica gel (PE:EA = 10:1). 1H NMR (400 MHz, CDCl3) δ 9.97 (s, 1H), 7.79 (d, J = 7.2 Hz, 2H), 7.33 (d, J = 7.6 Hz, 2H), 2.69 (t, J = 7.6 Hz, 2H), 1.67–1.58 (m, 2H), 1.44–1.31 (m, 2H), 0.93 (t, J = 7.2 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 192.0, 150.4, 134.4, 129.8, 129.0, 35.9, 33.2, 22.3, 13.8.
- Benzaldehyde (2e) [52]. Following the general procedure with phenylmethanamine (32.1 mg, 0.3 mmol), 2e was obtained as a colorless oil (22.3 mg, 70%). This target product was purified by column chromatography on silica gel (PE:EA = 10:1). 1H NMR (400 MHz, CDCl3) δ 10.02 (s, 1H), 7.93–7.83 (m, 2H), 7.65–7.59 (m, 1H), 7.57–7.44 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 192.4, 136.4, 134.5, 129.8, 129.0.
- 4-Fluorobenzaldehyde (2f) [52]. Following the general procedure with (4-fluorophenyl)methanamine (37.5 mg, 0.3 mmol), 2f was obtained as a colorless oil (27.5 mg, 74%). This target product was purified by column chromatography on silica gel (PE:EA = 10:1). 1H NMR (400 MHz, CDCl3) δ 9.99 (s, 1H), 7.96–7.88 (m, 2H), 7.29–7.20 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 190.5, 166.5 (d, J = 256.7 Hz), 132.8 (d, J = 9.4 Hz), 132.2 (d, J = 9.7 Hz), 116.4 (d, J = 22.3 Hz). 19F NMR (376 MHz, CDCl3) δ –102.39.
- 4-Chlorobenzaldehyde (2g) [27]. Following the general procedure with (4-chlorophenyl)methanamine (42.3 mg, 0.3 mmol), 2g was obtained as a white solid (29.8 mg, 71%). Following the general procedure with N-(4-chlorobenzyl)ethanamine (50.7 mg, 0.3 mmol), 2g was obtained as a white solid (29.4 mg, 70%). Following the general procedure with N-(4-chlorobenzyl)aniline (65.1 mg, 0.3 mmol), 2g was obtained as a white solid (23.1 mg, 55%). This target product was purified by column chromatography on silica gel (PE:EA = 10:1). 1H NMR (400 MHz, CDCl3) δ 9.98 (s, 1H), 7.82 (d, J = 8.4 Hz, 2H), 7.51 (d, J = 8.0 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 190.8, 140.9, 134.7, 130.9, 129.4.
- 4-Bromobenzaldehyde (2h) [27]. Following the general procedure with (4-bromophenyl)methanamine (55.8 mg, 0.3 mmol), 2h was obtained as a white solid (39.0 mg, 71%). This target product was purified by column chromatography on silica gel (PE:EA = 10:1). 1H NMR (400 MHz, CDCl3) δ 9.98 (s, 1H), 7.75 (d, J = 8.4 Hz, 2H), 7.69 (d, J = 8.4 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 191.0, 135.1, 132.4, 130.9, 129.8.
- 4-Iodobenzaldehyde (2i) [27]. Following the general procedure with (4-iodophenyl)methanamine (69.9 mg, 0.3 mmol), 2i was obtained as a white solid (44.5 mg, 64%). This target product was purified by column chromatography on silica gel (PE:EA = 10:1). 1H NMR (400 MHz, CDCl3) δ 9.95 (s, 1H), 7.91 (d, J = 8.0 Hz, 2H), 7.59 (d, J = 8.4 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 191.4, 138.4, 135.6, 130.8, 102.8.
- 4-(Methylsulfonyl)benzaldehyde (2j) [27]. Following the general procedure with (4-(methylsulfonyl)phenyl)methanamine (55.5 mg, 0.3 mmol), 2j was obtained as a white solid (41.4 mg, 75%). This target product was purified by column chromatography on silica gel (PE:EA = 10:1). 1H NMR (400 MHz, CDCl3) δ 10.13 (s, 1H), 8.12 (d, J = 8.4 Hz, 2H), 8.08 (d, J = 8.4 Hz, 2H), 3.09 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 190.7, 145.4, 139.7, 130.4, 128.2, 44.3.
- 4-Formylbenzonitrile (2k) [27]. Following the general procedure with 4-(aminomethyl)benzonitrile (39.6 mg, 0.3 mmol), 2k was obtained as a white solid (27.5 mg, 70%). This target product was purified by column chromatography on silica gel (PE:EA = 10:1). 1H NMR (400 MHz, CDCl3) δ 10.09 (s, 1H), 7.99 (d, J = 8.0 Hz, 2H), 7.84 (d, J = 8.0 Hz, 2H). 13C NMR (101 MHz, CDCl3) δ 190.6, 138.7, 132.9, 129.9, 117.7, 117.6.
- 4-Phenoxybenzaldehyde (2l) [27]. Following the general procedure with (4-phenoxyphenyl)methanamine (59.7 mg, 0.3 mmol), 2l was obtained as a white solid (23.8 mg, 40%). This target product was purified by column chromatography on silica gel (PE:EA = 10:1). 1H NMR (400 MHz, CDCl3) δ 9.92 (s, 1H), 7.91–7.79 (m, 2H), 7.48–7.36 (m, 2H), 7.28–7.18 (m, 1H), 7.13–7.02 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 190.7, 163.2, 155.1, 131.9, 131.2, 130.1, 124.9, 120.4, 117.5.
- 3-Methylbenzaldehyde (2m) [27]. Following the general procedure with m-tolylmethanamine (36.3 mg, 0.3 mmol), 2m was obtained as a colorless oil (19.8 mg, 55%). This target product was purified by column chromatography on silica gel (PE:EA = 10:1). 1H NMR (400 MHz, CDCl3) δ 9.98 (s, 1H), 7.71–7.62 (m, 2H), 7.47–7.36 (m, 2H), 2.43 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 192.6, 138.9, 136.4, 135.2, 130.0, 128.8, 127.2, 21.1.
- 3-Methoxybenzaldehyde (2n) [27]. Following the general procedure with (3-methoxyphenyl)methanamine (41.1 mg, 0.3 mmol), 2n was obtained as a colorless oil (20.4 mg, 50%). This target product was purified by column chromatography on silica gel (PE:EA = 10:1). 1H NMR (400 MHz, CDCl3) δ 9.97 (s, 1H), 7.50–7.42 (m, 2H), 7.41–7.36 (m, 1H), 7.21–7.14 (m, 1H), 3.86 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 192.2, 160.2, 137.8, 130.1, 123.6, 121.5, 112.1, 55.5.
- 3-Bromobenzaldehyde (2o) [27]. Following the general procedure with (3-bromophenyl)methanamine (55.5 mg, 0.3 mmol), 2o was obtained as a white solid (36.4 mg, 66%). Following the general procedure with (3–bromophenyl)methanamine 1-(3-bromophenyl)-N,N-dimethylmethanamine (64.2 mg, 0.3 mmol), 2o was obtained as a white solid (23.9 mg, 55%). This target product was purified by column chromatography on silica gel (PE:EA = 10:1). 1H NMR (400 MHz, CDCl3) δ 9.95 (s, 1H), 8.00 (s, 1H), 7.80 (d, J = 7.6 Hz, 1H), 7.74 (d, J = 7.6 Hz, 1H), 7.42 (t, J = 8.0 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 190.7, 138.0, 137.3, 132.3, 130.6, 128.3, 123.3.
- 3-Formylbenzonitrile (2p) [53]. Following the general procedure with 3-(aminomethyl)benzonitrile (39.6 mg, 0.3 mmol), 2p was obtained as a white solid (22.8 mg, 58%). This target product was purified by column chromatography on silica gel (PE:EA = 10:1). 1H NMR (400 MHz, CDCl3) δ 10.02 (s, 1H), 7.93–7.83 (m, 2H), 7.65–7.59 (m, 1H), 7.57–7.44 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 190.1, 137.2, 136.9, 133.4, 133.1, 130.2, 117.6, 113.6.
- 2-Methylbenzaldehyde (2q) [27]. Following the general procedure with o-tolylmethanamine (36.3 mg, 0.3 mmol), 2q was obtained as colorless oil (17.6 mg, 49%). This target product was purified by column chromatography on silica gel (PE:EA = 10:1). 1H NMR (400 MHz, CDCl3) δ 10.27 (d, J = 1.0 Hz, 1H), 7.83–7.76 (m, 1H), 7.51–7.44 (m, 1H), 7.36 (t, J = 7.6 Hz, 1H), 7.29–7.24 (m, 1H), 2.67 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 192.8, 140.6, 134.1, 133.6, 132.0, 131.7, 126.3, 19.5.
- 2-Chlorobenzaldehyde (2r) [27]. Following the general procedure with (2-chlorophenyl)methanamine (42.3 mg, 0.3 mmol), 2r was obtained as a white solid (24.8 mg, 59%). This target product was purified by column chromatography on silica gel (PE:EA = 10:1). 1H NMR (400 MHz, CDCl3) δ 10.49 (s, 1H), 7.92 (d, J = 8.0 Hz, 1H), 7.57–7.49 (m, 1H), 7.45 (d, J = 8.0 Hz, 1H), 7.39 (t, J = 7.2 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 189.8, 138.0, 135.1, 132.5, 130.6, 129.4, 127.3.
- 2-Bromobenzaldehyde (2s) [54]. Following the general procedure with (2-bromophenyl)methanamine (55.5 mg, 0.3 mmol), 2s was obtained as a white solid (31.6 mg, 57%). This target product was purified by column chromatography on silica gel (PE:EA = 10:1). 1H NMR (400 MHz, CDCl3) δ 10.35 (s, 1H), 7.98–7.85 (m, 1H), 7.71–7.60 (m, 1H), 7.49–7.34 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 191.8, 135.3, 133.9, 133.5, 129.8, 127.9, 127.1.
- 3,4-Difluorobenzaldehyde (2t) [27]. Following the general procedure with (3,4-difluorophenyl)methanamine (42.9 mg, 0.3 mmol), 2t was obtained as a colorless oil (30.7 mg, 72%). This target product was purified by column chromatography on silica gel (PE:EA = 10:1). 1H NMR (400 MHz, CDCl3) δ 9.93 (s, 1H), 7.75–7.64 (m, 2H), 7.34 (q, J = 8.4 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 189.4 (d, J = 2.1 Hz), 154.5 (dd, J = 259.1, 13.0 Hz), 151.0 (dd, J = 252.7, 13.3 Hz), 133.5 (dd, J = 4.0, 3.7 Hz), 127.29 (dd, J = 7.8, 3.6 Hz), 118.15 (d, J = 18.2 Hz), 117.64 (dd, J = 17.6, 2.1 Hz). 19F NMR (376 MHz, CDCl3) δ –126.89 (d, J = 20.4 Hz), –135.21 (d, J = 20.4 Hz).
- 3,4-Dimethylbenzaldehyde (2u) [27]. Following the general procedure with (3,4-dimethylphenyl)methanamine (40.5 mg, 0.3 mmol), 2u was obtained as a colorless oil (17.7 mg, 44%). This target product was purified by column chromatography on silica gel (PE:EA = 10:1). 1H NMR (400 MHz, CDCl3) δ 9.93 (s, 1H), 7.64 (s, 1H), 7.61 (d, J = 8.0 Hz, 1H), 7.28 (d, J = 8.0 Hz, 1H), 2.33 (d, J = 2.0 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 192.2, 144.3, 137.5, 134.6, 130.5, 130.2, 127.7, 20.2, 19.6.
- Acetophenone (2v) [27]. Following the general procedure with 1-phenylethan-1-amine (36.3 mg, 0.3 mmol), 2v was obtained as a colorless oil (23.8 mg, 66%). This target product was purified by column chromatography on silica gel (PE:EA = 10:1). 1H NMR (400 MHz, CDCl3) δ 7.96 (d, J = 8.0 Hz, 2H), 7.56 (t, J = 7.2 Hz, 1H), 7.46 (t, J = 7.2 Hz, 2H), 2.61 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 198.1, 137.1, 133.1, 128.5, 128.3, 26.6.
- Benzophenone (2w) [27]. Following the general procedure with diphenylmethanamine (54.9 mg, 0.3 mmol), 2w was obtained as a white solid (27.8 mg, 51%). This target product was purified by column chromatography on silica gel (PE:EA = 10:1). 1H NMR (400 MHz, CDCl3) δ 7.81 (d, J = 8.0 Hz, 4H), 7.59 (t, J = 7.6 Hz, 2H), 7.48 (t, J = 7.6 Hz, 4H). 13C NMR (101 MHz, CDCl3) δ 196.7, 137.6, 132.4, 130.0, 128.2.
- 3-Nitrobenzaldehyde (2x) [55]. Following the general procedure with N-methyl-1-(3-nitrophenyl)methanamine (49.8 mg, 0.3 mmol), 2x was obtained as a white solid (18.9 mg, 38%). This target product was purified by column chromatography on silica gel (PE:EA = 10:1). 1H NMR (400 MHz, CDCl3) δ 10.13 (s, 1H), 8.75–8.70 (m, 1H), 8.50 (d, J = 7.6 Hz, 1H), 8.24 (d, J = 7.6 Hz, 1H), 7.81–7.74 (m, 1H). 13C NMR (101 MHz, CDCl3) δ 189.7, 137.4, 134.6, 130.4, 128.6, 124.5.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kong, J.; Zhang, F.; Zhang, C.; Chang, W.; Liu, L.; Li, J. An efficient electrochemical oxidation of C(sp3)-H bond for the synthesis of arylketones. Mol. Catal. 2022, 530, 112633–112640. [Google Scholar] [CrossRef]
- Sun, Y.; Li, X.; Yang, M.; Xu, W.; Xie, J.; Ding, M. Highly selective electrocatalytic oxidation of benzyl C—H using water as safe and sustainable oxygen source. Green Chem. 2020, 22, 7543–7551. [Google Scholar] [CrossRef]
- Tang, C.; Qiu, X.; Cheng, Z.; Jiao, N. Molecular oxygen-mediated oxygenation reactions involving radicals. Chem. Soc. Rev. 2021, 50, 8067–8101. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Bai, F.; Liu, C.; Ma, X.; Gu, C.; Dai, B. Selective Electrochemical Oxygenation of Alkylarenes to Carbonyls. Org. Lett. 2021, 23, 7445–7449. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.-F.; Zhong, B.; He, J.-Y.; Zhu, C.; Xu, H. Sustainable electrochemical C(sp3)−H oxygenation using water as the oxygen source. Biorg. Med. Chem. 2022, 72, 116965–116973. [Google Scholar] [CrossRef] [PubMed]
- Mondal, J.; Trinh, Q.T.; Jana, A.; Ng, W.K.H.; Borah, P.; Hirao, H.; Zhao, Y. Size-Dependent Catalytic Activity of Palladium Nanoparticles Fabricated in Porous Organic Polymers for Alkene Hydrogenation at Room Temperature. ACS Appl. Mater. Interfaces 2016, 8, 15307–15319. [Google Scholar] [CrossRef] [PubMed]
- Pary, F.F.; Addanki Tirumala, R.T.; Andiappan, M.; Nelson, T.L. Copper(i) oxide nanoparticle-mediated C–C couplings for synthesis of polyphenylenediethynylenes: Evidence for a homogeneous catalytic pathway. Catal. Sci. Technol. 2021, 11, 2414–2421. [Google Scholar] [CrossRef]
- Albright, H.; Davis, A.J.; Gomez-Lopez, J.L.; Vonesh, H.L.; Quach, P.K.; Lambert, T.H.; Schindler, C.S. Carbonyl–Olefin Metathesis. Chem. Rev. 2021, 121, 9359–9406. [Google Scholar] [CrossRef]
- Holmes, M.; Schwartz, L.A.; Krische, M.J. Intermolecular Metal-Catalyzed Reductive Coupling of Dienes, Allenes, and Enynes with Carbonyl Compounds and Imines. Chem. Rev. 2018, 118, 6026–6052. [Google Scholar] [CrossRef]
- Milnes, K.K.; Pavelka, L.C.; Baines, K.M. Cycloaddition of carbonyl compounds and alkynes to (di)silenes and (di)germenes: Reactivity and mechanism. Chem. Soc. Rev. 2016, 45, 1019–1035. [Google Scholar] [CrossRef]
- Smith, A.M.R.; Hii, K.K. Transition Metal Catalyzed Enantioselective α-Heterofunctionalization of Carbonyl Compounds. Chem. Rev. 2010, 111, 1637–1656. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, Y.; Yasukawa, T.; Yoo, W.-J.; Kitanosono, T.; Kobayashi, S. Catalytic enantioselective aldol reactions. Chem. Soc. Rev. 2018, 47, 4388–4480. [Google Scholar] [CrossRef] [PubMed]
- Irrgang, T.; Kempe, R. Transition-Metal-Catalyzed Reductive Amination Employing Hydrogen. Chem. Rev. 2020, 120, 9583–9674. [Google Scholar] [CrossRef]
- O’Neil, L.G.; Bower, J.F. Electrophilic Aminating Agents in Total Synthesis. Angew. Chem. Int. Ed. 2021, 60, 25640–25666. [Google Scholar] [CrossRef] [PubMed]
- Philip, R.M.; Veetil Saranya, P.; Anilkumar, G. Nickel-Catalysed Amination of Arenes and Heteroarenes. Eur. J. Org. Chem. 2022, 2022, e202200184. [Google Scholar] [CrossRef]
- Bansode, A.H.; Suryavanshi, G. Metal-free hypervalent iodine/TEMPO mediated oxidation of amines and mechanistic insight into the reaction pathways. RSC Adv. 2018, 8, 32055–32062. [Google Scholar] [CrossRef] [PubMed]
- Bhukta, S.; Chatterjee, R.; Dandela, R. Metal-free oxidative radical arylation of styrene with anilines to access 2-arylacetophenones and selective oxidation of amine. J. Mol. Struct. 2023, 1279, 134995–135000. [Google Scholar] [CrossRef]
- Gaspa, S.; Porcheddu, A.; Valentoni, A.; Garroni, S.; Enzo, S.; De Luca, L. A Mechanochemical-Assisted Oxidation of Amines to Carbonyl Compounds and Nitriles. Eur. J. Org. Chem. 2017, 2017, 5519–5526. [Google Scholar] [CrossRef]
- Gong, J.-L.; Qi, X.; Wei, D.; Feng, J.-B.; Wu, X.-F. Oxidative cleavage of benzylic C–N bonds under metal-free conditions. Org. Biomol. Chem. 2014, 12, 7486–7488. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, Y.; Zhou, W.; Jiang, B.; Zhang, X.; Tian, G. Hydrogenated Cu2O\Au@CeO2 Z-scheme catalyst for photocatalytic oxidation of amines to imines. Catal. Sci. Technol. 2018, 8, 5535–5543. [Google Scholar] [CrossRef]
- Mandrekar, K.S.; Tilve, S.G. Molecular iodine mediated oxidative cleavage of the C–N bond of aryl and heteroaryl (dimethylamino)methyl groups into aldehydes. New J. Chem. 2021, 45, 4152–4155. [Google Scholar] [CrossRef]
- Nagarjun, N.; Jacob, M.; Varalakshmi, P.; Dhakshinamoorthy, A. UiO-66(Ce) metal-organic framework as a highly active and selective catalyst for the aerobic oxidation of benzyl amines. Mol. Catal. 2021, 499, 111277–111284. [Google Scholar] [CrossRef]
- Srogi, J.; Voltrova, S. Copper/Ascorbic Acid Dyad as a Catalytic System for Selective Aerobic Oxidation of Amines. Org. Lett. 2009, 11, 843–845. [Google Scholar] [CrossRef] [PubMed]
- Tashrifi, Z.; Khanaposhtani, M.M.; Larijani, B.; Mahdavi, M. Recent advances in the oxidative conversion of benzylamines. Tetrahedron 2021, 84, 131990–132008. [Google Scholar] [CrossRef]
- Togo, H.; Iinuma, M.; Moriyama, K. Simple and Practical Method for Preparation of [(Diacetoxy)iodo]arenes with Iodoarenes and m-Chloroperoxybenzoic Acid. Synlett 2012, 23, 2663–2666. [Google Scholar] [CrossRef]
- Singuru, R.; Trinh, Q.T.; Banerjee, B.; Govinda Rao, B.; Bai, L.; Bhaumik, A.; Reddy, B.M.; Hirao, H.; Mondal, J. Integrated Experimental and Theoretical Study of Shape-Controlled Catalytic Oxidative Coupling of Aromatic Amines over CuO Nanostructures. ACS Omega 2016, 1, 1121–1138. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Bai, W.; Hu, Z.; Yang, Z.; Xu, L. Visible light-induced metal-free chemoselective oxidative cleavage of benzyl C–heteroatom (N, S, Se) bonds utilizing organoboron photocatalysts. Chem. Commun. 2023, 59, 13344–13347. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Bao, W.; Tang, Z.; Guo, B.; Zhang, S.; Liu, H.; Huang, S.; Zhang, Y.; Rao, Y. Cercosporin-bioinspired selective photooxidation reactions under mild conditions. Green Chem. 2019, 21, 6073–6081. [Google Scholar] [CrossRef]
- Kumar, I.; Kumar, R.; Gupta, S.S.; Sharma, U. C70 Fullerene Catalyzed Photoinduced Aerobic Oxidation of Benzylamines to Imines and Aldehydes. J. Org. Chem. 2021, 86, 6449–6457. [Google Scholar] [CrossRef]
- Zhao, J.; Sun, H.; Lu, Y.; Li, J.; Yu, Z.; Zhu, H.; Ma, C.; Meng, Q.; Peng, X. A divergent photocatalysis strategy for selective aerobic oxidation of C(sp3)–H bonds promoted by disulfides. Green Chem. 2022, 24, 8503–8511. [Google Scholar] [CrossRef]
- Iqbal, N.; Cho, E.J. Formation of Carbonyl Compounds from Amines through Oxidative C—N Bond Cleavage using Visible Light Photocatalysis and Applications to N-PMB-Amide Deprotection. Adv. Synth. Catal. 2015, 357, 2187–2192. [Google Scholar] [CrossRef]
- Neerathilingam, N.; Bhargava Reddy, M.; Anandhan, R. Regioselective Synthesis of 2° Amides Using Visible-Light-Induced Photoredox-Catalyzed Nonaqueous Oxidative C–N Cleavage of N,N-Dibenzylanilines. J. Org. Chem. 2021, 86, 15117–15127. [Google Scholar] [CrossRef]
- Giraldi, V.; Marchini, M.; Di Giosia, M.; Gualandi, A.; Cirillo, M.; Calvaresi, M.; Ceroni, P.; Giacomini, D.; Cozzi, P.G. Acceleration of oxidation promoted by laccase irradiation with red light. New J. Chem. 2022, 46, 8662–8668. [Google Scholar] [CrossRef]
- Meng, L.; Su, J.; Zha, Z.; Zhang, L.; Zhang, Z.; Wang, Z. Direct Electrosynthesis of Ketones from Benzylic Methylenes by Electrooxidative C—H Activation. Chem. Eur. J. 2013, 19, 5542–5545. [Google Scholar] [CrossRef]
- Zhuang, W.; Zhang, J.; Ma, C.; Wright, J.S.; Zhang, X.; Ni, S.-F.; Huang, Q. Scalable Electrochemical Aerobic Oxygenation of Indoles to Isatins without Electron Transfer Mediators by Merging with an Oxygen Reduction Reaction. Org. Lett. 2022, 24, 4229–4233. [Google Scholar] [CrossRef]
- Addanki Tirumala, R.T.; Khatri, N.; Ramakrishnan, S.B.; Mohammadparast, F.; Khan, M.T.; Tan, S.; Wagle, P.; Puri, S.; McIlroy, D.N.; Kalkan, A.K.; et al. Tuning Catalytic Activity and Selectivity in Photocatalysis on Mie-Resonant Cuprous Oxide Particles: Distinguishing Electromagnetic Field Enhancement Effect from the Heating Effect. ACS Sustain. Chem. Eng. 2023, 11, 15931–15940. [Google Scholar] [CrossRef]
- Liu, C.; Liu, J.; Li, W.; Lu, H.; Zhang, Y. Recent advances in electrochemical C–H bond amination. Org. Chem. Front. 2023, 10, 5309–5330. [Google Scholar] [CrossRef]
- Xiong, P.; Xu, H.-C. Chemistry with Electrochemically Generated N-Centered Radicals. Acc. Chem. Res. 2019, 52, 3339–3350. [Google Scholar] [CrossRef]
- Yuan, Y.; Lei, A. Electrochemical Oxidative Cross-Coupling with Hydrogen Evolution Reactions. Acc. Chem. Res. 2019, 52, 3309–3324. [Google Scholar] [CrossRef]
- Dagar, N.; Sen, P.P.; Roy, S.R. Electrifying Sustainability on Transition Metal-Free Modes: An Eco-Friendly Approach for the Formation of C−N Bonds. ChemSusChem 2021, 14, 1229–1257. [Google Scholar] [CrossRef]
- Yuan, Y.; Yang, J.; Lei, A. Recent advances in electrochemical oxidative cross-coupling with hydrogen evolution involving radicals. Chem. Soc. Rev. 2021, 50, 10058–10086. [Google Scholar] [CrossRef]
- Yan, M.; Kawamata, Y.; Baran, P.S. Synthetic Organic Electrochemical Methods Since 2000: On the Verge of a Renaissance. Chem. Rev. 2017, 117, 13230–13319. [Google Scholar] [CrossRef]
- Guo, S.; Wu, Y.; Wang, C.; Gao, Y.; Li, M.; Zhang, B.; Liu, C. Electrocatalytic hydrogenation of quinolines with water over a fluorine-modified cobalt catalyst. Nat. Commun. 2022, 13, 5297–5307. [Google Scholar] [CrossRef]
- Qian, P.; Zha, Z.; Wang, Z. Recent Advances in C−H Functionalization with Electrochemistry and Various Iodine-Containing Reagents. ChemElectroChem 2020, 7, 2527–2544. [Google Scholar] [CrossRef]
- Bai, F.; Wang, N.; Bai, Y.; Ma, X.; Gu, C.; Dai, B.; Chen, J. NHPI-Mediated Electrochemical α-Oxygenation of Amides to Benzimides. J. Org. Chem. 2023, 88, 2985–2998. [Google Scholar] [CrossRef]
- Li, X.; Huang, J.; Xu, L.; Liu, J.; Wei, Y. Electrochemical Oxidative Dehydrogenative Coupling of Sulfoximines to Construct N-sulfenyl and N-phosphinyl Sulfoximines. Adv. Synth. Catal. 2023, 365, 4647–4653. [Google Scholar] [CrossRef]
- Liu, M.; Xu, L.; Wei, Y. Electrochemical utilization of methanol and methanol-d4 as a C1 source to access (deuterated) 2,3-dihydroquinazolin-4(1H)-one. Chin. Chem. Lett. 2022, 33, 1559–1562. [Google Scholar] [CrossRef]
- Tian, Q.; Zhang, J.; Xu, L.; Wei, Y. Synthesis of quinazolin-4(3H)-ones via electrochemical decarboxylative cyclization of α-keto acids with 2-aminobenzamides. Mol. Catal. 2021, 500, 111345–111349. [Google Scholar] [CrossRef]
- Huang, J.; Li, X.; Wei, Y.; Lei, Z.; Xu, L. Organoboron/iodide-catalyzed photoredox N-functionalization of NH-sulfoximines/sulfonimidamides. Chem. Commun. 2023, 59, 13643–13646. [Google Scholar] [CrossRef]
- Liu, G.; Liu, S.; Li, Z.; Chen, H.; Li, J.; Zhang, Y.; Shen, G.; Yang, B.; Hu, X.; Huang, X. Metal- and oxidant-free electrochemically promoted oxidative coupling of amines. RSC Adv. 2022, 12, 118–122. [Google Scholar] [CrossRef]
- Yu, W.H.; Zhou, C.H.; Tong, D.S.; Xu, T.N. Aerobic oxidation of 4-tert-butyltoluene over cobalt and manganese supported hexagonal mesoporous silicas as heterogeneous catalysts. J. Mol. Catal. A Chem. 2012, 365, 194–202. [Google Scholar] [CrossRef]
- Fan, Q.; Liu, D.; Xie, Z.; Le, Z.; Zhu, H.; Song, X. Visible-Light Photocatalytic Highly Selective Oxidation of Alcohols into Carbonyl Compounds by CsPbBr3 Perovskite. J. Org. Chem. 2023, 88, 14559–14570. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Snieckus, V. A Practical in situ Generation of the Schwartz Reagent. Reduction of Tertiary Amides to Aldehydes and Hydrozirconation. Org. Lett. 2013, 16, 390–393. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-X.; He, J.-T.; Wu, M.-C.; Liu, Z.-L.; Tang, K.; Xia, P.-J.; Chen, K.; Xiang, H.-Y.; Chen, X.-Q.; Yang, H. Photochemical Organocatalytic Aerobic Cleavage of C═C Bonds Enabled by Charge-Transfer Complex Formation. Org. Lett. 2022, 24, 3920–3925. [Google Scholar] [CrossRef]
- Fomenkov, D.I.; Budekhin, R.A.; Vil’, V.A.; Terent’ev, A.O. The Ozone and Hydroperoxide Teamwork: Synthesis of Unsymmetrical Geminal Bisperoxides from Alkenes. Org. Lett. 2023, 25, 4672–4676. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
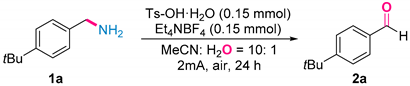 | ||
|---|---|---|
| Entry | Variations from the Standard Conditions | Yield |
| 1 | None | 79% |
| 2 | without current | N.R. |
| 3 | without TsOH·H2O | 35% |
| 4 | TFA instead of TsOH·H2O | 43% |
| 5 | AcOH instead of TsOH·H2O | 40% |
| 6 | PA instead of TsOH·H2O | 39% |
| 7 | C/C instead of Pt/Pt | 44% |
| 8 | 4 mA instead of 2 mA | 77% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, J.; Li, X.; Liu, P.; Wei, Y.; Liu, S.; Ma, X. Selective Oxidative Cleavage of Benzyl C–N Bond under Metal-Free Electrochemical Conditions. Molecules 2024, 29, 2851. https://doi.org/10.3390/molecules29122851
Huang J, Li X, Liu P, Wei Y, Liu S, Ma X. Selective Oxidative Cleavage of Benzyl C–N Bond under Metal-Free Electrochemical Conditions. Molecules. 2024; 29(12):2851. https://doi.org/10.3390/molecules29122851
Chicago/Turabian StyleHuang, Jiawei, Xiaoman Li, Ping Liu, Yu Wei, Shuai Liu, and Xiaowei Ma. 2024. "Selective Oxidative Cleavage of Benzyl C–N Bond under Metal-Free Electrochemical Conditions" Molecules 29, no. 12: 2851. https://doi.org/10.3390/molecules29122851
APA StyleHuang, J., Li, X., Liu, P., Wei, Y., Liu, S., & Ma, X. (2024). Selective Oxidative Cleavage of Benzyl C–N Bond under Metal-Free Electrochemical Conditions. Molecules, 29(12), 2851. https://doi.org/10.3390/molecules29122851