
First-Principles Investigation of Phosphorus-Doped Graphitic Carbon Nitride as Anchoring Material for the Lithium-Sulfur Battery

 , and
, and
Abstract

1. Introduction
2. Results and Discussion
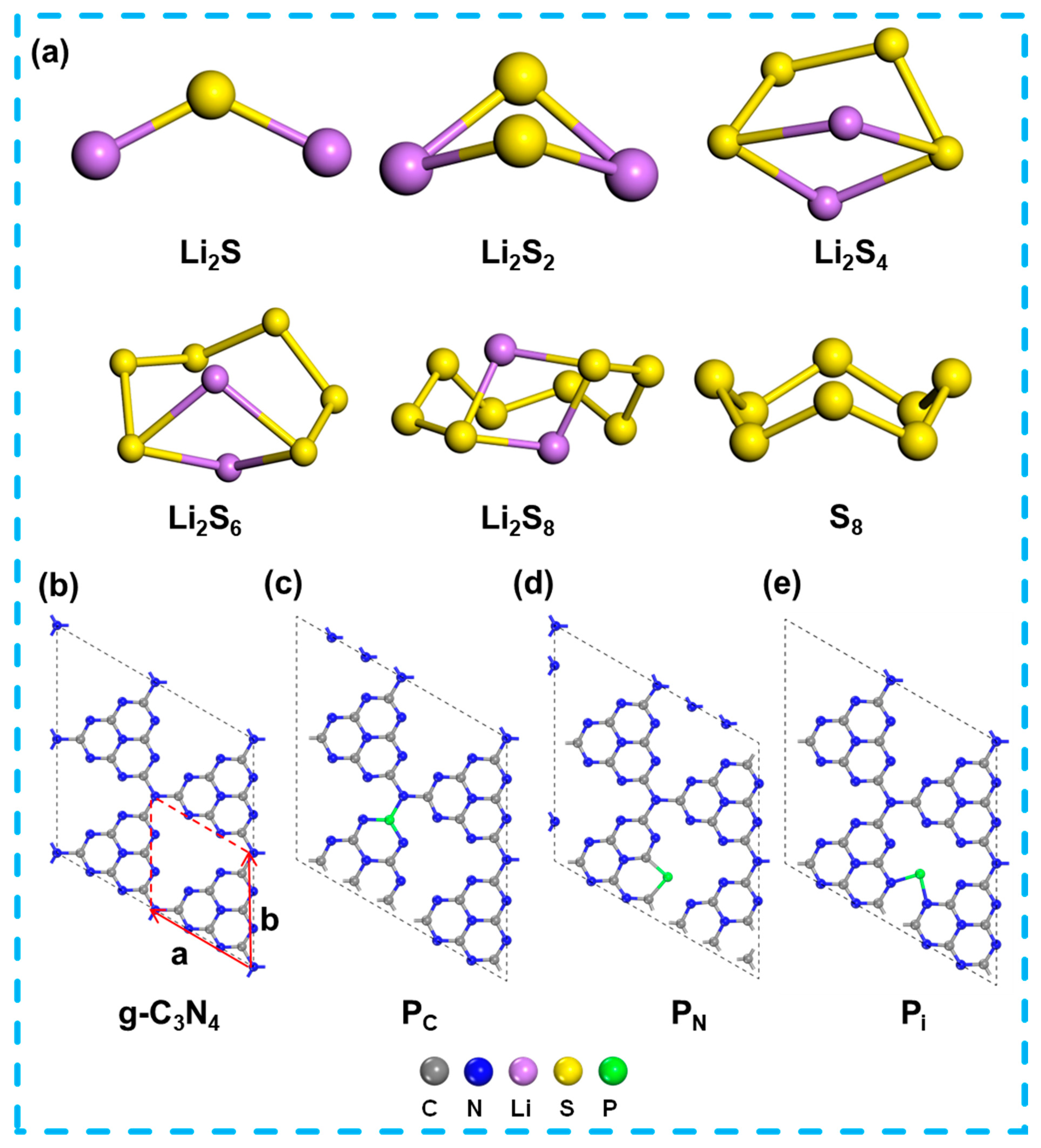
2.1. Structures for S8, LiPSs Species, g-C3N4 and P-Doping g-C3N4
2.2. Formation Energies of P-Doping g-C3N4
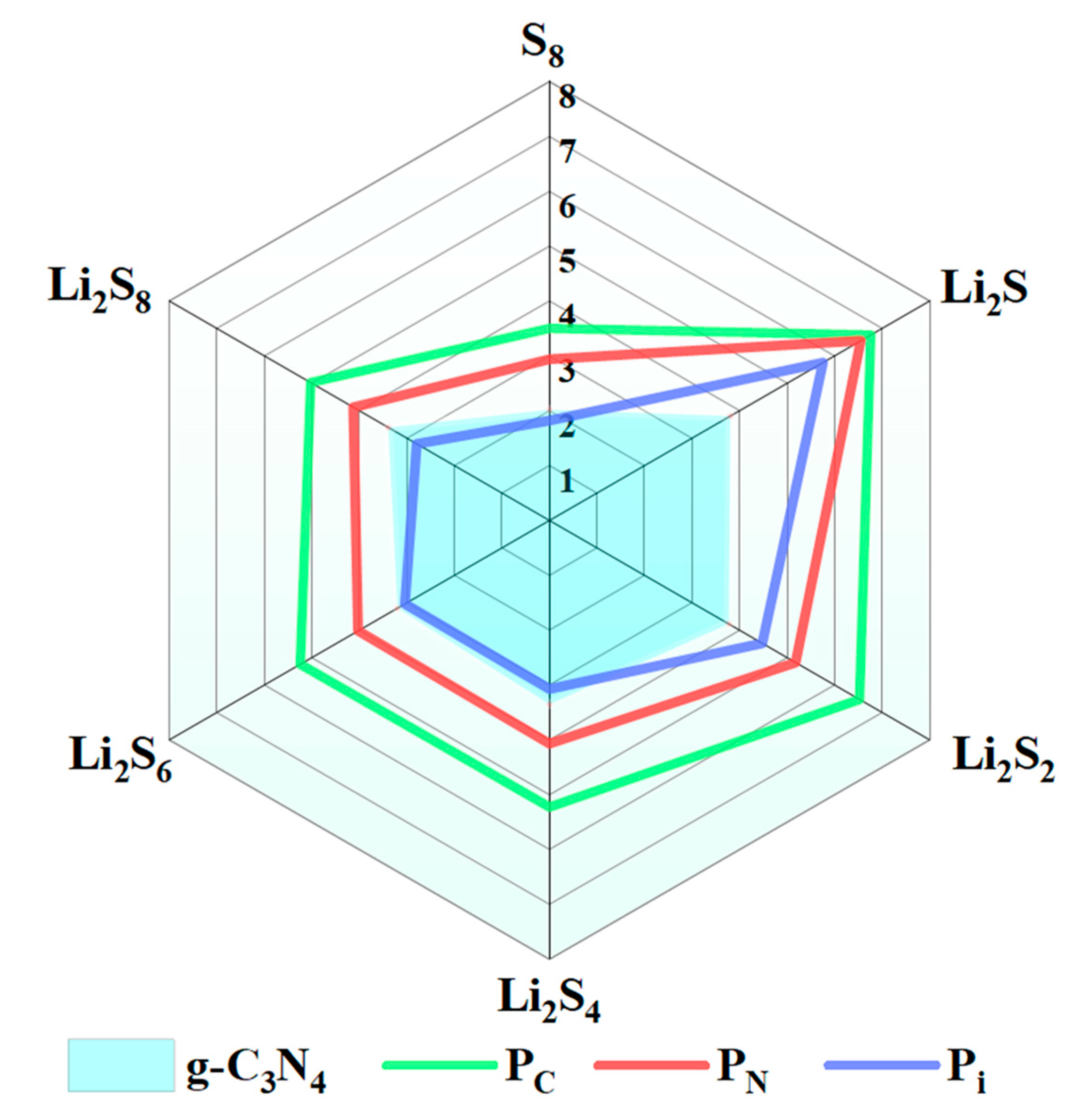
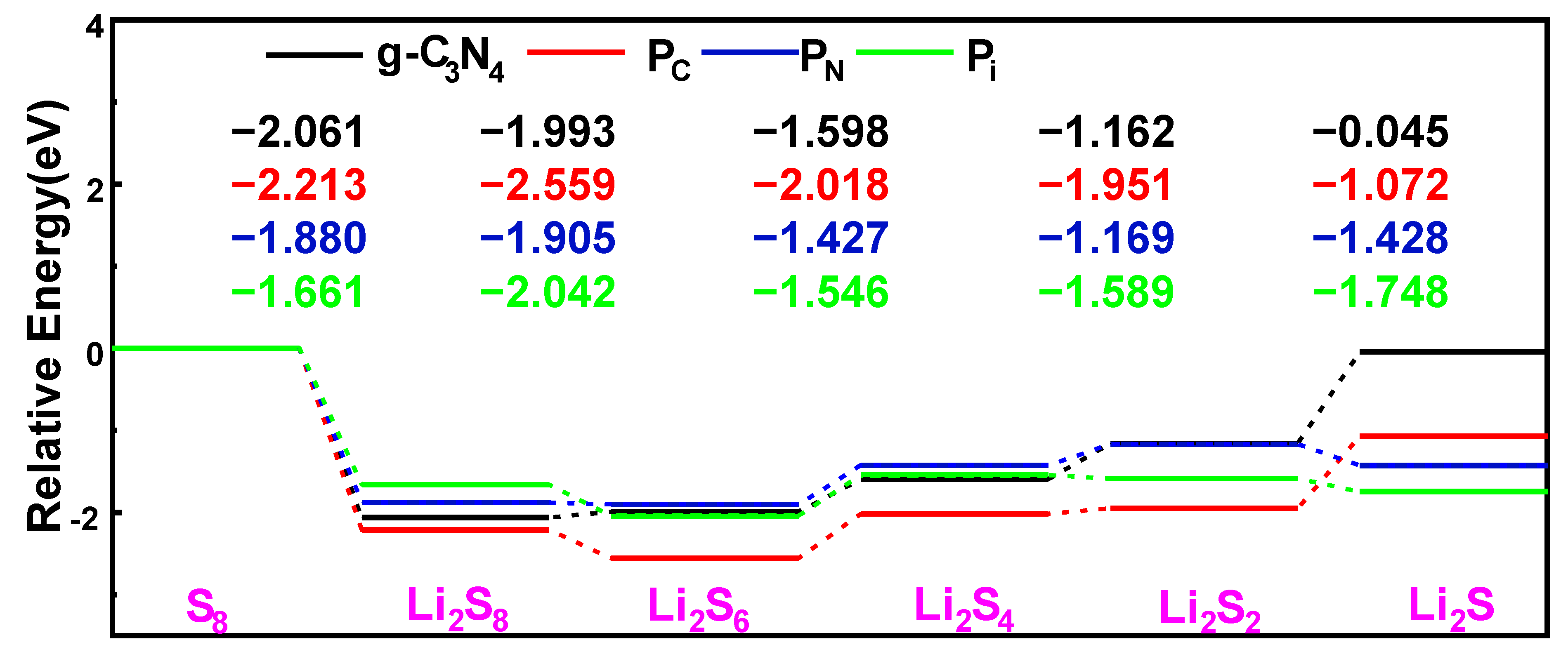
2.3. Anchoring Structure and Adsorption Energy
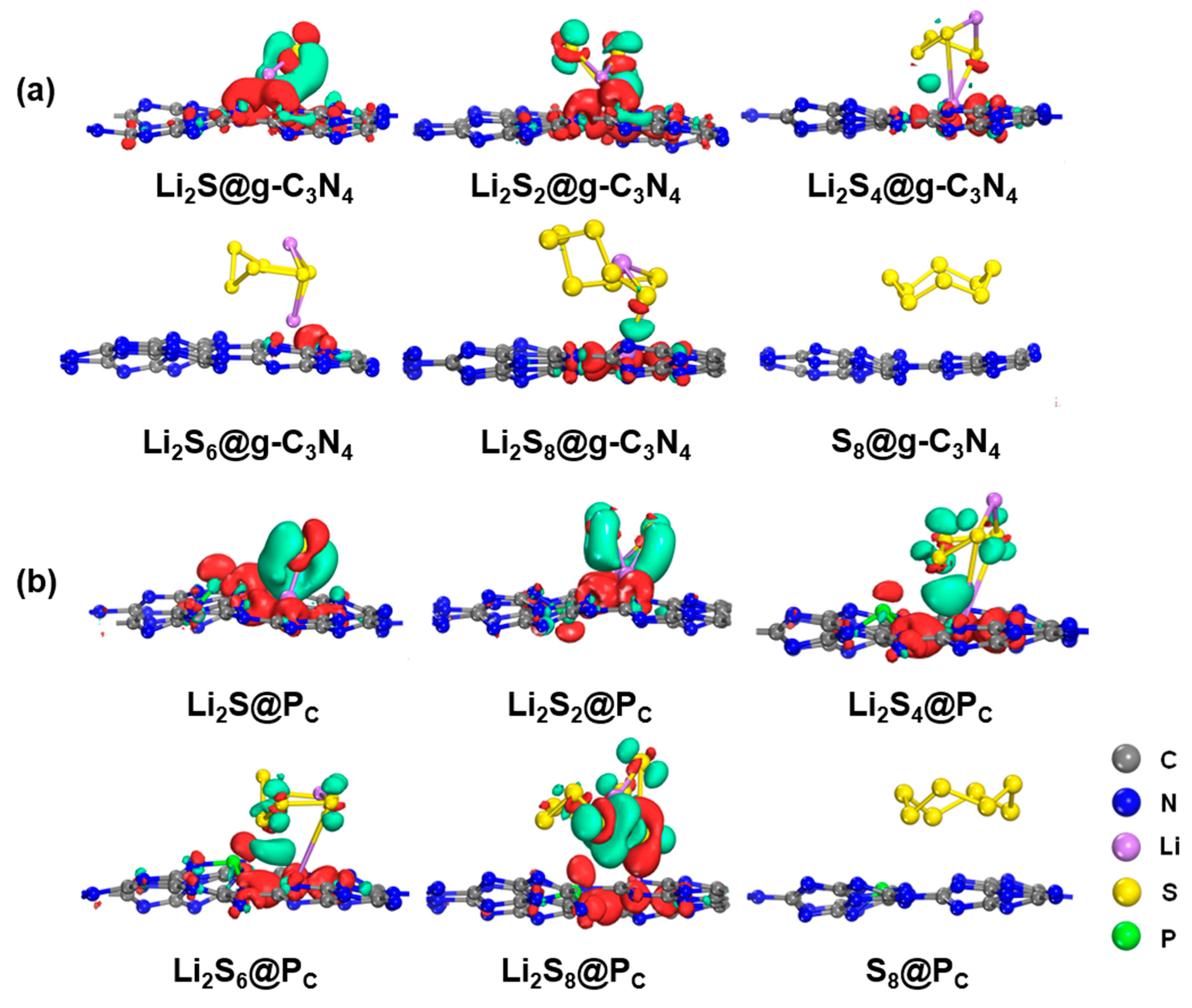
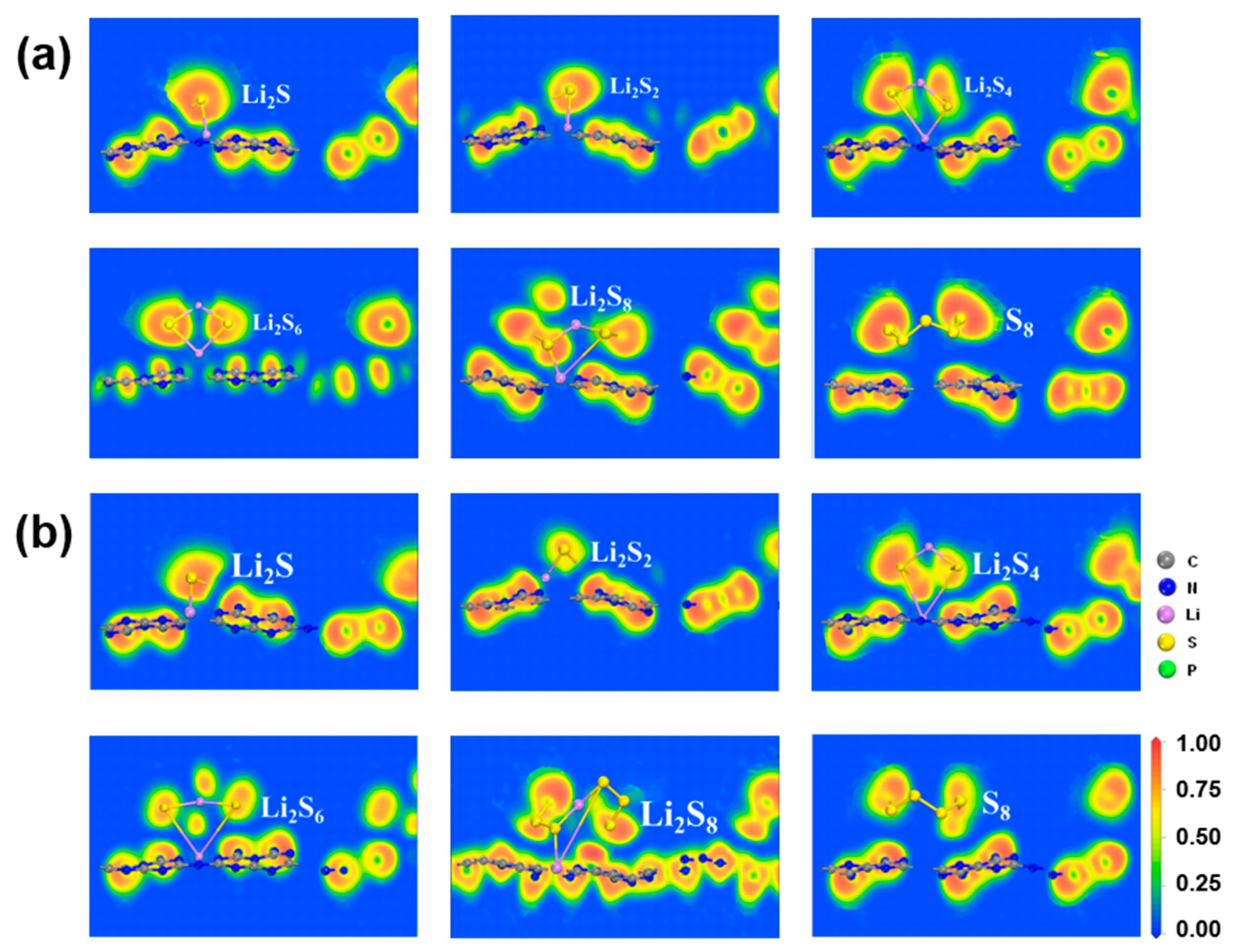
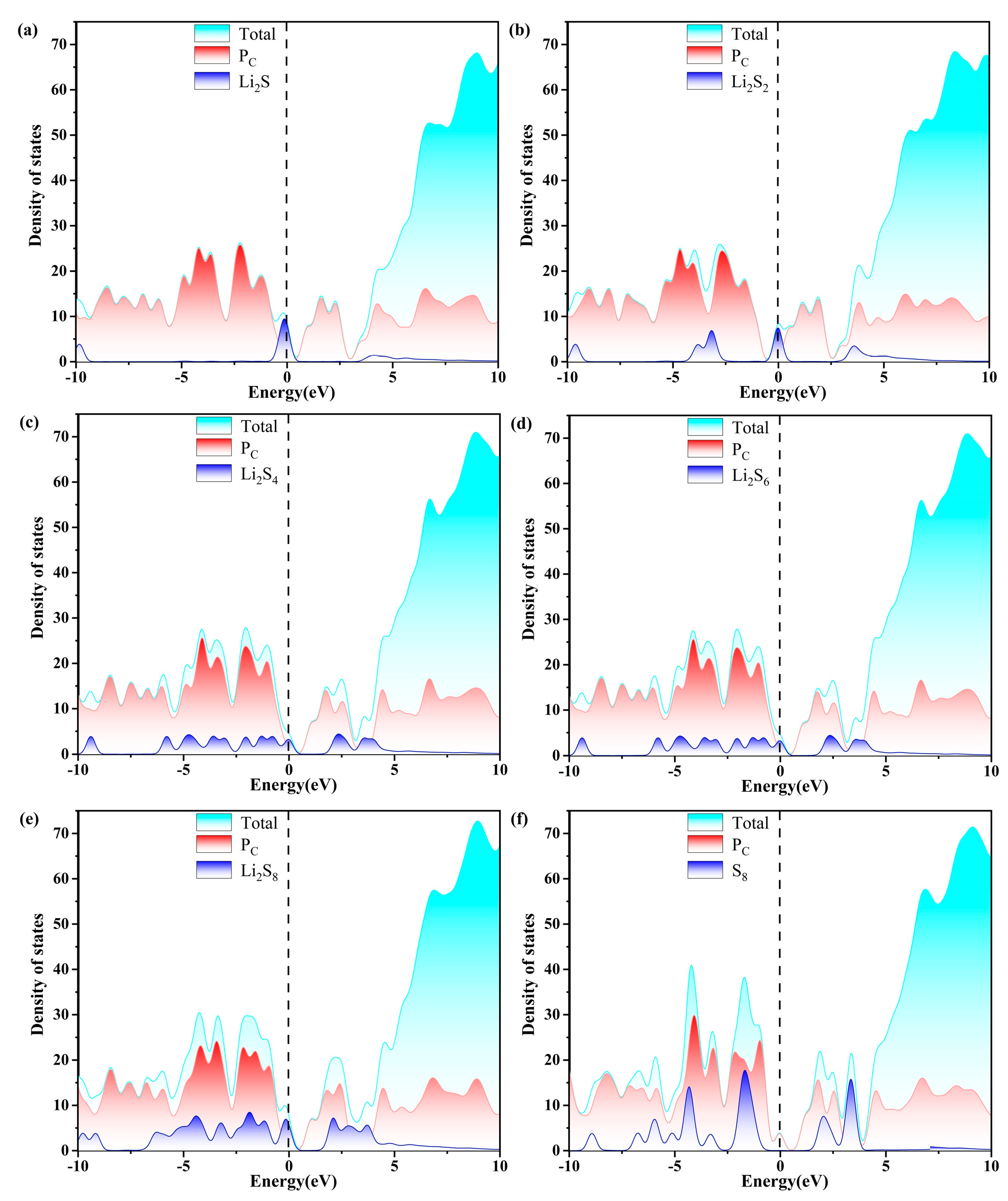
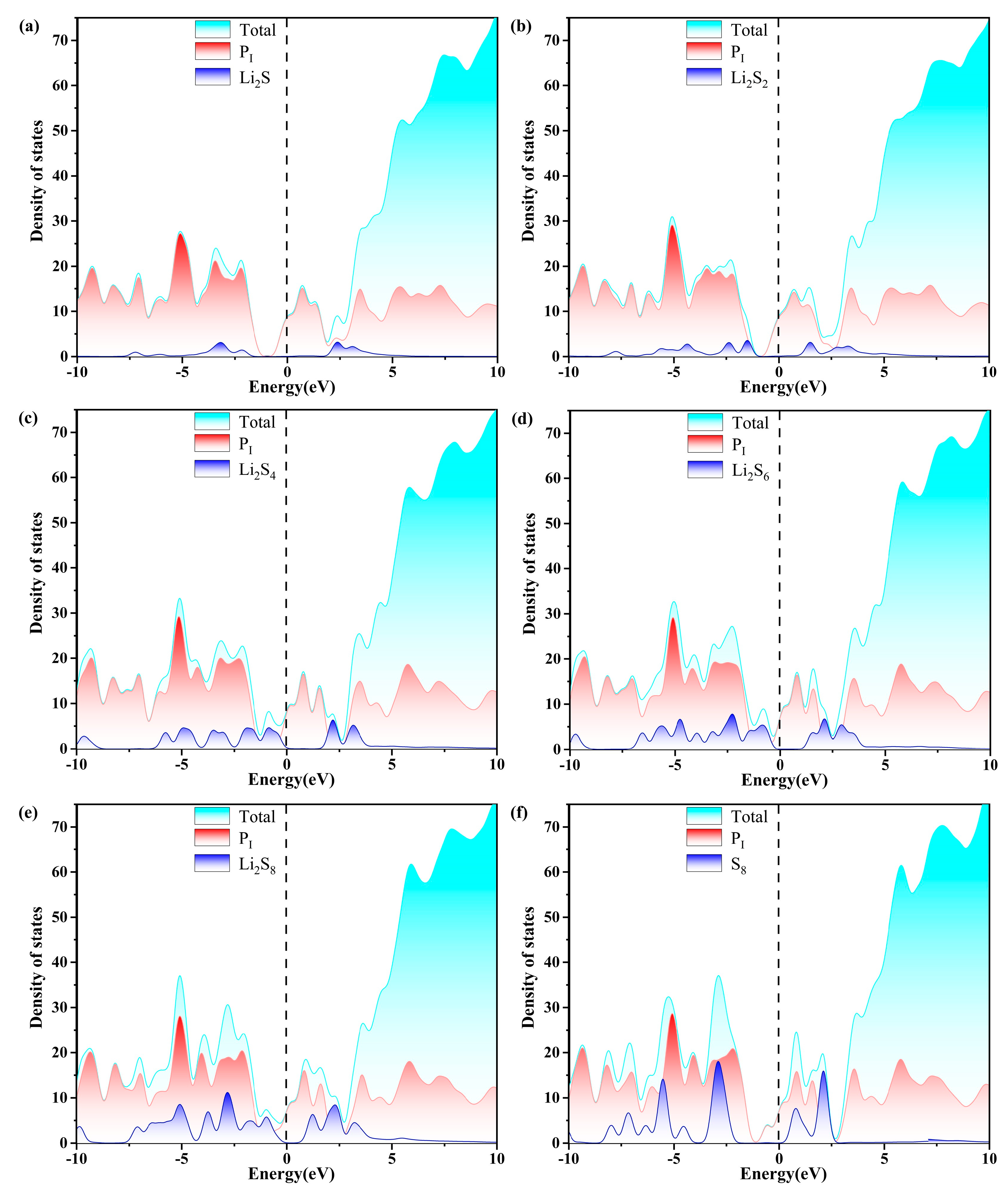
2.4. Charge Transfer and Electronic Properties
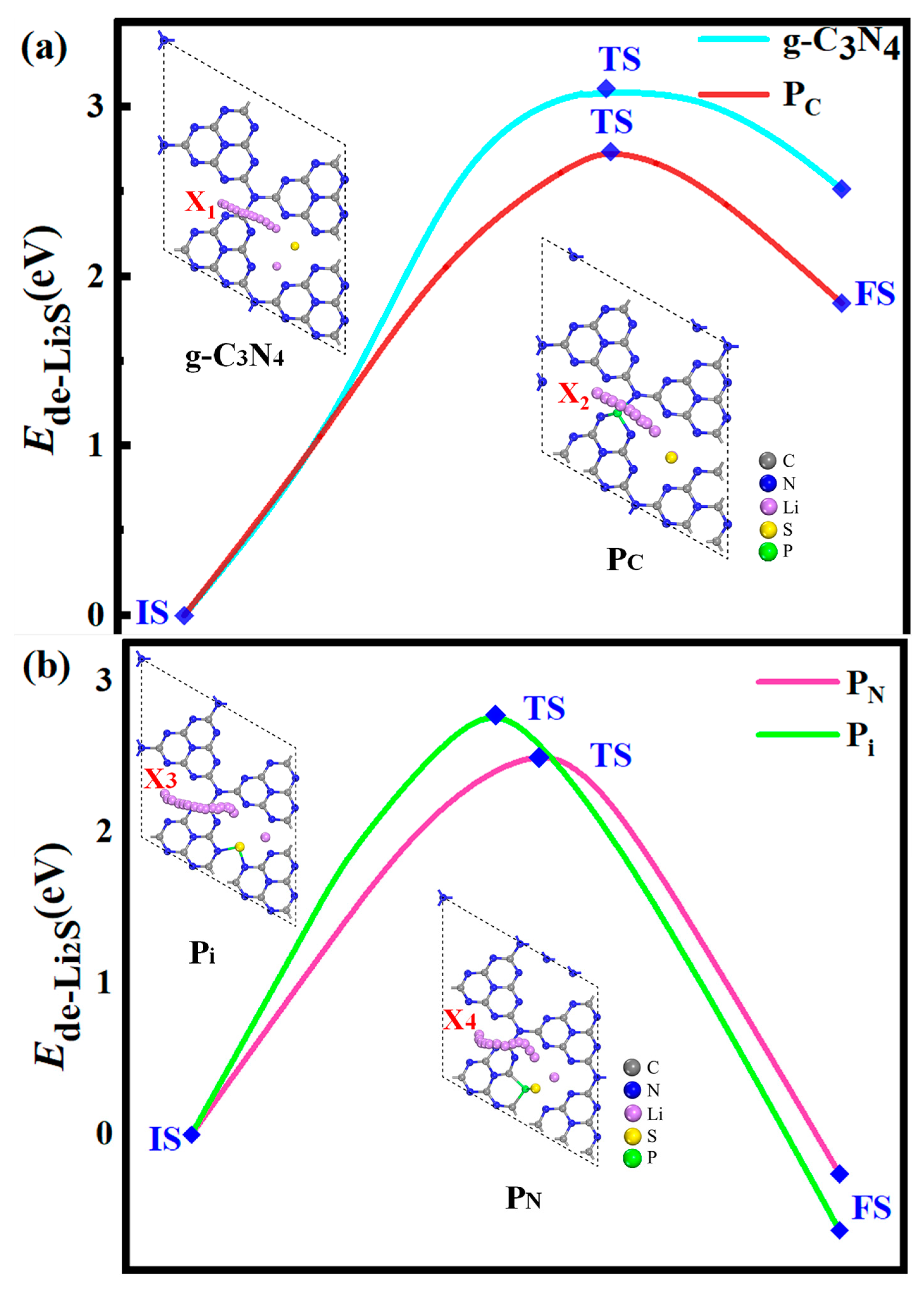
2.5. Energy Profiles and Decomposition of Li2S on the Surface of the Monolayer Substrates
3. Computational Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DFT | Density functional theory |
| GGA | Generalized gradient approximation |
| LDA-CAPZ | Local density approximation of Ceperley and Alder by Perdew and Zunger |
| TS | Tkatchenko-Scheffler |
| vdW | van der Waals |
| BFGS | Broyden–Fletcher–Goldfarb–Shanno |
| PDOS | Partial density of states |
| ELF | Electron localization function |
| LST/QST | Linear simultaneous transit/quadratic simultaneous transit |
References
- Xu, R.; Lu, J.; Amine, K. Progress in Mechanistic Understanding and Characterization Techniques of Li-S Batteries. Adv. Energy Mater. 2015, 5, 1500408. [Google Scholar] [CrossRef]
- Rosenman, A.; Markevich, E.; Salitra, G.; Aurbach, D.; Garsuch, A.; Chesneau, F. Review on Li-Sulfur Battery Systems: An Integral Perspective. Adv. Energy Mater. 2015, 5, 1500212. [Google Scholar] [CrossRef]
- Wild, M.; O’neill, L.; Zhang, T.; Purkayastha, R.; Minton, G.; Marinescu, M.; Offer, G. Lithium sulfur batteries, a mechanistic review. Energy Environ. Sci. 2015, 8, 3477–3494. [Google Scholar] [CrossRef]
- Ogoke, O.; Wu, G.; Wang, X.; Casimir, A.; Ma, L.; Wu, T.; Lu, J. Effective strategies for stabilizing sulfur for advanced lithium-sulfur batteries. J. Mater. Chem. A 2017, 5, 448–469. [Google Scholar] [CrossRef]
- Wang, T.; Dong, Q.; Li, C.; Wei, Z. Sulfur Cathode Electrocatalysis in Lithium-Sulfur Batteries: A Comprehensive Understanding. Acta Phys. Chim. Sin. 2024, 40, 2303061–2303073. [Google Scholar] [CrossRef]
- Fang, R.; Chen, K.; Yin, L.; Sun, Z.; Li, F.; Cheng, H. The Regulating Role of Carbon Nanotubes and Graphene in Lithium-Ion and Lithium–Sulfur Batteries. Adv. Mater. 2019, 31, 1800863–1800884. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zheng, Y.; Sun, Y.; Pan, H. Modulation of Potential-Limiting Steps in Lithium-Sulfur Batteries by Catalyst Synergy. Small 2024, 2309582–2309593. [Google Scholar] [CrossRef] [PubMed]
- Pei, F.; Lin, L.L.; Fu, A.; Mo, S.G.; Ou, D.H.; Fang, X.L.; Zheng, N.F. A Two-Dimensional Porous Carbon-Modified Separator for High-Energy-Density Li-S Batteries. Joule 2018, 2, 323–336. [Google Scholar] [CrossRef]
- Qin, X.Y.; Wu, J.X.; Xu, Z.L.; Chong, W.G.; Huang, J.Q.; Liang, G.M.; Li, B.H.; Kang, F.Y.; Kim, J.K. Electrosprayed multiscale porous carbon microspheres as sulfur hosts for long-life lithium-sulfur batteries. Carbon 2019, 141, 16–24. [Google Scholar] [CrossRef]
- Zhao, C.; Xu, G.L.; Yu, Z.; Zhang, L.C.; Hwang, I.; Mo, Y.X.; Ren, Y.X.; Cheng, L.; Sun, C.J.; Ren, Y.; et al. A high-energy and long-cycling lithium-sulfur pouch cell via a macroporous catalytic cathode with double-end binding sites. Nat. Nanotechnol. 2021, 16, 224. [Google Scholar] [CrossRef]
- Zheng, B.; Lin, X.; Zhang, X.; Wu, D.; Matyjaszewski, K. Emerging functional porous polymeric and carbonaceous materials for environmental treatment and energy storage. Adv. Funct. Mater. 2020, 30, 1907006–1907053. [Google Scholar] [CrossRef]
- Li, H.; Liu, D.; Zhu, X.X.; Qu, D.Y.; Xie, Z.Z.; Li, J.S.; Tang, H.L.; Zheng, D.; Qu, D.Y. Integrated 3D electrodes based on metal-nitrogen-doped graphitic ordered mesoporous carbon and carbon paper for high-loading lithium-sulfur batteries. Nano Energy 2020, 73, 104763–104774. [Google Scholar] [CrossRef]
- He, M.X.; Li, X.; Li, W.H.; Zheng, M.T.; Wang, J.J.; Ma, S.B.; Ma, Y.L.; Yin, G.P.; Zuo, P.J.; Sun, X.L. Immobilization and kinetic promotion of polysulfides by molybdenum carbide in lithium-sulfur batteries. Chem. Eng. J. 2021, 411, 128563–128566. [Google Scholar] [CrossRef]
- Jo, S.C.; Hong, J.W.; Choi, I.H.; Kim, M.J.; Kim, B.G.; Lee, Y.J.; Choi, H.Y.; Kim, D.; Kim, T.; Baeg, K.J.; et al. Multimodal Capturing of Polysulfides by Phosphorus-Doped Carbon Composites for Flexible High-Energy-Density Lithium-Sulfur Batteries. Small 2022, 18, 2270106–2270118. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Chao, D.L.; Deng, S.J.; Zhan, J.Y.; Fang, R.Y.; Xia, Y.; Wang, Y.D.; Wang, X.L.; Xia, X.H.; Tu, J.P. Confining Sulfur in Integrated Composite Scaffold with Highly Porous Carbon Fibers/Vanadium Nitride Arrays for High-Performance Lithium-Sulfur Batteries. Adv. Funct. Mater. 2018, 28, 1706391. [Google Scholar] [CrossRef]
- Park, J.; Yu, B.; Park, J.; Choi, J.; Kim, C.; Sung, Y.E.; Goodenough, J. Tungsten Disulfide Catalysts Supported on a Carbon Cloth Interlayer for High-Performance Li-S Battery. Adv. Energy Mater. 2017, 7, 1602567–1602572. [Google Scholar] [CrossRef]
- Wu, S.Y.; Li, X.; Zhang, Y.Z.; Guan, Q.H.; Wang, J.; Shen, C.Y.; Lin, H.Z.; Wang, J.T.; Wang, Y.L.; Zhan, L.; et al. Interface engineering of MXene-based heterostructures for lithium-sulfur batteries. Nano Res. 2023, 16, 9158–9178. [Google Scholar] [CrossRef]
- He, Y.B.; Chang, Z.; Wu, S.C.; Qiao, Y.; Bai, S.Y.; Jiang, K.Z.; He, P.; Zhou, H.S. Simultaneously Inhibiting Lithium Dendrites Growth and Polysulfides Shuttle by a Flexible MOF-Based Membrane in Li-S Batteries. Adv. Energy Mater. 2018, 8, 1802130–1802142. [Google Scholar] [CrossRef]
- Haseeb, H.H.; Li, Y.; Ayub, S.; Fang, Q.L.; Yu, L.J.; Xu, K.W.; Ma, F. Defective Phosphorene as a Promising Anchoring Material for Lithium-Sulfur Batteries. J. Phys. Chem. C 2020, 124, 2739–2746. [Google Scholar] [CrossRef]
- Zheng, Y.P.; Li, H.H.; Yuan, H.Y.; Fan, H.H.; Li, W.L.; Zhang, J.P. Understanding the anchoring effect of Graphene, BN, CN and CN monolayers for lithium-polysulfides in Li-S batteries. Appl. Surf. Sci. 2018, 434, 596–603. [Google Scholar] [CrossRef]
- Du, M.J.; Tian, X.Q.; Ran, R.; Zhou, W.; Liao, K.M.; Shao, Z.P. Tuning Nitrogen in Graphitic Carbon Nitride Enabling Enhanced Performance for Polysulfide Confinement in Li-S Batteries. Energy Fuels 2020, 34, 11557–11564. [Google Scholar] [CrossRef]
- Li, D.S.; Liu, J.; Wang, W.J.; Li, S.M.; Yang, G.L.; Wang, P.; Zhu, K.X.; Li, Z.J. Synthesis of porous N deficient graphitic carbon nitride and utilization in lithium-sulfur battery. Appl. Surf. Sci. 2021, 569, 151058. [Google Scholar] [CrossRef]
- Tong, Z.M.; Huang, L.; Liu, H.P.; Lei, W.; Zhang, H.J.; Zhang, S.W.; Jia, Q.L. Defective Graphitic Carbon Nitride Modified Separators with Efficient Polysulfide Traps and Catalytic Sites for Fast and Reliable Sulfur Electrochemistry. Adv. Funct. Mater. 2021, 31, 2010455. [Google Scholar] [CrossRef]
- Sun, W.H.; Song, Z.H.; Feng, Z.X.; Huang, Y.Q.; Xu, Z.J.; Lu, Y.C.; Zou, Q.L. Carbon-Nitride-Based Materials for Advanced Lithium-Sulfur Batteries. Nano-Micro Lett. 2022, 14, 222–243. [Google Scholar] [CrossRef] [PubMed]
- Fina, F.; Callear, S.K.; Carins, G.M.; Irvine, J.T.S. Structural Investigation of Graphitic Carbon Nitride via XRD and Neutron Diffraction. Chem. Mater. 2015, 27, 2612–2618. [Google Scholar] [CrossRef]
- Jun, Y.S.; Hong, W.H.; Antonietti, M.; Thomas, A. Mesoporous, 2D Hexagonal Carbon Nitride and Titanium Nitride/Carbon Composites. Adv. Mater. 2009, 21, 4270–4274. [Google Scholar] [CrossRef]
- Li, S.N.; Yang, S.B.; Shen, D.; Sun, W.; Shan, X.Y.; Dong, W.; Chen, Y.H.; Zhang, X.; Mao, Y.Q.; Tang, S.W. Polysulfide intercalation in bilayer-structured graphitic CN: A first-principles study. Phys. Chem. Chem. Phys. 2017, 19, 32708–32714. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.; Fischer, A.; Goettmann, F.; Antonietti, M.; Müller, J.-O.; Schlögl, R.; Carlsson, J.M. Graphitic carbon nitride materials: Variation of structure and morphology and their use as metal-free catalysts. J. Mater. Chem. 2008, 18, 4893–4908. [Google Scholar] [CrossRef]
- Zhang, Y.; Mori, T.; Ye, J.; Antonietti, M. Phosphorus-doped carbon nitride solid: Enhanced electrical conductivity and photocurrent generation. J. Am. Chem. Soc. 2010, 132, 6294–6295. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, S.B.; Chen, Y.H.; Li, S.N.; Tang, S.W.; Shen, D.; Dong, W.; Hao, D.Y. Effect of phosphorous-doped graphitic carbon nitride on electrochemical properties of lithium-sulfur battery. Ionics 2020, 26, 5491–5501. [Google Scholar] [CrossRef]
- Ma, T.Y.; Ran, J.R.; Dai, S.; Jaroniec, M.; Qiao, S.Z. Phosphorus-Doped Graphitic Carbon Nitrides Grown In Situ on Carbon-Fiber Paper: Flexible and Reversible Oxygen Electrodes. Angew. Chem. Int. Ed. 2015, 54, 4646–4650. [Google Scholar] [CrossRef] [PubMed]
- Bojdys, M.J.; Müller, J.O.; Antonietti, M.; Thomas, A. Ionothermal synthesis of crystalline, condensed, graphitic carbon nitride. Chem. Eur. J. 2008, 14, 8177–8182. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Hart, C.; Pang, Q.; Garsuch, A.; Weiss, T.; Nazar, L.F. A highly efficient polysulfide mediator for lithium-sulfur batteries. Nat. Commun. 2015, 6, 5682. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.L.; Wei, Z.Y.; Peng, L.L.; Zhang, L.Y.; Zohar, A.; Schoeppner, R.; Wang, P.Q.; Wan, C.Z.; Zhu, D.; Liu, H.T.; et al. Establishing reaction networks in the 16-electron sulfur reduction reaction. Nature 2024, 626, 98–104. [Google Scholar] [CrossRef]
- Kang, X.Y.; He, T.Q.; Zou, R.; Niu, S.T.; Ma, Y.X.; Zhu, F.L.; Ran, F. Size Effect for Inhibiting Polysulfides Shuttle in Lithium-Sulfur Batteries. Small 2024, 20, 2306503–2306534. [Google Scholar] [CrossRef] [PubMed]
- Gracia, J.; Kroll, P. Corrugated layered heptazine-based carbon nitride: The lowest energy modifications of C3N4 ground state. J. Mater. Chem. 2009, 19, 3013–3019. [Google Scholar] [CrossRef]
- Ma, X.; Wu, Y.; Lu, Y.; Xu, J.; Wang, Y.; Zhu, Y. Effect of compensated codoping on the photoelectrochemical properties of anatase TiO2 photocatalyst. J. Phys. Chem. C 2011, 115, 16963–16969. [Google Scholar] [CrossRef]
- Ma, X.; Lv, Y.; Xu, J.; Liu, Y.; Zhang, R.; Zhu, Y. A strategy of enhancing the photoactivity of g-C3N4 via doping of nonmetal elements: A first-principles study. J. Phys. Chem. C 2012, 116, 23485–23493. [Google Scholar] [CrossRef]
- Liu, J.J. Effect of phosphorus doping on electronic structure and photocatalytic performance of g-CN: Insights from hybrid density functional calculation. J. Alloys Compd. 2016, 672, 271–276. [Google Scholar] [CrossRef]
- Yang, S.; Gong, Y.; Zhang, J.; Zhan, L.; Ma, L.; Fang, Z.; Vajtai, R.; Wang, X.; Ajayan, P.M. Exfoliated Graphitic Carbon Nitride Nanosheets as Efficient Catalysts for Hydrogen Evolution Under Visible Light. Adv. Mater. 2013, 25, 2452–2456. [Google Scholar] [CrossRef]
- Ceperley, D.M.; Alder, B.J. Ground State of the Electron Gas by a Stochastic Method. Phys. Rev. Lett. 1980, 45, 566–569. [Google Scholar] [CrossRef]
- Perdew, J.P.; Zunger, A. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B 1981, 23, 5050–5079. [Google Scholar] [CrossRef]
- Pan, Y.; Li, Q.; Zhu, Y.; Li, Y.; Liu, H.; Cong, Y.; Wu, M. Homonuclear transition-metal dimers embedded monolayer C2N as promising anchoring and electrocatalytic materials for lithium-sulfur battery: First-principles calculations. Appl. Surf. Sci. 2023, 610, 155507–155515. [Google Scholar] [CrossRef]
- Yamsang, N.; Sittiwong, J.; Srifa, P.; Boekfa, B.; Sawangphruk, M.; Maihom, T.; Limtrakul, J. First-Principle study of lithium polysulfide adsorption on heteroatom doped graphitic carbon nitride for Lithium-Sulfur batteries. Appl. Surf. Sci. 2021, 565, 150378. [Google Scholar] [CrossRef]
- Mosquera-Lois, I.; Kavanagh, S.R.; Klarbring, J.; Tolborg, K.; Walsh, A. Imperfections are not 0 K: Free energy of point defects in crystals. Chem. Soc. Rev. 2023, 52, 5812–5826. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Maeda, K.; Thomas, A.; Takanabe, K.; Xin, G.; Carlsson, J.M.; Domen, K.; Antonietti, M. A metal-free polymeric photocatalyst for hydrogen production from water under visible light. Nat. Mater. 2009, 8, 76–80. [Google Scholar] [CrossRef]
- Mott, N. The mobility edge since 1967. J. Phys. C Solid State Phys. 1987, 20, 3075–3102. [Google Scholar] [CrossRef]
- Kundu, A.; Song, Y.; Galli, G. Influence of nuclear quantum effects on the electronic properties of amorphous carbon. Proc. Natl. Acad. Sci. USA 2022, 119, e2203083119. [Google Scholar] [CrossRef]
- Kundu, A.; Galli, G. Quantum Vibronic Effects on the Excitation Energies of the Nitrogen-Vacancy Center in Diamond. J. Phys. Chem. Lett. 2024, 15, 802–810. [Google Scholar] [CrossRef]
- Guo, S.; Tang, Y.; Xie, Y.; Tian, C.; Feng, Q.; Zhou, W.; Jiang, B. P-doped tubular g-C3N4 with surface carbon defects: Universal synthesis and enhanced visible-light photocatalytic hydrogen production. Appl. Catal. B Environ. 2017, 218, 664–671. [Google Scholar] [CrossRef]
- Liu, S.; Zhu, H.; Yao, W.; Chen, K.; Chen, D. One step synthesis of P-doped g-C3N4 with the enhanced visible light photocatalytic activity. Appl. Surf. Sci. 2018, 430, 309–315. [Google Scholar] [CrossRef]
- Chen, Y.S.; Li, S.; Dong, W.; Shen, D. Applications of first-principles in cathode materials of lithium-sulfide batteries. Chin. Nonf. Met. 2018, 28, 2310–2326. [Google Scholar] [CrossRef]
- Dong, W.; Zhu, X.; Shen, D.; Zhao, M.; Gu, H.; Yang, F.; Chang, Q.; Tang, S.; Hong, X.; Yang, S. Uncovering the lithium-embedded behavior and catalytic mechanism of g-C3N4 as a sulfur host of lithium-sulfur batteries in the initial discharge reaction. Diamond Relat. Mater. 2023, 140, 110534. [Google Scholar] [CrossRef]
- Lin, H.; Liu, G.; Zhu, L.; Zhang, Z.; Jin, R.; Huang, Y.; Gao, S. Flexible borophosphene monolayer: A potential Dirac anode for high-performance non-lithium ion batteries. Appl. Surf. Sci. 2021, 544, 148895. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic population analysis on LCAO–MO molecular wave functions. I. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef]
- Fonseca Guerra, C.; Handgraaf, J.W.; Baerends, E.J.; Bickelhaupt, F.M. Voronoi deformation density (VDD) charges: Assessment of the Mulliken, Bader, Hirshfeld, Weinhold, and VDD methods for charge analysis. J. Comput. Chem. 2004, 25, 189–210. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Martin, F.; Zipse, H. Charge distribution in the water molecule—A comparison of methods. J. Comput. Chem. 2004, 26, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Govind, N.; Petersen, M.; Fitzgerald, G.; King-Smith, D.; Andzelm, J. A generalized synchronous transit method for transition state location. Comput. Mater. Sci. 2003, 28, 250–258. [Google Scholar] [CrossRef]
- Segall, M.; Lindan, P.J.; Probert, M.A.; Pickard, C.J.; Hasnip, P.J.; Clark, S.; Payne, M. First-principles simulation: Ideas, illustrations and the CASTEP code. J. Phys. Condens. Matter 2002, 14, 2717–2744. [Google Scholar] [CrossRef]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I.J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Für Krist. Cryst. Mater. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 1990, 41, 7892. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Fischer, T.H.; Almlof, J. General methods for geometry and wave function optimization. J. Phys. Chem. C 1992, 96, 9768–9774. [Google Scholar] [CrossRef]
- Krukau, A.V.; Vydrov, O.A.; Izmaylov, A.F.; Scuseria, G.E. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys. 2006, 125, 224106–224110. [Google Scholar] [CrossRef]
- Cohen, A.J.; Mori-Sánchez, P.; Yang, W. Insights into current limitations of density functional theory. Science 2008, 321, 792–794. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | vdW Correction | a/Å | b/Å |
|---|---|---|---|
| LDA-CAPZ | no | 7.072 | 7.072 |
| GGA-PBE | no | 7.144 | 7.144 |
| GGA-PBE | Grimme | 7.144 | 7.144 |
| GGA-PBE | TS | 7.138 | 7.138 |
| Exp. a | - | 7.30 | - |
| Other theory b | OBS | 7.16 | - |
| Other theory c | - | 7.13 | - |
| P-Doping Positions | C1 | C2 | C3 | C4 | N1 | N2 | N3 | N4 | Interstitial (Pi) |
|---|---|---|---|---|---|---|---|---|---|
| Ef (eV) (2 × 2 × 1) | 1.06 | 1.81 | 1.07 | 1.78 | 2.31 | 1.34 | 1.34 | 5.03 | 0.81 |
| Ef (eV) (4 × 4 × 1) | 0.77 | 1.65 | 0.78 | 1.62 | 1.70 | 1.20 | 1.20 | 4.83 | 0.80 |
| Ef (eV) a | 0.73 | 1.52 | - | - | 1.93 | 1.33 | - | 3.55 | 0.78 |
| Eg Gap (eV) | g-C3N4 | P Substitute N Site (PN) | P Substitute C Site (PC) | P-Doping Interstitial Site (Pi) |
|---|---|---|---|---|
| GGA-PBE | 1.13 | 0.89 | 0.04 | 0.14 |
| HSE06 | 2.67 | 2.14 | 0.12 | 0.20 |
| Exp a | 2.70 | - | - | - |
| Molecules | LiPSs | g-C3N4 | PC | |||||
|---|---|---|---|---|---|---|---|---|
| S-S | Li-S | Li-S | Li-N | Ead | Li-S | Li-N | Ead | |
| Li2S | - | 2.108 | 2.264 | 2.400 | 3.809 | 2.342 | 1.992 | 6.760 |
| Li2S2 | 2.186 | 2.240 | 2.593 | 2.397 | 3.773 | 2.515 | 1.965 | 6.525 |
| Li2S4 | 2.093 | 2.386 | 3.040 | 2.351 | 3.350 | 2.336 | 2.250 | 5.225 |
| Li2S6 | 2.078 | 2.428 | 2.531 | 2.069 | 3.198 | 2.411 | 2.280 | 5.245 |
| Li2S8 | 2.075 | 2.419 | 2.781 | 2.065 | 3.392 | 2.473 | 2.299 | 5.026 |
| S8 | 2.060 | - | - | - | 2.021 | - | - | 3.502 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Liu, F.; Wei, S.; Xia, Y.; Li, X.; Liu, S.; Zhang, X.; Tang, S.; Shen, D.; Dong, W.; et al. First-Principles Investigation of Phosphorus-Doped Graphitic Carbon Nitride as Anchoring Material for the Lithium-Sulfur Battery. Molecules 2024, 29, 2746. https://doi.org/10.3390/molecules29122746
Chen Y, Liu F, Wei S, Xia Y, Li X, Liu S, Zhang X, Tang S, Shen D, Dong W, et al. First-Principles Investigation of Phosphorus-Doped Graphitic Carbon Nitride as Anchoring Material for the Lithium-Sulfur Battery. Molecules. 2024; 29(12):2746. https://doi.org/10.3390/molecules29122746
Chicago/Turabian StyleChen, Yuehui, Fengxia Liu, Shuang Wei, Yingkai Xia, Xiaodong Li, Shengnan Liu, Xu Zhang, Shuwei Tang, Ding Shen, Wei Dong, and et al. 2024. "First-Principles Investigation of Phosphorus-Doped Graphitic Carbon Nitride as Anchoring Material for the Lithium-Sulfur Battery" Molecules 29, no. 12: 2746. https://doi.org/10.3390/molecules29122746
APA StyleChen, Y., Liu, F., Wei, S., Xia, Y., Li, X., Liu, S., Zhang, X., Tang, S., Shen, D., Dong, W., & Yang, S. (2024). First-Principles Investigation of Phosphorus-Doped Graphitic Carbon Nitride as Anchoring Material for the Lithium-Sulfur Battery. Molecules, 29(12), 2746. https://doi.org/10.3390/molecules29122746