
Mild Iron-Catalyzed Oxidative Cross-Coupling of Quinoxalinones with Indoles


Abstract

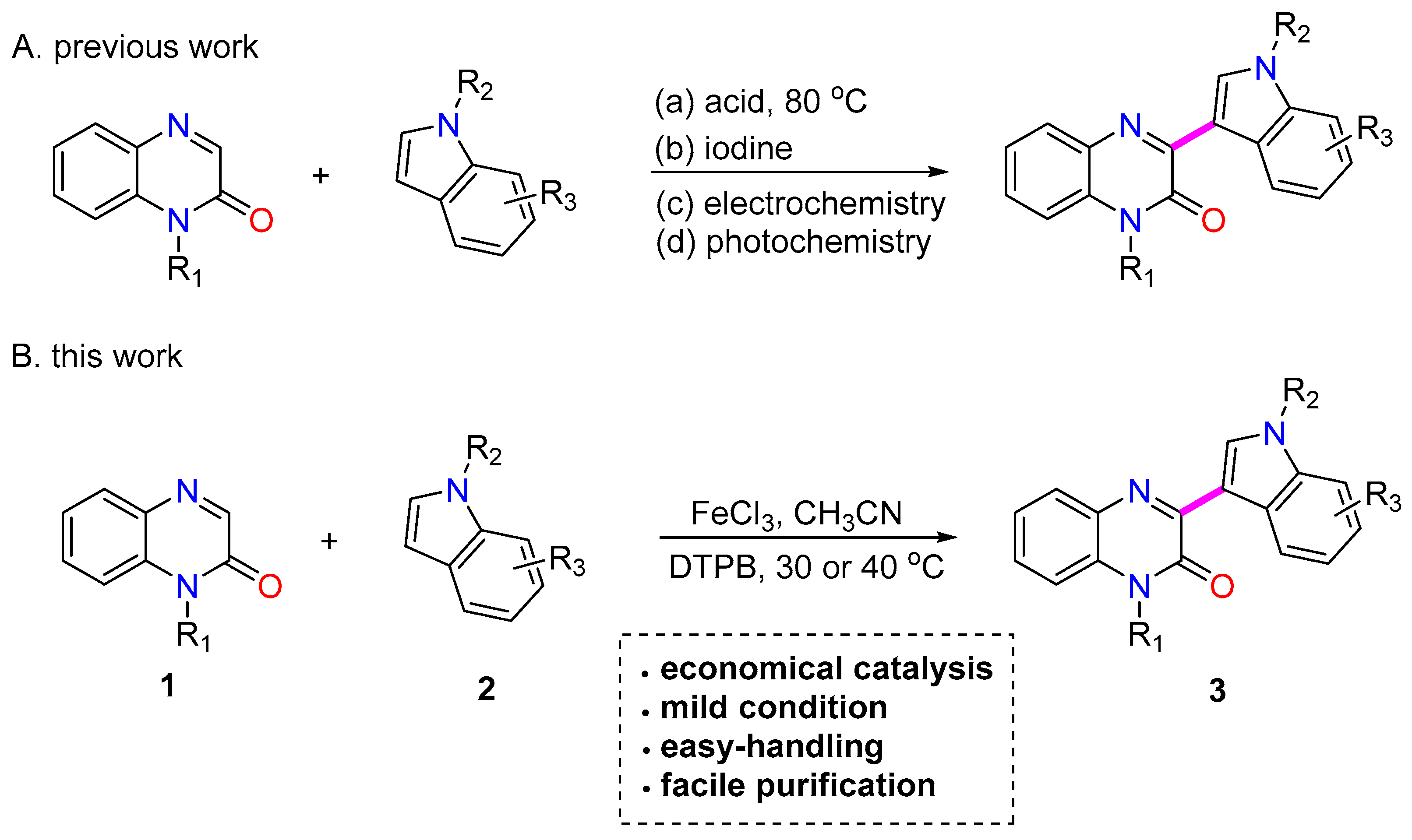
1. Introduction
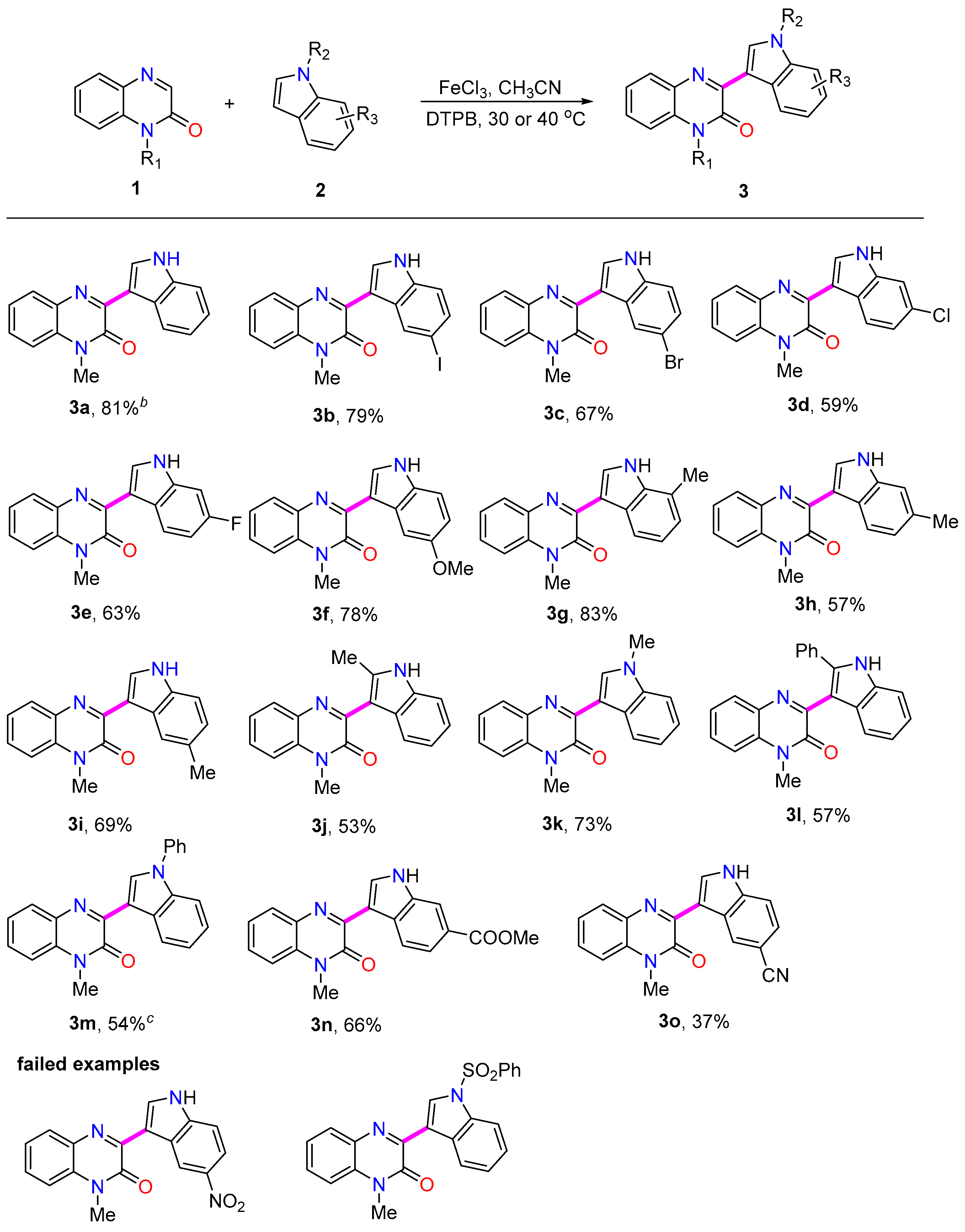
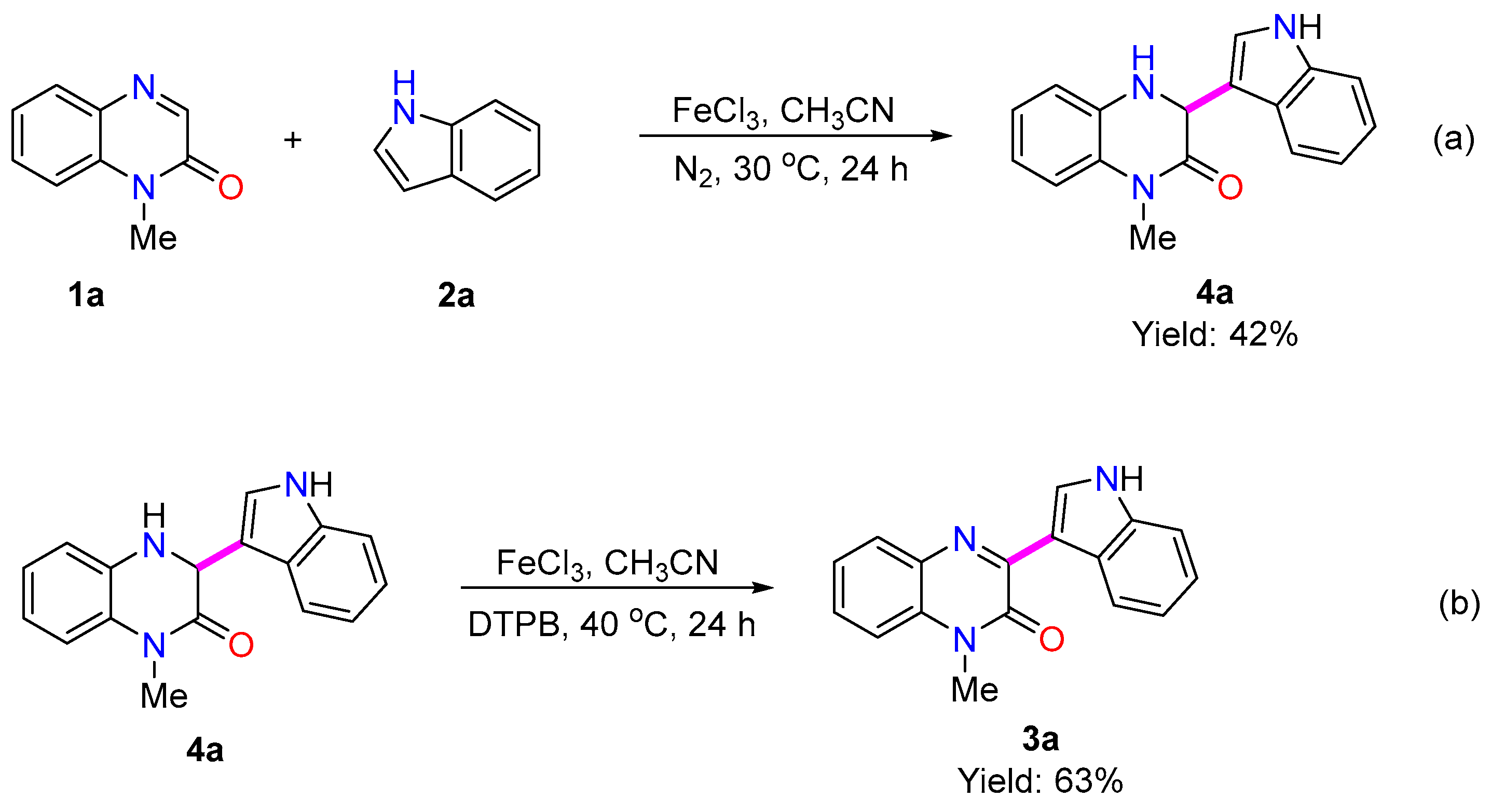
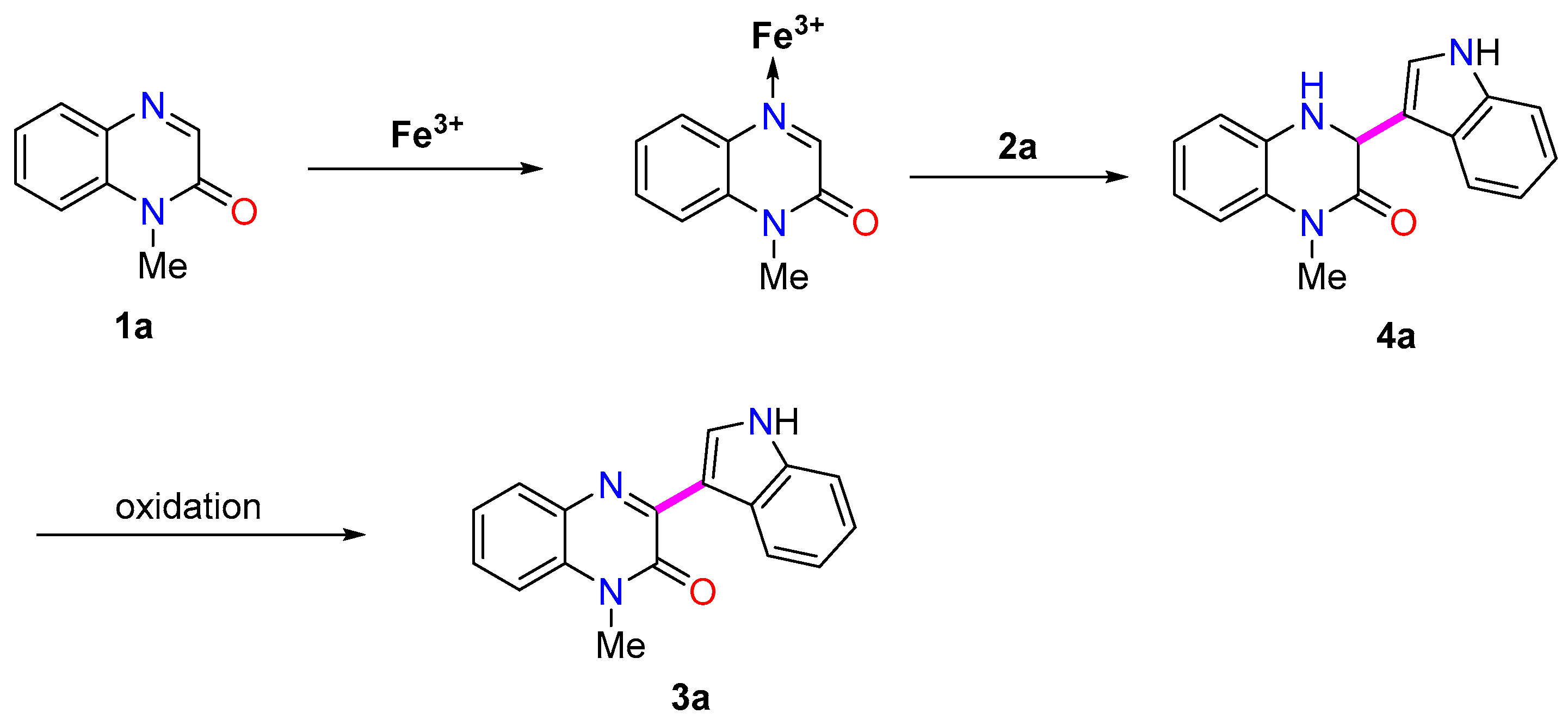
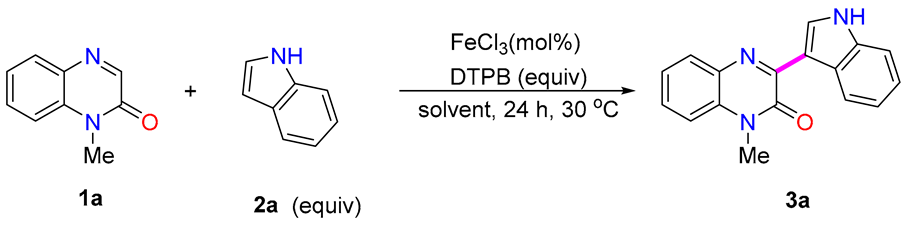
2. Results and Discussion
3. Materials and Methods
3.1. General Information
3.2. General Procedure for the Synthesis of Starting Materials (1a and 1c–1f) [19,20]
3.3. General Procedure for the Synthesis of Product (3a–3t)
3.4. Characterization Data of Products
- 3-(1H-indol-3-yl)-1-methylquinoxalin-2(1H)-one (3a):
- Isolated through filtration to produce yellow solid. Yield: 81%. 1H NMR (400 MHz, DMSO-d6) δ 11.79 (s, 1H), 9.03–8.85 (m, 2H), 7.92 (d, J = 7.9 Hz, 1H), 7.61–7.48 (m, 3H), 7.46–7.33 (m, 1H), 7.33–7.18 (m, 2H), 3.74 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 153.66, 150.62, 136.29, 133.18, 132.96, 131.49, 128.36, 128.25, 126.31, 123.42, 123.02, 122.53, 120.98, 114.42, 111.88, 111.41, 29.09. This is a known structure. These data are similar to the reported ones [14].
- 3-(5-iodo-1H-indol-3-yl)-1-methylquinoxalin-2(1H)-one (3b):
- Isolated through filtration to produce yellow solid. M.P.: >300 °C. Yield: 79%. 1H NMR (400 MHz, DMSO-d6) δ 11.93 (s, 1H), 9.23 (s, 1H), 8.89 (s, 1H), 7.85 (d, J = 7.9 Hz, 1H), 7.60–7.47 (m, 3H), 7.47–7.32 (m, 2H), 3.71 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 153.54, 150.22, 135.39, 133.80, 132.72, 131.55, 131.26, 130.51, 128.75, 128.54, 128.34, 123.53, 114.50, 114.36, 110.57, 85.43, 29.10. HRMS (ESI): m/z calcd for C17H12N3OI [M + Na]+: 423.9923. Found: 423.9923.
- 3-(5-bromo-1H-indol-3-yl)-1-methylquinoxalin-2(1H)-one (3c):
- Isolated through filtration to produce yellow solid. Yield: 67%. 1H NMR (400 MHz, DMSO-d6) δ 11.96 (s, 1H), 9.01 (s, 1H), 8.93 (s, 1H), 7.87 (d, J = 7.9 Hz, 1H), 7.72–6.40 (m, 5H), 3.71 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 153.55, 150.23, 135.04, 134.27, 132.73, 131.56, 128.57, 128.40, 127.99, 125.05, 125.00, 123.53, 114.51, 113.94, 113.78, 110.90, 29.11. This is a known structure. These data are similar to the reported ones [14].
- 3-(5-chloro-1H-indol-3-yl)-1-methylquinoxalin-2(1H)-one (3d):
- Isolated through filtration to produce yellow solid. Yield: 59%. 1H NMR (400 MHz, DMSO-d6) δ 11.86 (s, 1H), 8.92 (s, 1H), 8.85 (s, 1H), 7.89 (s, 1H), 7.53 (m, 3H), 7.37 (s, 1H), 7.23 (s, 1H), 3.71 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 153.53, 150.23, 136.80, 133.96, 132.78, 131.58, 128.52, 128.45, 127.09, 125.06, 124.26, 123.43, 121.11, 114.44, 111.58, 111.45, 29.08. This is a known structure. These data are similar to the reported ones [14].
- 3-(6-fluro-1H-indol-3-yl)-1-methylquinoxalin-2(1H)-one (3e):
- Isolated through filtration to produce yellow solid. M.P.: 261.0–263.0 °C. Yield: 63%. 1H NMR (400 MHz, DMSO-d6) δ 11.81 (s, 1H), 8.97–8.82 (m, 2H), 7.89 (d, J = 7.9 Hz, 1H), 7.52 (t, J = 2.8 Hz, 2H), 7.37 (s, 1H), 7.30 (d, J = 9.7 Hz, 1H), 7.07 (t, J = 9.3 Hz, 1H), 3.71 (d, J = 2.0 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 159.25 (d, J = 236.4 Hz), 153.55, 150.30, 136.40 (d, J = 12.5 Hz), 133.73 (d, J = 2.6 Hz), 132.82, 131.58, 128.43, 128.42, 124.14 (d, J = 9.6 Hz), 123.42, 123.01, 114.43, 111.45, 109.07 (d, J = 23.4 Hz), 98.06 (d, J = 25.7 Hz), 29.08. HRMS (ESI): m/z calcd for C17H12N3OF [M + H]+: 294.1043. Found: 294.1046.
- 3-(5-methoxy-1H-indol-3-yl)-1-methylquinoxalin-2(1H)-one (3f):
- Isolated through filtration to produce yellow solid. Yield: 78%. 1H NMR (400 MHz, DMSO-d6) δ 11.68 (s, 1H), 8.88 (s, 1H), 8.47 (s, 1H), 7.90 (d, J = 7.9 Hz, 1H), 7.53 (s, 2H), 7.47–7.33 (m, 2H), 6.89 (d, J = 8.7 Hz, 1H), 3.88 (s, 3H), 3.72 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 154.87, 153.67, 150.68, 133.52, 132.94, 131.42, 131.17, 128.31, 128.14, 126.99, 123.45, 114.44, 112.48, 112.08, 111.13, 105.06, 55.24, 29.09. This is a known structure. These data are similar to the reported ones [14].
- 3-(7-methyl-1H-indol-3-yl)-1-methylquinoxalin-2(1H)-one (3g):
- Isolated through filtration to produce yellow solid. M.P.: 289.0–291.0 °C. Yield: 83%. 1H NMR (400 MHz, DMSO-d6) δ 11.78 (s, 1H), 8.92 (s, 1H), 8.73 (d, J = 8.0 Hz, 1H), 7.91 (d, J = 7.9 Hz, 1H), 7.53 (d, J = 3.9 Hz, 2H), 7.46–7.34 (m, 1H), 7.14 (t, J = 7.6 Hz, 1H), 7.04 (d, J = 7.1 Hz, 1H), 3.73 (s, 3H), 2.53 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 153.70, 150.61, 135.71, 132.96, 132.86, 131.47, 128.36, 128.23, 126.08, 123.41, 123.17, 121.19, 120.93, 120.65, 114.41, 111.81, 29.08, 16.76. HRMS (ESI): m/z calcd for C18H15N3O [M + H]+: 290.1293. Found: 290.1298.
- 3-(6-methyl-1H-indol-3-yl)-1-methylquinoxalin-2(1H)-one (3h):
- Isolated through filtration to produce yellow solid. Yield: 57%. 1H NMR (400 MHz, DMSO-d6) δ 11.65 (s, 1H), 8.85 (d, J = 2.9 Hz, 1H), 8.74 (d, J = 8.1 Hz, 1H), 7.90 (d, J = 7.9 Hz, 1H), 7.53 (s, 1H), 7.50 (d, J = 9.1 Hz, 1H), 7.42–7.34 (m, 1H), 7.30 (s, 1H), 7.06 (d, J = 8.2 Hz, 1H), 3.72 (s, 3H), 2.44 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 153.64, 150.60, 136.71, 132.99, 132.73, 131.68, 131.46, 128.32, 128.15, 124.18, 123.39, 122.72, 122.63, 114.40, 111.70, 111.40, 29.07, 21.36. This is a known structure. These data are similar to the reported ones [14].
- 3-(5-methyl-1H-indol-3-yl)-1-methylquinoxalin-2(1H)-one (3i):
- Isolated through filtration to produce yellow solid. M.P.: >300 °C. Yield: 69%. 1H NMR (400 MHz, DMSO-d6) δ 11.67 (s, 1H), 8.88 (d, J = 2.6 Hz, 1H), 8.71 (s, 1H), 7.93 (d, J = 7.9 Hz, 1H), 7.54 (d, J = 2.2 Hz, 2H), 7.39 (d, J = 6.0 Hz, 2H), 7.07 (d, J = 8.2 Hz, 1H), 3.73 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 153.67, 150.67, 134.60, 133.22, 132.99, 131.43, 129.63, 128.35, 128.12, 126.57, 123.97, 123.38, 122.80, 114.40, 111.51, 111.02, 29.06, 21.66. HRMS (ESI): m/z calcd for C18H15N3O [M + H]+: 290.1293. Found: 290.1295.
- 3-(2-methyl-1H-indol-3-yl)-1-methylquinoxalin-2(1H)-one (3j):
- Isolated through silica gel column chromatography with ethyl acetate and petrol ether (1:4) to produce yellow solid. The yellow solid was washed with petrol ether. Yield: 69%. 1H NMR (400 MHz, DMSO-d6) δ 11.49 (s, 1H), 7.80 (d, J = 8.0 Hz, 1H), 7.75 (d, J = 7.9 Hz, 1H), 7.62–7.52 (m, 2H), 7.41–7.31 (m, 2H), 7.07 (t, J = 7.8 Hz, 1H), 7.03–6.97 (m, 1H), 3.71 (s, 3H), 2.55 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 154.01, 153.14, 139.29, 135.08, 132.81, 132.53, 129.02, 128.65, 127.96, 123.30, 120.83, 120.80, 119.49, 114.42, 110.59, 109.48, 29.28, 14.24. This is a known structure. These data are similar to the reported ones [14].
- 3-(1-methyl-1H-indol-3-yl)-1-methylquinoxalin-2(1H)-one (3k):
- Isolated through filtration to produce yellow solid. Yield: 73%. 1H NMR (400 MHz, DMSO-d6) δ 8.95–8.88 (m, 2H), 7.92 (d, J = 7.4 Hz, 1H), 7.56 (d, J = 7.2 Hz, 3H), 7.44–7.36 (m, 1H), 7.35–7.25 (m, 2H), 3.93 (s, 3H), 3.75 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 153.60, 150.32, 136.98, 136.87, 132.97, 131.49, 128.37, 128.31, 126.78, 123.48, 123.14, 122.63, 121.31, 114.48, 110.36, 110.25, 33.07, 29.11. This is a known structure. These data are similar to the reported ones [14].
- 3-(2-phenyl-1H-indol-3-yl)-1-methylquinoxalin-2(1H)-one (3l):
- Isolated through silica gel column chromatography with ethyl acetate and petrol ether (1:4) to produce yellow solid. Yield: 57%. 1H NMR (400 MHz, DMSO-d6) δ 11.84 (s, 1H), 7.78 (d, J = 7.9 Hz, 1H), 7.70–7.44 (m, 6H), 7.42–7.28 (m, 4H), 7.18 (t, J = 7.2 Hz, 1H), 7.07 (t, J = 7.5 Hz, 1H), 3.59 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 153.72, 153.56, 139.19, 135.96, 133.14, 133.05, 132.71, 129.74, 129.02, 128.57, 128.40, 127.86, 127.81, 123.35, 122.08, 120.33, 119.99, 114.61, 111.45, 109.13, 29.28. This is a known structure. These data are similar to the reported ones [14].
- 3-(1-phenyl-1H-indol-3-yl)-1-methylquinoxalin-2(1H)-one (3m):
- Isolated through filtration to produce yellow solid. Yield: 54%. 1H NMR (400 MHz, DMSO-d6) δ 9.09 (s, 1H), 9.03 (d, J = 7.4 Hz, 1H), 8.00 (d, J = 7.9 Hz, 1H), 7.73–7.57 (m, 7H), 7.56–7.48 (m, 1H), 7.42–7.31 (m, 2H), 3.76 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 153.68, 144.99, 138.26, 135.79, 134.97, 132.84, 131.72, 130.14, 129.01, 128.76, 127.67, 127.40, 124.52, 123.76, 123.65, 122.25, 114.66, 112.78, 110.74, 29.24. This is a known structure. These data are similar to the reported ones [18].
- methyl 3-(4-methyl-3-oxo-3,4-dihydroquinoxalin-2-yl)-1H-indole-6-carboxylate (3n):
- Isolated through filtration to produce yellow solid. Yield: 66%. 1H NMR (400 MHz, DMSO-d6) δ 12.10 (s, 1H), 9.09 (s, 1H), 8.94 (d, J = 8.4 Hz, 1H), 8.15 (s, 1H), 7.94 (d, J = 7.9 Hz, 1H), 7.83 (d, J = 8.5 Hz, 1H), 7.56 (s, 2H), 7.41 (d, J = 5.0 Hz, 1H), 3.89 (s, 3H), 3.74 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 166.97, 153.55, 150.16, 135.99, 135.65, 132.76, 131.60, 129.88, 128.59, 128.52, 123.43, 122.68, 121.46, 114.43, 113.64, 111.67, 51.93, 29.08. This is a known structure. These data are similar to the reported ones [18].
- 3-(4-methyl-3-oxo-3,4-dihydroquinoxalin-2-yl)-1H-indole-5-carbonitrile (3o):
- Isolated through filtration to produce yellow solid. M.P.: >300 °C. Yield: 37%. 1H NMR (400 MHz, DMSO-d6) δ 12.20 (s, 1H), 9.15 (s, 1H), 8.98 (s, 1H), 7.92 (d, J = 7.9 Hz, 1H), 7.64 (d, J = 8.4 Hz, 1H), 7.61–7.44 (m, 3H), 7.37 (t, J = 7.4 Hz, 1H), 3.66 (s, 3H). 13C NMR (101 MHz, DMSO-d6) δ 153.44, 149.85, 138.14, 135.00, 132.62, 131.64, 128.91, 128.76, 127.98, 125.95, 125.39, 123.52, 120.73, 114.48, 113.31, 111.77, 103.06, 29.11. HRMS (ESI): m/z calcd for C18H12N4O [M + Na]+: 323.0909. Found: 323.0909.
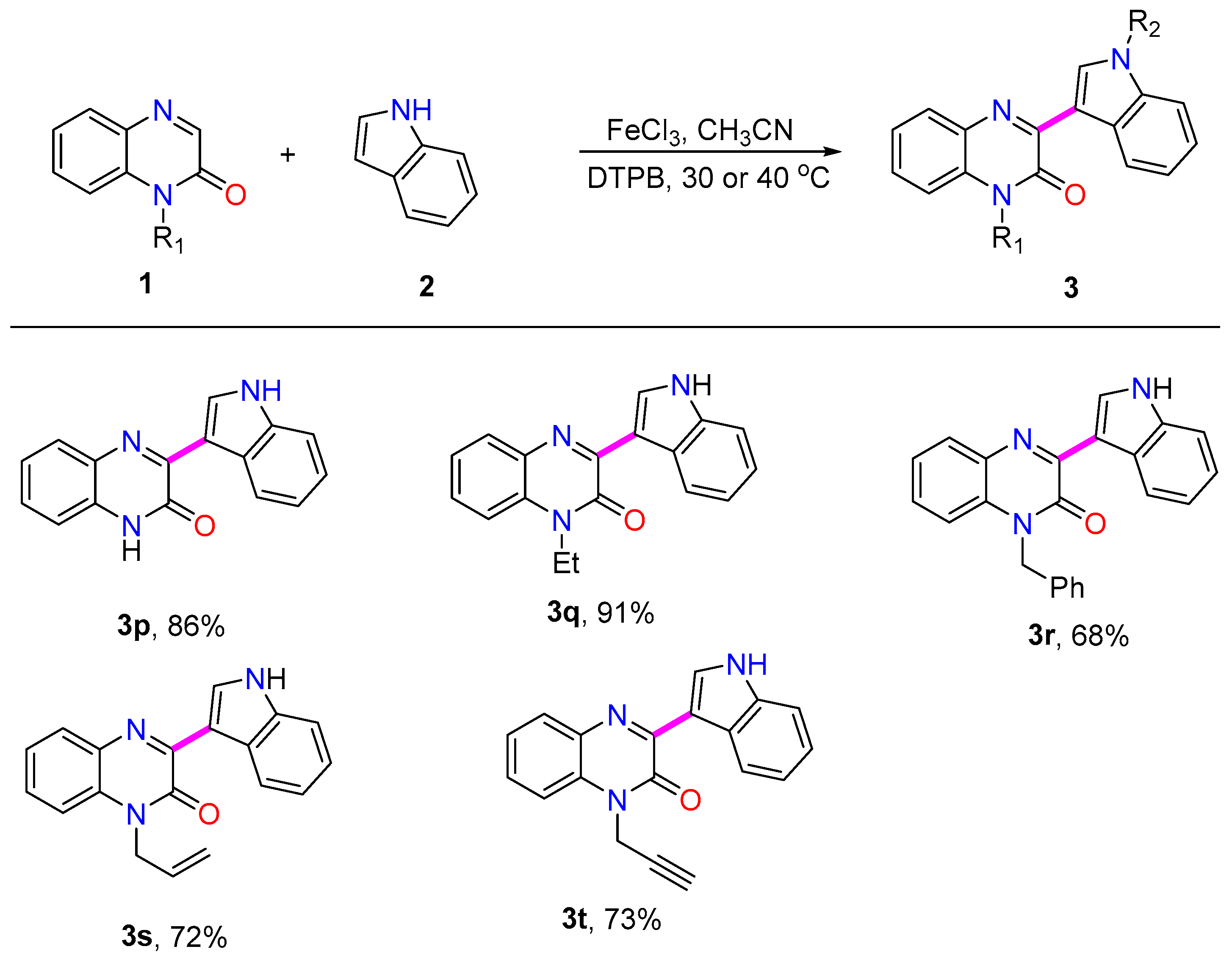
- 3-(1H-indol-3-yl)-1-methylquinoxalin-2(1H)-one (3p):
- Isolated through filtration to produce yellow solid. Yield: 86%. 1H NMR (400 MHz, DMSO-d6) δ 12.41 (s, 1H), 11.78 (s, 1H), 8.94 (s, 1H), 8.91–8.85 (m, 1H), 7.86 (d, J = 7.9 Hz, 1H), 7.54–7.47 (m, 1H), 7.43 (t, J = 7.6 Hz, 1H), 7.32 (d, J = 7.8 Hz, 2H), 7.27–7.20 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 154.41, 151.98, 136.28, 133.09, 132.65, 130.18, 127.99, 127.61, 126.20, 123.24, 122.99, 122.56, 121.00, 114.94, 111.90, 111.32. This is a known structure. These data are similar to the reported ones [17].
- 1-ethyl-3-(1H-indol-3-yl)quinoxalin-2(1H)-one (3q):
- Isolated through silica gel column chromatography with ethyl acetate and petrol ether (1:4) to produce red solid. M.P.: 175.0–176.5 °C. Yield: 91%. 1H NMR (400 MHz, DMSO-d6) δ 11.80 (s, 1H), 8.96–8.87 (m, 2H), 7.93 (d, J = 7.9 Hz, 1H), 7.63–7.49 (m, 3H), 7.38 (t, J = 7.5 Hz, 1H), 7.27–7.21 (m, 2H), 4.38 (q, J = 6.7, 2H), 1.30 (t, J = 6.9 Hz, 3H). 13C NMR (101 MHz, DMSO-d6) δ 153.17, 150.65, 136.29, 133.26, 133.21, 130.24, 128.73, 128.40, 126.33, 123.36, 123.01, 122.54, 121.00, 114.13, 111.89, 111.34, 36.89, 12.45. HRMS (ESI): m/z calcd for C18H15N3O [M + H]+: 290.1293. Found: 290.1295.
- 1-benzyl-3-(1H-indol-3-yl)quinoxalin-2(1H)-one (3r):
- Isolated through silica gel column chromatography with ethyl acetate and petrol ether (1:4) to produce orange solid. Yield: 68%. 1H NMR (400 MHz, DMSO-d6) δ 11.85 (s, 1H), 8.98–8.90 (m, 2H), 7.95 (d, J = 7.8 Hz, 1H), 7.56–7.50 (m, 1H), 7.43 (d, J = 3.2 Hz, 2H), 7.38–7.29 (m, 5H), 7.29–7.23 (m, 3H), 5.63 (s, 2H). 13C NMR (101 MHz, DMSO-d6) δ 153.84, 150.77, 136.33, 136.19, 133.39, 133.29, 130.61, 128.73, 128.64, 128.27, 127.26, 126.76, 126.33, 123.62, 123.02, 122.62, 121.10, 114.80, 111.94, 111.40, 44.98. This is a known structure. These data are similar to the reported ones [17].
- 1-allyl-3-(1H-indol-3-yl)quinoxalin-2(1H)-one (3s):
- Isolated through silica gel column chromatography with ethyl acetate and petrol ether (1:4) to produce orange solid. Yield: 72%. 1H NMR (400 MHz, DMSO-d6) δ 11.82 (s, 1H), 8.95–8.87 (m, 2H), 7.94 (d, J = 7.9 Hz, 1H), 7.55–7.44 (m, 3H), 7.38 (m, 1H), 7.28–7.21 (m, 2H), 6.08–5.94 (m, 1H), 5.20 (d, J = 10.5 Hz, 1H), 5.10 (d, J = 16.9 Hz, 1H), 5.01 (d, J = 2.4 Hz, 2H). 13C NMR (101 MHz, DMSO-d6) δ 153.41, 150.75, 136.40, 133.36, 133.26, 131.94, 130.63, 128.69, 128.42, 126.39, 123.69, 123.10, 122.75, 121.21, 117.06, 114.90, 112.05, 111.45, 44.05. This is a known structure. These data are similar to the reported ones [17].
- 3-(1H-indol-3-yl)-1-(prop-2-yn-1-yl)quinoxalin-2(1H)-one (3t):
- Isolated through silica gel column chromatography with ethyl acetate and petrol ether (1:4) to produce yellow solid. Yield: 73%. 1H NMR (400 MHz, DMSO-d6) δ 11.85 (s, 1H), 8.93–8.85 (m, 2H), 7.95 (d, J = 7.7 Hz, 1H), 7.60 (t, J = 9.0 Hz, 2H), 7.52 (d, J = 5.2 Hz, 1H), 7.43 (t, J = 7.5 Hz, 1H), 7.28–7.22 (m, 2H), 5.21 (s, 2H), 3.35 (s, 1H). 13C NMR (101 MHz, DMSO-d6) δ 152.78, 150.47, 136.32, 133.34, 133.19, 129.84, 128.60, 128.37, 126.22, 123.92, 122.97, 122.67, 121.14, 114.69, 111.96, 111.20, 78.37, 75.07, 31.34. This is a known structure. These data are similar to the reported ones [17].
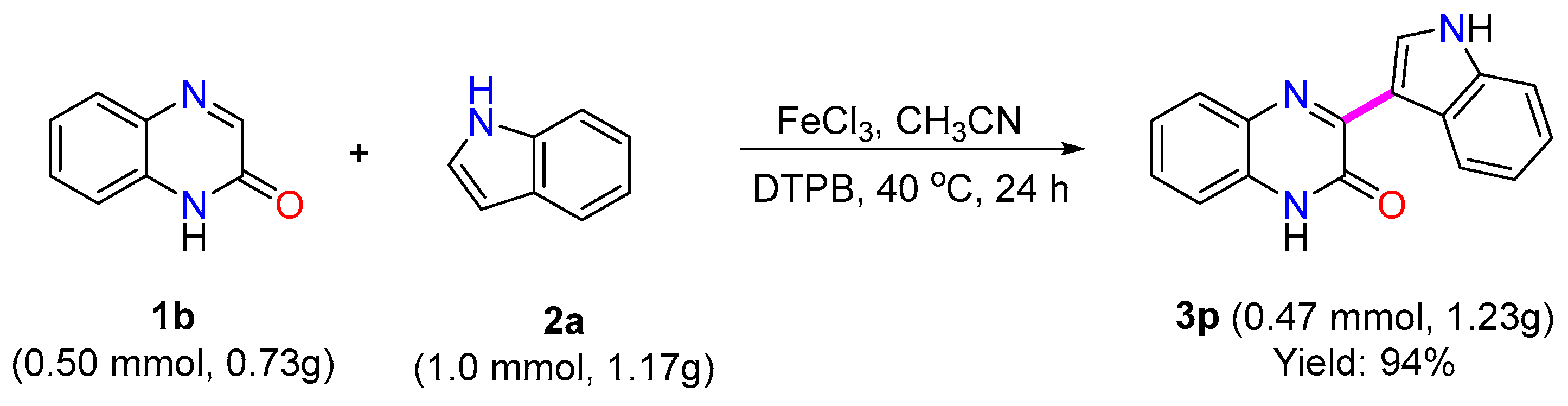
3.5. Gram Scale Reaction
4. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bauer, I.; Knölker, H.-J. Iron catalysis in organic synthesis. Chem. Rev. 2015, 115, 3170–3387. [Google Scholar] [CrossRef] [PubMed]
- Guðmundsson, A.; Bäckvall, J.E. On the use of iron in organic chemistry. Molecules 2020, 25, 1349. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.; Biswas, J.P.; Paul, S.; Paul, S.; Paik, A.; Maiti, D. Organic synthesis with the most abundant transition metal–iron: From rust to multitasking catalysts. Chem. Soc. Rev. 2021, 50, 243–472. [Google Scholar] [CrossRef]
- Casnati, A.; Lanzi, M.; Cera, G. Recent advances in asymmetric iron catalysis. Molecules 2020, 25, 3889. [Google Scholar] [CrossRef]
- de Groot, L.H.M.; Ilic, A.; Schwarz, J.; Wärnmark, K. Iron photoredox catalysis–past, present, and future. J. Am. Chem. Soc. 2023, 145, 9369–9388. [Google Scholar] [CrossRef]
- Carta, A.; Piras, S.; Loriga, G.; Paglietti, G. Chemistry, biological properties and SAR analysis of quinoxalinones. Mini-Rev. Med. Chem. 2006, 6, 1179–1200. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Huang, Z.-H.; Murray, M.-G.; Guo, X.-Y.; Liu, G. Quinoxalin-2(1H)-one derivatives as inhibitors against hepatitis C virus. J. Med. Chem. 2011, 54, 5747–5768. [Google Scholar] [CrossRef] [PubMed]
- El-Hawash, S.A.M.; Habib, N.S.; Kassem, M.A. Synthesis of some new quinoxalines and 1,2,4-triazolo [4,3-a]-quinoxalines for evaluation of in vitro antitumor and antimicrobial activities. Arch. Pharm. 2006, 339, 564–571. [Google Scholar] [CrossRef] [PubMed]
- Willardsen, J.A.; Dudley, D.A.; Cody, W.L.; Chi, L.; McClanahan, T.B.; Mertz, T.E.; Potoczak, R.E.; Narasimhan, L.S.; Holland, D.R.; Rapundalo, S.T.; et al. Design, synthesis, and biological activity of potent and selective inhibitors of blood coagulation factor Xa. J. Med. Chem. 2004, 47, 4089–4099. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.; Guo, C.; Ko, L.; Sun, B.; He, Y.; Li, Y. Pyrazino[2,3-g]quinoxaline-2,7-dione based π-conjugated polymers with affinity towards acids and semiconductor performance in organic thin film transistors. RSC Adv. 2016, 6, 22043–22051. [Google Scholar] [CrossRef]
- Aoki, K.; Obata, T.; Yamazaki, Y.; Mori, Y.; Hirokawa, H.; Koseki, J.; Hattori, T.; Niitsu, K.; Takeda, S.; Aburada, M.; et al. Potent platelet-derived growth factor-β receptor (PDGF-βR) inhibitors: Synthesis and structure–activity relationships of 7-[3-(cyclohexylmethyl)ureido]-3-{1-methyl-1H-pyrrolo[2,3-b]pyridin-3-yl}quinoxalin-2(1H)-one derivatives. Chem. Pharm. Bull. 2007, 55, 255–367. [Google Scholar] [CrossRef] [PubMed]
- Aoki, K.; Koseki, J.-I.; Takeda, S.; Aburada, M.; Miyamoto, K.-I. Convenient synthetic method for 3-(3-substituted indol-2-yl)quinoxalin-2-ones as VEGF Inhibitor. Chem. Pharm. Bull. 2007, 55, 922–925. [Google Scholar] [CrossRef] [PubMed]
- Sahu, N.K.; Mahajan, A.T.; Sharma, V.; Suhas, K.; Tripathi, P.; Mathur, M.; Jain, M.; Chaudhary, S. ‘Cephalandole A’ analogues as a new class of antioxidant agents: Design, microwave-assisted synthesis, bioevaluation, SAR and in silico studies. J. Mol. Struct. 2024, 1303, 137445. [Google Scholar] [CrossRef]
- Han, Y.-Y.; Wu, Z.-J.; Zhang, X.-M.; Yuan, W.-C. An efficient synthesis of 3-(indol-3-yl) quinoxalin-2-ones with TfOH-catalyzed Friedel–Crafts type coupling reaction in air. Tetrahedron Lett. 2010, 51, 2023–2028. [Google Scholar] [CrossRef]
- Noikham, M.; Kittikool, T.; Yotphan, S. Iodine-catalyzed oxidative cross-dehydrogenative coupling of quinoxalinones and indoles: Synthesis of 3-(indol-2-yl)quinoxalin-2-one under mild and ambient conditions. Synthesis 2018, 50, 2337–2346. [Google Scholar]
- Utepova, I.A.; Trestsova, M.A.; Chupakhin, O.N.; Charushin, V.N.; Rempel, A.A. Aerobic oxidative C–H/C–H coupling of azaaromatics with indoles and pyrroles in the presence of TiO2 as a photocatalyst. Green Chem. 2015, 17, 4401–4410. [Google Scholar] [CrossRef]
- Huang, J.; Wang, L.; Tang, X.-Y. Oxidative cross-coupling of quinoxalinones with indoles enabled by acidochromism. Org. Biomol. Chem. 2023, 21, 2709–2714. [Google Scholar] [CrossRef]
- Shen, M.; Li, L.; Zhou, Q.; Wang, J.; Wang, L. Visible-light-induced regio-selective oxidative coupling of quinoxalinones with pyrrole derivatives. Chin. J. Org. Chem. 2023, 43, 697–704. [Google Scholar]
- Ni, H.; Li, Y.; Shi, X.; Pang, Y.; Jin, C.; Zhao, F. Eosin Y as a direct hydrogen-atom transfer photocatalyst for the C3-H acylation of quinoxalin-2 (1H)-ones. Tetrahedron Lett. 2021, 68, 152915. [Google Scholar] [CrossRef]
- Ni, H.; Shi, X.; Li, Y.; Zhang, X.; Zhao, J.; Zhao, F. Metal-free C3–H acylation of quinoxalin-2 (1H)-ones with α-oxo-carboxylic acids. Org. Biomol. Chem. 2020, 18, 6558–6563. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | 2a (equiv) | FeCl3 (mol%) | DTPB (equiv) | Solvent | Yield (%) b |
| 1 | 1.5 | 10 | 2.0 | CH3CN | 81% |
| 2 | 1.0 | 10 | 2.0 | CH3CN | 74% |
| 3 | 2.0 | 10 | 2.0 | CH3CN | 88% |
| 4 | 3.0 | 10 | 2.0 | CH3CN | 86% |
| 5 | 2.0 | 1 | 2.0 | CH3CN | 17% |
| 6 | 2.0 | 5 | 2.0 | CH3CN | 82% |
| 7 | 2.0 | 15 | 2.0 | CH3CN | 81% |
| 8 | 2.0 | 10 | 1.5 | CH3CN | 83% |
| 9 | 2.0 | 10 | 2.5 | CH3CN | 86% |
| 10 | 2.0 | 10 | 2.0 | DMF | 12% |
| 11 | 2.0 | 10 | 2.0 | DCM | 52% |
| 12 c | 2.0 | 10 | 2.0 | CH3CN | 74% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ni, H.; Mao, H.; Huang, Y.; Lu, Y.; Liu, Z. Mild Iron-Catalyzed Oxidative Cross-Coupling of Quinoxalinones with Indoles. Molecules 2024, 29, 2649. https://doi.org/10.3390/molecules29112649
Ni H, Mao H, Huang Y, Lu Y, Liu Z. Mild Iron-Catalyzed Oxidative Cross-Coupling of Quinoxalinones with Indoles. Molecules. 2024; 29(11):2649. https://doi.org/10.3390/molecules29112649
Chicago/Turabian StyleNi, Hangcheng, Hui Mao, Ying Huang, Yi Lu, and Zhenxiang Liu. 2024. "Mild Iron-Catalyzed Oxidative Cross-Coupling of Quinoxalinones with Indoles" Molecules 29, no. 11: 2649. https://doi.org/10.3390/molecules29112649
APA StyleNi, H., Mao, H., Huang, Y., Lu, Y., & Liu, Z. (2024). Mild Iron-Catalyzed Oxidative Cross-Coupling of Quinoxalinones with Indoles. Molecules, 29(11), 2649. https://doi.org/10.3390/molecules29112649