Author Contributions
N.G., investigation, formal analysis, methodology, writing—original draft; D.K., synthesis; A.C., NMR experiments; F.M., molecular dynamics experiments, review and editing; D.T., resources, supervision, methodology, writing—review and editing; S.V., resources, synthesis supervision, methodology, writing—review and editing; T.M. conceptualization, resources, supervision, writing—original draft, writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Scheme 1.
Reagents and conditions: (i) thiosemicarbazide, EtOH, cat. AcOH, 80 °C, 4 h; (ii) methyl 2-chloroacetate, MeOH, CH3COONa, 65 °C, 6 h; (iii) (a) benzaldehyde, MeOH, cat. Piperidine, 65 °C, 48 h (for DKI39) and (b) anisaldehyde cat. piperidine, rt, 7d (for DKI40).
Scheme 1.
Reagents and conditions: (i) thiosemicarbazide, EtOH, cat. AcOH, 80 °C, 4 h; (ii) methyl 2-chloroacetate, MeOH, CH3COONa, 65 °C, 6 h; (iii) (a) benzaldehyde, MeOH, cat. Piperidine, 65 °C, 48 h (for DKI39) and (b) anisaldehyde cat. piperidine, rt, 7d (for DKI40).
Scheme 2.
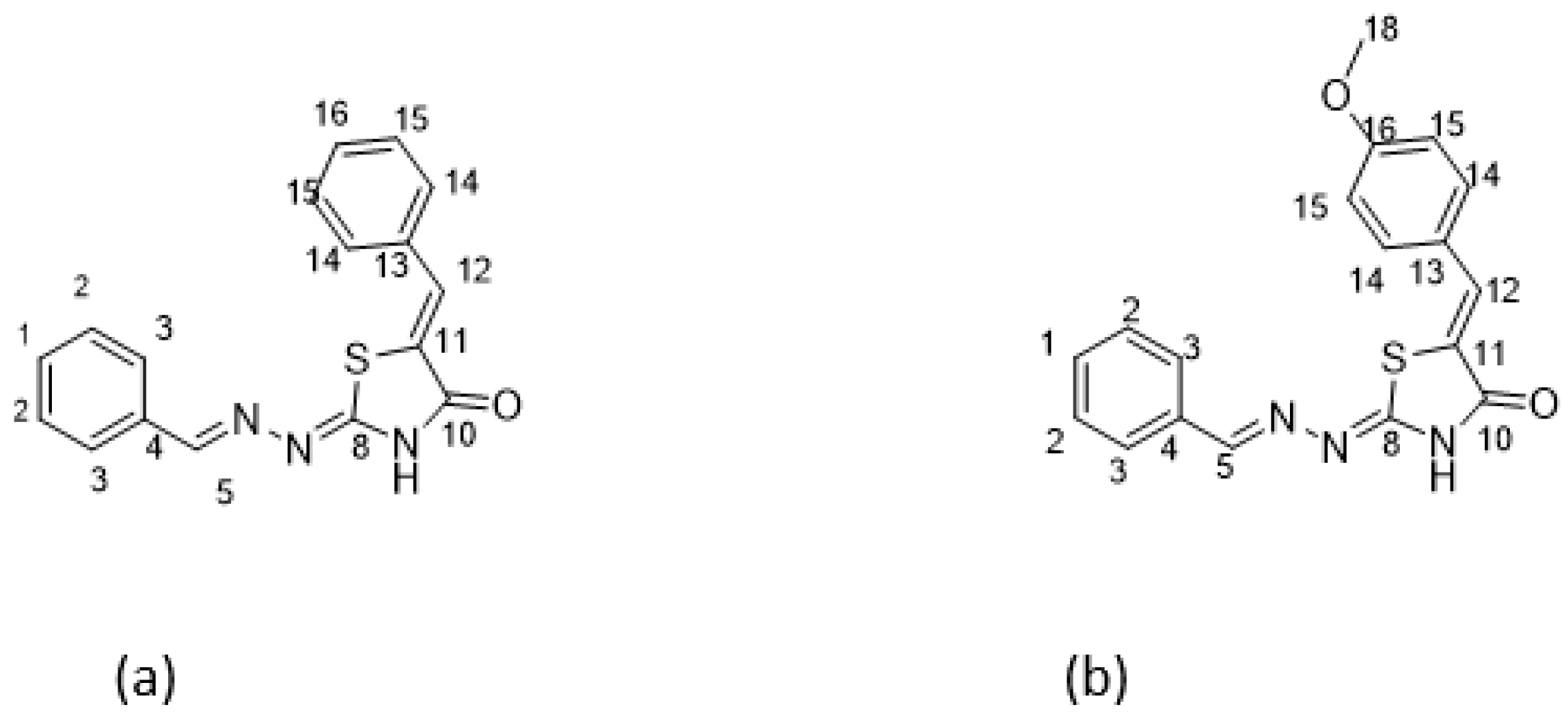
Structures of exo conformers of DKI39 (a) and DKI40 (b).
Scheme 2.
Structures of exo conformers of DKI39 (a) and DKI40 (b).
Scheme 3.
Optimized conformations derived from DFT calculations for the DKI39 endo compound. In the Figure the dihedral angles rotated by 180° are shown. For instance, in DKI39en_1, the dihedral angle τ1 was rotated and DKI39en_2 was obtained. In the orange circle, the double bond that indicates the endo nature of compound is shown.
Scheme 3.
Optimized conformations derived from DFT calculations for the DKI39 endo compound. In the Figure the dihedral angles rotated by 180° are shown. For instance, in DKI39en_1, the dihedral angle τ1 was rotated and DKI39en_2 was obtained. In the orange circle, the double bond that indicates the endo nature of compound is shown.
Scheme 4.
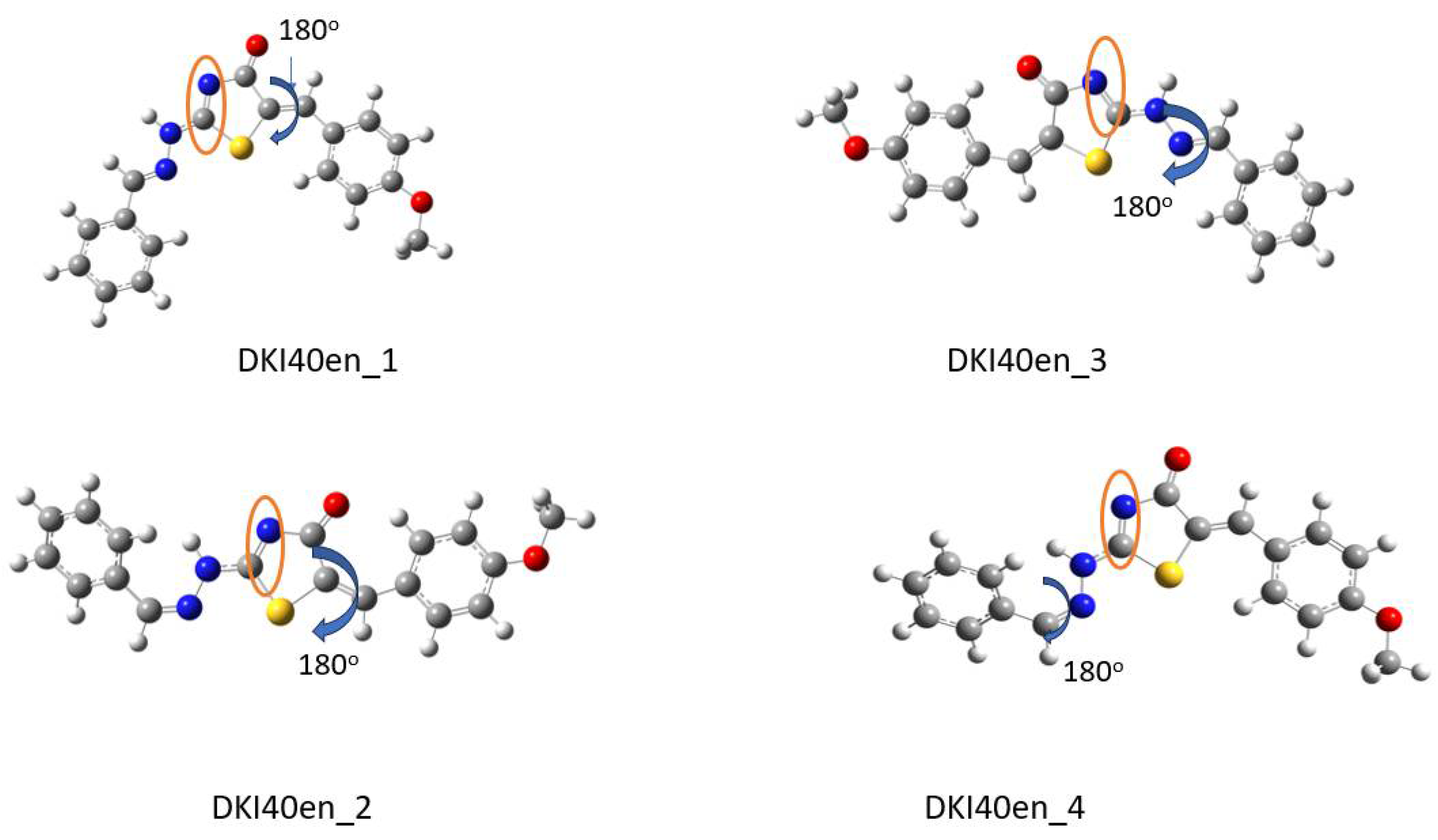
Optimized conformations derived from DFT calculations for the DKI40 endo compound. In the Figure, the dihedral angles rotated by 180° are shown. For instance, in DKI40en_1, the dihedral angle τ3′ was rotated and DKI39en_2 was obtained. In the orange circle, the double bond that indicates the endo nature of compound is shown.
Scheme 4.
Optimized conformations derived from DFT calculations for the DKI40 endo compound. In the Figure, the dihedral angles rotated by 180° are shown. For instance, in DKI40en_1, the dihedral angle τ3′ was rotated and DKI39en_2 was obtained. In the orange circle, the double bond that indicates the endo nature of compound is shown.
Scheme 5.
The global minima conformations for (a) DKI39 and (b) DKI40 endo compounds.
Scheme 5.
The global minima conformations for (a) DKI39 and (b) DKI40 endo compounds.
Scheme 6.
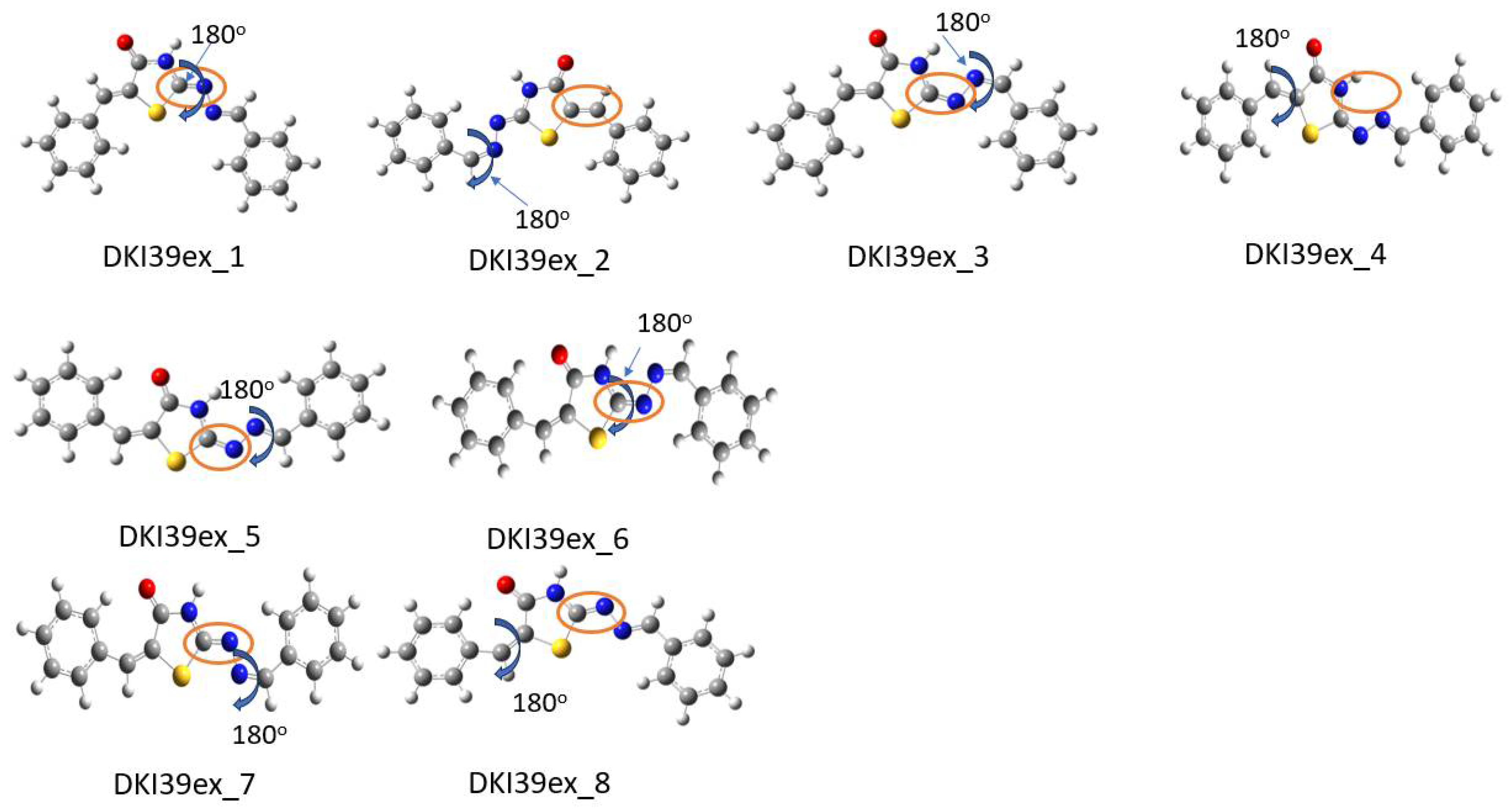
Optimized conformations derived from DFT calculations for DKI39 exo compound. In the Figure the dihedral angles rotated by 180° are shown. For instance, in DKI39ex_1, the dihedral angle τ3 was rotated and DKI39ex_2 was obtained. In the orange circle, the double bond that indicates the exo nature of compound is shown.
Scheme 6.
Optimized conformations derived from DFT calculations for DKI39 exo compound. In the Figure the dihedral angles rotated by 180° are shown. For instance, in DKI39ex_1, the dihedral angle τ3 was rotated and DKI39ex_2 was obtained. In the orange circle, the double bond that indicates the exo nature of compound is shown.
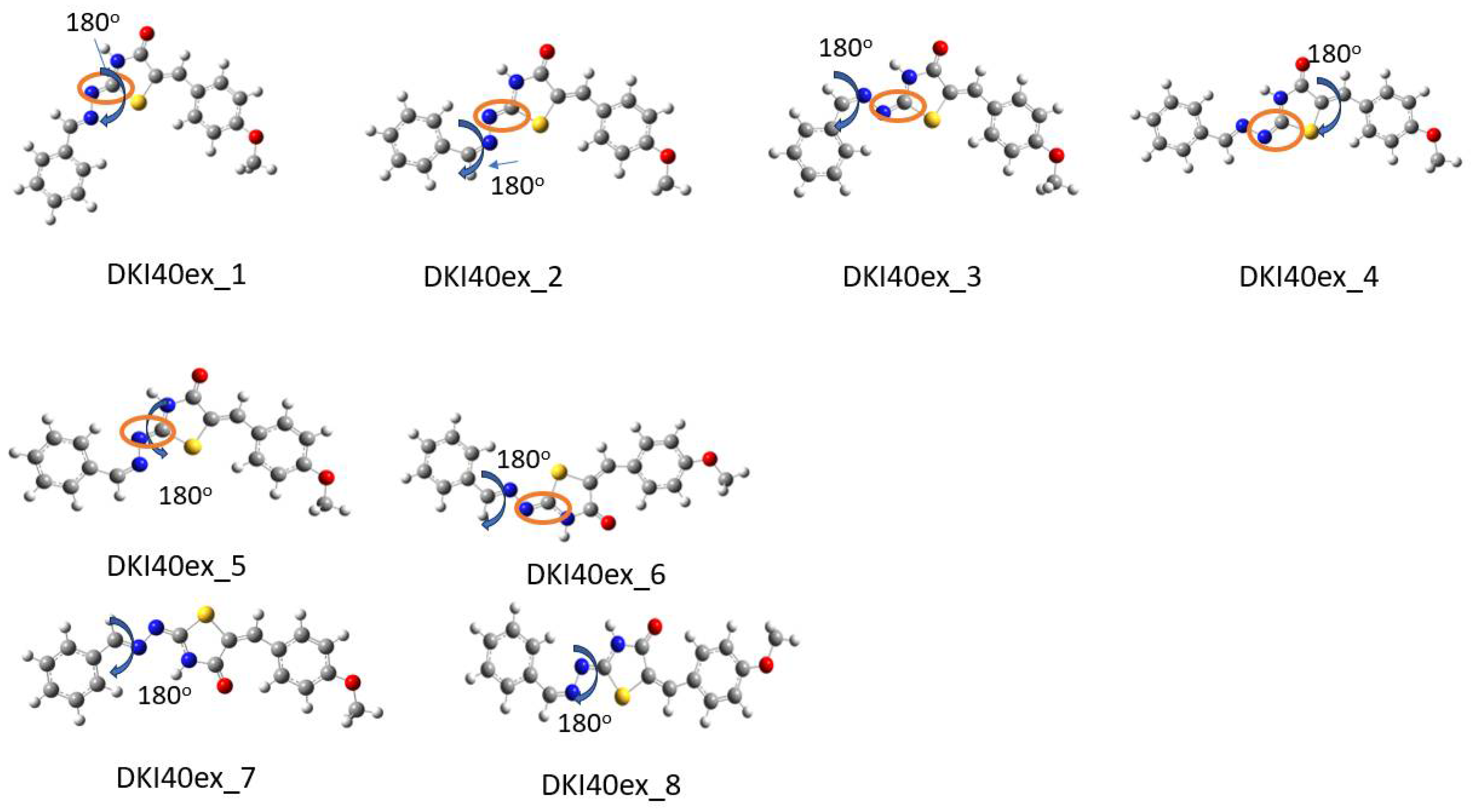
Scheme 7.
Optimized conformations derived from DFT calculations for the DKI40 exo compound. In the Figure, the dihedral angles rotated by 180° are shown. For instance, in DKI40ex_1, the dihedral angle τ2′ was rotated and DKI40ex_2 was obtained. In the orange circle the double bond that indicates the exo nature of compound is shown.
Scheme 7.
Optimized conformations derived from DFT calculations for the DKI40 exo compound. In the Figure, the dihedral angles rotated by 180° are shown. For instance, in DKI40ex_1, the dihedral angle τ2′ was rotated and DKI40ex_2 was obtained. In the orange circle the double bond that indicates the exo nature of compound is shown.
Scheme 8.
The lowest in energy conformations for (a) DKI39 and (b) DKI40 exo compounds.
Scheme 8.
The lowest in energy conformations for (a) DKI39 and (b) DKI40 exo compounds.
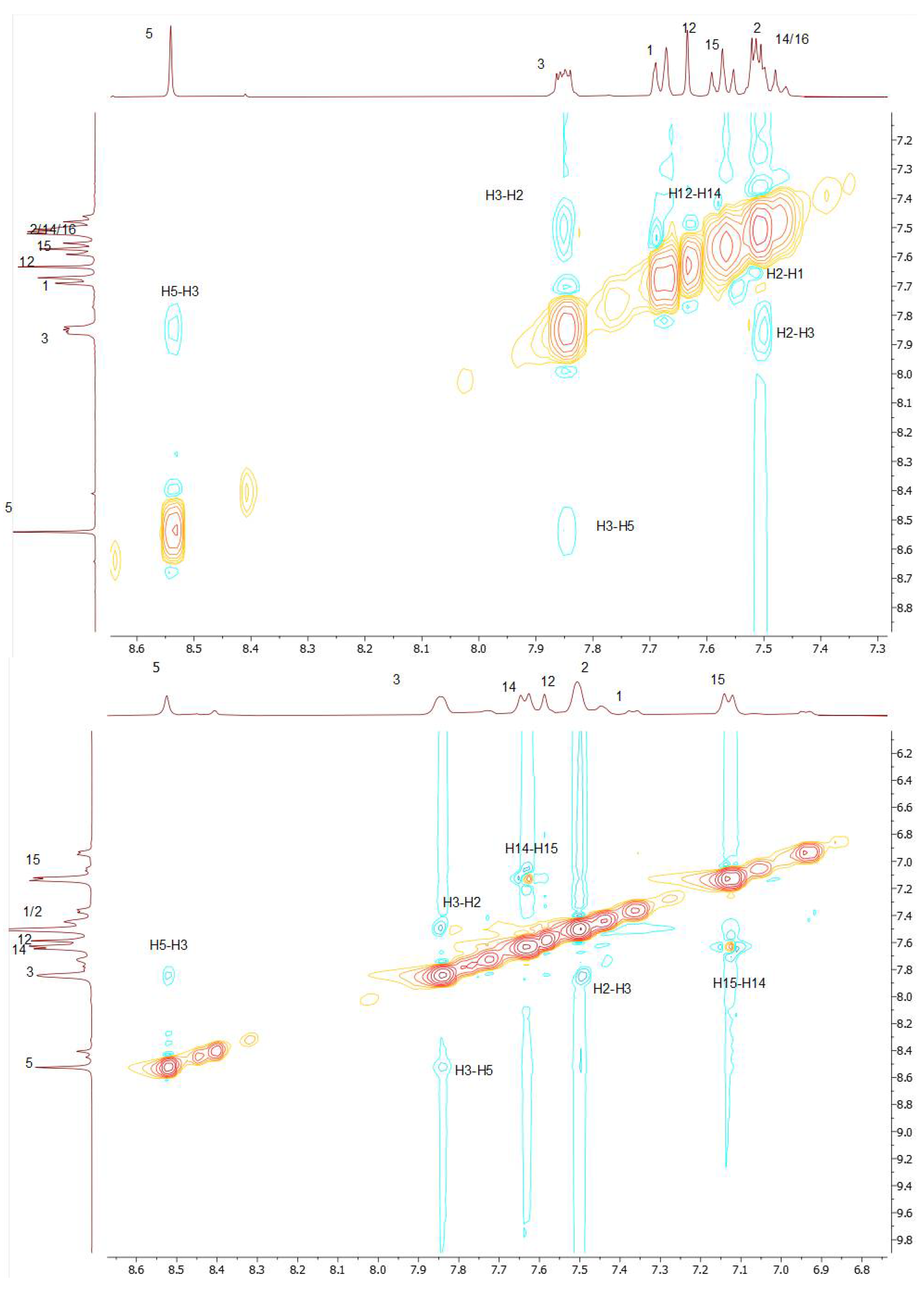
Figure 1.
NOE effects for DKI39 (top) and for DKI40 (bottom).
Figure 1.
NOE effects for DKI39 (top) and for DKI40 (bottom).
Figure 2.
Schematic representation of the isomerization energy barrier between cis-trans for DKI39 (y = energy in kcal/mol, x = conformation).
Figure 2.
Schematic representation of the isomerization energy barrier between cis-trans for DKI39 (y = energy in kcal/mol, x = conformation).
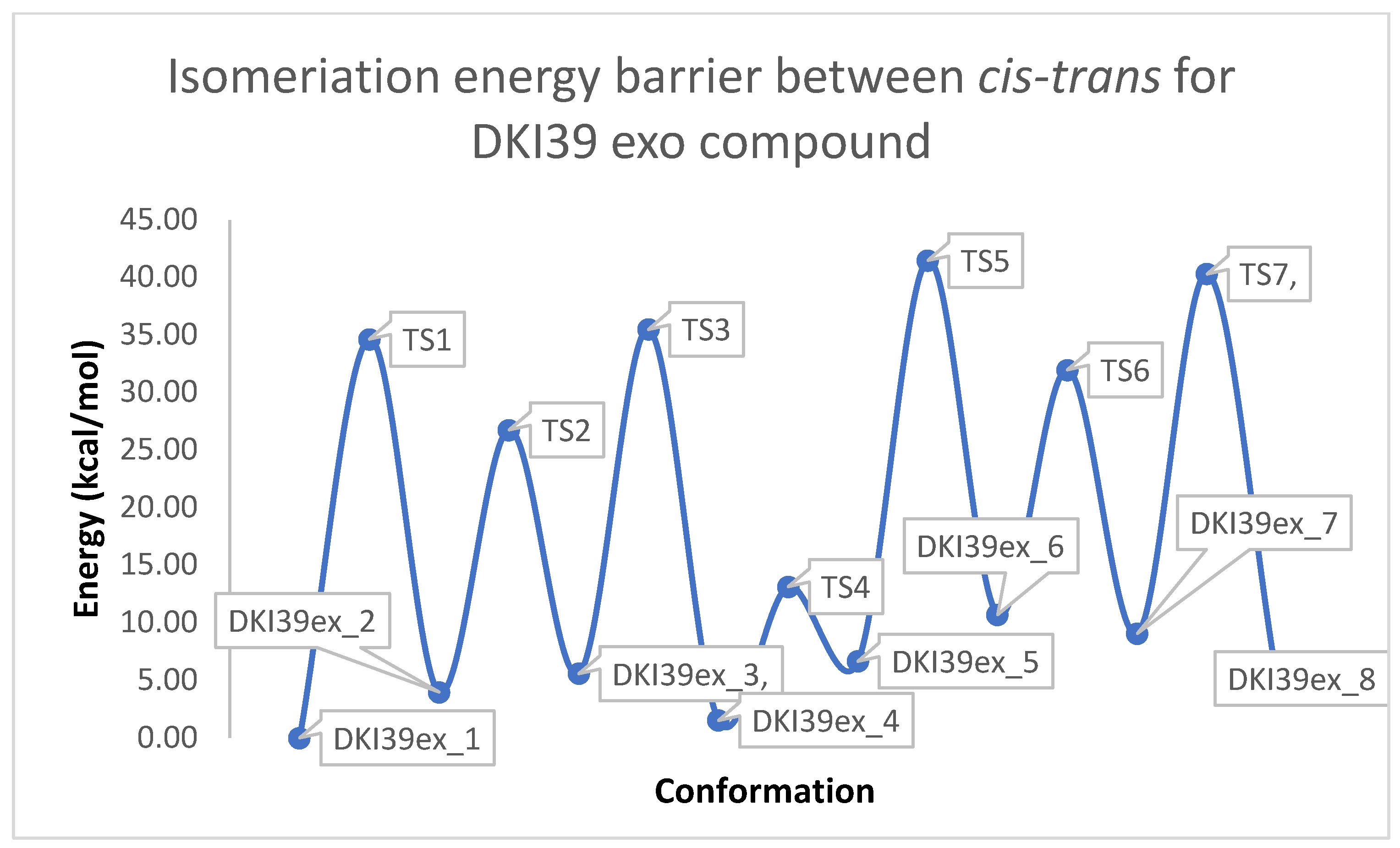
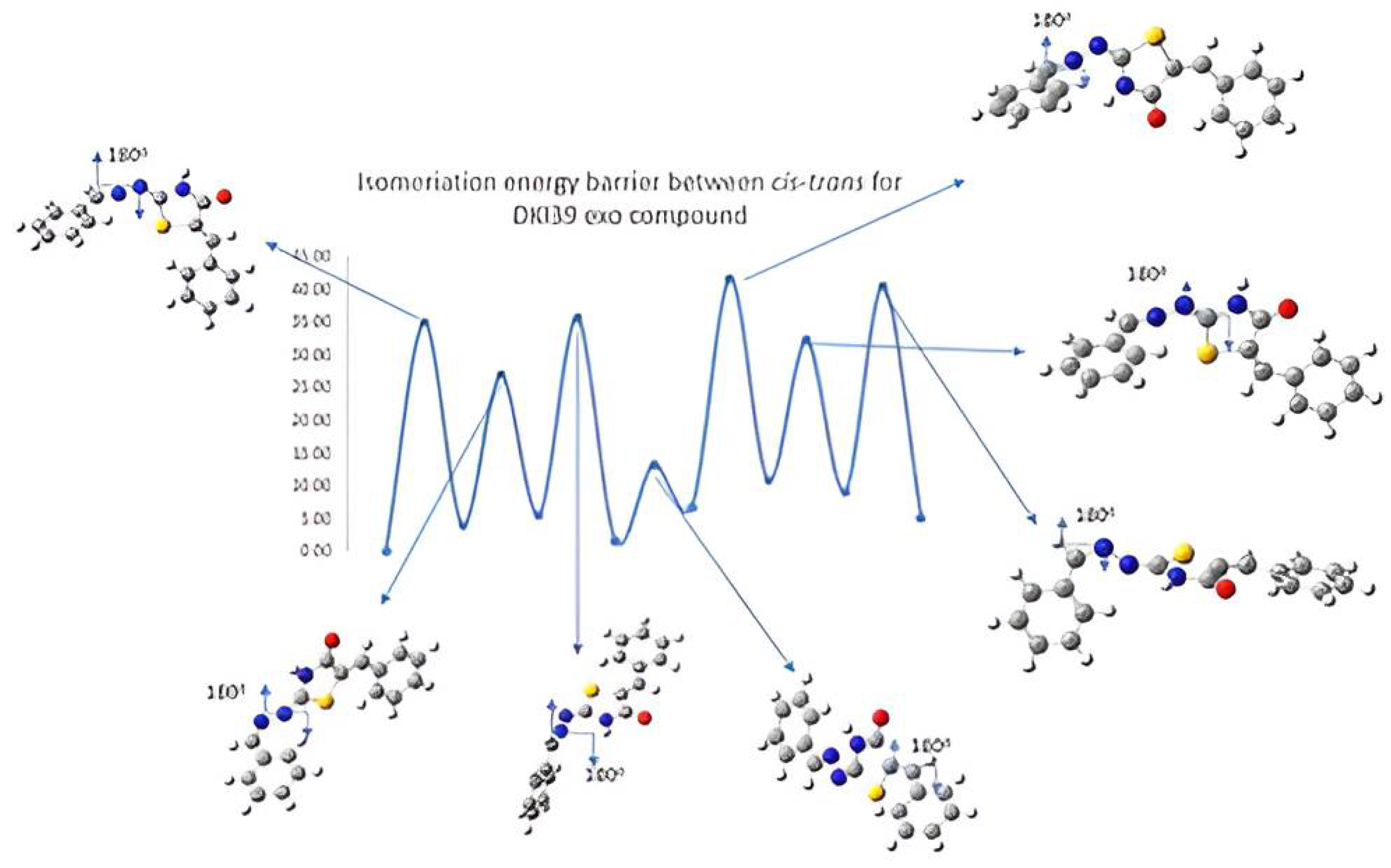
Figure 3.
Schematic representation of cis-trans kinetic isomerization for DKI39 exo compound (y = energy in kcal/mol, x = conformation). In each transition state, the dihedral angle, which is rotated by ~180°, is shown by an arrow.
Figure 3.
Schematic representation of cis-trans kinetic isomerization for DKI39 exo compound (y = energy in kcal/mol, x = conformation). In each transition state, the dihedral angle, which is rotated by ~180°, is shown by an arrow.
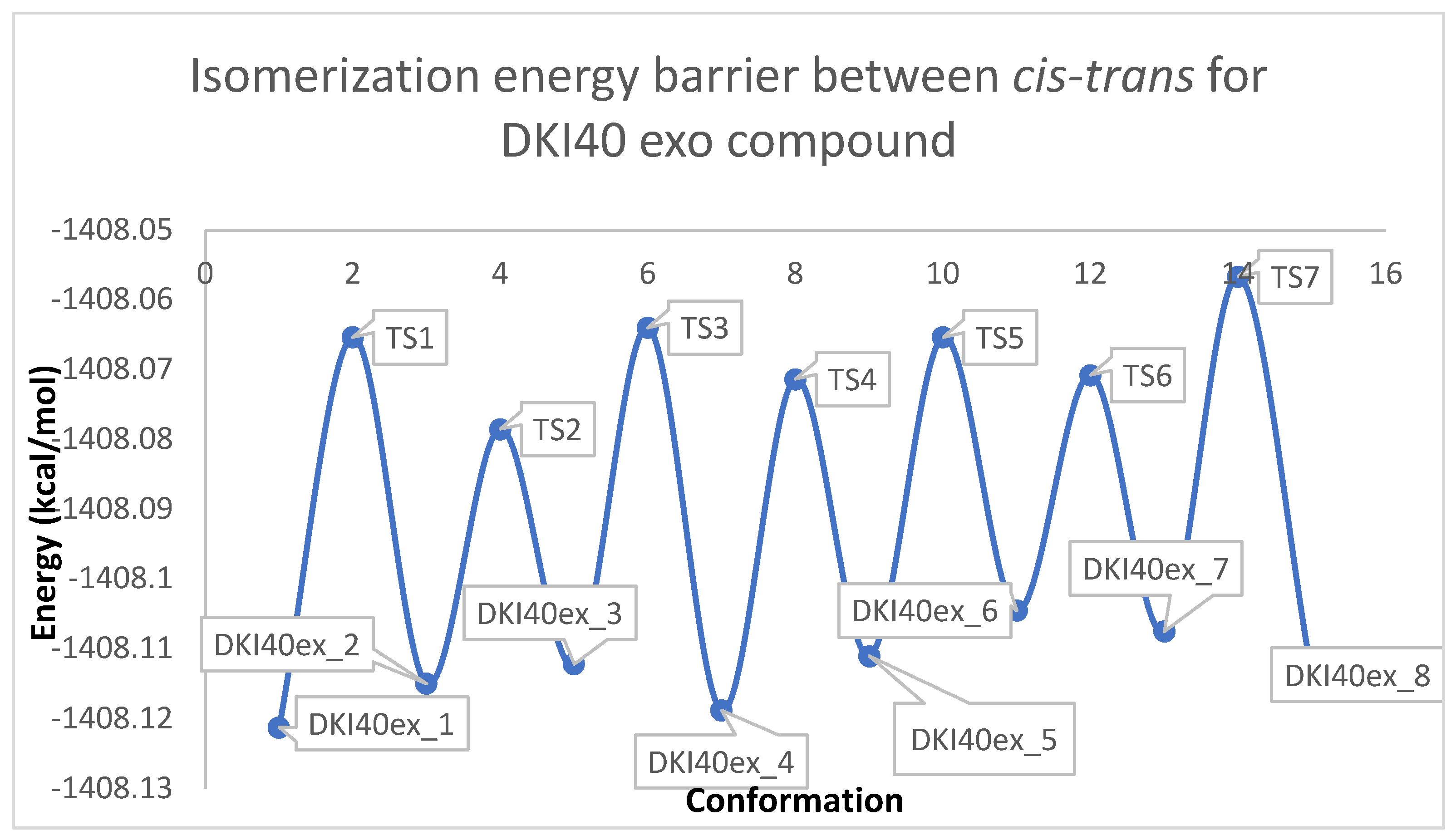
Figure 4.
Schematic representation of the isomerization energy barrier between cis −trans for DKI40 (y = energy in kcal/mol, x = conformation).
Figure 4.
Schematic representation of the isomerization energy barrier between cis −trans for DKI40 (y = energy in kcal/mol, x = conformation).
Figure 5.
Schematic representation of cis-trans kinetic isomerization for DKI40 exo compound (y = Energy in kcal/mol, x = conformation). In each transition state, the dihedral angle, which is rotated by ~180°, is shown by arrow.
Figure 5.
Schematic representation of cis-trans kinetic isomerization for DKI40 exo compound (y = Energy in kcal/mol, x = conformation). In each transition state, the dihedral angle, which is rotated by ~180°, is shown by arrow.
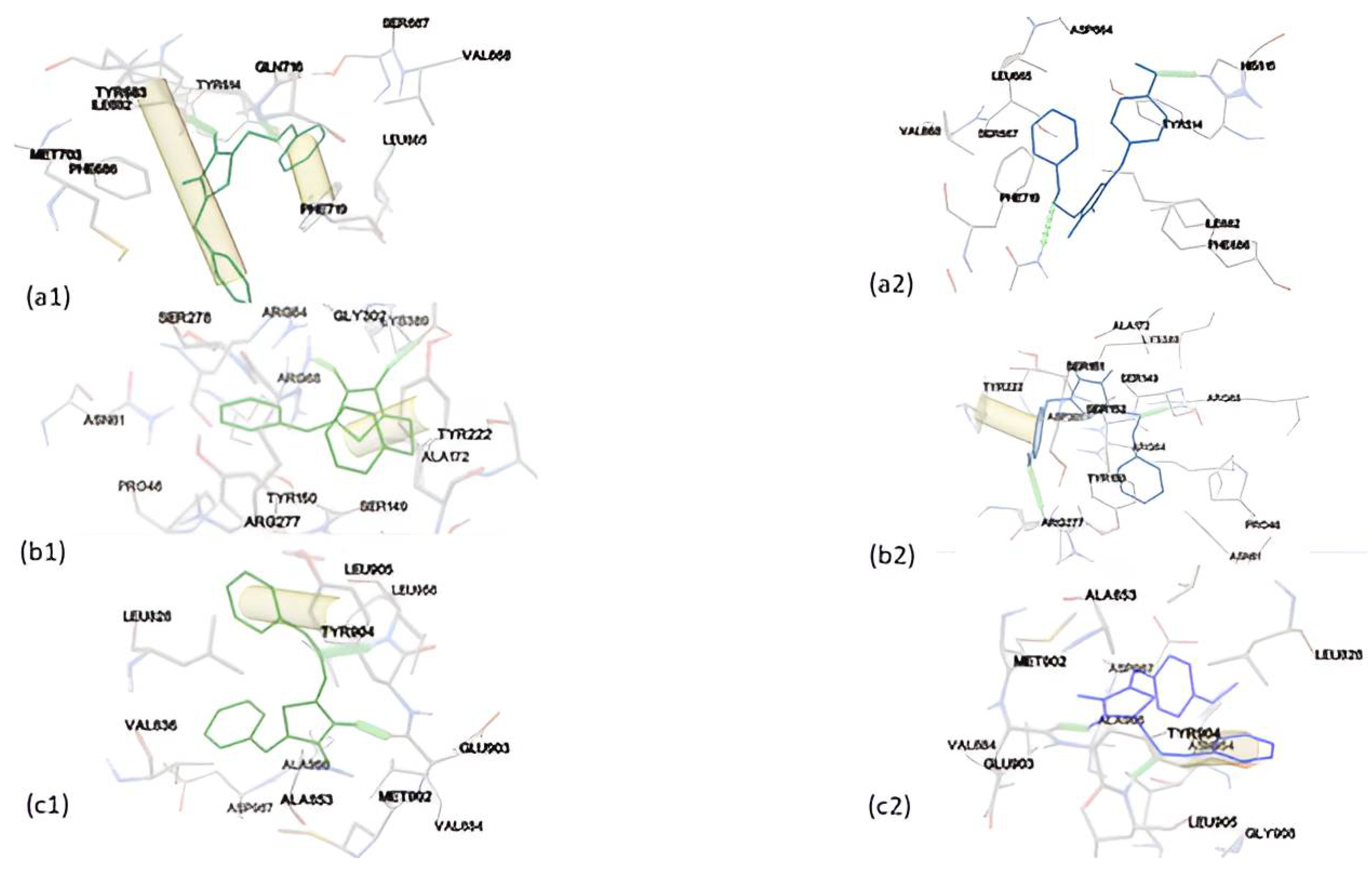
Figure 6.
Binding mode of DKI39 (left, green color) with (a) triazoloquinazolines, (b) mglur3, (c) Jak3, (d) Danio rerio HDAC6 CD2 and (e) acetylcholinesterase. Binding modes of DKI40 (right, blue color) with (a) triazoloquinazolines, (b) mglur3, (c) Jak3, (d) Danio rerio HDAC6 CD2 and (e) acetylcholinesterase.
Figure 6.
Binding mode of DKI39 (left, green color) with (a) triazoloquinazolines, (b) mglur3, (c) Jak3, (d) Danio rerio HDAC6 CD2 and (e) acetylcholinesterase. Binding modes of DKI40 (right, blue color) with (a) triazoloquinazolines, (b) mglur3, (c) Jak3, (d) Danio rerio HDAC6 CD2 and (e) acetylcholinesterase.
Figure 7.
Comparison of binding free energies during repeated simulations (3 replicas) of DKI39 and DKI40 complexes in aqueous solution.
Figure 7.
Comparison of binding free energies during repeated simulations (3 replicas) of DKI39 and DKI40 complexes in aqueous solution.
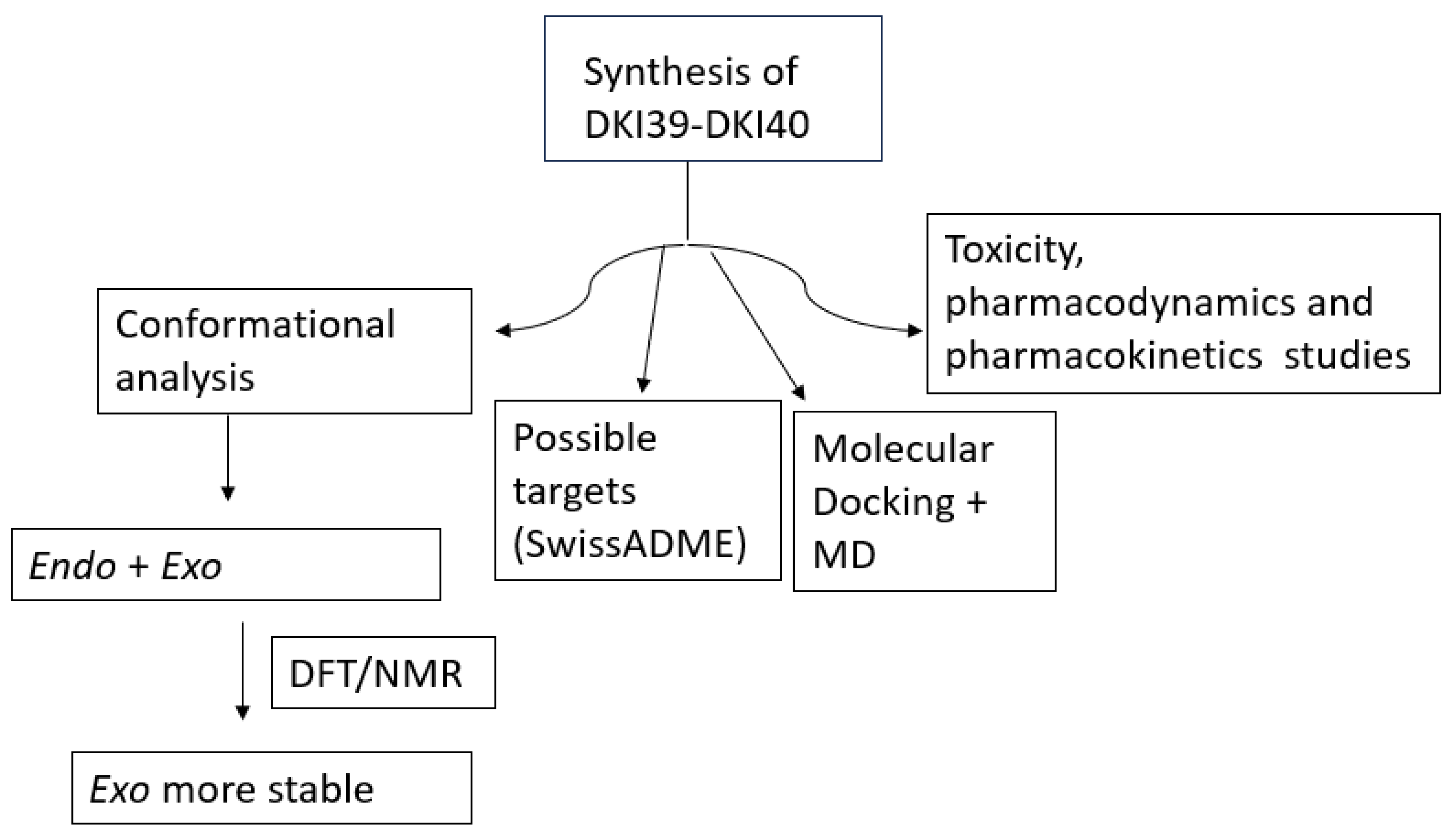
Scheme 9.
Summary of the scientific work described in the manuscript.
Scheme 9.
Summary of the scientific work described in the manuscript.
Table 1.
Assignment of the experimental 1H NMR spectra of DKI39 and DKI40 in DMSO-d6.
Table 1.
Assignment of the experimental 1H NMR spectra of DKI39 and DKI40 in DMSO-d6.
| Hydrogen DKI39 | Chemical Shift (ppm) | Hydrogen DKI39 | Chemical Shift (ppm) |
| 1 | 7.68 | 12 | 7.63 |
| 2 | 7.50 | 14 | 7.47 |
| 3 | 7.85 | 15 | 7.57 |
| 5 | 8.53 | 16 | 7.47 |
| Hydrogen DKI40 | Chemical Shift (ppm) | Hydrogen DKI40 | Chemical Shift (ppm) |
| 1 | 7.36 | 12 | 7.58 |
| 2 | 7.50 | 14 | 7.62 |
| 3 | 7.83 | 15 | 7.12 |
| 5 | 8.40 | 18 | 3.86 |
Table 2.
Relative energies (ΔΕ in kcal/mol) and dihedral angles (τ in degrees) of the minimized structures for DKI39 endo and DKI40 endo compounds a.
Table 2.
Relative energies (ΔΕ in kcal/mol) and dihedral angles (τ in degrees) of the minimized structures for DKI39 endo and DKI40 endo compounds a.
| DKI39 endo | ΔΕ | τ2 | τ1 | τ3 |
| DKI39en_1 | 0 | Z (180.0) | E (−180.0) | - |
| DKI39en_2 | 4.79 | Z (180.0) | Z (−2.2) | - |
| DKI39en_3 | 7.91 | E (0.8) | E (−179.9) | - |
| DKI39en_4 | 9.71 | E (−0.3) | Z (2.2) | - |
| DKI40 endo | ΔΕ | τ3′ | τ1′ | τ2′ |
| DKI40en_1 | 0 | Z (180.0) | E (180.0) | - |
| DKI40en_3 | 4.63 | E (0.1) | E (180.0) | - |
| DKI40en_4 | 5.00 | Z (−180.0) | Z (2.2) | - |
| DKI40en_2 | 9.39 | E (0.1) | Z (−2.03) | - |
Table 3.
Relative energy differences of the conformers with the respect to the global minimum, ΔΕ, and calculated dihedral angles (τ1, τ2, τ3) for DKI39 exo and DKI40 exo compounds a.
Table 3.
Relative energy differences of the conformers with the respect to the global minimum, ΔΕ, and calculated dihedral angles (τ1, τ2, τ3) for DKI39 exo and DKI40 exo compounds a.
| DKI39 exo | ΔΕ | τ2 | τ1 | τ3 |
| DKI39ex_1 | 0 | Z (180.0) | Z(−180.00) | E (−180.00) |
| DKI39ex_4 | 1.54 | Z (−180.0) | E (180.0) | E (−0.0) |
| DKI39ex_2 | 3.98 | Z (180.0) | Z (−0.2) | Z (180.0) |
| DKI39ex_8 | 5.17 | E (0.0) | Z (−180.0) | E (−180.0) |
| DKI39ex_3 | 5.59 | Z (180.0) | E (−1.5) | Z (−1.53) |
| DKI39ex_5 | 6.67 | E (0.16) | E (−180.0) | E (0.0) |
| DKI39ex_7 | 9.09 | E (0.0) | Z (−0.11) | Z (180.0) |
| DKI39ex_6 | 10.71 | E (0.1) | E (−0.0) | Z (−0.0) |
| DKI40 exo | ΔΕ | τ3′ | τ1′ | τ2′ |
| DKI40ex_1 | 0 | Z (180.0) | Z (180.0) | E (0.0) |
| DKI40ex_4 | 1.53 | Z (−0.0) | E (180.0) | E (−0.0) |
| DKI40ex_2 | 3.95 | Z (−180.0) | Z (−0.0) | Z (180.0) |
| DKI40ex_6 | 4.75 | E (−0.0) | Z (−180.0) | E (180.0) |
| DKI40ex_3 | 5.69 | Z (180.0) | E (−0.9) | Z (−0.9) |
| DKI40ex_5 | 6.40 | E (0.0) | E (180.0) | E (−0.0) |
| DKI40ex_8 | 8.63 | E (0.0) | Z (0.0) | Z (180.0) |
| DKI40ex_7 | 10.52 | E (−180.0) | E (−0.04) | Z (180.0) |
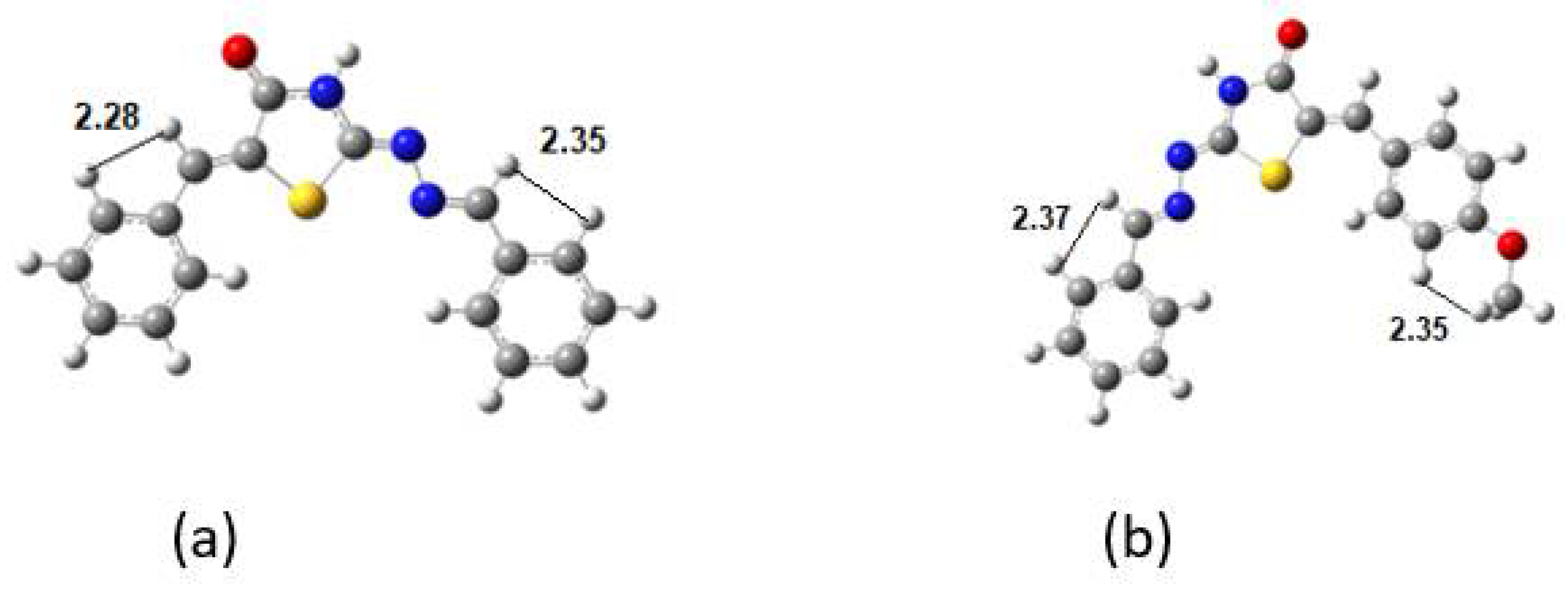
Table 4.
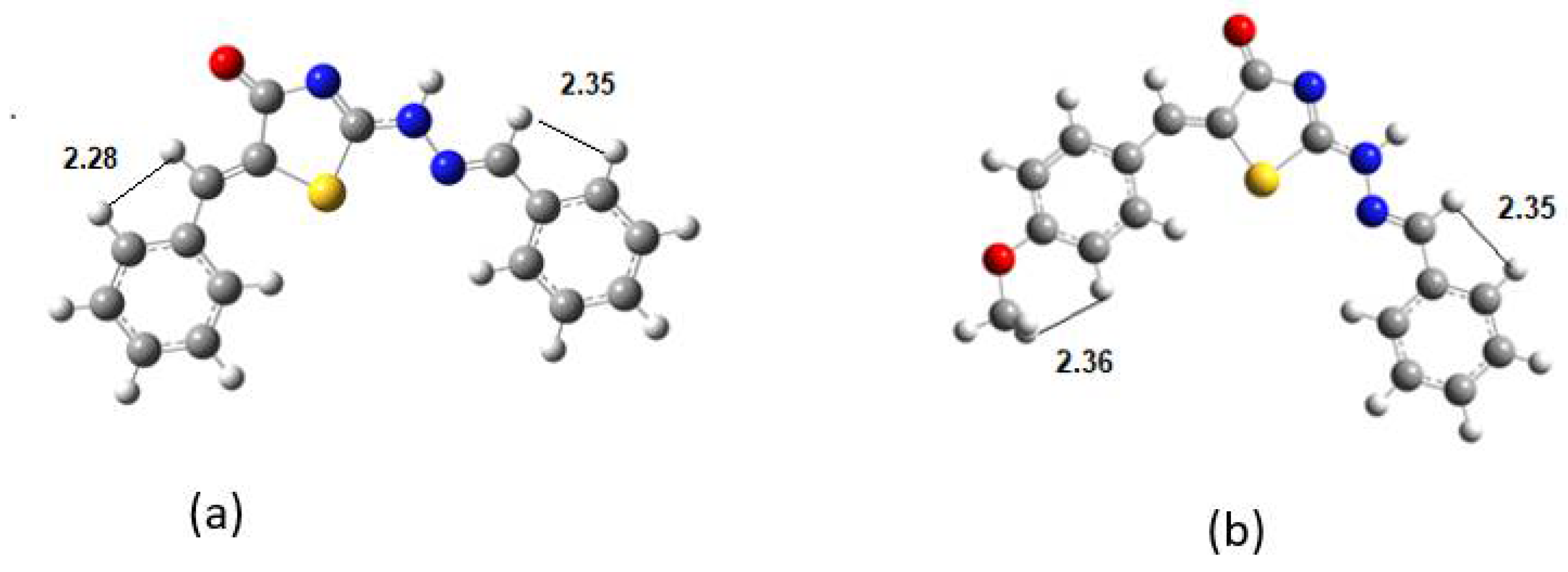
Selected H-H distances in Å at DFT.
Table 4.
Selected H-H distances in Å at DFT.
| DKI39b | H-H | DKI40a | H-H |
|---|
| H12-H14 | 2.280 | H18-H15 | 2.352 |
| H5-H3 | 2.354 | H5-H3 | 2.373 |
Table 5.
Molecular docking results for each compound with the macromolecules.
Table 5.
Molecular docking results for each compound with the macromolecules.
| Binding Energy (kcal/mol) |
|---|
| Macromolecules | DKI39 | DKI40 |
|---|
| 2Y0J (Triazoloquinazolines) | −8.22 | −7.82 |
| 5CNK (mglur3) | −9.16 | −6.19 |
| 5TTV (Jak3) | −8.95 | −8.83 |
| 8A8Z (Danio rerio HDAC6 CD2) | −8.23 | −7.94 |
| 4EY7 (Acetylcholinesterase) | −8.95 | −8.92 |
Table 6.
Drug-likeness of the compounds.
Table 6.
Drug-likeness of the compounds.
| Properties | Compound DK139 | Compound DKI40 |
|---|
| Molecular Weight | 307.37 | 337.40 |
| LogP | 2.66 | 3.08
|
| Rotable bonds | 3 | 4 |
| Hydrogen Bond Acceptors | 3 | 4 |
| Hydrogen Bond Donors | 1 | 1 |
| Surface Area | 79.12 (Å2) | 88.35 (Å2) |
| Water solubility | −4.54 (mol/L) | −4.61 (mol/L) |
Table 7.
ADME results according to preADMET.
Table 7.
ADME results according to preADMET.
| | Compound DKI39 | Compound DKI40 |
|---|
| BBB | 0.565933 | 0.304397 |
| Buffer_solubility_mg_L | 171.672 | 163.776 |
| Caco2 | 22.0759 | 21.9635 |
| CYP_2C19_inhibition | Non | Non |
| CYP_2C9_inhibition | Non | Non |
| CYP_2D6_inhibition | Non | Non |
| CYP_2D6_substrate | Non | Non |
| CYP_3A4_inhibition | Non | Non |
| CYP_3A4_substrate | Weakly | Weakly |
| HIA | 96.387179 | 96.785453 |
| MDCK | 11.204 | 2.28162 |
| Pgp_inhibition | Inhibitor | Inhibitor |
| Plasma_Protein_Binding | 94.011663 | 93.567531 |
| Pure_water_solubility_mg_L | 0.562912 | 0.382868 |
| Skin_Permeability | −2.82467 | −3.03751 |
Table 8.
Toxicity results for both compounds according to pKCSm.
Table 8.
Toxicity results for both compounds according to pKCSm.
| Properties | Compound DKI39 | Compound DKI40 |
|---|
| Toxicity | | |
| AMES toxicity | Yes | Yes |
| Max. tolerated dose (human) | 0.064 (log mg/kg/day) | 0.074 (log mg/kg/day) |
| Herg I inhibitor | No | No |
| Herg II inhibitor | Yes | No |
| Oral rat acute toxicity (LD50) | 2.318 (mol/kg) | 2.268 (mol/kg) |
| Oral rat chronic toxicity | 1.432 (log mg/kg_bw/day) | 1.723 (log mg/kg_bw/day) |
| Hepatotoxicity | No | No |
| Skin sensitization | No | No |


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}