3.1. Chemistry
All coupling reactions were carried out under the protection of argon, strictly in accordance with anhydrous and oxygen-free operation. All raw materials were commercial reagents or prepared using methods reported in the literature. The reactions were monitored using UV light, phosphomolybdic acid, potassium permanganate, or iodine bath. All compounds were purified via column chromatography, and the final products were purified using HPLC, acidified using 10% dilute hydrochloric acid solution, and freeze-dried using a lyophilizer. All types of Nuclear magnetic resonance spectrometer (NMR) data were acquired with a Bruker AV-400 (Bruker, Billerica, MA, USA) (400 MHz for 1H NMR and 101 MHz for 13C NMR). Chemical shifts (δ) are given in ppm, and all spectra were corrected with solvent peaks as follows: Chloroform-d 7.26 ppm H, 77.16 ppm C; DMSO-d6 2.50 ppm H, 39.52 ppm C. The unit used for the coupling constant (J) is hertz (Hz), and the divisions were abbreviated as follows: s, singlet; d, doublet; t, triplet; q, quartet; dd, doublet of doublet; dt, doublet of triplet; m, multiplet. Yield refers to the yield after purification. All compounds subjected to biological testing were more than 95% pure. Low-resolution ionization mass spectrometry (ESI-MS) was performed with Agilent 1260/6120 ES-LCMS system (Agilent, Santa Clara, CA, USA) or Shimadzu LCMS 2020 (Shimadzu, Kyoto, Japan). High-resolution mass spectra (HRMS) were collected with Thermo Fisher Scientific LTQ FTICR-MS or Thermo Scientific Q Exactive HF Orbitrap-FTMS (Thermo Fisher Scientific, Waltham, MA, USA).
3.1.1. N,3,4-Trimethoxy-N-methylbenzamide [30] (2a)
To a solution of 3,4-dimethoxybenzoic acid (10 g, 54.9 mmol) in CH2Cl2 (100 mL) in an ice bath, we added N,O-dimethylhydroxylamine hydrochloride (6.42 g, 65.9 mmol), HATU (25 g, 65.9 mmol), and TEA (38 mL, 275 mmol). The mixture was stirred at room temperature for 3 h and then concentrated under reduced pressure. The residue mixture was diluted with ethyl acetate (100 mL) and washed with saturated ammonium chloride, saturated sodium bicarbonate, and saturated brine solutions successively. The organic layers were dried with anhydrous sodium sulfate, concentrated, and purified via silica gel column chromatography (20% EtOAc in Pet. Ether), yielding 11.3 g (yield 91%) of light-yellow solid (2a). ESI-MS: m/z = 226.1 (M + H)+. 1H NMR (400 MHz, CDCl3) δ 7.38 (dd, J = 8.4, 2.0 Hz, 1H), 7.31 (d, J = 1.9 Hz, 1H), 6.86 (d, J = 8.4 Hz, 1H), 3.90 (d, J = 5.0 Hz, 6H), 3.57 (s, 3H), 3.35 (s, 3H).
3.1.2. Cyclopentyl(3,4-dimethoxyphenyl)methanone [31] (3a)
Cyclopentyl magnesium bromide (13.3 mL, 26.6 mmol, 2.0 M in THF) was added dropwise to a solution of 2a (2 g, 8.9 mmol) in THF (30 mL) at −78 °C. After 30 min, the low-temperature equipment was removed, and the mixture was stirred at rt for 2 h. The mixture was then quenched by dropwise addition of a saturated ammonium chloride solution at 0 °C and extracted with EtOAc, dried over Na2SO4, and concentrated under reduced pressure. The residue mixture was purified via silica gel column chromatography (5% EtOAc in Pet. Ether), yielding 1.2 g (yield 57.7%) of 3a in the form of a colorless oil. ESI-MS: m/z = 235.1 (M + H)+. 1H NMR (400 MHz, CDCl3) δ 7.63–7.55 (m, 2H), 6.90 (d, J = 8.4 Hz, 1H), 3.95 (d, J = 3.9 Hz, 6H), 3.70 (p, J = 7.9 Hz, 1H), 1.96–1.86 (m, 4H), 1.80–1.59 (m, 4H).
3.1.3. (1-Bromocyclopentyl)(3,4-dimethoxyphenyl)methanone [32] (4a)
Cupric bromide (476 mg, 2.13 mmol) was added to a solution of 3a (200 mg, 0.85 mmol) in EtOAc (5 mL). The mixture was refluxed at 80 °C for 2h. Then, the reaction mixture was directly concentrated and purified via silica gel column chromatography (5% EtOAc in Pet. Ether), yielding 4a (0.4 g, yield 86%) as a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.93 (dd, J = 8.5, 2.1 Hz, 1H), 7.69 (d, J = 2.1 Hz, 1H), 6.89 (d, J = 8.5 Hz, 1H), 3.95 (d, J = 7.3 Hz, 6H), 2.56–2.40 (m, 4H), 2.13–2.00 (m, 2H), 1.87–1.73 (m, 2H).
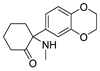
3.1.4. 2-(3,4-Dimethoxyphenyl)-2-(methylamino)cyclohexan-1-one (6)
KOH (90 mg, 1.6 mmol) was added to a solution of 4a (0.25 g, 0.8 mmol) in MeOH (5 mL). The mixture was stirred at rt for 5h, diluted with EtOAc (20 mL), washed with saturated aqueous NaCl, and dried over Na2SO4. It was then concentrated and purified using silica gel column chromatography (5% EtOAc in Pet. Ether) to attain 5a (0.13 g, yield 65%) in the form of a colorless oil. ESI-MS: m/z = 251.1 (M + H)+.
Methylamine aqueous solution (40%) was added to a solution of 5a (50 mg, 0.2 mmol) in decahydronaphthalene (2 mL) in a 15 mL sealed tube. The mixture was stirred at 170 °C for 6 h. After cooling, it was extracted with EtOAc and concentrated. The mixture was purified using prep-HPLC and then acidified with 10% HCl and lyophilized to obtain 6 (21 mg, 40%) as white solid. 1H NMR (400 MHz, CDCl3) δ 10.15 (s, 1H), 9.91 (s, 1H), 7.14 (d, J = 1.9 Hz, 1H), 7.00 (d, J = 8.2 Hz, 1H), 6.92 (d, J = 8.3 Hz, 1H), 3.98 (s, 3H), 3.91 (s, 3H), 3.04 (d, J = 13.4 Hz, 1H), 2.69–2.32 (m, 6H), 2.08–1.76 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 204.80, 150.32(2C), 122.25, 120.90, 71.94, 56.63, 55.98, 39.23, 32.61, 27.08, 27.03, 22.00. ESI-MS: m/z = 264.1 (M + H)+. HRMS m/z: calcd for C15H20NO3 264.1594; found, 264.1599.
3.1.5. 2-(2,3-Dihydrobenzo[b][1,4]dioxin-6-yl)-2-(methylamino)cyclohexan-1-one (7)
Compounds 7 and 8 were synthesized in a process similar to that described for the preparation of 1. 1H NMR (400 MHz, CDCl3) δ 10.14 (s, 1H), 9.69 (s, 1H), 6.98 (d, J = 19.6 Hz, 3H), 4.28 (s, 4H), 3.05 (d, J = 13.1 Hz, 1H), 2.66–2.33 (m, 6H), 2.03–1.64 (m, 4H). 13C NMR (101 MHz, CDCl3) δ 204.64, 145.04, 144.31, 122.98, 121.78, 118.68, 117.64, 71.56, 64.40, 64.25, 39.20, 33.03, 27.44, 27.08, 21.76.
ESI-MS: m/z = 262.1 (M + H)+. HRMS m/z: calcd for C15H20NO3 262.1438; found, 262.1445.
3.1.6. 2-(Benzo[d][1,3]dioxol-5-yl)-2-(methylamino)cyclohexan-1-one (8)
The result was a white solid. 1H NMR (400 MHz, DMSO-d6) δ 10.09 (dd, J = 11.5, 5.4 Hz, 1H), 9.31 (dd, J = 11.4, 5.5 Hz, 1H), 7.07 (d, J = 8.2 Hz, 1H), 7.01 (d, J = 1.8 Hz, 1H), 6.83 (dd, J = 8.2, 1.9 Hz, 1H), 6.12 (dd, J = 3.5, 0.8 Hz, 2H), 5.98 (s, 1H), 3.15–3.01 (m, 1H), 2.42–2.29 (m, 2H), 2.12 (q, J = 5.6 Hz, 4H), 2.03–1.76 (m, 2H), 1.69–1.51 (m, 2H). 13C NMR (101 MHz, DMSO-d6) δ 206.34, 148.98, 148.82, 124.11, 123.33, 109.49, 108.70, 102.39, 71.15, 31.74, 27.32, 26.94, 21.66. ESI-MS: m/z = 248.1 (M + H)+. HRMS m/z: calcd for C14H18NO3 248.1281; found, 248.1288.
3.1.7. 2-Phenylcycloheptan-1-one [33] (10b)
Pd2(dba)3 (292 mg, 0.32 mmol), Xantphos (370 mg, 0.64 mmol), and cesium carbonate (22.8 g, 70 mmol) were dissolved in 1,4-dioxane (50 mL). After the air was replaced with N2, bromobenzene (5 g, 32 mmol) and cycloheptanone (7.17 g, 64 mmol) were added. The mixture was stirred at 100 °C for 16 h. After cooling, it was poured into water (50 mL) and extracted with EtOAc (50 mL × 3). The combined organic layers were dried with anhydrous Na2SO4, concentrated, and purified via silica gel column chromatography (3% EtOAc in Pet. Ether) to obtain 10b (7.55 g, 100%) as yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.35–7.29 (m, 2H), 7.26–7.20 (m, 3H), 3.72 (dd, J = 11.4, 4.2 Hz, 1H), 2.74–2.65 (m, 1H), 2.57–2.48 (m, 1H), 2.19–2.11 (m, 1H), 2.09–1.91 (m, 4H), 1.71–1.59 (m, 1H), 1.52–1.40 (m, 2H). ESI-MS: m/z = 189.1 (M + H)+.
3.1.8. 2-Nitro-2-phenylcyclohexan-1-one [30] (11a)
Copper(II) acetate (211 mg, 1.16 mmol) and ceric ammonium nitrate (8 g, 14.5 mmol) were added to a solution of 2-phenylcyclohexan-1-one (2 g, 11.48 mmol) in DCE (20 mL) in a nitrogen atmosphere. The mixture was stirred at 80 °C for 16 h. The mixture was filtrated using celite, and the filtrate was concentrated and purified via silica gel column chromatography (5% EtOAc in Pet. Ether), yielding 11a (0.49 g, yield 33%) as yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.49–7.43 (m, 3H), 7.39–7.32 (m, 2H), 3.11–3.02 (m, 1H), 2.94–2.85 (m, 1H), 2.71–2.63 (m, 1H), 2.60–2.51 (m, 1H), 1.99–1.87 (m, 3H), 1.83–1.70 (m, 1H).
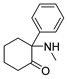
3.1.9. 2-Amino-2-phenylcyclohexan-1-one [34] (12a)
Zinc dust (1.09 g, 16.83 mmol) was added to a solution of 11a (1.23 g, 5.61 mmol) in HOAc (10 mL). The mixture was stirred at 80 °C for 10h; then, it was concentrated, and its pH was rendered basic with 10% sodium hydroxide solution. The mixture was filtrated, and the filtrate was extracted with EtOAc 3 times. The combined organic layers were concentrated under reduced pressure and purified using silica gel column chromatography (50% EtOAc in Pet. Ether), yielding 12a (0.72g, 67.8%) as colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.41–7.35 (m, 2H), 7.32–7.24 (m, 3H), 2.92–2.81 (m, 1H), 2.50–2.34 (m, 2H), 2.04–1.90 (m, 3H), 1.84–1.63 (m, 4H). ESI-MS: m/z = 190.1 (M + H)+.
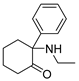
3.1.10. 2-(Ethylamino)-2-phenylcyclohexan-1-one hydrochloride (13)
To a solution of 12a (82 mg, 0.43 mmol) in MeOH (3 mL), we added acetaldehyde (29 μL, 0.52 mmol), HOAc (25 μL, 0.43 mmol), and NaBH3CN (41 mg, 0.65 mmol). The mixture was stirred at rt for 1.5 h and then extracted with EtOAc and washed with saturated brine. The combined organic layers were concentrated and purified using prep-HPLC and then acidified using 10% HCl and lyophilized to obtain 13 (56 mg, 60%) as white solid. 1H NMR (400 MHz, CDCl3) δ 7.37 (t, J = 7.5 Hz, 2H), 7.31–7.21 (m, 3H), 2.94–2.85 (m, 1H), 2.44–2.37 (m, 1H), 2.36–2.24 (m, 2H), 2.14 (s, 1H), 2.08–2.00 (m, 1H), 1.98–1.92 (m, 1H), 1.89–1.69 (m, 4H), 0.99 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 211.46, 139.38, 128.82 (2C), 127.43, 127.01 (2C), 69.79, 39.74, 36.58, 36.01, 27.70, 22.34, 15.65. ESI-MS: m/z = 218.1 (M + H)+. HRMS m/z: calcd for C14H20NO 218.1548; found, 218.1548.
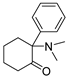
3.1.11. 2-(Dimethylamino)-2-phenylcyclohexan-1-one (14)
Compound 14 was synthesized from 12a and HCHO (37% in water, 2.5eq) in a process similar to that described for the preparation of 13. The result was a white solid. 1H NMR (400 MHz, CDCl3) δ 12.05 (s, 1H), 7.58–7.46 (m, 5H), 3.34 (d, J = 12.6 Hz, 1H), 2.85 (d, J = 4.3 Hz, 3H), 2.64 (d, J = 4.3 Hz, 3H), 2.59–2.52 (m, 2H), 2.51–2.41 (m, 1H), 2.02–1.88 (m, 2H), 1.86–1.71 (m, 1H), 1.63–1.47 (m, 1H). 13C NMR (101 MHz, CDCl3) δ 205.36, 130.89(2C), 130.15, 130.02(2C), 129.54, 77.87, 40.77, 40.30, 40.27, 33.11, 26.94, 21.76. ESI-MS: m/z = 218.2 (M + H)+. HRMS m/z: calcd for C14H20NO 218.1539; found, 215.1546.
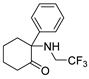
3.1.12. 2-Phenyl-2-((2,2,2-trifluoroethyl)amino)cyclohexan-1-one (15)
TEA (80 μL) and 2,2,2-trifluoroethyl trifluoromethanesulfonate (33 μL, 0.23 mmol) were added to a solution of 12a (36 mg, 0.19 mmol) in DMF (2 mL). The mixture was stirred at 90 °C for 12 h. After cooling, the mixture was extracted with EtOAc and washed with saturated brine. The combined organic layers were dried, concentrated, and purified using silica gel column chromatography (50% EtOAc in Pet. Ether), yielding 15 (25 mg, 49%) as colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.64–7.42 (m, 5H), 3.73–3.46 (m, 1H), 2.95–2.72 (m, 3H), 2.70–2.47 (m, 4H), 2.07–1.76 (m, 3H), 1.62–1.41 (m, 1H). 19F NMR (376 MHz, CDCl3) δ −67.42. 13C NMR (101 MHz, CDCl3) δ 206.57, 131.70, 130.45, 130.04, 122.75 (d, J = 279.4 Hz), 74.23, 45.22 (q, J = 36.3, 35.5 Hz), 40.32, 36.19, 27.43, 21.79. ESI-MS: m/z = 272.2 (M + H)+. HRMS m/z: calcd for C14H17F3NO 272.1257; found, 272.1264.
3.1.13. N-(2-Oxo-1-phenylcyclohexyl)acetamide [35] (16)
To a solution of 12a (36 mg, 0.19 mmol) in DCM, we added TEA (80 μL) and acetic anhydride (27 μL, 0.19 mmol). The mixture was stirred at rt for 2h and then concentrated and purified using prep-HPLC and lyophilized to obtain 16 (25 mg, 56.8%) as colorless oil. 1H NMR (400 MHz, CDCl3) δ 7.45–7.29 (m, 5H), 3.96–3.79 (m, 1H), 2.83 (s, 1H), 2.45–2.26 (m, 2H), 2.07–1.97 (m, 1H), 1.95–1.69 (m, 7H). 13C NMR (101 MHz, CDCl3) δ 208.66, 200.02, 137.32, 128.69(2C), 127.92, 127.54(2C), 77.23, 38.50, 35.57, 28.41, 24.07, 22.38. ESI-MS: m/z = 232.1 (M + H)+. HRMS m/z: calcd for C14H17NNaO2 254.1151; found, 254.1159.
3.1.14. 2,2,2-Trifluoro-N-(2-oxo-1-phenylcyclohexyl)acetamide (17)
Compound 17 was synthesized from 12a and trifluoroacetic anhydride in a process similar to that described for the preparation of 16. 1H NMR (400 MHz, CDCl3) δ 8.30 (s, 1H), 7.45–7.30 (m, 5H), 3.90–3.81 (m, 1H), 2.51–2.32 (m, 2H), 2.11–2.01 (m, 1H), 1.94–1.70 (m, 4H). 19F NMR (376 MHz, CDCl3) δ -76.14. ESI-MS: m/z = 286.1 (M + H)+. HRMS m/z: calcd for C14H14F3NNaO2 308.0869; found, 308.0875.
3.1.15. 2-(Methylamino)-2-phenylcyclohexan-1-one [34] (18)
To a solution of 12a (86 mg, 0.45 mmol) in THF (3 mL), we added di-tert-butyl dicarbonate (149 mg, 0.44 mmol) and TEA (0.1 mL, 0.9 mmol). The mixture was refluxed at 70 °C for 3 h and then concentrated and purified using silica gel column chromatography (10% EtOAc in Pet. Ether), yielding theyellow oil (92 mg, 70.7%). Then, the compound was dissolved in THF, and LiAlH4 (15 mg, 0.39 mmol) was added. The mixture was refluxed for 1 h, quenched using sodium hydroxide solution (10%), and extracted with EtOAc. The combined organic layers were concentrated under reduced pressure and dissolved in acetone (3 mL); then, Jones reagent (1 mL) was added. The mixture was stirred for 1 h at rt, and the pH was rendered basic using hydroxide solution (10%). The mixture was extracted with EtOAc and concentrated under reduced pressure and then purified using prep-HPLC to obtain 18 (55 mg, 69.6%) as white solid. 1H NMR (400 MHz, CDCl3) δ 7.39 (t, J = 7.6 Hz, 2H), 7.34–7.23 (m, 3H), 2.92 (dd, J = 14.1, 3.3 Hz, 1H), 2.70 (s, 1H), 2.45–2.39 (m, 1H), 2.37–2.28 (m, 1H), 2.07 (s, 3H), 2.00–1.80 (m, 3H), 1.80–1.67 (m, 2H). 13C NMR (101 MHz, CDCl3) δ 210.69, 137.58, 129.00, 127.92, 127.32, 70.15, 39.75, 34.96, 28.71, 27.71, 22.20. ESI-MS: m/z = 204.2 (M + H)+. HRMS m/z: calcd for C13H18NO 204.1383; found, 204.1389.
3.1.16. 2-Amino-2-phenylcycloheptanone (19)
Compound 19 was synthesized in a process similar to that described for the preparation of 12a. The result was a white solid. 1H NMR (400 MHz, CDCl3) δ 7.38–7.31 (m, 4H), 7.30–7.24 (m, 1H), 2.71–2.63 (m, 1H), 2.43–2.22 (m, 3H), 2.10 (s, 2H), 2.00–1.86 (m, 3H), 1.60–1.36 (m, 3H). 13C NMR (101 MHz, CDCl3) δ 214.25, 144.19, 128.91(2C), 127.55, 125.73(2C), 67.90, 40.52, 37.05, 30.49, 27.30, 24.01. ESI-MS: m/z = 204.2 (M + H)+. HRMS m/z: calcd for C13H18NO 204.1383; found, 204.1388.
3.1.17. 2-(Ethylamino)-2-phenylcycloheptan-1-one (20)
Compound 20 was synthesized in a process similar to that described for the preparation of 13. The result was a white solid. 1H NMR (400 MHz, CDCl3) δ 7.37 (t, J = 7.5 Hz, 2H), 7.31–7.21 (m, 3H), 2.94–2.85 (m, 1H), 2.44–2.37 (m, 1H), 2.36–2.24 (m, 2H), 2.14 (s, 1H), 2.08–2.00 (m, 1H), 1.98–1.92 (m, 1H), 1.89–1.69 (m, 4H), 0.99 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 211.89, 140.08, 128.63 (2C), 127.52, 127.20 (2C), 72.12, 40.01, 36.83, 31.30, 30.34, 27.21, 23.55, 15.49. ESI-MS: m/z = 232.2 (M + H)+. HRMS m/z: calcd for C15H22NO 232.1696; found, 232.1704.
3.1.18. 2-(Methylamino)-2-phenylcycloheptan-1-one (21)
Compound 21 was synthesized in a process similar to that described for the preparation of 18. The result was a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.39–7.31 (m, 2H), 7.30–7.23 (m, 3H), 2.58–2.49 (m, 1H), 2.38–2.24 (m, 2H), 2.20–2.08 (m, 2H), 1.96 (s, 3H), 1.93–1.81 (m, 3H), 1.59–1.39 (m, 3H). 13C NMR (101 MHz, CDCl3) δ 212.16, 139.71, 128.63 (2C), 127.46, 127.25 (2C), 71.99, 40.01, 30.44, 30.18, 28.82, 27.40, 23.61. ESI-MS: m/z = 218.2 (M + H)+. HRMS m/z: calcd for C14H20NO 218.1539; found, 218.1545.
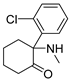
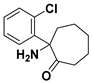
3.1.19. 2-Amino-2-(2-chlorophenyl)cycloheptanone (22)
Compound 22 was synthesized in a process similar to that described for the preparation of 12a. The result was a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.50 (d, J = 7.7 Hz, 1H), 7.37 (d, J = 7.7 Hz, 1H), 7.26 (dt, J = 23.1, 7.3 Hz, 2H), 2.75–2.61 (m, 2H), 2.47–2.35 (m, 1H), 2.10 (s, 2H), 1.97–1.85 (m, 1H), 1.82–1.54 (m, 6H). 13C NMR (101 MHz, CDCl3) δ 214.13, 142.09, 132.90, 131.29, 128.76, 127.98, 127.15, 68.38, 40.02, 38.26, 28.34, 23.82, 23.71. ESI-MS: m/z = 238.1 (M + H)+. HRMS m/z: calcd for C13H17ClNO 238.0993; found, 238.0995.
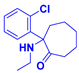
3.1.20. 2-(2-Chlorophenyl)-2-(ethylamino)cycloheptan-1-one hydrochloride (23)
Compound 23 was synthesized in a process similar to that described for the preparation of 13. The result was a white solid. 1H NMR (400 MHz, CDCl3) δ 10.74 (s, 1H), 8.80 (s, 1H), 8.17 (d, J = 7.7 Hz, 1H), 7.55–7.47 (m, 1H), 7.44–7.35 (m, 2H), 3.51–3.28 (m, 2H), 2.85–2.76 (m, 1H), 2.70–2.60 (m, 1H), 2.58–2.45 (m, 2H), 1.87–1.77 (m, 4H), 1.76–1.71 (m, 1H), 1.48 (t, J = 7.1 Hz, 3H), 1.41–1.29 (m, 1H). 13C NMR (101 MHz, CDCl3) δ 204.67, 133.48, 133.17, 132.17, 131.49, 130.88, 128.74, 75.02, 40.63, 40.11, 36.87, 29.61, 25.63, 23.57, 12.27. ESI-MS: m/z = 266.1 (M + H)+. HRMS m/z: calcd for C15H21ClNO 266.1306; found, 266.1315.
3.1.21. 2-(2-Chlorophenyl)-2-(methylamino)cycloheptan-1-one (24)
Compound 24 was synthesized in a process similar to that described for the preparation of 18. The result was a yellow oil. 1H NMR (400 MHz, CDCl3) δ 7.45 (dd, J = 7.8, 1.7 Hz, 1H), 7.36 (dd, J = 7.7, 1.6 Hz, 1H), 7.32–7.20 (m, 2H), 2.75–2.66 (m, 1H), 2.49 (ddd, J = 12.4, 10.5, 5.5 Hz, 1H), 2.32 (s, 1H), 2.21–2.11 (m, 2H), 2.02 (s, 3H), 1.89–1.66 (m, 4H), 1.64–1.54 (m, 1H), 1.50–1.37 (m, 1H). 13C NMR (101 MHz, CDCl3) δ 211.37, 138.69, 133.51, 131.25, 129.93, 128.66, 126.57, 73.01, 39.43, 34.00, 29.25, 28.35, 24.06, 23.73. ESI-MS: m/z = 252.1 (M + H)+. HRMS m/z: calcd for C14H19ClNO 252.1150; found, 252.1152.
3.1.22. 2-Amino-2-(naphthalen-1-yl)cycloheptan-1-one (25)
Compound 25 was synthesized in a process similar to that described for the preparation of 12a. The result was a yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.19 (dd, J = 6.4, 3.4 Hz, 1H), 7.87 (dd, J = 6.3, 3.3 Hz, 1H), 7.81 (d, J = 8.2 Hz, 1H), 7.57 (d, J = 7.3 Hz, 1H), 7.50–7.41 (m, 3H), 2.67–2.58 (m, 1H), 2.58–2.49 (m, 1H), 2.37–2.28 (m, 1H), 2.23–2.08 (m, 3H), 1.95–1.82 (m, 1H), 1.74–1.51 (m, 5H). 13C NMR (101 MHz, CDCl3) δ 216.15, 138.51, 134.96, 131.36, 129.38, 129.03, 126.37, 125.58, 125.05, 125.03, 124.49, 69.22, 40.55, 39.29, 28.52, 24.44, 23.62. ESI-MS: m/z = 254.2 (M + H)+. HRMS m/z: calcd for C17H20NO 254.1539; found, 254.1546.
3.1.23. 2-(Ethylamino)-2-(naphthalen-1-yl)cycloheptan-1-one (26)
Compound 26 was synthesized in a process similar to that described for the preparation of 13. The result was a yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.39 (d, J = 9.5 Hz, 1H), 7.88–7.78 (m, 2H), 7.60 (d, J = 7.4 Hz, 1H), 7.49–7.41 (m, 3H), 2.64 (dd, J = 14.2, 6.9 Hz, 1H), 2.45–2.32 (m, 2H), 2.22–2.10 (m, 2H), 1.98–1.89 (m, 1H), 1.84–1.75 (m, 1H), 1.74–1.52 (m, 6H), 0.92 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 213.41, 134.49, 132.25, 130.12, 129.03, 128.93, 126.41, 126.12, 125.56, 125.39, 124.72, 73.30, 38.89, 37.01, 35.85, 28.99, 25.12, 23.31, 15.89. ESI-MS: m/z = 282.2 (M + H)+. HRMS m/z: calcd for C19H24NO 282.1852; found, 282.1860.
3.1.24. 2-(Methylamino)-2-(naphthalen-1-yl)cycloheptan-1-one (27)
Compound 27 was synthesized in a process similar to that described for the preparation of 14. The result was a yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.34–8.25 (m, 1H), 7.89–7.78 (m, 2H), 7.62 (d, J = 7.3 Hz, 1H), 7.53–7.40 (m, 3H), 2.64 (dd, J = 14.5, 6.5 Hz, 1H), 2.40 (td, J = 11.5, 3.9 Hz, 1H), 2.20–2.13 (m, 1H), 2.08 (dd, J = 14.1, 10.4 Hz, 1H), 1.99 (s, 3H), 1.94 (s, 1H), 1.86–1.77 (m, 1H), 1.72–1.53 (m, 5H). 13C NMR (101 MHz, CDCl3) δ 213.56, 135.08, 134.53, 132.24, 129.11, 129.01, 126.65, 126.29, 125.57, 125.11, 124.74, 73.32, 38.87, 34.87, 29.15, 28.98, 25.05, 23.25. ESI-MS: m/z = 268.2 (M + H)+. HRMS m/z: calcd for C18H22NO 268.1696; found, 268.1698.
3.1.25. 2-(Dimethylamino)-2-(naphthalen-1-yl)cycloheptan-1-one (28)
Compound 28 was synthesized in a process similar to that described for the preparation of 14. The result was a yellow oil. 1H NMR (400 MHz, CDCl3) δ 9.01 (s, 1H), 7.81 (dd, J = 6.4, 3.3 Hz, 1H), 7.76 (d, J = 8.0 Hz, 1H), 7.45 (dt, J = 6.5, 3.3 Hz, 2H), 7.38 (t, J = 7.7 Hz, 1H), 7.34–7.29 (m, 1H), 2.61 (dd, J = 14.3, 10.2 Hz, 1H), 2.48 (d, J = 10.0 Hz, 2H), 2.36 (s, 7H), 1.82–1.72 (m, 1H), 1.69–1.56 (m, 2H), 1.55–1.40 (m, 3H). 13C NMR (101 MHz, CDCl3) δ 211.71, 137.36, 134.76, 132.46, 128.74 (2C), 127.80, 127.16, 125.50, 125.43, 124.42, 78.04, 44.00, 40.35 (2C), 32.85, 30.93, 25.76 (2C). ESI-MS: m/z = 282.2 (M + H)+. HRMS m/z: calcd for C19H24NO 282.1852; found, 282.1858.
3.1.26. 2-Phenyl-3,4-dihydronaphthalen-1(2H)-one [36] (29a)
Sodium tert-butoxide (1 g, 10.3 mmol) was added to a solution of 3,4-dihydronaphthalen-1(2H)-one (1 g, 6.85 mmol) in THF (30 mL). The mixture was stirred at 70 °C for 0.5 h before iodobenzene (0.92 mL, 8.22 mmol), Pd(dba)2 (79 mg, 0.14 mmol), and DtBPF (81 mg, 0.17 mmol) were added. The mixture was stirred at this temperature for 12h and then poured into water (50 mL) and extracted with ethyl acetate (40 mL*3). The combined organic layers were concentrated under reduced pressure and purified using silica gel column chromatography (3% EtOAc in Pet. Ether) to obtain 29a as white solid. 1H NMR (400 MHz, CDCl3) δ 8.11 (dd, J = 7.8, 1.2 Hz, 1H), 7.51 (td, J = 7.5, 1.4 Hz, 1H), 7.38–7.32 (m, 3H), 7.31–7.26 (m, 2H), 7.22–7.18 (m, 2H), 3.85–3.77 (m, 1H), 3.17–3.03 (m, 2H), 2.49–2.40 (m, 2H). ESI-MS: m/z = 223.20 (M + H)+.
3.1.27. 2-Bromo-2-phenyl-3,4-dihydronaphthalen-1(2H)-one (32a)
Compound 32a was synthesized in a process similar to that described for the preparation of 4a. The compound was a yellow solid. 1H NMR (400 MHz, CDCl3) δ 8.20 (d, J = 7.9 Hz, 1H), 7.56 (d, J = 7.5 Hz, 2H), 7.50 (t, J = 7.4 Hz, 1H), 7.40–7.28 (m, 4H), 7.26–7.20 (m, 1H), 3.26–3.17 (m, 1H), 3.01–2.90 (m, 2H), 2.90–2.82 (m, 1H). 13C NMR (101 MHz, CDCl3) δ 190.75, 142.63, 139.03, 133.91, 130.70, 129.24, 128.62, 128.49, 128.40 (2C), 127.98 (2C), 127.18, 69.71, 40.37, 28.02.
3.1.28. 2-Azido-2-phenyl-3,4-dihydronaphthalen-1(2H)-one (33a)
To a solution of 32a (410 mg, 1.36 mmol) in DMSO (10 mL), we added sodium azide (93 mg, 1.43 mmol). The mixture was stirred at rt for 12h and then poured into water (10 mL) and extracted with ethyl acetate (10 mL*3). The combined organic layers were concentrated under reduced pressure and purified using silica gel column chromatography (5% EtOAc in Pet. Ether) to obtain 33a as white solid. 1H NMR (400 MHz, CDCl3) δ 8.21 (dd, J = 7.9, 1.1 Hz, 1H), 7.50 (td, J = 7.5, 1.3 Hz, 1H), 7.42–7.29 (m, 6H), 7.17 (d, J = 7.6 Hz, 1H), 2.92 (dt, J = 17.2, 4.1 Hz, 1H), 2.78–2.67 (m, 1H), 2.61 (dt, J = 13.7, 4.0 Hz, 1H), 2.38 (ddd, J = 13.7, 11.9, 4.5 Hz, 1H).13C NMR (101 MHz, CDCl3) δ 196.22, 143.26, 135.90, 134.18, 131.86, 129.03(2C), 128.89, 128.79, 128.15, 127.16, 127.08(2C), 71.60, 35.42, 26.14.
3.1.29. 2-Amino-2-phenyl-3,4-dihydronaphthalen-1(2H)-one (31a)
We added 5% palladium/C catalyst (30 mg) to a solution of 33a (300 mg, 1.14 mmol) in MeOH (10 mL). The mixture was stirred in a hydrogen atmosphere for 2h. The mixture was filtrated using celite, and the filtrate was concentrated and purified using silica gel column chromatography, yielding 31a (200 mg, 74%) as yellow oil. 1H NMR (400 MHz, CDCl3) δ 8.18 (dd, J = 7.8, 1.2 Hz, 1H), 7.46 (td, J = 7.5, 1.5 Hz, 1H), 7.38–7.32 (m, 1H), 7.32–7.21 (m, 5H), 7.15 (d, J = 7.6 Hz, 1H), 2.92–2.82 (m, 1H), 2.80–2.64 (m, 2H), 2.35–2.24 (m, 1H), 2.14 (s, 2H). 13C NMR (101 MHz, CDCl3) δ 201.59, 143.77, 141.42, 133.61, 132.35, 128.81, 128.65(2C), 127.85, 127.72, 126.83, 126.43(2C), 63.44, 37.22, 26.62. ESI-MS: m/z = 238.20 (M + H)+.
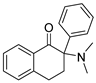
3.1.30. 2-(Dimethylamino)-2-phenyl-3,4-dihydronaphthalen-1(2H)-one hydrochloride (34)
Compound 34 was synthesized in a process similar to that described for the preparation of 14. The result was a white solid. 1H NMR (400 MHz, CDCl3) δ 8.14 (d, J = 7.8 Hz, 1H), 7.41 (t, J = 7.3 Hz, 1H), 7.35–7.25 (m, 6H), 7.08 (d, J = 7.6 Hz, 1H), 2.89–2.82 (m, 1H), 2.77–2.54 (m, 3H), 2.47 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 196.37, 142.62, 134.28, 133.57, 132.80, 129.44, 129.18 (2C), 128.69, 128.65 (2C), 127.90, 127.14, 73.59, 40.27 (2C), 31.75, 26.91. ESI-MS: m/z = 266.25 (M + H)+. HRMS m/z: calcd for C18H20NO 266.1539; found, 266.1542.
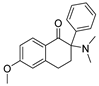
3.1.31. 2-(Dimethylamino)-6-methoxy-2-phenyl-3,4-dihydronaphthalen-1(2H)-one (35)
Compound 35 was synthesized in a process similar to that described for the preparation of 14. The result was a white solid. 1H NMR (400 MHz, CDCl3) δ 8.11 (d, J = 8.8 Hz, 1H), 7.35–7.23 (m, 5H), 6.83 (dd, J = 8.8, 2.4 Hz, 1H), 6.52 (d, J = 2.2 Hz, 1H), 3.80 (s, 3H), 2.85–2.47 (m, 4H), 2.43 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 198.93, 163.49, 145.47, 138.13, 130.03, 128.35 (2C), 127.81, 127.67, 113.43, 112.01, 71.39, 55.39, 48.93, 40.44, 32.06, 27.70. ESI-MS: m/z = 296.25 (M + H)+. HRMS m/z: calcd for C19H22NO2 296.1645; found, 296.1649.
3.1.32. 2-(2-Chlorophenyl)-2-(dimethylamino)-3,4-dihydronaphthalen-1(2H)-one (36)
Compound 36 was synthesized in a process similar to that described for the preparation of 14. The result was a white solid. 1H NMR (400 MHz, CDCl3) δ 8.11 (d, J = 7.9 Hz, 1H), 7.42 (td, J = 7.5, 1.5 Hz, 1H), 7.33 (q, J = 7.6 Hz, 2H), 7.21–7.10 (m, 4H), 3.17 (ddd, J = 14.1, 6.0, 4.3 Hz, 1H), 2.97 (dt, J = 17.1, 5.1 Hz, 1H), 2.66 (ddd, J = 17.0, 9.5, 4.2 Hz, 1H), 2.59 (s, 6H), 2.45 (ddd, J = 13.9, 9.4, 4.4 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 197.84, 142.82, 137.47, 134.07, 134.02, 132.81, 131.99, 130.96, 128.64, 128.30, 128.12, 126.63, 126.32, 71.99, 40.19 (2C), 32.71, 27.20.ESI-MS: m/z = 300.2 (M + H)+. HRMS m/z: calcd for C18H19ClNO 300.1150; found, 300.1146.
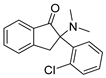
3.1.33. 2-(2-Chlorophenyl)-2-(dimethylamino)-2,3-dihydro-1H-inden-1-one Hydrochloride (37)
Compound 37 was synthesized in a process similar to that described for the preparation of 14. The result was a white solid. 1H NMR (400 MHz, CDCl3) δ 12.41 (s, 1H), 7.96 (d, J = 7.7 Hz, 1H), 7.88 (m, 1H), 7.77 (t, J = 7.3 Hz, 1H), 7.59–7.47 (m, 3H), 7.44–7.34 (m, 2H), 4.45 (d, J = 19.2 Hz, 1H), 3.91 (d, J = 19.0 Hz, 1H), 2.90 (d, J = 60.9 Hz, 6H). 13C NMR (101 MHz, CDCl3) δ 198.87, 150.41, 137.75, 135.53, 132.45, 132.07, 131.20, 131.11, 129.22, 127.69, 126.53, 124.98, 74.78, 41.89, 40.66, 40.05. ESI-MS: m/z = 286.20 (M + H)+. HRMS m/z: calcd for C17H17ClNO 286.0993; found, 286.0998.
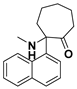
3.1.34. 6-(Dimethylamino)-6-phenyl-6,7,8,9-tetrahydro-5H-benzo[7]annulen-5-one Hydrochloride (38)
Compound 38 was synthesized in a process similar to that described for the preparation of 14. The result was a white solid. 1H NMR (400 MHz, CDCl3) δ 12.28 (s, 1H), 7.90 (d, J = 7.6 Hz, 1H), 7.59–7.47 (m, 4H), 7.43 (t, J = 7.5 Hz, 1H), 7.26 (s, 2H), 7.15 (d, J = 7.5 Hz, 1H), 3.51–3.42 (m, 1H), 2.83 (d, J = 3.6 Hz, 3H), 2.72 (d, J = 4.1 Hz, 3H), 2.50–2.26 (m, 3H), 1.92–1.80 (m, 1H), 1.73 (s, 1H). 13C NMR (101 MHz, CDCl3) δ 202.21, 139.67, 137.49, 134.68, 130.56, 130.48, 130.16 (2C), 129.80 (4C), 127.57, 77.24, 41.88, 39.71, 30.73, 29.22, 22.17. ESI-MS: m/z = 280.2 (M + H)+. HRMS m/z: calcd for C19H22NO 280.1696; found, 280.1692.
3.1.35. 2-(Methylamino)-2-phenyl-3,4-dihydronaphthalen-1(2H)-one (41)
TEA (87 μL, 0.63 mmol) and di-tert-butyl decarbonate (70 mg, 0.32 mmol) were added to a solution of 31a (30 mg, 0.13 mmol) in DCM (3 mL). The mixture was stirred at rt for 10 h and directly concentrated and purified using silica gel column chromatography to attain 39a (40 mg, 91%) as yellow oil. Then, 39a (40 mg, 0.12) was dissolved in THF (3 mL), and NaH (10 mg, 0.24 mmol) and MeI (15 μL, 0.24 mmol) were added. The mixture was stirred at rt for 1h and then quenched with water and extracted with EtOAc. The combined organic layers were concentrated and dissolved in DCM (2 mL). Then, HCl/dioxane solution (1 mL, 4 M) was added, and the mixture was stirred at rt for 1 h. Sodium hydroxide solution (2.5 N) was added to change the pH so that it was basic. The mixture was extracted with EtOAc, and the combined organic was concentrated and then purified using HPLC and acidified to attain 41 (10 mg, 30%) as white solid. 1H NMR (400 MHz, CDCl3) δ 10.29 (d, J = 22.7 Hz, 2H), 8.12 (d, J = 7.7 Hz, 1H), 7.61–7.52 (m, 2H), 7.46 (t, J = 7.1 Hz, 1H), 7.42–7.30 (m, 4H), 7.09 (d, J = 7.6 Hz, 1H), 3.39–3.27 (m, 1H), 3.11–2.92 (m, 2H), 2.86–2.74 (m, 1H), 2.61 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 193.39, 142.23, 134.51, 131.44, 130.11, 129.79, 129.51 (2C), 128.69 (2C), 128.64, 128.35, 127.20, 69.48, 31.43, 29.49, 25.94. ESI-MS: m/z = 252.25 (M + H)+. HRMS m/z: calcd for C17H18NO 252.1383; found, 252.1385.
3.1.36. 6-Methoxy-2-(methylamino)-2-phenyl-3,4-dihydronaphthalen-1(2H)-one (42)
Compound 42 was synthesized in a process similar to that described for the preparation of 41. The result was a white solid. 1H NMR (400 MHz, CDCl3) δ 10.17 (d, J = 39.4 Hz, 2H), 8.10 (d, J = 8.8 Hz, 1H), 7.63–7.53 (m, 2H), 7.44–7.34 (m, 3H), 6.85 (dd, J = 8.8, 2.2 Hz, 1H), 6.53 (d, J = 2.0 Hz, 1H), 3.81 (s, 3H), 3.28 (d, J = 12.9 Hz, 1H), 3.07–2.86 (m, 2H), 2.81–2.68 (m, 1H), 2.60 (t, J = 5.2 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 191.90, 164.56, 144.92, 130.88, 130.24, 129.99, 129.44 (2C), 128.68 (2C), 124.85, 114.18, 112.27, 69.20, 55.54, 31.42, 29.58, 26.29. ESI-MS: m/z = 282.20 (M + H)+. HRMS m/z: calcd for C18H20NO2 282.1489; found, 282.1494.
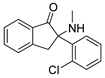
3.1.37. 2-(2-Chlorophenyl)-2-(methylamino)-2,3-dihydro-1H-inden-1-one (43)
Compound 43 was synthesized in a process similar to that described for the preparation of 41. The result was a white solid. 1H NMR (400 MHz, CDCl3) δ 11.34 (s, 1H), 10.10 (s, 1H), 7.90 (d, J = 7.6 Hz, 1H), 7.67 (t, J = 7.3 Hz, 1H), 7.59 (d, J = 7.2 Hz, 1H), 7.50–7.39 (m, 3H), 7.37–7.28 (m, 2H), 4.23 (d, J = 17.8 Hz, 1H), 3.86 (d, J = 17.6 Hz, 1H), 2.73 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 198.84, 149.89, 136.85, 134.66, 133.12, 131.78, 131.69, 130.75, 130.38, 128.78, 127.54, 126.35, 125.23, 69.47, 40.69, 29.51. ESI-MS: m/z = 272.15 (M + H)+. HRMS m/z: calcd for C16H15ClNO 272.0837; found, 272.0845.






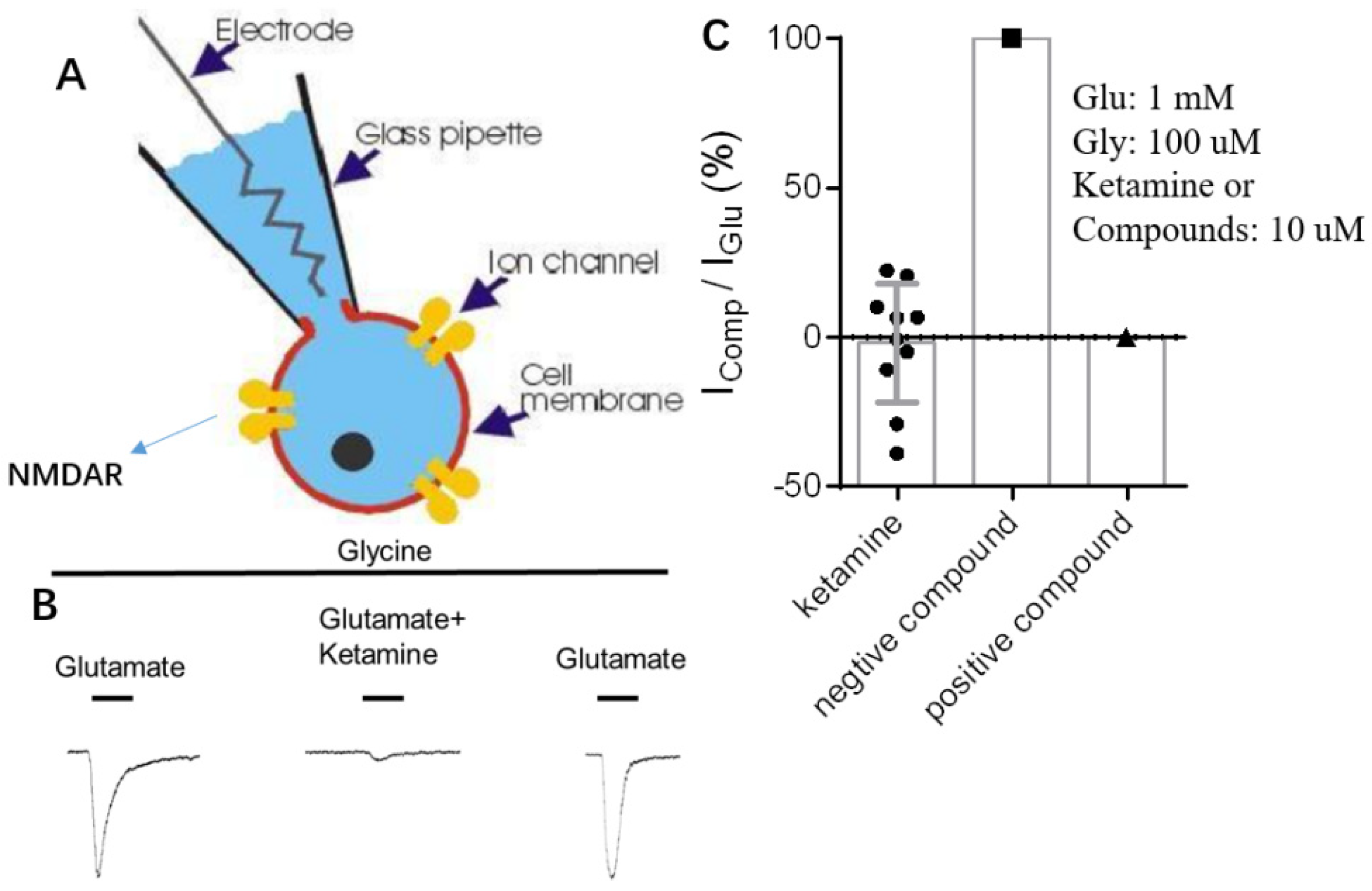





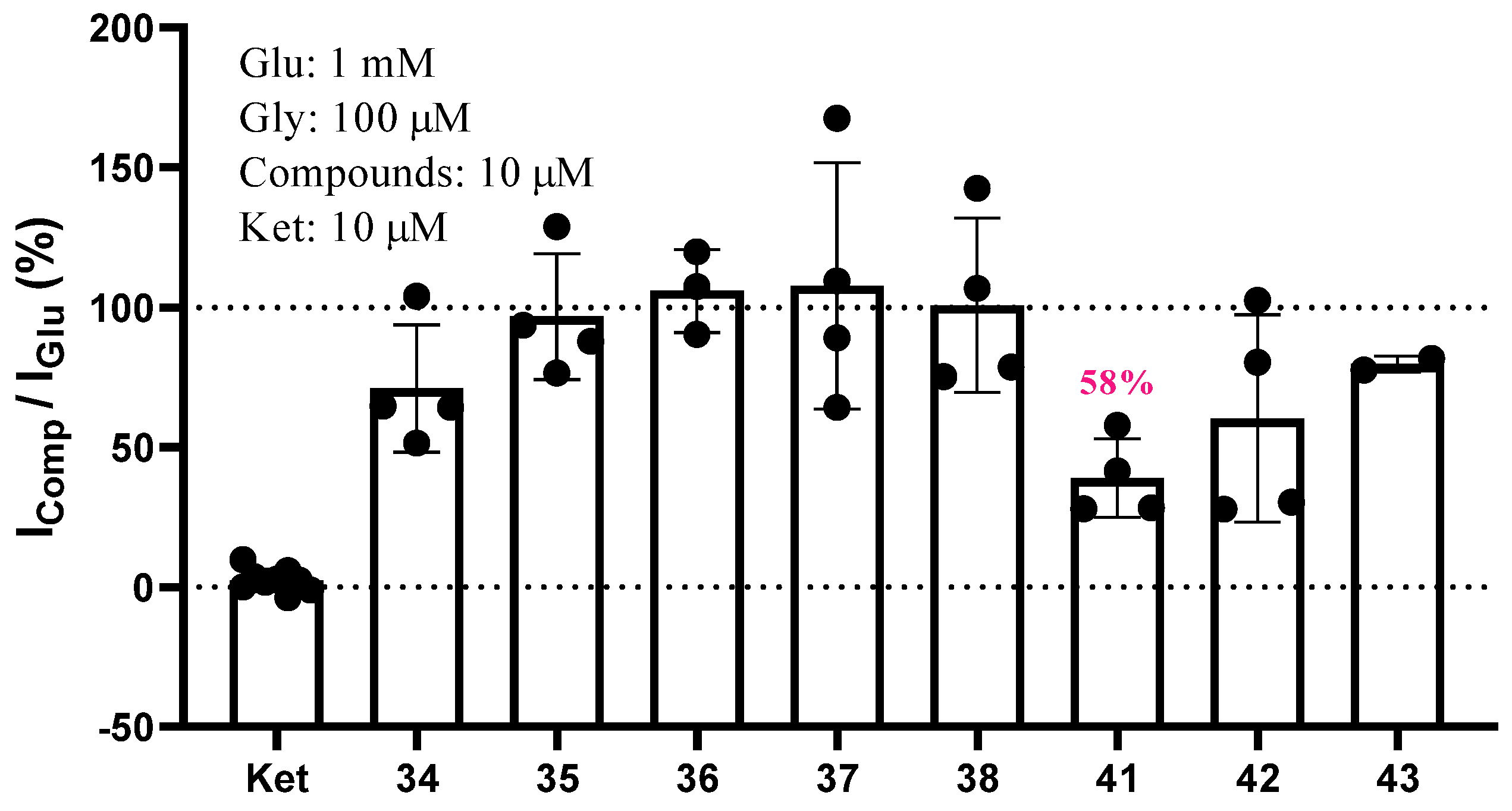

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}