
Microwave Synthesis of Au Nanoparticles in the Presence of Tetrahydrothiophenocucurbituril



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
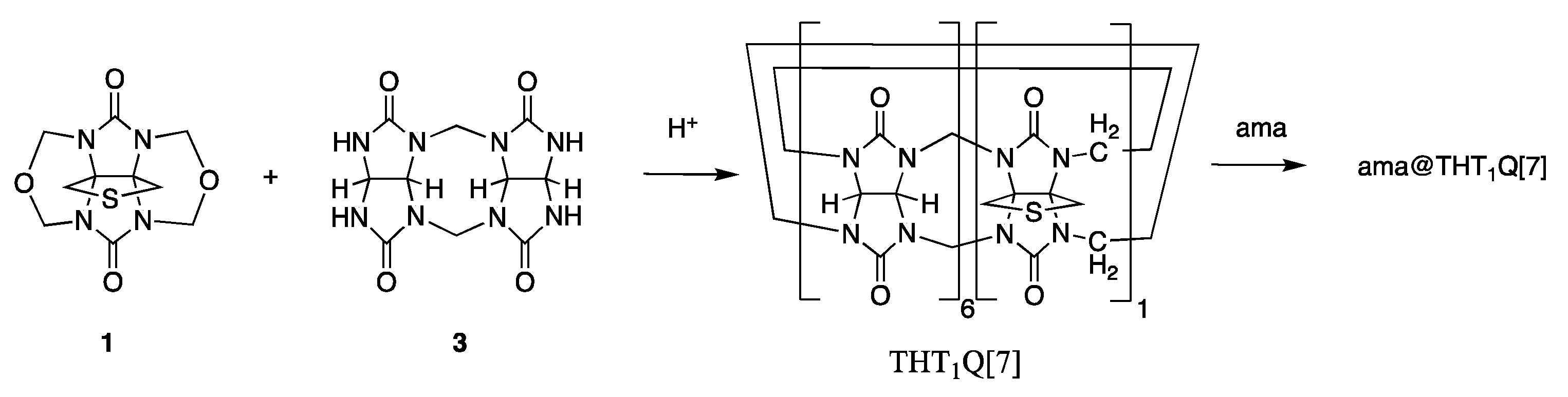
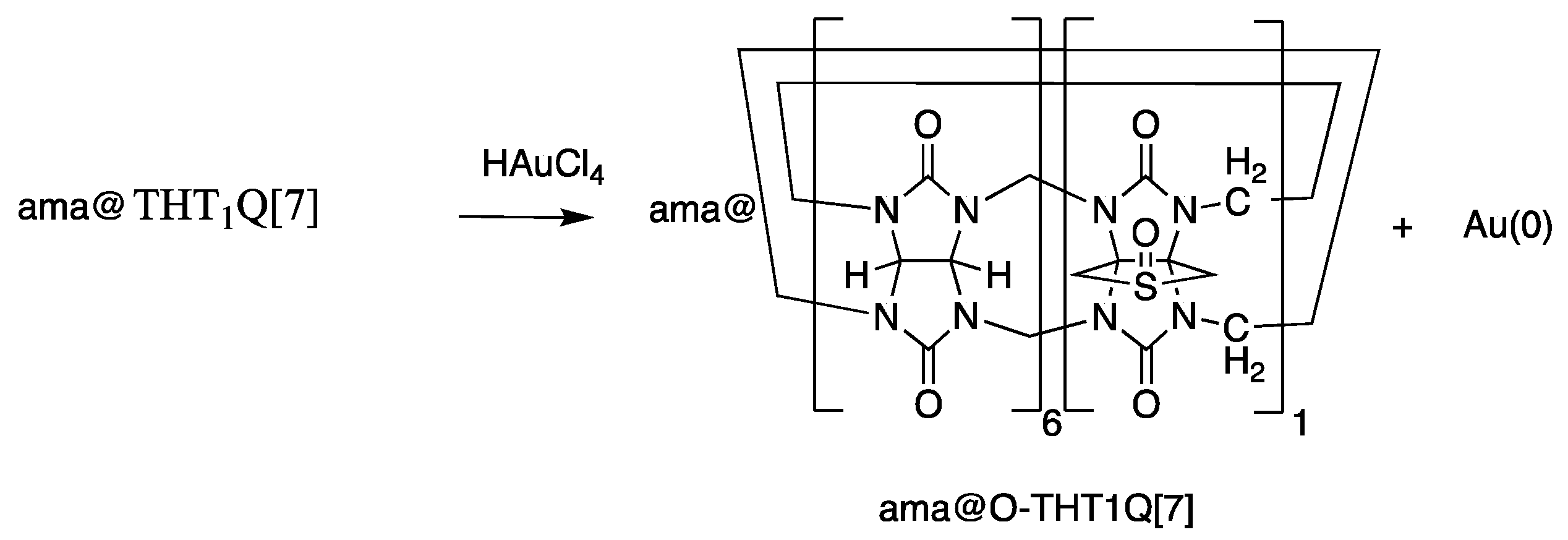
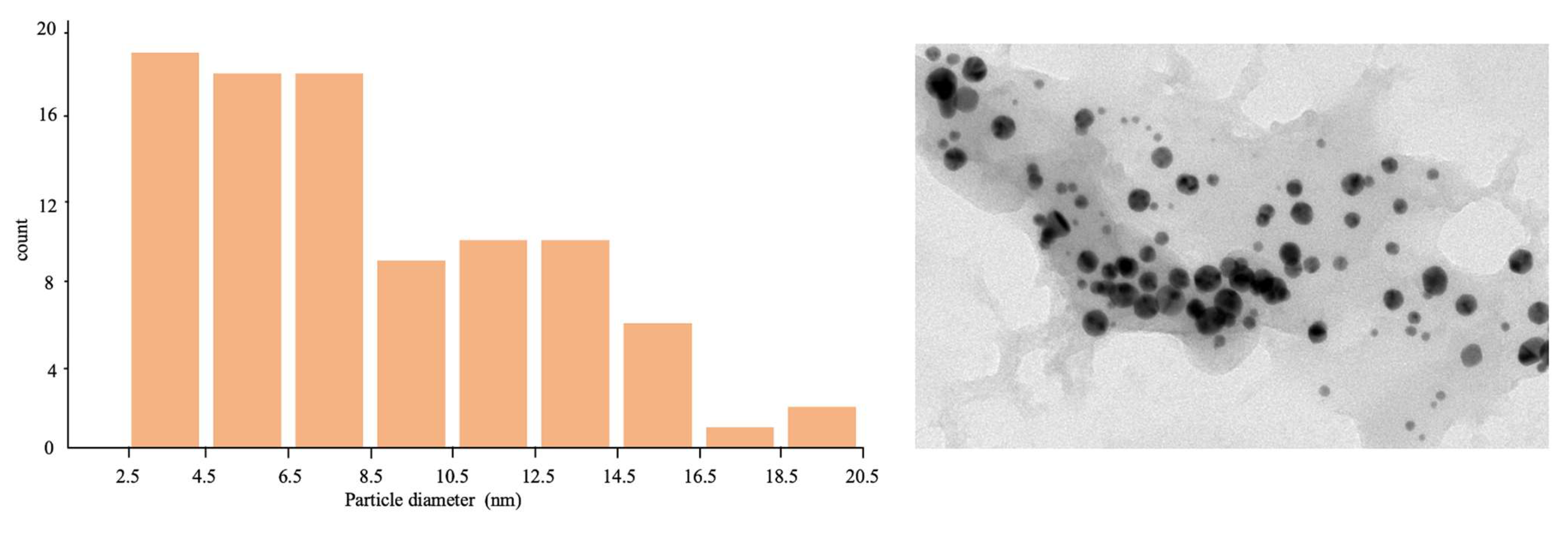
2.1. AuNP-ama@THT1Q[7]
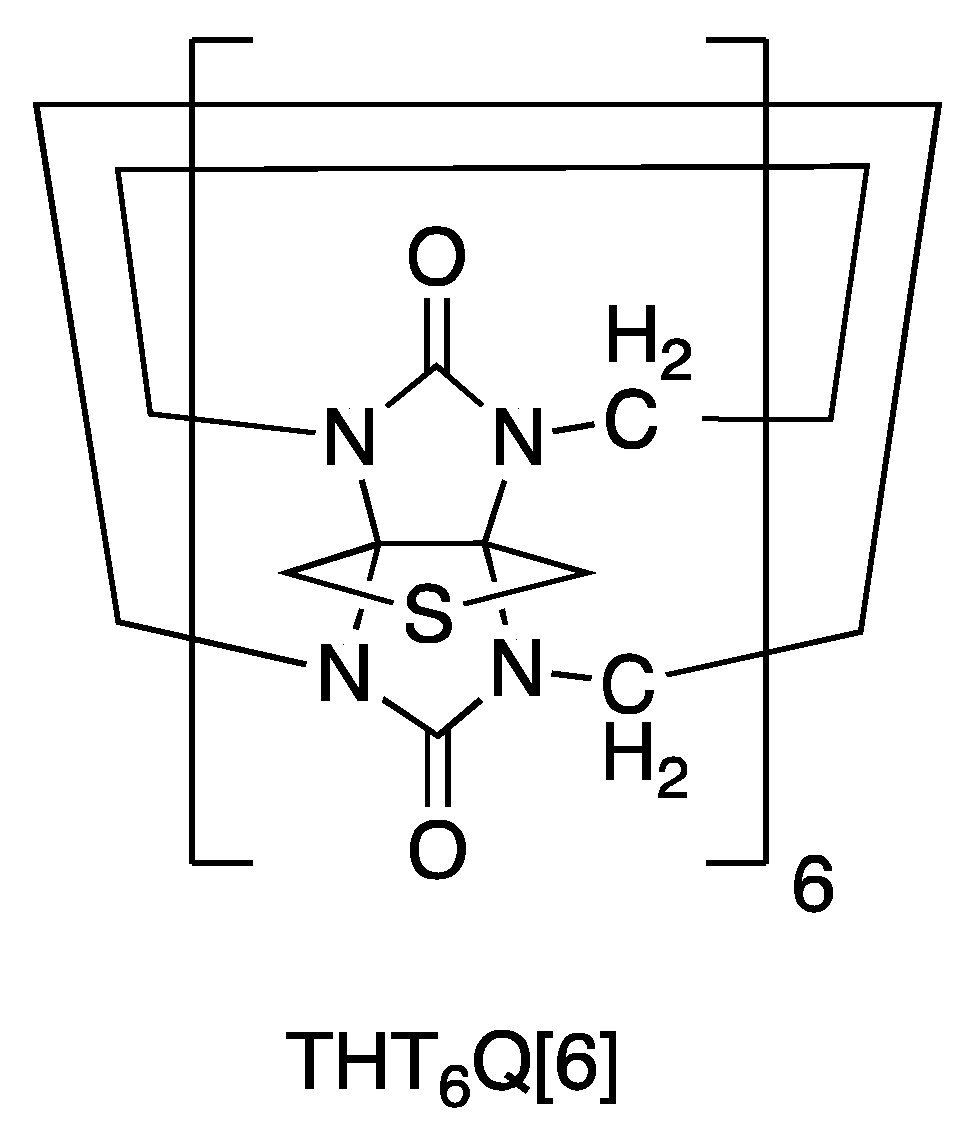
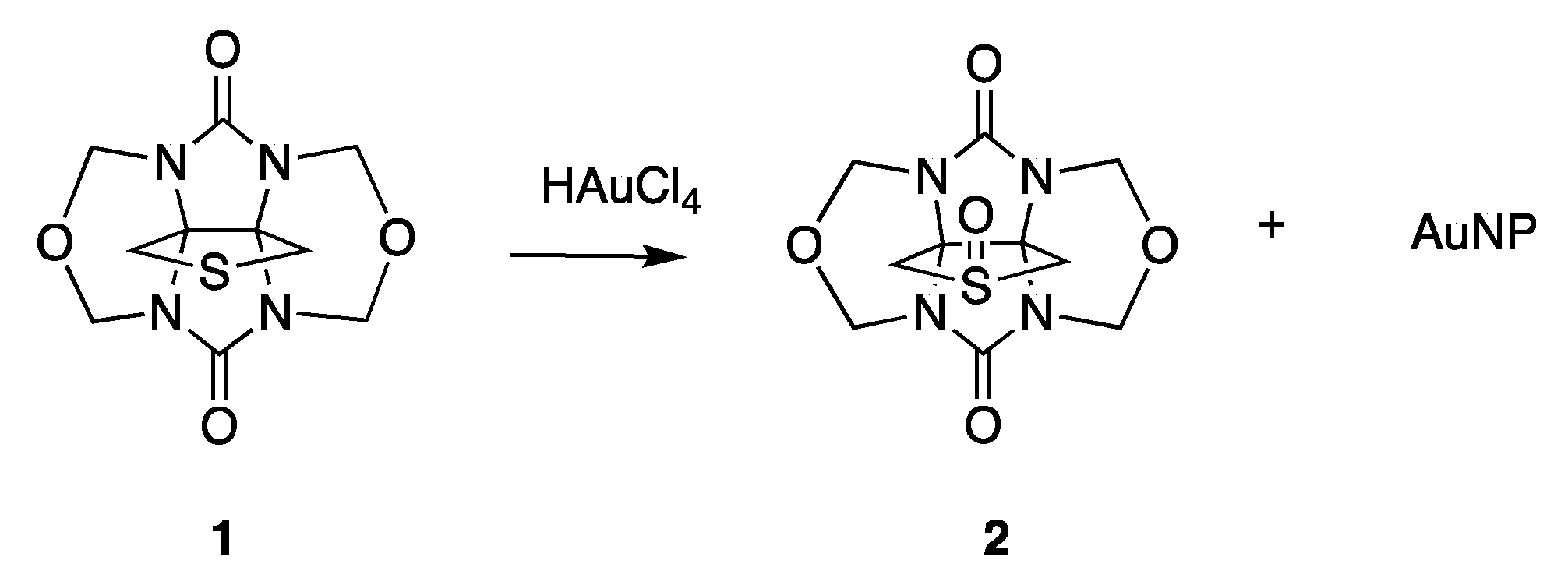
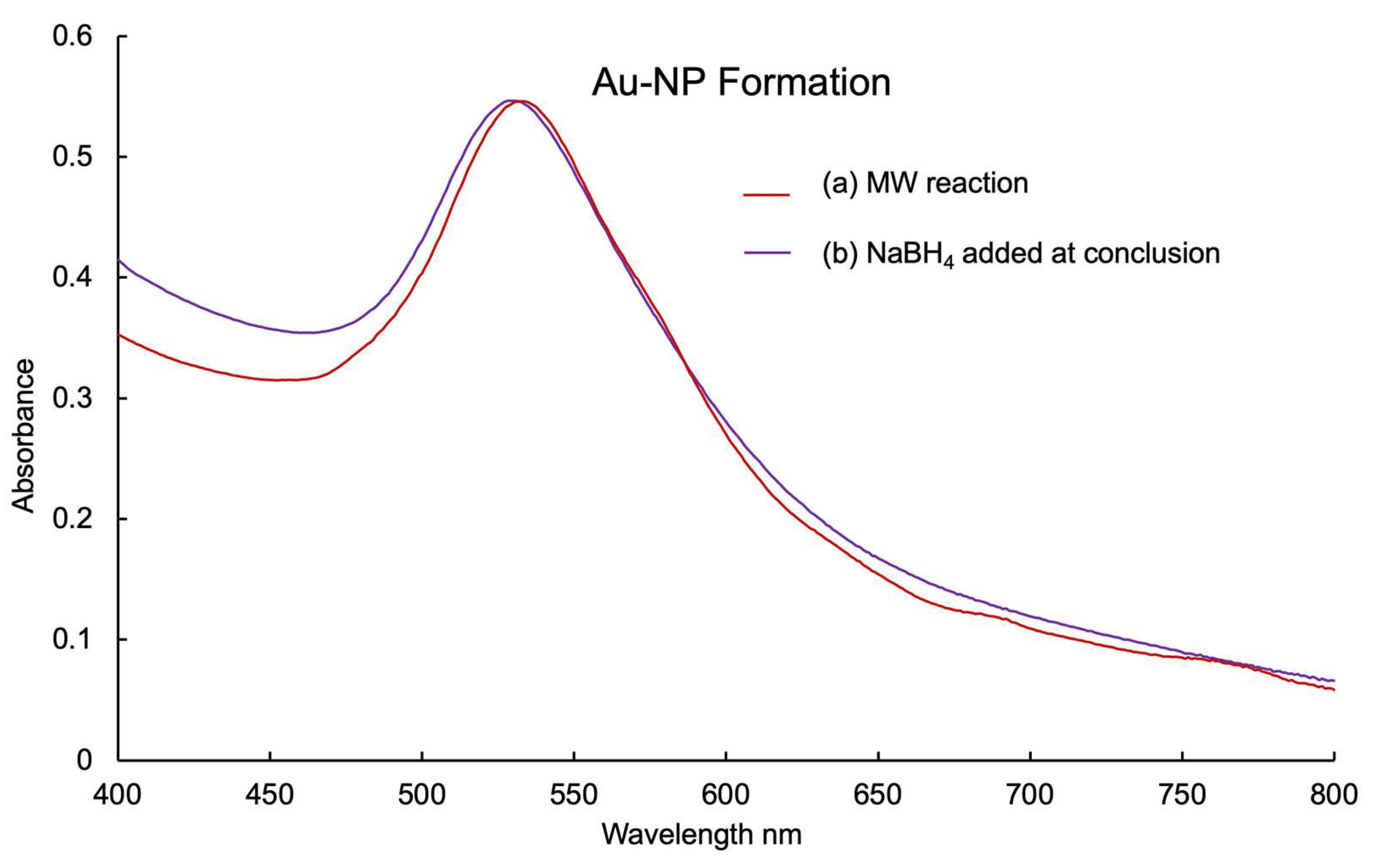
2.2. AuNP-THT6Q[6]
3. Materials and Methods
3.1. Synthesis of AuNP-THTglycoluril
3.2. Synthesis of Amantadinylammonium@tetrahydrothiophenoQ[n]Cl (ama@THT1Q[7]Cl)
3.3. Synthesis of Amantadinylammonium@tetrahydrothiopheno-1-oxideQ[n] (ama@O-THT1Q[7]Cl)
3.4. Reaction Procedure for the Microwave Reactor
3.4.1. Preparation of AuNP-ama@THT1Q[7]Cl
- A.
- Mole ratio 1:1 (THTQ:Au(III))
- B.
- Mole ratio 2:1 (THTQ:Au(III))
- C.
- Mole ratio 3:1 (THTQ:Au(III))
- D.
- A repeat of method C with ama@THT1Q[7]PF6
3.4.2. Preparation AuNP-THT6Q[6]
- A.
- Reaction in pure water
- B.
- Reaction in a 50 mM Ca(OAc)2 solution
- C.
- Reaction of cpa@THT6Q[6]Cl in pure water
3.5. Purification of AuNP-THTmQ[n]
3.6. Sulfoxide Affinity for AuNPs
3.7. Control Reaction with Q[7] and ama@Q[7]
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dreaden, E.C.; Alkilany, A.M.; Huang, X.; Murphy, C.J.; El-Sayed, M.A. The golden age: Gold nanoparticles for biomedicine. Chem. Soc. Rev. 2012, 41, 2740–2779. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Yang, M.; Pang, B.; Vara, M.; Xia, Y. Gold Nanomaterials at Work in Biomedicine. Chem. Rev. 2015, 115, 10410–10488. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Gao, X.; Liu, D.; Chen, X. Gold Nanoparticles for In Vitro Diagnostics. Chem. Rev. 2015, 115, 10575–10636. [Google Scholar] [CrossRef] [PubMed]
- Sapsford, K.E.; Algar, W.R.; Berti, L.; Gemmill, K.B.; Casey, B.J.; Oh, E.; Stewart, M.H.; Medintz, I.L. Functionalizing Nanoparticles with Biological Molecules: Developing Chemistries that Facilitate Nanotechnology. Chem. Rev. 2013, 113, 1904–2074. [Google Scholar] [CrossRef] [PubMed]
- Mirkin, C.A.; Letsinger, R.L.; Mucic, R.C.; Storhoff, J.J. A DNA-based method for rationally assembling nanoparticles into macroscopic materials. Nature 1996, 382, 607–609. [Google Scholar] [CrossRef] [PubMed]
- Yeh, Y.-C.; Creran, B.; Rotello, V.M. Gold nanoparticles: Preparation, properties, and applications in bionanotechnology. Nanoscale 2012, 4, 1871. [Google Scholar] [CrossRef] [PubMed]
- Kreyling, W.G.; Abdelmonem, A.M.; Ali, Z.; Alves, F.; Geiser, M.; Haberl, N.; Hartmann, R.; Hirn, S.; Jimenez de Aberasturi, D.; Kantner, K.; et al. In vivo integrity of polymer-coated gold nanoparticles. Nat. Nanotechnol. 2015, 10, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Zhao, X.; Yuan, Y.; Miao, F.; Li, W.; Ji, S.; Huang, X.; Chen, X.; Jiang, T.; Weitz, D.A.; et al. Microfluidic Synthesis of Multi-mode Au@CoFeB-Rg3 Nanomedicines and Their Cytotoxicity and Anti-Tumor Effects. Chem. Mater. 2020, 32, 5044–5056. [Google Scholar] [CrossRef]
- Ojea-Jiménez, I.; Bastús, N.G.; Puntes, V. Influence of the Sequence of the Reagents Addition in the Citrate-Mediated Synthesis of Gold Nanoparticles. J. Phys. Chem. C 2011, 115, 15752–15757. [Google Scholar] [CrossRef]
- Liu, J.; Qin, G.; Raveendran, P.; Ikushima, Y. Facile “Green” Synthesis, Characterization, and Catalytic Function of b-D-Glucose-Stabilized Au Nanocrystals. Chem. Eur. J. 2006, 12, 2131–2138. [Google Scholar] [CrossRef]
- Martin, M.N.; Basham, J.I.; Chando, P.; Eah, S.K. Charged gold nanoparticles in non-polar solvents: 10-min synthesis and 2D self-assembly. Langmuir 2010, 26, 7410–7417. [Google Scholar] [CrossRef]
- Mallick, K.; Wang, Z.L.; Pal, T. Seed-mediated successive growth of gold particles accomplished by UV irradiation: A photochemical approach for size-controlled synthesis. J. Photochem. Photobiol. A Chem. 2001, 140, 75–80. [Google Scholar] [CrossRef]
- Niidome, Y.; Hori, A.; Sato, T.; Yamada, S. Enormous size growth of thiol-passivated gold nanoparticles induced by near-IR laser light. Chem. Lett. 2000, 29, 310–311. [Google Scholar] [CrossRef]
- Zhang, J.; Du, J.; Han, B.; Liu, Z.; Jiang, T.; Zhang, Z. Sonochemical formation of single-crystalline gold nanobelts. Angew. Chem. Int. Ed. 2006, 45, 1116–1119. [Google Scholar] [CrossRef]
- Su, C.-H.; Wu, P.-L.; Yeh, C.-S. Sonochemical synthesis of well-dispersed gold nanoparticles at the ice temperature. J. Phys. Chem. B 2003, 107, 14240–14243. [Google Scholar] [CrossRef]
- Shen, M.; Du, Y.; Hua, N.; Yang, P. Microwave irradiation synthesis and self-assembly of alkylamine-stabilized gold nanoparticles. Powder Technol. 2006, 162, 64–72. [Google Scholar] [CrossRef]
- Steven, D.; Perrault, S.D.; Chan, W.C.W. Synthesis and Surface Modification of Highly Monodispersed, Spherical Gold nanoparticles 50–200 nm. J Amer. Chem. Soc. 2009, 131, 17042–17043. [Google Scholar]
- Kumar, D.; Mutreja, I.; Sykes, P. Seed mediated synthesis of highly mono-dispersed gold nanoparticles in the presence of hydroquinone. Nanotechnology 2016, 27, 355601. [Google Scholar] [CrossRef]
- Wang, R.; Yang, W.; Song, Y.; Shen, X.; Wang, J.; Zhong, X.; Li, S.; Song, Y. A General Strategy for Nanohybrids Synthesis via Coupled Competitive Reactions Controlled in a Hybrid Process. Sci. Rep. 2015, 5, 9189. [Google Scholar] [CrossRef]
- Corma, A.; Garc, H.; Montes-Navajas, P.; Primo, A.; Calvino, J.J.; Trasobares, S. Gold nanoparticles in organic capsules: A supramolecular assembly of gold nanoparticles and cucurbituril. Chem. Eur. J. 2007, 13, 6359–6364. [Google Scholar] [CrossRef]
- Premkumar, T.; Geckeler, K.E. Cucurbit[7]uril as a tool in the green synthesis of gold nanoparticles. Chem.–Asian J. 2010, 5, 2468–2476. [Google Scholar] [CrossRef] [PubMed]
- Tao, C.; An, Q.; Zhu, W.; Yang, H.; Li, W.; Lin, C.; Xu, D.; Li, G. Cucurbit[n]urils as a SERS hot-spot nanocontainer through bridging gold nanoparticles. Chem. Commun. 2011, 47, 9867–9869. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.W.; Coulston, R.J.; Biedermann, F.; Mahajan, S.; Baumberg, J.J.; Scherman, O.A. In Situ SERS Monitoring of Photochemistry within a Nanojunction Reactor. Nano Lett. 2013, 13, 5985–5990. [Google Scholar] [CrossRef] [PubMed]
- Yan, B.; Tonga, G.Y.; Hou, S.; Fedick, P.W.; Yeh, Y.-C.; Alfonso, F.S.; Mizuhara, T.; Vachet, R.W.; Rotello, V.M. Mass spectrometric detection of nanoparticle host–guest interactions in cells. Anal. Chem. 2014, 86, 6710–6714. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Feng, Q.; Yang, X.; Liu, S.; Xu, C.; Huang, L.; Chen, M.; Liang, F.; Cheng, Y. Near infrared light triggered cucurbit[7]uril-stabilized gold nanostars as a supramolecular nanoplatform for combination treatment of cancer. Bioconjug. Chem. 2018, 29, 2855–2866. [Google Scholar] [CrossRef] [PubMed]
- Jana, B.; Kim, S.; Choi, H.; Jin, S.; Kim, K.; Kim, M.; Lee, H.; Lee, K.H.; Lee, J.; Parke, M.-H.; et al. Supramolecular protection-mediated one-pot synthesis of cationic gold nanoparticles. J. Ind. Eng. Chem. 2020, 81, 303–308. [Google Scholar] [CrossRef]
- Mahajan, S.; Lee, T.-C.; Biedermann, F.; Hugall, J.T.; Baumberg, J.J.; Scherman, O.A. Raman and SERS spectroscopy of cucurbit[n]urils. Phys. Chem. Chem. Phys. 2010, 12, 10429–10433. [Google Scholar] [CrossRef]
- Kasera, S.; Biedermann, F.; Baumberg, J.J.; Scherman, O.A.; Mahajan, S. Quantitative SERS Using the Sequestration of Small Molecules Inside Precise Plasmonic Nanoconstructs. Nano Lett. 2012, 12, 5924–5928. [Google Scholar] [CrossRef]
- Lanterna, A.; Pino, E.; Doménech-Carbó, A.; González-Béjar, M.; Pérez-Prieto, J. Enhanced catalytic electrochemical reduction of dissolved oxygen with ultraclean cucurbituril[7]-capped gold nanoparticles. Nanoscale 2014, 6, 9550–9553. [Google Scholar] [CrossRef]
- Kim, C.; Tonga, G.Y.; Yan, B.; Kim, C.S.; Kim, S.T.; Park, M.-H.; Zhu, Z.; Duncan, B.; Creran, B.; Rotello, V.M. Regulating exocytosis of nanoparticles via host–guest chemistry. Org. Biomol. Chem. 2015, 13, 2474–2479. [Google Scholar] [CrossRef]
- Lee, T.-C.; Scherman, O.A. Formation of dynamic aggregates in water by cucurbit[5]uril capped with gold nanoparticles. Chem. Commun. 2010, 46, 2438–2440. [Google Scholar] [CrossRef]
- Lee, T.C.; Scherman, O.A. A facile synthesis of dynamic supramolecular aggregates of cucurbit[n]uril (n = 5–8) capped with gold nanoparticles in aqueous media. Chem.–A Eur. J. 2012, 18, 1628–1633. [Google Scholar] [CrossRef] [PubMed]
- Al Muqarrabun, L.M.R.; Atthar, A.S.; Kumar, C.P.; Mandadapu, V.; Abdulrahman, A.; Iranmanesh, H.; Beves, J.E.; Day, A.I. Gold and Silver Chains from Tetrahydrothiophenocucurbit[6]uril as Au or Ag-Nanoparticles. J. Org. Chem. 2023, 88, 12208–12215. [Google Scholar] [CrossRef]
- El Kurdi, R.; Patra, D. Capping of supramolecular curcubit[7]uril facilitates formation of Au nanorods during pre-reduction by curcumin, Colloids and Surf. A Physicochem. Eng. Asp. 2018, 553, 97–104. [Google Scholar] [CrossRef]
- Smith, A.M.; Marbella, L.E.; Johnston, K.A.; Hartmann, M.J.; Crawford, S.E.; Kozycz, L.M.; Seferos, D.S.; Millstone, J.E. Quantitative Analysis of Thiolated Ligand Exchange on Gold Nanoparticles Monitored by 1H NMR Spectroscopy. Anal. Chem. 2015, 87, 2771–2778. [Google Scholar] [CrossRef]
- Uson, R.; Laguna, A.; Laguna, M.; Briggs, D.A.; Murray, H.H.; Fackler, J.P., Jr. Inorganic Syntheses; Kaesz, H.D., Ed.; Wiley: Hoboken, NJ, USA, 1989; Volume 25, Chapter 17. [Google Scholar]
- Zheng, L.; Zhu, J.; Zhang, Y.; Zhu, Q.; Xue, S.; Tao, Z.; Zhang, J.; Zhou, X.; Wei, Z.; Long, L.; et al. Opposing substitution in cucurbit[6]urils forms ellipsoid cavities—The symmetrical dicyclohexanocucurbit[6]uril is no exception highlighted by inclusion and exclusion complexes. Supramol. Chem. 2008, 20, 709–716. [Google Scholar]
- Lui, S.; Ruspic, C.; Mukhopadhyay, P.; Chakrabarti, S.; Zavalij, P.Y.; Isaacs, L. The cucurbit[n]uril family: Prime components for self-sorting systems. J. Amer. Chem. Soc. 2005, 127, 15959–15967. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Atthar, A.S.; Saha, S.; Abdulrahman, A.; Day, A.I. Microwave Synthesis of Au Nanoparticles in the Presence of Tetrahydrothiophenocucurbituril. Molecules 2024, 29, 168. https://doi.org/10.3390/molecules29010168
Atthar AS, Saha S, Abdulrahman A, Day AI. Microwave Synthesis of Au Nanoparticles in the Presence of Tetrahydrothiophenocucurbituril. Molecules. 2024; 29(1):168. https://doi.org/10.3390/molecules29010168
Chicago/Turabian StyleAtthar, Asma S., Shreya Saha, Ahmed Abdulrahman, and Anthony I. Day. 2024. "Microwave Synthesis of Au Nanoparticles in the Presence of Tetrahydrothiophenocucurbituril" Molecules 29, no. 1: 168. https://doi.org/10.3390/molecules29010168
APA StyleAtthar, A. S., Saha, S., Abdulrahman, A., & Day, A. I. (2024). Microwave Synthesis of Au Nanoparticles in the Presence of Tetrahydrothiophenocucurbituril. Molecules, 29(1), 168. https://doi.org/10.3390/molecules29010168