
Qualitative and Quantitative Determination of Decoquinate in Chicken Tissues by Gas Chromatography Tandem Mass Spectrometry
 ,
, 

Abstract
1. Introduction
2. Results and Discussion
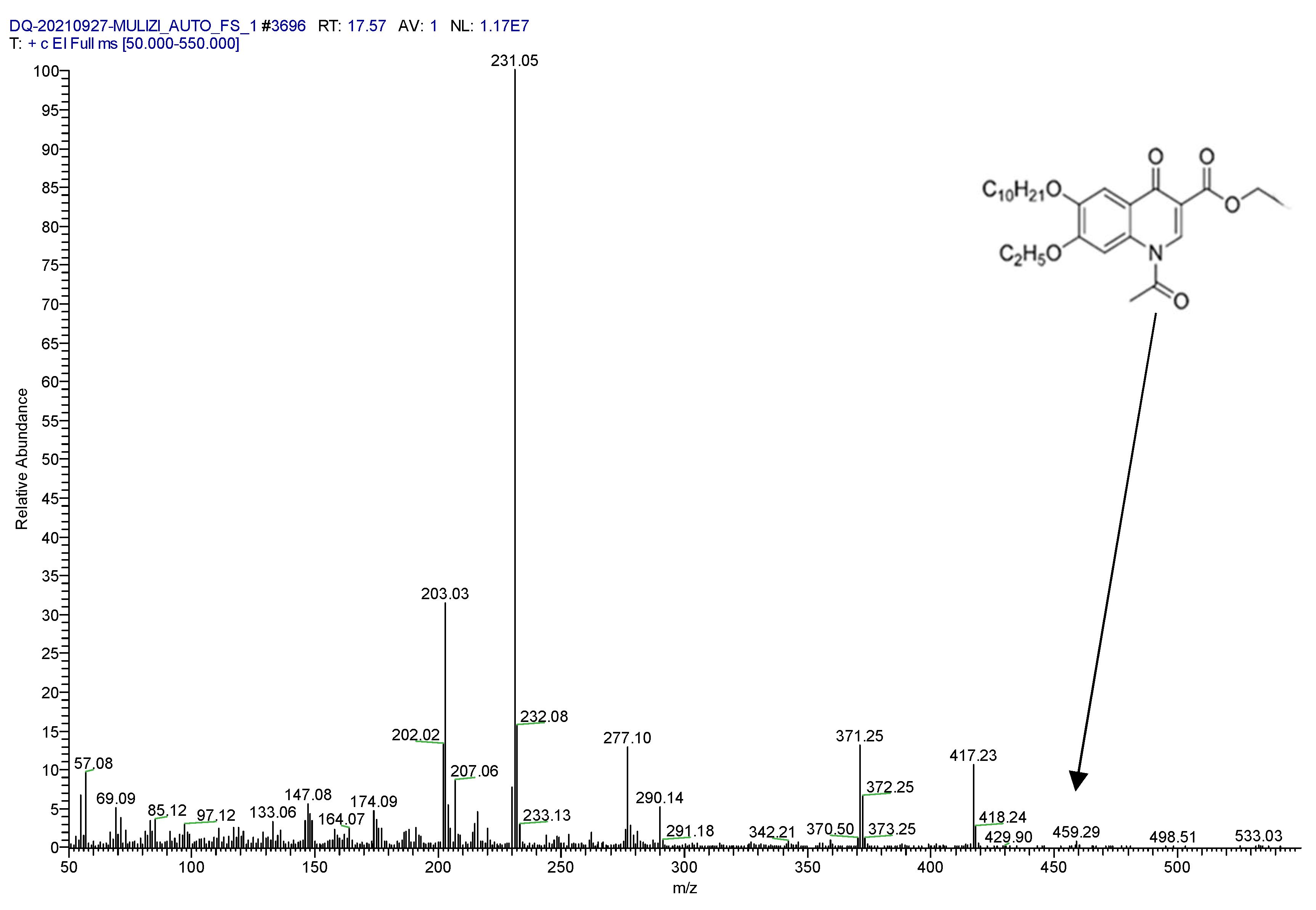
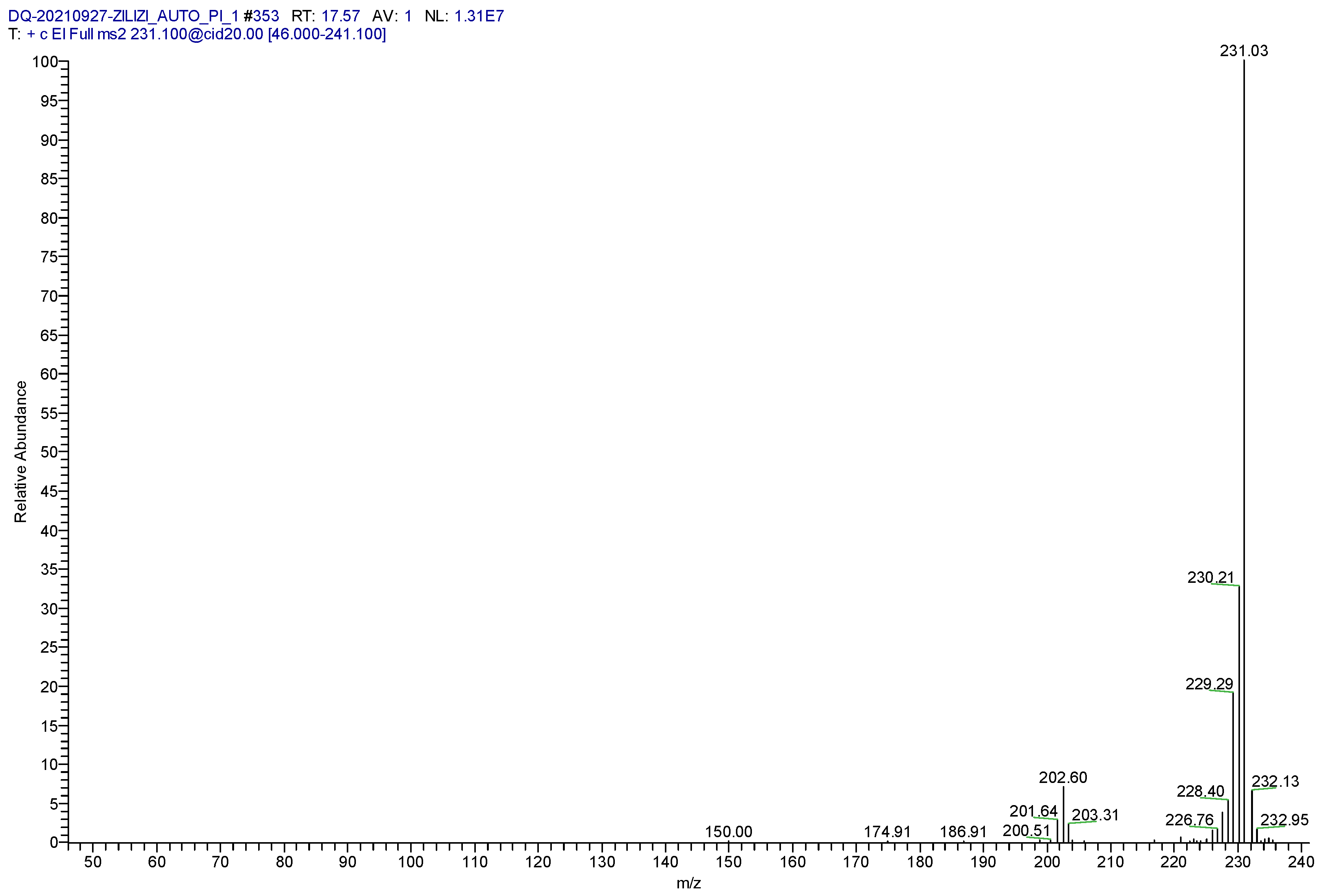
2.1. Confirmation of Derivative Products
2.2. Determination of Parent and Product Ions
2.3. Optimization of Derivative Conditions
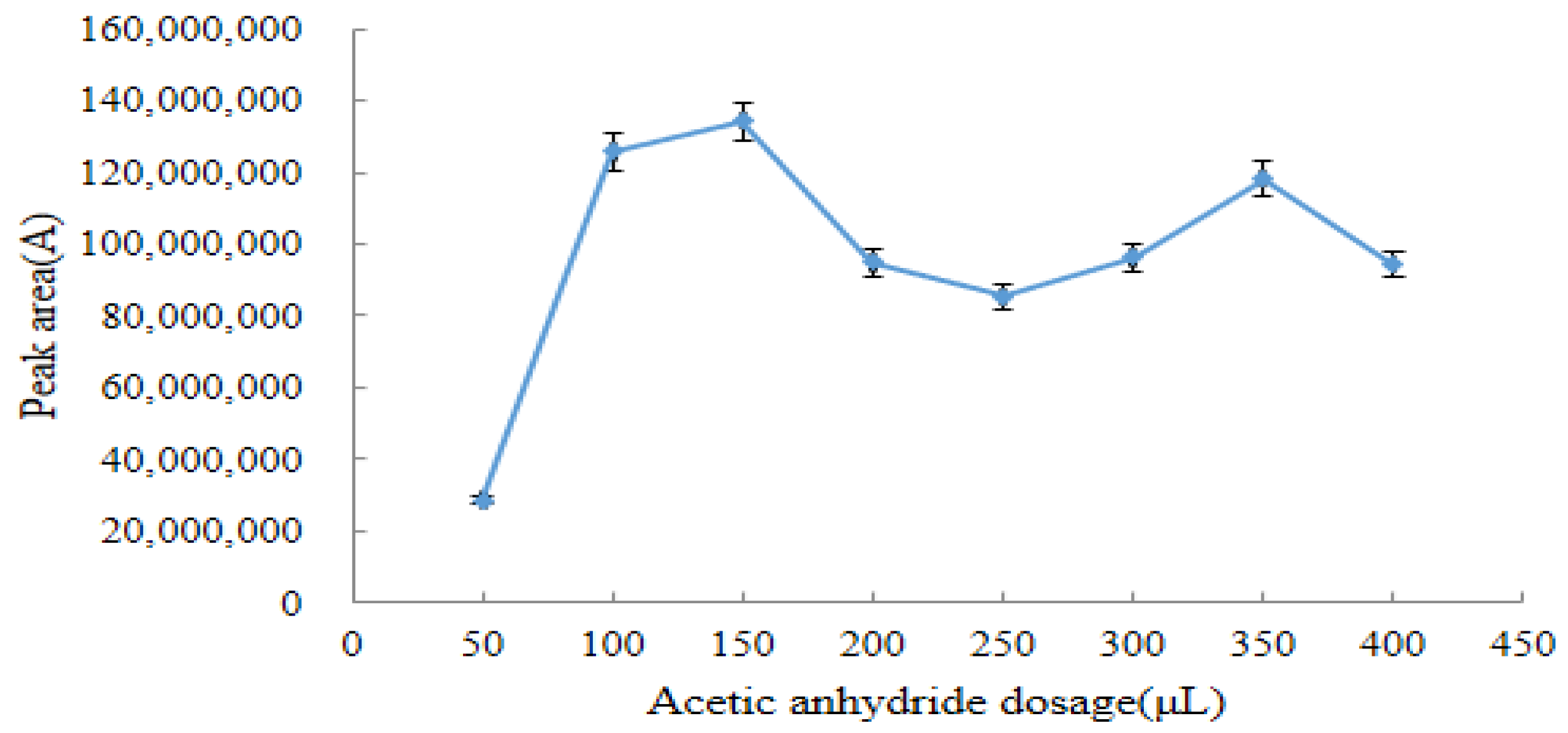
2.3.1. Optimum Dosage of Acetic Anhydride
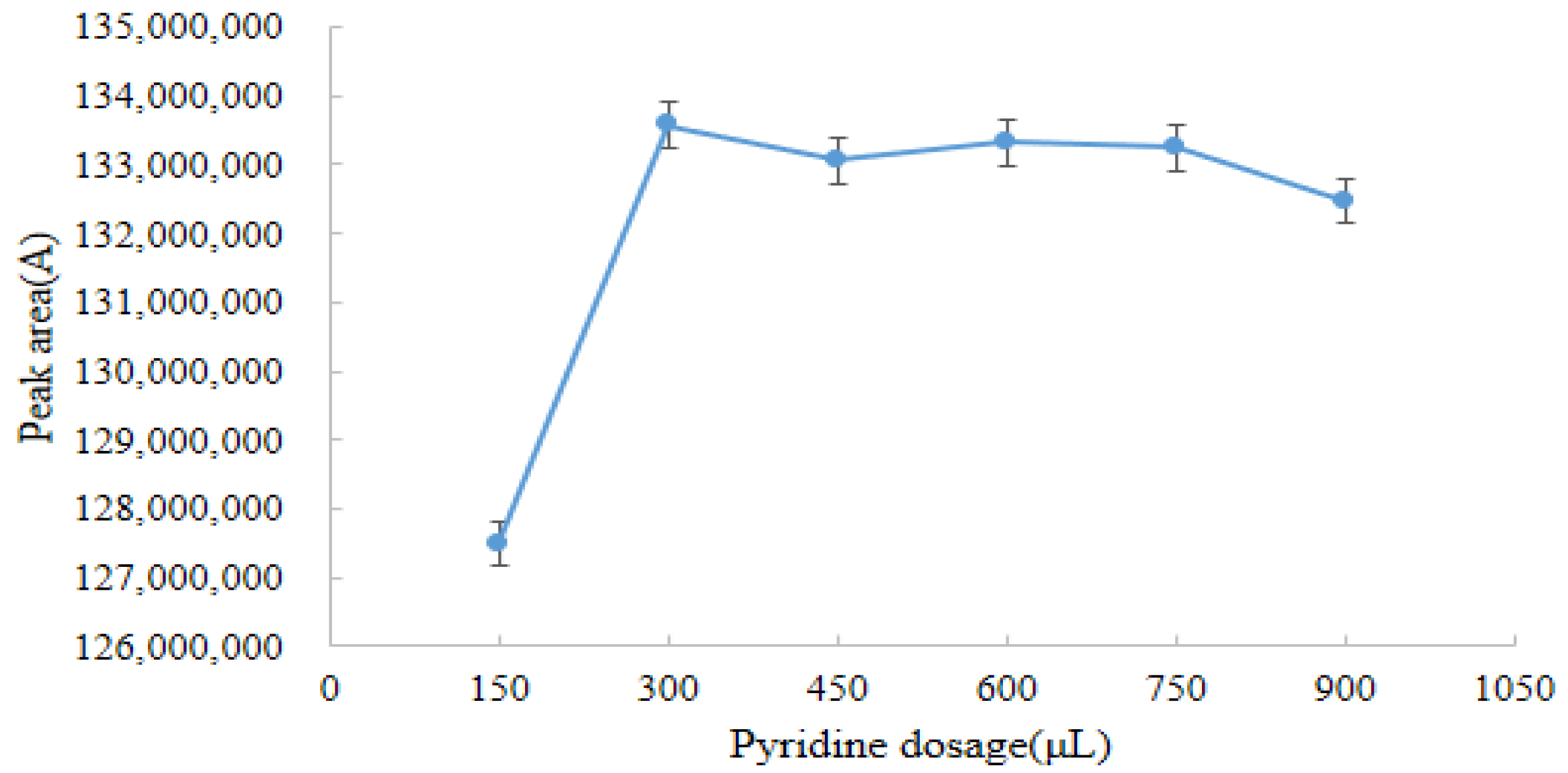
2.3.2. Optimum Pyridine Dosage
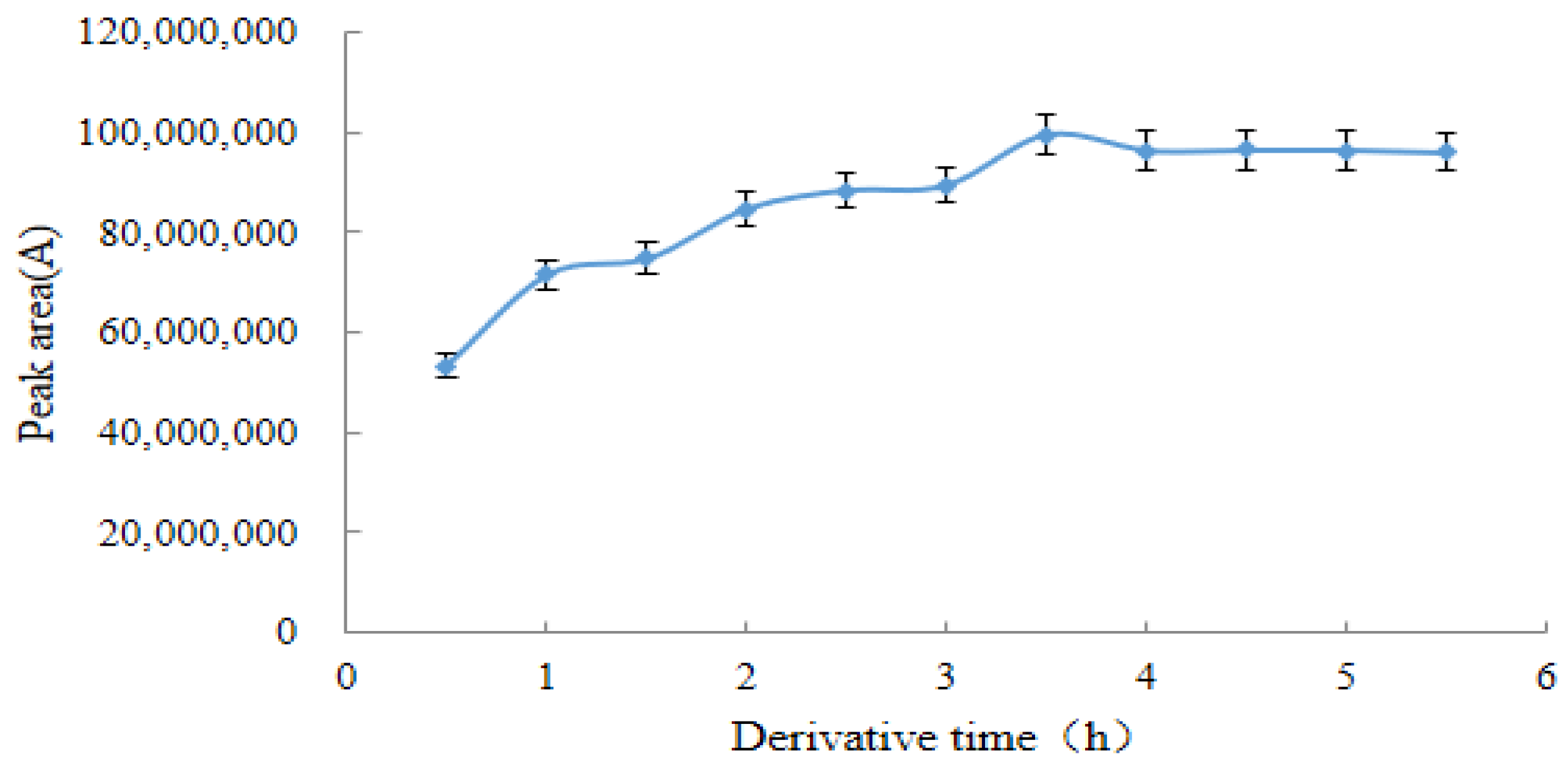
2.3.3. Optimal Derivation Time
2.4. Sample Pretreatment Optimization
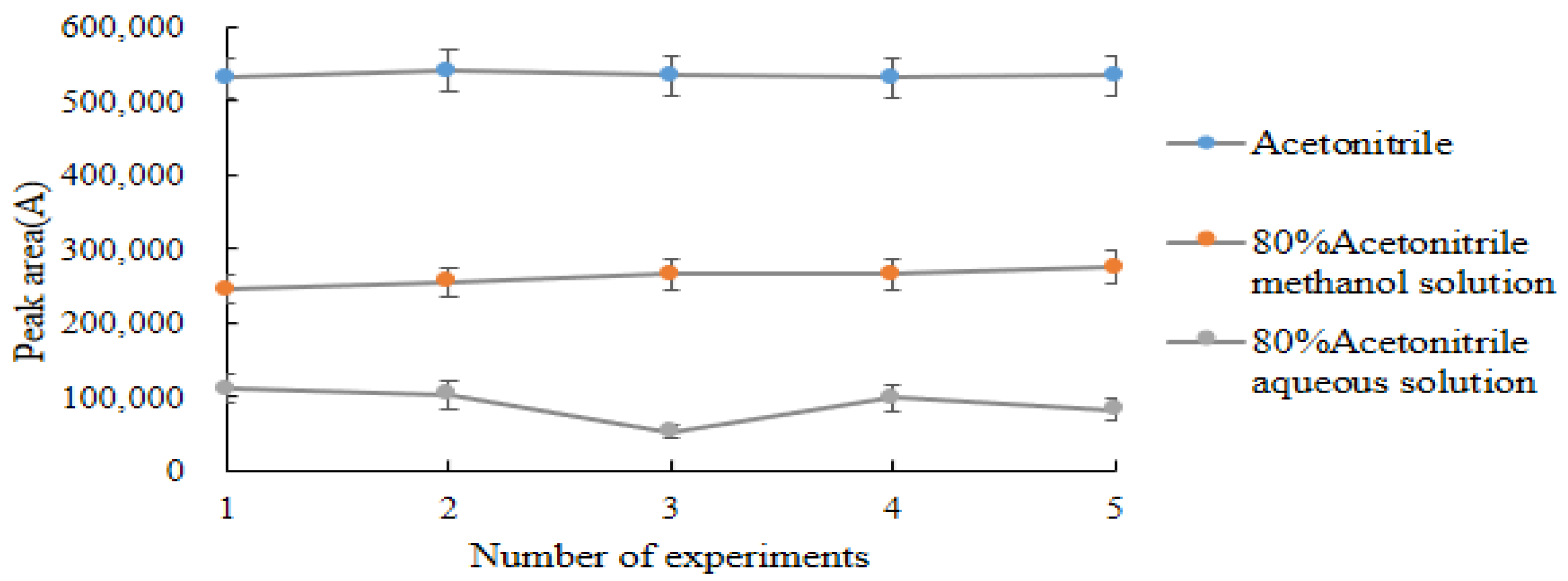
2.4.1. Selection of Extraction Reagents
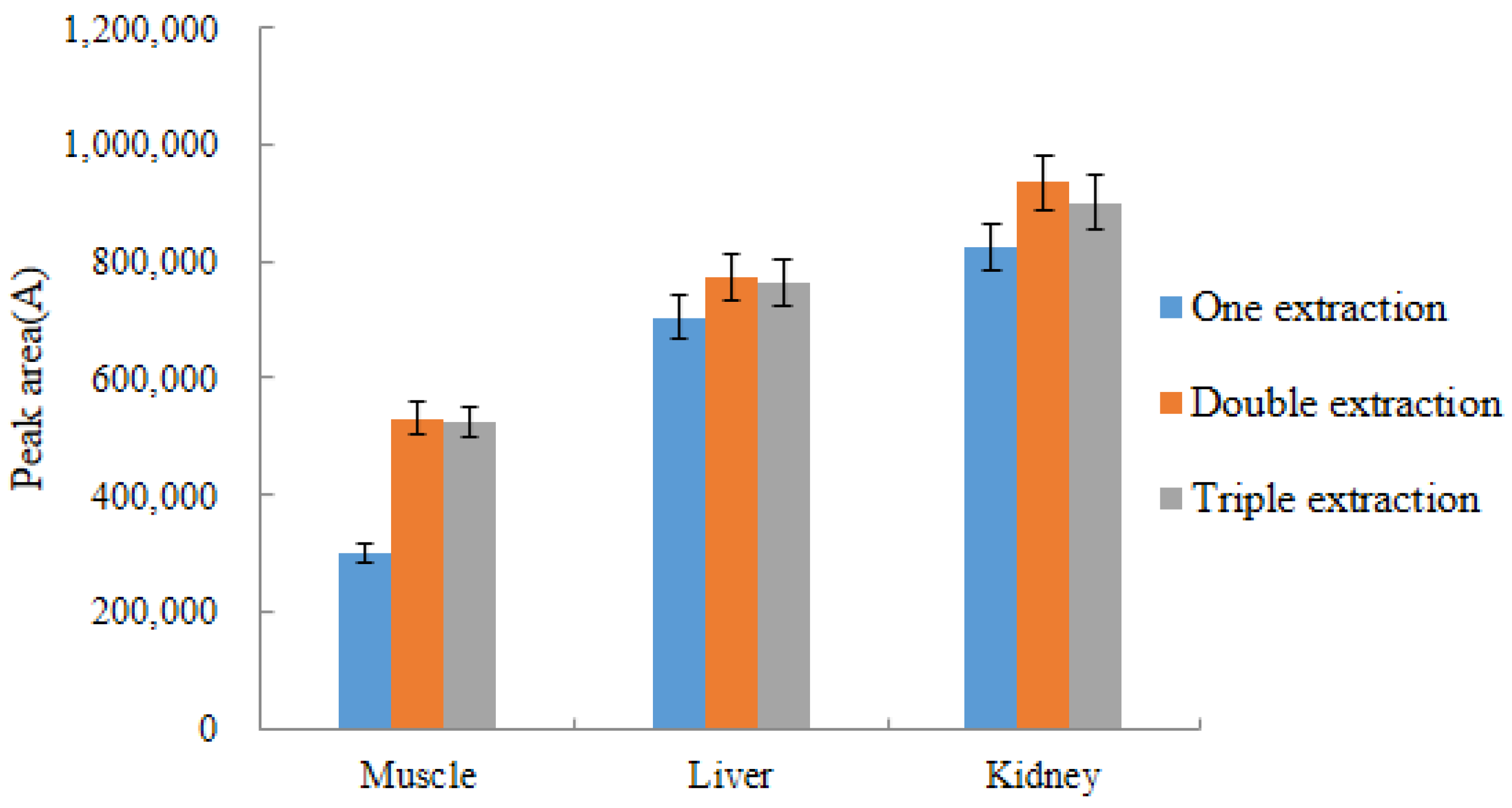
2.4.2. Selection of Extraction Times
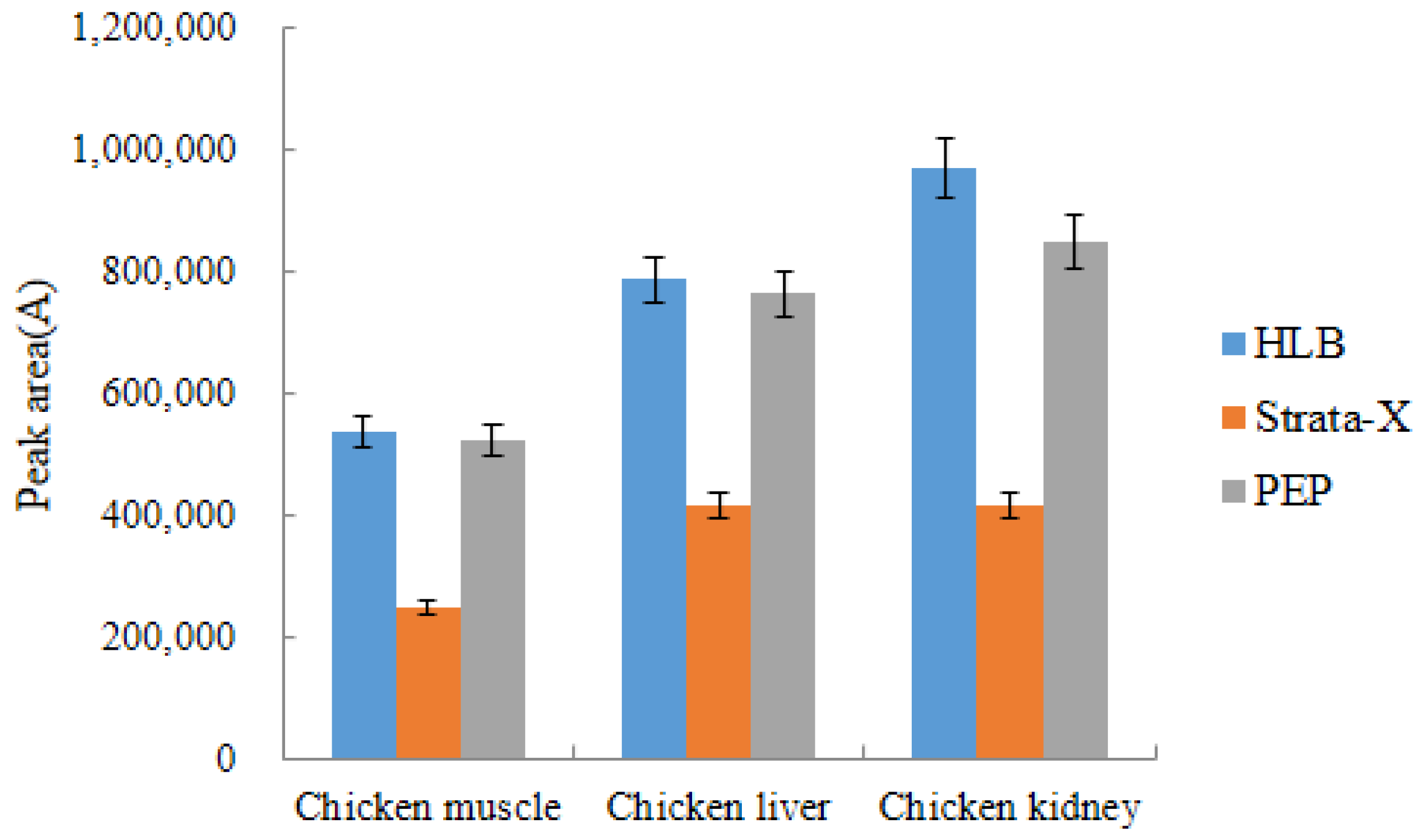
2.4.3. Selection of Solid Phase Extraction Column
2.5. GC-MS/MS Analysis Optimization
2.6. Method Validation
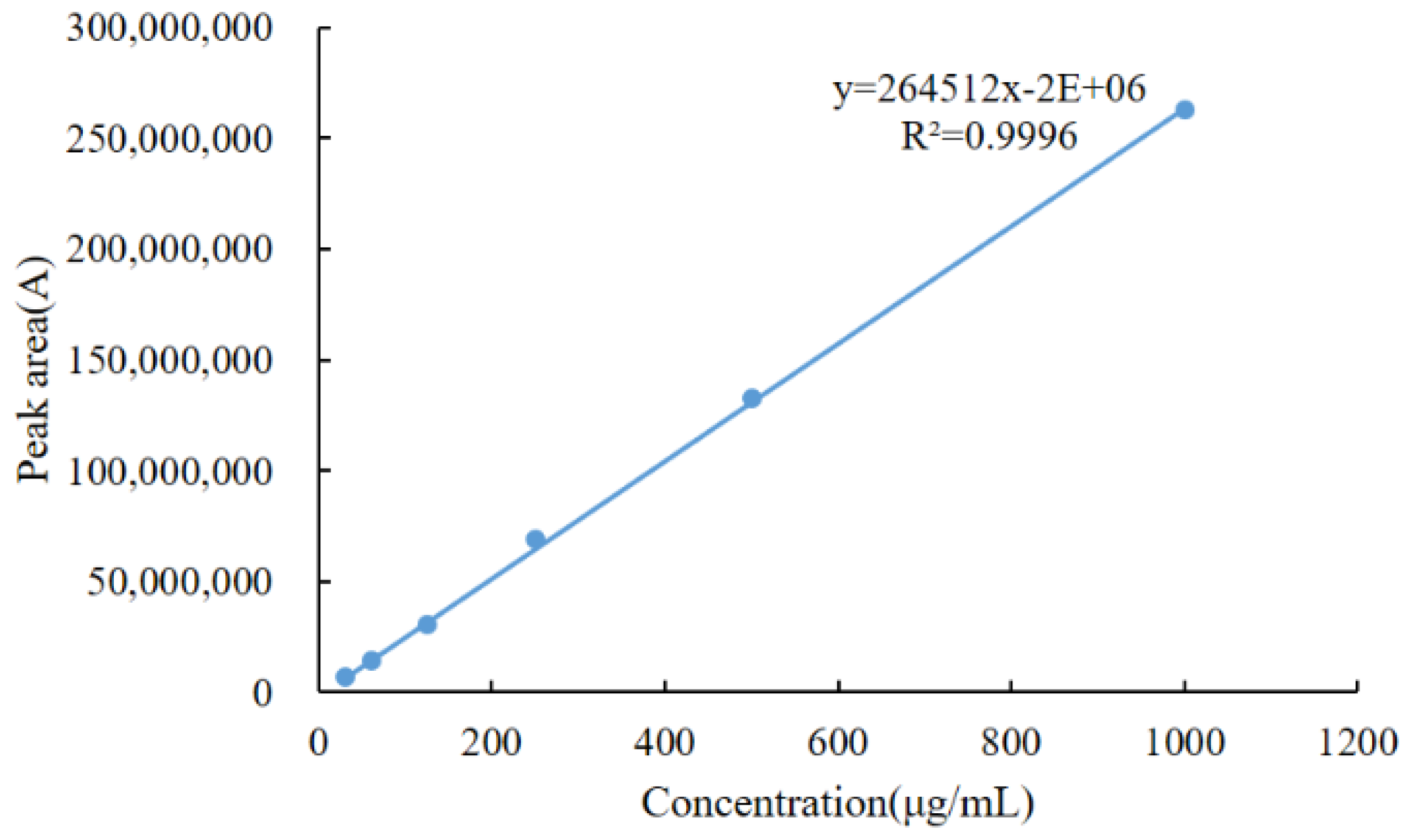
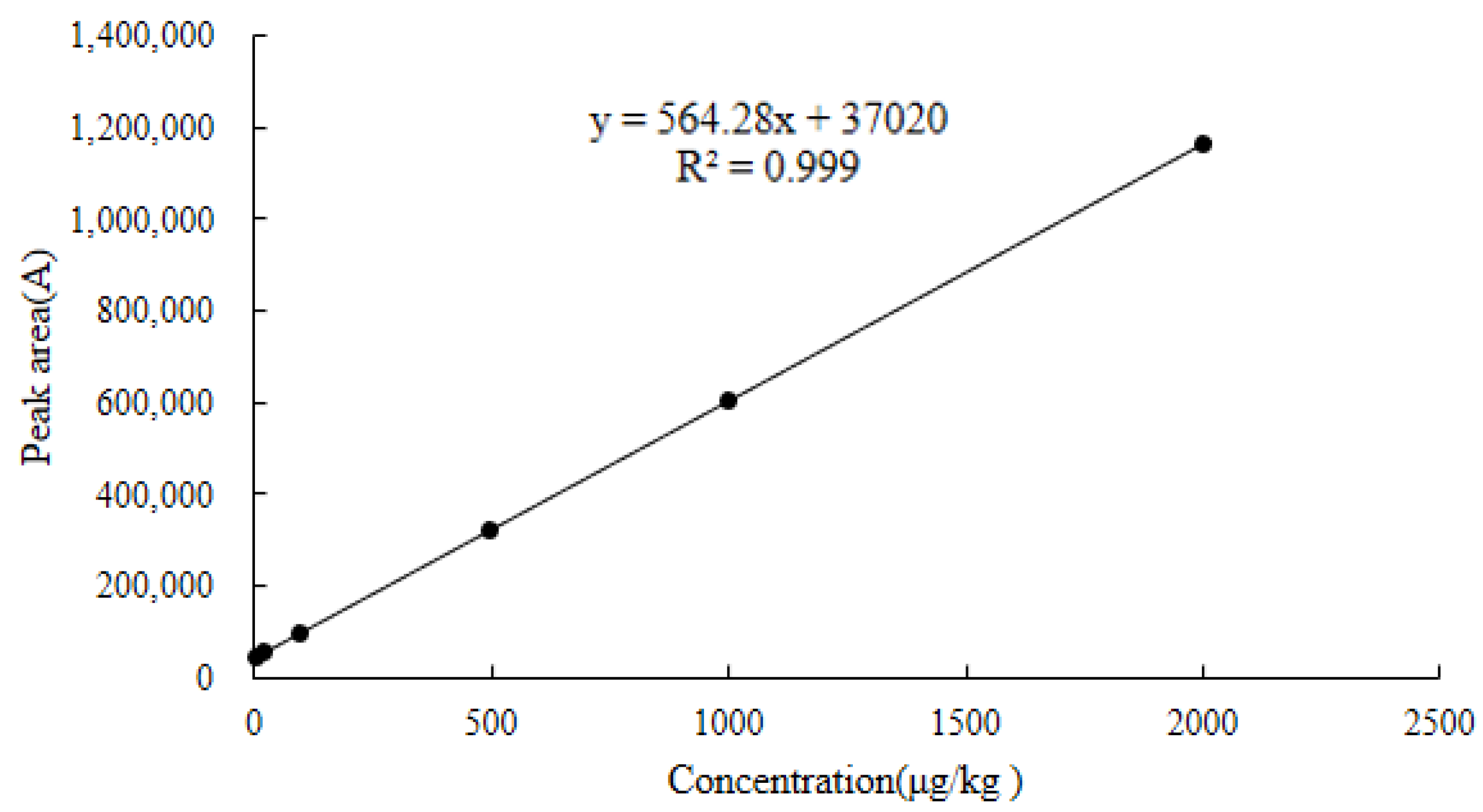
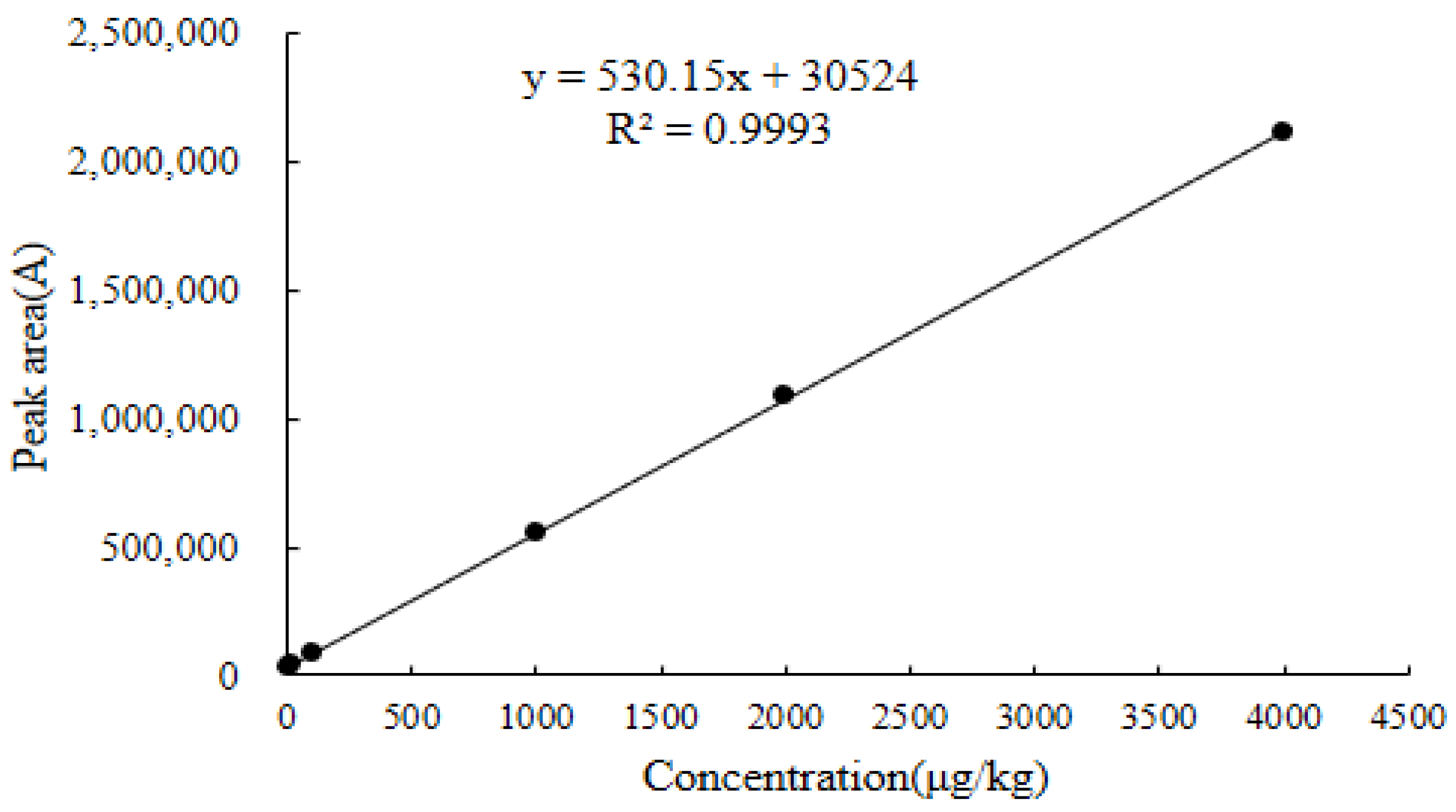
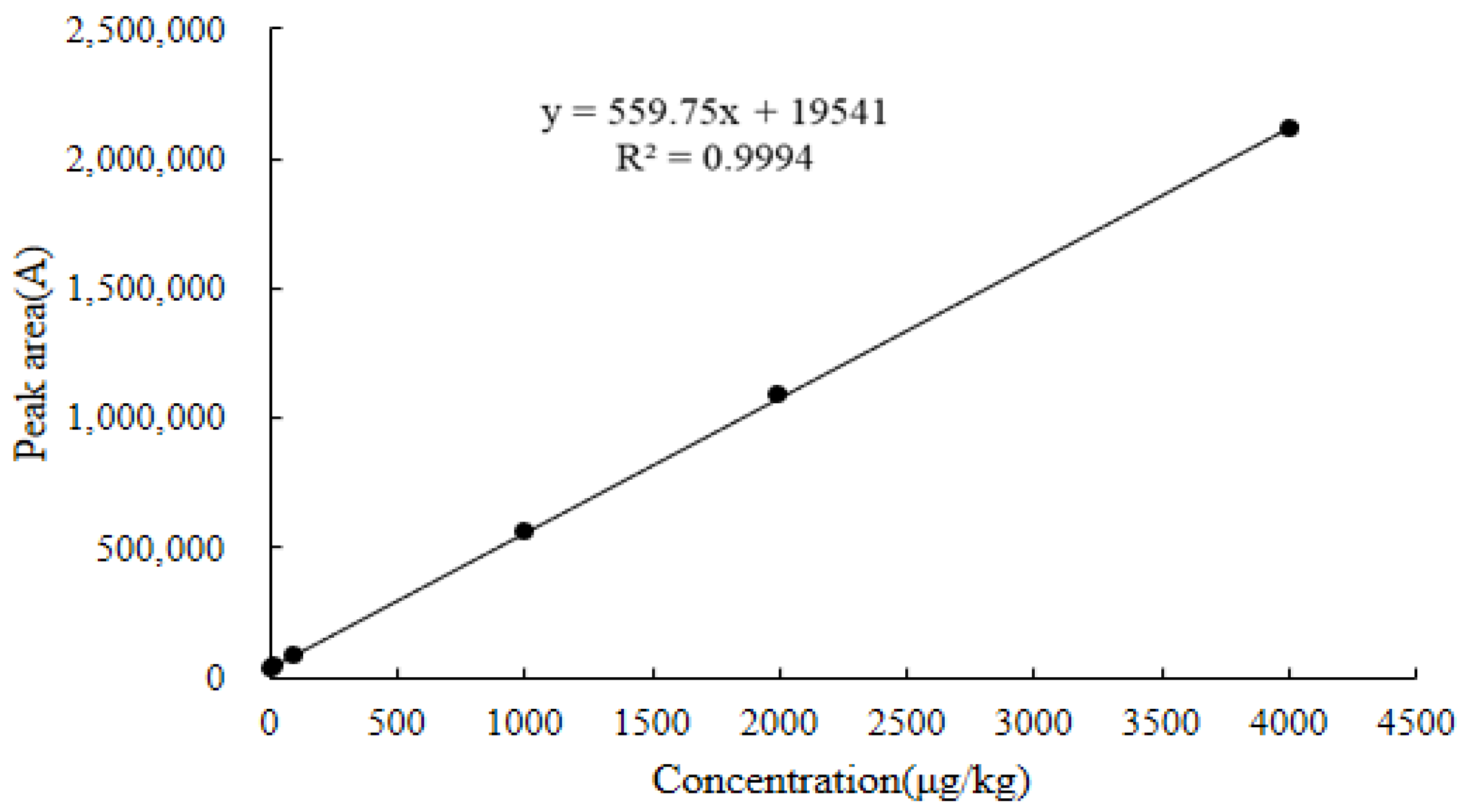
2.6.1. Linearity
2.6.2. LOD and LOQ
2.6.3. Matrix Effect
2.6.4. CCα and CCβ
2.6.5. Recovery and Precision
2.7. Stability of Standard Solutions and Derivatives
2.8. Comparison with Other Methods
2.9. Real Sample Analysis
3. Materials and Methods
3.1. Materials and Reagents
3.2. Standard Stock Solutions and Working Solutions
3.3. Preparation of the Samples
3.3.1. Breed of Test Animals and Sample Collection
3.3.2. Sample Extraction
3.3.3. Purification and Concentration
3.4. Derivatization Reaction
3.5. GC–MS/MS Analysis
3.6. Quality Parameters
3.6.1. Linearity
3.6.2. LOD and LOQ
3.6.3. Matrix Effect
3.6.4. CCα and CCβ
3.6.5. Recovery and Precision
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Tian, D.; Liu, X.; Li, X.; Xu, L.; Yan, R.; Song, X. Eimeria Maxima Rhomboid-Like Protein 5 Provided Partial Protection against Homologous Challenge in Forms of Recombinant Protein and DNA Plasmid in Chickens. Vaccines 2021, 10, 32. [Google Scholar] [CrossRef]
- Souza, T.S.; Moreira, D.R.M.; Marcelino, H.R. Chemical and pharmacological properties of decoquinate: A review of its pharmaceutical potential and future perspectives. Pharmaceutics. 2022, 14, 1383. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Wei, S.; Lu, M.; Li, J. Progress on anti-protozoan effects of decoquinate. Prog. Vet. Med. 2019, 40, 4. [Google Scholar] [CrossRef]
- Chen, X.; Hu, J.; Fang, K.; Gui, G.; Dai, J. Research progress of anti-coccidiosis drug-decoquinate. Anhui Agric. Sci. Bull. 2010, 16, 184–186. [Google Scholar] [CrossRef]
- Foote, S.J.; Thompson, J.K.; Cowman, A.F.; Kemp, D.J. Amplification of the multidrug resistance gene in some chloroquine-resistant isolates of P. falciparum. Cell 1989, 57, 921–930. [Google Scholar] [CrossRef] [PubMed]
- Baneth, G. Perspectives on canine and feline hepatozoonosis. Vet. Parasitol. 2011, 181, 3–11. [Google Scholar] [CrossRef]
- Lindsay, D.S.; Butler, J.M.; Blagburn, B.L. Efficacy of decoquinate against neospora caninum tachyzoites in cell cultures. Vet. Parasitol. 1997, 68, 35–40. [Google Scholar] [CrossRef]
- Taylor, M.A.; Bartram, D.J. The history of decoquinate in the control of coccidial infections in ruminants. J. Vet. Pharmacol. Ther. 2012, 35, 417–427. [Google Scholar] [CrossRef]
- Zhang, H. Five kinds of non-sulfanilamide antigen insect drugs and their clinical application in veterinary medicine. Chin. J. Anim. Husb. Vet. Med. 2019, 1, 153. Available online: https://kns.cnki.net/kcms/detail/detail.aspx?FileName=XMKX201901144&DbName=CJFQ2019 (accessed on 15 February 2023).
- Jiang, Y. Study on the Preparation for Decoquinate Solid Dispertion and Its Effect against Rabbit Coccidian; Henan University of Science and Technology: Luoyang, China, 2013. [Google Scholar]
- Moore, D.A.; Atwill, E.R.; Kirk, J.H.; Brahmbhatt, D.; Herrera, A.L.; Hou, L.; Singer, M.D.; Miller, T.D. Prophylactic use of decoquinate for infections with Cryptosporidium parvum in experimentally challenged neonatal calves. J. Am. Vet. Med. Assoc. 2003, 223, 839–845. [Google Scholar] [CrossRef]
- GB 31650-2019; National Food Safety Standard-Maximum Residue Limits for Veterinary Drugs in Foods. Chinese Ministry of Agriculture: Beijing, China, 2019.
- Ministry of Health, Labour and Welfare. Maximum Residue Limits (MRLs) List of Agricultural Chemicals in Foods; The Japan Food Chemical Research Foundation: Tokyo, Japan, 2015. [Google Scholar]
- US Food and Drug Administration. CFR-Code of Federal Regulations Title 21 Part 556 Tolerances for Residue of New Animal Drugs in Food; US Food and Drug Administration: Rockville, MD, USA, 2014. [Google Scholar]
- The European Medicines Agency. Commission Regulation (EU) No. 37/2010 of 22 December 2009 on Pharmacologically Active Substances and Their Classifification Regarding Maximum Residue Limits in Foodstuffs of Animal Origin; European Union: Amsterdam, The Netherlands, 2010. [Google Scholar]
- Xiao, W.; Fang, Y.; Chang, T.; Bu, S. HPLC detection method for decoquinate residues in edible tissues of chicken. J. Yangzhou Univ. Agric. Life Sci. Ed. 2014, 35, 41–45. [Google Scholar] [CrossRef]
- Preez, J.L.D.; Aucamp, M.E.; Burger, C.; Gerber, M.; Viljoen, J.M.; Zyl, L.V.; Plessis, J.D. Development and validation of the simultaneous determination of artemisone, clofazimine and decoquinate with HPLC. Die Pharm. 2018, 73, 139–142. [Google Scholar] [CrossRef]
- Chen, Q.; Long, X.; Kuang, G.; Li, Y. Determination of decoquinate in feed by high performance liquid chromatography. China Feed 2014, 13, 37–38+42. [Google Scholar] [CrossRef]
- Hobson-Frohock, A. Determination of decoquinate in poultry feed. Analyst 1982, 107, 1195–1199. [Google Scholar] [CrossRef] [PubMed]
- Chen, R. Study on the Analytical Methods and Elimination of Decoquinate Residue in Chicken Tissues. Ph.D. Thesis, Fujian Agriculture and Forestry University, Fuzhou, China, 2011. [Google Scholar] [CrossRef]
- Nász, S.; Debreczeni, L.; Rikker, T.; Eke, Z. Development and validation of a liquid chromatographic-tandem mass spectrometric method for determination of eleven coccidiostats in milk. Food Chem. 2012, 133, 536–543. [Google Scholar] [CrossRef]
- Olejnik, M.; Szprengier-Juszkiewicz, T. Deposition and depletion of decoquinate in eggs after administration of cross-contaminated feed. Food Addit. Contam. Part A Chem. Anal. Control. Expo. Risk Assess. 2015, 32, 1124–1128. [Google Scholar] [CrossRef] [PubMed]
- Pietruk, K.; Olejnik, M.; Posyniak, A. Coccidiostats in milk: Development of a multi-residue method and transfer of salinomycin and lasalocid from contaminated feed. Food Addit. Contam. Part A Chem. Anal. Control. Expo. Risk Assess. 2018, 35, 1508–1518. [Google Scholar] [CrossRef]
- Dasenaki, M.E.; Thomaidis, N.S. Multi-residue methodology for the determination of 16 coccidiostats in animal tissues and eggs by hydrophilic interaction liquid chromatography-Tandem mass spectrometry. Food Chem. 2019, 275, 668–680. [Google Scholar] [CrossRef]
- Špánik, I.; Machyňáková, A. Recent applications of gas chromatography with high-resolution mass spectrometry. J. Sep. Sci. 2018, 41, 163–179. [Google Scholar] [CrossRef]
- Beteck, R.M.; Coertzen, D.; Smit, F.J.; Birkholtz, L.M.; Haynes, R.K.; N’Da, D.D. Straightforward conversion of decoquinate into inexpensive tractable new derivatives with significant antimalarial activities. Bioorganic Med. Chem. Lett. 2016, 26, 3006–3009. [Google Scholar] [CrossRef]
- Wang, Z.; Cai, C.; Chang, X.; Fei, C.; Qiu, M.; Jiang, S.; Xue, F.; Zhang, L. Residue depletion of decoquinate in chicken tissues after oral administration. J. Vet. Pharmacol. Ther. 2013, 2, 116–121. [Google Scholar] [CrossRef]
- Chen, R.; Chen, F.; Huang, Y.; Yu, D. Determination of decoquinate in chicken meat by high performance liquid chromatography-tandem mass spectrometry. Chin. J. Chromatogr. 2010, 28, 997–1000. Available online: https://www.chrom-china.com/EN/10.3724/SP.J.1123.2010.00997 (accessed on 15 February 2023).
- Kim, B.; Ham, H.; Lee, J.J.; Cheong, N.Y.; Myung, S.W. Determination of coccidiostats (amprolium and decoquinate) in cattle and chicken’s muscle using high performance liquid chromatography. Bull. Korean Chem. Soc. 2012, 33, 559–563. [Google Scholar] [CrossRef]
- Feás, X.; Vázquez, B.; Iglesias, A.; Jesús Cantalapiedra, J.; Fente, C.A.; Cepeda, A. Sensitive determination of decoquinate in milk by high-performance liquid chromatographic coupled to laser-induced fluorescence detection. Food Anal. Methods 2010, 3, 138–143. [Google Scholar] [CrossRef]
- Wei, E.; Zhang, Q.; Wang, J.; Qu, Z.; Zhao, S.; Zou, M. Determination of content of decoquinate in chitosan-alginate microcapsules by HPLC. Southwest China J. Agric. Sci. 2013, 26, 1486–1489. [Google Scholar] [CrossRef]
- Yue, Z.; Kang, H.; Chen, X.; Xiao, C.; Hou, L.; Luo, Z.; Zhao, F.; Shen, J. Simultaneous determination of 20 coccidiostat residues in chicken by liquid chromatography-tandem mass spectrometry. Chin. J. Anal. Chem. 2012, 40, 1262–1266. [Google Scholar] [CrossRef]
- Matus, J.L.; Boison, J.O. A multi-residue method for 17 anticoccidial drugs and ractopamine in animal tissues by liquid chromatography-tandem mass spectrometry and time-of-flight mass spectrometry. Drug Test. Anal. 2016, 8, 465–476. [Google Scholar] [CrossRef]
- Lin, L.; Kang, H.; Xiao, C.; Shen, J.; Yi, G. Determination of nequinate and decoquinate residues in chicken tissues and bovine tissues by ultra performance liquid chromatography-tandem mass spectrometry. J. Food Saf. Qual. 2014, 5, 3371–3378. [Google Scholar] [CrossRef]
- Wang, Z. Determination of Residues of Decoquinate in Food of Animal Origin; Nanjing Agricultural University: Nanjing, China, 2011. [Google Scholar] [CrossRef]
- Li, Y.; Wang, X. Determination of normal used anticoccidial drug residues in chicken or eggs by liquid chromatography combined with tandem-mass spectrometry. J. Shanghai Jiaotong Univ. Agric. Sci. 2011, 6, 16–23. [Google Scholar] [CrossRef]
- Vinci, F.; Guadagnuolo, G.; Danese, V.; Salini, M.; Serpe, L.; Gallo, P. In-house validation of a liquid chromatography/electrospray tandem mass spectrometry method for confirmation of chloramphenicol residues in muscle according to decision 2002/657/EC. Rapid Commun. Mass Spectrom. 2005, 19, 3349–3355. [Google Scholar] [CrossRef]
- Lee, K.M.; Yarbrough, D.; Kozman, M.M.; Herrman, T.J.; Park, J.; Wang, R.; Kurouski, D. A rapid and convenient screening method for detection of restricted monensin, decoquinate, and lasalocid in animal feed by applying SERS and chemometrics. Food Chem. Toxicol. 2020, 144, 111633. [Google Scholar] [CrossRef]
- Wang, B.; Pang, M.; Xie, X.; Xie, K.; Zhang, Y.; Cui, L.; Zhao, X.; Wang, Y.; Shi, H.; Guo, Y.; et al. Quantification of piperazine in chicken and pig tissues by gas chromatography-electron ionization tandem mass spectrometry employing pre-column derivatization with acetic anhydride. J. Chromatogr. A 2017, 1519, 9–18. [Google Scholar] [CrossRef]
- He, L.; Liu, X.; Zeng, Z. Solutions to matrix-induced response enhancement in pesticide residue analysis by gas chromatography. Chin. J. Chromatogr. 2008, 1, 98–104. [Google Scholar] [CrossRef]
- SANTE/11945/2015; Guidance document on analytical quality control and method validation procedures for pesticides residues analysis in food and feed european commission document no SANTE/11945/2015. European Union: Brussels, Belgium, 2015.
- The European Communities. Commission Decision 2002/657/EC of 12 August 2002 Implementing Council Directive 96/23/EC Concerning the Performance of Analytical Methods and the Interpretation of Results; European Communities: Brussels, Belgium, 2002. [Google Scholar]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
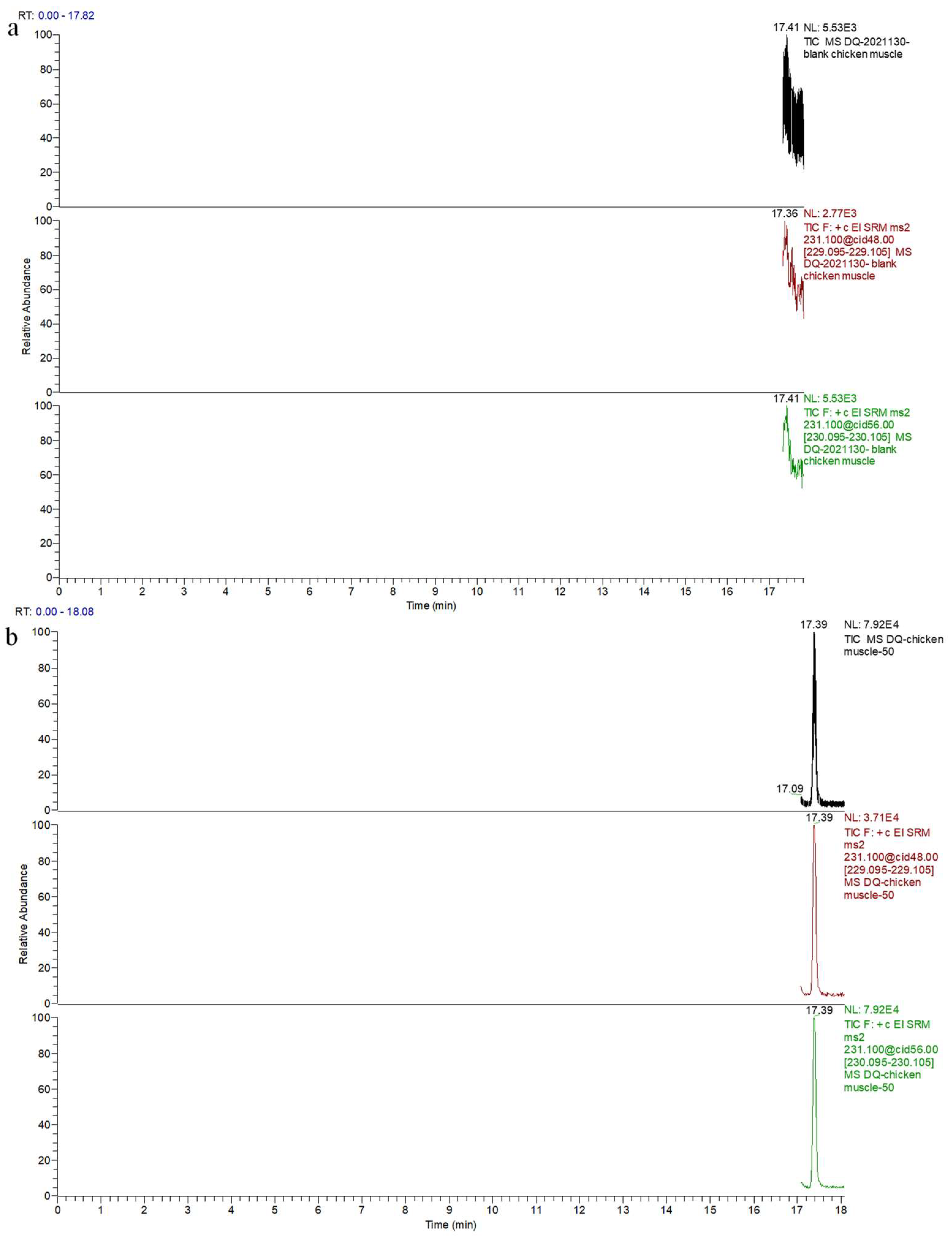{kind=link}
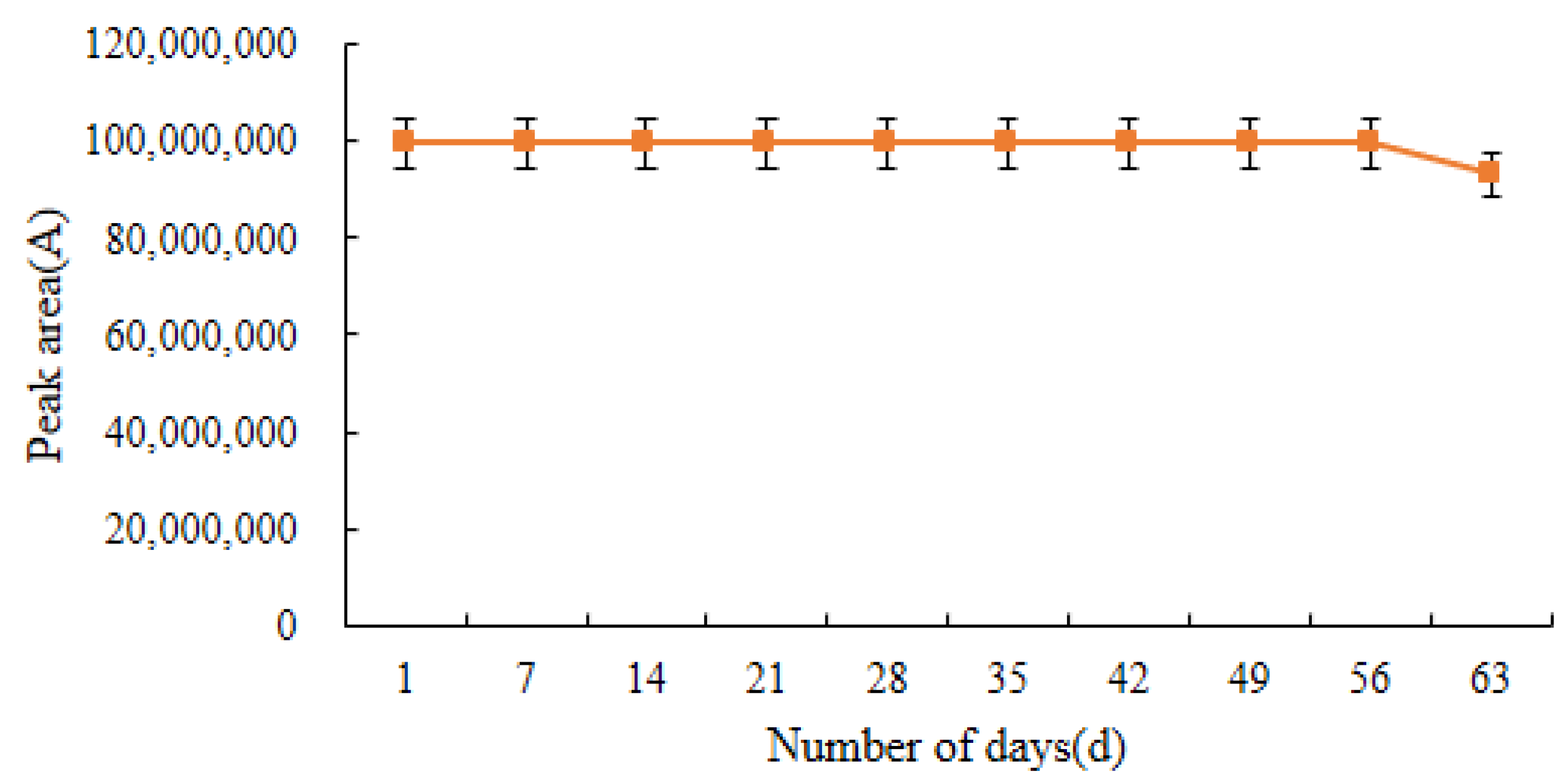{kind=link}
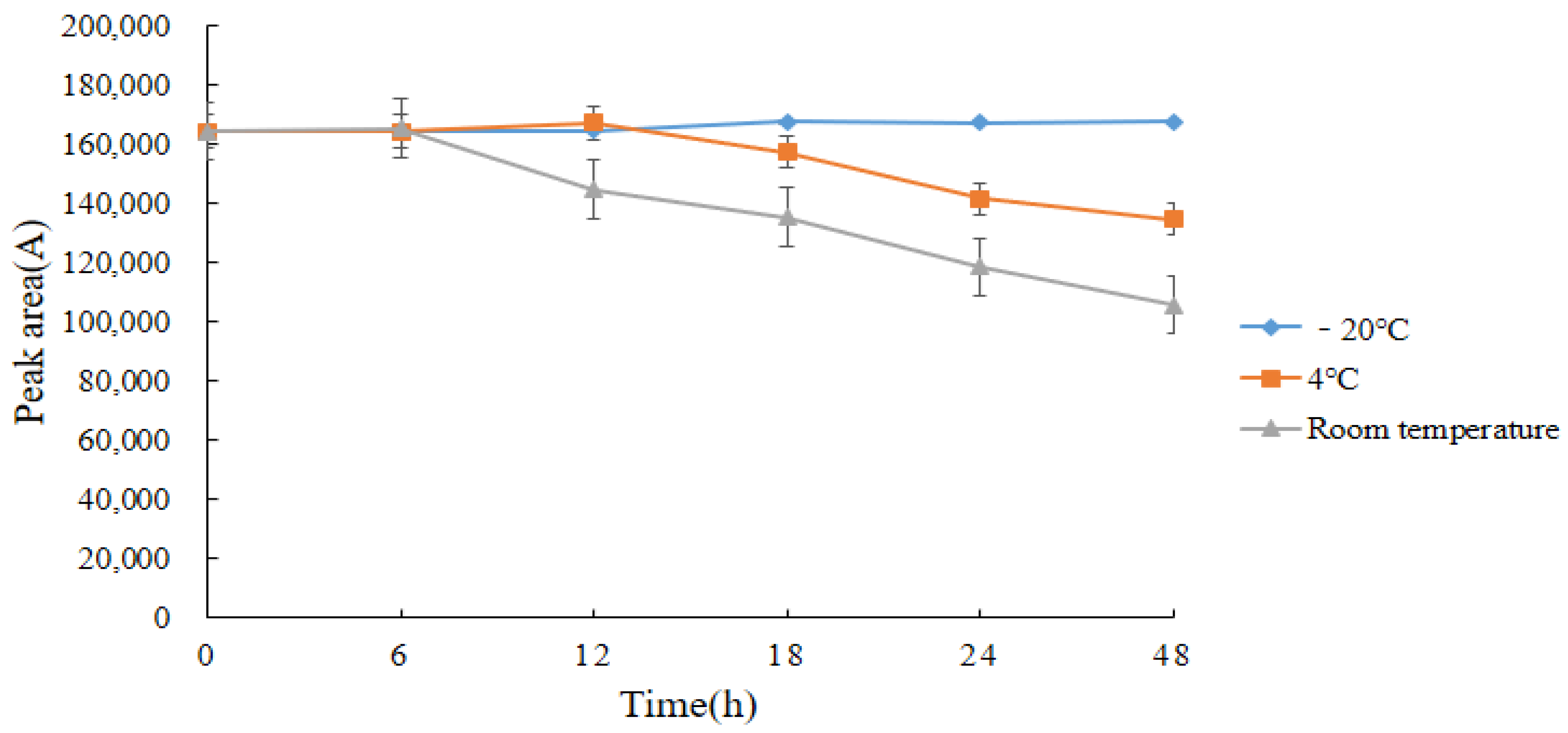{kind=link}
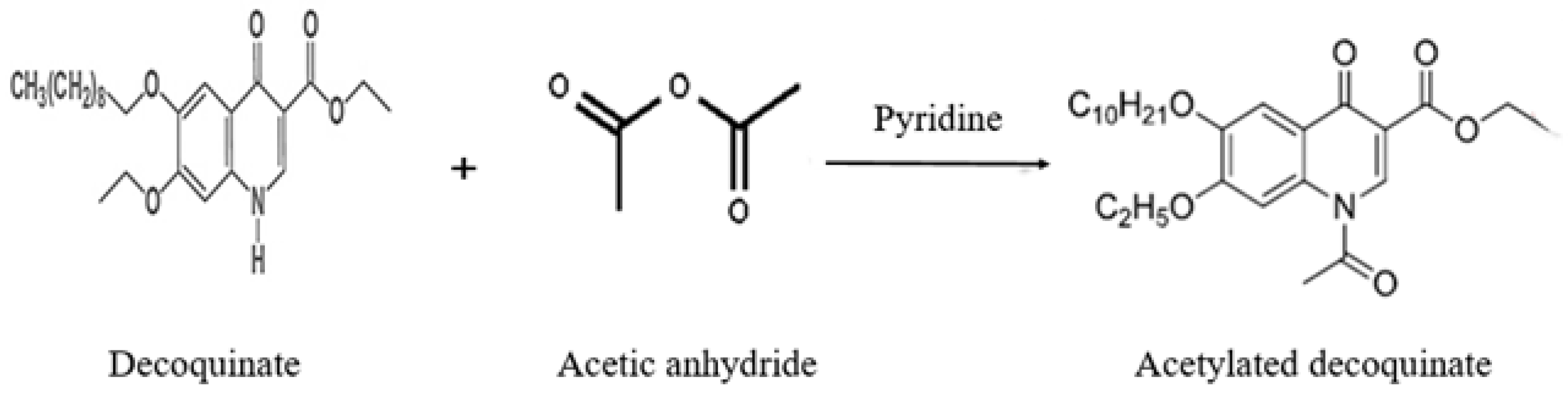{kind=link}
| Animal Species | Matrix | China (μg/kg) | Japan (μg/kg) | USA (μg/kg) | EU (μg/kg) |
|---|---|---|---|---|---|
| Chicken | Muscle | 1000 | 100 | 1000 | - |
| Liver | 2000 | 100 | 2000 | - | |
| Kidney | 2000 | 100 | 2000 | - | |
| Fat | 2000 | 2000 | 2000 | - | |
| Other edible viscera | 2000 | 2000 | 2000 | - | |
| Cattle | Muscle | - | 1000 | 1000 | - |
| Fat | - | 2000 | 2000 | - | |
| Liver | - | 2000 | 2000 | - | |
| Kidney | - | 2000 | 2000 | - | |
| Other edible viscera | - | 2000 | 2000 | - | |
| Other terrestrial mammals | Muscle | - | 1000 | 1000 (Goat only) | - |
| Liver | - | 2000 | 2000 (Goat only) | - | |
| Kidney | - | 2000 | 2000 (Goat only) | - | |
| Other edible viscera | 2000 | 2000 (Goat only) | - |
| Target Compound | Molecular Weight | Retention Time (min) | Mass Transitions (m/z) | Collision Energy (eV) |
|---|---|---|---|---|
| Acetylated decoquinate | 459.57 | 17.40 | 231.1 > 229.1 231.1 > 230.1 * | 48 56 |
| Matrix | Extraction Method | ||
|---|---|---|---|
| Acetonitrile | Acetonitrile:Ethyl Acetate (1:1, v/v) | 4% Acetic Acid–Acetonitrile Solution | |
| Chicken muscle | 87.03 ± 1.02 | 55.14 ± 2.81 | 83.26 ± 1.83 |
| Chicken liver | 86.02 ± 3.21 | 41.84 ± 0.63 | 84.32 ± 1.44 |
| Chicken kidney | 85.29 ± 0.76 | 55.80 ± 1.14 | 83.67 ± 0.55 |
| Initial Temperature (°C) | Heating Rate (°C/min) | Temperature (°C) | Hold Time (min) |
|---|---|---|---|
| 100.0 | - | 100.0 | 1.0 |
| 100.0 | 30.0 | 220.0 | 1.0 |
| 220.0 | 30.0 | 290.0 | 13.0 |
| Matrix | Linear Regression Equation | Determination Coefficient (R2) | Linearity Range (µg/kg) | LOD (µg/kg) | LOQ (µg/kg) | CCα | CCβ |
|---|---|---|---|---|---|---|---|
| Chicken muscle | y = 564.28x + 37,020 | 0.9990 | 4.9–3200 | 2.2 | 4.9 | 1032.0 | 1034.1 |
| Chicken liver | y = 530.15x + 30,524 | 0.9993 | 8.2–4800 | 4.3 | 8.2 | 2042.0 | 2049.0 |
| Chicken kidney | y = 559.75x + 19,541 | 0.9994 | 6.3–4800 | 3.7 | 6.3 | 2047.0 | 2054.1 |
| Matrix | Solvent Calibration Curve Equation | Matrix Calibration Curve Equation | Matrix Effect |
|---|---|---|---|
| Muscle | y = 668.43x + 53,066, R2 = 0.9995 | y = 564.28x + 37,020, R2 = 0.9990 | −15.58 |
| Liver | y = 659.69x + 80,573, R2 = 0.9991 | y = 530.15x + 30,524, R2 = 0.9993 | −19.64 |
| Kidney | y = 668.78x + 82,749, R2 = 0.9992 | y = 559.75x + 19,541, R2 = 0.9994 | −16.30 |
| Matrix | Added Level (μg/kg) | Recovery (%) | RSD (%) | Intraday RSD (%) | Interday RSD (%) |
|---|---|---|---|---|---|
| Chicken muscle | 4.9 | 77.38 ± 3.78 | 4.88 | 5.09 | 6.94 |
| 500.0 | 86.48 ± 3.74 | 4.32 | 4.98 | 5.71 | |
| 1000.0 α | 85.61 ± 1.28 | 1.50 | 2.14 | 2.27 | |
| 2000.0 | 88.33 ± 1.45 | 1.64 | 3.02 | 3.44 | |
| Chicken liver | 8.2 | 78.33 ± 2.14 | 2.73 | 5.56 | 6.62 |
| 1000.0 | 84.78 ± 2.20 | 2.59 | 3.41 | 5.07 | |
| 2000.0 α | 85.15 ± 1.17 | 1.37 | 2.26 | 3.47 | |
| 4000.0 | 89.65 ± 3.63 | 4.05 | 4.91 | 5.30 | |
| Chicken kidney | 6.3 | 79.15 ± 3.43 | 4.33 | 5.74 | 8.06 |
| 1000.0 | 87.95 ± 1.01 | 1.15 | 2.10 | 2.47 | |
| 2000.0 α | 88.59 ± 1.03 | 1.16 | 1.63 | 3.20 | |
| 4000.0 | 89.10 ± 2.71 | 3.04 | 4.09 | 4.53 |
| Matrix | Analytical Method | Chromatographic Conditions | LODs (μg/kg) | LOQs (μg/kg) | Recovery Rate (%) |
|---|---|---|---|---|---|
| Chicken liver [33] | HPLC-UV | Agilent Eclipse XDB-C18 (4.6 × 250 mm, 5 μm) Mobile phase: Acetonitrile-ethyl acetate (1:1, V/V) | 100 | 200 | 72.9~96.8 |
| DQ capsule [31] | HPLC-UV | Agilent ZORBAX 80A Extend-C18 (25 × 4.6 mm, 5 μm) Mobile phase: acetonitrile-0.1% formic acid aqueous solution (75:25, V/V) | - | 50 | ≥98 |
| Chicken muscle [36] | LC-MS/MS | XTerra MS (C18: 2.1 × 100.0 mm, 3.5 μm) Mobile phase: A: 0.1% formic acid aqueous solution; B: acetonitrile | 1.0 | 2.5 | 85.3~104.9 |
| Milk [24] | HPLC-MS/MS | Agilent Zorbax Eclipse Plus C18 RRHD (50.0 × 2.1 mm, 1.8 µm) Mobile phase: A: 0.01 moL ammonium formate buffer (pH 4.0); B: acetonitrile | 0.78 | 5.0 | ≥98.3 |
| Eggs [23] | LC-MS/MS | Zorbax 80 SB-C18 (2.1 × 150 mm, 5 μm) Mobile phase: A: methyl alcohol; B: acetonitrile; C: 0.01 mol/L ammonium formate buffer (pH 4.0) | 0.09 | 0.36 | 80.0~120.0 |
| Chicken muscle [37] | LC-MS/MS | Agilent Poroshell 120 EC-C18 (2.1 × 100 mm,2.7 μm) Mobile phase: A: methyl alcohol; B: 1% acetic acid and 5 mmoL ammonia buffer | 0.008 | 0.027 | 74~112 |
| Fodder [8] | HPLC-FLD | C18 (250 × 4.6 mm, 5 μm) Mobile phase: 0.005 mol/L calcium chloride solution- methyl alcohol (15:85, V/V) | - | 500 | 82.5~99.0 |
| Bovine liver [34] | UHPLC-MS/MS | Acquity BEH C18 (50.0 × 2.1 mm i.d., 1.7 μm) Mobile phase: A: 0.005 moL/L aqueous ammonium acetate solution (containing 0.05% formic acid); B: acetonitrile | 5 | 10 | 78~102 |
| Fodder [38] | SERS | - | 381 | - | |
| Chicken muscle [32] | HPLC-MS | Acquity BEH C18 (50.0 × 2.1 mm, 1.7 μm) Mobile phases: A: 0.005 mol/L ammonium acetate solution (contained 0.05% formic acid); B: methyl alcohol | 5 | 50 | ≥84.8 |
| Chicken muscle, liver, and kidney (this study) | GC-MS/MS | TG-5MS AMINE (30.0 m × 0.25 mm × 0.25 μm) Mobile phase: high purity helium (99.999%, 60 psi) | 2.2~4.3 | 4.9~8.2 | 77.38~89.65 |
| Instrument Content | Experimental Conditions |
|---|---|
| Chromatographic column | TG-5MS (30.0 m × 0.25 μm × 0.25 mm i.d.) |
| Carrier gas | Helium (99.999%, 60 psi) |
| Carrier gas mode | Constant current mode |
| Carrier gas column velocity | 1.0 mL/min |
| Carrier gas saves time and flow | Time: 2 min, flow rate: 20.0 mL/min |
| Injection port temperature | 280 °C |
| Shunt mode | Undivided injection |
| Shunt flow | 50.0 mL/min |
| Non shunting time | 1.0 min |
| Injection volume | 1.0 μL |
| Temperature programmed | Initial temperature 100 °C, maximum temperature 290 °C |
| Instrument Content | Experimental Conditions |
|---|---|
| Ionization mode | Electron bombardment ion source (EI) |
| Electron beam energy | 70 eV |
| Collision gas | High purity argon (>99.999%, 40 psi) |
| Ion source temperature | 280 °C |
| Transmission line temperature | 280 °C |
| Solvent delay | 3.0 min |
| Acquisition data mode | SCAN mode is qualitative Auto SRM mode is quantitative |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Tang, Y.; Lu, Y.; Guo, Y.; Xie, K.; Guan, F.; Gao, P.; Zhu, Y.; Dong, Y.; Zhang, T.; et al. Qualitative and Quantitative Determination of Decoquinate in Chicken Tissues by Gas Chromatography Tandem Mass Spectrometry. Molecules 2023, 28, 3875. https://doi.org/10.3390/molecules28093875
Liu S, Tang Y, Lu Y, Guo Y, Xie K, Guan F, Gao P, Zhu Y, Dong Y, Zhang T, et al. Qualitative and Quantitative Determination of Decoquinate in Chicken Tissues by Gas Chromatography Tandem Mass Spectrometry. Molecules. 2023; 28(9):3875. https://doi.org/10.3390/molecules28093875
Chicago/Turabian StyleLiu, Shuyu, Yayun Tang, Yang Lu, Yawen Guo, Kaizhou Xie, Fanxun Guan, Pengfei Gao, Yali Zhu, Yuhao Dong, Tao Zhang, and et al. 2023. "Qualitative and Quantitative Determination of Decoquinate in Chicken Tissues by Gas Chromatography Tandem Mass Spectrometry" Molecules 28, no. 9: 3875. https://doi.org/10.3390/molecules28093875
APA StyleLiu, S., Tang, Y., Lu, Y., Guo, Y., Xie, K., Guan, F., Gao, P., Zhu, Y., Dong, Y., Zhang, T., Zhang, G., Dai, G., & Xie, X. (2023). Qualitative and Quantitative Determination of Decoquinate in Chicken Tissues by Gas Chromatography Tandem Mass Spectrometry. Molecules, 28(9), 3875. https://doi.org/10.3390/molecules28093875