
Skeleton Synthesis of a Plant-Derived Radioprotective Alkaloid Born to Produce a Novel Fused Heterocycle

 ,
,
Abstract
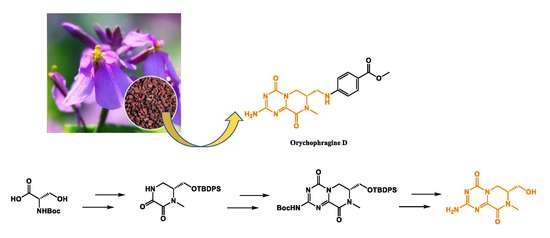
1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Reagents and Instruments
4.2. Experimental Procedures
4.2.1. N-(Tert-butoxycarbonyl)-O-(tetrahydro-2H-pyran-2-yl)-L-serine (6)
4.2.2. Tert-butyl((2S)-1-amino-1-oxo-3-((tetrahydro-2H-pyran-2-yl)oxy)propan-2-yl)carbamate (7)
4.2.3. (2.R)-N2-Methyl-3-((tetrahydro-2H-pyran-2-yl)oxy)propane-1,2-diamine (8)
4.2.4. (6.R)-1-Methyl-6-(((tetrahydro-2H-pyran-2-yl)oxy)methyl)piperazine-2,3-dione (9)
4.2.5. (R)-6-(Hydroxymethyl)-1-methylpiperazine-2,3-dione (10)
4.2.6. (R)-6-(((Tert-butyldiphenylsilyl)oxy)methyl)-1-methylpiperazine-2,3-dione (11)
4.2.7. (R)-6-(((Tert-butyldiphenylsilyl)oxy)methyl)-1-methyl-3-thioxopiperazin-2-one (12)
4.2.8. Tert-butyl ((Z)-amino(((R,Z)-5-(((tert-butyldiphenylsilyl)oxy)methyl)-4-methyl-3-oxopiperazin-2-ylidene)amino)methylene)carbamate (13)
4.2.9. Tert-butyl (R)-(7-(((tert-butyldiphenylsilyl)oxy)methyl)-8-methyl-4,9-dioxo-6,7,8,9-tetrahydro-4H-pyrazino [1,2-a][1,3,5]triazin-2-yl)carbamate (14)
4.2.10. Tert-butyl (R)-(7-(hydroxymethyl)-8-methyl-4,9-dioxo-6,7,8,9-tetrahydro-4H-pyrazino [1,2-a][1,3,5]triazin-2-yl)carbamate (15)
4.2.11. (R)-2-Amino-7-(hydroxymethyl)-8-methyl-7,8-dihydro-4H-pyrazino [1,2-a][1,3,5]triazine-4,9(6H)-dione (1)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Roberts, M.F.; Wink, M. Alkaloids: Biochemistry, Ecology, and Medicinal Applications; Springer: Berlin/Heidelberg, Germany, 1998. [Google Scholar]
- Cushnie, T.P.T.; Cushnie, B.; Lamb, A.J. Alkaloids: An overview of their antibacterial, antibiotic-enhancing and antivirulence activities. Int. J. Antimicrob. Agents 2014, 44, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Ettefagh, K.A.; Burns, J.T.; Junio, H.A.; Kaatz, G.W.; Cech, N.B. Goldenseal (Hydrastis canadensis L.) extracts synergistically enhance the antibacterial activity of berberine via efflux pump inhibition. Planta Med. 2011, 77, 835–840. [Google Scholar] [CrossRef] [PubMed]
- Yohannes, A.; Eyalarasan, K.; Eyob, L.; Ghebrengus, E.; Weldemariam, L.; Yohannes, T.; Yemane, A. Antibacterial and Antifungal Activities of easily grown Eritrean Black Pepper. Int. J. Eng. Res. Technol. 2018, 8, 81–83. [Google Scholar]
- Pérez, S.; Tvaroška, I. Carbohydrate-protein interactions: Molecular modeling insights. Adv. Carbohydr. Chem. Biochem. 2014, 71, 9–136. [Google Scholar] [PubMed]
- Kurek, J. Alkaloids—Their Importance in Nature and Human Life; Books on Demand: Norderstedt, Germany, 2019. [Google Scholar]
- Christiansen, J.L.; Jørnsgård, B.; Buskov, S.; Olsen, C.E. Effect of drought stress on content and composition of seed alkaloids in narrow-leafed lupin, Lupinus angustifolius L. Eur. J. Agron. 1997, 7, 307–314. [Google Scholar] [CrossRef]
- Szabó, B.; Tyihák, E.; Szabó, G.; Botz, L. Mycotoxin and drought stress induced change of alkaloid content of Papaver somniferum plantlets. Acta Bot. Hung. 2003, 45, 409–417. [Google Scholar] [CrossRef]
- Bhambhani, S.; Kondhare, K.R.; Giri, A.P. Diversity in Chemical Structures and Biological Properties of Plant Alkaloids. Molecules 2021, 26, 3374. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.-J.; Li, B.; Cui, H.-M.; Chen, L.; Tian, Y.; Liu, S.-J.; Li, B.-W.; Li, M.; Xia, Z.-M.; Chen, X.-X.; et al. Orychophragines A–C, Three Biologically Active Alkaloids from Orychophragmus violaceus. Org. Lett. 2018, 20, 656–659. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.J.; Dong, J.X.; Liu, S.F.; Li, B.; Tian, Y.; Li, M. Application of Orychophragine D in the Preparation of Anti-Radiation Drugs. CN113262229A, 20 May 2021. [Google Scholar]
- Ramos-Tomillero, I.; Rodríguez, H.M.; Albericio, F. Trends to Acid-Labile Cys Protecting Groups: Thp as an Efficient and Non-Aromatic Cys Protecting Group for Fmoc Chemistry. In Proceedings of the 24th American Peptide Symposium 2015, Orlando, FL, USA, 20–25 June 2015. [Google Scholar] [CrossRef]
- Yu, S.; Zhu, S.; Pan, X.; Yang, J.; Ma, D. Reinvestigation on total synthesis of kaitocephalin and its isomers. Tetrahedron 2011, 67, 1673–1680. [Google Scholar] [CrossRef]
- Yu, X.; Dai, Y.; Yang, T.; Gagné, M.R.; Gong, H. Facile synthesis of salmochelin S1, S2, MGE, DGE, and TGE. Tetrahedron 2011, 67, 144–151. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||
|---|---|---|---|
| Lewis Acid | Solvent | Temperature (°C) | Yield |
| ZnCl2 | 1.4-dioxane | 25 | NR * |
| ZnCl2 | DMF | 25 | NR |
| ZnCl2 | acetonitrile | 25 | NR |
| ZnCl2 | toluene | 25 | NR |
| ZnCl2 | 1.4-dioxane | 90 | 53% |
| ZnCl2 | DMF | 90 | 65% |
| ZnCl2 | acetonitrile | 90 | 16% |
| ZnCl2 | toluene | 90 | 39% |
| CuCl2 | DMF | 90 | 48% |
| CuCl2 | 1.4-dioxane | 90 | 34% |
| CuCl2 | toluene | 90 | 20% |
| Zn(AcO)2 | DMF | 90 | 70% |
| Zn(AcO)2 | 1.4-dioxane | 90 | 55% |
| Zn(AcO)2 | toluene | 90 | 42% |
| HgCl2 | DMF | 90 | 78% |
| HgCl2 | 1.4-dioxane | 90 | 71% |
| HgCl2 | toluene | 90 | 40% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, S.; Gu, H.; Liang, K.; Wei, Z.; Li, B.; Tian, Y.; Li, R.; Zhang, G.; Liu, S. Skeleton Synthesis of a Plant-Derived Radioprotective Alkaloid Born to Produce a Novel Fused Heterocycle. Molecules 2023, 28, 3829. https://doi.org/10.3390/molecules28093829
Liu S, Gu H, Liang K, Wei Z, Li B, Tian Y, Li R, Zhang G, Liu S. Skeleton Synthesis of a Plant-Derived Radioprotective Alkaloid Born to Produce a Novel Fused Heterocycle. Molecules. 2023; 28(9):3829. https://doi.org/10.3390/molecules28093829
Chicago/Turabian StyleLiu, Sifan, Huiling Gu, Kai Liang, Zhenzhen Wei, Bin Li, Ying Tian, Ruihong Li, Guangjie Zhang, and Shuchen Liu. 2023. "Skeleton Synthesis of a Plant-Derived Radioprotective Alkaloid Born to Produce a Novel Fused Heterocycle" Molecules 28, no. 9: 3829. https://doi.org/10.3390/molecules28093829
APA StyleLiu, S., Gu, H., Liang, K., Wei, Z., Li, B., Tian, Y., Li, R., Zhang, G., & Liu, S. (2023). Skeleton Synthesis of a Plant-Derived Radioprotective Alkaloid Born to Produce a Novel Fused Heterocycle. Molecules, 28(9), 3829. https://doi.org/10.3390/molecules28093829