

Multiple Strategies to Develop Small Molecular KRAS Directly Bound Inhibitors


{kind=link}
{kind=link}
{kind=link}
{kind=link}
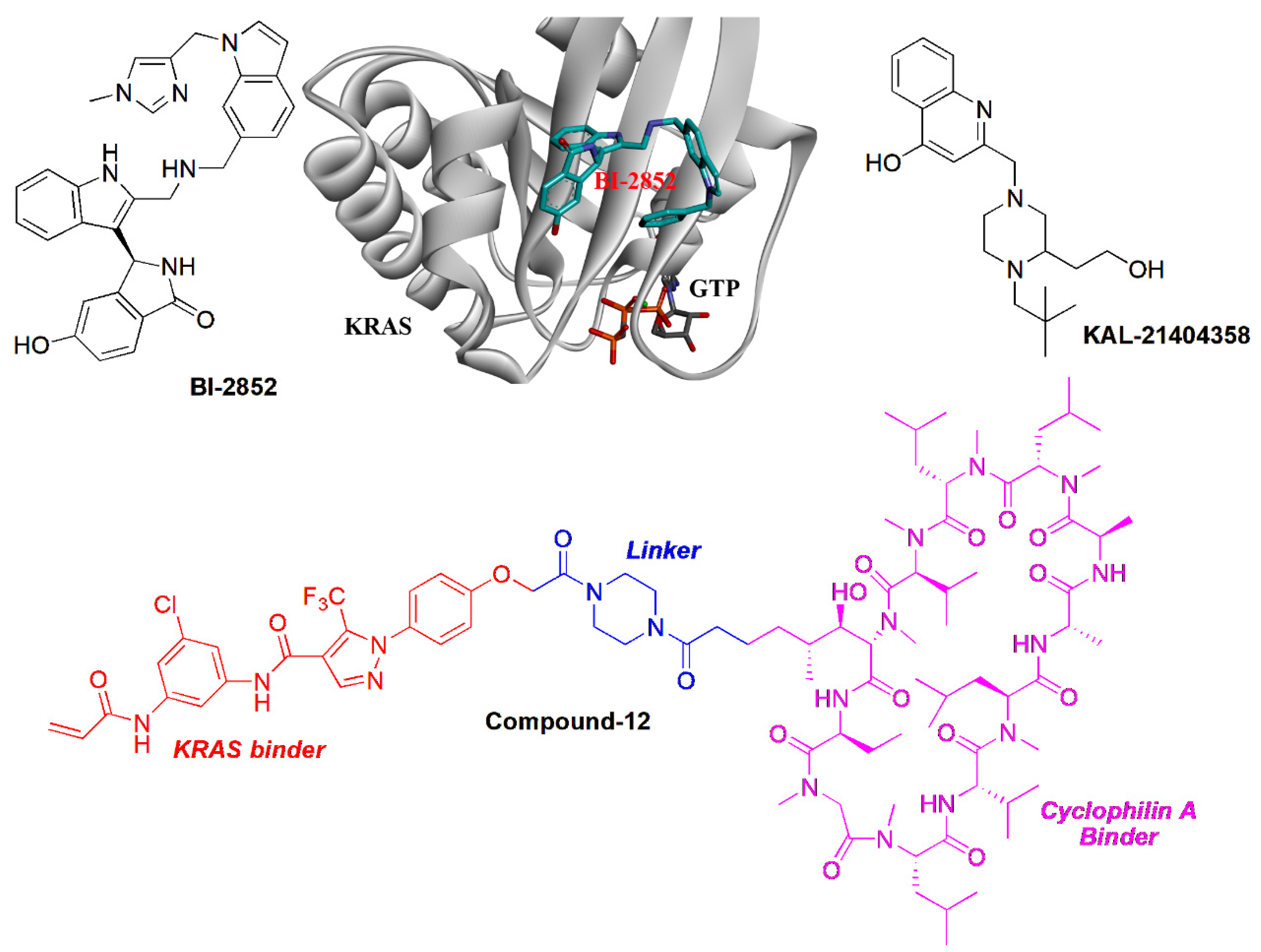{kind=link}
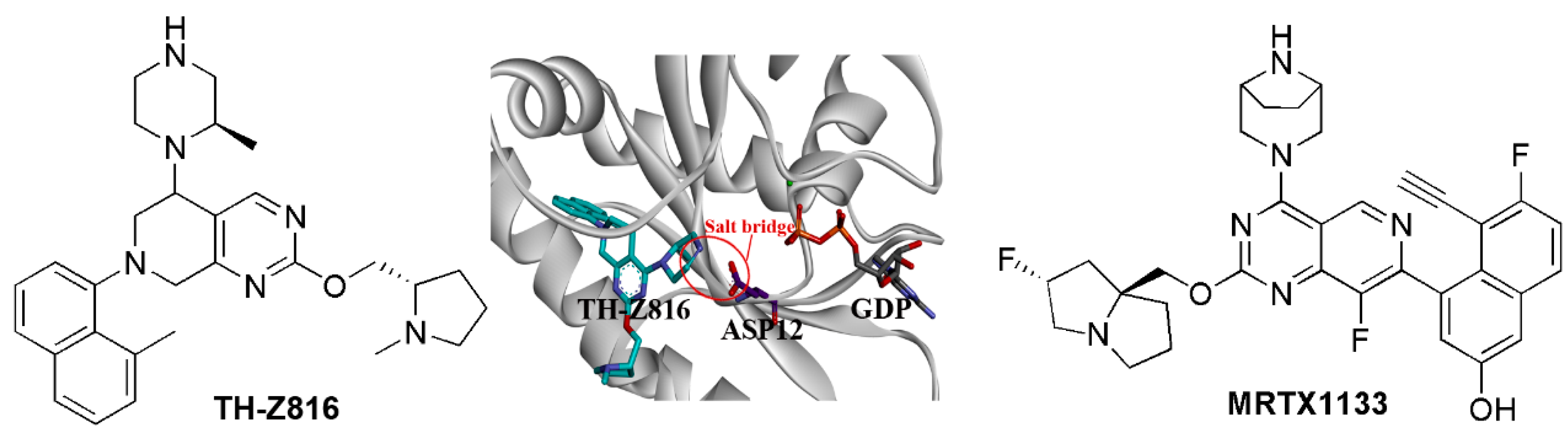{kind=link}
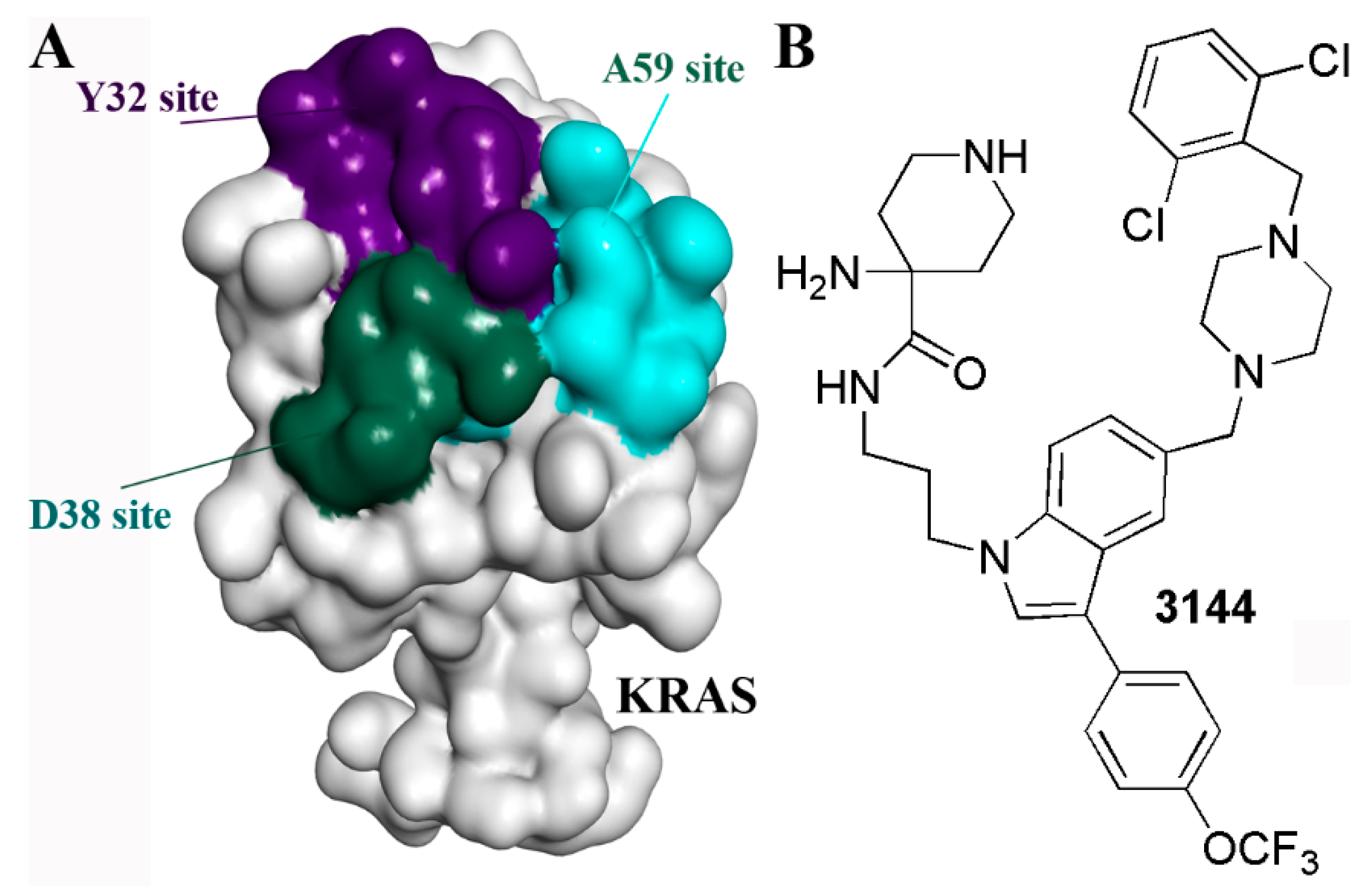{kind=link}
Abstract
1. Introduction
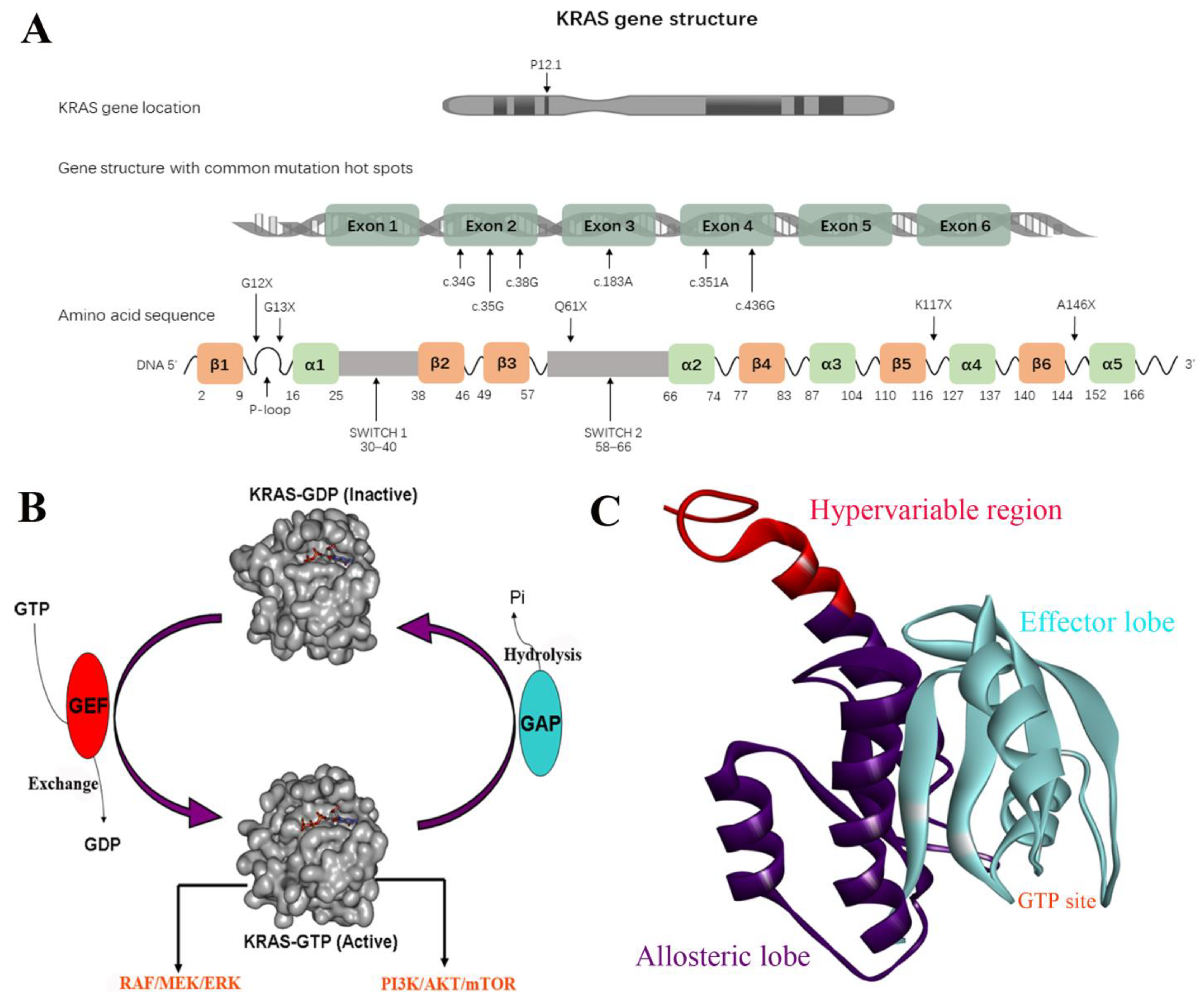
2. Structural Basis of KRAS Function
3. KRAS Mutations in Cancer
4. Challenge in Directly Targeting Oncogenic KRAS
5. Strategies Directly Targeting Mutated KRAS
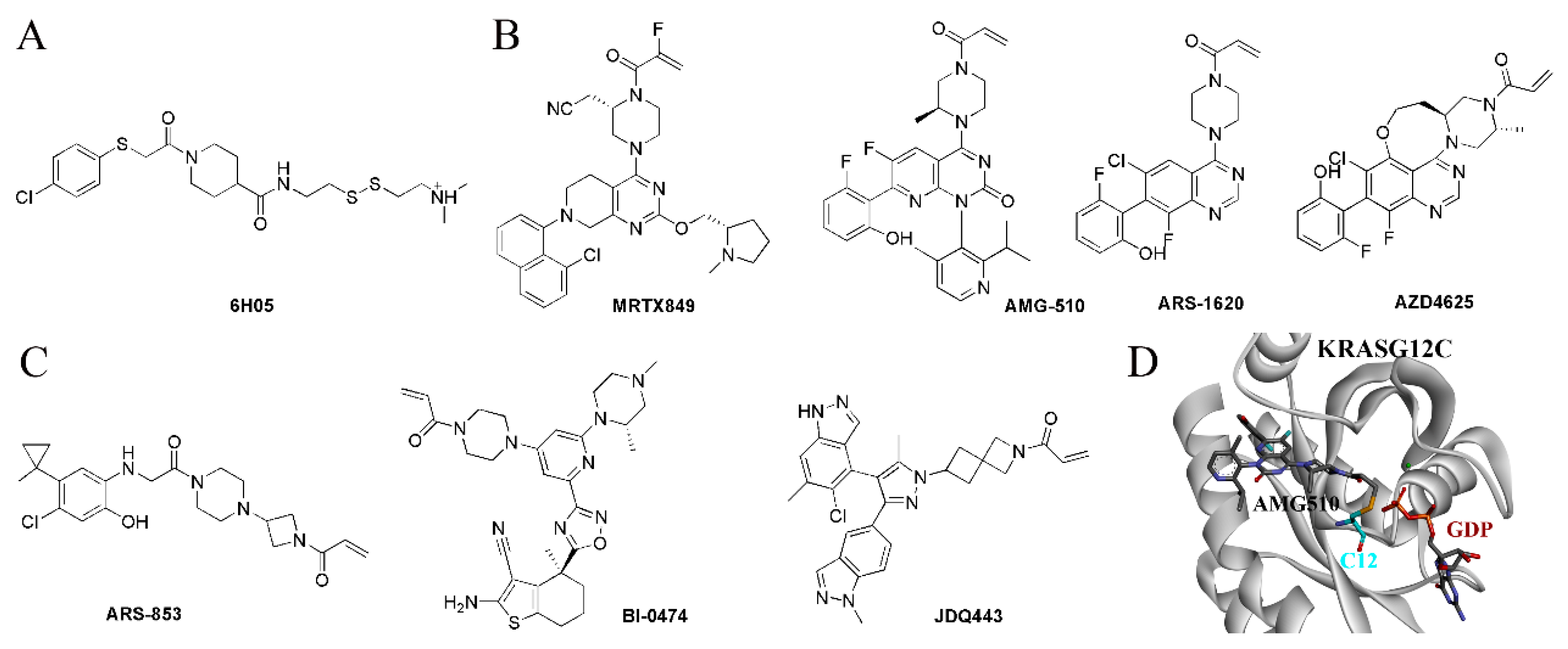
5.1. Covalent Binding Strategy
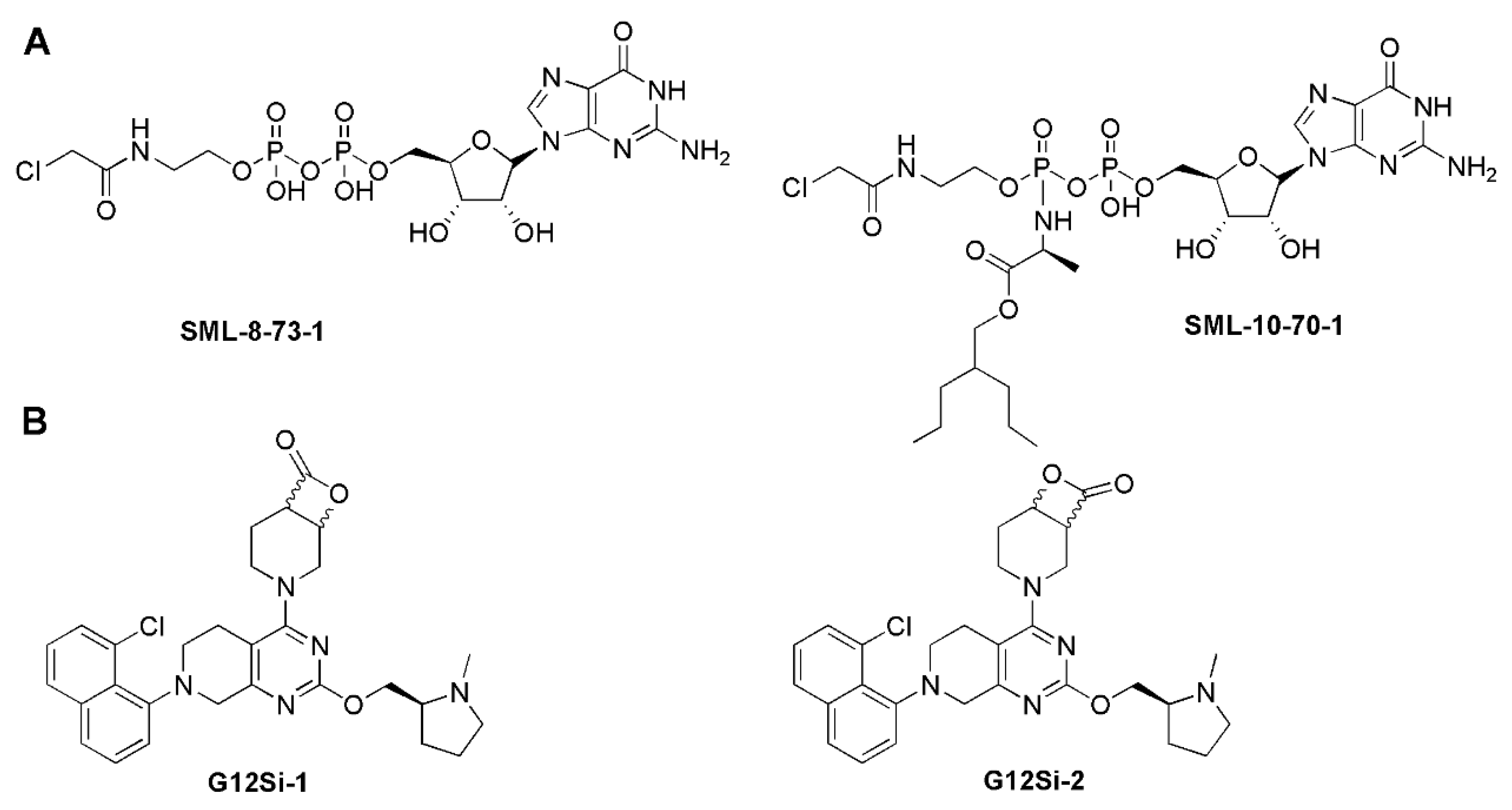
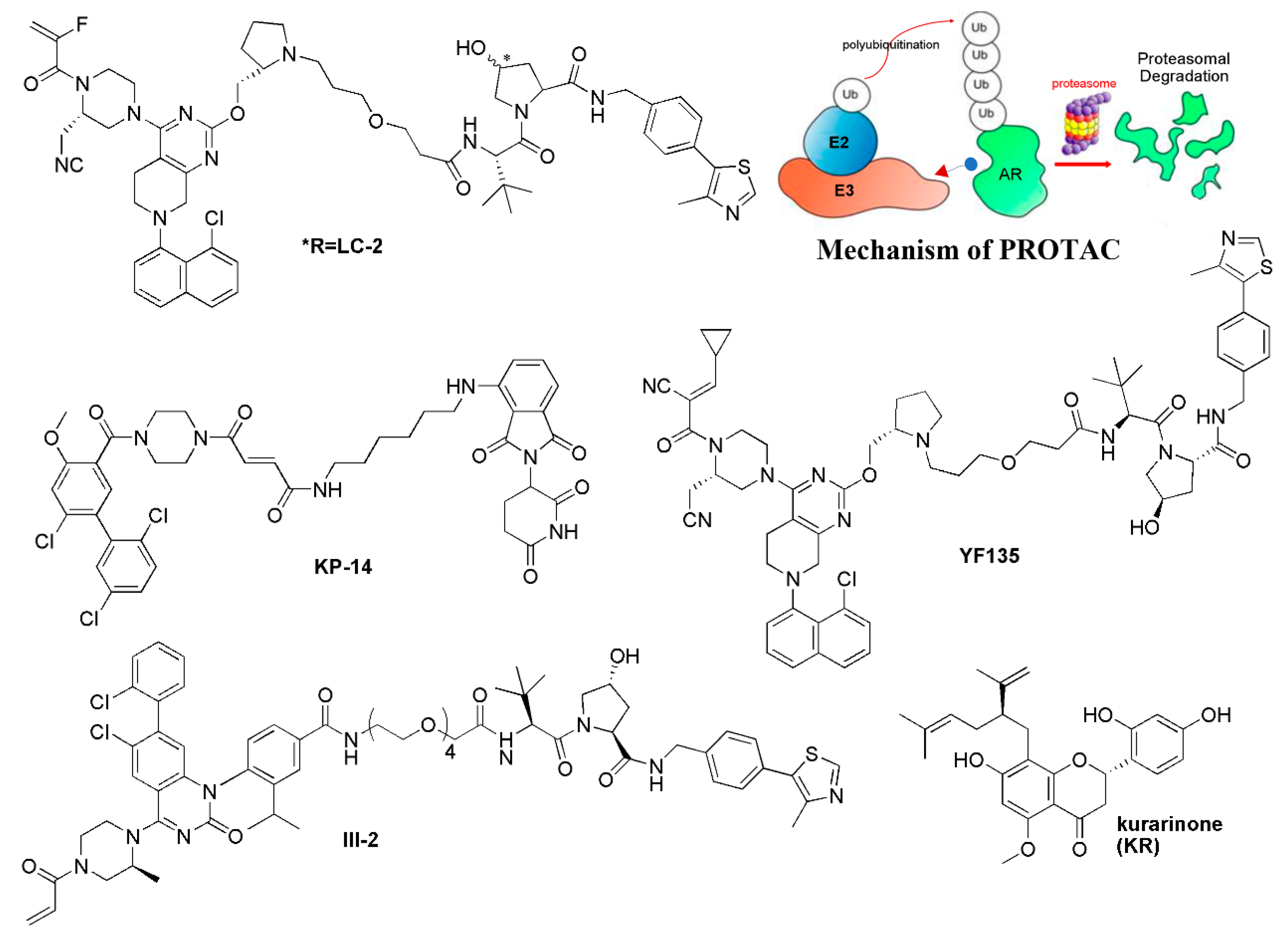
5.2. Targeted Protein Degradation Strategy
5.3. Targeting the Dimerization of Oncogenic KRAS Strategy
5.4. Blocking KRAS-G12D with B-Raf Interaction
5.5. Salt Bridge Strategy
5.6. Multivalent Strategy
6. Discussion and Perspective
Funding
Conflicts of Interest
References
- Prior, I.A.; Lewis, P.D.; Mattos, C. A Comprehensive Survey of Ras Mutations in Cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [PubMed]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Ostrem, J.M.L.; Shokat, K.M. Direct small-molecule inhibitors of KRAS: From structural insights to mechanism-based design. Nat. Rev. Drug Discov. 2016, 15, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef]
- Gimple, R.C.; Wang, X. RAS: Striking at the Core of the Oncogenic Circuitry. Front. Oncol. 2019, 9, 965. [Google Scholar] [CrossRef]
- Hamarsheh, S.; Groß, O.; Brummer, T.; Zeiser, R. Immune modulatory effects of oncogenic KRAS in cancer. Nat. Commun. 2020, 11, 5439. [Google Scholar] [CrossRef]
- Pereira, F.; Ferreira, A.; Reis, C.A.; Sousa, M.J.; Oliveira, M.J.; Preto, A. KRAS as a Modulator of the Inflammatory Tumor Microenvironment: Therapeutic Implications. Cells 2022, 11, 398. [Google Scholar] [CrossRef]
- Asimgil, H.; Ertetik, U.; Çevik, N.C.; Ekizce, M.; Doğruöz, A.; Gökalp, M.; Arık-Sever, E.; Istvanffy, R.; Friess, H.; Ceyhan, G.O.; et al. Targeting the undruggable oncogenic KRAS: The dawn of hope. J. Clin. Investig. 2022, 7, e153688. [Google Scholar] [CrossRef]
- Parikh, K.; Banna, G.; Liu, S.V.; Friedlaender, A.; Desai, A.; Subbiah, V.; Addeo, A. Drugging KRAS: Current perspectives and state-of-art review. J. Hematol. Oncol. 2022, 15, 152. [Google Scholar] [CrossRef]
- Wang, H.; Chi, L.; Yu, F.; Dai, H.; Gao, C.; Si, X.; Wang, Z.; Liu, L.; Zheng, J.; Shan, L.; et al. Annual review of KRAS inhibitors in 2022. Eur. J. Med. Chem. 2023, 249, 115124. [Google Scholar] [CrossRef]
- Wang, X.; Allen, S.; Blake, J.F.; Bowcut, V.; Briere, D.M.; Calinisan, A.; Dahlke, J.R.; Fell, J.B.; Fischer, J.P.; Gunn, R.J.; et al. Identification of MRTX1133, a Noncovalent, Potent, and Selective KRAS(G12D) Inhibitor. J. Med. Chem. 2022, 65, 3123–3133. [Google Scholar] [CrossRef] [PubMed]
- Punekar, S.R.; Velcheti, V.; Neel, B.G.; Wong, K.-K. The current state of the art and future trends in RAS-targeted cancer therapies. Nat. Rev. Clin. Oncol. 2022, 19, 637–655. [Google Scholar] [CrossRef] [PubMed]
- Bery, N.; Miller, A.; Rabbitts, T. A potent KRAS macromolecule degrader specifically targeting tumours with mutant KRAS. Nat. Commun. 2020, 11, 3233. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Park, S.; Gunassekaran, G.R.; Jeon, M.; Cho, Y.-E.; Baek, M.-C.; Park, J.Y.; Shim, G.; Oh, Y.-K.; Kim, I.-S.; et al. A peptide probe enables photoacoustic-guided imaging and drug delivery to lung tumors in K-rasLA2 mutant mice. Cancer Res. 2019, 79, 4271–4282. [Google Scholar] [CrossRef]
- Mullard, A. The KRAS crowd targets its next cancer mutations. Nat. Rev. Drug Discov. 2023, 22, 167–171. [Google Scholar] [CrossRef]
- Moreau-Gachelin, F.; Camonis, J.; de Gunzburg, J.; Goud, B. Armand Tavitian (1931–2020): From oncogenes to the Ras superfamily. Med. Sci. 2020, 36, 810–812. [Google Scholar] [CrossRef]
- Jančík, S.; Drábek, J.; Radzioch, D.; Hajdúch, M. Clinical Relevance of KRAS in Human Cancers. J. Biomed. Biotechnol. 2010, 2010, 150960. [Google Scholar] [CrossRef]
- Bourne, H.R.; Sanders, D.A.; McCormick, F. The GTPase superfamily: Conserved structure and molecular mechanism. Nature 1991, 349, 117–127. [Google Scholar] [CrossRef]
- Rojas, J.M.; Oliva, J.L.; Santos, E. Mammalian Son of Sevenless Guanine Nucleotide Exchange Factors: Old Concepts and New Perspectives. Genes Cancer 2011, 2, 298–305. [Google Scholar] [CrossRef]
- Bowtell, D.; Fu, P.; Simon, M.; Senior, P. Identification of murine homologues of the Drosophila son of sevenless gene: Potential activators of ras. Proc. Natl. Acad. Sci. USA 1992, 89, 6511–6515. [Google Scholar] [CrossRef]
- Boriack-Sjodin, P.A.; Margarit, S.M.; Bar-Sagi, D.; Kuriyan, J. The structural basis of the activation of Ras by Sos. Nature 1998, 394, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Burns, M.C.; Sun, Q.; Daniels, R.N.; Camper, D.; Kennedy, J.P.; Phan, J.; Olejniczak, E.T.; Lee, T.; Waterson, A.G.; Rossanese, O.W.; et al. Approach for targeting Ras with small molecules that activate SOS-mediated nucleotide exchange. Proc. Natl. Acad. Sci. USA 2014, 111, 3401–3406. [Google Scholar] [CrossRef] [PubMed]
- Eblen, S.T. Extracellular-Regulated Kinases: Signaling from Ras to ERK Substrates to Control Biological Outcomes. In Advances in Cancer Research; Elsevier: New York, NY, USA, 2018; Volume 138, pp. 99–142. [Google Scholar] [CrossRef]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef]
- Hunter, J.C.; Manandhar, A.; Carrasco, M.A.; Gurbani, D.; Gondi, S.; Westover, K.D. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol. Cancer Res. 2015, 13, 1325–1335. [Google Scholar] [CrossRef]
- Ihle, N.T.; Byers, L.A.; Kim, E.S.; Saintigny, P.; Lee, J.J.; Blumenschein, G.R.; Tsao, A.; Liu, S.; Larsen, J.E.; Wang, J.; et al. Effect of KRAS Oncogene Substitutions on Protein Behavior: Implications for Signaling and Clinical Outcome. JNCI J. Natl. Cancer Inst. 2012, 104, 228–239. [Google Scholar] [CrossRef]
- Chen, H.; Smaill, J.B.; Liu, T.; Ding, K.; Lu, X. Small-Molecule Inhibitors Directly Targeting KRAS as Anticancer Therapeutics. J. Med. Chem. 2020, 63, 14404–14424. [Google Scholar] [CrossRef]
- Hall, B.E.; Bar-Sagi, D.; Nassar, N. The structural basis for the transition from Ras-GTP to Ras-GDP. Proc. Natl. Acad. Sci. USA 2002, 99, 12138–12142. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Jang, H.; Muratcioglu, S.; Gursoy, A.; Keskin, O.; Nussinov, R.; Zhang, J. Ras Conformational Ensembles, Allostery, and Signaling. Chem. Rev. 2016, 116, 6607–6665. [Google Scholar] [CrossRef]
- Kato, K.; Cox, A.D.; Hisaka, M.M.; Graham, S.M.; Buss, J.E.; Der, C.J. Isoprenoid addition to Ras protein is the critical modification for its membrane association and transforming activity. Proc. Natl. Acad. Sci. USA 1992, 89, 6403–6407. [Google Scholar] [CrossRef]
- Nan, X.; Tamgüney, T.M.; Collisson, E.A.; Lin, L.-J.; Pitt, C.; Galeas, J.; Lewis, S.; Gray, J.W.; McCormick, F.; Chu, S. Ras-GTP dimers activate the Mitogen-Activated Protein Kinase (MAPK) pathway. Proc. Natl. Acad. Sci. USA 2015, 112, 7996–8001. [Google Scholar] [CrossRef]
- Greenhough, A.; Patsos, H.A.; Williams, A.C.; Paraskeva, C. The cannabinoid delta(9)-tetrahydrocannabinol inhibits RAS-MAPK and PI3K-AKT survival signalling and induces BAD-mediated apoptosis in colorectal cancer cells. Int. J. Cancer. 2007, 121, 2172–2180. [Google Scholar] [CrossRef] [PubMed]
- Downward, J. Ras signalling and apoptosis. Curr. Opin. Genet. Dev. 1998, 8, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Awaji, M.; Saxena, S.; Wu, L.; Prajapati, D.R.; Purohit, A.; Varney, M.L.; Kumar, S.; Rachagani, S.; Ly, Q.P.; Jain, M.; et al. CXCR2 signaling promotes secretory cancer-associated fibroblasts in pancreatic ductal adenocarcinoma. FASEB J. 2020, 34, 9405–9418. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Fang, W.; Lin, Z.; Peng, P.; Wang, J.; Zhan, J.; Hong, S.; Huang, J.; Liu, L.; Sheng, J.; et al. KRAS mutation-induced upregulation of PD-L1 mediates immune escape in human lung adenocarcinoma. Cancer Immunol. Immunother. 2017, 66, 1175–1187. [Google Scholar] [CrossRef] [PubMed]
- Lastwika, K.J.; Wilson, W., 3rd; Li, Q.K.; Norris, J.; Xu, H.; Ghazarian, S.R.; Kitagawa, H.; Kawabata, S.; Taube, J.M.; Yao, S.; et al. Control of PD-L1 Expression by Oncogenic Activation of the AKT-mTOR Pathway in Non-Small Cell Lung Cancer. Cancer Res. 2016, 76, 227–238. [Google Scholar] [CrossRef]
- Liu, C.; Zheng, S.; Jin, R.; Wang, X.; Wang, F.; Zang, R.; Xu, H.; Lu, Z.; Huang, J.; Lei, Y.; et al. The superior efficacy of anti-PD-1/PD-L1 immunotherapy in KRAS-mutant non-small cell lung cancer that correlates with an inflammatory phenotype and increased immunogenicity. Cancer Lett. 2020, 470, 95–105. [Google Scholar] [CrossRef]
- Zhu, C.; Guan, X.; Zhang, X.; Luan, X.; Song, Z.; Cheng, X.; Zhang, W.; Qin, J.-J. Targeting KRAS mutant cancers: From druggable therapy to drug resistance. Mol. Cancer 2022, 21, 159. [Google Scholar] [CrossRef]
- Hofmann, M.H.; Gerlach, D.; Misale, S.; Petronczki, M.; Kraut, N. Expanding the Reach of Precision Oncology by Drugging All KRAS Mutants. Cancer Discov. 2022, 12, 924–937. [Google Scholar] [CrossRef]
- Vatansever, S.; Erman, B.; Gümüş, Z.H. Oncogenic G12D mutation alters local conformations and dynamics of K-Ras. Sci. Rep. 2019, 9, 11730. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, S.; Wang, W.; Pang, L.; Zhang, Q.; Liu, X. Mutation-Induced Impacts on the Switch Transformations of the GDP- and GTP-Bound K-Ras: Insights from Multiple Replica Gaussian Accelerated Molecular Dynamics and Free Energy Analysis. J. Chem. Inf. Model. 2021, 61, 1954–1969. [Google Scholar] [CrossRef]
- Judd, J.; Karim, N.A.; Khan, H.; Naqash, A.R.; Baca, Y.; Xiu, J.; VanderWalde, A.M.; Mamdani, H.; Raez, L.E.; Nagasaka, M.; et al. Characterization of KRAS Mutation Subtypes in Non–small Cell Lung Cancer. Mol. Cancer Ther. 2021, 20, 2577–2584. [Google Scholar] [CrossRef]
- Zhang, M.; Hu, W.; Hu, K.; Lin, Y.; Feng, Z.; Yun, J.-P.; Gao, N.; Zhang, L. Association of KRAS mutation with tumor deposit status and overall survival of colorectal cancer. Cancer Causes Control 2020, 31, 683–689. [Google Scholar] [CrossRef] [PubMed]
- Fisher, G.H.; Wellen, S.L.; Klimstra, D.; Lenczowski, J.M.; Tichelaar, J.W.; Lizak, M.J.; Whitsett, J.A.; Koretsky, A.; Varmus, H.E. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev. 2001, 15, 3249–3262. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Wang, Y.; Li, X. Targeting the untargetable KRAS in cancer therapy. Acta Pharm. Sin. B 2019, 9, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, J.; Cromm, P.M.; Zimmermann, G.; Grossmann, T.N.; Waldmann, H. Small-molecule modulation of Ras signaling. Nat. Chem. Biol. 2014, 10, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Marcus, K.; Mattos, C. Direct Attack on RAS: Intramolecular Communication and Mutation-Specific Effects. Clin. Cancer Res. 2015, 21, 1810–1818. [Google Scholar] [CrossRef]
- Bradshaw, J.M.; McFarland, J.M.; Paavilainen, V.O.; Bisconte, A.; Tam, D.; Phan, V.T.; Romanov, S.; Finkle, D.; Shu, J.; Patel, V.; et al. Prolonged and tunable residence time using reversible covalent kinase inhibitors. Nat. Chem. Biol. 2015, 11, 525–531. [Google Scholar] [CrossRef]
- Huang, F.; Han, X.; Xiao, X.; Zhou, J. Covalent Warheads Targeting Cysteine Residue: The Promising Approach in Drug Development. Molecules 2022, 27, 7728. [Google Scholar] [CrossRef]
- Ni, D.; Li, X.; He, X.; Zhang, H.; Zhang, J.; Lu, S. Drugging K-Ras(G12C) through covalent inhibitors: Mission possible? Pharmacol. Ther. 2019, 202, 1–17. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- O’leary, K. KRAS inhibitors gather momentum. Nat. Med. 2023. [Google Scholar] [CrossRef]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef] [PubMed]
- Lanman, B.A.; Parsons, A.T.; Zech, S.G. Addressing Atropisomerism in the Development of Sotorasib, a Covalent Inhibitor of KRAS G12C: Structural, Analytical, and Synthetic Considerations. Acc. Chem. Res. 2022, 55, 2892–2903. [Google Scholar] [CrossRef] [PubMed]
- Kettle, J.G.; Bagal, S.K.; Bickerton, S.; Bodnarchuk, M.S.; Boyd, S.; Breed, J.; Carbajo, R.J.; Cassar, D.J.; Chakraborty, A.; Cosulich, S.; et al. Discovery of AZD4625, a Covalent Allosteric Inhibitor of the Mutant GTPase KRAS(G12C). J. Med. Chem. 2022, 65, 6940–6952. [Google Scholar] [CrossRef] [PubMed]
- Janes, M.R.; Zhang, J.; Li, L.-S.; Hansen, R.; Peters, U.; Guo, X.; Chen, Y.; Babbar, A.; Firdaus, S.J.; Darjania, L.; et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018, 172, 578–589.e17. [Google Scholar] [CrossRef]
- Fell, J.B.; Fischer, J.P.; Baer, B.R.; Blake, J.F.; Bouhana, K.; Briere, D.M.; Brown, K.D.; Burgess, L.E.; Burns, A.C.; Burkard, M.R.; et al. Identification of the Clinical Development Candidate MRTX849, a Covalent KRASG12C Inhibitor for the Treatment of Cancer. J. Med. Chem. 2020, 63, 6679–6693. [Google Scholar] [CrossRef]
- Bröker, J.; Waterson, A.G.; Smethurst, C.; Kessler, D.; Böttcher, J.; Mayer, M.; Gmaschitz, G.; Phan, J.; Little, A.; Abbott, J.R.; et al. Fragment Optimization of Reversible Binding to the Switch II Pocket on KRAS Leads to a Potent, In Vivo Active KRASG12C Inhibitor. J. Med. Chem. 2022, 65, 14614–14629. [Google Scholar] [CrossRef]
- Lorthiois, E.; Gerspacher, M.; Beyer, K.S.; Vaupel, A.; Leblanc, C.; Stringer, R.; Weiss, A.; Wilcken, R.; Guthy, D.A.; Lingel, A.; et al. JDQ443, a Structurally Novel, Pyrazole-Based, Covalent Inhibitor of KRAS(G12C) for the Treatment of Solid Tumors. J. Med. Chem. 2022, 65, 16173–16203. [Google Scholar] [CrossRef]
- Patricelli, M.P.; Janes, M.R.; Li, L.-S.; Hansen, R.; Peters, U.; Kessler, L.V.; Chen, Y.; Kucharski, J.M.; Feng, J.; Ely, T.; et al. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer Discov. 2016, 6, 316–329. [Google Scholar] [CrossRef]
- Lim, S.M.; Westover, K.D.; Ficarro, S.B.; Harrison, R.A.; Choi, H.G.; Pacold, M.E.; Carrasco, M.; Hunter, J.; Kim, N.D.; Xie, T.; et al. Therapeutic targeting of oncogenic K-Ras by a covalent catalytic site inhibitor. Angew. Chem. Int. Ed. Engl. 2014, 53, 199–204. [Google Scholar] [CrossRef]
- Zhang, Z.; Guiley, K.Z.; Shokat, K.M. Chemical acylation of an acquired serine suppresses oncogenic signaling of K-Ras(G12S). Nat. Chem. Biol. 2022, 18, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Rohweder, P.J.; Ongpipattanakul, C.; Basu, K.; Bohn, M.-F.; Dugan, E.J.; Steri, V.; Hann, B.; Shokat, K.M.; Craik, C.S. A covalent inhibitor of K-Ras(G12C) induces MHC class I presentation of haptenated peptide neoepitopes targetable by immunotherapy. Cancer Cell 2022, 40, 1060–1069.e7. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Xin, M.; Zhang, S. Progress of small molecules for targeted protein degradation: PROTACs and other technologies. Drug Dev. Res. 2023, 84, 337–394. [Google Scholar] [CrossRef] [PubMed]
- Bond, M.J.; Chu, L.; Nalawansha, D.A.; Li, K.; Crews, C.M. Targeted Degradation of Oncogenic KRAS(G12C) by VHL-Recruiting PROTACs. ACS Cent. Sci. 2020, 6, 1367–1375. [Google Scholar] [CrossRef]
- Kargbo, R.B. PROTAC-Mediated Degradation of KRAS Protein for Anticancer Therapeutics. ACS Med. Chem. Lett. 2019, 11, 5–6. [Google Scholar] [CrossRef]
- Li, L.; Wu, Y.; Yang, Z.; Xu, C.; Zhao, H.; Liu, J.; Chen, J.; Chen, J. Discovery of KRas G12C-IN-3 and Pomalidomide-based PROTACs as degraders of endogenous KRAS G12C with potent anticancer activity. Bioorg. Chem. 2021, 117, 105447. [Google Scholar] [CrossRef]
- Yang, F.; Wen, Y.; Wang, C.; Zhou, Y.; Zhou, Y.; Zhang, Z.M.; Liu, T.; Lu, X. Efficient targeted oncogenic KRAS(G12C) degradation via first reversible-covalent PROTAC. Eur. J. Med. Chem. 2022, 230, 114088. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, T.; Sun, M.; Li, P.; Lai, M.; Xie, L.; Chen, J.; Ding, J.; Xie, H.; Zhou, J.; et al. Design, synthesis and biological evaluation of KRAS(G12C)-PROTACs. Bioorg. Med. Chem. 2023, 78, 117153. [Google Scholar] [CrossRef]
- Kwon, M.; Oh, T.; Jang, M.; Kim, G.H.; Kim, J.H.; Ryu, H.W.; Oh, S.R.; Jang, J.H.; Ahn, J.S.; Ko, S.K. Kurarinone induced p53-independent G0/G1 cell cycle arrest by degradation of K-RAS via WDR76 in human colorectal cancer cells. Eur. J. Pharmacol. 2022, 923, 174938. [Google Scholar] [CrossRef]
- Choi, J.K.; Cho, H.; Moon, B.S. Small Molecule Destabilizer of beta-Catenin and Ras Proteins Antagonizes Growth of K-Ras Mutation-Driven Colorectal Cancers Resistant to EGFR Inhibitors. Target Oncol. 2020, 15, 645–657. [Google Scholar] [CrossRef]
- Lin, Y.-J.; Haigis, K.M. Brother’s Keeper: Wild-Type Mutant K-Ras Dimers Limit Oncogenesis. Cell 2018, 172, 645–647. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Enomoto, M.; Gebregiworgis, T.; Gasmi-Seabrook, G.M.C.; Ikura, M.; Marshall, C.B. Oncogenic KRAS G12D mutation promotes dimerization through a second, phosphatidylserine-dependent interface: A model for KRAS oligomerization. Chem. Sci. 2021, 12, 12827–12837. [Google Scholar] [CrossRef] [PubMed]
- Khan, I.; Spencer-Smith, R.; O’bryan, J.P. Targeting the α4–α5 dimerization interface of K-RAS inhibits tumor formation in vivo. Oncogene 2018, 38, 2984–2993. [Google Scholar] [CrossRef]
- Kessler, D.; Gmachl, M.; Mantoulidis, A.; Martin, L.J.; Zoephel, A.; Mayer, M.; Gollner, A.; Covini, D.; Fischer, S.; Gerstberger, T.; et al. Drugging an undruggable pocket on KRAS. Proc. Natl. Acad. Sci. USA 2019, 116, 15823–15829. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Zhang, Y.; Bos, P.H.; Chambers, J.M.; Dupont, M.M.; Stockwell, B.R. K-RasG12D Has a Potential Allosteric Small Molecule Binding Site. Biochemistry 2019, 58, 2542–2554. [Google Scholar] [CrossRef]
- Zhang, Z.; Shokat, K.M. Bifunctional Small-Molecule Ligands of K-Ras Induce Its Association with Immunophilin Proteins. Angew. Chem. Int. Ed. Engl. 2019, 58, 16314–16319. [Google Scholar] [CrossRef]
- Mao, Z.; Xiao, H.; Shen, P.; Yang, Y.; Xue, J.; Yang, Y.; Shang, Y.; Zhang, L.; Li, X.; Zhang, Y.; et al. KRAS(G12D) can be targeted by potent inhibitors via formation of salt bridge. Cell Discov. 2022, 8, 5. [Google Scholar] [CrossRef]
- Kargbo, R.B. Targeting KRAS G12D Mutant for the Potential Treatment of Pancreatic Cancer. ACS Med. Chem. Lett. 2021, 12, 1643–1645. [Google Scholar] [CrossRef]
- Hallin, J.; Bowcut, V.; Calinisan, A.; Briere, D.M.; Hargis, L.; Engstrom, L.D.; Laguer, J.; Medwid, J.; Vanderpool, D.; Lifset, E.; et al. Anti-tumor efficacy of a potent and selective non-covalent KRAS(G12D) inhibitor. Nat. Med. 2022, 28, 2171–2182. [Google Scholar] [CrossRef]
- Kim, Y.; Hyun, J.Y.; Shin, I. Multivalent glycans for biological and biomedical applications. Chem. Soc. Rev. 2021, 50, 10567–10593. [Google Scholar] [CrossRef]
- Welsch, M.E.; Kaplan, A.; Chambers, J.M.; Stokes, M.E.; Bos, P.H.; Zask, A.; Zhang, Y.; Sanchez-Martin, M.; Badgley, M.A.; Huang, C.S.; et al. Multivalent Small-Molecule Pan-RAS Inhibitors. Cell 2017, 168, 878–889.e29. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Zhang, J.; Gao, Y.; Li, Y.; Li, Y. Strategies for targeting undruggable targets. Expert Opin. Drug Discov. 2021, 17, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kang, R.; Tang, D. The KRAS-G12C inhibitor: Activity and resistance. Cancer Gene Ther. 2021, 29, 875–878. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Li, B.T.; Dy, G.K.; Price, T.J.; Falchook, G.S.; Wolf, J.; Italiano, A.; Schuler, M.; Borghaei, H.; Barlesi, F.; et al. Sotorasib for Lung Cancers with KRAS p.G12C Mutation. N. Engl. J. Med. 2021, 384, 2371–2381. [Google Scholar] [CrossRef]
- Skoulidis, F.; Goldberg, M.E.; Greenawalt, D.M.; Hellmann, M.D.; Awad, M.M.; Gainor, J.F.; Schrock, A.B.; Hartmaier, R.J.; Trabucco, S.E.; Gay, L.; et al. STK11/LKB1 Mutations and PD-1 Inhibitor Resistance in KRAS-Mutant Lung Adenocarcinoma. Cancer Discov. 2018, 8, 822–835. [Google Scholar] [CrossRef]
- Tanaka, N.; Lin, J.J.; Li, C.; Ryan, M.B.; Zhang, J.; Kiedrowski, L.A.; Michel, A.G.; Syed, M.U.; Fella, K.A.; Sakhi, M.; et al. Clinical Acquired Resistance to KRAS(G12C) Inhibition through a Novel KRAS Switch-II Pocket Mutation and Polyclonal Alterations Converging on RAS-MAPK Reactivation. Cancer Discov. 2021, 11, 1913–1922. [Google Scholar] [CrossRef]
- Feng, S.; Callow, M.G.; Fortin, J.-P.; Khan, Z.; Bray, D.; Costa, M.; Shi, Z.; Wang, W.; Evangelista, M. A saturation mutagenesis screen uncovers resistant and sensitizing secondary KRAS mutations to clinical KRAS G12C inhibitors. Proc. Natl. Acad. Sci. USA 2022, 119, e2120512119. [Google Scholar] [CrossRef]
- Zhou, Y.; Zou, Y.; Yang, M.; Mei, S.; Liu, X.; Han, H.; Zhang, C.-D.; Niu, M.-M. Highly Potent, Selective, Biostable, and Cell-Permeable Cyclic d-Peptide for Dual-Targeting Therapy of Lung Cancer. J. Am. Chem. Soc. 2022, 144, 7117–7128. [Google Scholar] [CrossRef]
- Zhang, Z.; Gao, R.; Hu, Q.; Peacock, H.; Peacock, D.M.; Dai, S.; Shokat, K.M.; Suga, H. GTP-State-Selective Cyclic Peptide Ligands of K-Ras(G12D) Block Its Interaction with Raf. ACS Central Sci. 2020, 6, 1753–1761. [Google Scholar] [CrossRef]
- Li, F.-Y.; Zhang, Z.-F.; Voss, S.; Wu, Y.-W.; Zhao, Y.-F.; Li, Y.-M.; Chen, Y.-X. Inhibition of K-Ras4B-plasma membrane association with a membrane microdomain-targeting peptide. Chem. Sci. 2019, 11, 826–832. [Google Scholar] [CrossRef]
- Lim, S.; Khoo, R.; Juang, Y.-C.; Gopal, P.; Zhang, H.; Yeo, C.; Peh, K.M.; Teo, J.; Ng, S.; Henry, B.; et al. Exquisitely Specific anti-KRAS Biodegraders Inform on the Cellular Prevalence of Nucleotide-Loaded States. ACS Cent. Sci. 2020, 7, 274–291. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, F.M. Harnessing Antibody-Mimetic Selectivity for Activation-State-Specific Targeted Degradation of Endogenous K-Ras. ACS Cent. Sci. 2021, 7, 222–224. [Google Scholar] [CrossRef] [PubMed]
- Röth, S.; Macartney, T.J.; Konopacka, A.; Chan, K.-H.; Zhou, H.; Queisser, M.A.; Sapkota, G.P. Targeting Endogenous K-RAS for Degradation through the Affinity-Directed Protein Missile System. Cell Chem. Biol. 2020, 27, 1151–1163.e6. [Google Scholar] [CrossRef] [PubMed]
- Péczka, N.; Orgován, Z.; Ábrányi-Balogh, P.; Keserű, G.M. Electrophilic warheads in covalent drug discovery: An overview. Expert Opin. Drug Discov. 2022, 17, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Nam, G.-H.; Kwon, M.; Jung, H.; Ko, E.; Kim, S.A.; Choi, Y.; Song, S.J.; Kim, S.; Lee, Y.; Kim, G.B.; et al. Statin-mediated inhibition of RAS prenylation activates ER stress to enhance the immunogenicity of KRAS mutant cancer. J. Immunother. Cancer 2021, 9, e002474. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, X.; Ji, Y.; Zhou, J. Multiple Strategies to Develop Small Molecular KRAS Directly Bound Inhibitors. Molecules 2023, 28, 3615. https://doi.org/10.3390/molecules28083615
Zhou X, Ji Y, Zhou J. Multiple Strategies to Develop Small Molecular KRAS Directly Bound Inhibitors. Molecules. 2023; 28(8):3615. https://doi.org/10.3390/molecules28083615
Chicago/Turabian StyleZhou, Xile, Yang Ji, and Jinming Zhou. 2023. "Multiple Strategies to Develop Small Molecular KRAS Directly Bound Inhibitors" Molecules 28, no. 8: 3615. https://doi.org/10.3390/molecules28083615
APA StyleZhou, X., Ji, Y., & Zhou, J. (2023). Multiple Strategies to Develop Small Molecular KRAS Directly Bound Inhibitors. Molecules, 28(8), 3615. https://doi.org/10.3390/molecules28083615