Abstract
A rhodium(II)-catalyzed reaction of cyclic nitronates (5,6-dihydro-4H-1,2-oxazine N-oxides) with vinyl diazoacetates proceeds as a [3+3]-annulation producing bicyclic unsaturated nitroso acetals (4a,5,6,7-tetrahydro-2H-[1,2]oxazino[2,3-b][1,2]oxazines). Optimization of reaction conditions revealed the use of Rh(II) octanoate as the preferred catalyst in THF at room temperature, which allows the preparation of target products in good yields and excellent diastereoselectivity. Under basic conditions, namely, the combined action of DBU and alcohol, these nitroso acetals undergo ring contraction of an unsaturated oxazine ring into the corresponding pyrrole. Both transformations can be performed in a one-pot fashion, thus constituting a quick approach to oxazine-annulated pyrroles from available starting materials, such as nitroalkenes, olefins, and diazo compounds.
1. Introduction
1,2-Oxazine derivatives represent an important class of nitrogen–oxygen heterocycles which are widely used in organic chemistry [1,2,3,4,5,6,7,8]. Reductive cleavage of the N–O bond makes 1,2-oxazine derivatives useful precursors to aminoalcohols and functionalized carbonyl compounds. Moreover, 1,2-oxazines are versatile substrates for the preparation of other important heterocyclic motifs, such as pyrrolidine, pyrrole, or furan. The recent growth of the research interest in the chemistry of 1,2-oxazines is promoted by the discovery of bioactive natural products possessing 1,2-oxazine core (Scheme 1, (1)) [9,10,11,12,13,14,15,16,17,18]. Such compounds as trichodermamides or phyllantidine became attractive targets for both total synthesis and preparation of analogues in pursuit of increased bioactivity [19,20,21,22,23,24,25,26].
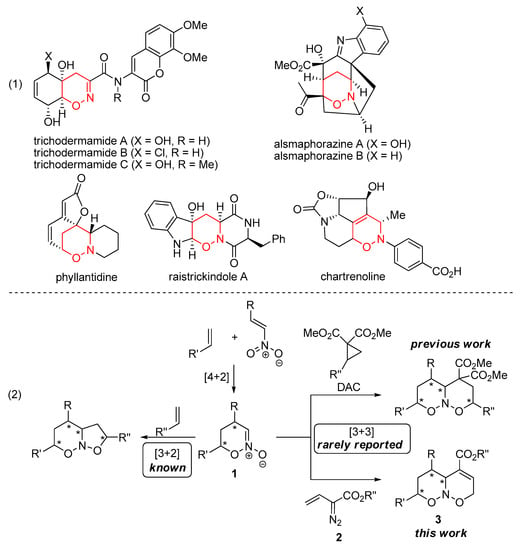
Scheme 1.
Representative bioactive 1,2-oxazine derivatives (1) and synthesis of 1,2-oxazine derivatives via cycloaddition of cyclic nitronates 1 (2). *—chiral center.
One of the synthetic routes to 1,2-oxazines is based on various transformations of six-membered cyclic nitronates, 5,6-dihydro-4H-1,2-oxazine N-oxides 1 (Scheme 1, (2)). These substrates are readily accessible via the [4+2]-cycloaddition between such simple starting materials as conjugated nitroalkenes and olefins [27,28]. It allows a quick and stereoselective construction of an oxazine ring, while further transformations of the N-oxide moiety are used for the desired modifications of the assembled heterocyclic core. One of the most known and most used reactivity patterns of nitronates is the [3+2]-cycloaddition [28,29,30,31]. It leads to the assembly of annulated 6,5-bicyclic nitroso acetals, possessing six- and five-membered rings. For instance, this strategy was extensively exploited by Denmark and coworkers in the total synthesis of pyrrolizidine and indolizidine alkaloids [32,33,34,35,36,37,38,39]. In contrast to [3+2]-cycloaddition, other cycloaddition/annulation reactions of nitronates remain underexplored. Recently, we reported a formal [3+3]-cycloaddition reaction of cyclic nitronates 1 with donor–acceptor cyclopropanes (DACs) [40,41]. DACs reacted as three-carbon components and allowed a smooth diastereoselective preparation of nitroso acetals, possessing two saturated oxazine rings. To expand the scope of polycyclic frameworks, which can be available from nitronates 1, we pursued other three-carbon annulation partners. In continuation of these studies, we turned our attention to the vinyl diazoacetates 2, which recommended themselves as useful 1,3-dipole equivalents in annulations leading to various carbocycles (cyclobutenes, cyclopentenes, etc.) and heterocycles (pyrazoles, pyrroles, isoxazolines, quinolines, etc.) [42,43]. Herein, we report the [3+3]-annulation of nitronates 1 with vinyl diazoacetates 2 for the synthesis of 6,6-bicyclic nitroso acetals 3 and their further transformation into pyrrole-annulated oxazine derivatives.
2. Results and Discussion
We initiated our study with optimization of the reaction between model nitronate 1a and vinyl diazoacetate 2a (Table 1). Variations of catalyst, solvent, temperature, and reagent ratio were performed. Notably, only one diastereomer of adduct 3a was detected in all experiments. Among the solvents tested, the best results were achieved in THF. In other ethereal solvents (diethyl ether, glyme, 1,4-dioxane), a decrease in yield was observed (Entries 2–4), while in acetonitrile, dichloromethane, and hexane, starting nitronate 1a was not fully consumed (Entries 6–8). As compared to rhodium(II) octanoate, rhodium(II) catalysts (acetate, trifluoroacetate, tetramethyl-1,3-benzenedipropionate) gave worse results (Entries 10–12), while other metal catalysts that are often used in carbenoid reactions [42,44,45,46,47,48,49,50,51,52] did not produce the target product at all (Entries 13–17). Variations of reagent ratios (Entries 18–20), concentration (Entry 21), and reaction temperatures (Entries 22–23) did not lead to improvement in yield. Therefore, the use of 2 equiv. of vinyl diazoacetate and 2 mol.% of rhodium(II) octanoate in THF at r.t. (Entry 1) was chosen for subsequent studies.

Table 1.
Optimization of reaction conditions 1.
With the optimized conditions in hand, an evaluation of the substrate scope was performed (Scheme 2). Diazoacetates possessing various ester groups were successfully involved in the reaction. Alkyl (Me, 3b; Et, 3c), benzyl (3d), p-nitrobenzyl (3e), and phenyl (3f) esters gave corresponding products in good yields. However, better results were achieved for electron-accepting 2,2,2-trichloroethyl (3a) and p-nitrobenzyl (3e) moieties, which is in line with the previously reported advantages of these substituents in C–H functionalization and cyclopropanation [53,54,55,56]. Poor yields and/or conversions were observed for vinyl diazoacetates with bulky tert-butyl (2g) or BHT (2,6-di-tert-butyl-4-methylphenyl, 2h) ester groups [57]. Unfortunately, complex mixtures were observed when employing substituted (β-phenyl or γ-phenyl) vinyl diazoacetates. Regarding the scope of nitronates 1, the reaction tolerates various aryl groups at the C4 of starting nitronate. Meta-(3i)- and para-(3a,g,h)-substituted substrates gave products 3 smoothly, while for ortho-substituent (3j) a diminished yield was attained, presumably, due to steric hindrance. Importantly, no influence of the electronic effect of aromatic substituent was observed as electron-accepting (NO2, 3h), halogen (3g,j), as well as pharmaceutically attractive dihydroxylated aromatics [58,59] (3k) gave comparable outcomes. Apart from aryl-, 5-benzoyloxy-(3l), 5-alkyl-(3m), and 7-ethoxy-(3o) products were obtained, thus demonstrating the possibility of incorporation of different functionalities in the target structures. Poor yield was attained for cyclohexane-annulated product 3p, while a complex product mixture was observed for nitronate 1q. When more sterically encumbered camphene-derived (1r) or 3-methyl-substituted nitronate (1s) was involved, the reaction was decelerated and starting materials were observed even after 4 days.
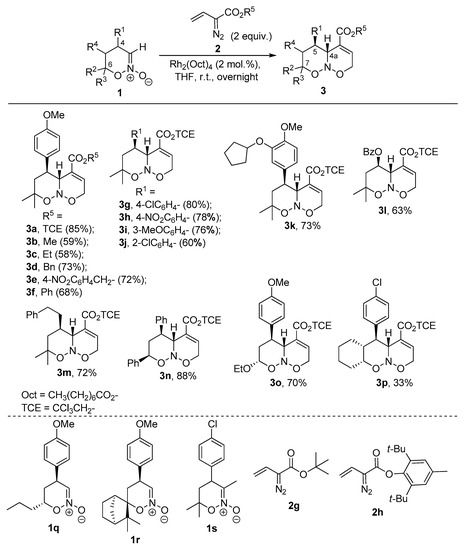
Scheme 2.
Synthesis of bicyclic nitroso acetals 3.
Structures of the obtained products were supported by their NMR (1D and 2D) and HRMS data. Gratifyingly, in all examined cases, the annulation was found to be stereoselective producing only one diastereomer of products 3. The relative configuration of stereocenters was also deduced on the basis of the coupling constant between C(4a)–H and C(5)–H. Its high value of 9–11 Hz evidenced about (pseudo)axial positions of both protons. This conclusion was supported by the X-ray analysis for products 3b and 3p (Figure 1). Notably, the cis-junction of two oxazine rings was observed, which can be favored by anomeric interaction within O–N–O moiety [60]. However, we should mention the conformational lability of products 3, as many signals in their NMR spectra were broadened. Especially notable was product 3p possessing three contiguous annulated six-membered rings. In its case, a lot of the 13C signals were of low intensity and were assigned only with the aim of 1H-13C HSQC spectra (see Supplementary Materials). Mentioned effects can be attributed to both inversion of six-membered rings and to nitrogen inversion [41,61].
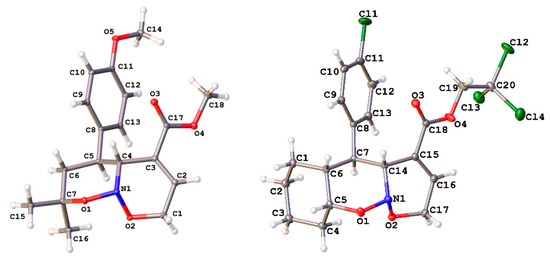
Figure 1.
General view of the compounds 3b (left) and 3p (right) in the representation of non-hydrogen atoms as thermal ellipsoids at a 30% probability level.
Based on the obtained and the literature [42,43,62] data, the following mechanism for the annulation was proposed (Scheme 3). In the first step, electrophilic vinyl carbenoid species A is generated from vinyl diazo compound 2 and a rhodium catalyst. Nucleophilic attack of the oxygen atom of N-oxide at the unhindered =CH2 terminus in A produces zwitterion B that undergoes ring closure with the expulsion of the catalyst and formation of target cycloadduct (path a). Observed stereochemistry can be explained by an approach of the bulky vinyl-rhodium moiety from the face of the dipole opposite to substituent R1. Hence, low yields in the case of product 3p and the messy reaction for nitronate 1q can be attributed to the presence of a relatively bulky substituent at C(6) of the nitronate, which together with the substituent at C(4) may block both sides of the dipole, thus preventing the annulation. Interestingly, the observed [3+3]-annulation with nitronates differs from the reaction of vinyl diazoacetates with nitrones, which often proceeds via a [3+2]-pathway and subsequent carbene reactions (cf. path b) [63,64,65,66], while the respective [3+3]-pathway was rarely observed [62].
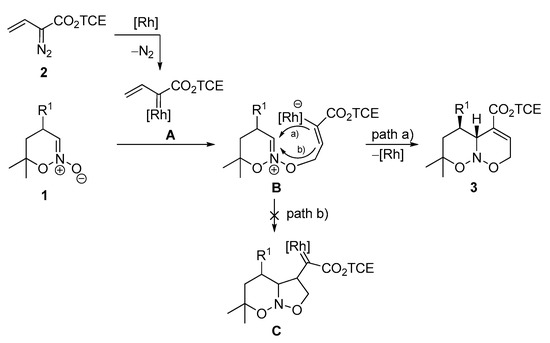
Scheme 3.
Proposed reaction mechanism for the [3+3]-annulation of 1 and 2.
Aiming at the subsequent functionalization of product 3b, we attempted a Michael addition of dimethyl malonate across the C=C double bond. However, none of the desired transformation was observed, albeit ring contraction occurred providing pyrrolo[1,2-b]oxazine derivative 4b. This process has some precedents in the literature [67,68,69,70,71,72,73,74,75], with the closest analogue observed by Kerr et al. for non-annulated oxazines under the treatment with DBU in MeCN [67]. The proposed mechanism starts with the deprotonation at C(2), which is facilitated by the conjugation of the formed anion with C=C–CO2 moiety in D (Scheme 4). Subsequent cleavage of the N–O bond, formation of an aldehyde group, and recyclization produce hydroxypyrroline E, upon which dehydration furnishes the target pyrrole ring (path a). Some optimization of reaction conditions was performed (Scheme 5) and a combination of base (DBU) and protic additive (alcohol) was found optimal, instead of the use of a base alone (cf. yields for DBU and DBU/TCEOH). We attribute it to two main reasons. First, N–O bonds in nitroso acetals are also susceptible to basic cleavage (e.g., Scheme 4, path b) [76,77], which can lead to the formation of side products. The use of alcohol promotes the elimination of the hydroxyl group from intermediate E via hydrogen bonding, thus facilitating pyrrole formation (path a). Secondly, the presence of alcohol diminishes the amount of carboxylic acid 5, which originates from the hydrolysis of product 4a by water. Particularly, relatively large amounts of product 5 were observed when a strong base, namely, sodium tert-butylate was used. Similarly, in the excess of methanol, transesterification was observed (Scheme 6).
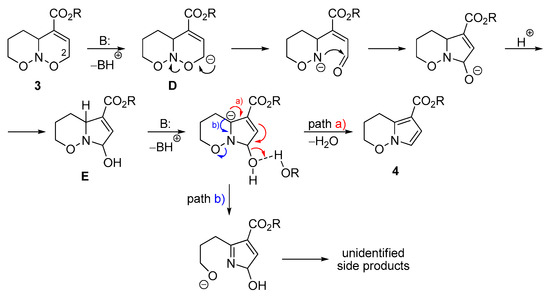
Scheme 4.
Proposed mechanism for the ring contraction of 3 and formation of pyrrolooxazines 4. Substituents are omitted for clarity.
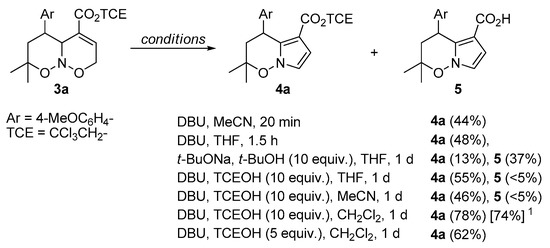
Scheme 5.
Transformations of nitroso acetal 3a in basic conditions (see Supplementary Materials for full details). Yields were determined by 1H NMR with an internal standard (dimethyl terephthalate). 1 Isolated yield.
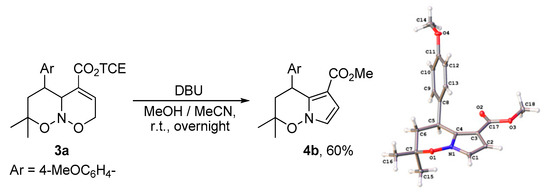
Scheme 6.
Formation of pyrrole 4b from nitroso acetal 3a by sequential ring contraction/transesterification.
The literature provides limited data on the preparation of pyrrolo[1,2-b]oxazine derivatives [78,79], especially possessing an unsaturated pyrrole core [80,81]. Therefore, we elucidated the scope of the found transformation. Since the reagents used for the [3+3]-annulation should not interfere with the ring contraction, we decided to make a one-pot protocol for the synthesis of pyrrolooxazines 4 from oxazine N-oxides 1 and vinyl diazoacetates 2. For this purpose, the reaction mixture was evaporated after the annulation step and redissolved in CH2Cl2, followed by the addition of other reagents. This worked well providing target products 4 in reasonable yields for the whole two-step sequence (Scheme 7).
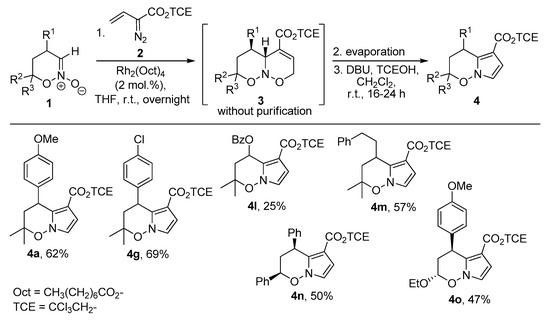
Scheme 7.
One-pot synthesis of pyrrolo[1,2-b]oxazines 4.
3. Materials and Methods
3.1. General Experiment
All reactions were performed in oven-dried (150 °C) glassware. Most of the chemicals were acquired from commercial sources (Sigma-Aldrich, St. Louis, MO, USA; Acros Organics, Geel, Belgium; Alfa Aesar, Heysham, UK; ABCR, Karlsruhe, Germany; and P&M Invest, Moscow, Russia) with purities >95% and used as received. Petroleum ether (PE) and ethyl acetate were distilled. THF, CH3CN, CH2Cl2, and DBU were distilled from CaH2. Brine refers to the saturated aqueous solution of NaCl. TLC was performed on silica coated on aluminum with UV254 indicator. Visualization was accomplished with UV and/or anisaldehyde/H2SO4/EtOH stain. Column chromatography was performed on silica (0.04–0.063 mm, 60 Å). NMR spectra were recorded at 300 K (unless otherwise mentioned) on Bruker AM300 (Bruker, Karlsruhe, Germany), Fourier 300HD (Bruker, Karlsruhe, Germany), and Avance NEO (Bruker, Karlsruhe, Germany) spectrometers at the following spectrometer frequencies: 300 MHz (1H NMR) and 75 MHz (13C NMR). Multiplicities are assigned as s (singlet), d (doublet), t (triplet), q (quartet), m (multiplet), br (broad), and app (apparent). Assignment (including rel. configuration) was made using 2D NMR spectra for selected products. For other products, the assignment was made by analogy. High-resolution mass spectra were acquired on Bruker micrOTOF (Bruker, Bremen, Germany) spectrometer using electrospray ionization (ESI). Melting points were determined on a Koffler melting point apparatus and are uncorrected. Starting nitroalkenes [82,83,84,85,86], nitronates 1 [40,87,88,89], and vinyl diazoacetates 2 [55,90,91,92,93,94,95,96,97] were prepared according to the literature (see Supplementary Materials for detailed procedures).
3.2. X-ray Crystallography
X-ray diffraction data for 3b, 3p, and 4b were collected at 100 K with a Bruker Quest D8 CMOS diffractometer (Bruker, Karlsruhe, Germany), using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å, ω-scans). Structures were solved using Intrinsic Phasing with the ShelXT [98] structure solution program in Olex2 [99] and then refined with the XL refinement package [100] using Least-Squares minimization against F2 in the anisotropic approximation for non-hydrogen atoms. Positions of other hydrogen atoms were calculated, and they were refined in the isotropic approximation within the riding model. Crystal data and structure refinement parameters are given in Table S1 (see Supplementary Materials). The crystallographic information for compounds 3b, 3p, and 4b was deposited in the Cambridge Crystallographic Data Centre (CCDC 2242307-2242309) and can be obtained free of charge via https://www.ccdc.cam.ac.uk/structures/ (accessed on 25 February 2023).
3.3. Synthetic Procedures for Products 3–5
3.3.1. General Procedure for the Synthesis of Nitroso Acetals 3 (GP-1)
To the 0.3 M solution of nitronate 1 (1 equiv.) in THF, Rh2(Oct)4 (0.02 equiv.) was added under an argon atmosphere at r.t. and the mixture was stirred for 5 min. Then THF solution of vinyl diazoacetate 2 (2 equiv. rel. to nitronate 1, C = 0.5 M) was added dropwise (approx. 5–10 min.) using a syringe with stirring. The resulting solution was left overnight and then evaporated. The residue was preadsorbed onto Celite® or silica gel and subjected to column chromatography on silica gel (eluent: PE/EtOAc or PE/CH2Cl2) to give target nitroso acetal 3.
2,2,2-Trichloroethyl (4aR*,5S*)-5-(4-methoxyphenyl)-7,7-dimethyl-4a,5,6,7-tetrahydro-2H-[1,2]oxazino[2,3-b][1,2]oxazine-4-carboxylate 3a. Nitroso acetal 3a was obtained from nitronate 1a (24 mg, 0.10 mmol) and vinyl diazoacetate 2a (50 mg, 0.21 mmol) according to GP-1. Column chromatography (eluent: PE/EtOAc, 10:1) afforded 39 mg (85%) of the target nitroso acetal 3a as colorless oil, which solidified upon storage in a fridge (ca. 4 °C). Rf = 0.30 (PE/EtOAc, 5:1, UV, anisaldehyde). m.p. = 112–114 °C (EtOAc). 1H NMR (300 MHz, CDCl3): δ 1.38 (s, 3H, Me(7)), 1.63 (s, 3H, Me(7)), 1.92 (dd, J = 13.3, 3.1 Hz, 1H, CH2eq(6)), 2.24 (app t, J = 12.9 Hz, 1H, CH2ax(6)), 3.50 (ddd, J = 13.4, 10.5, 3.1 Hz, 1H, CH(5)), 3.80 (s, 3H, OMe), 4.05–4.09 (m, 2H, OCH2aCCl3 and CH(4a)), 4.39 (d, J = 11.9 Hz, 1H, OCH2bCCl3), 4.55 (dd, J = 18.1, 3.5 Hz, 1H, CH2a(2)), 4.80 (br d, J = 18.1 Hz, 1H, CH2b(2)), 6.83 (d, J = 8.7 Hz, 2H, CHAr), 6.95 (dd, J = 3.2, 1.5 Hz, 1H, CH(3)), 7.18 (d, J = 8.7 Hz, 2H, CHAr). 13C NMR (75 MHz, DEPT, CDCl3): δ 28.7 (Me(7)), 30.8 (Me(7)), 36.3 (CH(5)), 41.2 (CH2(6)), 55.3 (OMe), 65.1 (CH(4a)), 68.5 (CH2(2)), 73.8 (OCH2CCl3), 79.1 (C(7)), 94.7 (CCl3), 113.7 (CHAr), 129.6 (CHAr), 130.4 and 132.0 (C(4) and CHAr), 138.6 (=CH(3)), 158.7 (CAr–OMe), 162.8 (C=O). HRMS (ESI): m/z calcd. for C19H23Cl3NO5+ [M + H]+: 450.0636, found: 450.0649.
Methyl (4aR*,5S*)-5-(4-methoxyphenyl)-7,7-dimethyl-4a,5,6,7-tetrahydro-2H-[1,2]oxazino[2,3-b][1,2]oxazine-4-carboxylate 3b. Nitroso acetal 3b was obtained from nitronate 1a (24 mg, 0.10 mmol) and vinyl diazoacetate 2b (50 mg, 0.21 mmol) according to GP-1. Column chromatography (eluent: PE/EtOAc, 10:1) afforded 20 mg (59%) of the target nitroso acetal 3b as colorless oil, which solidified upon storage in a fridge (ca. 4 °C). Rf = 0.59 (PE/EtOAc, 1:1, UV, anisaldehyde). m.p. = 131–133 °C (PE/CH2Cl2, 3:1). 1H NMR (300 MHz, COSY, CDCl3): δ 1.37 (s, 3H, Meeq(7)), 1.62 (s, 3H, Meax(7)), 1.90 (dd, J = 13.3, 3.1 Hz, 1H, CH2eq(6)), 2.24 (app t, J = 13.1 Hz, 1H, CH2ax(6)), 3.19 (s, 3H, CO2Me), 3.47 (ddd, J = 12.8, 10.5, 2.7 Hz, 1H, CH(5)), 3.80 (s, 3H, OMe), 4.01 (dd, J = 10.5, 1.5 Hz, 1H, CH(4a)), 4.49 (dd, J = 17.8, 3.3 Hz, 1H, CH2a(2)), 4.75 (br d, J = 17.8 Hz, 1H, CH2b(2)), 6.77 (dd, J = 3.3, 1.4 Hz, 1H, CH(3)), 6.84 (d, J = 8.7 Hz, 2H, CHAr), 7.16 (d, J = 8.7 Hz, 2H, CHAr). Characteristic NOESY interactions: CH(5)/CH2eq(6); CH2ax(6)/CH(4a); CH2ax(6)/CHAr; CH(4a)/CHAr; Meax(7)/CH(5). 13C NMR (75 MHz, DEPT, HSQC, HMBC, CDCl3): δ 28.6 (Me(7)), 30.9 (Me(7)), 36.3 (CH(5)), 41.1 (CH2(6)), 51.5 (CO2Me), 55.3 (OMe), 65.4 (CH(4a)), 68.5 (CH2(2)), 78.9 (C(7)), 113.6 (CHAr), 129.4 (CHAr), 131.6 and 132.3 (C(4) and CHAr), 136.4 (=CH(3)), 158.6 (CAr–OMe), 165.4 (C=O). HRMS (ESI): m/z calcd. for C18H24NO5+ [M + H]+: 334.1649, found: 334.1645. The crystallographic information for compound 3b was deposited in the Cambridge Crystallographic Data Centre (CCDC 2242307).
4-Ethyl (4aR*,5S*)-5-(4-methoxyphenyl)-7,7-dimethyl-4a,5,6,7-tetrahydro-2H-[1,2]oxazino[2,3-b][1,2]oxazine-4-carboxylate 3c. Nitroso acetal 3c was obtained from nitronate 1a (21 mg, 0.09 mmol) and vinyl diazoacetate 2c (25 mg, 0.18 mmol) according to GP-1. Column chromatography (eluent: PE/EtOAc, 10:1, then 8:1) afforded 18 mg (58%) of the target nitroso acetal 3c as colorless oil, which solidified upon storage in a fridge (ca. 4 °C). Rf = 0.30 (PE/EtOAc, 3:1, UV, anisaldehyde). m.p. = 132–134 °C (PE/CH2Cl2, 3:1). 1H NMR (300 MHz, CDCl3): δ 1.02 (t, J = 7.1 Hz, 3H, OCH2CH3), 1.37 (s, 3H, Me(7)), 1.62 (s, 3H, Me(7)), 1.90 (dd, J = 13.2, 3.0 Hz, 1H, CH2eq(6)), 2.23 (app t, J = 13.1 Hz, 1H, CH2ax(6)), 3.39–3.35 (m, 2H, CH(5) and OCH2aCH3), 3.72–3.83 (m, 1H, OCH2bCH3), 3.79 (s, overlapped, 3H, OMe), 4.03 (dd, J = 10.6, 1.4 Hz, 1H, CH(4a)), 4.50 (dd, J = 17.7, 3.4 Hz, 1H, CH2a(2)), 4.75 (br d, J = 17.7 Hz, 1H, CH2b(2)), 6.75 (d, J = 3.1, 1.4 Hz, 1H, CH(3)), 6.83 (d, J = 8.7 Hz, 2H, CHAr), 7.16 (d, J = 8.7 Hz, 2H, CHAr). 13C NMR (75 MHz, DEPT, CDCl3): δ 13.9 (OCH2CH3), 28.6 (Me(7)), 30.9 (Me(7)), 36.3 (CH(5)), 41.2 (CH2(6)), 55.3 (OMe), 60.6 (OCH2CH3), 65.3 (CH(4a)), 68.4 (CH2(2)), 79.0 (C(7)), 113.6 (CHAr), 129.5 (CHAr), 131.9 and 132.3 (CAr and C(4)), 135.9 (=CH(3)), 158.7 (CAr–O), 165.0 (C=O). HRMS (ESI): m/z calcd. for C19H26NO5+ [M + H]+: 348.1805, found: 348.1805.
Benzyl (4aR*,5S*)-5-(4-methoxyphenyl)-7,7-dimethyl-4a,5,6,7-tetrahydro-2H-[1,2]oxazino[2,3-b][1,2]oxazine-4-carboxylate 3d. Nitroso acetal 3d was obtained from nitronate 1a (29 mg, 0.13 mmol) and vinyl diazoacetate 2d (47 mg, 0.25 mmol) according to GP-1. Column chromatography (eluent: PE/EtOAc, 15:1) afforded 37 mg (73%) of the target nitroso acetal 3d as slightly yellow oil. Analytically pure material was obtained after crystallization from PE/CH2Cl2 (1:1) as white powder. Rf = 0.62 (PE/EtOAc, 1:1, UV, anisaldehyde). m.p. = 110–112 °C (PE/CH2Cl2, 1:1). 1H NMR (300 MHz, CDCl3): δ 1.36 (s, 3H, Me(7)), 1.62 (s, 3H, Me(7)), 1.90 (dd, J = 13.3, 3.1 Hz, 1H, CH2eq(6)), 2.22 (app t, J = 13.1 Hz, 1H, CH2ax(6)), 3.49 (ddd, J = 13.3, 10.5, 3.1 Hz, 1H, CH(5)), 3.80 (s, 3H, OMe), 4.07 (app d, J = 10.3 Hz, 1H, CH(4a)), 4.38 (d, J = 12.5 Hz, 1H, OCH2aPh), 4.50 (dd, J = 17.8, 3.5 Hz, 1H, CH2a(2)), 4.73–4.82 (m, 2H, CH2b(2) and OCH2bPh), 6.78–6.81 (m, 3H, CHAr and CH(3)), 7.14–7.19 (m, 4H, CHAr), 7.32–7.38 (m, 3H, CHAr). 13C NMR (75 MHz, DEPT, CDCl3): δ 28.7 (Me(7)), 30.9 (Me(7)), 36.3 (CH(5)), 41.2 (CH2(6)), 55.3 (OMe), 65.3 (CH(4a)), 66.2 (OCH2Ph), 68.5 (CH2(2)), 79.0 (C(7)), 113.7 (CHAr), 128.0 (CHPh), 128.1 (CHPh), 128.4 (CHPh), 129.5 (CHAr), 131.6 and 132.2 (CAr and C(4)), 135.6 (CPh), 136.6 (=CH(3)), 158.6 (CAr–O), 164.7 (C=O). HRMS (ESI): m/z calcd. for C24H28NO5+ [M + H]+: 410.1962, found: 410.1964.
4-Nitrobenzyl (4aR*,5S*)-5-(4-methoxyphenyl)-7,7-dimethyl-4a,5,6,7-tetrahydro-2H-[1,2]oxazino[2,3-b][1,2]oxazine-4-carboxylate 3e. Nitroso acetal 3e was obtained from nitronate 1a (35 mg, 0.15 mmol) and vinyl diazoacetate 2e (73 mg, 0.30 mmol) according to GP-1. Column chromatography (eluent: PE/EtOAc, 7:1, then 5:1) afforded 48 mg (72%) of the target nitroso acetal 3e as colorless oil, which solidified upon storage in a fridge (ca. 4 °C). Rf = 0.52 (PE/EtOAc, 1:1, UV, anisaldehyde). m.p. = 140–142 °C (EtOAc). 1H NMR (300 MHz, CDCl3): δ 1.37 (s, 3H, Me(7)), 1.62 (s, 3H, Me(7)), 1.90 (dd, J = 13.3, 2.9 Hz, 1H, CH2eq(6)), 2.21 (app t, J = 13.1 Hz, 1H, CH2ax(6)), 3.49 (ddd, J = 13.3, 10.5, 3.1 Hz, 1H, CH(5)), 3.76 (s, 3H, OMe), 4.06 (app d, J = 10.6 Hz, 1H, CH(4a)), 4.46 (d, J = 13.5 Hz, 1H, OCH2aAr), 4.53 (dd, J = 18.0, 3.5 Hz, 1H, CH2a(2)), 4.78 (d, J = 18.0 Hz, 1H, CH2b(2)), 4.93 (d, J = 13.5 Hz, 1H, OCH2bAr), 6.73 (d, J = 8.6 Hz, 2H, CHArOMe), 6.87 (br s, 1H, CH(3)), 7.13 (d, J = 8.6 Hz, 2H, CHArOMe), 7.29 (d, J = 8.7 Hz, 2H, CHArNO2), 8.20 (d, J = 8.7 Hz, 2H, CHArNO2). 13C NMR (75 MHz, DEPT, HSQC, CDCl3): δ 28.6 (Me(7)), 30.8 (Me(7)), 36.3 (CH(5)), 41.2 (CH2(6)), 55.2 (OMe), 64.8 (OCH2Ar), 65.2 (CH(4a)), 68.5 (CH2(2)), 79.1 (C(7)), 113.6 (CHArOMe), 123.7 (CHArNO2), 128.2 (CHArNO2), 129.5 (CHArOMe), 131.1 and 132.2 (CArOMe and C(4)), 137.7 (=CH(3)), 142.9 (CArNO2), 147.6 (CAr–NO2), 158.6 (CAr–O), 164.4 (C=O). HRMS (ESI): m/z calcd. for C24H27N2O7+ [M + H]+: 455.1813, found: 455.1807.
Phenyl (4aR*,5S*)-5-(4-methoxyphenyl)-7,7-dimethyl-4a,5,6,7-tetrahydro-2H-[1,2]oxazino[2,3-b][1,2]oxazine-4-carboxylate 3f. Nitroso acetal 3f was obtained from nitronate 1a (30 mg, 0.13 mmol) and vinyl diazoacetate 2f (47 mg, 0.25 mmol) according to GP-1. Column chromatography (eluent: PE/EtOAc, 20:1, then 9:1) afforded 68 mg of crude nitroso acetal, which was crystallized from PE/CH2Cl2, 1:1 to give 34 mg (68%) of the target nitroso acetal 3f as white powder. Rf = 0.30 (PE/EtOAc, 3:1, UV, anisaldehyde). m.p. = 175–176 °C (PE/CH2Cl2, 1:1). 1H NMR (300 MHz, CDCl3): δ 1.39 (s, 3H, Me(7)), 1.66 (s, 3H, Me(7)), 1.95 (dd, J = 13.2, 3.1 Hz, 1H, CH2eq(6)), 2.26 (app t, J = 13.1 Hz, 1H, CH2ax(6)), 3.58 (ddd, J = 13.3, 10.5, 3.0 Hz, 1H, CH(5)), 3.79 (s, 3H, OMe), 4.18 (dd, J = 10.5, 1.9 Hz, 1H, CH(4a)), 4.59 (dd, J = 18.0, 3.4 Hz, 1H, CH2a(2)), 4.85 (dt, J = 18.0, 1.9 Hz, 1H, CH2b(2)), 6.54–6.59 (m, 2H, CHPh), 6.85 (d, J = 8.7 Hz, 2H, CHAr), 7.01 (dd, J = 3.4, 1.9 Hz, 1H, CH(3)), 7.12–7.18 (m, 1H, CHPh), 7.22–7.28 (m, 4H, CHAr and CHPh). 13C NMR (75 MHz, DEPT, CDCl3): δ 28.7 (Me(7)), 30.9 (Me(7)), 36.4 (CH(5)), 41.4 (CH2(6)), 55.3 (OMe), 65.2 (CH(4a)), 68.6 (CH2(2)), 79.2 (C(7)), 114.1 (CHAr), 121.3 (CHPh), 126.6 (CHPh), 129.0 and 129.6 (CHAr and CHPh), 131.2 and 132.2 (CAr and C(4)), 138.1 (=CH(3)), 150.2 (CPh–O), 158.8 (CAr–O), 163.1 (C=O). HRMS (ESI): m/z calcd. for C23H26NO5+ [M + H]+: 396.1805, found: 396.1806.
2,2,2-Trichloroethyl (4aR*,5S*)-5-(4-chlorophenyl)-7,7-dimethyl-4a,5,6,7-tetrahydro-2H-[1,2]oxazino[2,3-b][1,2]oxazine-4-carboxylate 3g. Nitroso acetal 3g was obtained from nitronate 1b (35 mg, 0.15 mmol) and vinyl diazoacetate 2a (72 mg, 0.30 mmol) according to GP-1. Column chromatography (eluent: PE/CH2Cl2, 1:1, then CH2Cl2) afforded 54 mg (80%) of the target nitroso acetal 3g as colorless oil, which solidified upon storage in a fridge (ca. 4 °C). Rf = 0.48 (PE/EtOAc, 3:1, UV, anisaldehyde). m.p. = 121–123 °C (PE/CH2Cl2, 3:1). 1H NMR (300 MHz, CDCl3): δ 1.38 (s, 3H, Me(7)), 1.63 (s, 3H, Me(7)), 1.93 (dd, J = 13.2, 3.0 Hz, 1H, CH2eq(6)), 2.24 (app t, J = 13.2 Hz, 1H, CH2ax(6)), 3.55 (ddd, J = 13.3, 10.5, 3.1 Hz, 1H, CH(5)), 4.06–4.12 (m, overlapped, 1H, CH(4a)), 4.13 (d, overlapped, J = 11.9 Hz, 1H, OCH2aCCl3), 4.42 (d, J = 11.9 Hz, 1H, OCH2bCCl3), 4.56 (dd, J = 18.2, 3.5 Hz, 1H, CH2a(2)), 4.81 (app d, J = 18.2 Hz, 1H, CH2b(2)), 7.00 (dd, J = 3.5, 1.7 Hz, 1H, CH(3)), 7.20–7.28 (m, 4H, CHAr). 13C NMR (75 MHz, DEPT, CDCl3): δ 28.6 (Me(7)), 30.8 (Me(7)), 36.8 (CH(5)), 40.9 (CH2(6)), 64.7 (CH(4a)), 68.5 (CH2(2)), 73.8 (OCH2CCl3), 78.9 (C(7)), 94.6 (CCl3), 128.5 (CHAr), 130.0 (CHAr and C(4)), 133.0 (CAr), 138.5 (CAr), 139.3 (=CH(3)), 162.7 (C=O). HRMS (ESI): m/z calcd. for [C18H20Cl4NO4+ [M + H]+: 454.0141, found: 456.0145.
2,2,2-Trichloroethyl (4aR*,5S*)-7,7-dimethyl-5-(4-nitrophenyl)-4a,5,6,7-tetrahydro-2H-[1,2]oxazino[2,3-b][1,2]oxazine-4-carboxylate 3h. Nitroso acetal 3h was obtained from nitronate 1c (27 mg, 0.11 mmol) and vinyl diazoacetate 2a (55 mg, 0.23 mmol) according to GP-1. Column chromatography (eluent: PE/EtOAc, 20:1, then 15:1) afforded 38 mg (78%) of the target nitroso acetal 3h as colorless oil. Rf = 0.36 (PE/EtOAc, 3:1, UV, anisaldehyde). NB: Even on short standing in solution in CDCl3, decomposition of compound 3h was observed. 1H NMR (300 MHz, CDCl3): δ 1.40 (s, 3H, Me(7)), 1.64 (s, 3H, Me(7)), 1.96 (dd, J = 13.2, 2.6 Hz, 1H, CH2eq(6)), 2.30 (app t, J = 13.1 Hz, 1H, CH2ax(6)), 3.71 (br t, J = 12.3 Hz, 1H, CH(5)), 4.14 (br d, J = 10.7 Hz, 1H, CH(4a)), 4.22 (d, J = 11.9 Hz, 1H, OCH2aCCl3), 4.33 (d, J = 11.9 Hz, 1H, OCH2bCCl3), 4.59 (dd, J = 18.3, 3.2 Hz, 1H, CH2a(2)), 4.83 (app d, J = 18.3 Hz, 1H, CH2b(2)), 7.09 (br s, 1H, CH(3)), 7.46 (d, J = 8.6 Hz, 2H, CHAr), 8.15 (d, J = 8.7 Hz, 2H, CHAr). 13C NMR (75 MHz, DEPT, CDCl3): δ 28.6 (Me(7)), 30.8 (Me(7)), 37.5 (CH(5)), 40.8 (CH2(6)), 64.1 (CH(4a)), 68.6 (CH2(2)), 73.8 (OCH2CCl3), 78.7 (C(7)), 94.5 (CCl3), 123.4 (CHAr), 129.1 (C(4)), 129.6 (CHAr), 140.4 (=CH(3) and CAr), 147.1 (CAr), 162.5 (C=O). HRMS (ESI): m/z calcd. for C18H20Cl4NO4+ [M + H]+: 454.0141, found: 456.0145.
2,2,2-Trichloroethyl (4aR*,5S*)-5-(3-methoxyphenyl)-7,7-dimethyl-4a,5,6,7-tetrahydro-2H-[1,2]oxazino[2,3-b][1,2]oxazine-4-carboxylate 3i. Nitroso acetal 3i was obtained from nitronate 1d (33 mg, 0.14 mmol) and vinyl diazoacetate 2a (68 mg, 0.28 mmol) according to GP-1. Column chromatography (eluent: PE/EtOAc, 20:1, then 15:1) afforded 47 mg (76%) of the target nitroso acetal 3i as slightly yellow oil, which solidified upon storage in a fridge (ca. 4 °C). Rf = 0.30 (PE/EtOAc, 5:1, UV, anisaldehyde). m.p. = 107–109 °C (PE/CH2Cl2, 2:1). 1H NMR (300 MHz, CDCl3): δ 1.38 (s, 3H, Me(7)), 1.63 (s, 3H, Me(7)), 1.95 (dd, J = 13.3, 3.1 Hz, 1H, CH2eq(6)), 2.28 (app t, J = 13.1 Hz, 1H, CH2ax(6)), 3.54 (ddd, J = 13.3, 10.5, 3.1 Hz, 1H, CH(5)), 3.81 (s, 3H, OMe), 4.03 (d, J = 11.9 Hz, 1H, OCH2aCCl3), 4.10 (d, J = 10.5 Hz, 1H, CH(4a)), 4.40 (d, J = 11.9 Hz, 1H, OCH2bCCl3), 4.56 (dd, J = 18.1, 3.5 Hz, 1H, CH2a(2)), 4.81 (app d, J = 18.1 Hz, 1H, CH2b(2)), 6.76–6.81 (m, 2H, CHAr), 6.87 (d, J = 7.7 Hz, 1H, CHAr), 6.97 (dd, J = 3.5, 1.7 Hz, 1H, CH(3)), 7.22 (app t, J = 7.7 Hz, 1H, CHAr). 13C NMR (75 MHz, DEPT, CDCl3): δ 28.6 (Me(7)), 30.8 (Me(7)), 37.2 (CH(5)), 40.7 (CH2(6)), 55.2 (OMe), 64.9 (CH(4a)), 68.5 (CH2(2)), 73.7 (OCH2CCl3), 79.0 (C(7)), 94.7 (CCl3), 112.5 (CHAr), 114.5 (CHAr), 120.9 (CHAr), 129.4 (CHAr), 130.4 (C(4)), 138.6 (=CH(3)), 141.6 (CAr), 159.6 (CAr–OMe), 162.8 (C=O). HRMS (ESI): m/z calcd. for C19H23Cl3NO5+ [M + H]+: 450.0636, found: 450.0642.
2,2,2-Trichloroethyl (4aR*,5S*)-5-(2-chlorophenyl)-7,7-dimethyl-4a,5,6,7-tetrahydro-2H-[1,2]oxazino[2,3-b][1,2]oxazine-4-carboxylate 3j. Nitroso acetal 3j was obtained from nitronate 1e (28 mg, 0.12 mmol) and vinyl diazoacetate 2a (57 mg, 0.24 mmol) according to GP-1. Column chromatography (eluent: PE/EtOAc, 40:1) afforded 32 mg (60%) of the target nitroso acetal 3j as colorless oil, which solidified upon storage in a fridge (ca. 4 °C). Rf = 0.51 (PE/EtOAc, 3:1, UV, anisaldehyde). m.p. = 147–149 °C (PE/CH2Cl2, 2:1). 1H NMR (300 MHz, CDCl3): δ 1.38 (s, 3H, Me(7)), 1.67 (br s, 3H, Me(7)), 1.88 (dd, J = 13.2, 2.7 Hz, 1H, CH2eq(6)), 2.22 (app t, J = 13.0 Hz, 1H, CH2ax(6)), 4.10–4.16 (m, 2H, CH(4a) and OCH2aCCl3), 4.36–4.46 (m, 2H, CH(5) and OCH2bCCl3), 4.60 (dd, J = 18.2, 3.3 Hz, 1H, CH2a(2)), 4.81 (app d, J = 18.6 Hz, 1H, CH2b(2)), 7.00 (br s, 1H, CH(3)), 7.13–7.19 (m, 1H, CHAr), 7.27–7.32 (m, 2H, CHAr), 7.13–7.44–7.47 (m, 1H, CHAr). 13C NMR (75 MHz, DEPT, CDCl3): δ 28.7 (Me(7)), 30.7 (Me(7)), 32.1 (br, CH(5)), 41.2 (CH2(6)), 64.5 (CH(4a)), 68.5 (CH2(2)), 73.7 (OCH2CCl3), 78.9 (C(7)), 94.7 (CCl3), 126.9 (CHAr), 128.1 (CHAr), 129.1 (C(4)), 129.5 (CHAr), 129.8 (CHAr), 134.4 (CAr), 137.6 (CAr), 139.6 (=CH(3)), 162.8 (C=O). HRMS (ESI): m/z calcd. for C18H20Cl4NO4+ [M + H]+: 454.0141, found: 454.0136.
2,2,2-Trichloroethyl (4aR*,5S*)-5-(3-(cyclopentyloxy)-4-methoxyphenyl)-7,7-dimethyl-4a,5,6,7-tetrahydro-2H-[1,2]oxazino [2,3-b][1,2]oxazine-4-carboxylate 3k. Nitroso acetal 3k was obtained from nitronate 1f (36 mg, 0.11 mmol) and vinyl diazoacetate 2a (54 mg, 0.22 mmol) according to GP-1. Column chromatography (eluent: PE/EtOAc, 20:1, then 15:1) afforded 44 mg (73%) of the target nitroso acetal 3k as colorless oil. Rf = 0.46 (PE/EtOAc, 3:1, UV, anisaldehyde). 1H NMR (300 MHz, CDCl3): δ 1.38 (s, 3H, Me(7)), 1.63 (br s, 3H, Me(7) and CH2cycl), 1.80–2.01 (m, 7H, 3 × CH2cycl and CH2eq(5)), 2.21 (app t, J = 13.1 Hz, 1H, CH2ax(6)), 3.46 (ddd, J = 13.3, 10.5, 3.1 Hz, 1H, CH(5)), 3.83 (s, 3H, OMe), 4.07 (dd, J = 10.5, 1.9 Hz, 1H, CH(4a)), 4.16 (d, J = 12.0 Hz, 1H, OCH2aCCl3), 4.37 (d, J = 12.0 Hz, 1H, OCH2bCCl3), 4.56 (dd, J = 18.0, 3.3 Hz, 1H, CH2a(2)), 4.77–4.83 (m, 2H, CH2b(2) and CHcycl–O), 6.75–6.78 (m, 3H, CHAr), 6.94 (dd, J = 3.3, 1.4 Hz, 1H, CH(3)). 13C NMR (75 MHz, DEPT, CDCl3): δ 24.1 and 24.2 (2 × CH2cycl), 28.7 (Me(7)), 30.9 (Me(7)), 32.88 and 32.92 (2 × CH2cycl), 36.7 (CH(5)), 41.1 (CH2(6)), 56.2 (OMe), 65.1 (CH(4a)), 68.5 (CH2(2)), 73.7 (OCH2CCl3), 79.1 (C(7)), 80.5 (CHcycl–O), 94.8 (CCl3), 111.8 (CHAr), 115.5 (CHAr), 120.6 (CHAr), 130.6 (C(4)), 132.4 (CAr), 138.2 (=CH(3)), 147.6 (CAr–O), 149.2 (CAr–O), 162.6 (C=O). HRMS (ESI): m/z calcd. for C24H31Cl3NO6+ [M + H]+: 534.1211, found: 534.1196.
2,2,2-Trichloroethyl (4aS*,5R*)-5-(benzoyloxy)-7,7-dimethyl-4a,5,6,7-tetrahydro-2H-[1,2]oxazino[2,3-b][1,2]oxazine-4-carboxylate 3l. Nitroso acetal 3l was obtained from nitronate 1g (34 mg, 0.14 mmol) and vinyl diazoacetate 2a (68 mg, 0.28 mmol) according to GP-1. Column chromatography (eluent: PE/EtOAc, 10:1) afforded 48 mg of crude nitroso acetal 3l as colorless oil. Then, column chromatography (eluent: PE/CH2Cl2, 1:1, then CH2Cl2) afforded 40 mg (63%) of the target nitroso acetal 3l as colorless oil. Rf = 0.50 (PE/EtOAc, 3:1, UV, anisaldehyde). 1H NMR (300 MHz, 320 K, COSY, CDCl3): δ 1.42 (s, 3H, Me), 1.63 (br s, 3H, Me), 1.98 (dd, J = 12.8, 10.3 Hz, 1H, CH2a(6)), 2.20–2.40 (br m, 1H, CH2b(6)), 4.29 (d, J = 9.3 Hz, 1H, CH(4a)), 4.52–4.85 (m, 4H, OCH2CCl3 and CH2(2)), 5.80–5.91 (br m, 1H, CH(5)), 7.11–7.22 (m, 1H, CH(3)), 7.46 (app t, J = 7.5 Hz, 2H, CHPh), 7.58 (app tt, J = 7.4, 2.1 Hz, 1H, CHPh), 8.05 (app d, J = 7.2 Hz, 2H, CHPh). 13C NMR (75 MHz, 320 K, DEPT, HSQC, HMBC, CDCl3): δ 29.3 (Me), 30.4 (br, Me), 40.4 (br, CH2(6)), 62.0 (CH(4a)), 67.9 (CH(5)), 74.1 (OCH2CCl3), 81.2 (CH(7)), 94.7 (CCl3), 128.4 (CHPh), 129.8 (CHPh and CPh), 133.2 (CHPh), 140.0 (=CH(3)), 162.9 (C=O), 165.8 (C=O). CH2(2) and C(4) were not observed due to broadening/low intensity. 1H NMR (300 MHz, 343 K, COSY, DMSO-d6): δ 1.35 (s, 3H, Meeq), 1.50 (br s, 3H, Meax), 1.95 (dd, J = 13.0, 9.7 Hz, 1H, CH2ax(6)), 2.15–2.27 (br m, 1H, CH2eq(6)), 4.15 (d, J = 8.3 Hz, 1H, CH(4a)), 4.62–4.65 (m, 2H, CH2(2)), 4.77 (s, 2H, OCH2CCl3), 5.77 (app td, J = 9.1, 4.1 Hz, 1H, CH(5)), 7.20 (br s, 1H, CH(3)), 7.53 (app t, J = 7.6 Hz, 2H, CHPh), 7.67 (app tt, J = 7.4, 1.3 Hz, 1H, CHPh), 7.95 (app d, J = 7.1 Hz, 2H, CHPh). Characteristic NOESY interactions: Meax/CH(5); CH2eq(6)/CH(5); CH2ax(6)/CH(4a); Meeq/CH2ax(6). 13C NMR (75 MHz, 343 K, DEPT, HSQC, HMBC, DMSO-d6): δ 29.6 (2×Me), 39.8 (CH2(6)), 61.9 (CH(4a)), 67.3 (br, CH2(2)), 68.3 (CH(5)), 74.0 (OCH2CCl3), 80.6 (br, CH(7)), 95.5 (CCl3), 126.8 (br, C(4)), 129.1 (CHPh), 129.7 (CHPh), 130.0 (CPh), 133.9 (CHPh), 141.4 (=CH(3)), 163.1 (C=O), 165.5 (C=O). HRMS (ESI): m/z calcd. for C19H21Cl3NO6+ [M + H]+: 464.0429, found: 464.0437.
2,2,2-Trichloroethyl (4aR*,5R*)-7,7-dimethyl-5-phenethyl-4a,5,6,7-tetrahydro-2H-[1,2]oxazino[2,3-b][1,2]oxazine-4-carboxylate 3m. Nitroso acetal 3m was obtained from nitronate 1h (31 mg, 0.13 mmol) and vinyl diazoacetate 2a (63 mg, 0.26 mmol) according to GP-1. Column chromatography (eluent: PE/EtOAc, 30:1, then 20:1) afforded 43 mg (72%) of the target nitroso acetal 3m as colorless oil, which solidified upon storage in a fridge (ca. 4 °C). Rf = 0.47 (PE/EtOAc, 3:1, UV, anisaldehyde). m.p. = 136–138 °C (EtOAc). 1H NMR (300 MHz, COSY, CDCl3): δ 1.32 (s, 3H, Me(7)), 1.53 (s, 3H, Me(7)), 1.53–1.59 (m, 2H, PhCH2CH2a and CH2ax(6)), 1.63–1.75 (m, 1H, PhCH2CH2b), 2.03 (dd, J = 13.3, 2.7 Hz, 1H, CH2eq(6)), 2.36–2.52 (m, 2H, CH(5) and PhCH2a), 2.79 (ddd, J = 13.8, 10.6, 5.3 Hz, 1H, PhCH2b), 3.87 (app d, J = 10.2 Hz, 1H, CH(4a)), 4.49 (dd, J = 18.4, 3.1 Hz, 1H, CH2a(2)), 4.71–4.81 (m, 2H, OCH2aCCl3 and CH2b(2)), 4.87 (d, J = 12.0 Hz, 1H, OCH2bCCl3), 7.10–7.31 (m, 6H, CH(3) and CHPh). 13C NMR (75 MHz, DEPT, HSQC, CDCl3): δ 28.9 (Me(7)), 30.4 (CH(5)), 30.8 (Me(7)), 32.6 (PhCH2), 33.6 (PhCH2CH2), 40.5 (CH2(6)), 64.5 (CH(4a)), 68.4 (br, CH2(2)), 74.1 (OCH2CCl3), 78.8 (C(7)), 94.8 (CCl3), 125.9 (CHPh), 128.3 (CHPh), 128.4 (CHPh), 129.8 (C(4)), 140.5 (=CH(3)), 141.8 (CAr), 163.6 (C=O). HRMS (ESI): m/z calcd. for C20H25Cl3NO4+ [M + H]+: 448.0844, found: 448.0847.
2,2,2-Trichloroethyl (4aR*,5S*,7R*)-5,7-diphenyl-4a,5,6,7-tetrahydro-2H-[1,2]oxazino[2,3-b][1,2]oxazine-4-carboxylate 3n. Nitroso acetal 3n was obtained from nitronate 1i (30 mg, 0.12 mmol) and vinyl diazoacetate 2a (58 mg, 0.24 mmol) according to GP-1. Column chromatography (eluent: PE/EtOAc, 10:1) afforded 49 mg (88%) of the target nitroso acetal 3n as colorless oil. Analytically pure material was obtained after crystallization from PE/CH2Cl2, 3:1 as white powder. Rf = 0.51 (PE/EtOAc, 3:1, UV, anisaldehyde). m.p. = 128–129 °C (PE/CH2Cl2, 3:1). 1H NMR (300 MHz, COSY, CDCl3): δ 2.22 (app dt, J = 13.5, 3.2 Hz, 1H, CH2eq(6)), 2.48 (app q, J = 12.6 Hz, 1H, CH2ax(6)), 3.65 (ddd, J = 12.5, 10.7, 3.4 Hz, 1H, CH(5)), 3.97 (d, J = 11.9 Hz, 1H, OCH2aCCl3), 4.33 (dd, J = 10.7, 1.9 Hz, 1H, CH(4a)), 4.42 (d, J = 11.9 Hz, 1H, OCH2bCCl3), 4.70 (dd, J = 18.1, 3.4 Hz, 1H, CH2a(2)), 4.96 (dt, J = 18.1, 1.8 Hz, 1H, CH2b(2)), 5.62 (dd, J = 11.8, 2.5 Hz, 1H, CH(7)), 7.02 (dd, J = 3.4, 1.8 Hz, 1H, CH(3)), 7.23–7.51 (m, 10H, 2 × Ph). Characteristic NOESY interactions: CH(7)–CH2eq(6), CH(7)–CH(5), CH(4a)–CH2ax(6), CH(5)–CH2eq(6). 13C NMR (75 MHz, DEPT, HSQC, CDCl3): δ 36.7 (CH2(6)), 41.6 (CH(5)), 64.6 (CH(4a)), 68.8 (CH2(2)), 73.7 (OCH2CCl3), 74.1 (CH(7)), 94.7 (CCl3), 126.8 (CHPh), 127.4 (CHPh), 128.4 (CHPh), 128.48 (CHPh), 128.51 (CHPh), 128.6 (CHPh), 130.5 (C(4)), 138.7 (=CH(3)), 139.2 (CPh), 139.8 (CPh), 162.7 (C=O). HRMS (ESI): m/z calcd. for C22H21Cl3NO4+ [M + H]+: 468.0531, found: 468.0527.
2,2,2-Trichloroethyl (4aR*,5S*,7S*)-7-ethoxy-5-(4-methoxyphenyl)-4a,5,6,7-tetrahydro-2H-[1,2]oxazino[2,3-b][1,2]oxazine-4-carboxylate 3o. Nitroso acetal 3o was obtained from nitronate 1j (26 mg, 0.10 mmol) and vinyl diazoacetate 2a (51 mg, 0.21 mmol) according to GP-1. Column chromatography (eluent: PE/EtOAc, 20:1, then 10:1) afforded 34 mg (70%) of the target nitroso acetal 3o as colorless oil. Rf = 0.45 (PE/EtOAc, 3:1, UV, anisaldehyde). 1H NMR (300 MHz, COSY, CDCl3): δ 1.27 (t, J = 7.1 Hz, 3H, OCH2CH3), 2.11 (ddd, J = 13.4, 3.3, 1.2 Hz, 1H, CH2eq(6)), 2.45 (td, J = 13.4, 4.1 Hz, 1H, CH2ax(6)), 3.55–3.75 (m, 2H, CH(5) and OCH2aCH3), 3.79 (s, 3H, OMe), 4.05–4.19 (m, 3H, OCH2aCCl3, CH(4a), and OCH2bCH3), 4.37 (d, J = 11.9 Hz, 1H, OCH2bCCl3), 4.55 (dd, J = 18.1, 3.5 Hz, 1H, CH2a(2)), 4.79 (app dt, J = 18.1, 1.8 Hz, 1H, CH2b(2)), 5.13 (app d, J = 3.2 Hz, 1H, O–CH(7)–O), 6.82 (d, J = 8.7 Hz, 2H, CHAr), 6.96 (dd, J = 3.0, 1.6 Hz, 1H, CH(3)), 7.18 (d, J = 8.7 Hz, 2H, CHAr). Characteristic NOESY interactions: CH(7)/CH2ax(6); [CH(5) and OCH2aCH3]/CH2eq(6); CH2ax(6)/[OCH2aCCl3, CH(4a), and OCH2aCH3]; CH2ax(6)/CHAr. 13C NMR (75 MHz, DEPT, HSQC, HMBC, CDCl3): δ 14.9 (OCH2CH3), 33.7 (CH(5)), 34.0 (CH2(6)), 55.3 (OMe), 63.9 (OCH2CH3), 65.3 (CH(4a)), 68.7 (CH2(2)), 73.8 (OCH2CCl3), 94.7 (CCl3), 100.2 (O–CH(7)–O), 113.7 (CHAr), 129.5 (CHAr), 130.3 (C(4)), 131.7 (CAr), 138.8 (=CH(3)), 158.7 (CAr–OMe), 162.7 (C=O). HRMS (ESI): m/z calcd. for C19H23Cl3NO6+ [M + H]+: 466.0585, found: 466.0599.
2,2,2-Trichloroethyl (4aR*,5S*,5aR*,9aR*)-5-(4-chlorophenyl)-4a,5,5a,6,7,8,9,9a-octahydro-2H-benzo[e][1,2]oxazino[2,3-b][1,2]oxazine-4-carboxylate 3p. Nitroso acetal 3p was obtained from nitronate 1k (30 mg, 0.11 mmol) and vinyl diazoacetate 2a (56 mg, 0.23 mmol) according to GP-1 (reaction time = 2 days). Column chromatography (eluent: PE/EtOAc, 40:1) afforded 21 mg of crude nitroso acetal as oil, which was crystallized from PE/CH2Cl2, 3:1 to give 8 mg of target nitroso acetal 3p as white powder. Column chromatography (eluent: PE/CH2Cl2, 1:1.5, then 1:2) of filtrate afforded 10 mg of target nitroso acetal 3p as colorless oil. Total yield: 18 mg (33%). Rf = 0.47 (PE/EtOAc, 3:1, UV, anisaldehyde). m.p. = 153–155 °C (PE/CH2Cl2, 3:1). 1H NMR (300 MHz, COSY, CDCl3, 320 K): δ 1.15–1.38 (m, 4H, CH2abcd), 1.57–1.65 (m, 1H, CH2e), 1.70–1.79 (m, 1H, CH2f), 1.91–2.01 (m, 1H, CH2a(9)), 2.31–2.55 (m, 2H, CH(5a) and CH2b(9)), 3.60 (app t, J = 9.4 Hz, 1H, CH(5)–Ar), 4.22–4.45 (m, 4H, CH(4a)–N, CH(5a)–O, and OCH2CCl3), 4.59–4.74 (m, 2H, CH2(2)–O), 6.99 (br s, 1H, CH(3)=), 7.28 (br s, 4H, CHAr). 13C NMR (75 MHz, HSQC, CDCl3, 320 K): δ 21.5 (br, CH2), 24.7 (br, CH2), 27.2 (CH2), 31.3 (CH2(9)), 38.5 (CH(5a)), 40.0 (br, CH(5)), 64.5 (br, CH(4a)), 67.1 (br, CH2(2)), 73.9 (OCH2CCl3), 78.8 (br, CH(9a)–O), 94.7 (CCl3), 128.5 (CHAr), 129.8 (br, C(4)), 130.5 (br, CHAr), 132.8 (CAr), 138.4 (=CH(3)), 162.9 (C=O). CAr could not be unambiguously identified due to broadening/low intensity/possible overlapping. HRMS (ESI): m/z calcd. for C20H22Cl4NO4+ [M + H]+: 480.0297, found: 480.0286. The crystallographic information for compound 3p was deposited in the Cambridge Crystallographic Data Centre (CCDC 2242309).
3.3.2. General Procedure for the Synthesis of Pyrrolooxazines 4 (GP-2)
To the 0.3 M solution of nitronate 1 (1 equiv.) in THF, Rh2(Oct)4 (0.02 equiv.) was added under an argon atmosphere at r.t. and the mixture was stirred for 5 min. Then THF solution of vinyl diazoacetate 2 (2 equiv. rel. to nitronate 1, C = 0.5M) was added dropwise (appr. 5–10 min) using a syringe with stirring. The resulting solution was stirred overnight and evaporated. The residue was dissolved in freshly distilled CH2Cl2 (5 mL/1 mmol of starting nitronate 1). Then 2,2,2-trichloroethanol (10 equiv.) and DBU (2 equiv.) were consequently added under an argon atmosphere. The resulting solution was maintained for 16–24 h (TLC monitoring) and transferred into EtOAc (20 mL)/H2O (10 mL). The organic layer was washed with NaHSO4 (0.5 M in H2O, 10 mL). Then aqueous layer was washed with EtOAc (10 mL) and the combined layer was washed with brine (15 mL), dried (Na2SO4), and evaporated. The residue was preadsorbed onto Celite® and subjected to column chromatography on silica gel (eluent: PE/EtOAc or PE/CH2Cl2) to give target pyrrolooxazines 4. NB: Even small residual amounts of 2,2,2-trichloroethanol can affect the Rf values of target pyrroles 4 and chromatography rate.
2,2,2-Trichloroethyl 4-(4-methoxyphenyl)-2,2-dimethyl-3,4-dihydro-2H-pyrrolo[1,2-b][1,2]oxazine-5-carboxylate 4a.
1. To a stirring solution of nitroso acetal 3a (46 mg, 0.10 mmol) in CH2Cl2 (0.50 mL), 2,2,2-trichloroethanol (96 μL, 149 mg, 1.00 mmol) and DBU (30 μ, 30 mg, 0.20 mmol) were consequently added under an argon atmosphere. The resulting solution was stirred for 24 h and transferred into EtOAc (20 mL)/H2O (10 mL). The organic layer was washed with NaHSO4 (0.5 M in H2O, 10 mL). Then aqueous layer was washed with EtOAc (10 mL) and the combined layer was washed with brine (15 mL), dried (Na2SO4), and evaporated. The residue was preadsorbed onto Celite® and subjected to column chromatography on silica gel (eluent: PE/EtOAc, 30:1) to give 32 mg (74%) of the target pyrrolooxazine 4a as colorless oil, which solidified upon storage in a fridge (ca. 4 °C).
2. (One-pot procedure) Pyrrolooxazine 4a was obtained from nitronate 1a (26 mg, 0.11 mmol) and vinyl diazoacetate 2a (54 mg, 0.22 mmol) according to GP-2. Column chromatography (eluent: PE/EtOAc, 50:1, then 40:1) afforded 29 mg (62%) of the target pyrrolooxazine 4a as colorless oil. Rf = 0.30 (PE/EtOAc, 5:1, UV, anisaldehyde). m.p. = 93–95 °C (PE/CH2Cl2, 3:1). 1H NMR (300 MHz, CDCl3): δ 1.29 (s, 3H, Me), 1.41 (s, 3H, Me), 2.07 (dd, J = 14.1, 9.2 Hz, 1H, CH2a(3)), 2.32 (dd, J = 14.1, 8.6 Hz, 1H, CH2b(3)), 3.79 (s, 3H, OMe), 4.50 (d, J = 12.0 Hz, 1H, CO2CH2aCCl3), 4.61 (app t, J = 8.8 Hz, 1H, CH(4)), 4.79 (d, J = 12.0 Hz, 1H, CO2CH2aCCl3), 6.65 (d, J = 3.2 Hz, 1H, CH(6 or 7)), 6.72 (d, J = 3.2 Hz, 1H, CH(7 or 6)), 6.82 (d, J = 8.7 Hz, 2H, CHAr), 7.10 (d, J = 8.7 Hz, 2H, CHAr). 13C NMR (75 MHz, DEPT, CDCl3): δ 22.5 (Me), 27.1 (Me), 35.6 (CH(4)), 43.3 (CH2(3)), 55.2 (OMe), 73.1 (OCH2CCl3), 82.2 (C(2)), 95.8 (CCl3), 105.7 (C(5)), 106.5 (CH(6 or 7)), 113.8 (CHAr), 114.3 (CH(7 or 6)), 128.1 (CHAr), 129.9 (C(4a)), 136.5 (CAr), 158.0 (CAr–OMe), 161.9 (C=O). HRMS (ESI): m/z calcd. for C19H21Cl3NO4+ [M + H]+: 432.0531, found: 432.0520.
2,2,2-Trichloroethyl 4-(4-chloroxyphenyl)-2,2-dimethyl-3,4-dihydro-2H-pyrrolo[1,2-b][1,2]oxazine-5-carboxylate 4g. Pyrrolooxazine 4g was obtained from nitronate 1b (27 mg, 0.11 mmol) and vinyl diazoacetate 2a (54 mg, 0.22 mmol) according to GP-2. Column chromatography (eluent: PE/EtOAc, 40:1) afforded 33 mg (69%) of the target pyrrolooxazine 4g as white powder. Rf = 0.42 (PE/EtOAc, 5:1, UV, anisaldehyde). m.p. = 111–113 °C (PE/CH2Cl2, 3:1). 1H NMR (300 MHz, CDCl3): δ 1.29 (s, 3H, Me), 1.42 (s, 3H, Me), 2.07 (dd, J = 14.1, 9.4 Hz, 1H, CH2a(3)), 2.33 (dd, J = 14.1, 8.7 Hz, 1H, CH2b(3)), 4.53 (d, J = 12.0 Hz, 1H, CO2CH2aCCl3), 4.63 (app t, J = 9.0 Hz, 1H, CH(4)), 4.77 (d, J = 12.0 Hz, 1H, CO2CH2bCCl3), 6.66 (d, J = 3.2 Hz, 1H, CH(6 or 7)), 6.74 (d, J = 3.2 Hz, 1H, CH(7 or 6)), 7.12 (d, J = 8.5 Hz, 2H, CHAr), 7.26 (d, J = 8.5 Hz, 2H, CHAr). 13C NMR (75 MHz, DEPT, CDCl3): δ 22.4 (Me), 27.1 (Me), 35.9 (CH(4)), 43.0 (CH2(3)), 73.1 (OCH2CCl3), 82.1 (C(2)), 95.7 (CCl3), 105.9 (C(5)), 106.6 (CH(6 or 7)), 114.6 (CH(7 or 6)), 128.5 (CHAr), 128.6 (CHAr), 128.8 and 132.0 (C(4a) and CAr), 142.8 (CAr), 161.8 (C=O). HRMS (ESI): m/z calcd. for C18H17Cl4NNaO3+ [M + Na]+: 457.9855, found: 457.9862.
2,2,2-Trichloroethyl 4-(benzoyloxy)-2,2-dimethyl-3,4-dihydro-2H-pyrrolo[1,2-b][1,2]oxazine-5-carboxylate 4l. Pyrrolooxazine 4l was obtained from nitronate 1g (27 mg, 0.11 mmol) and vinyl diazoacetate 2a (54 mg, 0.22 mmol) according to GP-2. Column chromatography (eluent: PE/CH2Cl2, 1:1, then 1:1.5) afforded 12 mg (25%) of the target pyrrolooxazine 4l as orange oil. Rf = 0.19 (PE/CH2Cl2, 1:2, UV, anisaldehyde). 1H NMR (300 MHz, CDCl3): δ 1.45 (s, 3H, Me), 1.51 (s, 3H, Me), 2.32 (dd, J = 15.3, 3.0 Hz, 1H, CH2a(3)), 2.50 (dd, J = 15.3, 5.9 Hz, 1H, CH2b(3)), 4.61 (d, J = 12.0 Hz, 1H, CO2CH2aCCl3), 4.94 (d, J = 12.0 Hz, 1H, CO2CH2bCCl3), 6.64 (dd, J = 5.9, 3.0 Hz, 1H, CH(4)), 6.72 (d, J = 3.2 Hz, 1H, CH(6 or 7)), 6.79 (d, J = 3.2 Hz, 1H, CH(7 or 6)), 7.42 (app t, J = 7.6 Hz, 2H, CHBz), 7.56 (d, J = 7.4, 1.3 Hz, 1H, CHBz), 7.98–8.02 (m, 2H, CHBz). 13C NMR (75 MHz, DEPT, CDCl3): δ 23.9 (Me), 26.5 (Me), 38.2 (CH2(3)), 61.3 (CH(4)), 73.4 (OCH2CCl3), 81.1 (C(2)), 95.5 (CCl3), 107.2 (CH(6 or 7)), 107.9 (C(5)), 115.2 (CH(7 or 6)), 123.5 (CAr), 128.4 (CHAr), 129.7 (CHAr), 130.0 (C(4a)), 133.1 (CHAr), 161.7 (C=O), 165.5 (C=O). HRMS (ESI): m/z calcd. for C19H18Cl3NNaO5+ [M + Na]+: 468.0143, found: 468.0131.
2,2,2-Trichloroethyl 2,2-dimethyl-4-phenethyl-3,4-dihydro-2H-pyrrolo[1,2-b][1,2]oxazine-5-carboxylate 4m. Pyrrolooxazine 4m was obtained from nitronate 1h (26 mg, 0.11 mmol) and vinyl diazoacetate 2a (54 mg, 0.22 mmol) according to GP-2. Column chromatography (eluent: PE/EtOAc, 70:1) afforded 27 mg (57%) of the target pyrrolooxazine 4m as colorless oil. Rf = 0.48 (PE/EtOAc, 5:1, UV, anisaldehyde). 1H NMR (300 MHz, COSY, CDCl3): δ 1.15 (s, 3H, Me), 1.49 (s, 3H, Me), 1.91–2.04 (m, 2H, PhCH2CH2a and CH2a(3)), 2.13 (dd, J = 13.9, 8.7 Hz, 1H, CH2b(3)), 2.39–2.50 (m, 1H, PhCH2CH2b), 2.65–2.75 (m, 2H, PhCH2), 3.58 (app qd, J = 8.7, 4.0 Hz, 1H, CH(4)), 4.87–4.95 (m, 2H, OCH2CCl3), 6.61–6.64 (m, 2H, CH(6) and CH(7)), 7.17–7.23 (m, 2H, CHPh), 7.27–7.32 (m, 3H, CHPh). 13C NMR (75 MHz, DEPT, HSQC, CDCl3): δ 23.0 (Me), 27.6 (Me), 29.6 (CH(4)), 32.9 (PhCH2), 36.9 (PhCH2CH2), 39.0 (CH2(3)), 73.6 (OCH2CCl3), 82.7 (C(2)), 95.8 (CCl3), 104.9 (C(5)), 106.5 (CH(6 or 7)), 114.4 (CH(7 or 6)), 125.9 and 128.4 (all CHPh), 132.2 (C(4a)), 141.9 (CPh), 162.6 (C=O). HRMS (ESI): m/z calcd. for C20H23Cl3NO3+ [M + H]+: 430.0738, found: 430.0740.
2,2,2-Trichloroethyl (2R*,4S*)-2,4-diphenyl-3,4-dihydro-2H-pyrrolo[1,2-b][1,2]oxazine-5-carboxylate 4n. Pyrrolooxazine 4n was obtained from nitronate 1i (28 mg, 0.11 mmol) and vinyl diazoacetate 2a (54 mg, 0.22 mmol) according to GP-2. Column chromatography (eluent: PE/EtOAc, 70:1, then 50:1) afforded 25 mg (50%) of the target pyrrolooxazine 4n as colorless oil. Rf = 0.38 (PE/EtOAc, 5:1, UV, anisaldehyde). 1H NMR (300 MHz, CDCl3): δ 2.41 (ddd, J = 14.4, 11.8, 9.8 Hz, 1H, CH2a(3)), 2.76 (ddd, J = 14.4, 8.9, 1.9 Hz, 1H, CH2b(3)), 4.36 (d, J = 12.0 Hz, 1H, CO2CH2aCCl3), 4.85 (d, J = 12.0 Hz, 1H, CO2CH2bCCl3), 4.90 (app t, J = 9.2 Hz, 1H, CH(4)), 5.31 (dd, J = 11.8, 1.9 Hz, 1H, CH(2)), 6.69 (d, J = 3.3 Hz, 1H, CH(6 or 7)), 6.85 (d, J = 3.3 Hz, 1H, CH(7 or 6)), 7.20–7.34 (m, 5H, Ph), 7.40–7.46 (m, 5H, Ph). 13C NMR (75 MHz, DEPT, CDCl3): δ 38.9 (CH(4)), 39.7 (CH2(3)), 72.8 (OCH2CCl3), 84.2 (CH(2)), 95.8 (CCl3), 106.2 (C(5)), 106.7 (CH(6 or 7)), 114.0 (CH(7 or 6)), 126.5 (CHPh), 126.86 (CHPh), 126.93 (CHPh), 128.6 (CHPh), 128.9 (CHPh), 129.4 (CHPh), 129.5 (C(4a)), 136.7 (CPh), 144.5 (CPh), 161.8 (C=O). HRMS (ESI): m/z calcd. for C22H18Cl3NNaO3+ [M + Na]+: 472.0244, found: 472.0234.
2,2,2-Trichloroethyl (2S*,4S*)-2-ethoxy-4-(4-methoxyphenyl)-3,4-dihydro-2H-pyrrolo[1,2-b][1,2]oxazine-5-carboxylate 4o. Pyrrolooxazine 4o was obtained from nitronate 1j (18 mg, 0.07 mmol) and vinyl diazoacetate 2a (34 mg, 0.14 mmol) according to GP-2. Column chromatography (eluent: PE/EtOAc, 50:1, then 30:1) afforded 15 mg (47%) of the target pyrrolooxazine 4o as orange oil. Rf = 0.31 (PE/EtOAc, 5:1, UV, anisaldehyde). 1H NMR (300 MHz, CDCl3): δ 1.22 (t, J = 7.1 Hz, 3H, OCH2CH3), 2.30–2.47 (m, 2H, CH2(3)), 3.67 (dq, J = 9.7, 7.1 Hz, 1H, OCH2aCH3), 3.79 (s, 3H, OMe), 3.91 (dq, J = 9.7, 7.1 Hz, 1H, OCH2bCH3), 4.54 (d, J = 12.0 Hz, 1H, CO2CH2aCCl3), 4.60 (app t, overlapped, J = 7.2 Hz, 1H, CH(4)), 4.81 (d, overlapped, J = 12.0 Hz, 1H, CO2CH2bCCl3), 5.35 (app t, J = 4.0 Hz, 1H, O–CH(7)–O), 6.66 (d, J = 3.3 Hz, 1H, CH(6 or 7)), 6.73 (d, J = 3.3 Hz, 1H, CH(7 or 6)), 6.83 (d, J = 8.7 Hz, 2H, CHAr), 7.09 (d, J = 8.6 Hz, 2H, CHAr). 13C NMR (75 MHz, DEPT, CDCl3): δ 15.0 (OCH2CH3), 34.2 (CH(4)), 36.2 (CH2(3)), 55.2 (OMe), 65.2 (OCH2CH3), 73.2 (OCH2CCl3), 95.7 (CCl3), 102.1 (O–CH(7)–O), 105.6 (C(5)), 106.6 (CH(6 or 7)), 113.9 (CHAr), 114.1 (CH(7 or 6)), 128.2 (CHAr), 130.8 (C(4a)), 135.6 (CAr), 158.1 (CAr–OMe), 162.0 (C=O). HRMS (ESI): m/z calcd. for C19H21Cl3NO5+ [M + H]+: 470.0299, found: 470.0295.
3.3.3. Preparation of Pyrrolooxazines 4b, 5
Methyl 4-(4-methoxyphenyl)-2,2-dimethyl-3,4-dihydro-2H-pyrrolo[1,2-b][1,2]oxazine-5-carboxylate 4b. To a stirring solution of nitroso acetal 3a (31 mg, 0.07 mmol) in a mixture of MeOH (0.34 mL) and MeCN (0.17 mL), DBU (20 μL, 20 mg, 0.13 mmol) was added dropwise under an argon atmosphere at r.t. The resulting solution was left overnight and transferred into EtOAc (20 mL)/H2O (10 mL). The organic layer was washed with NaHSO4 (0.5 M in H2O, 10 mL). Then aqueous layer was washed with EtOAc (10 mL) and the combined layer was washed with brine (15 mL), dried (Na2SO4), and evaporated. The residue was preadsorbed onto Celite® and subjected to column chromatography on silica gel (eluent: PE/EtOAc 20:1, then 12:1) to give 13 mg (60%) of the target pyrrolooxazine 4b as colorless oil, which solidified upon storage in a fridge (ca. 4 °C). Rf = 0.29 (PE/EtOAc, 3:1, UV, anisaldehyde). mp = 93–95 °C (PE/CH2Cl2, 3:1). 1H NMR (300 MHz, COSY, CDCl3): δ 1.25 (s, 3H, Me), 1.41 (s, 3H, Me), 2.05 (dd, J = 14.1, 9.5 Hz, 1H, CH2a(3)), 2.28 (dd, J = 14.1, 8.7 Hz, 1H, CH2b(3)), 3.51 (s, 3H, CO2Me), 3.79 (s, 3H, OMe), 4.54 (app t, J = 9.1 Hz, 1H, CH(4)), 6.53 (d, J = 3.2 Hz, 1H, CH(6 or 7)), 6.68 (d, J = 3.2 Hz, 1H, CH(7 or 6)), 6.83 (d, J = 8.7 Hz, 2H, CHAr), 7.10 (d, J = 8.7 Hz, 2H, CHAr). 13C NMR (75 MHz, DEPT, HSQC, HMBC, CDCl3): δ 22.2 (Me), 27.2 (Me), 35.8 (CH(4)), 43.6 (CH2(3)), 50.5 (CO2Me), 55.2 (CAr–OMe), 82.0 (C(2)), 105.9 (CH(6 or 7)), 107.4 (C(5)), 113.8 (CHAr), 113.9 (CH(7 or 6)), 128.0 (CHAr), 128.7 (C(4a)), 137.2 (CAr), 157.9 (CAr–OMe), 164.5 (C=O). HRMS (ESI): m/z calcd. for C18H22NO4+ [M + H]+: 316.1543, found: 316.1550. The crystallographic information for compound 4b was deposited in the Cambridge Crystallographic Data Centre (CCDC 2242308).
4-(4-Methoxyphenyl)-2,2-dimethyl-3,4-dihydro-2H-pyrrolo[1,2-b][1,2]oxazine-5-carboxylic acid 5. To a stirring solution of nitroso acetal 3a (35 mg, 0.08 mmol) in THF (0.39 mL), t-BuONa (15 mg, 0.16 mmol) was added under an argon atmosphere at r.t. The resulting solution was stirred for 0.5 h and transferred into EtOAc (20 mL)/H2O (10 mL). The organic layer was washed with NaHSO4 (0.5 M in H2O, 10 mL). Then aqueous layer was washed with EtOAc (10 mL) and the combined layer was washed with brine (15 mL), dried (Na2SO4), and evaporated. The residue was preadsorbed onto Celite® and subjected to column chromatography on silica gel (eluent: PE/EtOAc, 20:1, then 5:1, then 3:1) to give 2 mg (6%) of pyrrolooxazine 4a and 5.5 mg (23%) of the target pyrrolooxazine acid 5 as an oil, which solidified upon storage in a fridge (ca. 4 °C) as white powder. Rf = 0.28 (PE/EtOAc, 1:1, UV, anisaldehyde). m.p. = 185–187 °C (PE/CH2Cl2, 3:1). 1H NMR (300 MHz, CDCl3): δ 1.22 (s, 3H, Me), 1.41 (s, 3H, Me), 2.09 (dd, J = 14.2, 8.7 Hz, 1H, CH2a(3)), 2.30 (dd, J = 14.2, 8.9 Hz, 1H, CH2b(3)), 3.79 (s, 3H, OMe), 4.58 (app t, J = 8.8 Hz, 1H, CH(4)), 6.54 (d, J = 3.2 Hz, 1H, CH(6 or 7)), 6.68 (d, J = 3.2 Hz, 1H, CH(7 or 6)), 6.81 (d, J = 8.7 Hz, 2H, CHAr), 7.09 (d, J = 8.7 Hz, 2H, CHAr). 13C NMR (75 MHz, DEPT, CDCl3): δ 22.5 (Me), 27.2 (Me), 35.5 (CH(4)), 43.4 (CH2(3)), 55.2 (OMe), 82.5 (C(2)), 106.4 (C(5)), 106.5 (CH(6 or 7)), 113.8 (CHAr), 114.3 (CH(7 or 6)), 128.3 (CHAr), 130.3 (C(4a)), 136.8 (CAr), 157.9 (CAr–OMe), 168.3 (C=O). HRMS (ESI): m/z calcd. for C17H20NO4+ [M + H]+: 302.1384, found: 302.1390.
4. Conclusions
In conclusion, an approach was developed for the synthesis of bicyclic nitroso acetals (4a,5,6,7-tetrahydro-2H-[1,2]oxazino[2,3-b][1,2]oxazines) via an Rh(II)-catalyzed annulation of six-membered cyclic nitronates with vinyl diazoacetates. Target products were obtained in good yields with excellent diastereoselectivity. These nitroso acetals undergo smooth ring contraction under the action of the DBU/alcohol system. Combined with a [3+3]-annulation in a one-pot protocol, it represents a quick approach to the assembly of pyrrolooxazines and complements already known approaches to this relatively rare heterocyclic core.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/molecules28073025/s1, Table S1: Preparation of starting compounds, crystal data, optimization, Tables S2 and S3: Tables for the synthesis of pyrrolooxazines 4a and 4b, Copies of NMR spectra.
Author Contributions
Conceptualization, Y.A.A. and A.A.T.; methodology, Y.A.A. and A.A.T.; validation, Y.A.A., Y.V.N. and A.A.T.; formal analysis, Y.A.A., Y.V.N. and A.A.T.; investigation, Y.A.A. and Y.V.N.; data curation, Y.A.A.; writing—original draft preparation, Y.A.A. and A.A.T.; writing—review and editing, A.A.T. and S.L.I.; supervision, A.A.T.; project administration, A.A.T.; funding acquisition, A.A.T. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Russian Science Foundation (grant 21-73-10011). X-ray diffraction data were collected using the equipment of the Center for Molecular Composition Studies of INEOS RAS with financial support from the Ministry of Science and Higher Education of the Russian Federation (Contract/agreement No. 075-03-2023-642).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in this article and in the Supplementary Materials.
Acknowledgments
The authors thank N.G. Kolotyrkina and A.O. Chizhov (N. D. Zelinsky Institute of Organic Chemistry) for registration of HRMS.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are not available from the authors.
References
- Majireck, M.M.; Bennett, J.M. 1,2-Oxazines and their benzo derivatives. In Comprehensive Heterocyclic Chemistry IV; Weinreb, S.M., Black, D.S.C., Cossy, J., Stevens, C.V., Eds.; Elsevier Ltd.: Amsterdam, The Netherlands, 2022; Volume 8, pp. 283–415. [Google Scholar] [CrossRef]
- Ghosh, P.; Mondal, S.L.; Baidya, M. Ascending of cycloaddition strategy for N–O heterocycles. Synthesis 2022, 54, 1043–1054. [Google Scholar] [CrossRef]
- Sukhorukov, A.Y.; Ioffe, S.L. Chemistry of six-membered cyclic oxime ethers. Application in the synthesis of bioactive compounds. Chem. Rev. 2011, 111, 5004–5041. [Google Scholar] [CrossRef] [PubMed]
- Utecht, G.; Jasinski, M. 3,6-Dihydro-2H-1,2-oxazines (microreview). Chem. Heterocycl. Compd. 2016, 52, 143–145. [Google Scholar] [CrossRef]
- Brulikova, L.; Harrison, A.; Miller, M.J.; Hlavac, J. Stereo- and regioselectivity of the hetero-Diels–Alder reaction of nitroso derivatives with conjugated dienes. Beilstein J. Org. Chem. 2016, 12, 1949–1980. [Google Scholar] [CrossRef] [PubMed]
- Bouche, L.; Reissig, H.-U. Synthesis of novel carbohydrate mimetics via 1,2-oxazines. Pure Appl. Chem. 2012, 84, 23–36. [Google Scholar] [CrossRef]
- Bodnar, B.S.; Miller, M.J. The nitrosocarbonyl hetero-Diels-Alder reaction as a useful tool for organic syntheses. Angew. Chem. Int. Ed. 2011, 50, 5630–5647. [Google Scholar] [CrossRef] [PubMed]
- Pfrengle, F.; Reissig, H.-U. Amino sugars and their mimetics via 1,2-oxazines. Chem. Soc. Rev. 2010, 39, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Saito, T.; Zakarian, A. Approaches to dihydrooxazine ring systems and application in the synthesis of bioactive natural products. Chem. Heterocycl. Compd. 2012, 48, 11–16. [Google Scholar] [CrossRef]
- Wehlauch, R.; Gadermann, K. Securinega alkaloids: Complex structures, potent bioactivities, and efficient total syntheses. Asian J. Org. Chem. 2017, 6, 1146–1159. [Google Scholar] [CrossRef]
- Ohta, S.; Oshimo, S.; Ohta, E.; Nehira, T.; Omura, H.; Uy, M.M.; Ishihara, Y. Asaroidoxazines from the roots of Asarum asaroides induce apoptosis in human neuroblastoma cells. J. Nat. Prod. 2020, 83, 3050–3057. [Google Scholar] [CrossRef]
- Liu, C.; Yang, C.; Zeng, Y.; Shi, J.; Li, L.; Li, W.; Jiao, R.; Tan, R.; Ge, H. Chartrenoline, a novel alkaloid isolated from a marine Streptomyces chartreusis NA02069. Chin. Chem. Lett. 2019, 30, 44–46. [Google Scholar] [CrossRef]
- Li, J.; Hu, Y.; Hao, X.; Tan, J.; Li, F.; Qiao, X.; Chen, S.; Xiao, C.; Chen, M.; Peng, Z.; et al. Raistrickindole A, an anti-HCV oxazinoindole alkaloid from Penicillium raistrickii IMB17-034. J. Nat. Prod. 2019, 82, 1391–1395. [Google Scholar] [CrossRef]
- Wu, Z.-L.; Huang, X.-J.; Xu, M.-T.; Ma, X.; Li, L.; Shi, L.; Wang, W.-J.; Jiang, R.-W.; Ye, W.-C.; Wang, Y. Flueggeacosines A–C, dimeric securinine-type alkaloid analogues with neuronal differentiation activity from Flueggea suffruticosa. Org. Lett. 2018, 20, 7703–7707. [Google Scholar] [CrossRef]
- Kim, Y.; In, Y.; Ishida, T.; Onaka, H.; Igarashi, Y. Biosynthetic origin of alchivemycin A, a new polyketide from Streptomyces and absolute configuration of alchivemycin B. Org. Lett. 2013, 15, 3514–3517. [Google Scholar] [CrossRef]
- Koyama, K.; Hirasawa, Y.; Nugroho, A.E.; Hosoya, T.; Hoe, T.C.; Chan, K.-L.; Morita, H. Alsmaphorazines A and B, novel indole alkaloids from Alstonia pneumatophora. Org. Lett. 2010, 12, 4188–4191. [Google Scholar] [CrossRef]
- Shao, C.-L.; Wang, C.-Y.; Gu, Y.-C.; Cai, J.-W.; Deng, D.-S.; She, Z.-G.; Lin, Y.-C. Revised structure of penicillazine and preparation, bioactivities of penicillazine derivatives. Lett. Org. Chem. 2009, 6, 387–391. [Google Scholar] [CrossRef]
- Garo, E.; Starks, C.M.; Jensen, P.R.; Fenical, W.; Lobkovsky, E.; Clardy, J. Trichodermamides A and B, cytotoxic modified dipeptides from the marine-derived fungus Trichoderma virens. J. Nat. Prod. 2003, 66, 423–426. [Google Scholar] [CrossRef]
- Mfuh, A.M.; Zhang, Y.; Stephens, D.E.; Vo, A.X.T.; Arman, H.D.; Larionov, O.V. Concise total synthesis of trichodermamides A, B and C enabled by an efficient construction of the 1,2-oxazadecaline core. J. Am. Chem. Soc. 2015, 137, 8050–8053. [Google Scholar] [CrossRef] [PubMed]
- Lambert, K.M.; Cox, J.B.; Liu, L.; Jackson, A.C.; Yruegas, S.; Wiberg, K.B.; Wood, J.L. Total synthesis of (±)-phyllantidine: Development and mechanistic evaluation of a novel ring expansion for installation of embedded nitrogen-oxygen bonds. Angew. Chem. Int. Ed. 2020, 59, 9757–9766. [Google Scholar] [CrossRef]
- Doens, D.; Valiente, P.A.; Mfuh, A.M.; Vo, A.X.T.; Tristan, A.; Carreno, L.; Quijada, M.; Nguyen, V.T.; Perry, G.; Larionov, O.V.; et al. Identification of inhibitors of CD36-amyloid beta binding as potential agents for Alzheimer’s disease. ACS Chem. Neurosci. 2017, 8, 1232–1241. [Google Scholar] [CrossRef] [PubMed]
- Jans, P.E.; Mfuh, A.M.; Arman, H.D.; Shaffer, C.V.; Larionov, O.V.; Mooberry, S.L. Cytotoxicity and mechanism of action of the marine-derived fungal metabolite trichodermamide B and synthetic analogues. J. Nat. Prod. 2017, 80, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.L.; Sae-Lao, P.; Toh, H.H.M.; Csokas, D.; Bates, R.W. The total synthesis of raistrickindole A. J. Org. Chem. 2022, 87, 16111–16114. [Google Scholar] [CrossRef]
- Li, T.-Z.; Liu, S.-J.; Sun, Y.-W.; Deng, S.; Tan, W.; Jiao, Y.; Zhang, Y.-C.; Shi, F. Regio- and enantioselective (3+3) cycloaddition of nitrones with 2-indolylmethanols enabled by cooperative organocatalysis. Angew. Chem. Int. Ed. 2021, 60, 2355–2363. [Google Scholar] [CrossRef] [PubMed]
- Herrera, L.; Stephens, D.E.; D’Avila, A.; George, K.G.; Arman, H.; Zhang, Y.; Perry, G.; Lleonart, R.; Larionov, O.V.; Fernandez, P.L. Insights into the structural patterns of the antileishmanial activity of bi- and tricyclic N-heterocycles. Org. Biomol. Chem. 2016, 14, 7053–7060. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Zollner, T.; Gebhardt, P.; Mollmann, U.; Miller, M.J. Preparation and biological evaluation of novel leucomycin analogs derived from nitroso Diels-Alder reactions. Org. Biomol. Chem. 2010, 8, 691–697. [Google Scholar] [CrossRef]
- Tabolin, A.A.; Sukhorukov, A.Y.; Ioffe, S.L.; Dilman, A.D. Recent advances in the synthesis and chemistry of nitronates. Synthesis 2017, 49, 3255–3268. [Google Scholar] [CrossRef]
- Denmark, S.E.; Thorarensen, A. Tandem [4+2]/[3+2] cycloadditions of nitroalkenes. Chem. Rev. 1996, 96, 137–166. [Google Scholar] [CrossRef]
- Ioffe, S.L. Nitronates. In Nitrile Oxides, Nitrones, and Nitronates in Organic Synthesis, Novel Strategies in Synthesis, 2nd ed.; Feuer, H., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008; pp. 541–604. [Google Scholar]
- Baiazitov, R.Y.; Denmark, S.E. Tandem [4+2]/[3+2] Cycloadditions. In Methods and Applications of Cycloaddition Reactions in Organic Syntheses; Nishiwaki, N., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014; pp. 471–550. [Google Scholar]
- Denmark, S.E.; Cottell, J.J. Nitronates. In The Chemistry of Heterocyclic Compounds: Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry toward Heterocycles and Natural Products; Padwa, A., Pearson, W.H., Eds.; Wiley-Interscience: New York, NY, USA, 2002; pp. 83–167. [Google Scholar]
- Denmark, S.E.; Cottell, J.J. Synthesis of (+)-1-epiaustraline. J. Org. Chem. 2001, 66, 4276–4284. [Google Scholar] [CrossRef] [PubMed]
- Denmark, S.E.; Hurd, A.R. Synthesis of (+)-casuarine. J. Org. Chem. 2000, 65, 2875–2886. [Google Scholar] [CrossRef]
- Denmark, S.E.; Martinborough, E.A. Enantioselective total syntheses of (+)-castanospermine, (+)-6-epicastanospermine, (+)-australine, and (+)-3-epiaustraline. J. Am. Chem. Soc. 1999, 121, 3046–3056. [Google Scholar] [CrossRef]
- Denmark, S.E.; Herbert, B. Synthesis of (1R,2R,3R,7R,7aR)-hexahydro-3-(hydroxymethyl)-1H-pyrrolizine-1,2,7-triol: 7-epiaustraline. J. Am. Chem. Soc. 1998, 120, 7357–7358. [Google Scholar] [CrossRef]
- Denmark, S.E.; Hurd, A.R.; Sacha, H.J. Tandem [4+2]/[3+2] cycloadditions of nitroalkenes. 13. The synthesis of (−)-detoxinine. J. Org. Chem. 1997, 62, 1668–1674. [Google Scholar] [CrossRef]
- Denmark, S.E.; Marcin, L.R. Asymmetric construction of a quaternary carbon center by tandem [4+2]/[3+2] cycloaddition of a nitroalkene. The total synthesis of (−)-mesembrine. J. Org. Chem. 1997, 62, 1675–1686. [Google Scholar] [CrossRef]
- Denmark, S.E.; Thorarensen, A. Tandem [4+2]/[3+2] cycloadditions of nitroalkenes. 11. The synthesis of (+)-crotanecine. J. Am. Chem. Soc. 1997, 119, 125–137. [Google Scholar] [CrossRef]
- Denmark, S.E.; Thorarensen, A.; Middleton, D.S. Tandem [4+2]/[3+2] cycloadditions of nitroalkenes. 9. Synthesis of (−)-rosmarinecine. J. Am. Chem. Soc. 1996, 118, 8266–8277. [Google Scholar] [CrossRef]
- Gorbacheva, E.O.; Tabolin, A.A.; Novikov, R.A.; Khomutova, Y.A.; Nelyubina, Y.V.; Tomilov, Y.V.; Ioffe, S.L. Six-membered cyclic nitronates as 1,3-dipoles in formal [3+3]-cycloaddition with donor-acceptor cyclopropanes. Synthesis of new type of bicyclic nitrosoacetals. Org. Lett. 2013, 15, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Tabolin, A.A.; Gorbacheva, E.O.; Novikov, R.A.; Khoroshutina, Y.A.; Nelyubina, Y.V.; Ioffe, S.L. Synthesis and chemical transformations of six/six-membered bicyclic nitroso acetals. Russ. Chem. Bull. 2016, 65, 2243–2259. [Google Scholar] [CrossRef]
- Cheng, Q.-Q.; Yu, Y.; Yedoyan, J.; Doyle, M.P. Vinyldiazo reagents and metal catalysts: A versatile toolkit for heterocycle and carbocycle construction. ChemCatChem 2018, 10, 488–496. [Google Scholar] [CrossRef]
- Xu, X.-F.; Doyle, M.P. Divergent pathways of β,γ-unsaturated α-diazocarbonyl compounds catalyzed by dirhodium and Lewis acids catalysts separately or in combination. Chin. Chem. Lett. 2015, 26, 227–232. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, Z.; Sun, J. Asymmetric [3+1]-cycloaddition reaction via diazo discrimination. Org. Lett. 2021, 23, 7613–7617. [Google Scholar] [CrossRef] [PubMed]
- Barluenga, J.; Riesgo, L.; Lopez, L.A.; Rubio, E.; Tomas, M. Discrimination of diazo compounds toward carbenoids: Copper(I)-catalyzed synthesis of substituted cyclobutenes. Angew. Chem. Int. Ed. 2009, 48, 7569–7572. [Google Scholar] [CrossRef] [PubMed]
- Dong, K.; Xu, X.; Doyle, M.P. Copper(I)-catalyzed highly enantioselective [3+3]-cycloaddition of γ-alkyl enoldiazoacetates with nitrones. Org. Chem. Front. 2020, 7, 1653–1657. [Google Scholar] [CrossRef]
- Barluenga, J.; Lonzi, G.; Riesgo, L.; Lopez, L.A.; Tomas, M. Pyridine activation via copper(I)-catalyzed annulation toward indolizines. J. Am. Chem. Soc. 2010, 132, 13200–13202. [Google Scholar] [CrossRef]
- Thompson, J.L.; Davies, H.M.L. Enhancement of cyclopropanation chemistry in the silver-catalyzed reactions of aryldiazoacetates. J. Am. Chem. Soc. 2007, 129, 6090–6091. [Google Scholar] [CrossRef]
- Ueda, J.; Harada, S.; Nakayama, H.; Nemoto, T. Silver-catalyzed regioselective hydroamination of alkenyl diazoacetates to synthesize γ-amino acid equivalents. Org. Biomol. Chem. 2018, 16, 4675–4682. [Google Scholar] [CrossRef]
- Xu, G.; Liu, K.; Dai, Z.; Sun, J. Gold/silver-catalyzed controllable regioselective vinylcarbene insertion into O–H bonds. Org. Biomol. Chem. 2017, 15, 2345–2348. [Google Scholar] [CrossRef] [PubMed]
- Pagar, V.V.; Liu, R.-S. Gold-catalyzed α-furanylations of quinoline N-oxides with alkenyldiazo carbonyl species. Org. Biomol. Chem. 2015, 13, 6166–6169. [Google Scholar] [CrossRef]
- Zhou, P.-X.; Zhou, Z.-Z.; Chen, Z.-S.; Ye, Y.-Y.; Zhao, L.-B.; Yang, Y.-F.; Xia, X.-F.; Luo, J.-Y.; Liang, Y.-M. Palladium-catalyzed insertion of α-diazocarbonyl compounds for the synthesis of cyclic amino esters. Chem. Commun. 2013, 49, 561–563. [Google Scholar] [CrossRef] [PubMed]
- Guptill, D.M.; Davies, H.M.L. 2,2,2-Trichloroethyl aryldiazoacetates as robust reagents for the enantioselective C–H functionalization of methyl ethers. J. Am. Chem. Soc. 2014, 136, 17718–17721. [Google Scholar] [CrossRef]
- Negretti, S.; Cohen, C.M.; Chang, J.J.; Guptill, D.M.; Davies, H.M.L. Enantioselective dirhodium(II)-catalyzed cyclopropanations with trimethylsilylethyl and trichloroethyl aryldiazoacetates. Tetrahedron 2015, 71, 7415–7420. [Google Scholar] [CrossRef]
- Shved, A.S.; Tabolin, A.A.; Novikov, R.A.; Nelyubina, Y.V.; Timofeev, V.P.; Ioffe, S.L. Six-membered cyclic nitroso acetals: Synthesis and studies of the nitrogen inversion process of N-silyloxy-3,6-dihydro-2H-1,2-oxazines. Eur. J. Org. Chem. 2016, 2016, 5569–5578. [Google Scholar] [CrossRef]
- Ueda, Y.; Roberge, G.; Vinet, V. A simple method of preparing trimethylsilyl- and tert-butyldimethylsilyl-enol ethers of α-diazoacetoacetates and their use in the synthesis of a chiral precursor to thienamycin analogs. Can. J. Chem. 1984, 62, 2936–2940. [Google Scholar] [CrossRef]
- Davies, H.M.L.; Hu, B.; Saikali, E.; Bruzinski, P.R. Carbenoid versus vinylogous reactivity in rhodium(II)-stabilized vinylcarbenoids. J. Org. Chem. 1994, 59, 4535–4541. [Google Scholar] [CrossRef]
- Crocetti, L.; Floresta, G.; Cilibrizzi, A.; Giovannoni, M.P. An overview of PDE4 inhibitors in clinical trials: 2010 to early 2022. Molecules 2022, 27, 4964. [Google Scholar] [CrossRef] [PubMed]
- Dyke, H.J.; Montana, J.G. Update on the therapeutic potential of PDE4 inhibitors. Expert Opin. Investig. Drugs 2002, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Nelyubina, Y.V.; Lyssenko, K.A. Probing stereoelectronic interactions in an O–N–O unit by the atomic energies: Experimental and theoretical electron density study. J. Phys. Chem. A 2013, 117, 3084–3092. [Google Scholar] [CrossRef]
- Tishkov, A.A.; Lesiv, A.V.; Khomutova, Y.A.; Strelenko, Y.A.; Nesterov, I.D.; Antipin, M.Y.; Ioffe, S.L.; Denmark, S.E. 2-Silyloxy-1,2-oxazines, a new type of acetals of conjugated nitroso alkenes. J. Org. Chem. 2003, 68, 9477–9480. [Google Scholar] [CrossRef]
- Zheng, H.; Faghihi, I.; Doyle, M.P. Copper(I)-catalyzed highly enantioselective [3+3]-cycloaddition of β-aryl/alkyl vinyl diazoacetates with nitrones. Helvetica Chim. Acta 2021, 104, e2100081. [Google Scholar] [CrossRef]
- Wang, X.; Abrahams, Q.M.; Zavalij, P.Y.; Doyle, M.P. Highly regio- and stereoselective dirhodium vinylcarbene induced nitrone cycloaddition with subsequent cascade carbenoid aromatic cycloaddition/N–O cleavage and rearrangement. Angew. Chem. Int. Ed. 2012, 51, 5907–5910. [Google Scholar] [CrossRef]
- Qin, C.; Davies, H.M.L. Rh2(R-TPCP)4-catalyzed enantioselective [3+2]-cycloaddition between nitrones and vinyldiazoacetates. J. Am. Chem. Soc. 2013, 135, 14516–14519. [Google Scholar] [CrossRef] [PubMed]
- Pagar, V.V.; Liu, R.-S. Gold-catalyzed cycloaddition reactions of ethyl diazoacetate, nitrosoarenes, and vinyldiazo carbonyl compounds: Synthesis of isoxazolidine and benzo[b]azepine derivatives. Angew. Chem. Int. Ed. 2015, 54, 4923–4926. [Google Scholar] [CrossRef]
- Stefkova, K.; Guerzoni, M.G.; van Ingen, Y.; Richards, E.; Melen, R.L. B(C6F5)3-catalyzed diastereoselective and divergent reactions of vinyldiazo esters with nitrones: Synthesis of highly functionalized diazo compounds. Org. Lett. 2023, 25, 500–505. [Google Scholar] [CrossRef]
- Humenny, W.J.; Kyriacou, P.; Sapeta, K.; Karadeolian, A.; Kerr, M.A. Multicomponent synthesis of pyrroles from cyclopropanes: A one-pot palladium(0)-catalyzed dehydrocarbonylation/dehydration. Angew. Chem. Int. Ed. 2012, 51, 11088–11091. [Google Scholar] [CrossRef] [PubMed]
- Firl, J.; Kresze, G. Heterocyclen durch Diensynthese, III. Pyrrole, α-Pyrone und Pyridone aus Diels-Alder-Addukten von Nitrosoverbindungen. Chem. Ber. 1966, 99, 3695–3706. [Google Scholar] [CrossRef]
- Firl, J. Heterocyclen durch Diensynthese, Pyridylpyrrole aus Diels-Alder-Addukten von Nitrosoverbindungen. Chem. Ber. 1968, 101, 218–225. [Google Scholar] [CrossRef]
- Kefalas, P.; Grierson, D.S. Synthesis of pyrrolo-castanospermine analogs: A novel fluoride ion induced rearrangement of a 3-trialkylsilyl substituted dihydro-2H-1,2-oxazine to a pyrrole. Tetrahedron Lett. 1993, 34, 3555–3558. [Google Scholar] [CrossRef]
- Eberlin, L.; Carboni, B.; Whiting, A. Regioisomeric and substituent effects upon the outcome of the reaction of 1-borodienes with nitrosoarene compounds. J. Org. Chem. 2015, 80, 6574–6583. [Google Scholar] [CrossRef]
- Yasukawa, N.; Kuwata, M.; Imai, T.; Monguchi, Y.; Sajiki, H.; Sawama, Y. Copper-catalyzed pyrrole synthesis from 3,6-dihydro-1,2-oxazines. Green Chem. 2018, 20, 4409–4413. [Google Scholar] [CrossRef]
- Jasinski, M.; Watanabe, T.; Reissig, H.-U. Samarium diiodide promoted reduction of 3,6-dihydro-2H-1,2-oxazines: Competition of 1,4-amino alcohol formation and ring contraction to pyrrole derivatives. Eur. J. Org. Chem. 2013, 2013, 605–610. [Google Scholar] [CrossRef]
- Scheiner, P.; Chapman, O.L.; Lassila, J.D. The photolysis of dihydro-l,2-oxazines. J. Org. Chem. 1969, 34, 813–816. [Google Scholar] [CrossRef]
- Givens, R.S.; Choo, D.J.; Merchant, S.N.; Stitt, R.P.; Matuszewski, B. Photochemistry of 3,6-dihydro-1,2-oxazines: A versatile route to substituted pyrroles. Tetrahedron Lett. 1982, 23, 1327–1330. [Google Scholar] [CrossRef]
- Rudchenko, V.F. Synthesis, reactions, and properties of oxygen-nitrogen-oxygen systems. Chem. Rev. 1993, 93, 725–739. [Google Scholar] [CrossRef]
- Lukoyanov, A.A.; Tabolin, A.A.; Nelyubina, Y.V.; Ioffe, S.L.; Sukhorukov, A.Y. Deoxygenative arylation of 5,6-dihydro-4H-1,2-oxazine-N-oxides with arynes. J. Org. Chem. 2022, 87, 6838–6851. [Google Scholar] [CrossRef]
- Kumar, P.; Kumar, R.; Banerjee, P. Accessing dihydro-1,2-oxazine via Cloke−Wilson-type annulation of cyclopropyl carbonyls: Application toward the diastereoselective synthesis of pyrrolo [1,2-b][1,2]oxazine. J. Org. Chem. 2020, 85, 6535–6550. [Google Scholar] [CrossRef]
- Jasinski, M.; Moreno-Clavijo, E.; Reissig, H.-U. Synthesis of a series of enantiopure polyhydroxylated bicyclic N-heterocycles from an L-erythrose-derived nitrone and alkoxyallenes. Eur. J. Org. Chem. 2014, 2014, 442–454. [Google Scholar] [CrossRef]
- Mondal, S.K.; Ali, S.A.; Manna, S.K.; Mandal, A.; Senapati, B.K.; Hossain, M.; Samanta, S. Palladium-catalyzed intramolecular cyclization: Access to rare pentacyclic N-fused heterocycles. ChemistrySelect 2017, 2, 9312–9318. [Google Scholar] [CrossRef]
- Diab, S.; Noel-Duchesneau, L.; Sanselme, M.; Kondo, Y.; De Paolis, M.; Chataigner, I. High pressure elicits unexpected transformations of plain nitroaromatics with 4-(cyclohex-1-en-1-yl)morpholine. Eur. J. Org. Chem. 2018, 2018, 2048–2052. [Google Scholar] [CrossRef]
- Xu, C.; Du, J.; Ma, L.; Li, G.; Tao, M.; Zhang, W. Tertiary amine functionalized polyacrylonitrile fiber catalyst for the synthesis of tetrahydrothiophenes. Tetrahedron 2013, 69, 4749–4757. [Google Scholar] [CrossRef]
- Ramirez-Osuna, M.; Chavez, D.; Hernandez, L.; Mollins, E.; Somanathan, R.; Aguirre, G. Synthesis of analogs of amathamide A and their preliminary antimicrobial activity. Molecules 2005, 10, 295–301. [Google Scholar] [CrossRef]
- Jakubec, P.; Cockfield, D.M.; Hynes, P.S.; Cleator, E.; Dixon, D.J. Enantio- and diastereoselective Michael additions of C-succinimidyl esters to nitro olefins using cinchonine-derived bifunctional organocatalysts. Tetrahedron Asymm. 2011, 22, 1147–1155. [Google Scholar] [CrossRef]
- Wen, L.; Tang, F.; Ge, C.; Wang, X.; Han, Z.; Wu, J. Practical large-scale preparation of (R)-rolipram using chiral nickel catalyst. Synth. Commun. 2012, 42, 3288–3295. [Google Scholar] [CrossRef]
- Fadeeva, A.A.; Ioffe, S.L.; Tabolin, A.A. Chlorination of conjugated nitroalkenes with PhICl2 and SO2Cl2 for the synthesis of α-chloronitroalkenes. Synthesis 2020, 52, 2679–2688. [Google Scholar] [CrossRef]
- Smirnov, V.O.; Ioffe, S.L.; Tishkov, A.A.; Khomutova, Y.A.; Nesterov, I.D.; Antipin, M.Y.; Smit, W.A.; Tartakovsky, V.A. New C-C coupling reaction of cyclic nitronates with carbon nucleophiles. Umpolung of the conventional reactivity of nitronates. J. Org. Chem. 2004, 69, 8485–8488. [Google Scholar] [CrossRef] [PubMed]
- Ushakov, P.Y.; Tabolin, A.A.; Ioffe, S.L.; Sukhorukov, A.Y. In situ generated magnesium cyanide as an efficient reagent for nucleophilic cyanation of nitrosoalkenes and parent nitronates. Eur. J. Org. Chem. 2019, 2019, 1888–1892. [Google Scholar] [CrossRef]
- Mikhaylov, A.A.; Dilman, A.D.; Khomutova, Y.A.; Arkhipov, D.E.; Korlyukov, A.A.; Ioffe, S.L. Stereoselective amine addition to six-membered cyclic nitronates promoted by silyl triflate. Eur. J. Org. Chem. 2013, 2013, 5670–5677. [Google Scholar] [CrossRef]
- Davies, H.M.L.; Hougland, P.W.; Cantrell, W.R., Jr. Convenient Synthesis of Vinyldiazomethanes from α-Diazo-β-Keto Esters and Related Systems. Synth. Commun. 1992, 22, 971–978. [Google Scholar] [CrossRef]
- Wu, J.-Q.; Yang, Z.; Zhang, S.-S.; Jiang, C.-Y.; Li, Q.; Huang, Z.-S.; Wang, H. From indoles to carbazoles: Tandem Cp*Rh(III)-catalyzed C–H activation/Brønsted acid-catalyzed cyclization reactions. ACS Catal. 2015, 5, 6453–6457. [Google Scholar] [CrossRef]
- Korte, F.; Wusten, F. Über den Enolgehalt von Acetessigsäureestern mit verschiedenen Esteralkyl-Komponenten. Justus Liebigs Ann. Chem. 1961, 647, 18–22. [Google Scholar] [CrossRef]
- Dolle, F.; Hinnen, F.; Valette, H.; Fuseau, C.; Duval, R.; Peglion, J.-L.; Crouzel, C. Synthesis of two optically active calcium channel antagonists labelled with carbon-11 for in vivo cardiac PET imaging. Bioorg. Med. Chem. Lett. 1997, 5, 749–764. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J.; Li, B.; Dikarev, E.; Autschbach, J.; Davies, H.M.L. Combined experimental and computational studies of heterobimetallic Bi−Rh paddlewheel carboxylates as catalysts for metal carbenoid transformations. J. Org. Chem. 2009, 74, 6564–6571. [Google Scholar] [CrossRef]
- Chinthapally, K.; Massaro, N.P.; Ton, S.; Gardner, E.D.; Sharma, I. Trapping rhodium vinylcarbenoids with aminochalcones for the synthesis of medium-sized azacycles. Tetrahedron Lett. 2019, 60, 151253. [Google Scholar] [CrossRef]
- Rossbach, J.; Baumeister, J.; Harms, K.; Koert, U. Regio- and diastereoselective crotylboration of vic-tricarbonyl compounds. Eur. J. Org. Chem. 2013, 2013, 662–665. [Google Scholar] [CrossRef]
- Sarabia, F.J.; Li, Q.; Ferreira, E.M. Cyclopentene annulations of alkene radical cations with vinyl diazo species using photocatalysis. Angew. Chem. Int. Ed. 2018, 57, 11015–11019. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).