
The Unusual Architecture of RNA-Dependent RNA Polymerase (RdRp)’s Catalytic Chamber Provides a Potential Strategy for Combination Therapy against COVID-19

 , , , ,
, , , ,  , ,
, , 
Abstract
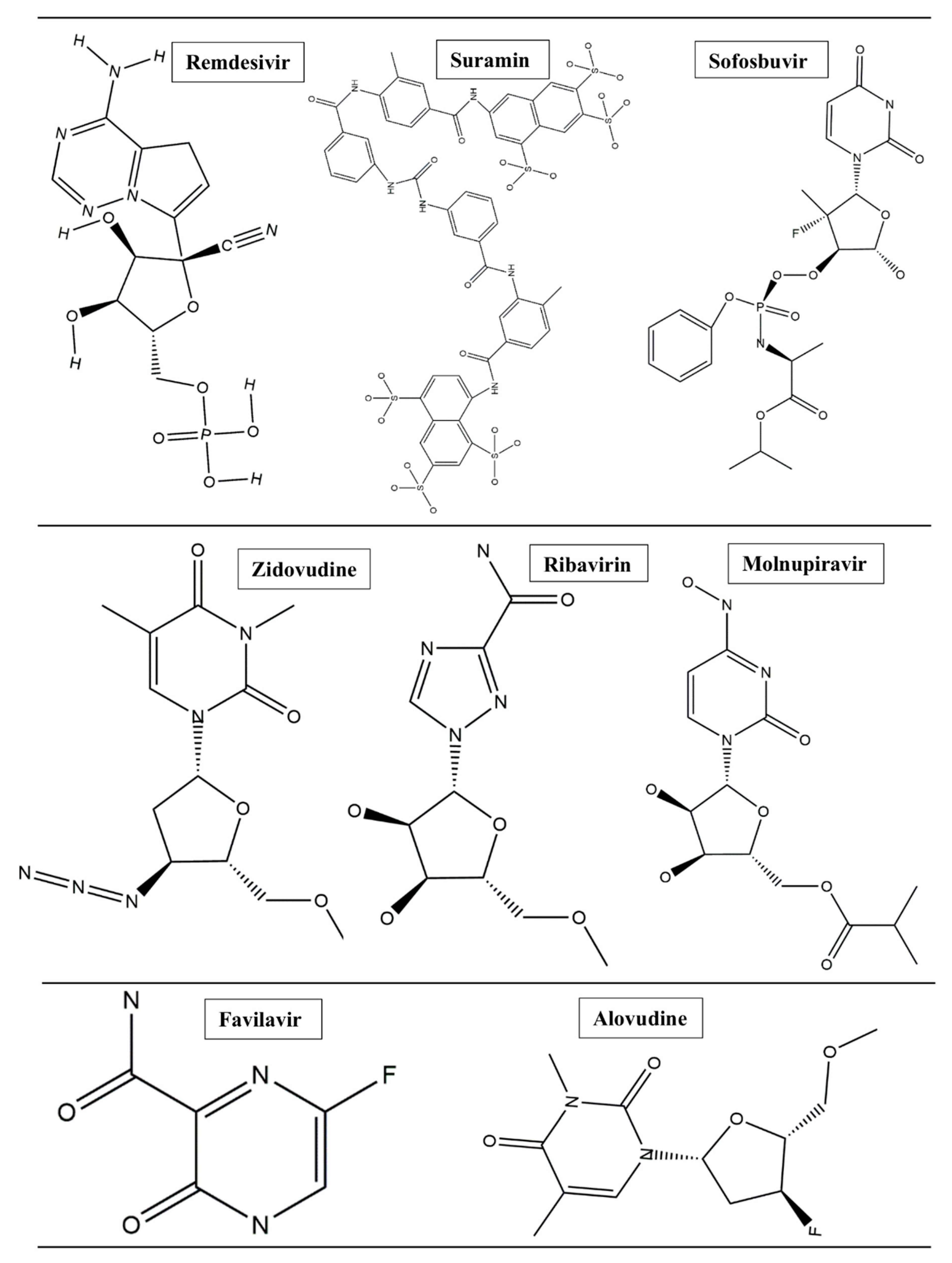
1. Introduction
2. Methods and Results
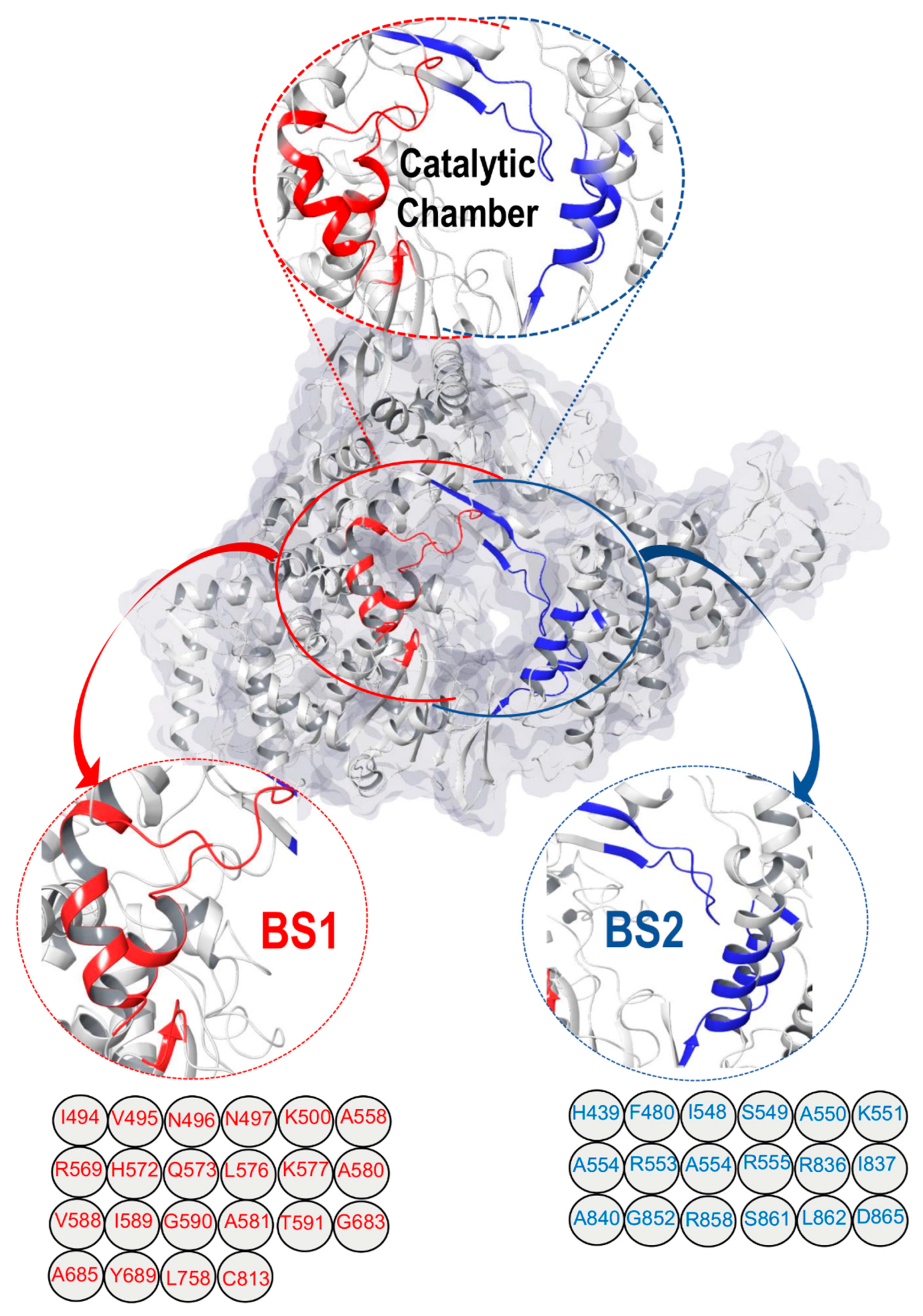
2.1. SARS-CoV-2 RdRp Catalytic Chamber
Mapping and Characterization of the Two Binding Pockets, BS1 and BS2
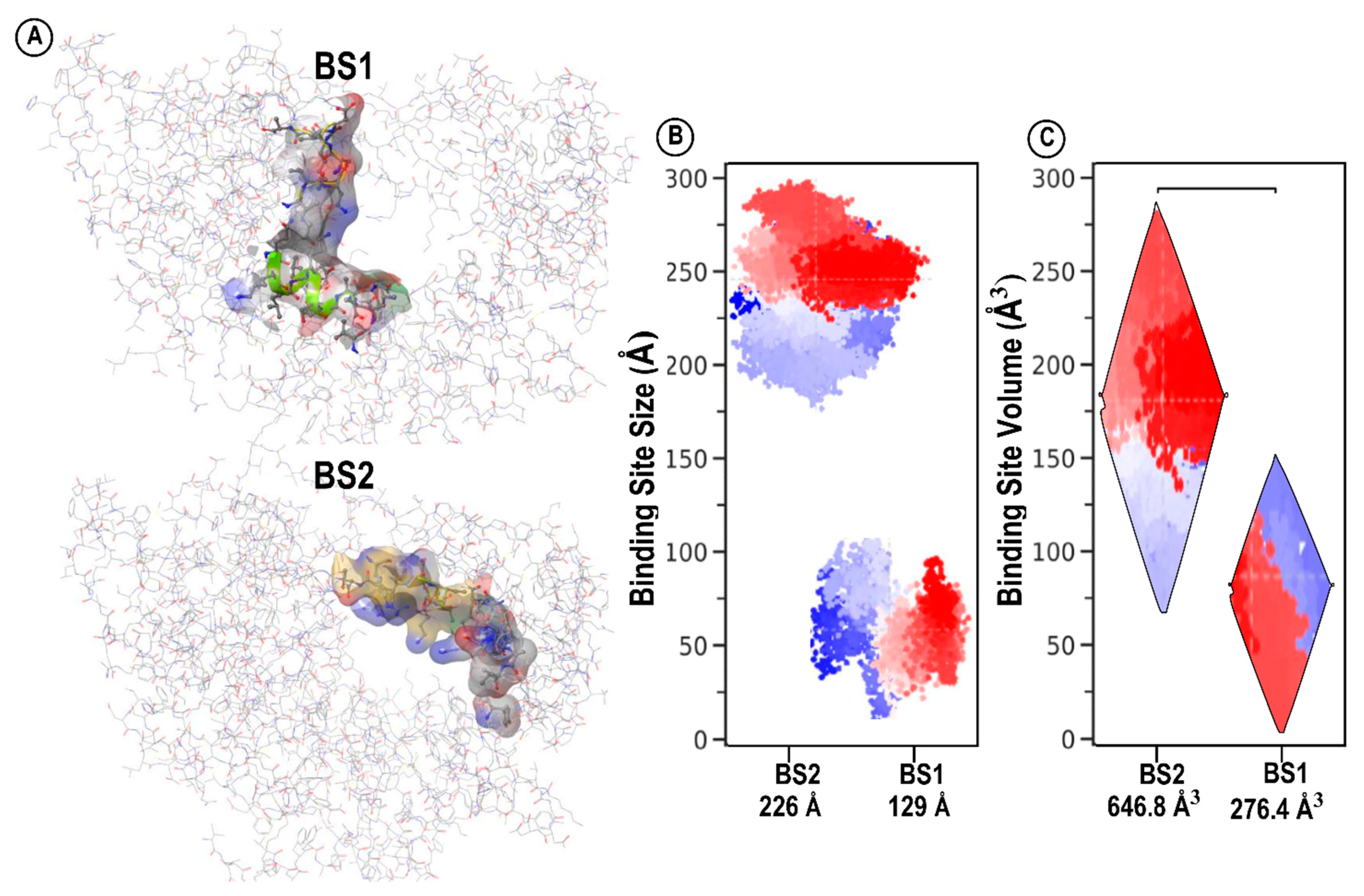
3. Structural Architecture of BS1 and BS2
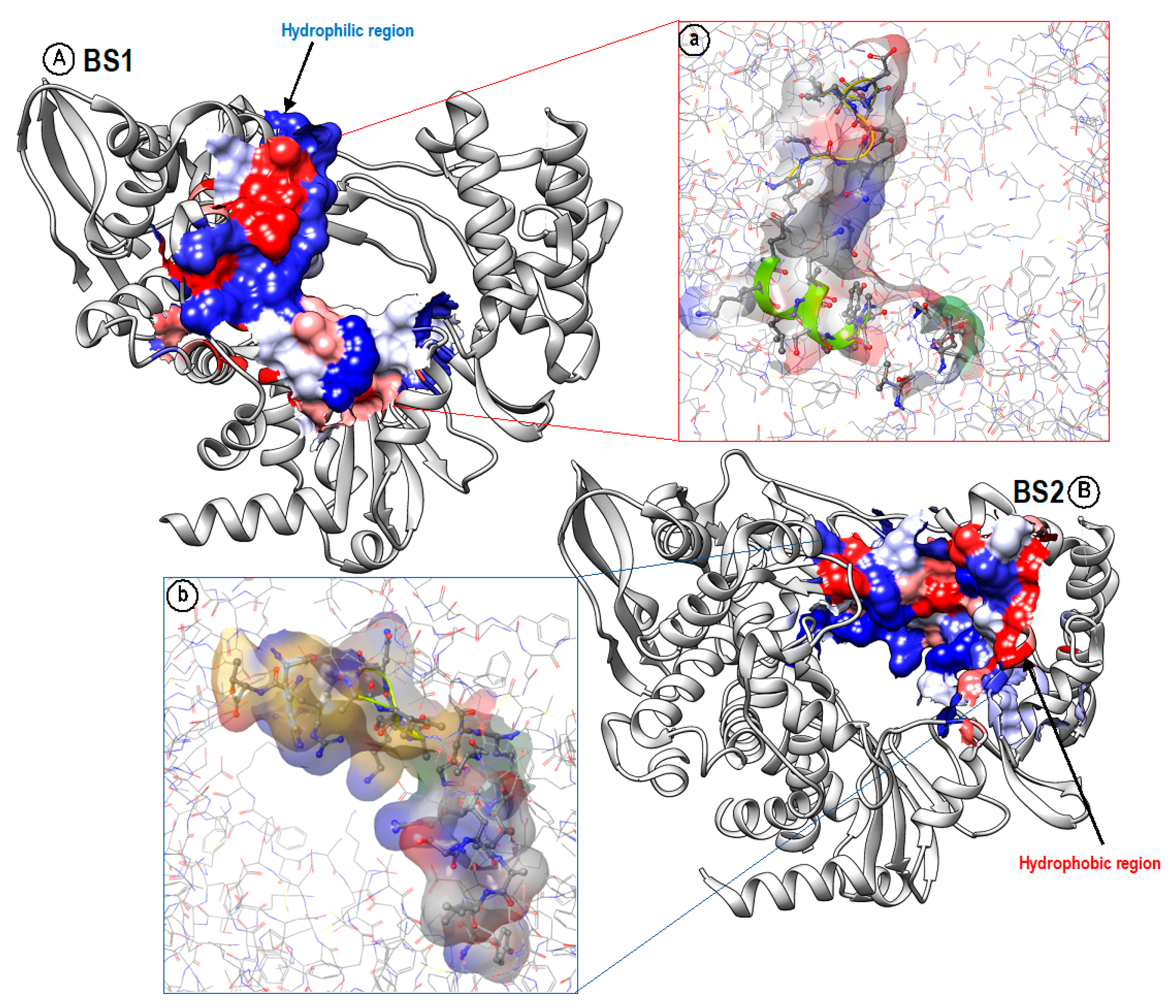
4. Hydrophobicity/Hydrophilicity Profiles of BS1 and BS2
5. BS1 and BS2 Binding Pocket Per-Residue Contribution Using Suramin as a Prototype
6. System Preparation and Molecular Dynamic Simulation
7. Dynamic Conformational Stability and Fluctuations
8. Binding Free Energy Calculations
9. Assessment of Comparative Binding Energies
10. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dey, S.K.; Rahman, M.; Siddiqi, U.R.; Howlader, A. Analyzing the epidemiological outbreak of COVID-19: A visual exploratory data analysis approach. J. Med. Virol. 2020, 92, 632–638. [Google Scholar] [CrossRef]
- Harapan, H.; Itoh, N.; Yufika, A.; Winardi, W.; Keam, S.; Te, H.; Megawati, D.; Hayati, Z.; Wagner, A.L.; Mudatsir, M. Coronavirus disease 2019 (COVID-19): A literature review. J. Infect. Public Health 2020, 13, 667–673. [Google Scholar] [CrossRef]
- Kirchdoerfer, R.N.; Ward, A.B. Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nat. Commun. 2019, 10, 2342. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 2020, 368, 779–782. [Google Scholar] [CrossRef]
- Hasan, A.H.; Hussen, N.H.; Shakya, S.; Jamalis, J.; Pratama, M.R.F.; Chander, S.; Kharkwal, H.; Murugesan, S. In silico discovery of multi-targeting inhibitors for the COVID-19 treatment by molecular docking, molecular dynamics simulation studies, and ADMET predictions. Struct. Chem. 2022, 33, 1645–1665. [Google Scholar] [CrossRef]
- Peng, Q.; Peng, R.; Yuan, B.; Zhao, J.; Wang, M.; Wang, X.; Wang, Q.; Sun, Y.; Fan, Z.; Qi, J.; et al. Structural and Biochemical Characterization of the nsp12-nsp7-nsp8 Core Polymerase Complex from SARS-CoV-2. Cell Rep. 2020, 31, 107774. [Google Scholar] [CrossRef]
- Yin, W.; Mao, C.; Luan, X.; Shen, D.-D.; Shen, Q.; Su, H.; Wang, X.; Zhou, F.; Zhao, W.; Gao, M.; et al. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 2020, 368, 1499–1504. [Google Scholar] [CrossRef] [PubMed]
- Lung, J.; Lin, Y.; Yang, Y.; Chou, Y.; Shu, L.; Cheng, Y.; Liu, H.T.; Wu, C. The potential chemical structure of anti-SARS-CoV-2 RNA-dependent RNA polymerase. J. Med. Virol. 2020, 92, 693–697. [Google Scholar] [CrossRef] [PubMed]
- Koulgi, S.; Jani, V.; Uppuladinne, M.V.N.; Sonavane, U.; Joshi, R. Remdesivir-bound and ligand-free simulations reveal the probable mechanism of inhibiting the RNA dependent RNA polymerase of severe acute respiratory syndrome coronavirus 2. RSC Adv. 2020, 10, 26792–26803. [Google Scholar] [CrossRef]
- Gaurav, A.; Al-Nema, M. Polymerases of Coronaviruses: Structure, Function, and Inhibitors; Elsevier Inc.: Singapore, 2018; ISBN 9780128154229. [Google Scholar]
- Yin, W.; Luan, X.; Li, Z.; Zhou, Z.; Wang, Q.; Gao, M.; Wang, X.; Zhou, F.; Shi, J.; You, E.; et al. Structural basis for inhibition of the SARS-CoV-2 RNA polymerase by suramin. Nat. Struct. Mol. Biol. 2021, 28, 319–325. [Google Scholar] [CrossRef]
- Wiedemar, N.; Hauser, D.A.; Mäser, P. 100 Years of Suramin. Antimicrob. Agents Chemother. 2020, 64, e01168-19. [Google Scholar] [CrossRef]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Chien, M.; Anderson, T.K.; Jockusch, S.; Tao, C.; Li, X.; Kumar, S.; Russo, J.J.; Kirchdoerfer, R.N.; Ju, J. Nucleotide Analogues as Inhibitors of SARS-CoV-2 Polymerase, a Key Drug Target for COVID-19. J. Proteome Res. 2020, 19, 4690–4697. [Google Scholar] [CrossRef] [PubMed]
- Kloostra, S. Sitemap. Joomla! 3 SEO and Performance; Apress: New York, NY, USA, 2015. [Google Scholar]
- Thomsen, R.; Christensen, M.H. MolDock: A New Technique for High-Accuracy Molecular Docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The Protein Data Bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera? A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Kusumaningrum, S.; Budianto, E.; Kosela, S.; Sumaryono, W.; Juniarti, F. The molecular docking of 1,4-naphthoquinone derivatives as inhibitors of Polo-like kinase 1 using Molegro Virtual Docker. J. Appl. Pharm. Sci. 2014, 4, 47–53. [Google Scholar] [CrossRef]
- Allouche, A. Software News and Updates Gabedit—A Graphical User Interface for Computational Chemistry Soft-wares. J. Comput. Chem. 2012, 32, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Ahmed, A.; Fatima, A.; Shakya, S.; Rahman, Q.I.; Ahmad, M.; Javed, S.; AlSalem, H.S.; Ahmad, A. Crystal Structure, Topology, DFT and Hirshfeld Surface Analysis of a Novel Charge Transfer Complex (L3) of Anthraquinone and 4-{[(anthracen-9-yl)meth-yl] amino}-benzoic Acid (L2) Exhibiting Photocatalytic Properties: An Experimental and Theoretical Approach. Molecules 2022, 27, 1724. [Google Scholar] [CrossRef]
- Alhomrani, M.; Alsanie, W.F.; Alamri, A.S.; Alyami, H.; Habeeballah, H.; Alkhatabi, H.A.; Felimban, R.I.; Haynes, J.M.; Shakya, S.; Raafat, B.M.; et al. Enhancing the Antipsychotic Effect of Risperidone by Increasing Its Binding Affinity to Serotonin Receptor via Picric Acid: A Molecular Dynamics Simulation. Pharmaceuticals 2022, 15, 285. [Google Scholar] [CrossRef]
- Case, D.A.; Walker, R.C.; Cheatham, T.E.; Simmerling, C.; Roitberg, A.; Merz, K.M.; Luo, R.; Darden, T. Amber 2018. Univ. Calif. San Fr. 2018, 2018, 1–923. [Google Scholar]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Grest, G.S.; Kremer, K. Molecular dynamics simulation for polymers in the presence of a heat bath. Phys. Rev. A 1986, 33, 3628–3631. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Roe, D.R.; Cheatham, T.E., III. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef] [PubMed]
- Seifert, E. OriginPro 9.1: Scientific Data Analysis and Graphing Software—Software Review. J. Chem. Inf. Model. 2014, 54, 1552. [Google Scholar] [CrossRef]
- Elamin, G.; Aljoundi, A.; Soliman, M.E. A synergistic multitargeted of BET and HDAC: An intra-molecular mechanism of communication in treatment of Waldenström macroglobulinemia. Mol. Simul. 2021, 48, 197–208. [Google Scholar] [CrossRef]
- Aljoundi, A.; El Rashedy, A.; Soliman, M.E. Distinguishing the optimal binding mechanism through reversible and irreversible inhibition analysis of HSP72 protein in cancer therapy. Comput. Biol. Med. 2021, 132, 104301. [Google Scholar] [CrossRef]
- Elamin, G.; Aljoundi, A.; Soliman, M.E.S. Co-Binding of JQ1 and Venetoclax Exhibited Synergetic Inhibitory Effect for Cancer Therapy; Potential Line of Treatment for the Waldenström Macroglobulinemia Lymphoma. Chem. Biodivers. 2022, 19, e202100845. [Google Scholar] [CrossRef] [PubMed]
- Aljoundi, A.; El Rashedy, A.; Soliman, M.E.S. Comparison of irreversible inhibition targeting HSP72 protein: The resurgence of covalent drug developments. Mol. Simul. 2021, 47, 1093–1103. [Google Scholar] [CrossRef]
- Alamri, A.S.; Alhomrani, M.; Alsanie, W.F.; Alyami, H.; Shakya, S.; Habeeballah, H.; Alamri, A.; Alzahrani, O.; Alzahrani, A.S.; Alkhatabi, H.A.; et al. Enhancement of Haloperidol Binding Affinity to Dopamine Receptor via Forming a Charge-Transfer Complex with Picric Acid and 7,7,8,8-Tetracyanoquinodimethane for Improvement of the Antipsychotic Efficacy. Molecules 2022, 27, 3295. [Google Scholar] [CrossRef] [PubMed]
- Alsanie, W.F.; Alamri, A.S.; Alyami, H.; Alhomrani, M.; Shakya, S.; Habeeballah, H.; Alkhatabi, H.A.; Felimban, R.I.; Alzahrani, A.S.; Alhabeeb, A.A.; et al. Increasing the Efficacy of Seproxetine as an Antidepressant Using Charge–Transfer Complexes. Molecules 2022, 27, 3290. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the Performance of the MM/PBSA and MM/GBSA Methods. 1. The Accuracy of Binding Free Energy Calculations Based on Molecular Dynamics Simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef]
- Homeyer, N.; Gohlke, H. Free Energy Calculations by the Molecular Mechanics Poisson−Boltzmann Surface Area Method. Mol. Inform. 2012, 31, 114–122. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site | Site Score | Size | Volume A3 | Dscore | Exposure | Enclosure | Contact | Phobic | Philic | Balance | Don/acc |
|---|---|---|---|---|---|---|---|---|---|---|---|
| BS1 | 1.030 | 129 | 276.458 | 0.943 | 0.522 | 0.743 | 1.027 | 0.252 | 1.357 | 0.186 | 0.370 |
| BS2 | 0.976 | 226 | 646.898 | 0.940 | 0.695 | 0.662 | 0.771 | 0.286 | 1.219 | 0.235 | 0.866 |
| BS1/Suramin Complex | BS2/Suramin Complex | ||
|---|---|---|---|
 |  | ||
| BS1 per-residue energy contribution | BS2 per-residue energy contribution | ||
| Residue | Energy (kcal/mol) | Residue | Energy (kcal/mol) |
| Asn496 | −22.51 | Arg555 | −30.26 |
| Ile494 | −22.51 | Arg836 | −19.47 |
| Arg569 | −19.44 | Arg553 | −13.50 |
| Lys577 | −16.14 | LIe548 | −11.90 |
| Gly590 | −15.77 | Ser549 | −11.19 |
| Lys500 | −14.60 | Lys551 | −11.01 |
| Gln573 | −8.99 | His439 | −10.08 |
| Ala558 | −7.89 | Gly852 | −8.93 |
| Ile589 | −7.01 | Arg858 | −5.84 |
| Leu576 | −6.80 | Ala550 | −5.81 |
| Asn497 | −6.48 | Ala840 | −5.48 |
| Ala685 | −4.61 | Phe480 | −5.18 |
| Systems | Estimated Averages (Å) | ||
|---|---|---|---|
| Ligand | RMSD | RMSF | SASA |
| Remdesivir-BS1 | 1.58 | 1.06 | 14,011.99 |
| Sofosbuvir-BS1 | 2.04 | 1.12 | 14,510.63 |
| Alovudine-BS1 | 1.34 | 1.03 | 13,772.77 |
| Molnupiravir-BS1 | 1.65 | 1.09 | 13,800.19 |
| Zidovudine-BS1 | 1.72 | 1.08 | 13,706.09 |
| Favilavir-BS1 | 1.57 | 1.17 | 14,271.97 |
| Ribavirin-BS1 | 1.53 | 1.04 | 14,116.37 |
| Suramin-BS1 | 1.62 | 1.20 | 13,911.23 |
| Systems | Estimated Averages (Å) | ||
|---|---|---|---|
| Ligand | RMSD | RMSF | SASA |
| Remdesivir-BS2 | 1.87 | 1.14 | 11,018.38 |
| Sofosbuvir-BS2 | 2.09 | 1.35 | 11,193.50 |
| Alovudine-BS2 | 1.80 | 1.06 | 10,655.36 |
| Molnupiravir-BS2 | 2.13 | 1.21 | 11,180.05 |
| Zidovudine-BS2 | 2.12 | 1.22 | 11,245.66 |
| Favilavir-BS2 | 1.64 | 1.20 | 10,948.94 |
| Ribavirin- BS2 | 1.68 | 1.11 | 11,244.42 |
| Suramin-BS2 | 1.78 | 1.24 | 11,142.55 |
| Systems | Energy Components | ||||
|---|---|---|---|---|---|
| (kcal/mol) | |||||
| Ligand | ΔEvdw | ΔEele | ΔGgas | ΔGsol | ΔGbind |
| Remdesivir-BS1 | −41.9522 | −335.8092 | −377.7613 | 325.1969 | −52.5645 |
| Remdesivir-BS2 | −42.7075 | −90.9436 | −133.6512 | 109.1042 | −24.5469 |
| Sofosbuvir-BS1 | −34.6272 | −15.0141 | −49.6413 | 25.2979 | −24.3434 |
| Sofosbuvir-BS2 | −32.0037 | −19.3497 | −51.3534 | 33.6064 | −17.7470 |
| Alovudine-BS1 | −21.7154 | −8.8406 | −30.5559 | 13.5672 | −16.9888 |
| Alovudine-BS2 | −19.2905 | 3.337 | −15.9536 | 5.5781 | −10.3754 |
| Molnupiravir-BS1 | −22.4816 | −34.6077 | −57.0892 | 43.4611 | −13.6282 |
| Molnupiravir-BS2 | −26.1253 | −24.0320 | −56.1573 | 35.6699 | −14.4874 |
| Zidovudine-BS1 | −24.3850 | −31.0102 | −57.3952 | 40.5715 | −14.8237 |
| Zidovudine-BS2 | −15.3793 | −193.9331 | −209.3124 | 198.1934 | −11.1190 |
| Favilavir-BS1 | −7.5696 | −37.4859 | −79.4035 | 90.7847 | −11.3812 |
| Favilavir-BS2 | −7.3014 | −28.6874 | −72.1635 | 85.046 | −12.8825 |
| Ribavirin-BS1 | −8.5964 | −20.8937 | −63.3037 | 76.6451 | −13.3414 |
| Ribavirin-BS2 | −7.7187 | −27.1752 | −69.8784 | 83.4503 | −13.5719 |
| Suramin-BS1 | −8.6775 | −48.0019 | −89.7680 | 102.9012 | −13.1331 |
| Suramin-BS2 | −7.7503 | −51.9245 | −94.8926 | 107.5886 | −12.6959 |
| All energies are in kcal/mol. | ΔEele = electrostatic energy | ΔEvdw = van der Waals energy | ΔGbind = total binding free energy | ΔGsol = solvation free energy | ΔG = gas phase free energy |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Metwally, K.; Abo-Dya, N.E.; Alahmdi, M.I.; Albalawi, M.Z.; Yahya, G.; Aljoundi, A.; Salifu, E.Y.; Elamin, G.; Ibrahim, M.A.A.; Sayed, Y.; et al. The Unusual Architecture of RNA-Dependent RNA Polymerase (RdRp)’s Catalytic Chamber Provides a Potential Strategy for Combination Therapy against COVID-19. Molecules 2023, 28, 2806. https://doi.org/10.3390/molecules28062806
Metwally K, Abo-Dya NE, Alahmdi MI, Albalawi MZ, Yahya G, Aljoundi A, Salifu EY, Elamin G, Ibrahim MAA, Sayed Y, et al. The Unusual Architecture of RNA-Dependent RNA Polymerase (RdRp)’s Catalytic Chamber Provides a Potential Strategy for Combination Therapy against COVID-19. Molecules. 2023; 28(6):2806. https://doi.org/10.3390/molecules28062806
Chicago/Turabian StyleMetwally, Kamel, Nader E. Abo-Dya, Mohammed Issa Alahmdi, Maha Z. Albalawi, Galal Yahya, Aimen Aljoundi, Elliasu Y. Salifu, Ghazi Elamin, Mahmoud A. A. Ibrahim, Yasien Sayed, and et al. 2023. "The Unusual Architecture of RNA-Dependent RNA Polymerase (RdRp)’s Catalytic Chamber Provides a Potential Strategy for Combination Therapy against COVID-19" Molecules 28, no. 6: 2806. https://doi.org/10.3390/molecules28062806
APA StyleMetwally, K., Abo-Dya, N. E., Alahmdi, M. I., Albalawi, M. Z., Yahya, G., Aljoundi, A., Salifu, E. Y., Elamin, G., Ibrahim, M. A. A., Sayed, Y., Fanucchi, S., & Soliman, M. E. S. (2023). The Unusual Architecture of RNA-Dependent RNA Polymerase (RdRp)’s Catalytic Chamber Provides a Potential Strategy for Combination Therapy against COVID-19. Molecules, 28(6), 2806. https://doi.org/10.3390/molecules28062806