
Silylated-Acetylated Cyclodextrins as Chiral Sensors for the Enantiodiscrimination of Fluorinated Anesthetics

 , , ,
, , ,
Abstract
1. Introduction
2. Results and Discussion
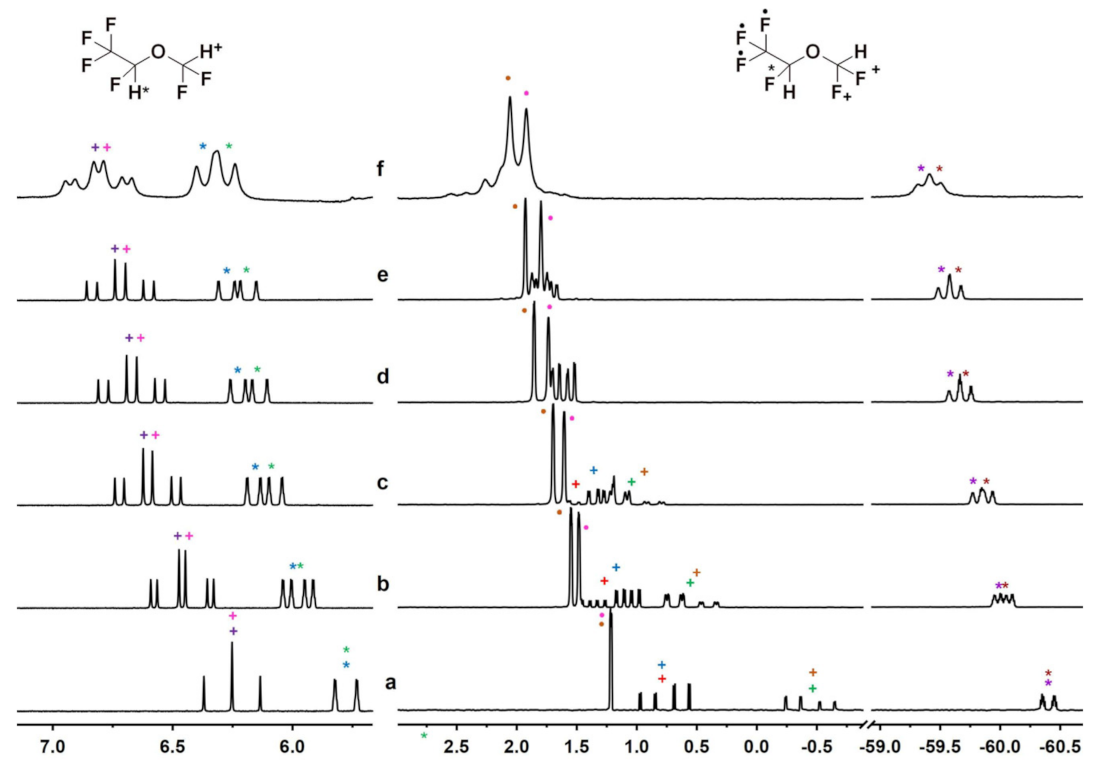
2.1. NMR Enantiodiscrimination Experiments of Isoflurane (ISO)
2.2. NMR Enantiodiscrimination Experiments with the Other Substrates
2.2.1. Desflurane (DSF)
2.2.2. Halothane (HAL)
2.2.3. Enflurane (ENF)
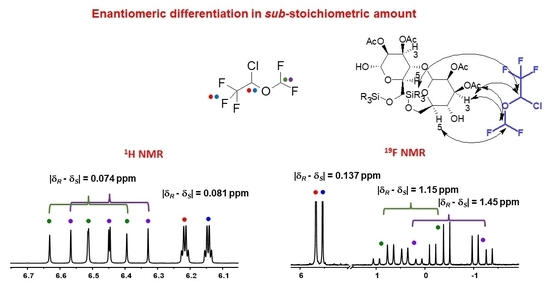
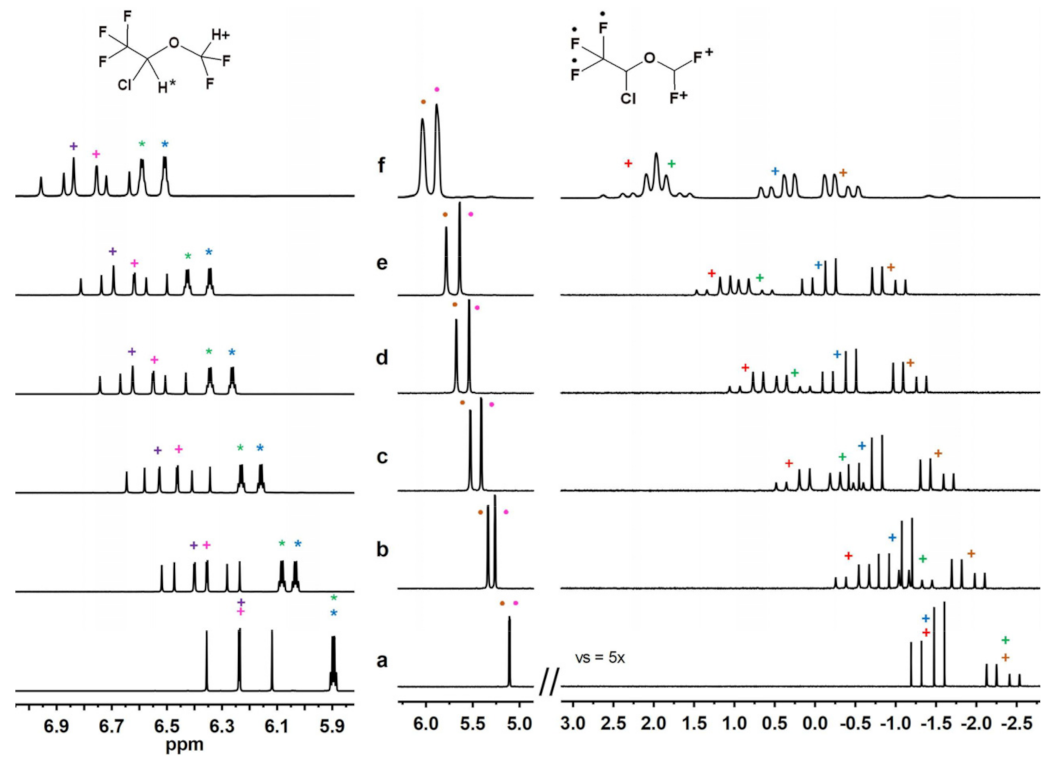
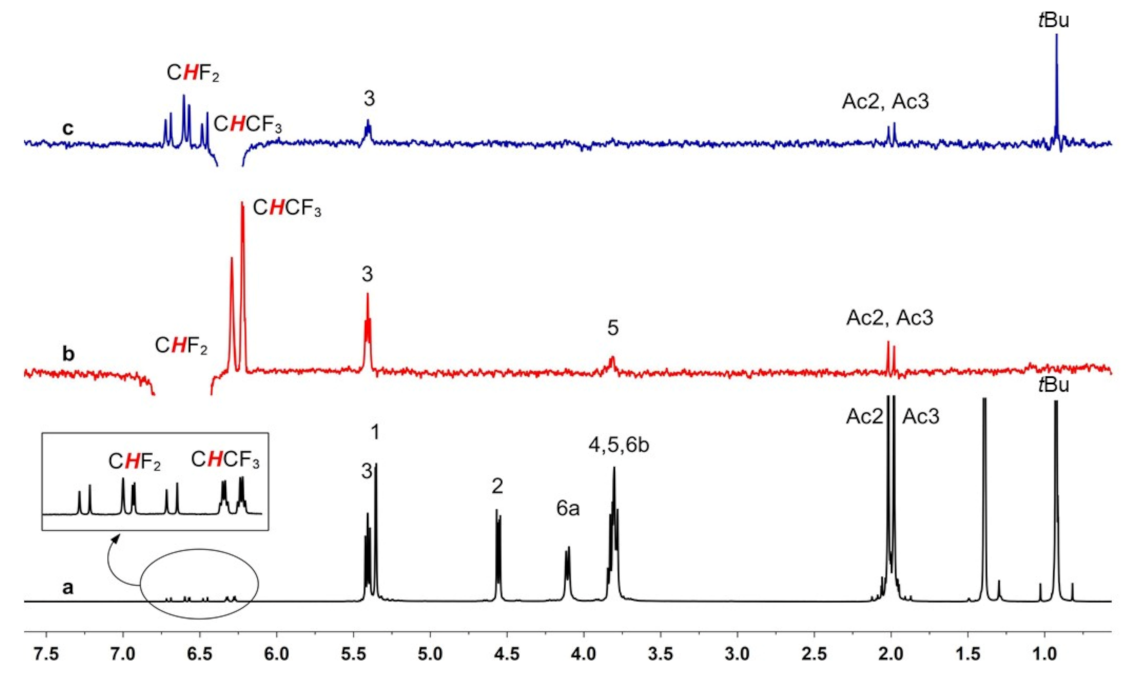
2.2.4. Experiments with DSF, HAL, ENF and Compound B in Sub-Stoichiometric Conditions
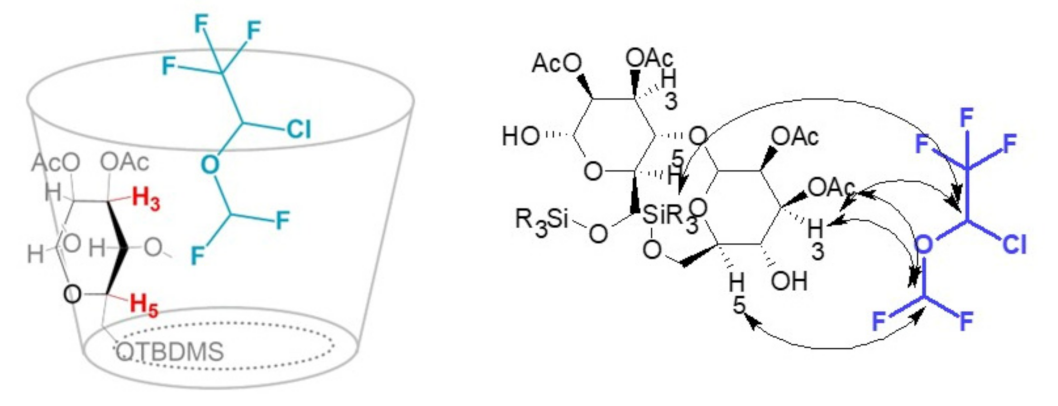
2.3. Interaction Mechanism
3. Materials and Methods
3.1. Materials
3.2. 1H and 19F Characterization of the Fluorinated Substrates
3.2.1. Isoflurane
3.2.2. Desflurane
3.2.3. Halothane
3.2.4. Enflurane
3.2.5. Compound B
3.3. Synthesis of Silylated-Acetylated Cyclodextrins
3.4. Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Crini, G.; Fourmentin, S.; Fenyvesi, É.; Torri, G.; Fourmentin, M.; Morin-Crini, N. Cyclodextrins, from molecules to applications. Environ. Chem. Lett. 2018, 16, 1361–1375. [Google Scholar] [CrossRef]
- Řezanka, M. Synthesis of substituted cyclodextrins. Environ. Chem. Lett 2019, 17, 49–63. [Google Scholar] [CrossRef]
- Wankar, J.; Kotla, N.G.; Gera, S.; Rasala, S.; Pandit, A.; Rochev, Y.A. Recent Advances in Host–Guest Self-Assembled Cyclodextrin Carriers: Implications for Responsive Drug Delivery and Biomedical Engineering. Adv. Funct. Mater. 2020, 30, 1909049. [Google Scholar] [CrossRef]
- Bai, C.C.; Tian, B.R.; Zhao, T.; Huang, Q.; Wang, Z.Z. Cyclodextrin-Catalyzed Organic Synthesis: Reactions, Mechanisms, and Applications. Molecules 2017, 22, 1475. [Google Scholar] [CrossRef] [PubMed]
- Macaev, F.; Boldescu, V. Cyclodextrins in Asymmetric and Stereospecific Synthesis. Symmetry 2015, 7, 1699–1720. [Google Scholar] [CrossRef]
- Chankvetadze, B. Application of enantioselective separation techniques to bioanalysis of chiral drugs and their metabolites. TrAC Trends Anal. Chem. 2021, 143, 116332. [Google Scholar] [CrossRef]
- Gunjal, P.; Singh, K.S.; Kumar, R.; Kumar, R.; Gulati, M. Role of Chromatograph-based Analytical Techniques in Quantification of Chiral Compounds: An Update. Curr. Anal. Chem. 2021, 17, 355–373. [Google Scholar] [CrossRef]
- Guo, C.; Xiao, Y. Negatively charged cyclodextrins: Synthesis and applications in chiral analysis-A review. Carbohydr. Polym. 2021, 256, 117517. [Google Scholar] [CrossRef]
- Hancu, G.; Papp, L.A.; Tóth, G.; Kelemen, H. The Use of Dual Cyclodextrin Chiral Selector Systems in the Enantioseparation of Pharmaceuticals by Capillary Electrophoresis: An Overview. Molecules 2021, 26, 2261. [Google Scholar] [CrossRef]
- Juvancz, Z.; Bodane-Kendrovics, R.; Szente, L.; Maklari, D. Cyclodextrins as Dominant Chiral Selective Agents in the Capillary Separation Techniques. Period. Polytechn. Chem. Eng. 2021, 65, 580–594. [Google Scholar] [CrossRef]
- Balzano, F.; Uccello-Barretta, G.; Aiello, F. Chapter 9—Chiral Analysis by NMR Spectroscopy: Chiral Solvating Agents. In Chiral Analysis, 2nd ed.; Polavarapu, P.L., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 367–427. [Google Scholar]
- Wang, S.-Y.; Li, L.; Xiao, Y.; Wang, Y. Recent advances in cyclodextrins-based chiral-recognizing platforms. TrAC Trends Anal. Chem. 2019, 121, 115691. [Google Scholar] [CrossRef]
- Wenzel, T.J.; Wenzel, T.J. Differentiation of Chiral Compounds Using NMR Spectroscopy, 2nd ed.; Wiley: Hoboken, NJ, USA, 2018. [Google Scholar]
- Koehler, J.E.H.; Hohla, M.; Richters, M.; Koenig, W.A. Cyclodextrin Derivatives as Chiral Selectors—Investigation of the Interaction with (R,S)-Methyl-2-chloropropionate by Enantioselective Gas Chromatography, NMR Spectroscopy, and Molecular Dynamics Simulation. Angew Chem. Int. Ed. Engl. 1992, 31, 319–320. [Google Scholar] [CrossRef]
- Schmidt, R.; Roeder, M.; Oeckler, O.; Simon, A.; Schurig, V. Separation and absolute configuration of the enantiomers of a degradation product of the new inhalation anesthetic sevoflurane (Dedicated to Professor Nakanishi on the occasion of his 75th birthday). Chirality 2000, 12, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Schurig, V. Use of derivatized cyclodextrins as chiral selectors for the separation of enantiomers by gas chromatography. Ann. Pharm. Fr. 2010, 68, 82–98. [Google Scholar] [CrossRef] [PubMed]
- Schurig, V. Salient Features of Enantioselective Gas Chromatography: The Enantiomeric Differentiation of Chiral Inhalation Anesthetics as a Representative Methodological Case in Point. In Differentiation of Enantiomers I; Schurig, V., Ed.; Springer: Cham, Switzerland, 2013; Volume 340, pp. 153–207. [Google Scholar] [CrossRef]
- Schurig, V.; Kreidler, D. Gas-chromatographic enantioseparation of unfunctionalized chiral hydrocarbons: An overview. Methods Mol. Biol. 2013, 970, 45–67. [Google Scholar] [CrossRef] [PubMed]
- Sicoli, G.; Kreidler, D.; Czesia, H.; Hopf, H.; Schurig, V. Gas Chromatographic Enantioseparation of Unfunctionalized Chiral Alkanes: A Challenge in Separation Science (Overview, State of the Art, and Perspectives). Chirality 2009, 21, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Uccello-Barretta, G.; Balzano, F.; Pertici, F.; Jicsinszky, L.; Sicoli, G.; Schurig, V. External vs. Internal Interactions in the Enantiodiscrimination of Fluorinated α-Amino Acid Derivatives by Heptakis [2,3-di-O-acetyl-6-O-(tert-butyldimethylsilyl)]-β-cyclodextrin, a Powerful Chiral Solvating Agent for NMR Spectroscopy. Eur. J. Org. Chem. 2008, 2008, 1855–1863. [Google Scholar] [CrossRef]
- Uccello-Barretta, G.; Sicoli, G.; Balzano, F.; Schurig, V.; Salvadori, P. Highly efficient NMR enantiodiscrimination of 1,1,1,3,3-pentafluoro-2-(fluoromethoxy)-3-methoxypropane, a chiral degradation product of sevoflurane, by heptakis(2,3-di-O-acetyl-6-O-tert-butyldimethylsilyl)-β-cyclodextrin. Tetrahedr. Asymm. 2006, 17, 2504–2510. [Google Scholar] [CrossRef]
- Balzano, F.; Uccello-Barretta, G.; Sicoli, G.; Vanni, L.; Recchimurzo, A.; Aiello, F. Chiral Discrimination Mechanisms by Silylated-Acetylated Cyclodextrins: Superficial Interactions vs. Inclusion. Int. J. Mol. Sci. 2022, 23, 13169. [Google Scholar] [CrossRef]
- Klika, K.D. Use of sub-stoichiometric amounts of chiral auxiliaries for enantiodifferentiation by NMR. Caveats and potential utility. Tetrahedr. Asymm. 2009, 20, 1099–1102. [Google Scholar] [CrossRef]
- Recchimurzo, A.; Balzano, F.; Uccello-Barretta, G.; Gherardi, L. Bis-Thiourea Chiral Sensor for the NMR Enantiodiscrimination of N-Acetyl and N-Trifluoroacetyl Amino Acid Derivatives. J. Org. Chem. 2022, 87, 11968–11978. [Google Scholar] [CrossRef]
- Recchimurzo, A.; Maccabruni, F.; Uccello-Barretta, G.; Balzano, F. Quinine as a highly responsive chiral sensor for the H-1 and F-19 NMR enantiodiscrimination of N-trifluoroacetyl amino acids with free carboxyl functions. Analyst 2022, 147, 1669–1677. [Google Scholar] [CrossRef] [PubMed]
- Salgado, A.; Chankvetadze, B. Applications of nuclear magnetic resonance spectroscopy for the understanding of enantiomer separation mechanisms in capillary electrophoresis. J. Chromatogr. A 2016, 1467, 95–144. [Google Scholar] [CrossRef] [PubMed]
- Uccello-Barretta, G.; Vanni, L.; Balzano, F. Nuclear magnetic resonance approaches to the rationalization of chromatographic enantiorecognition processes. J. Chromatogr. A 2010, 1217, 928–940. [Google Scholar] [CrossRef] [PubMed]
- Benkovics, G.; Malanga, M.; Cutrone, G.; Béni, S.; Vargas-Berenguel, A.; Casas-Solvas, J.M. Facile synthesis of per(6-O-tertbutyldimethylsilyl)-α-, β-, and γ-cyclodextrin as protected intermediates for the functionalization of the secondary face of the macrocycles. Nat. Protoc. 2021, 16, 965–987. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Concentration (mM) | Molar Ratio ISO/AcSiCD | |δR − δS| | |||
|---|---|---|---|---|---|
| 1H | 19F | ||||
| AcSiαCD | CHCF3 | CHF2 | CF3 | CHF2 | |
| 5 | 6:1 | - | - | 0.006 | 0.010/0.006 |
| 10 | 3:1 | - | - | 0.007 | 0.022/0.010 |
| 15 | 2:1 | - | - | 0.007 | 0.032/0.015 |
| 30 | 1:1 | - | - | nd * | 0.054/0.025 |
| AcSiβCD | |||||
| 4.3 | 7:1 | 0.006 | 0.006 | 0.020 | 0.019/0.008 |
| 8.6 | 3.5:1 | 0.007 | 0.010 | 0.033 | 0.039/0.013 |
| 15 | 2:1 | 0.007 | 0.013 | 0.044 | 0.059/0.016 |
| 30 | 1:1 | 0.013 | 0.019 | 0.071 | 0.121/0.018 |
| AcSiγCD | |||||
| 3.75 | 8:1 | 0.049 | 0.045 | 0.075 | 0.653/0.535 |
| 7.5 | 4:1 | 0.071 | 0.065 | 0.116 | 1.12/0.894 |
| 11.25 | 3:1 | 0.081 | 0.074 | 0.137 | 1.45/1.15 |
| 15 | 2:1 | 0.081 | 0.075 | 0.143 | 1.66/1.31 |
| 30 | 1:1 | 0.083 | 0.082 | 0.165 | 2.08/1.98 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Recchimurzo, A.; Balzano, F.; Uccello Barretta, G.; Gherardi, L.; Malanga, M.; Aiello, F. Silylated-Acetylated Cyclodextrins as Chiral Sensors for the Enantiodiscrimination of Fluorinated Anesthetics. Molecules 2023, 28, 2804. https://doi.org/10.3390/molecules28062804
Recchimurzo A, Balzano F, Uccello Barretta G, Gherardi L, Malanga M, Aiello F. Silylated-Acetylated Cyclodextrins as Chiral Sensors for the Enantiodiscrimination of Fluorinated Anesthetics. Molecules. 2023; 28(6):2804. https://doi.org/10.3390/molecules28062804
Chicago/Turabian StyleRecchimurzo, Alessandra, Federica Balzano, Gloria Uccello Barretta, Luca Gherardi, Milo Malanga, and Federica Aiello. 2023. "Silylated-Acetylated Cyclodextrins as Chiral Sensors for the Enantiodiscrimination of Fluorinated Anesthetics" Molecules 28, no. 6: 2804. https://doi.org/10.3390/molecules28062804
APA StyleRecchimurzo, A., Balzano, F., Uccello Barretta, G., Gherardi, L., Malanga, M., & Aiello, F. (2023). Silylated-Acetylated Cyclodextrins as Chiral Sensors for the Enantiodiscrimination of Fluorinated Anesthetics. Molecules, 28(6), 2804. https://doi.org/10.3390/molecules28062804