

Unraveling Binding Mechanism and Stability of Urease Inhibitors: A QM/MM MD Study


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction

2. Results and Discussion
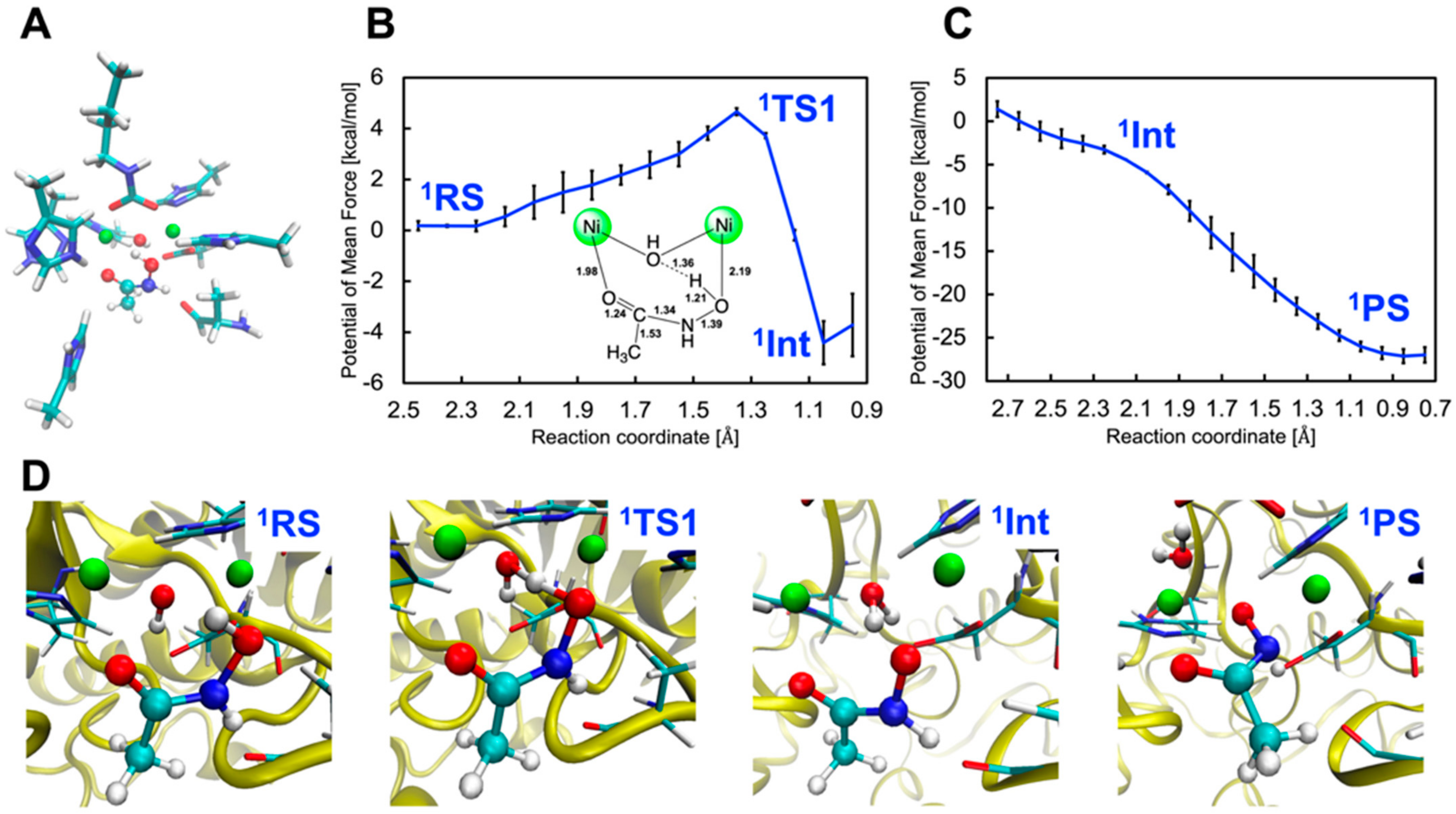
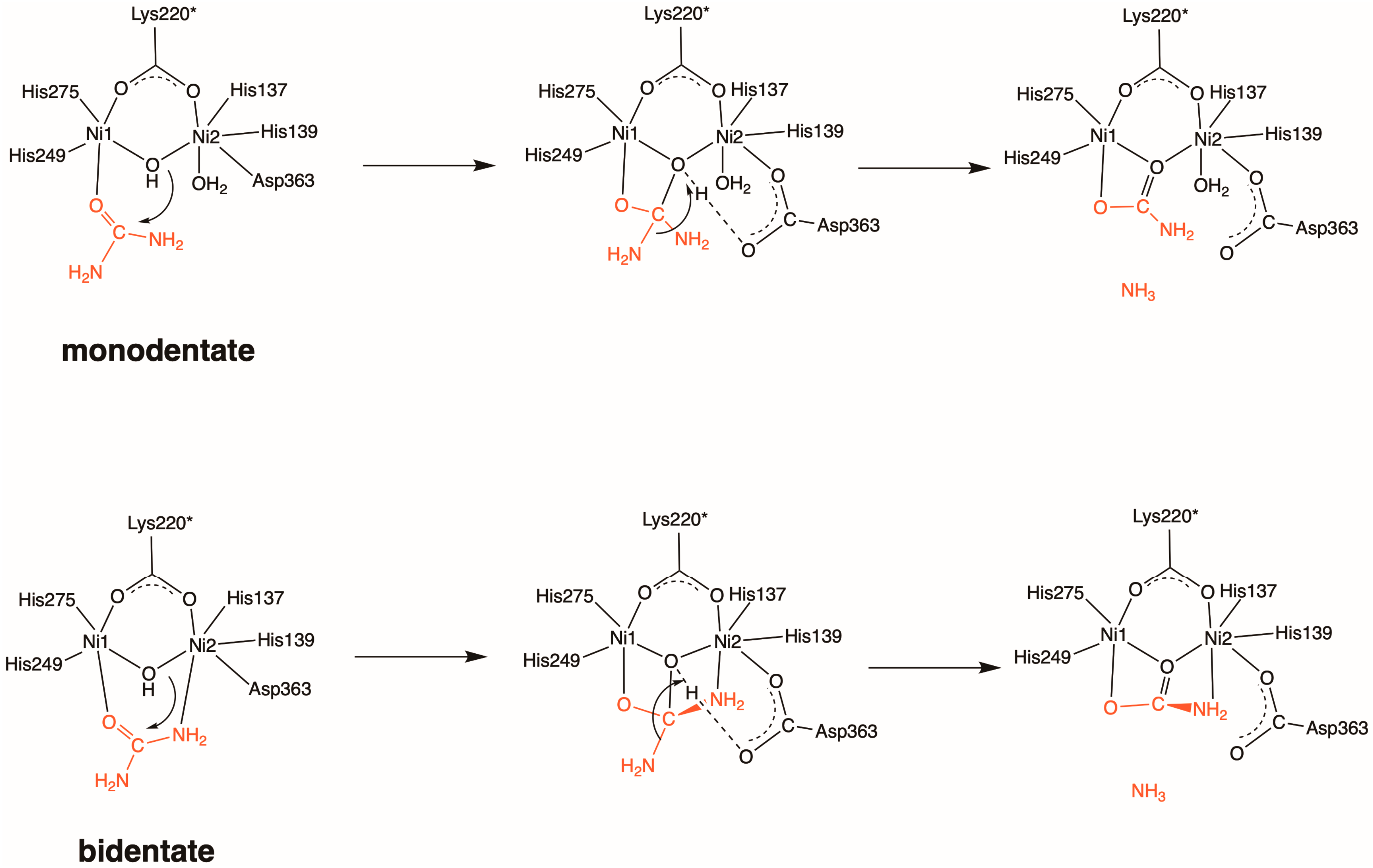
2.1. Inhibition by AHA (1)

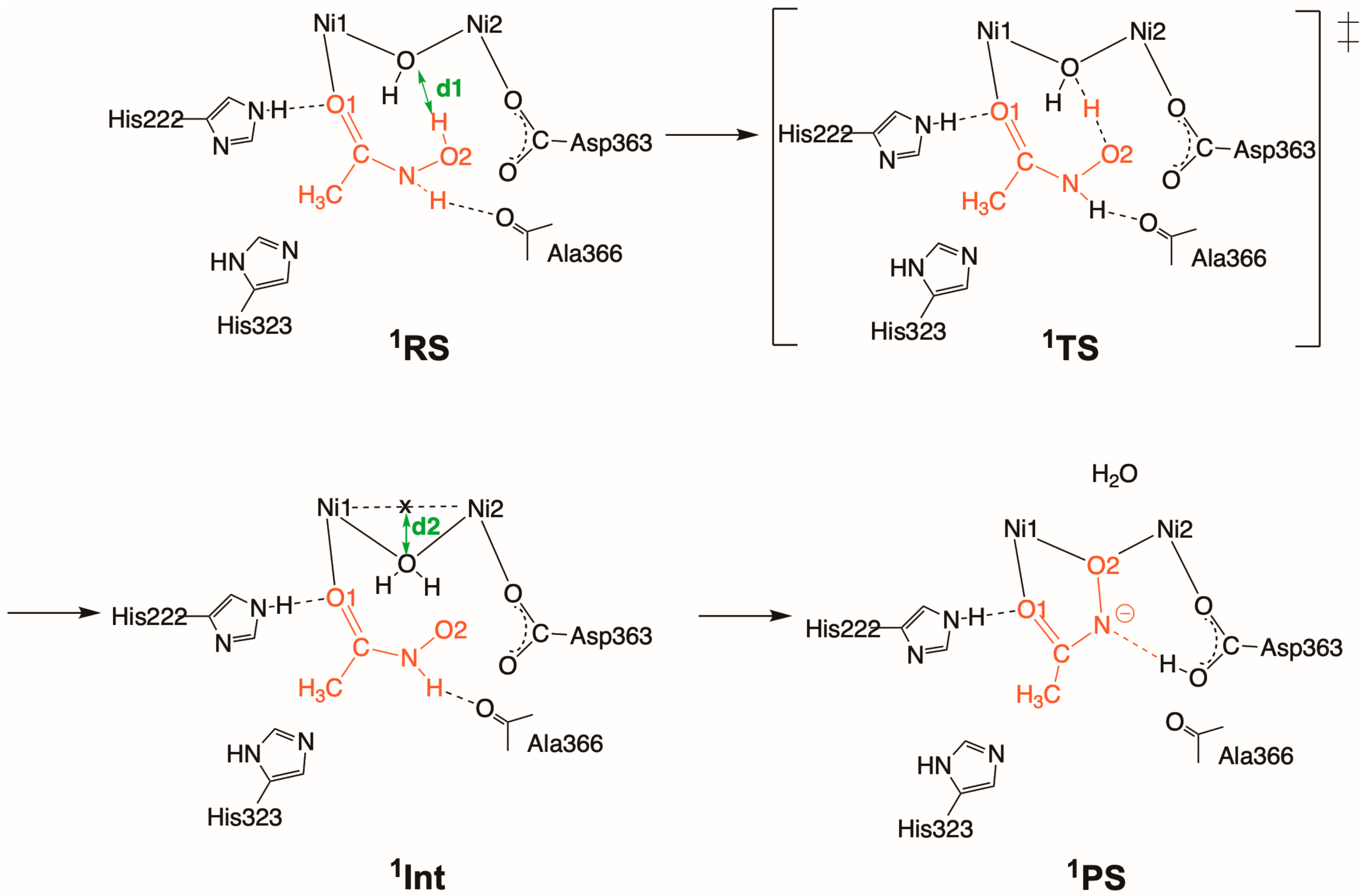
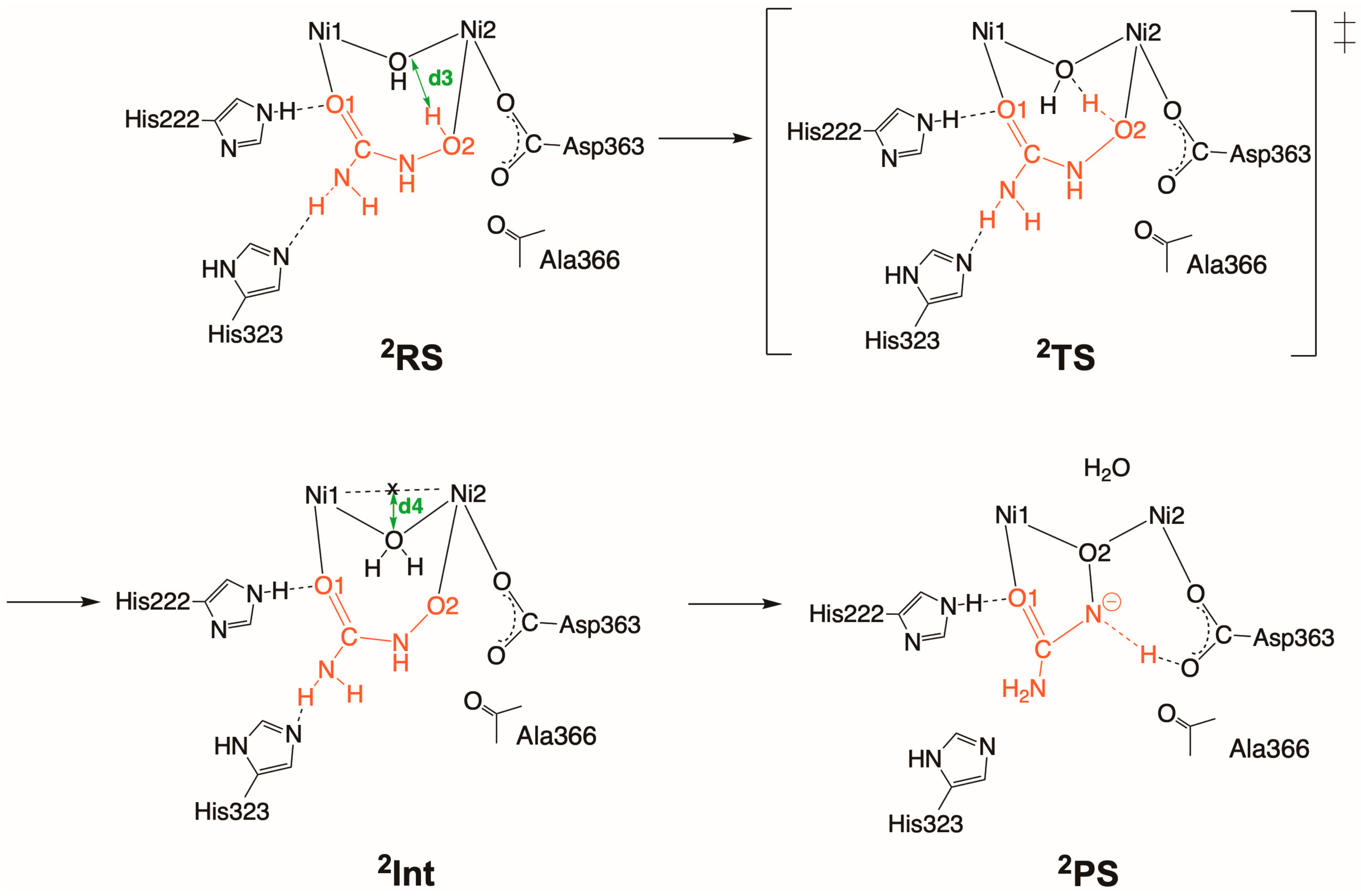
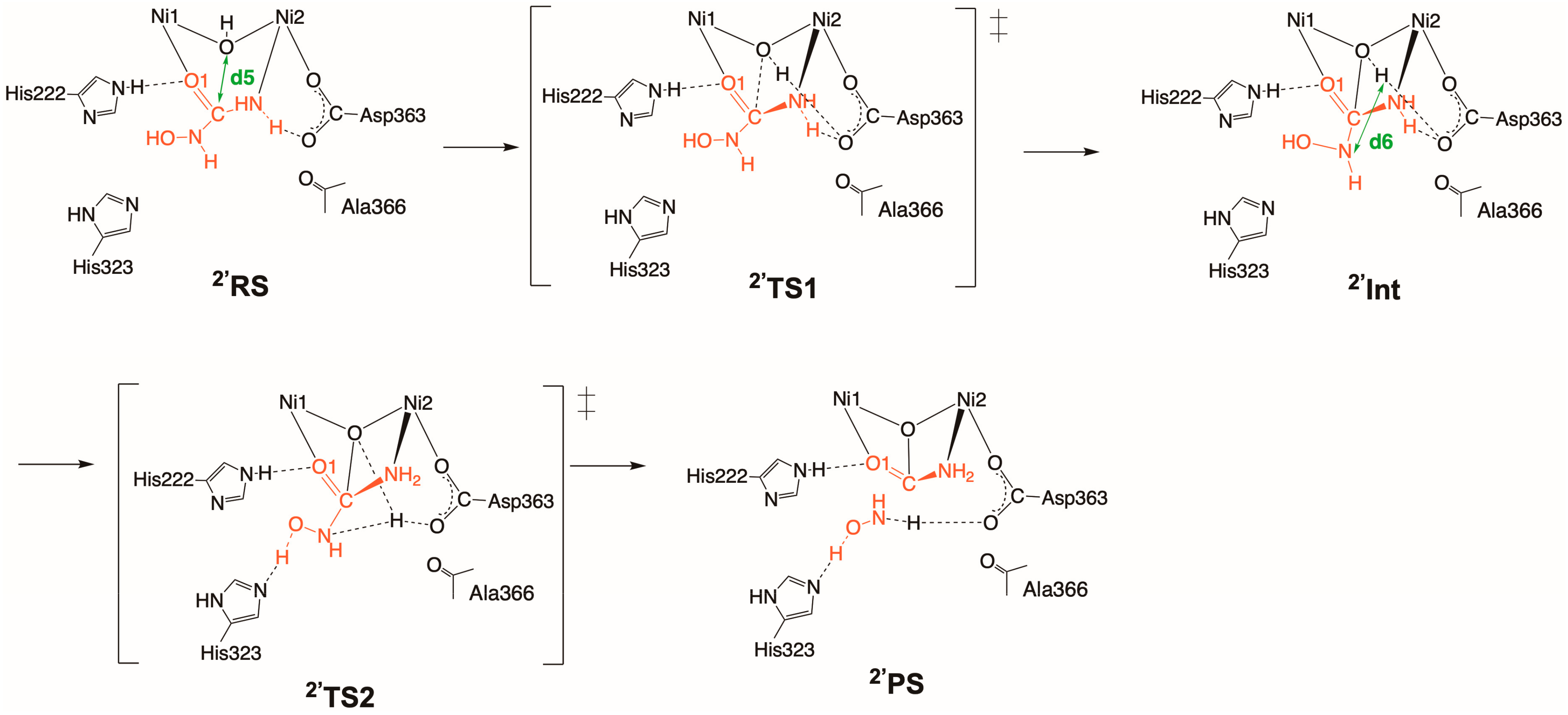
2.2. Inhibition by HU (2 and 2′)
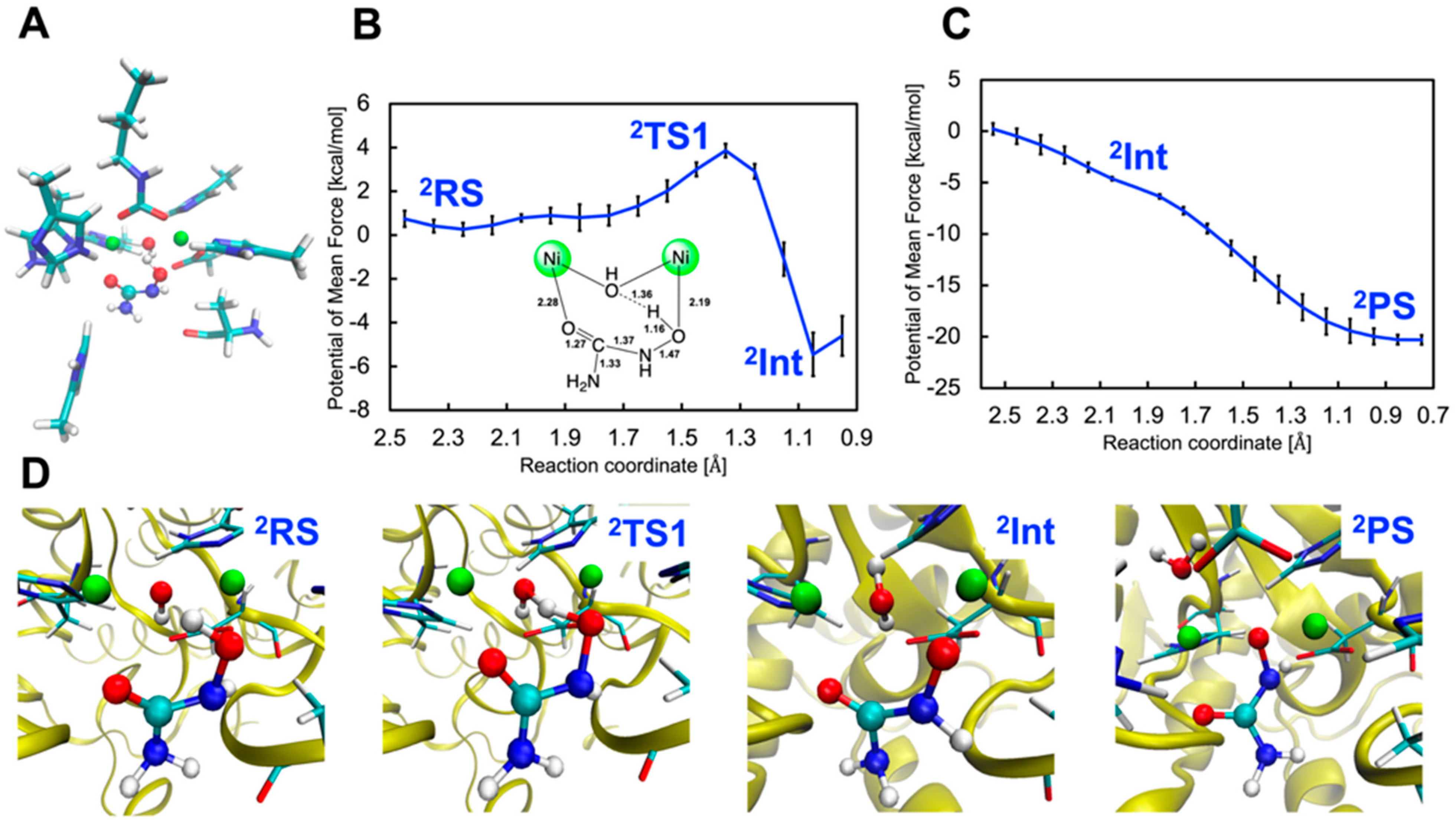
2.2.1. Binding Mode 2
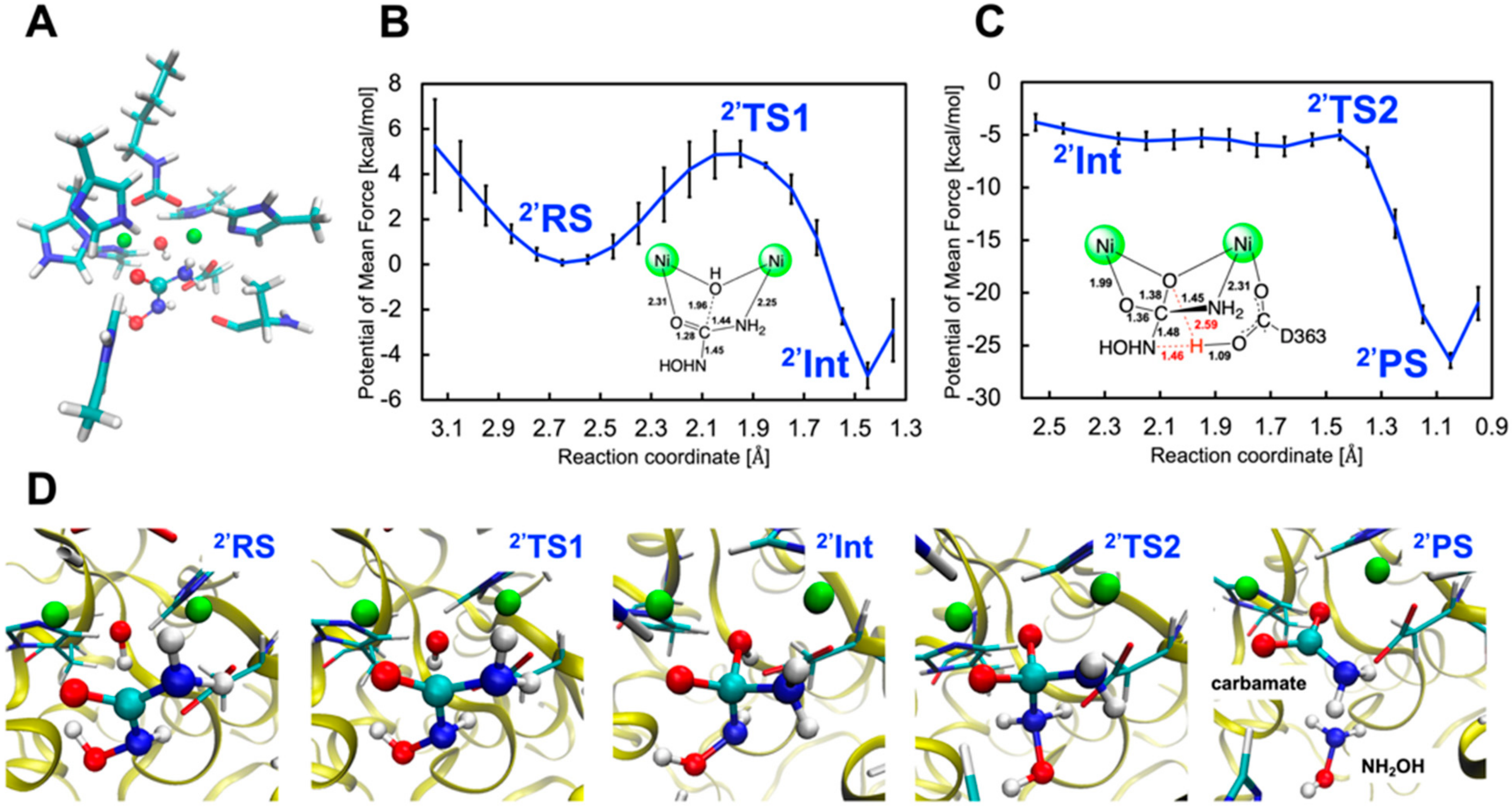
2.2.2. Binding Mode 2′

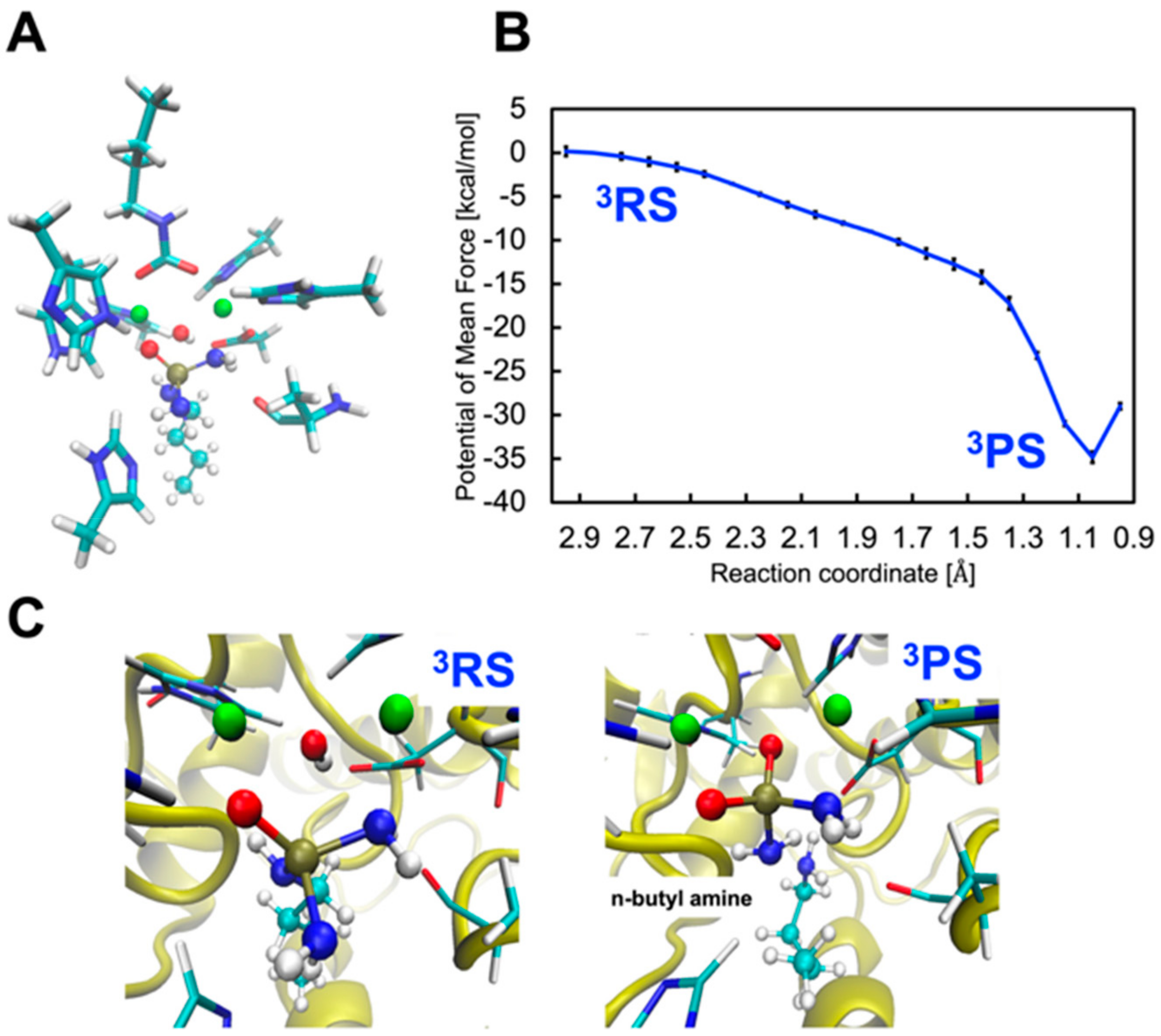
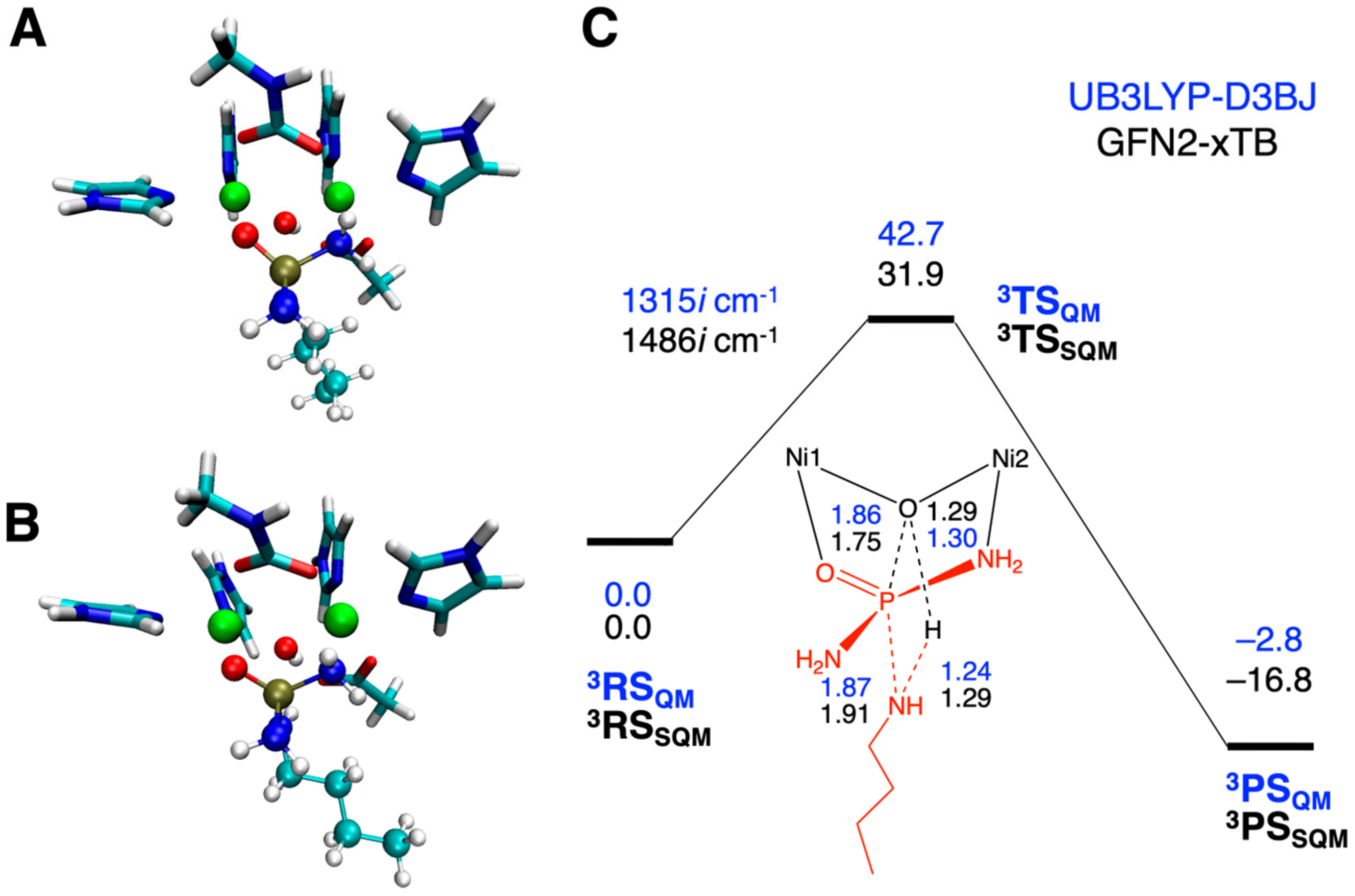
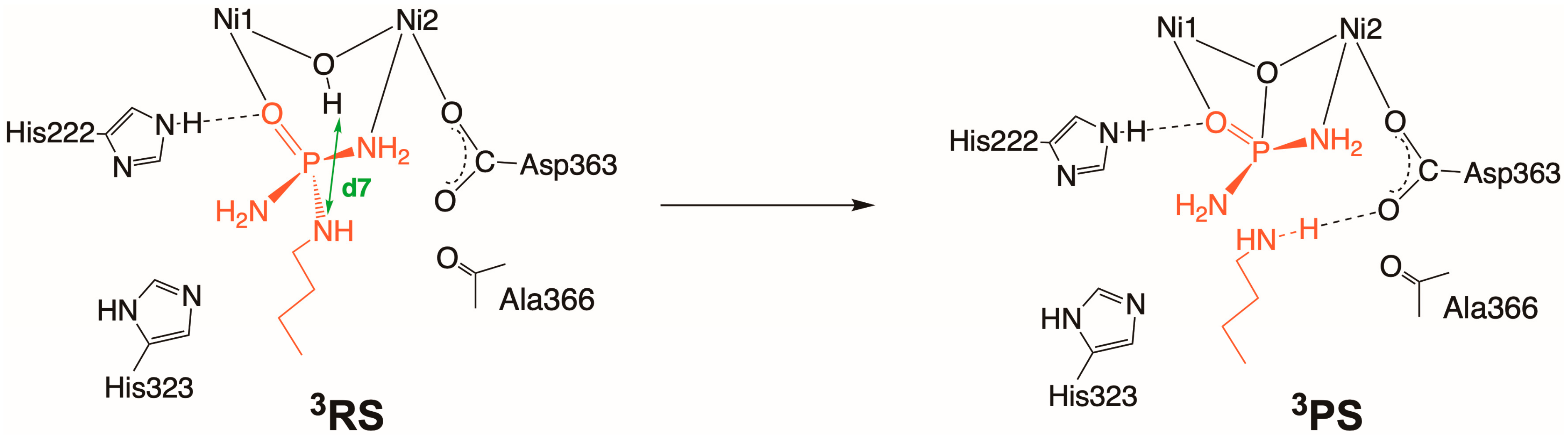
2.3. Inhibition by NBPTO (3)
3. Methods
3.1. Model Preparation and Classical MD Simulations
3.2. QM/MM MD Simulations
3.3. QM-Only Cluster Model Approach
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Klimczyk, M.; Siczek, A.; Schimmelpfennig, L. Improving the Efficiency of Urea-based Fertilization Leading to Reduction in Ammonia Emission. Sci. Total Environ. 2021, 771, 145483. [Google Scholar] [CrossRef] [PubMed]
- Mazzei, L.; Musiani, F.; Ciurli, S. The Structure-based Reaction Mechanism of Urease, a Nickel Dependent Enzyme: Tale of a Long Debate. J. Biol. Inorg. Chem. 2020, 25, 829–845. [Google Scholar] [CrossRef] [PubMed]
- Modolo, L.V.; da-Silva, C.J.; Brandão, D.S.; Chaves, I.S. A Minireview on What We Have Learned about Urease Inhibitors of Agricultural Interest Since Mid-2000s. J. Adv. Res. 2018, 13, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, J.C. The Emerging Role of Urease as a General Microbial Virulence Factor. PLoS Pathog. 2014, 10, e1004062. [Google Scholar] [CrossRef] [PubMed]
- Paulot, F.; Jacob, D.J. Hidden Cost of U.S. Agricultural Exports: Particulate Matter from Ammonia Emissions. Environ. Sci. Technol. 2014, 48, 903–908. [Google Scholar] [CrossRef] [PubMed]
- Crowe, S.E. Helicobacter pylori infection. N. Engl. J. Med. 2019, 380, 1158–1165. [Google Scholar] [CrossRef]
- Alipour, M. Molecular Mechanism of Helicobacter pylori-Inducted Gastric Cancer. J. Gastrointest. Canc. 2021, 52, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Imran, M.; Waqar, S.; Ogata, K.; Ahmed, M.; Noreen, Z.; Javed, S.; Bibi, N.; Bokhari, H.; Amjad, A.; Muddassar, M. Identification of Novel Bacterial Urease Inhibitors through Molecular Shape and Structure Based Virtual Screening Approaches. RSC Adv. 2020, 10, 16061–16070. [Google Scholar] [CrossRef]
- Fishbein, W.N.; Carbon, P.P. Urease Catalysis: I. Stoichiometry, Specificity, and Kinetics of a Second Substrate: Hydroxyurea. J. Biol. Chem. 1965, 240, 2402–2406. [Google Scholar] [CrossRef]
- Fishbein, W.N.; Carbon, P.P. Urease Catalysis: II. Inhibition of the Enzyme by Hydroxyurea, Hydroxylamine, and Acetohydroxamic Acid. J. Biol. Chem. 1965, 240, 2407–2414. [Google Scholar] [CrossRef]
- Dixon, N.E.; Gazzola, C.; Watters, J.J.; Blakeley, R.L.; Zerner, B. Inhibition of Jack Bean Urease (EC 3.5.1.5) by Acetohydroxamic Acid and by Phosphoramidate. Equivalent Weight for Urease. J. Am. Chem. Soc. 1975, 97, 4130–4131. [Google Scholar] [CrossRef]
- Griffith, D.P.; Gibson, J.R.; Clinton, C.W.; Musher, D.M. Acetohydroxamic Acid: Clinical Studies of a Urease Inhibitor in Patients With Staghorn Renal Calculi. J. Urol. 1978, 119, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Stemmler, A.J.; Kampf, J.W.; Kirk, M.L.; Pecoraro, V.L. A Model for the Inhibition of Urease by Hydroxamates. J. Am. Chem. Soc. 1995, 117, 6368–6369. [Google Scholar] [CrossRef]
- Benini, S.; Rypniewski, W.R.; Wilson, K.S.; Miletti, S.; Ciurli, S.; Mangani, S. A New Proposal for Urease Mechanism Based on the Crystal Structures of the Native and Inhibited Enzyme from Bacillus pasteurii: Why Urea Hydrolysis Costs Two Nickels. Structure 1999, 7, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Benini, S.; Rypniewski, W.R.; Wilson, K.S.; Miletti, S.; Ciurli, S.; Mangani, S. The Complex of Bacillus pasteurii Urease with Acetohydroxamate Anion from X-ray Data at 1.55 Å Resolution. J. Biol. Inorg. Chem. 2000, 5, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Mazzei, L.; Cianci, M.; Contaldo, U.; Ciurli, S. Insights into Urease Inhibition by N-(n-Butyl) Phosphoric Triamide through an Integrated Structural and Kinetic Approach. J. Agric. Food Chem. 2019, 67, 2127–2138. [Google Scholar] [CrossRef]
- Kot, M.; Zaborska, W.; Orlinska, K. Inhibition of Jack Bean Urease by N-(n- butyl)thiophosphorictriamide and N-(n-butyl)phosphorictriamide: Determination of the Inhibition Mechanism. J. Enzyme Inhib. 2001, 16, 507–516. [Google Scholar] [CrossRef]
- Mazzei, L.; Cianci, M.; Benini, S.; Ciurli, S. The Structure of the Elusive Urease-Urea Complex Unveils the Mechanism of a Paradigmatic Nickel-Dependent Enzyme. Angew. Chem. Int. Ed. 2019, 58, 7415–7419. [Google Scholar] [CrossRef]
- Svane, S.; Sigurdarson, J.J.; Finkenwirth, F.; Eitinger, T.; Karring, H. Inhibition of Urease Activity by Different Compounds Provides Insight into the Modulation and Association of Bacterial Nickel Import and Ureolysis. Sci. Rep. 2020, 10, 8503. [Google Scholar] [CrossRef]
- Pedrood, K.; Azizian, H.; Montazer, M.N.; Moazzam, A.; Asadi, M.; Montazeri, H.; Biglar, M.; Zamani, M.; Larijani, B.; Zomorodian, K.; et al. Design and Synthesis of New N-thioacylated Ciprofloxacin Derivatives as Urease Inhibitors with Potential Antibacterial Activity. Sci. Rep. 2022, 12, 13827. [Google Scholar] [CrossRef]
- Chen, W.; Li, Y.; Cui, Y.; Zhang, X.; Zhu, H.-L.; Zeng, Q. Synthesis, Molecular Docking and Biological Evaluation of Schiff Base Transition Metal Complexes as Potential Urease Inhibitors. Eur. J. Med. Chem. 2010, 45, 4473–4478. [Google Scholar] [CrossRef]
- Cui, Y.M.; Li, Y.; Cai, Y.J.; Chen, W.; Zhu, H.L. Synthesis, Molecular Docking, and Activity of Schiff-base Copper(II) Complex with N-n-butylsalicylaldiminate as Helicobacter Pylori Urease Inhibitor. J. Coord. Chem. 2011, 64, 610–616. [Google Scholar] [CrossRef]
- Moghadam, E.; Al-Sadi, A.; Talebi, M.; Amanlou, M.; Shongwe, M.; Amini, M.; Abdel-Jalil, R. Piperazine-based Semicarbazone Derivatives as Potent Urease Inhibitors: Design, Synthesis, and Bioactivity Screening. Lett. Drug Des. Discov. 2022, 19, 1111–1120. [Google Scholar]
- Tanaka, T.; Kawase, M.; Tani, S. α-Hydroxyketones as Inhibitors of Urease. Bioorg. Med. Chem. 2004, 12, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Milo, S.; Heylen, R.A.; Glancy, J.; Williams, G.T.; Patenall, B.L.; Hathaway, H.J.; Thet, N.T.; Allinson, S.L.; Laabei, M.; Jenkins, A.T.A. A small-molecular Inhibitor against Proteus Mirabilis Urease to Treat Catheter-associated Urinary Tract Infections. Sci. Rep. 2021, 11, 3726. [Google Scholar] [CrossRef] [PubMed]
- Jabri, E.; Carr, M.B.; Hausinger, R.P.; Karplus, P.A. The Crystal Structure of Urease from Klebsiella aerogenes. Science 1995, 268, 998–1004. [Google Scholar] [CrossRef]
- Karplus, P.A.; Pearson, M.A.; Hausinger, R.P. 70 Years of Crystalline Urease: What Have We Learned? Acc. Chem. Res. 1997, 30, 330–337. [Google Scholar] [CrossRef]
- Suárez, D.; Díaz, N.; Merz, K.M. Ureases: Quantum Chemical Calculations on Cluster Models. J. Am. Chem. Soc. 2003, 125, 15324–15337. [Google Scholar] [CrossRef]
- Estiu, G.; Merz, K.M. Competitive Hydrolytic and Elimination Mechanisms in the Urease Catalyzed Decomposition of Urea. J. Phys. Chem. B 2007, 111, 10263–10274. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, H.; Nordlander, E. Computational Modeling of the Mechanism of Urease. Bioinorg. Chem. Appl. 2010, 2010, 364891. [Google Scholar] [CrossRef]
- Saito, T.; Takano, Y. QM/MM Molecular Dynamics Simulations Revealed Catalytic Mechanism of Urease. J. Phys. Chem. B 2022, 126, 2087–2097. [Google Scholar] [CrossRef]
- Åqvist, J.; Kazemi, M.; Isaksen, G.V.; Brandsdal, B.O. Entropy and Enzyme Catalysis. Acc. Chem. Res. 2017, 50, 199–207. [Google Scholar] [CrossRef]
- Marlier, J.F.; Robins, L.I.; Tucks, K.A.; Rawlings, J.; Anderson, M.A.; Cleland, W.W. A Kinetic and Isotope Investigation of the Urease-Catalyzed Hydrolysis of Hydroxyurea. Biochemistry 2010, 49, 8213–8219. [Google Scholar] [CrossRef] [PubMed]
- Marlier, J.F.; Fogle, E.J.; Cleand, W.W. A Heavy-Atom Isotope Effect and Kinetics Investigation of the Hydrolysis of Semicarbazide by Urease from Jack Bean (Canavalia ensiformis). Biochemistry 2008, 47, 11158–11163. [Google Scholar] [CrossRef] [PubMed]
- Søndergaard, C.R.; Olsson, M.H.M.; Rostkowski, M.; Jensen, J.H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Maruno, Y.; Shoji, M.; Koizumi, K.; Nishiyama, Y.; Kitagawa, Y.; Kawakami, T.; Okumura, M.; Yamaguchi, K. Theoretical Studies on Magnetic Interactions between Ni (II) Ions in Urease. Polyhedron 2005, 24, 2778–2783. [Google Scholar] [CrossRef]
- Kitagawa, Y.; Saito, T.; Ito, M.; Nakanishi, Y.; Shoji, M.; Koizumi, K.; Yamanaka, S.; Kawakami, T.; Okumura, M.; Yamaguchi, K. Geometry Optimization Method Based on Approximate Spin Projection and its Application to F2, CH2, CH2OO, and Active Site of Urease. Int. J. Quantum Chem. 2007, 107, 3094–3102. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional Exchange-energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Breneman, C.M.; Wiberg, K.B. Determining Atom-centered Monopoles from Molecular Electrostatic Potentials. The Need for High Sampling Density in Formamide Conformational Analysis. J. Comput. Chem. 1990, 11, 361–373. [Google Scholar] [CrossRef]
- Neese, F. The ORCA Program System. WIREs Comput. Mol. Sci. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Li, P.; Roberts, B.P.; Chakravorty, D.K.; Merz, K.M., Jr. Rational Design of Particle Mesh Ewald Compatible Lennard-Jones Parameters for + 2 Metal Cations in Explicit Solvent. J. Chem. Theory Comput. 2013, 9, 2733–2748. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field: A Force Field for Drug-like Molecules Compatible with the CHARMM All-atom Additive Biological Force Fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald: An N·log (N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Nelson, M.T.; Humphrey, W.; Gursoy, A.; Dalke, A.; Kalé, L.V.; Skeel, R.D.; Schulten, K. NAMD: A Parallel, Object-Oriented Molecular Dynamics Program. Int. J. High Perform. Comput. Appl. 1996, 10, 251–268. [Google Scholar] [CrossRef]
- Huang, J.; MacKerell, A.D., Jr. CHARMM36 All-atom Additive Protein Force Field: Validation Based on Comparison to NMR Data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [PubMed]
- Melo, M.C.; Bernardi, R.C.; Rudack, T.; Scheurer, M.; Riplinger, C.; Phillips, J.C.; Maia, J.D.; Rocha, G.B.; Ribeiro, J.V.; Stone, J.E.; et al. NAMD Goes Quantum: An Integrative Suite for Hybrid Simulations. Nat. Methods 2018, 15, 351–354. [Google Scholar] [CrossRef]
- Bannwarth, C.; Ehlert, S.; Grimme, S. GFN2-xTB–An Accurate and Broadly Parametrized Self-Consistent Tight-Binding Quantum Chemical Method with Multipole Electrostatics and Density-Dependent Dispersion Contributions. J. Chem. Theory Comput. 2019, 15, 1652–1671. [Google Scholar] [CrossRef]
- Bursch, M.; Neugebauer, H.; Grimme, S. Structure Optimisation of Large Transition-Metal Complexes with Extended Tight-Binding Methods. Angew. Chem. Int. Ed. 2019, 58, 11078–11087. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. [Google Scholar] [CrossRef]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods VI: More modifications to the NDDO approximations and re-optimization of parameters. J. Mol. Model. 2013, 19, 1–32. [Google Scholar] [CrossRef]
- Singh, U.C.; Kollman, P.A. A Combined Ab initio Quantum Mechanical and Molecular Mechanical Method for Carrying out Simulations on Complex Molecular Systems: Applications to the CH3Cl + Cl− Exchange Reaction and Gas Phase Protonation of Polyethers. J. Comput. Chem. 1986, 7, 718–730. [Google Scholar] [CrossRef]
- Bussi, G.; Laio, A.; Parrinello, M. Equilibrium Free Energies from Nonequilibrium Metadynamics. Physi. Rev. Lett. 2006, 96, 090601. [Google Scholar] [CrossRef] [PubMed]
- Ensing, B.; Laio, A.; Parrinello, M.; Klein, M. A Recipe for the Computation of the Free Energy Barrier and the Lowest Free Energy Path of Concerted Reactions. J. Phys. Chem. B 2005, 109, 6676–6687. [Google Scholar] [CrossRef]
- Wang, B.; Cao, Z.; Rovira, C.; Song, J.; Shaik, S. Fenton-Derived OH Radicals Enable the MPnS Enzyme to Convert 2-Hydroxyethylphosphonate to Methylphosphonate: Insights from Ab Initio QM/MM MD Simulations. J. Am. Chem. Soc. 2019, 141, 9284–9291. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M.; Görlich, S.; Longbotham, J.E.; Scrutton, N.S.; Hay, S.; Moliner, V.; Tuñón, I. Convergence of Theory and Experiment on the Role of Preorganization, Quantum Tunneling, and Enzyme Motions into Flavoenzyme-Catalyzed Hydride Transfer. ACS Catal. 2017, 7, 3190–3198. [Google Scholar] [CrossRef] [PubMed]
- Dohm, S.; Bursch, M.; Hansen, A.; Grimme, S. Semiautomated Transition State Localization for Organometallic Complexes with Semiempirical Quantum Chemical Methods. J. Chem. Theory Comput. 2020, 16, 2002–2012. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suenaga, S.; Takano, Y.; Saito, T. Unraveling Binding Mechanism and Stability of Urease Inhibitors: A QM/MM MD Study. Molecules 2023, 28, 2697. https://doi.org/10.3390/molecules28062697
Suenaga S, Takano Y, Saito T. Unraveling Binding Mechanism and Stability of Urease Inhibitors: A QM/MM MD Study. Molecules. 2023; 28(6):2697. https://doi.org/10.3390/molecules28062697
Chicago/Turabian StyleSuenaga, Shunya, Yu Takano, and Toru Saito. 2023. "Unraveling Binding Mechanism and Stability of Urease Inhibitors: A QM/MM MD Study" Molecules 28, no. 6: 2697. https://doi.org/10.3390/molecules28062697
APA StyleSuenaga, S., Takano, Y., & Saito, T. (2023). Unraveling Binding Mechanism and Stability of Urease Inhibitors: A QM/MM MD Study. Molecules, 28(6), 2697. https://doi.org/10.3390/molecules28062697