
Catalytic Asymmetric α-Functionalization of α-Branched Aldehydes
 and
and
Abstract

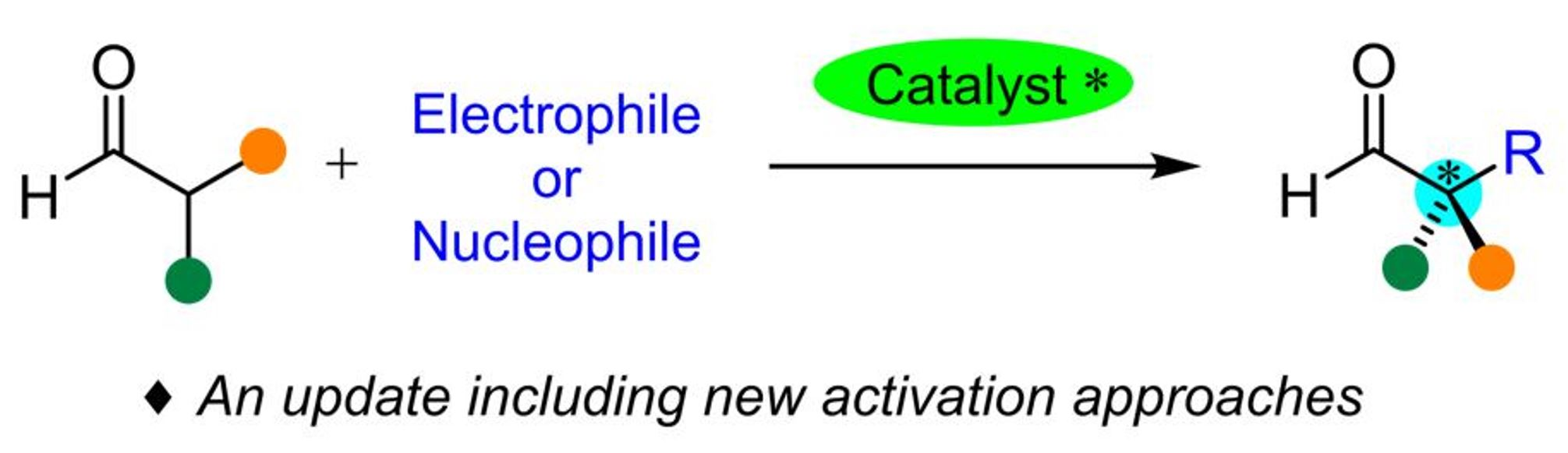{kind=link}
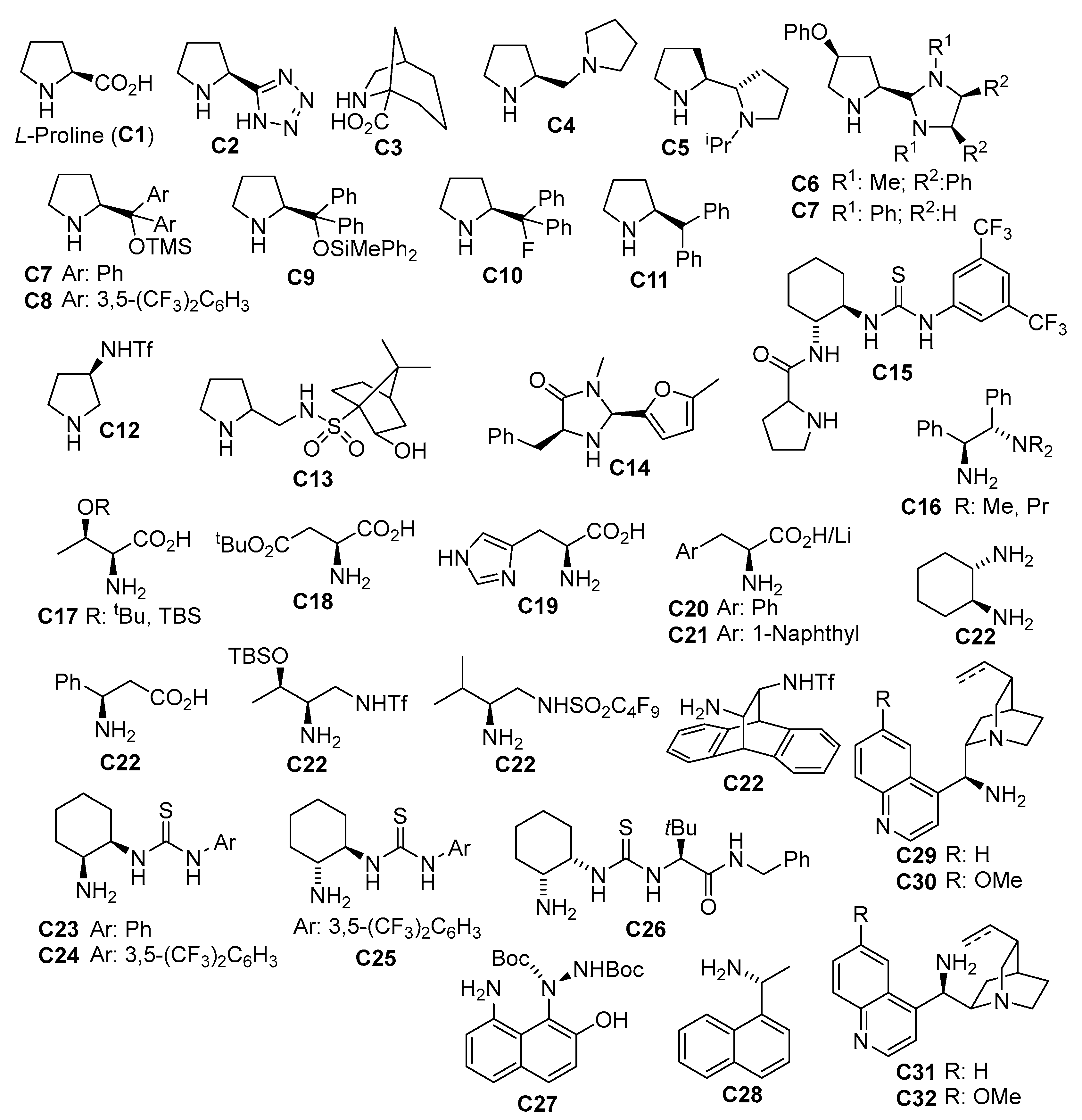{kind=link}
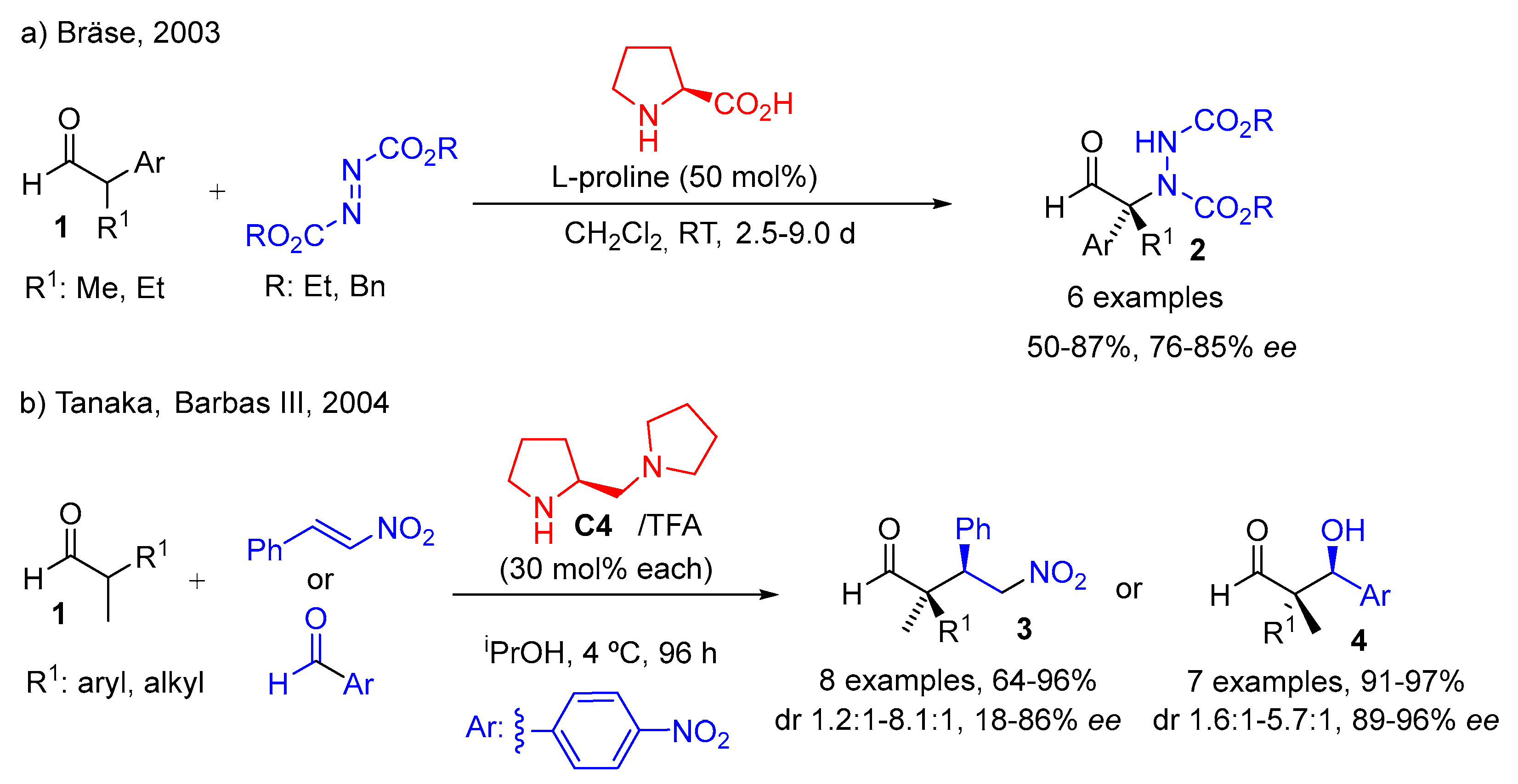{kind=link}
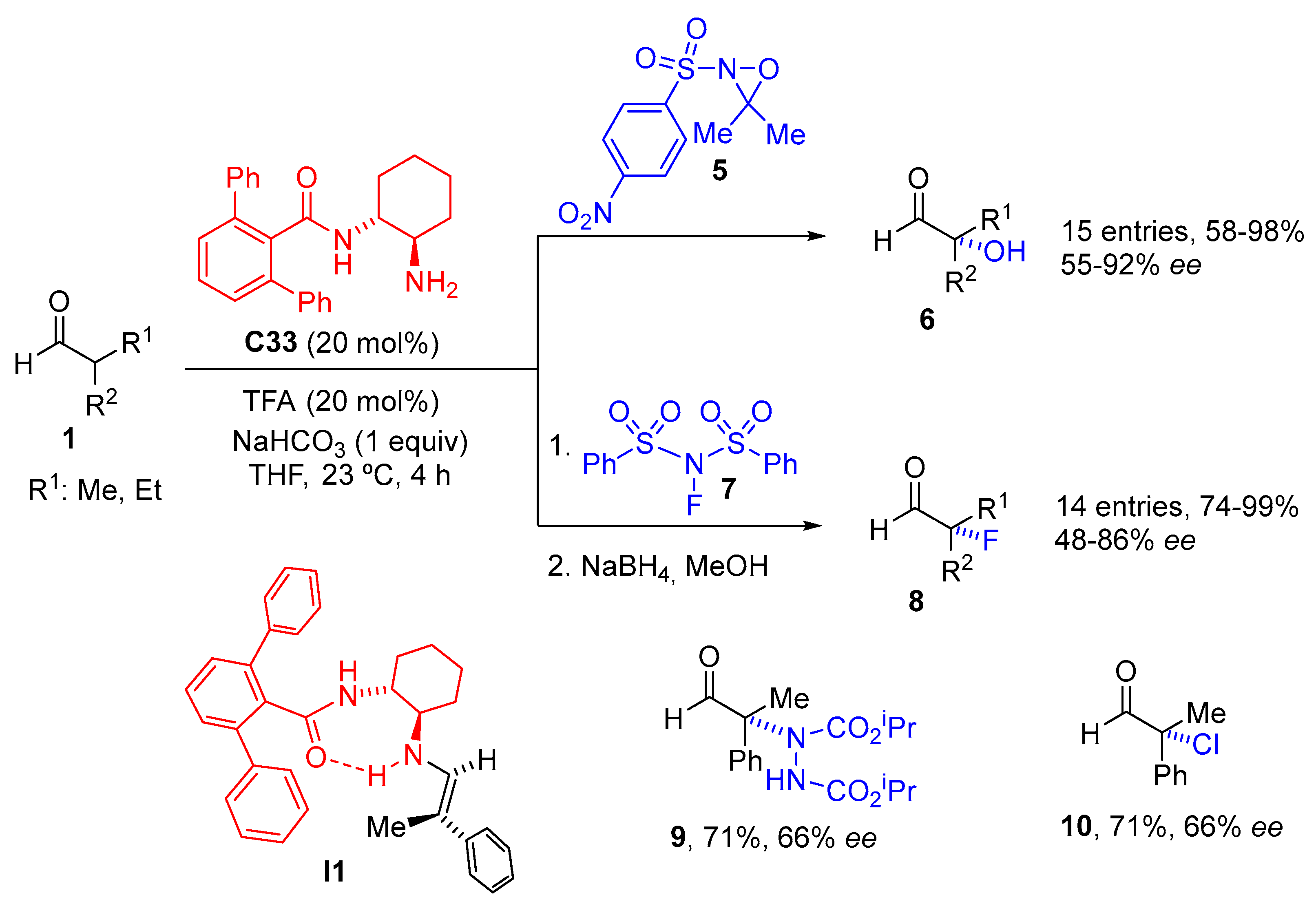{kind=link}
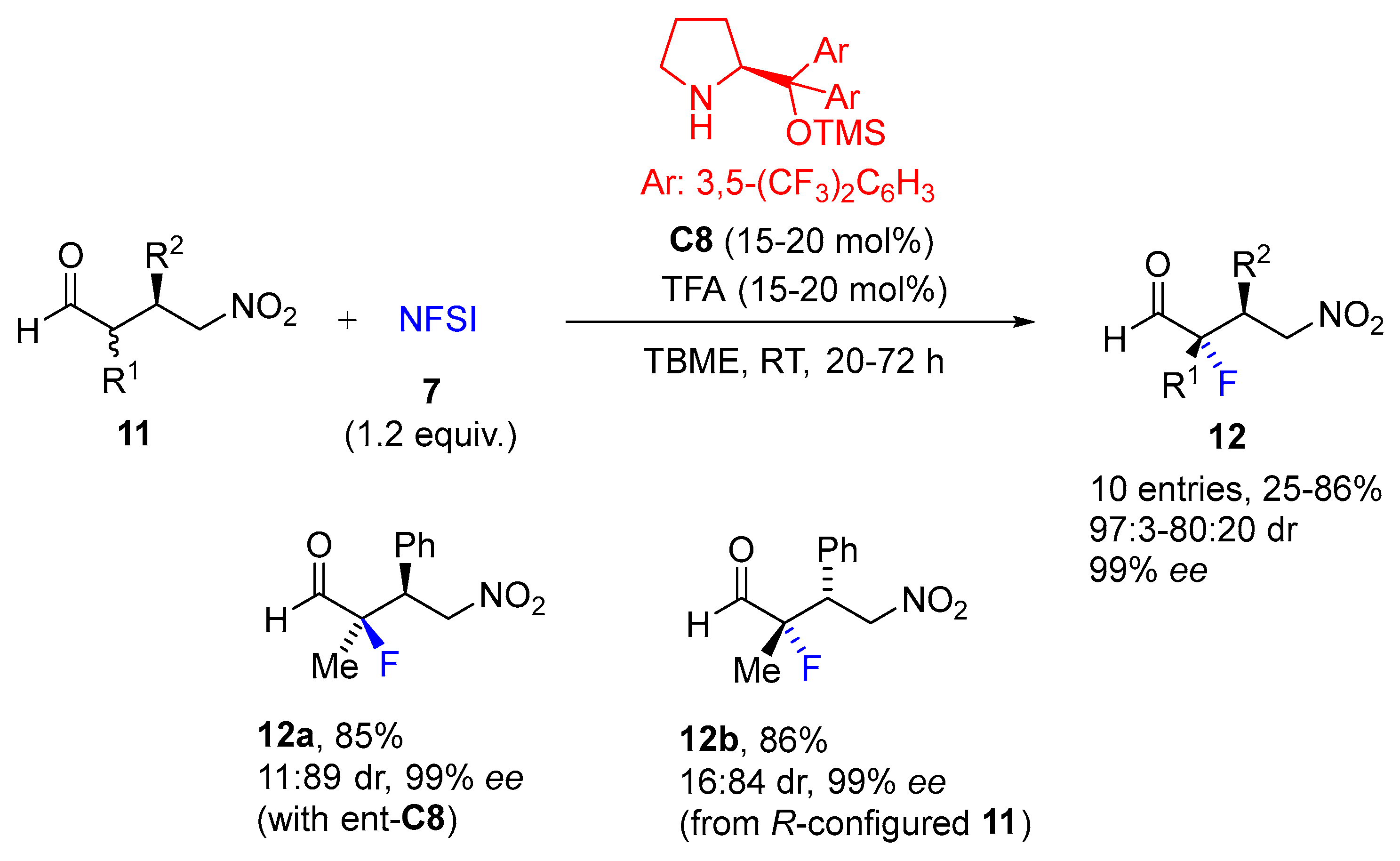{kind=link}
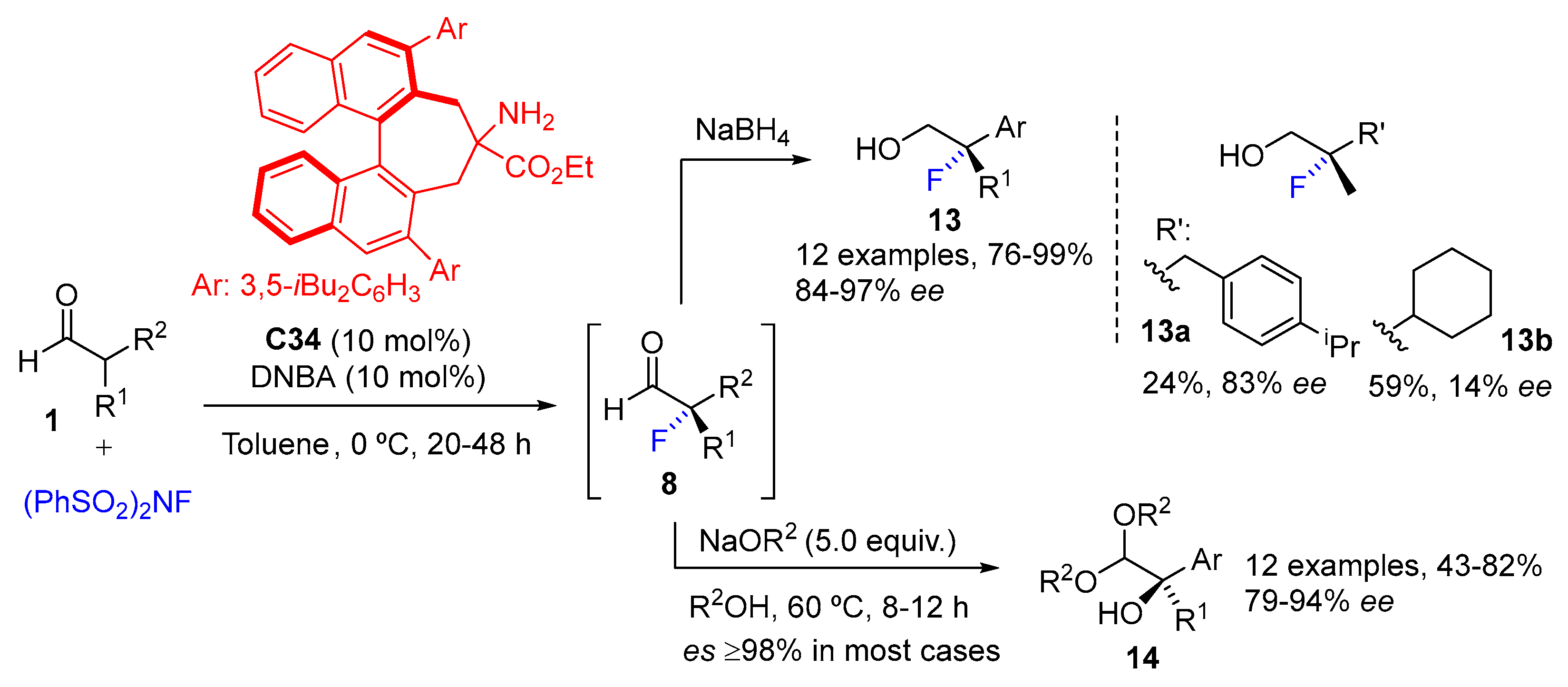{kind=link}
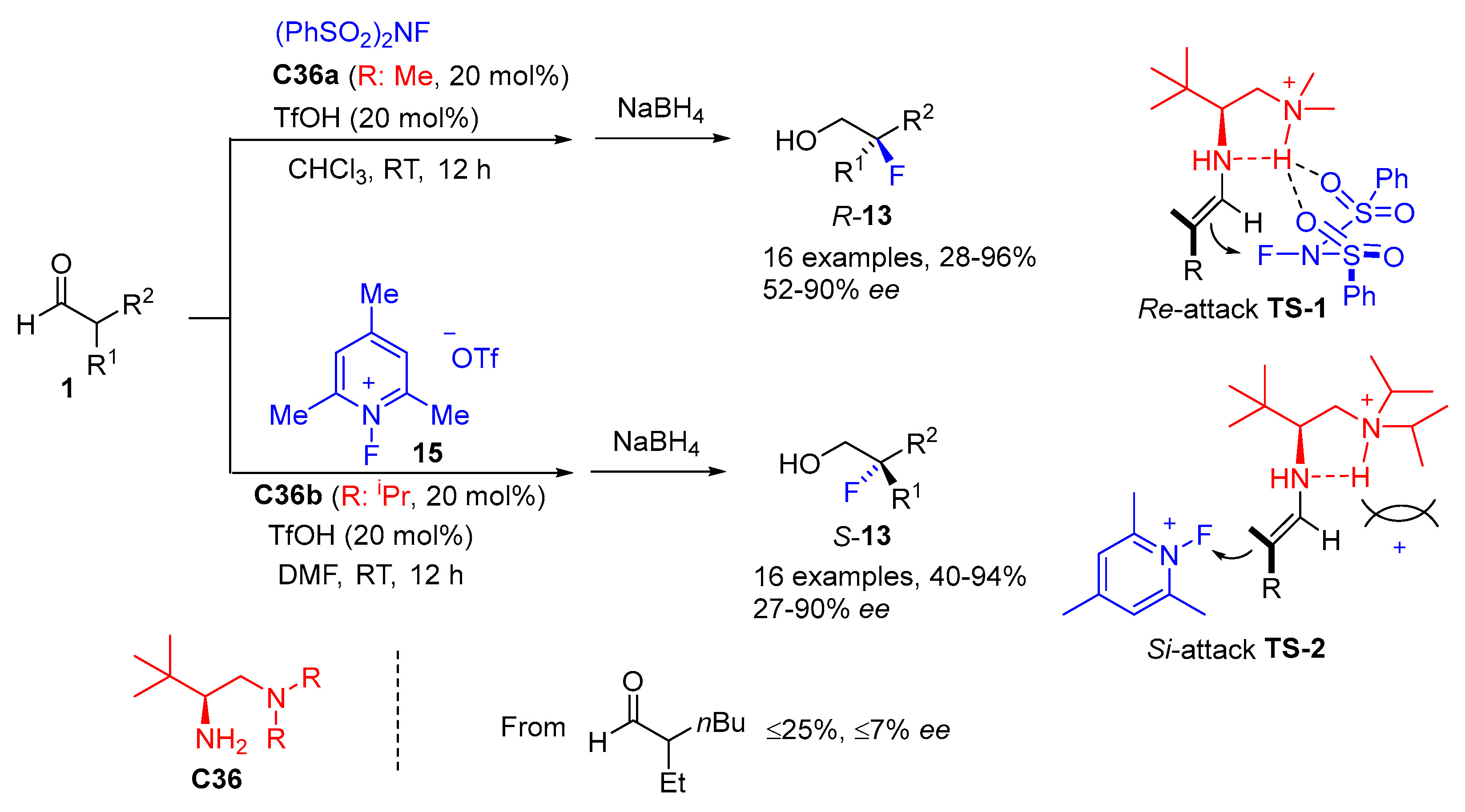{kind=link}
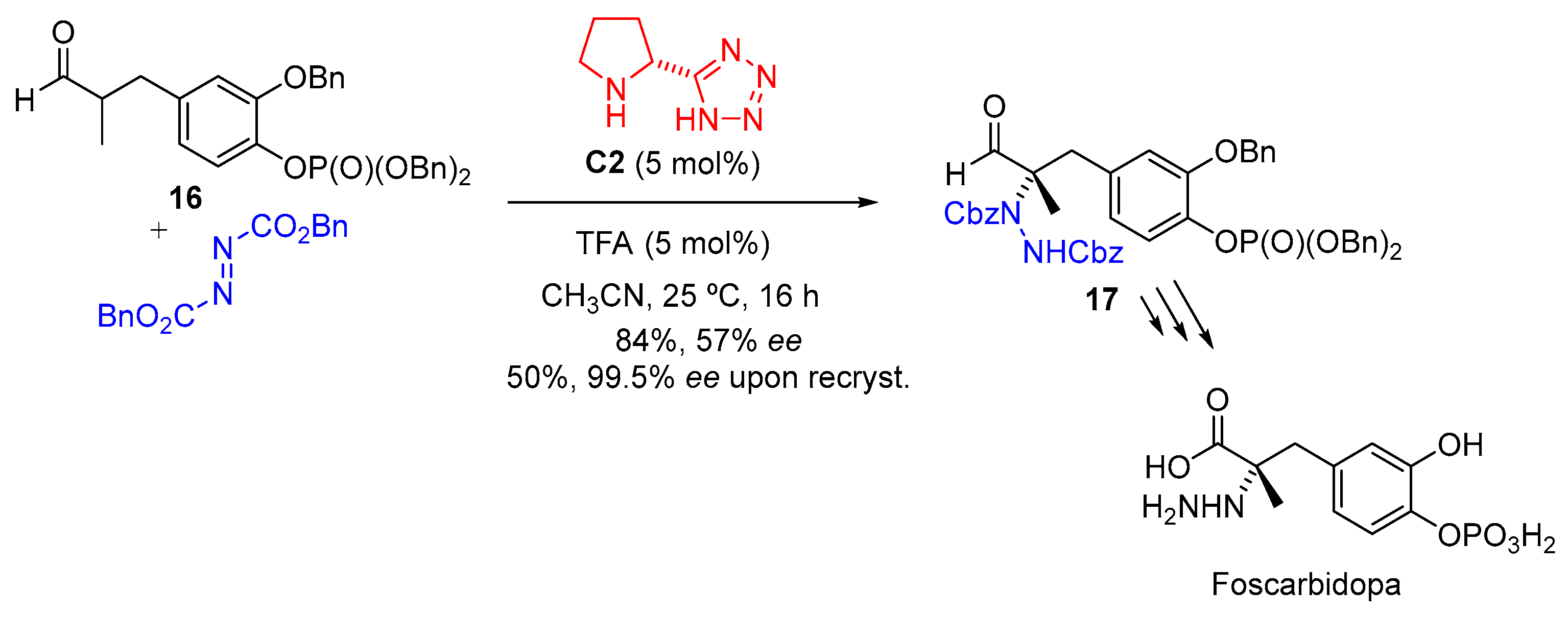{kind=link}
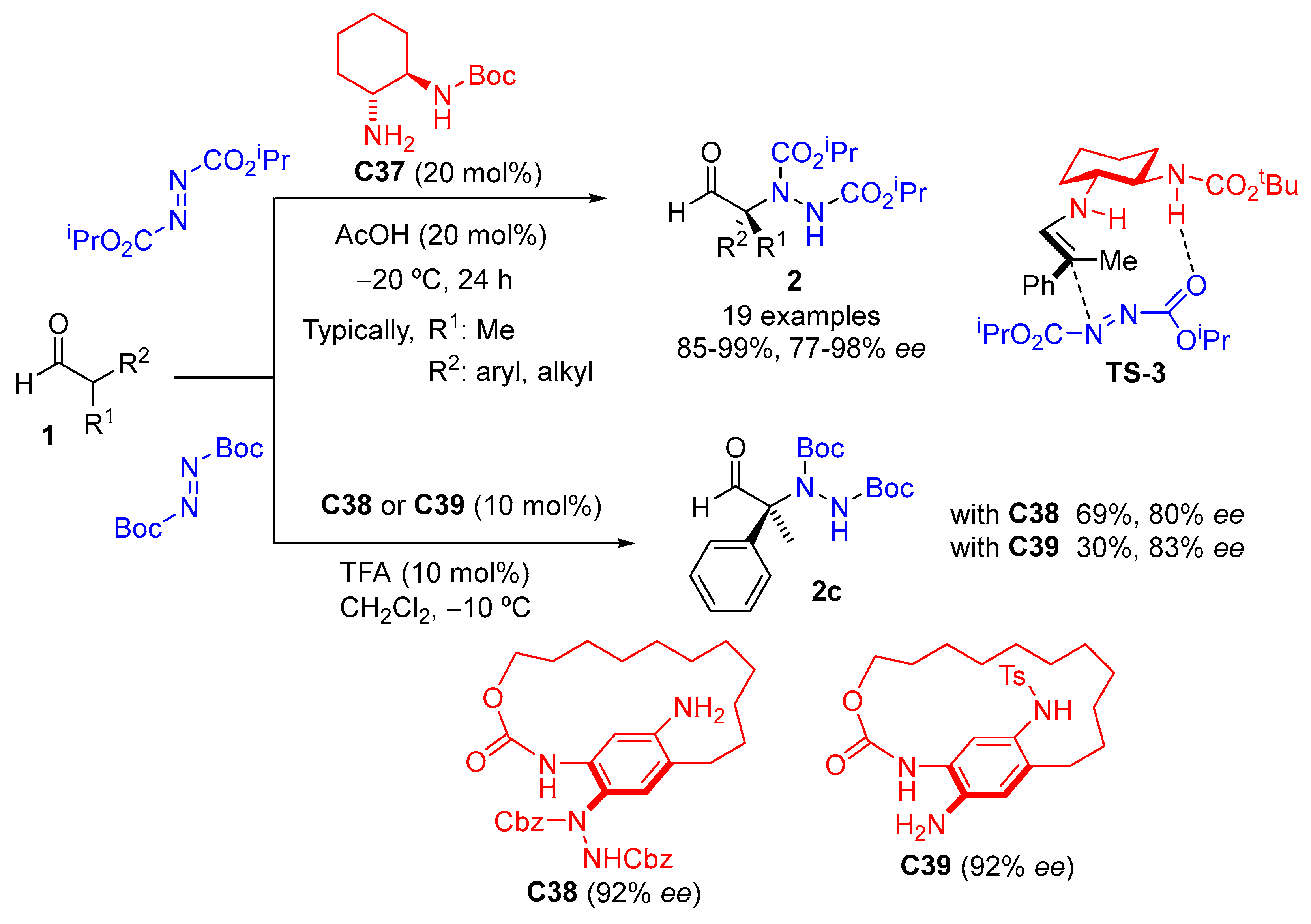{kind=link}
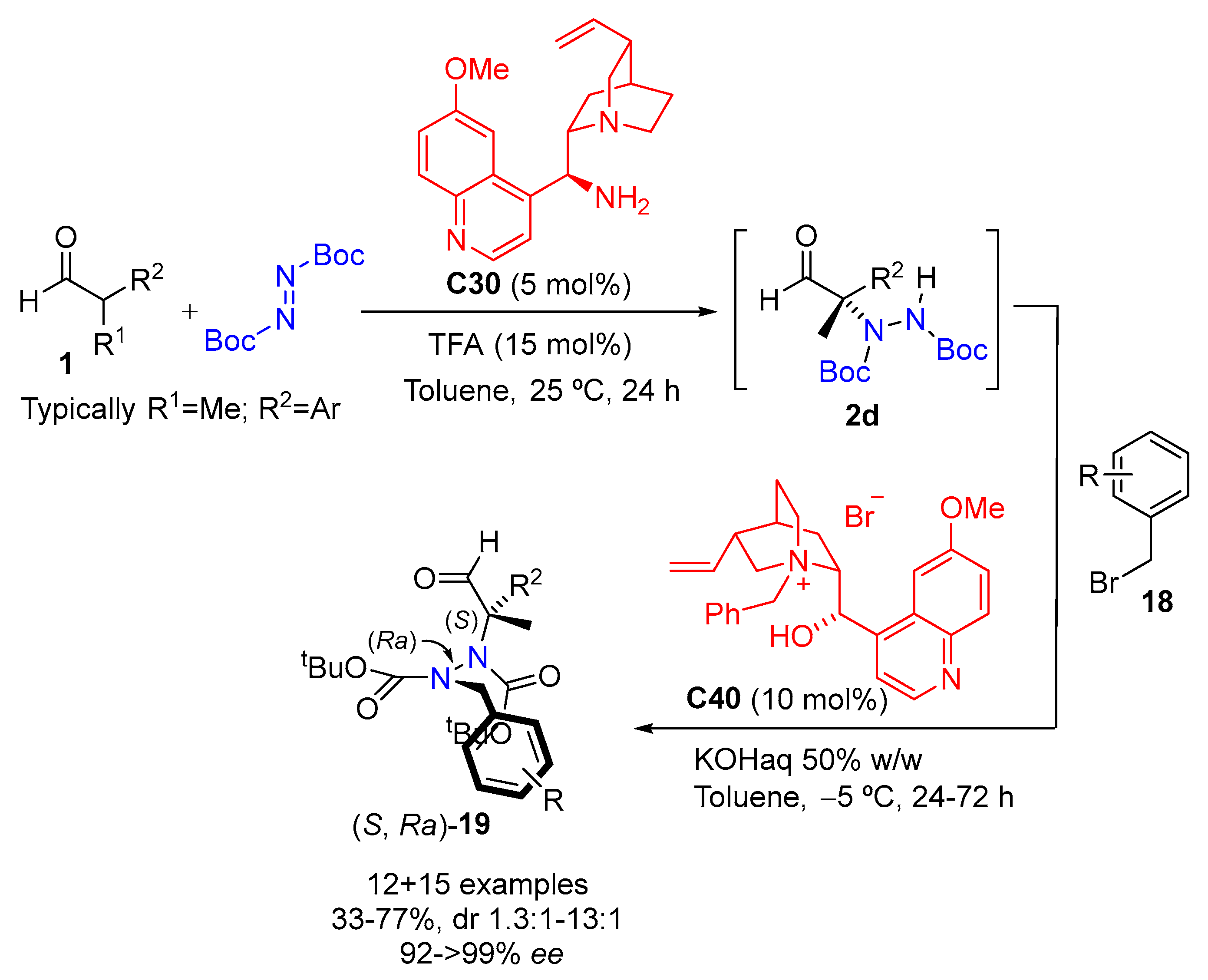{kind=link}
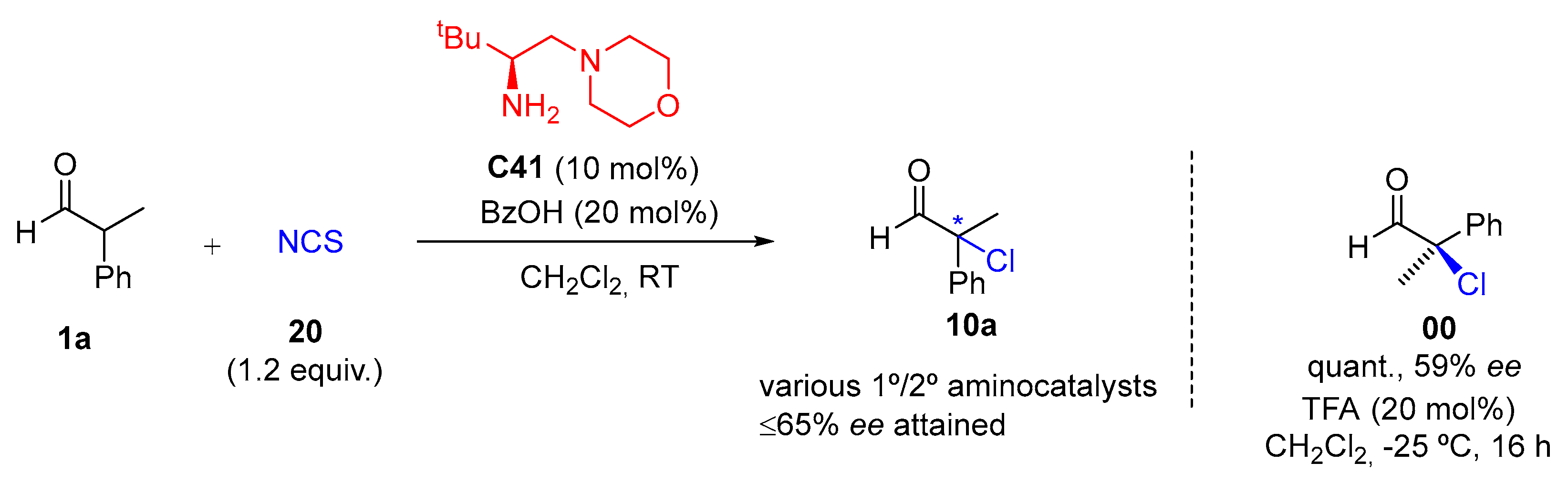{kind=link}
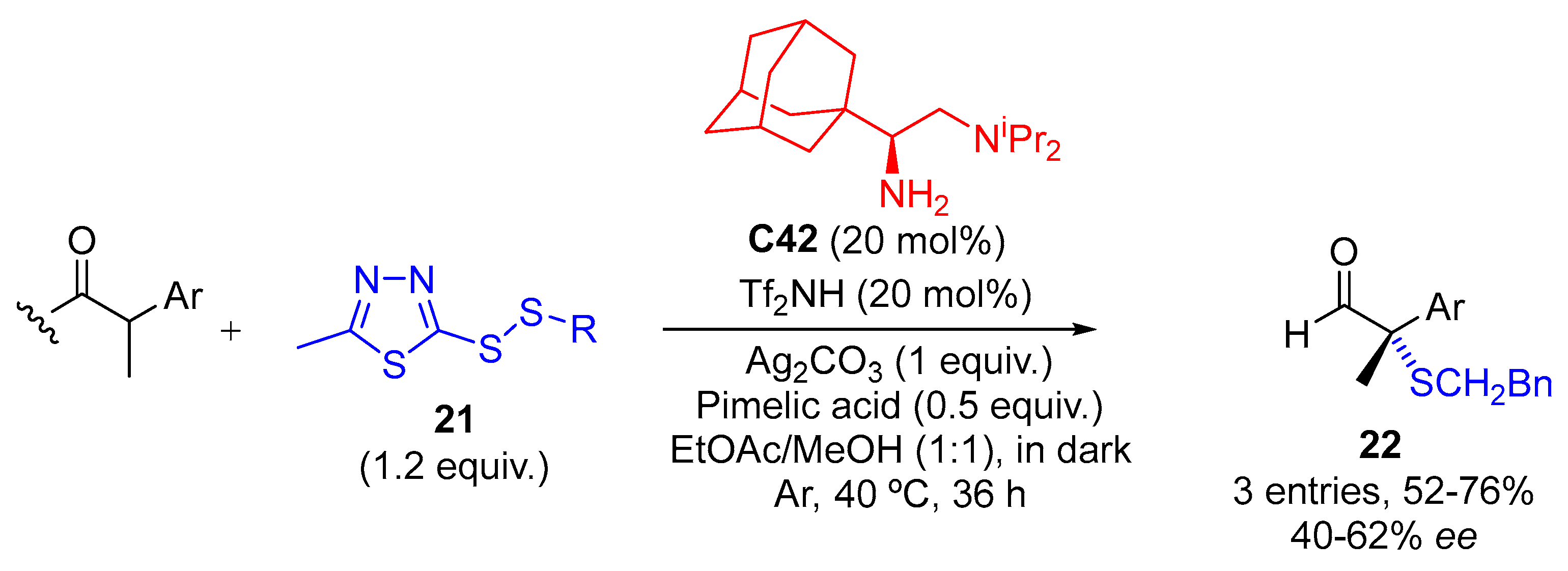{kind=link}
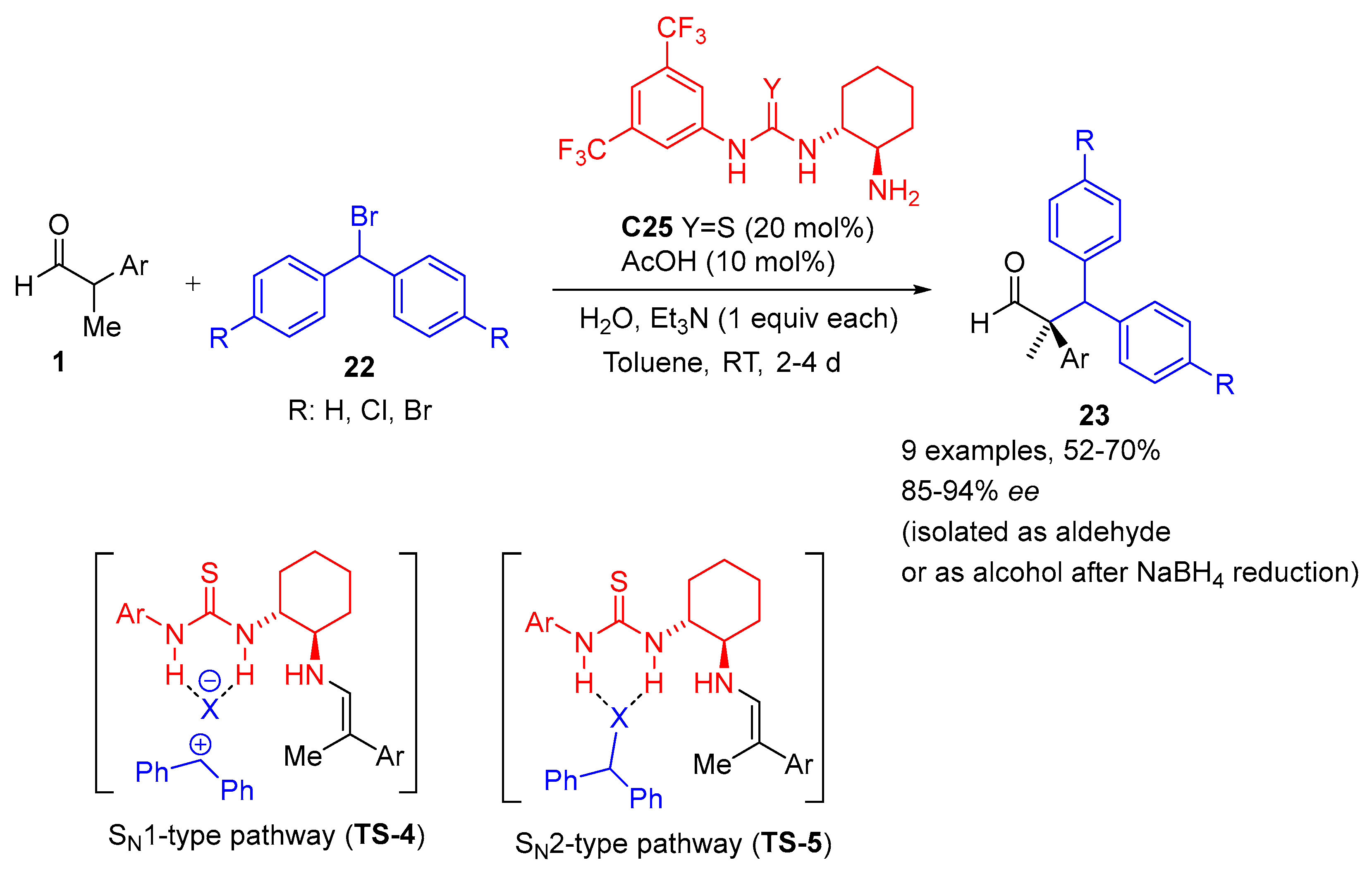{kind=link}
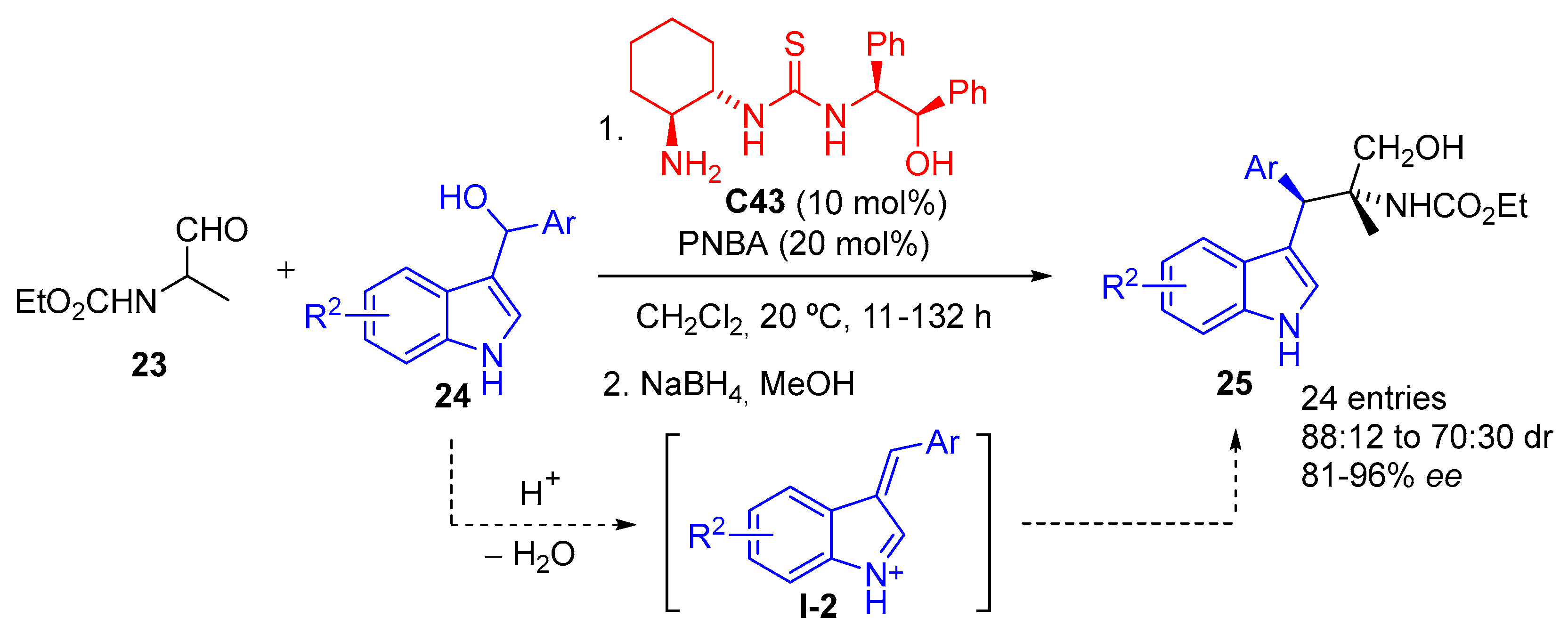{kind=link}
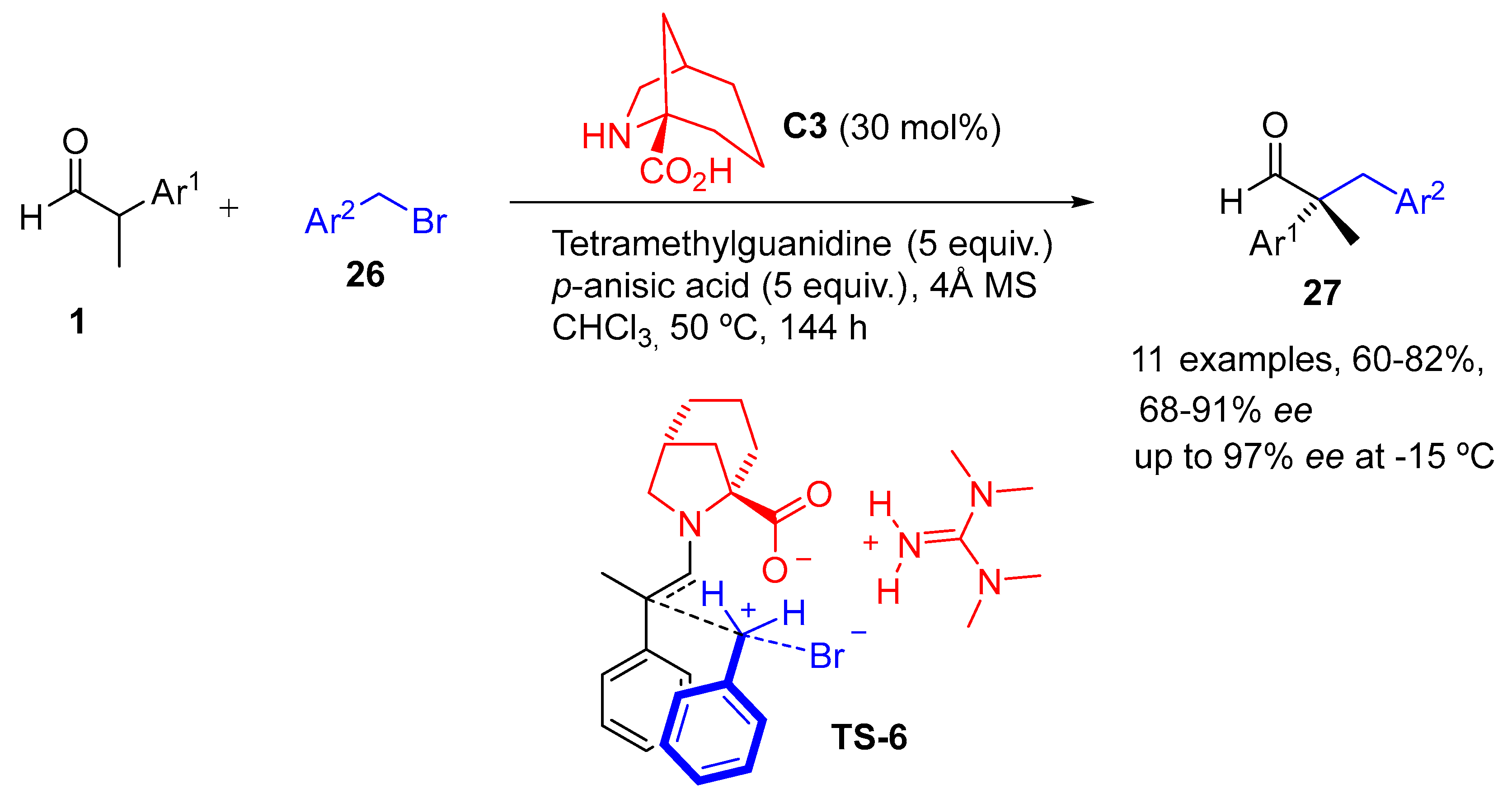{kind=link}
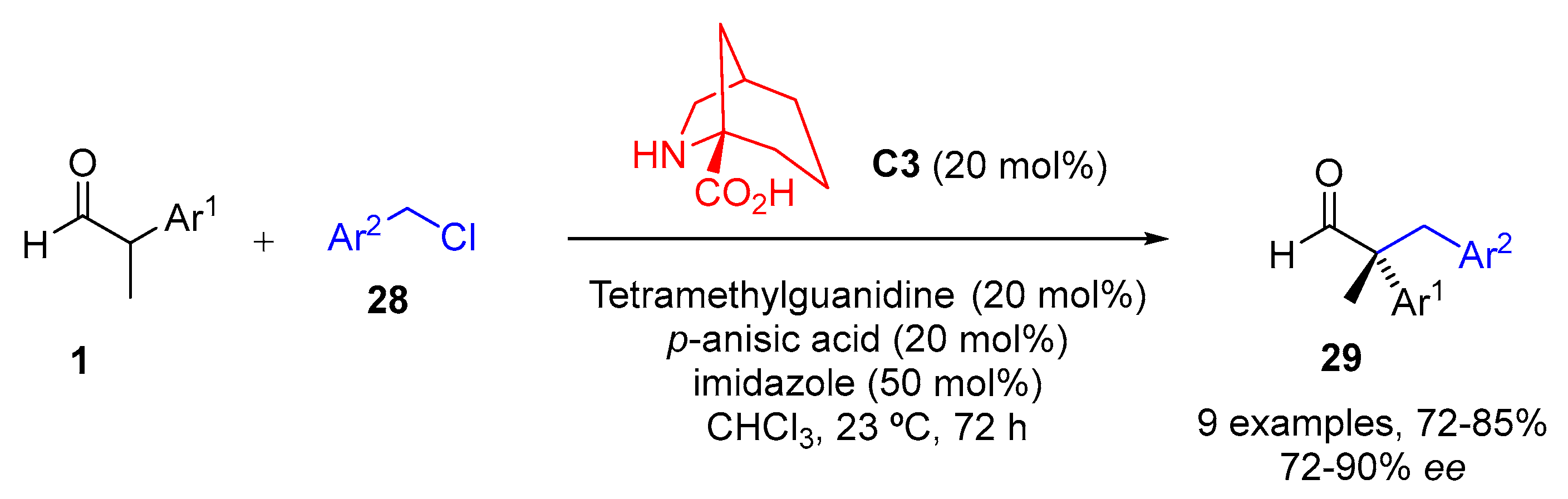{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
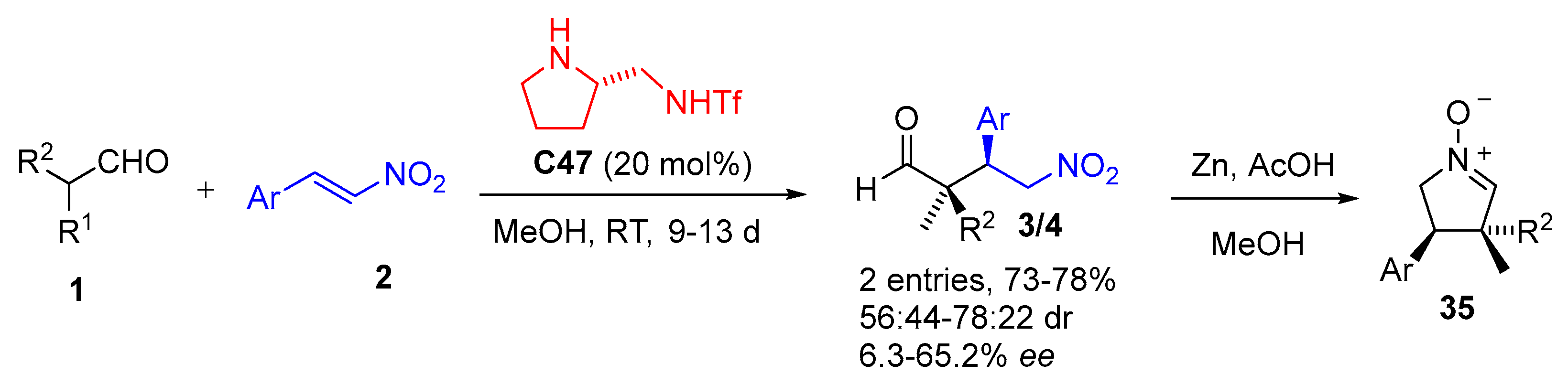{kind=link}
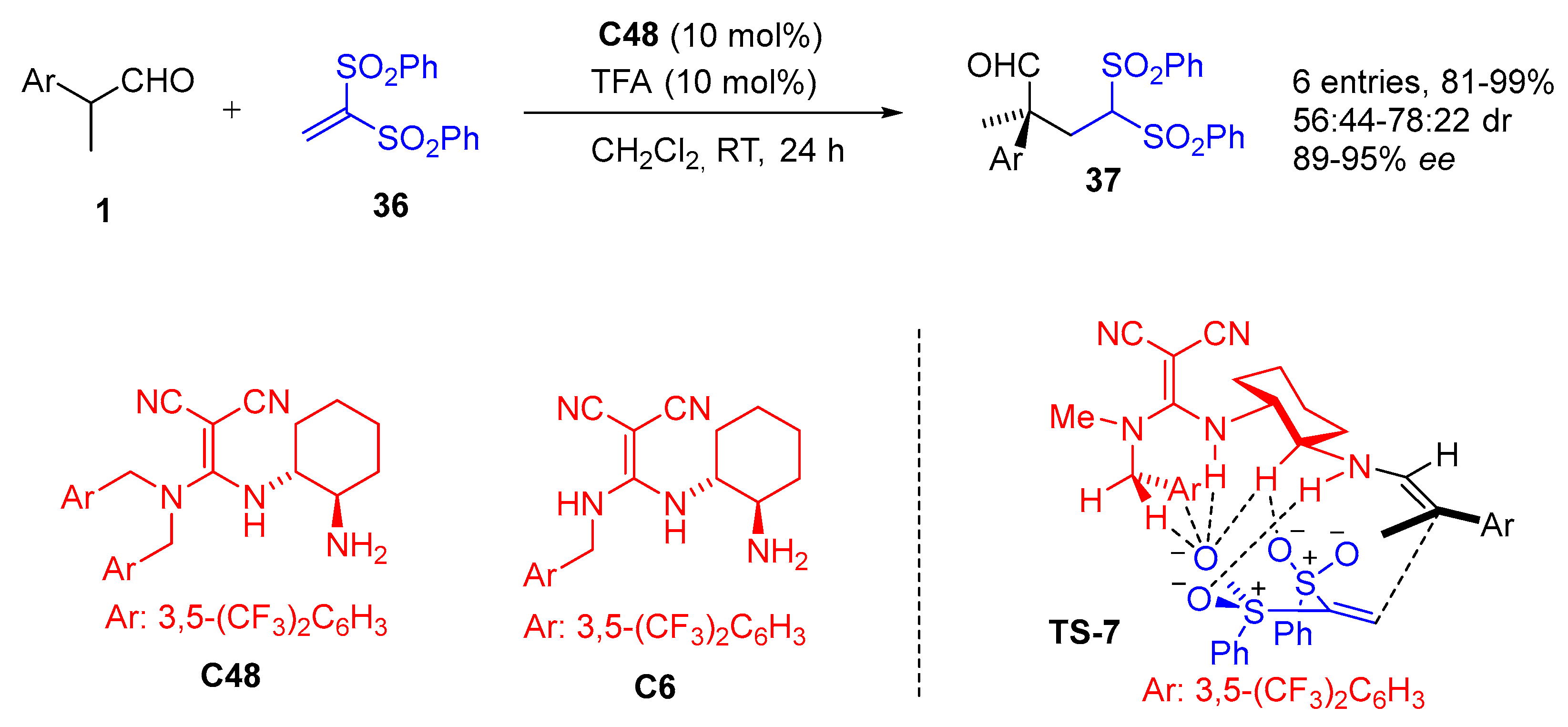{kind=link}
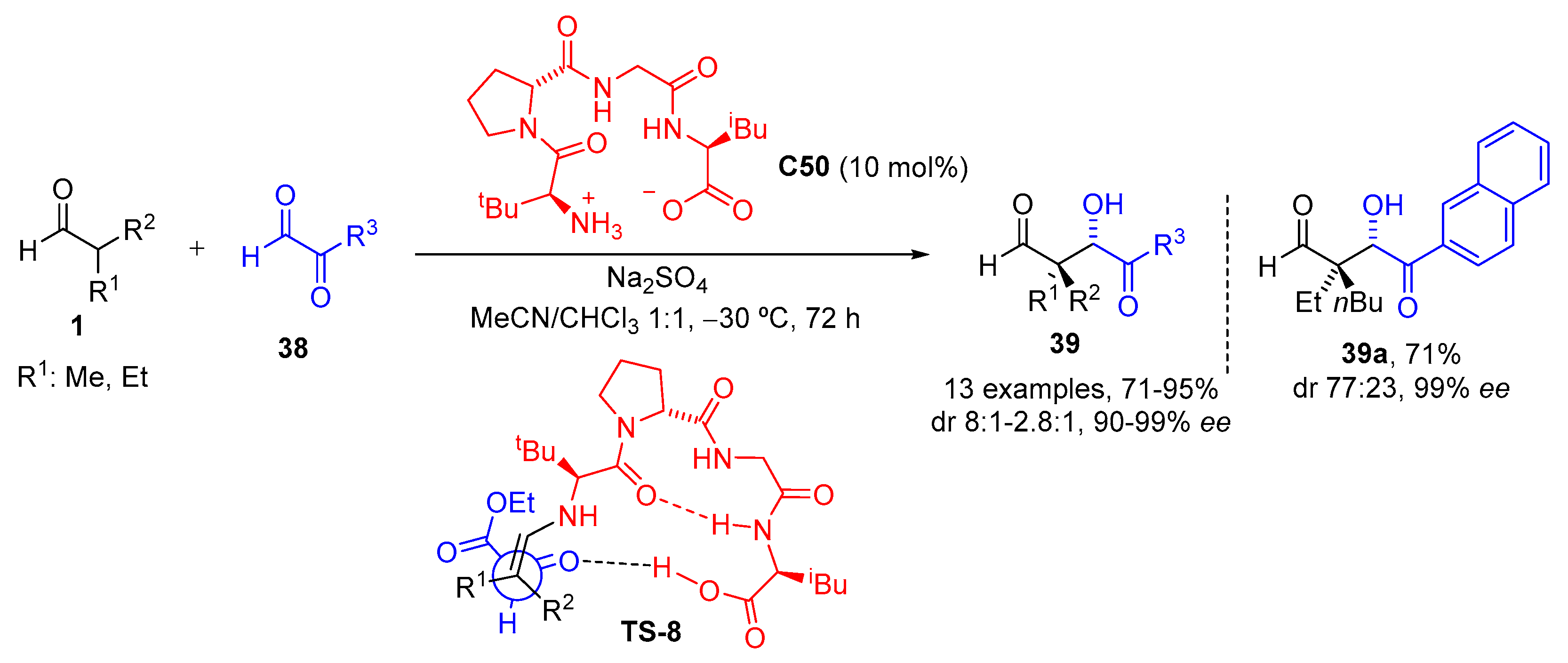{kind=link}
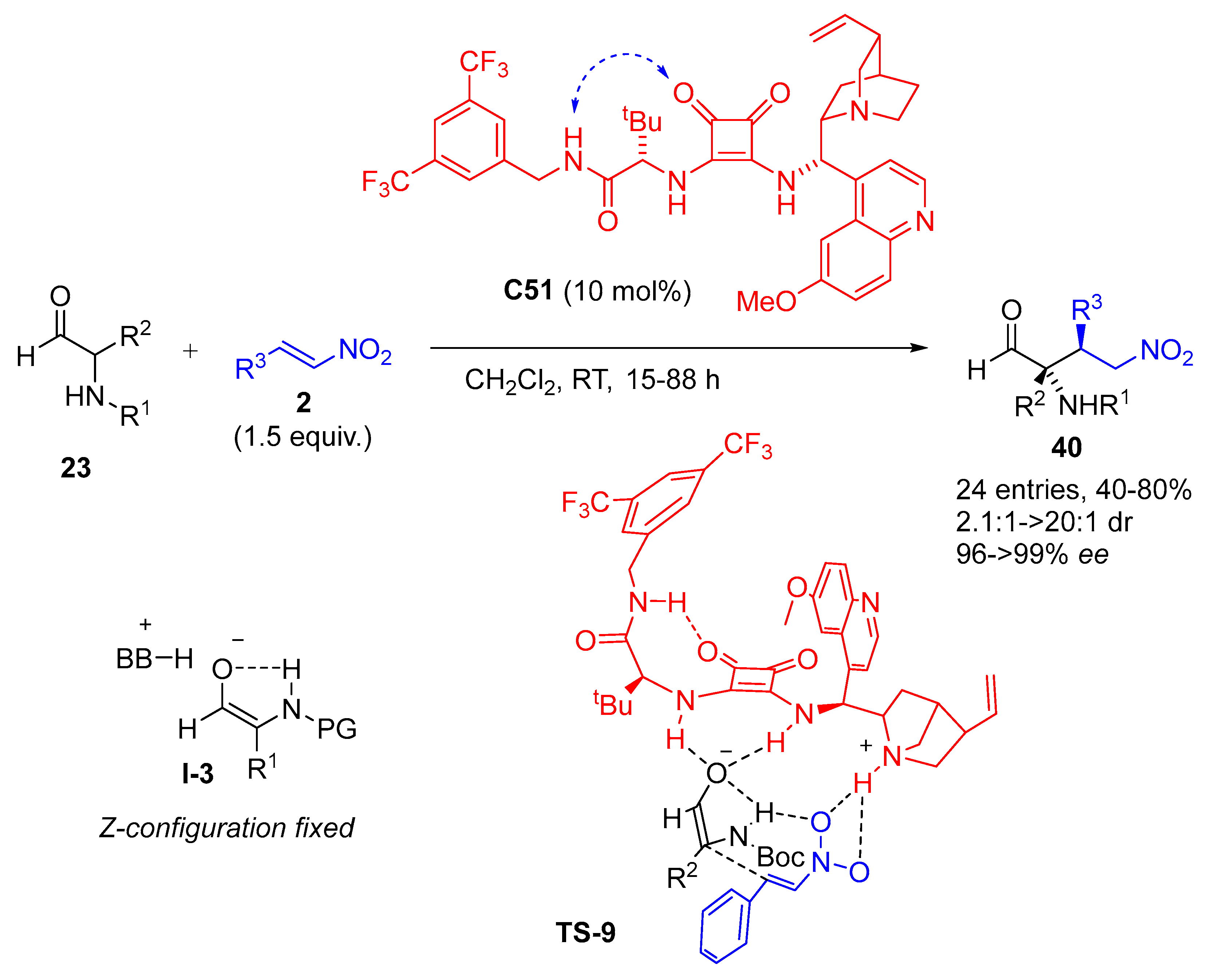{kind=link}
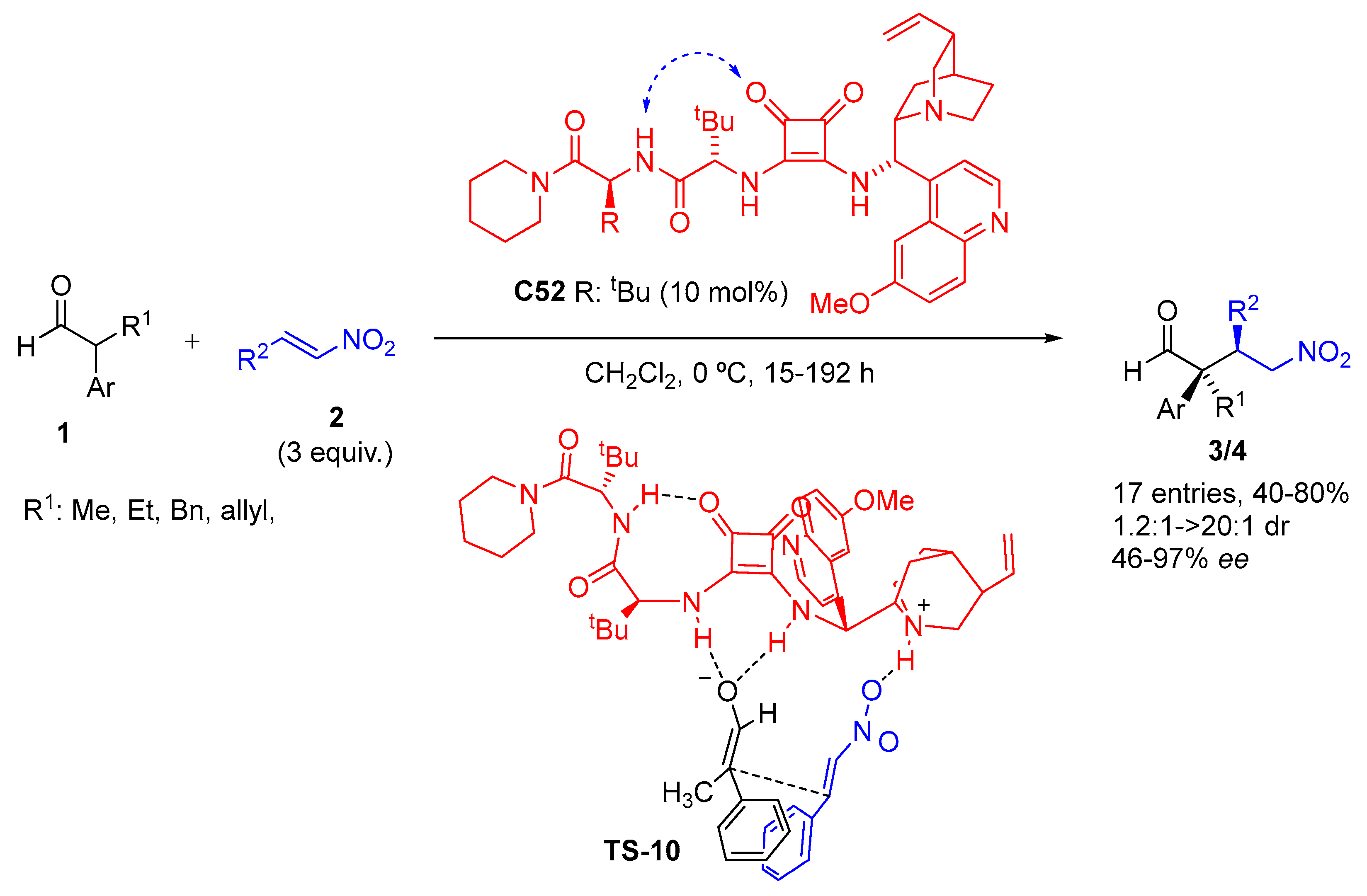{kind=link}
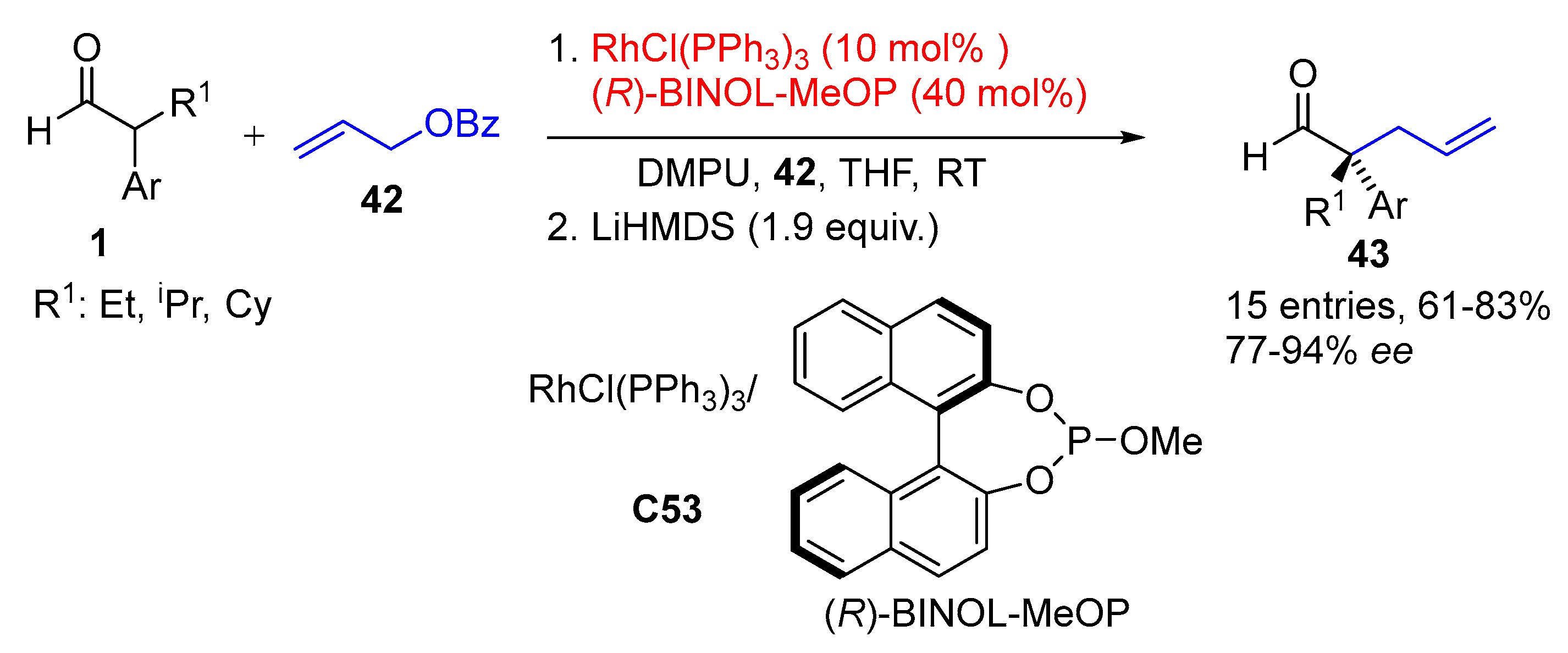{kind=link}
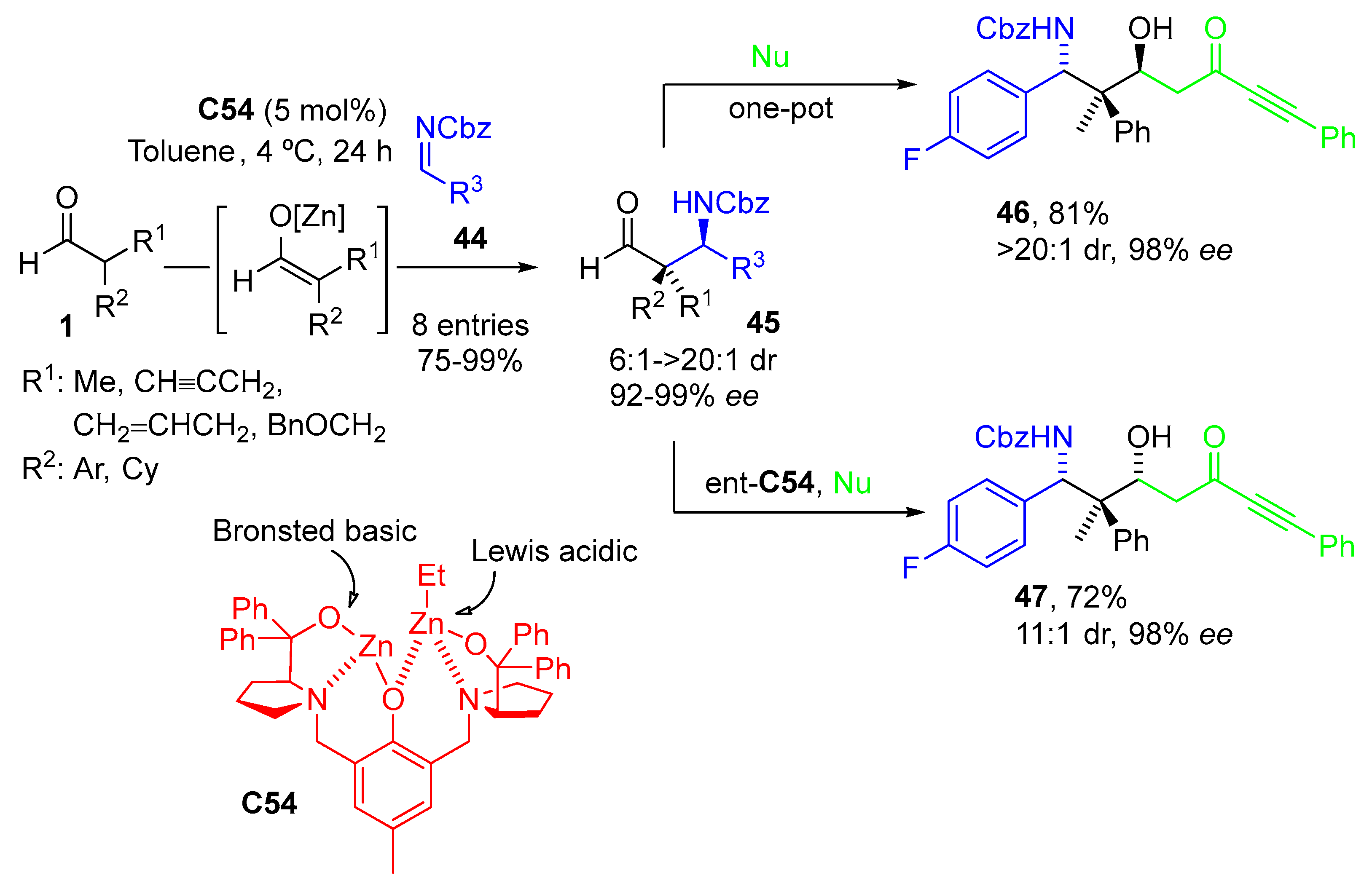{kind=link}
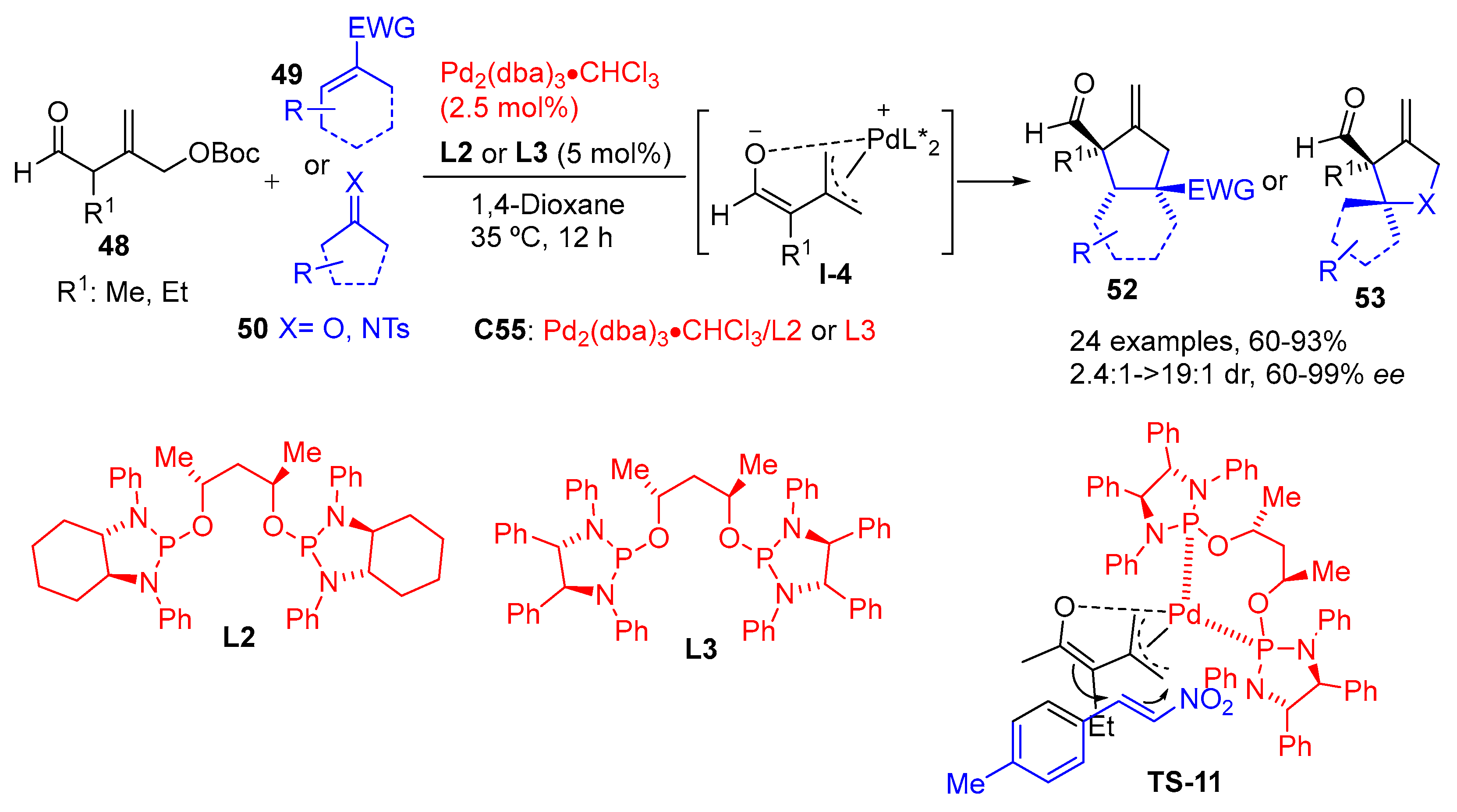{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
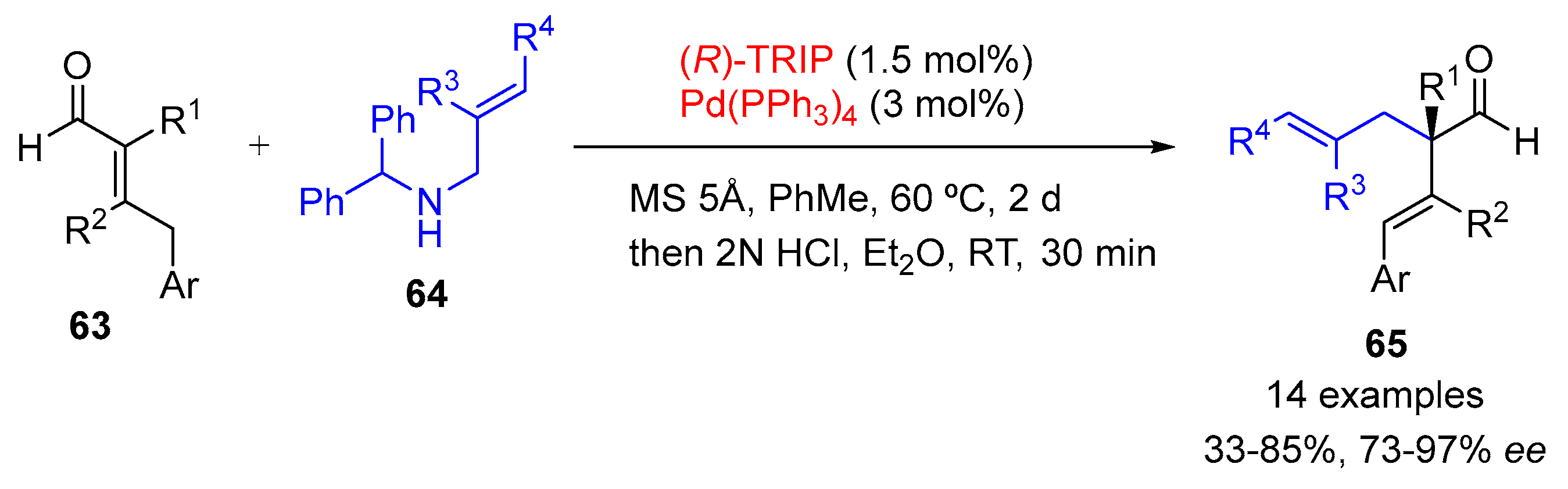{kind=link}
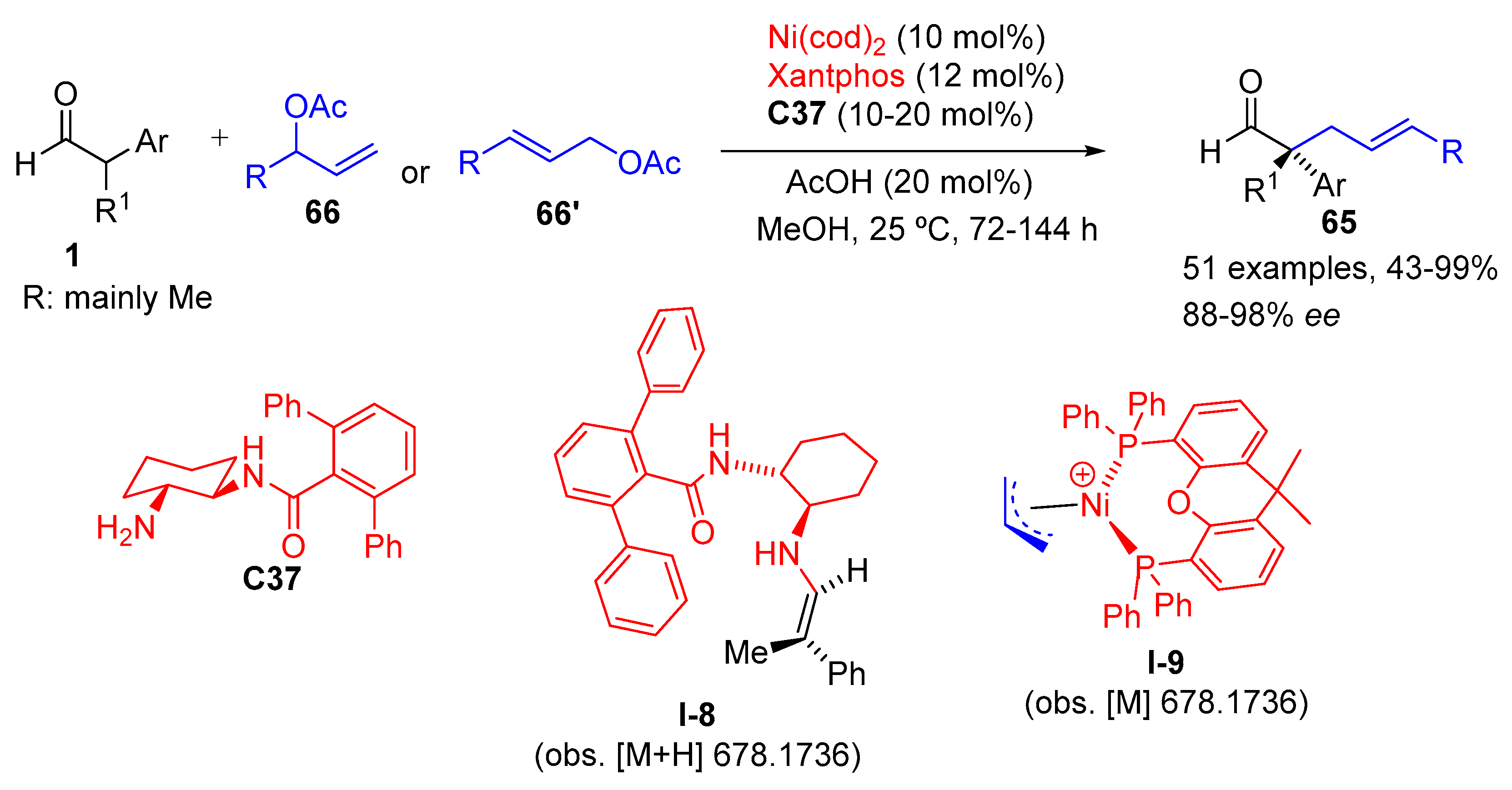{kind=link}
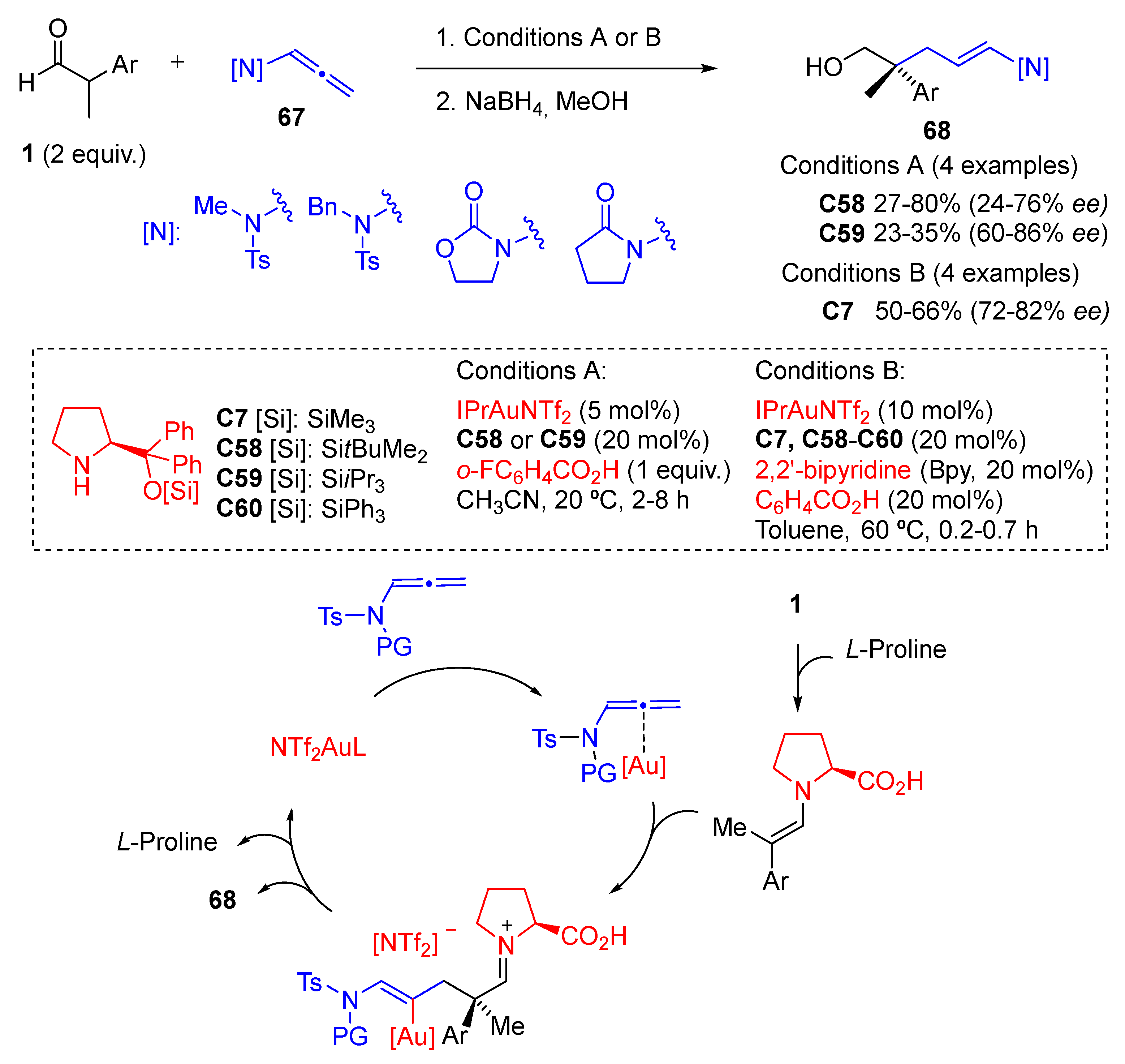{kind=link}
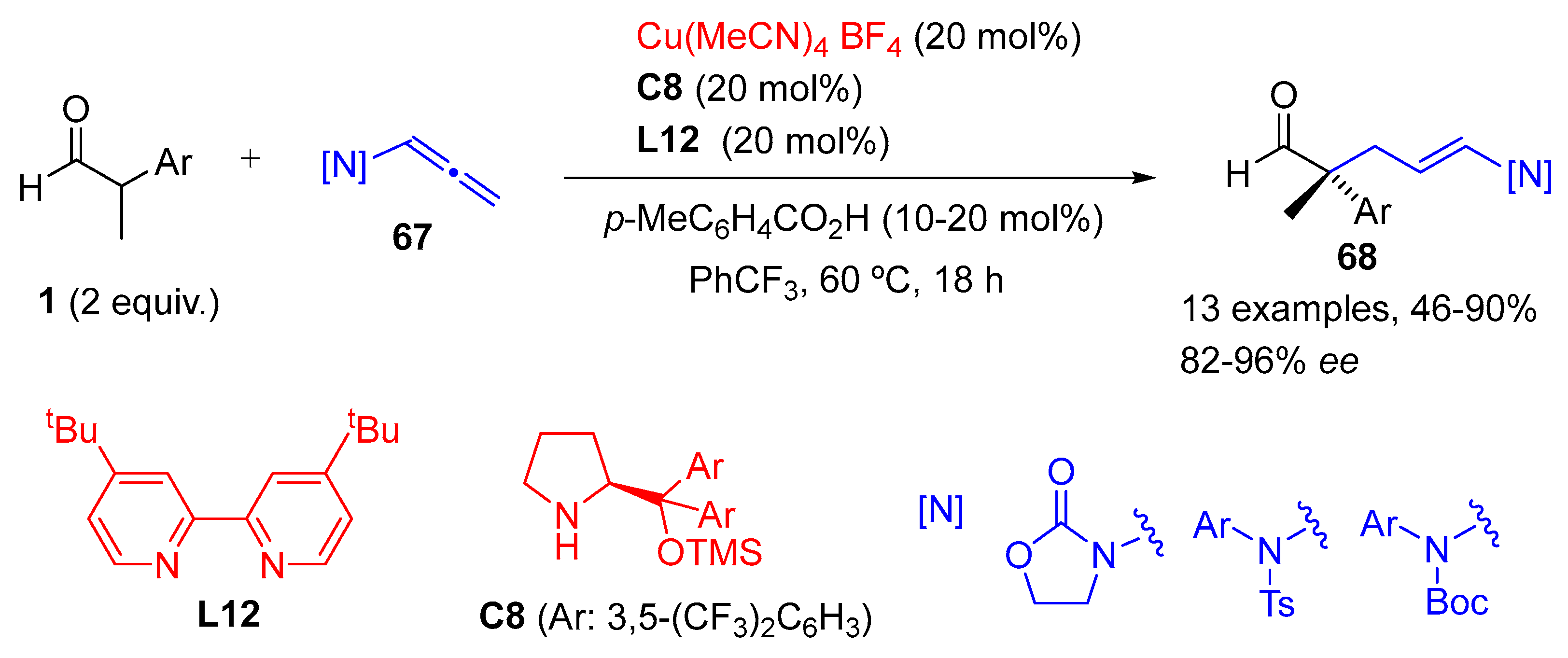{kind=link}
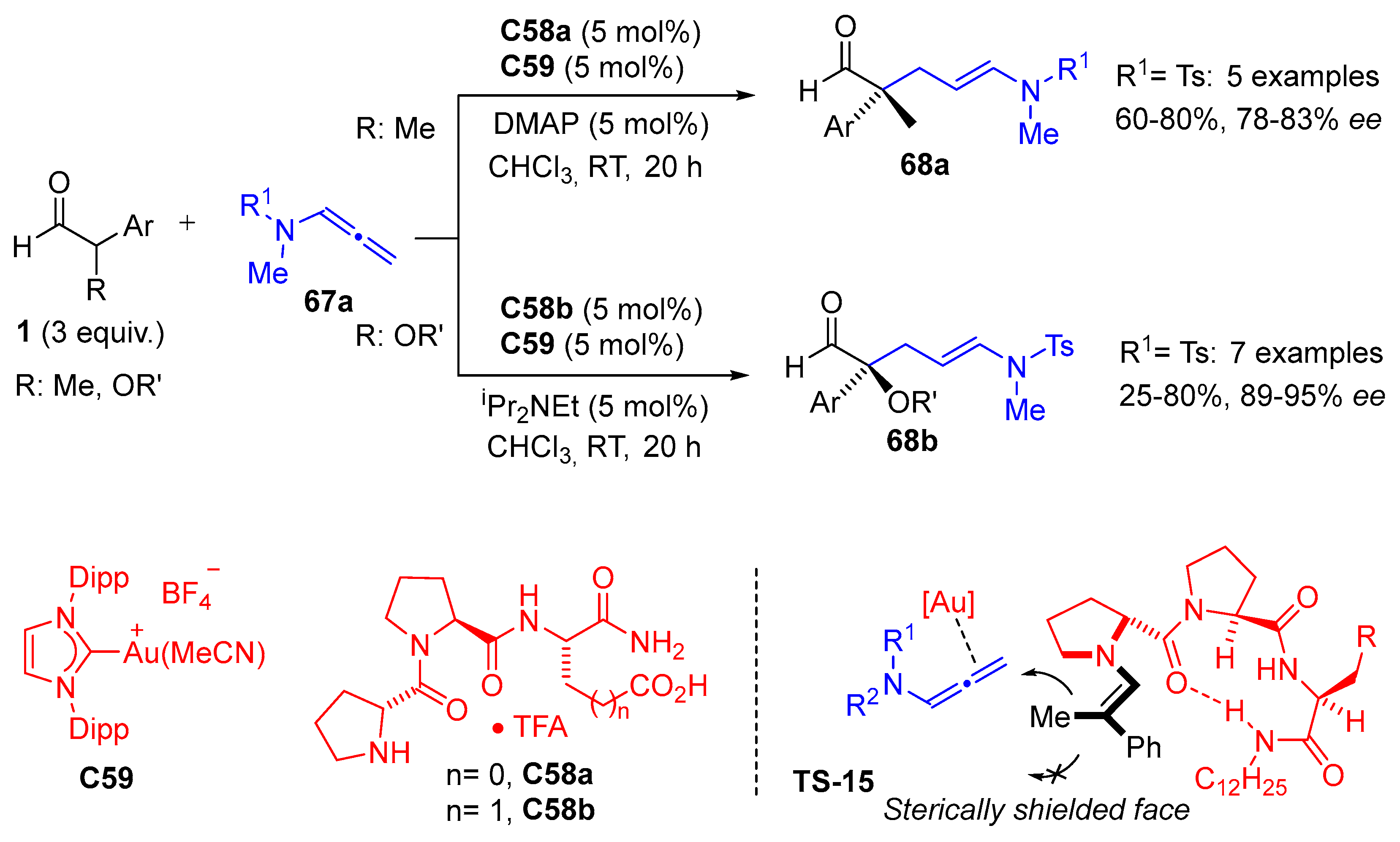{kind=link}
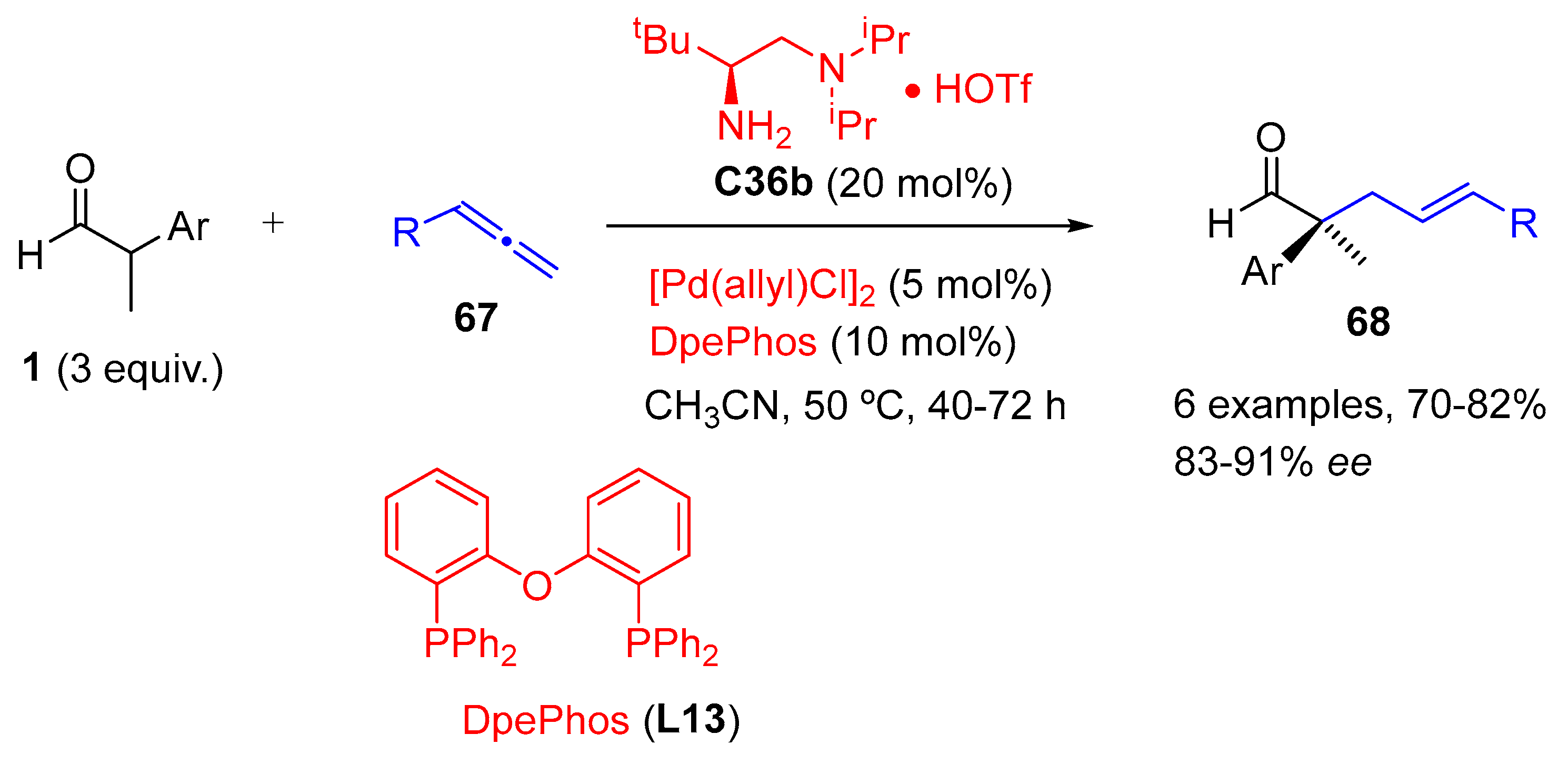{kind=link}
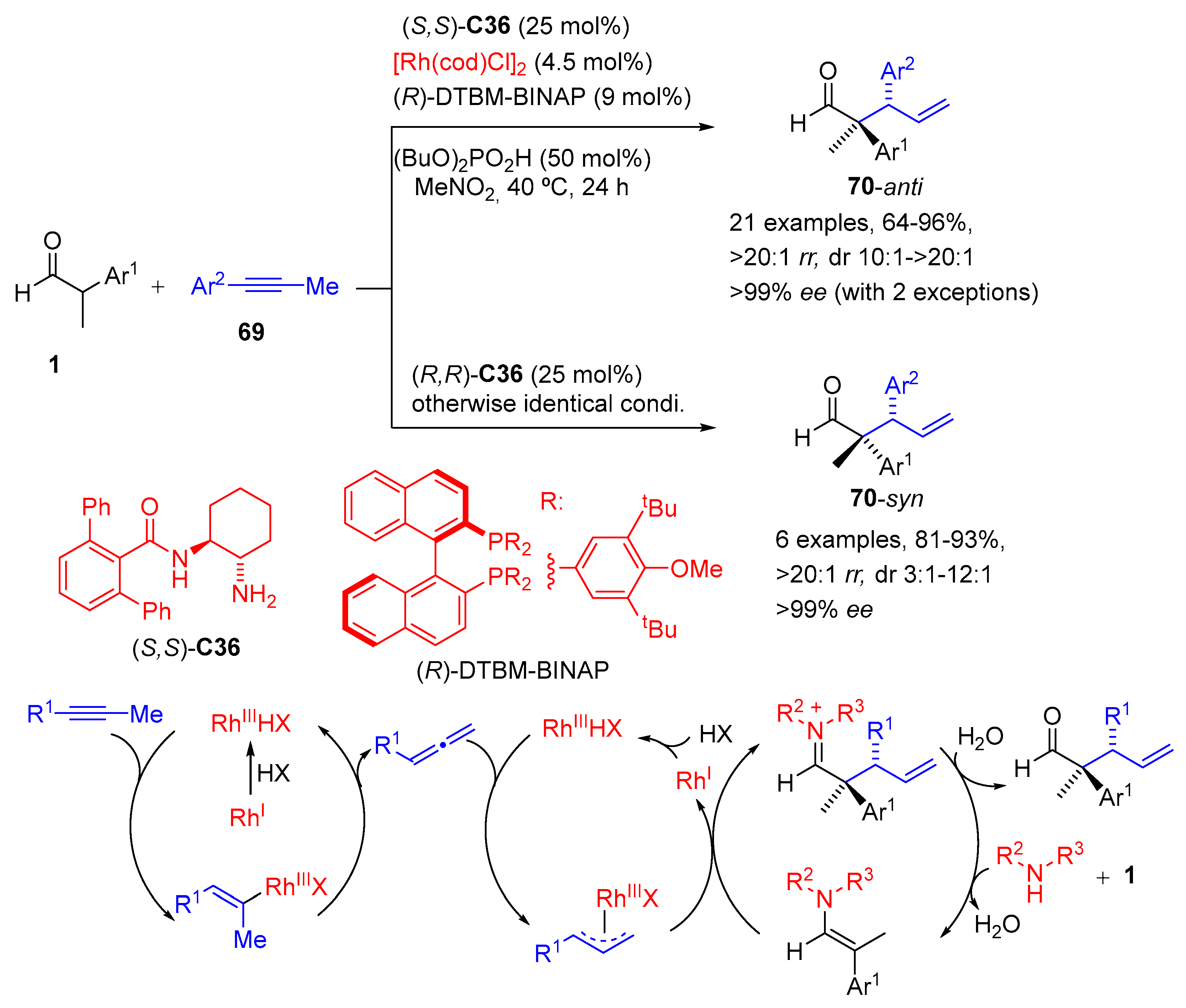{kind=link}
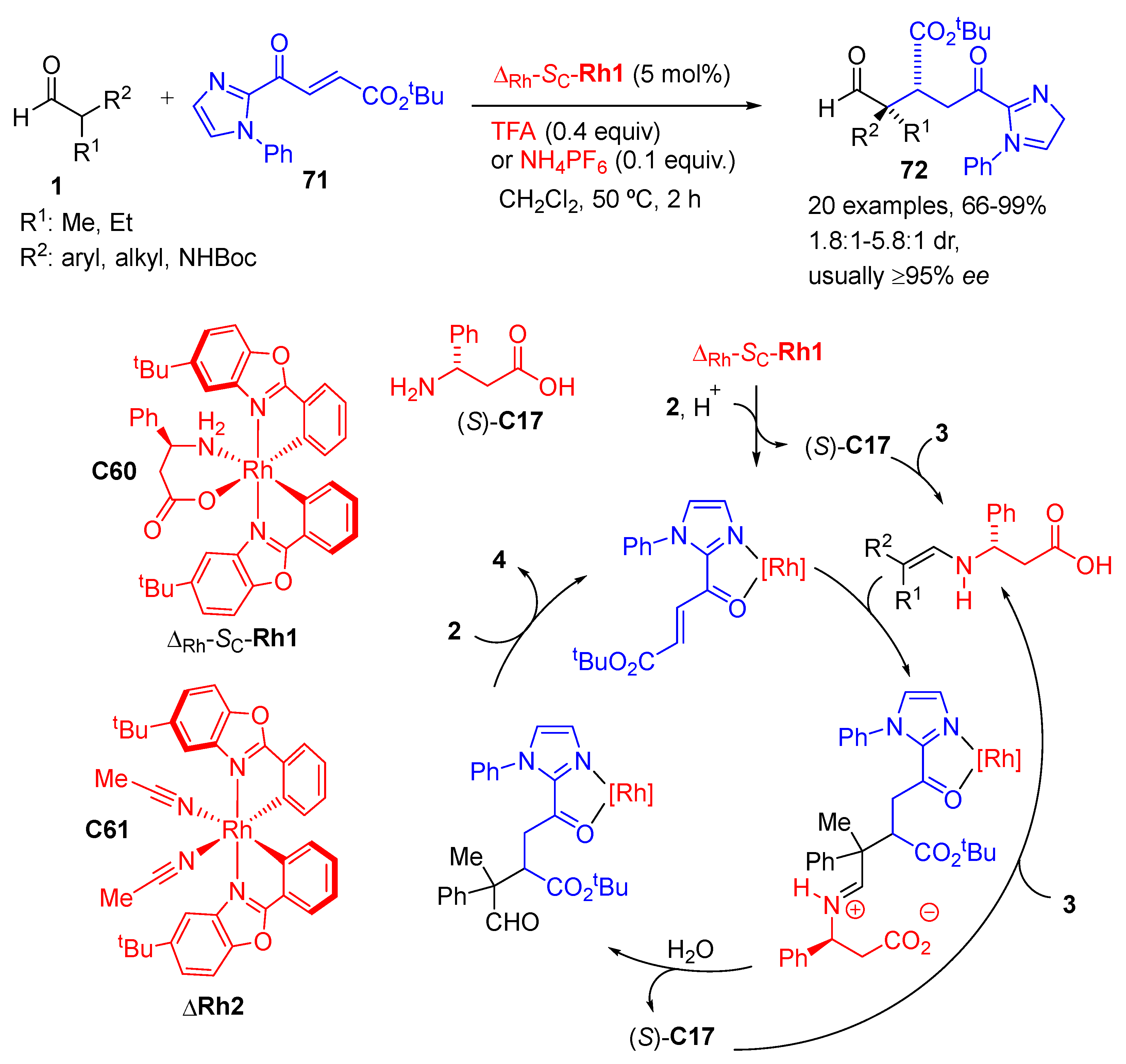{kind=link}
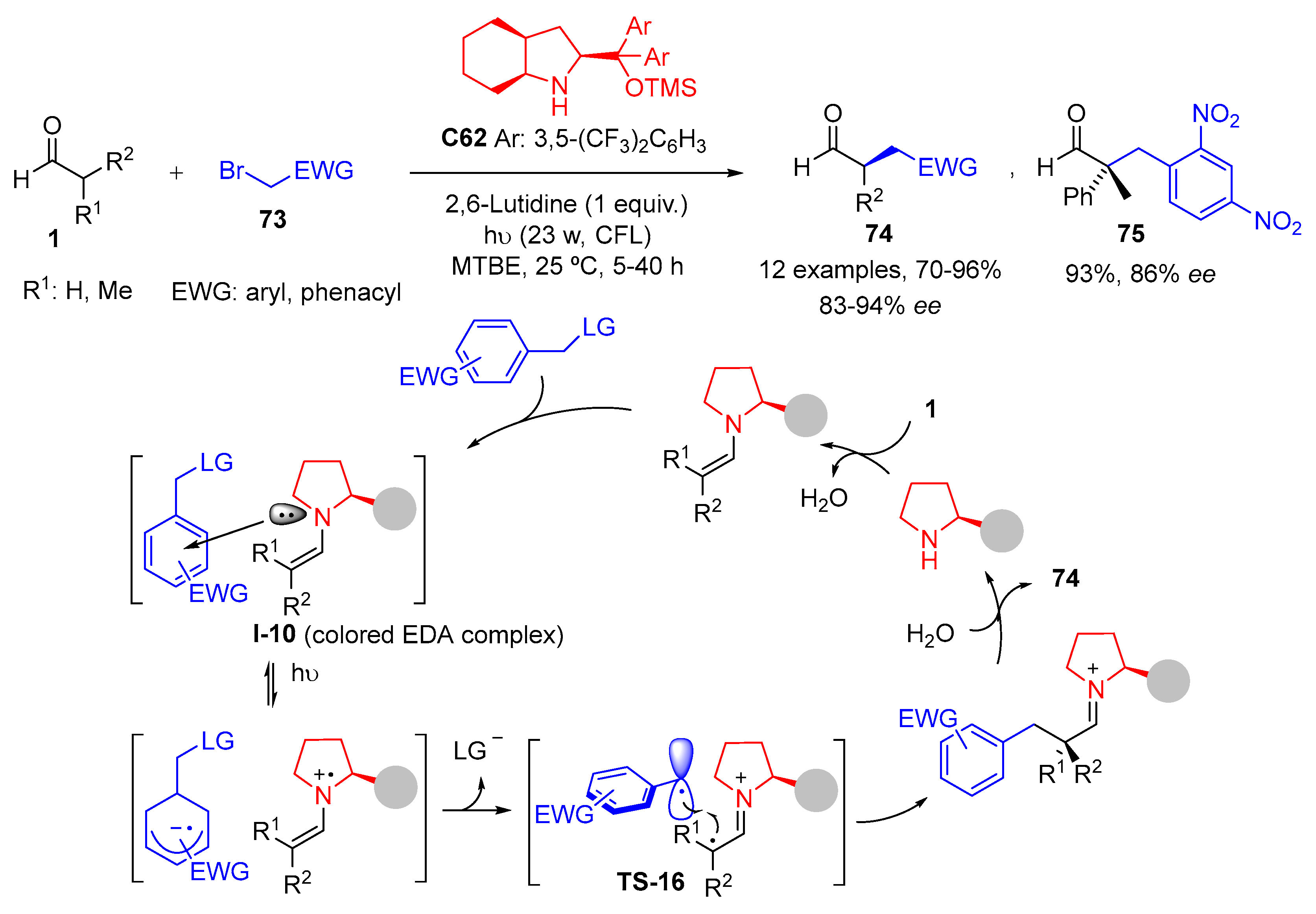{kind=link}
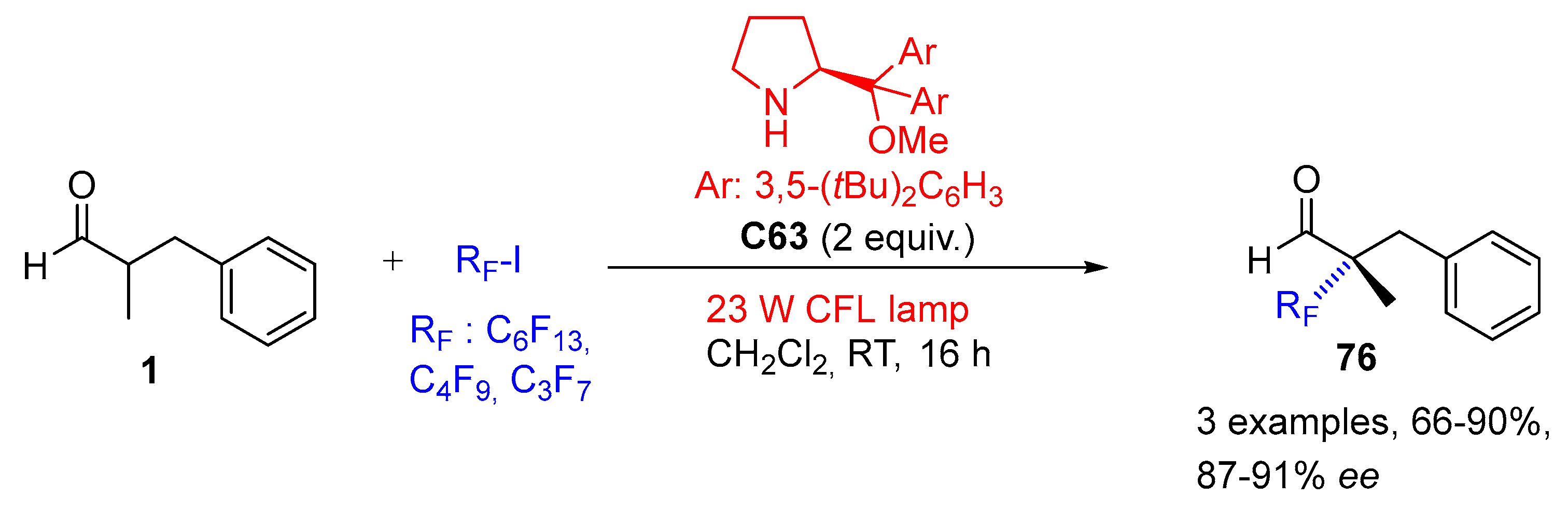{kind=link}
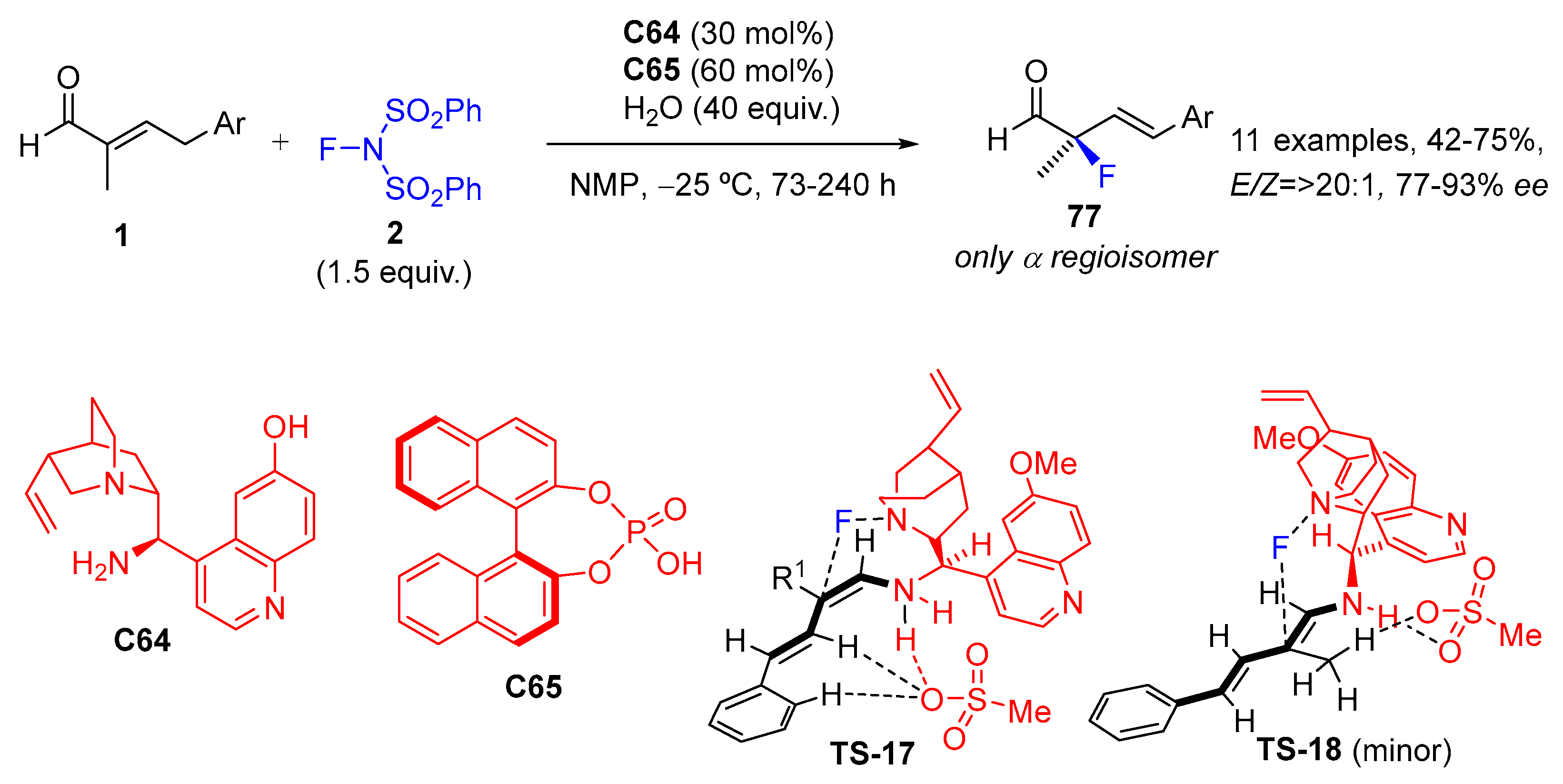{kind=link}
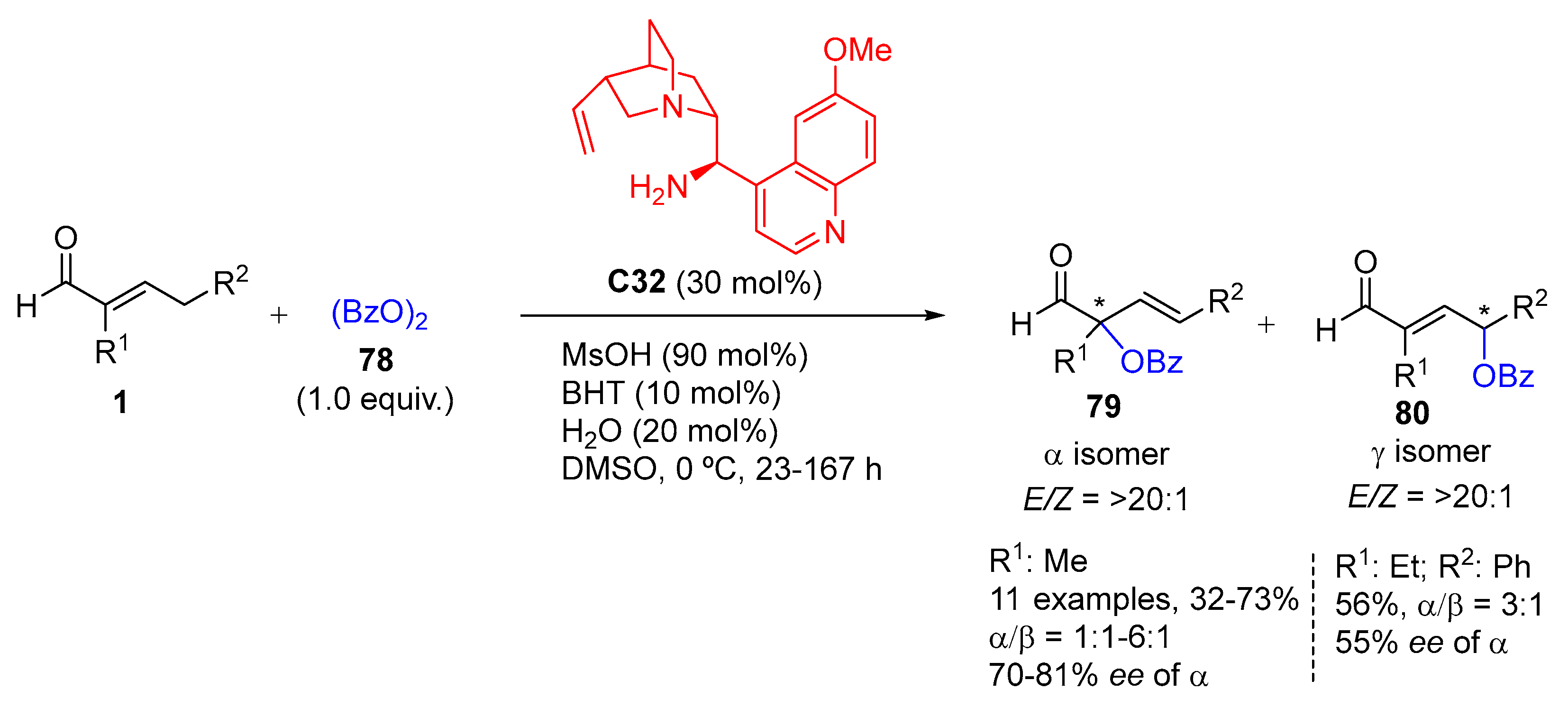{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
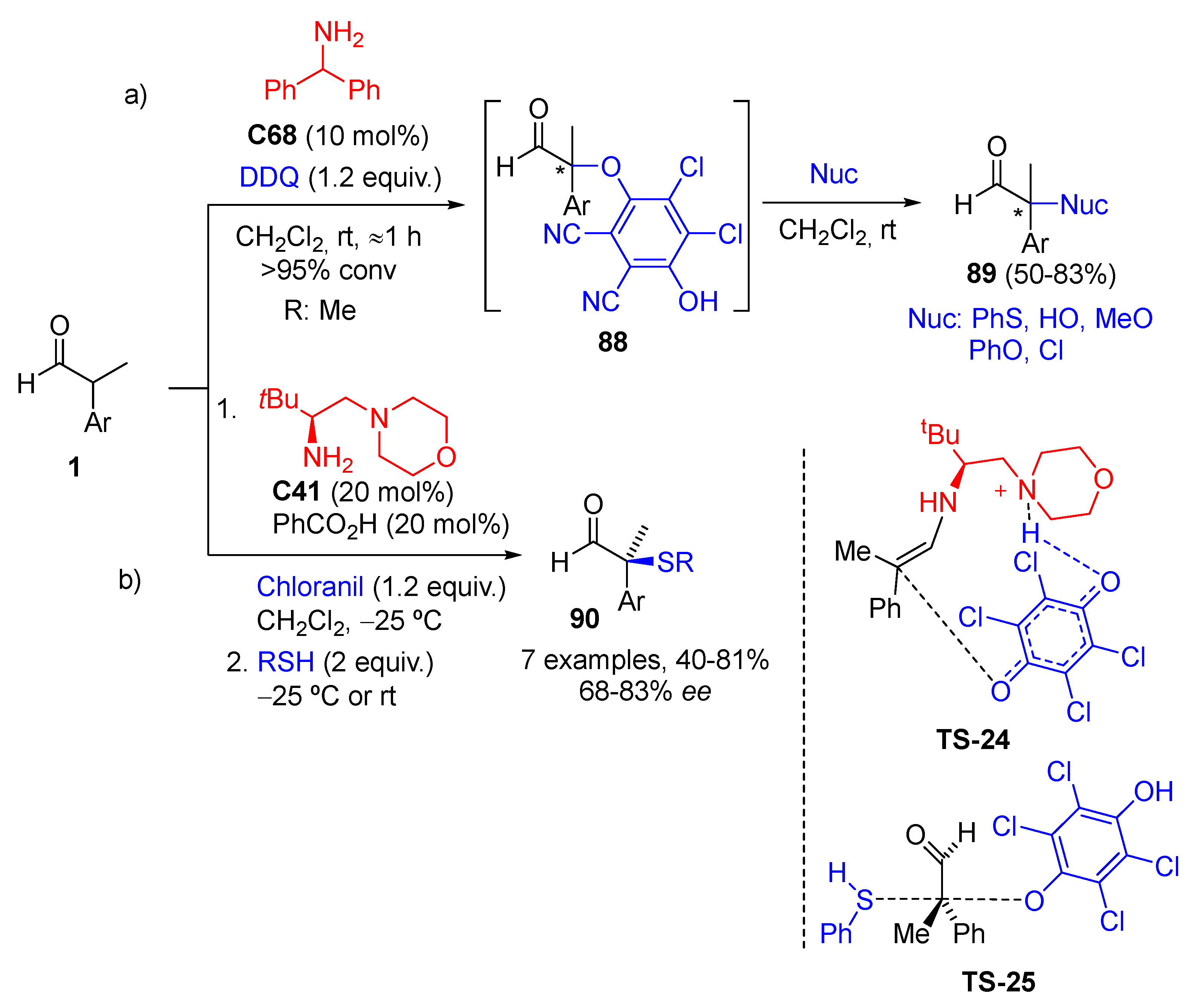{kind=link}
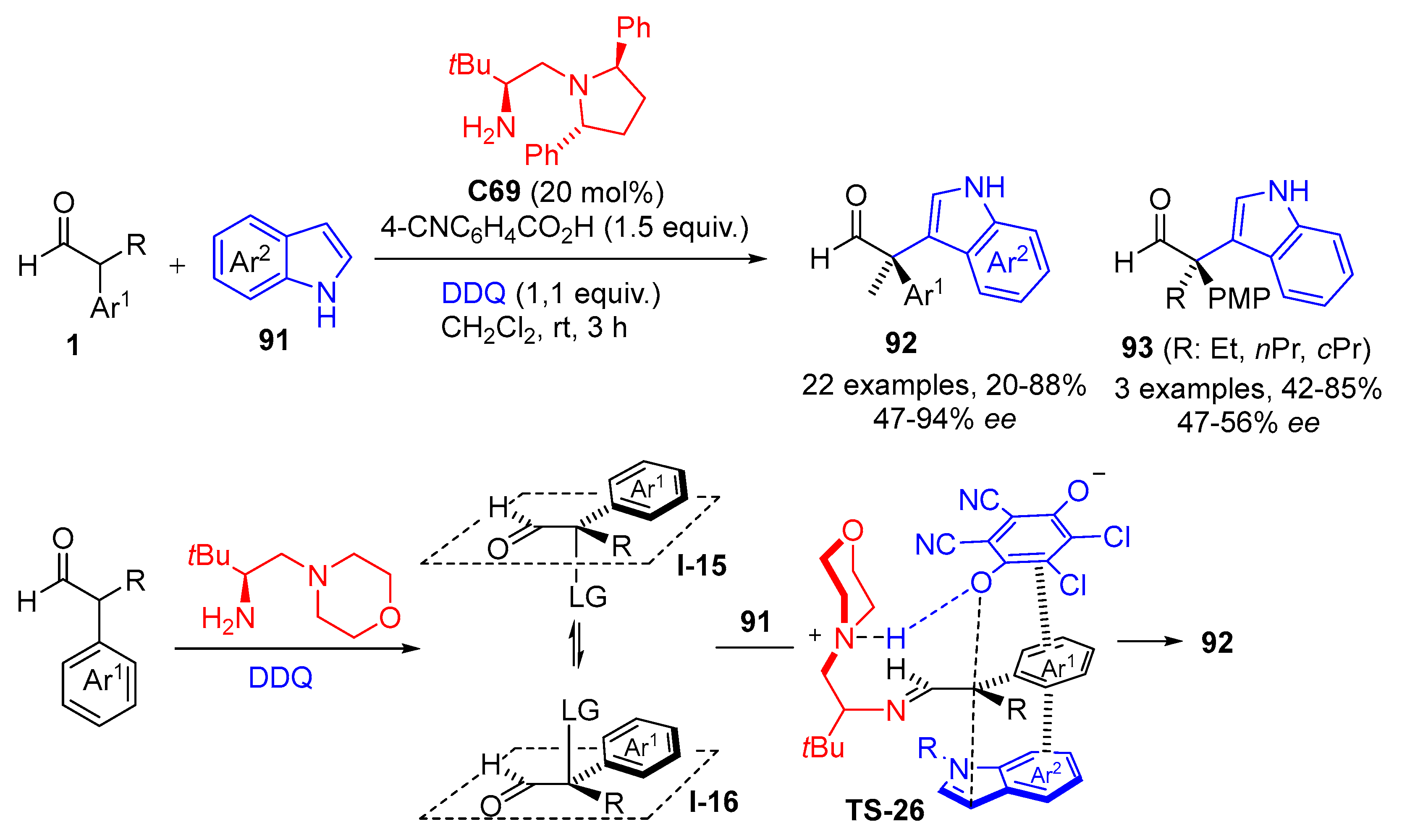{kind=link}
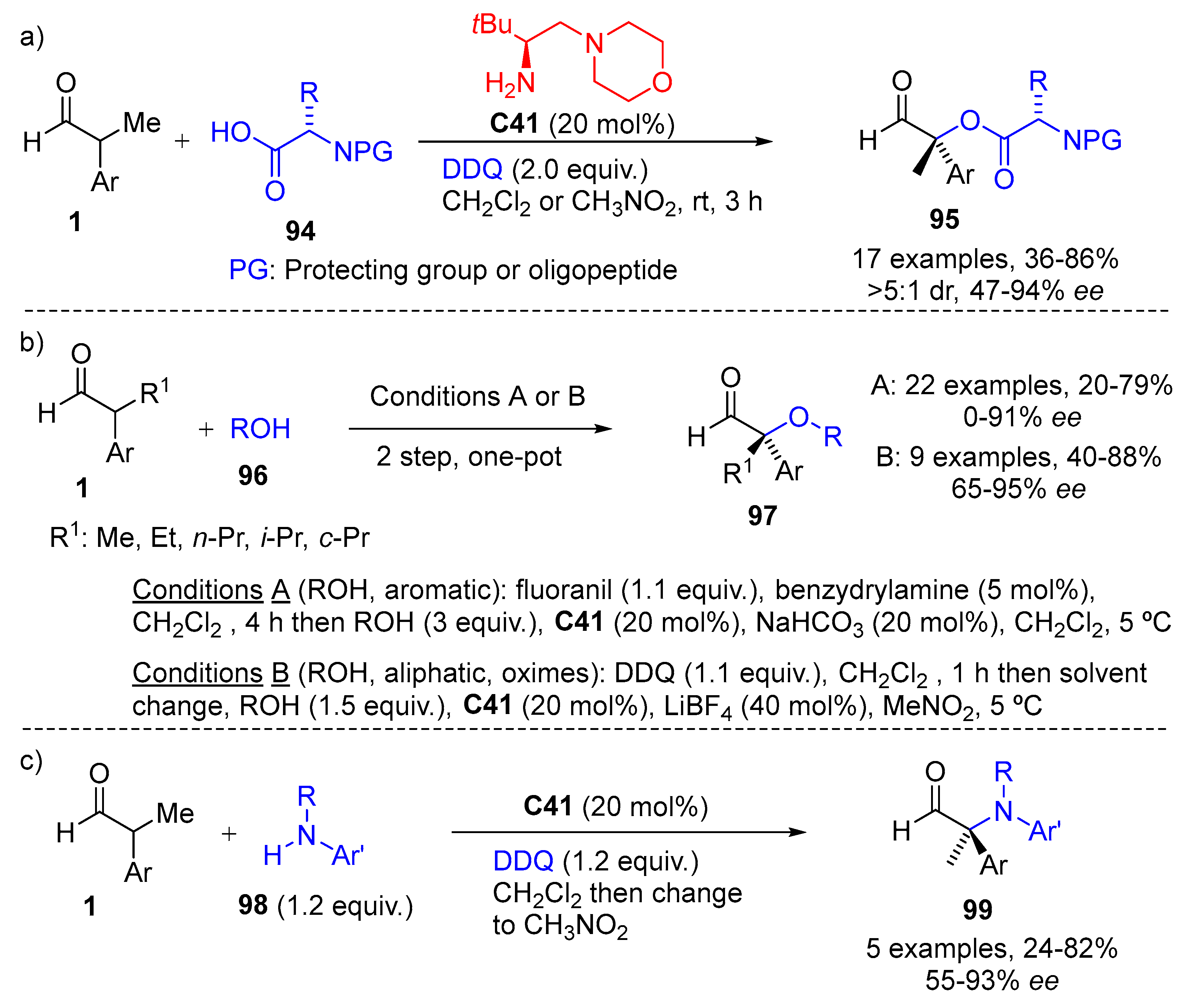{kind=link}
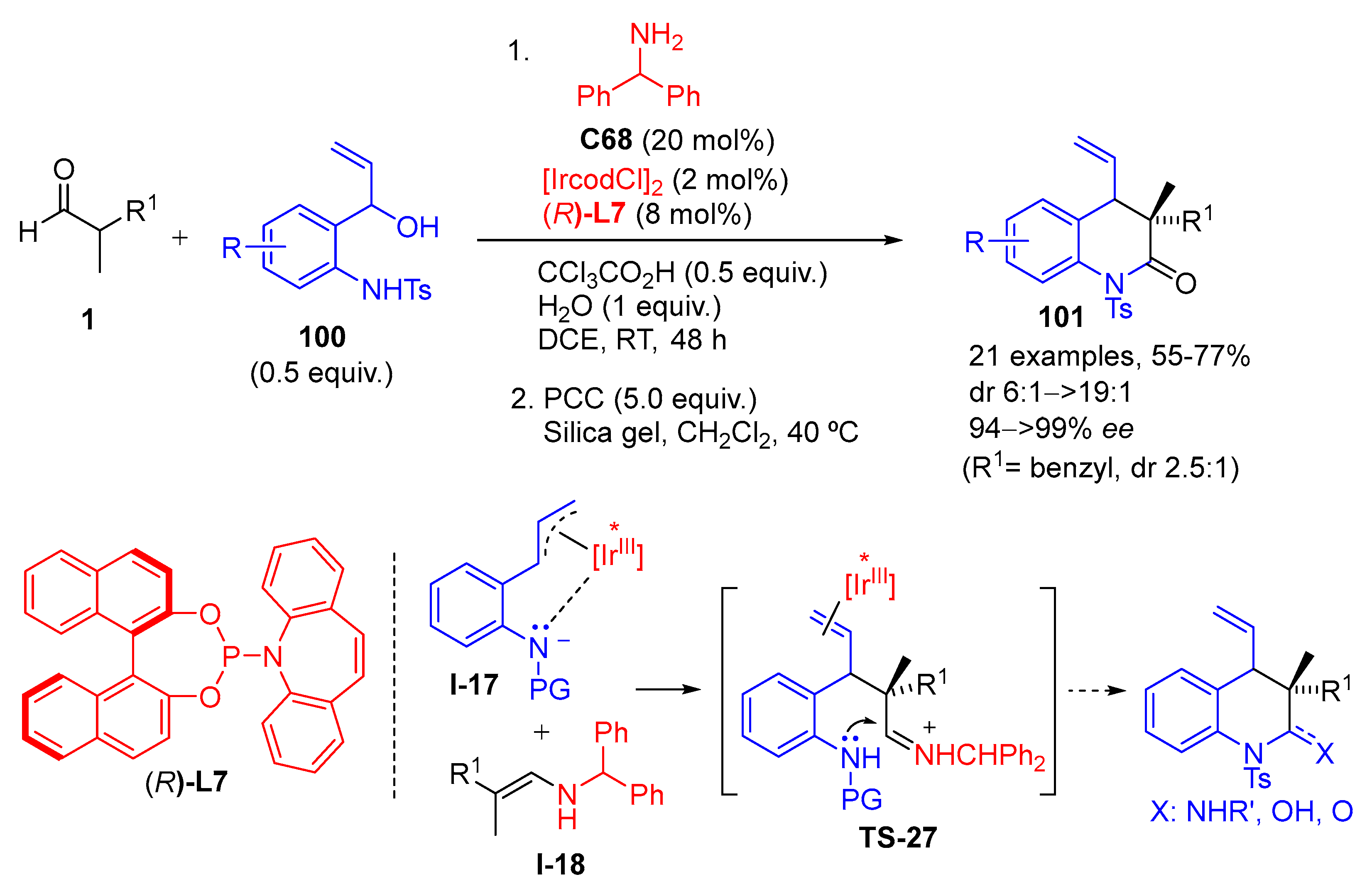{kind=link}
{kind=link}
1. Introduction
2. Methods Based on Enamine Activation
- (i)
- α-Heterofunctionalization (oxylation, amination, fluorination, chlorination, and thiolation)
- (ii)
- α-alkylation reactions
- (iii)
- 1,2- and 1,4-addition reactions
3. Methods Based on Brønsted Base Activation
4. Methods Based on Metal-Centered Activation Catalysis
5. Methods Based on Dual Activation Involving Amine and Metal Co-Catalysis
6. Methods Based on the Merging of Enamine Activation and Photoactivation
7. Methods Based on Dual Enamine Activation/Brønsted Acid Catalysis
8. Methods Based on Combined Use of α-Amino Acid Catalysts and a Base Cocatalyst
9. Enamine Mediated α-Functionalization via Reactivity Umpolung
10. Miscellaneous
11. Conclusions and Prospects
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bella, M.; Gasperi, T. Organocatalytic Formation of Quaternary Stereocenters. Synthesis 2009, 2009, 1583–1614. [Google Scholar] [CrossRef]
- Zhou, F.; Liu, Y.-L.; Zhou, J. Catalytic Asymmetric Synthesis of Oxindoles Bearing a Tetrasubstituted Stereocenter at the C-3 Position. Adv. Synth. Catal. 2010, 352, 1381–1407. [Google Scholar] [CrossRef]
- Das, J.P.; Marek, I. Enantioselective Synthesis of All-Carbon Quaternary Stereogenic Centers in Acyclic Systems. Chem. Commun. 2011, 47, 4593–4623. [Google Scholar] [CrossRef] [PubMed]
- Hong, A.; Stoltz, B.M. The Construction of All-Carbon Quaternary Stereocenters by Use of Pd-Catalyzed Asymmetric Allylic Alkylation Reactions in Total Synthesis. Eur. J. Org. Chem. 2013, 2013, 2745–2759. [Google Scholar] [CrossRef] [PubMed]
- Quasdorf, K.W.; Overman, L.E. Catalytic Enantioselective Synthesis of Quaternary Carbon Stereocentres. Nature 2014, 516, 181–191. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Han, S.-J.; Liu, W.-B.; Stoltz, B.M. Catalytic Enantioselective Construction of Quaternary Stereocenters: Assembly of Key Building Blocks for the Synthesis of Biologically Active Molecules. Acc. Chem. Res. 2015, 48, 740–751. [Google Scholar] [CrossRef]
- Büschleb, M.; Dorich, S.; Hanessian, S.; Tao, D.; Schenthal, K.B.; Overman, L.E. Synthetic Strategies toward Natural Products Containing Contiguous Stereogenic Quaternary Carbon Atoms. Angew. Chem. Int. Ed. 2016, 55, 4156–4186. [Google Scholar] [CrossRef]
- Ling, T.; Rivas, F. All-Carbon Quaternary Centers in Natural Products and Medicinal Chemistry: Recent Advances. Tetrahedron 2016, 72, 6729–6777. [Google Scholar] [CrossRef]
- Feng, J.; Holmes, M.; Krische, M.J. Acyclic Quaternary Carbon Stereocenters via Enantioselective Transition Metal Catalysis. Chem. Rev. 2017, 117, 12564–12580. [Google Scholar] [CrossRef]
- Pierrot, D.; Marek, I. Synthesis of Enantioenriched Vicinal Tertiary and Quaternary Carbon Stereogenic Centers within an Acyclic Chain. Angew. Chem. Int. Ed. 2020, 59, 36–49. [Google Scholar] [CrossRef]
- Hajos, Z.G.; Parrish, D.R. Asymmetric Synthesis of Optically Active Polycyclic Organic Compounds. Ger. Pat. DE 1971, 2102623. [Google Scholar]
- Eder, U.; Sauer, G.R.; Wiechert, R. Optically Active 1,5–Indanone and 1,6–Naphthalenedione Derivatives. Ger. Pat. DE 1971, 2014757. [Google Scholar]
- Hajos, Z.G.; Parrish, D.R. Asymmetric Synthesis of Bicyclic Intermediates of Natural Product Chemistry. J. Org. Chem. 1974, 39, 1615–1621. [Google Scholar] [CrossRef]
- List, B.; Lerner, R.A.; Barbas, C.F., III. Proline-Catalyzed Direct Asymmetric Aldol Reactions. J. Am. Chem. Soc. 2000, 122, 2395–2396. [Google Scholar] [CrossRef]
- Northrup, A.B.; MacMillan, D.W.C. The First Direct and Enantioselective Cross-Aldol Reaction of Aldehydes. J. Am. Chem. Soc. 2002, 124, 6798–6799. [Google Scholar] [CrossRef]
- List, B. Enamine Catalysis Is a Powerful Strategy for the Catalytic Generation and Use of Carbanion Equivalents. Acc. Chem. Res. 2004, 37, 548–557. [Google Scholar] [CrossRef]
- Mukherjee, S.; Yang, J.W.; Hoffmann, S.; List, B. Asymmetric Enamine Catalysis. Chem. Rev. 2007, 107, 5471–5569. [Google Scholar] [CrossRef]
- Pihko, P.M.; Majander, I.; Erkkilä, A. Enamine Catalysis. Top. Curr. Chem. 2010, 291, 29–76. [Google Scholar] [CrossRef]
- Melchiorre, P. Cinchona-based Primary Amine Catalysis in the Asymmetric Functionalization of Carbonyl Compounds. Angew. Chem. Int. Ed. 2012, 51, 9748–9770. [Google Scholar] [CrossRef]
- Albrecht, L.; Jiang, H.; Jørgensen, K.A. Hydrogen-Bonding in Aminocatalysis: From Proline and Beyond. Chem. Eur. J. 2013, 20, 358–368. [Google Scholar] [CrossRef]
- Wang, L.; Liu, J. Recent Advances in Asymmetric Reactions Catalyzed by Proline and Its Derivatives. Synthesis 2017, 49, 960–972. [Google Scholar] [CrossRef]
- Reyes-Rodríguez, G.J.; Rezayee, N.M.; Vidal-Albalat, A.; Jørgensen, K.A. Prevalence of Diarylprolinol Silyl Ethers as Catalysts in Total Synthesis and Patents. Chem. Rev. 2019, 119, 4221–4260. [Google Scholar] [CrossRef] [PubMed]
- Vega-Penñaloza, A.; Paria, S.; Bonchio, M.; Dell’Amico, L.; Companyó, X. Profiling the Privileges of Pyrrolidine-Based Catalysts in Asymmetric Synthesis: From Polar to Light-Driven Radical Chemistry. ACS Catal. 2019, 9, 6058–6072. [Google Scholar] [CrossRef]
- Shao, Z.; Zhang, H. Combining Transition Metal Catalysis and Organocatalysis: A Broad New Concept for Catalysis. Chem. Soc. Rev. 2009, 38, 2745–2755. [Google Scholar] [CrossRef] [PubMed]
- Allen, A.E.; Macmillan, D.W.C. Synergistic Catalysis: A Powerful Synthetic Strategy for New Reaction Development. Chem. Sci. 2012, 3, 633–658. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.; Shao, Z. Combining Transition Tetal Catalysis and Organocatalysis—An Update. Chem. Soc. Rev. 2013, 42, 1337–1378. [Google Scholar] [CrossRef]
- Peters, R. Cooperative Catalysis: Designing Efficient Catalysts for Synthesis; Wiley-VCH: Wheinheim, Germany, 2015. [Google Scholar]
- Afewerki, S.; Córdova, A. Combinations of Aminocatalysts and Metal Catalysts: A Powerful Cooperative Approach in Selective Organic Synthesis. Chem. Rev. 2016, 116, 13512–13570. [Google Scholar] [CrossRef]
- Gong, L.-Z. Asymmetric Organo-Metal Catalysis, Concepts, Principles, and Applications; Wiley-VCH: Wheinheim, Germany, 2022. [Google Scholar]
- Chen, D.-F.; Gong, L.-Z. Organo/Transition-Metal Combined Catalysis Rejuvenates Both in Asymmetric Synthesis. J. Am. Chem. Soc. 2022, 144, 2415–2437. [Google Scholar] [CrossRef]
- Chakraborty, N.; Das, B.; Rajbongshi, K.K.; Patel, B.K. Combined Power of Organo- and Transition Metal Catalysis in Organic Synthesis. Eur. J. Org. Chem. 2022, 2022, e202200273. [Google Scholar] [CrossRef]
- Del Vecchio, A.; Sinibaldi, A.; Nori, V.; Giorgianni, G.; Di Carmine, G.; Pesciaioli, F. Synergistic Strategies in Aminocatalysis. Chem. Eur. J. 2022, 28, e202200818. [Google Scholar] [CrossRef]
- Nair, V.V.; Arunprasath, D.; Pandidurai, S.; Sekar, G. Synergistic Dual Amine/Transition Metal Catalysis: Recent Advances. Eur. J. Org. Chem. 2022, 2022, e202200244. [Google Scholar] [CrossRef]
- Roy, S.; Paul, H.; Chatterjee, I. Light-Mediated Aminocatalysis: The Dual-Catalytic Ability Enabling New Enantioselective Route. Eur. J. Org. Chem. 2022, 2022, e202200446. [Google Scholar] [CrossRef]
- Desmarchelier, A.; Coeffard, V.; Moreau, X.; Greck, C. Asymmetric Organocatalytic Functionalization of α,α-Disubstituted Aldehydes through Enamine Activation. Tetrahedron 2014, 70, 2491–2513. [Google Scholar] [CrossRef]
- Gladiali, S.; Pinna, L. Completely Regioselective Hydroformylation of Methyl n-Acetamidoacrylate by Chiral Rhodium Phosphine Catalysts. Tetrahedron: Asymmetry 1990, 1, 693–696. [Google Scholar] [CrossRef]
- Lee, C.W.; Alper, H. Influence of 1,4-Bis(Diphenylphosphino)-Butane on the Hydroformylation of α,β-Unsaturated Esters Catalyzed by Zwitterionic, Cationic and Neutral Rhodium(I) Complexes. The Asymmetric Hydroformylation of α-Methylene-γ-Butyrolactone. J. Org. Chem. 1995, 60, 499–503. [Google Scholar] [CrossRef]
- Wang, X.; Buchwald, S.L. Synthesis of Optically Pure 2-Trifluoromethyl Lactic Acid by Asymmetric Hydroformylation. J. Org. Chem. 2013, 78, 3429–3433. [Google Scholar] [CrossRef]
- Fanfoni, L.; Diab, L.; Smejkal, T.; Breit, B. Efficient Synthesis of New Fluorinated Building Blocks by means of Hydroformylation. Chimia 2014, 68, 371–377. [Google Scholar] [CrossRef]
- Eshon, J.; Foarta, F.; Landis, C.R.; Schomaker, J.M. α-Tetrasubstituted Aldehydes through Electronic and Strain-Controlled Branch-Selective Stereoselective Hydroformylation. J. Org. Chem. 2018, 83, 10207–10220. [Google Scholar] [CrossRef]
- Vogt, H.; Vanderheiden, S.; Bräse, S. Proline-Catalysed Asymmetric Amination of α,α-Disubstituted Aldehydes: Synthesis of Configurationally Stable Enantioenriched α-Aminoaldehydes. Chem. Commun. 2003, 2448–2449. [Google Scholar] [CrossRef]
- Baumann, T.; Vogt, H.; Bräse, S. The Proline-Catalyzed Asymmetric Amination of Branched Aldehydes. Eur. J. Org. Chem. 2007, 2007, 266–282. [Google Scholar] [CrossRef]
- Hartmann, C.E.; Baumann, T.; Bächle, M.; Bräse, S. Asymmetric Synthesis of Deuterated and Fluorinated Aromatic α,α-Disubstituted Amino Acid Derivatives. Tetrahedron: Asymmetry 2010, 21, 1341–1349. [Google Scholar] [CrossRef]
- Mase, N.; Thayumanavan, R.; Tanaka, F.; Barbas, C.F., III. Direct Asymmetric Organocatalytic Michael Reactions of α,α-Disubstituted Aldehydes with β-Nitrostyrenes for the Synthesis of Quaternary Carbon-Containing Products. Org. Lett. 2004, 6, 2527–2530. [Google Scholar] [CrossRef] [PubMed]
- Mase, N.; Tanaka, F.; Barbas, C.F., III. Synthesis ofβ-Hydroxyaldehydes with Stereogenic Quaternary Carbon Centers by Direct Organocatalytic Asymmetric Aldol Reactions. Angew. Chem. Int. Ed. 2004, 43, 2420–2423. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Aceña, J.L.; Soloshonok, V.A.; Izawa, K.; Liu, H. Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II–III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev. 2016, 116, 422–518. [Google Scholar] [CrossRef]
- Mei, H.; Han, J.; White, S.; Graham, D.J.; Izawa, K.; Sato, T.; Fustero, S.; Meanwell, N.A.; Soloshonok, V.A. Tailor-Made Amino Acids and Fluorinated Motifs as Prominent Traits in the Modern Pharmaceuticals. Chem. Eur. J. 2020, 26, 11349–11390. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Kiss, L.; Mei, H.; Remete, A.M.; Ponikvar-Svet, M.; Sedgwick, D.M.; Roman, R.; Fustero, S.; Moriwaki, H.; Soloshonok, V.A. Chemical Aspects of Human and Environmental Overload with Fluorine. Chem. Rev. 2021, 121, 4678–4742. [Google Scholar] [CrossRef]
- Witten, M.R.; Jacobsen, E.N. A Simple Primary Amine Catalyst for Enantioselective α-Hydroxylations and α-Fluorinations of Branched Aldehydes. Org. Lett. 2015, 17, 2772–2775. [Google Scholar] [CrossRef] [PubMed]
- Emma, M.G.; Lombardo, M.; Trombini, C.; Quintavalla, A. The Organocatalytic α-Fluorination of Chiral γ-Nitroaldehydes: The Challenge of Facing the Construction of a Quaternary Fluorinated Stereocenter. Eur. J. Org. Chem. 2016, 2016, 3223–3232. [Google Scholar] [CrossRef]
- Shibatomi, K.; Kitahara, K.; Okimi, T.; Abe, Y.; Iwasa, S. Enantioselective Fluorination of α-Branched Aldehydes and Subsequent Conversion to α-Hydroxyacetals via Stereospecific C–F Bond Cleavage. Chem. Sci. 2016, 7, 1388–1392. [Google Scholar] [CrossRef]
- Cui, L.; You, Y.; Mi, X.; Luo, S. Asymmetric Fluorination of α-Branched Aldehydes by Chiral Primary Amine Catalysis: Reagent-Controlled Enantioselectivity Switch. J. Org. Chem. 2018, 83, 4250–4256. [Google Scholar] [CrossRef]
- Huters, A.D.; Stambuli, J.; Klix, R.C.; Matulenko, M.A.; Chan, V.S.; Simanis, J.; Hill, D.R.; Reddy, R.E.; Towne, T.B.; Bellettini, J.R.; et al. Scalable Asymmetric Syntheses of Foslevodopa and Foscarbidopa Drug Substances for the Treatment of Parkinson’s Disease. J. Org. Chem. 2022, 87, 1986–1995. [Google Scholar] [CrossRef] [PubMed]
- Torregrosa-Chinillach, A.; Carral-Menoyo, A.; Gómez-Bengoa, E.; Chinchilla, R. Organocatalytic Enantioselective α-Nitrogenation of α,α-Disubstituted Aldehydes in the Absence of a Solvent. J. Org. Chem. 2022, 87, 14507–14513. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Shao, Y.; Chen, Y.; Xue, X.; Yang, X. Enantioselective Synthesis of Planar-Chiral Macrocycles through Asymmetric Electrophilic Aromatic Amination. Angew. Chem. Int. Ed. 2022, 61, e202201064. [Google Scholar] [CrossRef]
- Portolani, C.; Centonze, G.; Luciani, S.; Pellegrini, A.; Righi, P.; Mazzanti, A.; Ciogli, A.; Sorato, A.; Bencivenni, G. Synthesis of Atropisomeric Hydrazides by One-Pot Sequential Enantio- and Diastereoselective Catalysis. Angew. Chem. Int. Ed. 2022, 61, e202209895. [Google Scholar] [CrossRef]
- Mohr, L.; McCulley, C.H.; Blom, J.; Lamhauge, J.N.; Jørgensen, K.A. Investigation of the Organocatalytic Chlorination of 2-Phenylpropanal. Chem. Eur. J. 2021, 27, 17465–17475. [Google Scholar] [CrossRef]
- Shi, M.; Zhang, Q.; Gao, J.; Mi, X.; Luo, S. Catalytic Asymmetric α-Alkylsulfenylation with a Disulfide Reagent. Angew. Chem. Int. Ed. 2022, 61, e202209044. [Google Scholar] [CrossRef]
- Brown, A.R.; Kuo, W.-H.; Jacobsen, E.N. Enantioselective Catalytic α-Alkylation of Aldehydes via an SN1 Pathway. J. Am. Chem. Soc. 2010, 132, 9286–9288. [Google Scholar] [CrossRef]
- Cozzi, P.G.; Benfatti, F.; Zoli, L. Organocatalytic Asymmetric Alkylation of Aldehydes by SN1-Type Reaction of Alcohols. Angew. Chem. Int. Ed. 2009, 48, 1313–1316. [Google Scholar] [CrossRef]
- Guo, Z.-L.; Xue, J.-H.; Fu, L.-N.; Zhang, S.-E.; Guo, Q.-X. The Direct Asymmetric Alkylation of α-Amino Aldehydes with 3-Indolylmethanols by Enamine Catalysis. Org. Lett. 2014, 16, 6472–6475. [Google Scholar] [CrossRef]
- List, B.; Čorić, I.; Grygorenko, O.O.; Kaib, P.S.J.; Komarov, I.; Lee, A.; Leutzsch, M.; Pan, S.C.; Tymtsunik, A.; Van Gemmeren, M. The Catalytic Asymmetric α-Benzylation of Aldehydes. Angew. Chem. Int. Ed. 2014, 53, 282–285. [Google Scholar] [CrossRef]
- Lee, A. Chiral Amine-catalyzed α-Benzylation of α-Branched Aldehydes. Bull. Korean Chem. Soc. 2015, 36, 2585–2586. [Google Scholar] [CrossRef]
- Yoshida, M. Organocatalytic Asymmetric α-Allylation and Propargylation of α-Branched Aldehydes with Alkyl Halides. J. Org. Chem. 2021, 86, 10921–10927. [Google Scholar] [CrossRef] [PubMed]
- Tsogoeva, S.B. Recent Advances in Asymmetric Organocatalytic 1,4-Conjugate Additions. Eur. J. Org. Chem. 2007, 2007, 1701–1716. [Google Scholar] [CrossRef]
- Córdova, A. (Ed.) Catalytic Asymmetric Conjugate Reactions; Wiley-VCH Verlag GmbH & Co.: Weinheim, Germany, 2010. [Google Scholar]
- Vicario, J.L.; Badía, D.; Carrillo, L.; Reyes, E. Organocatalytic Enantioselecive Conjugate Addition Reactions; RSC Publishing: Cambridge, UK, 2010. [Google Scholar]
- Zhang, Y.; Wang, W. Recent Advances in Organocatalytic Asymmetric Michael Reactions. Catal. Sci. Technol. 2012, 2, 42–53. [Google Scholar] [CrossRef]
- Desimoni, G.; Faita, G.; Quadrelli, P. Enantioselective Catalytic Reactions with N-Acyliden Penta-atomic Aza-heterocycles. Heterocycles as Masked Bricks to Build Chiral Scaffolds. Chem. Rev. 2015, 115, 9922–9980. [Google Scholar] [CrossRef]
- Singh, G.S.; Yeboah, E.M. Recent Applications of Cinchona Alkaloid-based Catalysts in Asymmetric Addition Reactions. Rep. Org. Chem. 2016, 6, 47–75. [Google Scholar] [CrossRef]
- Hayashi, M.; Matsubara, R. Recent Topics on Catalytic Asymmetric 1,4-Addition. Tetrahedron Lett. 2017, 58, 1793–1805. [Google Scholar] [CrossRef]
- Yoshida, M.; Ukigai, H.; Shibatomi, K.; Hara, S. Organocatalytic Asymmetric Michael Addition of α-Branched Aldehydes to Vinyl Ketones: Synthesis of 5-Ketoaldehydes Possessing a Stereo-controlled all-Carbon Quaternary Stereogenic Center. Tetrahedron Lett. 2015, 56, 3890–3893. [Google Scholar] [CrossRef]
- Nájera, C.; Gómez-Bengoa, E.; Vízcaíno-Milla, P.; Sansano, J.M.; Fiser, B. Primary Amine–2-Aminopyrimidine Chiral Organocatalysts for the Enantioselective Conjugate Addition of Branched Aldehydes to Maleimides. Synthesis 2015, 47, 2199–2206. [Google Scholar] [CrossRef]
- Porta, R.; Benaglia, M.; Coccia, F.; Cozzi, F.; Puglisi, A. Solid Supported 9-Amino-9-deoxy-epi-quinine as Efficient Organocatalyst for Stereoselective Reactions in Batch and Under Continuous Flow Conditions. Adv. Synth. Catal. 2015, 357, 377–383. [Google Scholar] [CrossRef]
- Wang, W.; Wang, J.; Li, H. Direct, Highly Enantioselective Pyrrolidine Sulfonamide Catalyzed Michael Addition of Aldehydes to Nitrostyrenes. Angew. Chem. Int. Ed. 2005, 44, 1369–1371. [Google Scholar] [CrossRef]
- Szcześniak, P.; Staszewska-Krajewska, O.; Furman, B.; Mlynarski, J. Asymmetric Synthesis of Cyclic NitronesviaOrganocatalytic Michael Addition of Aldehydes to Nitroolefins and Subsequent Reductive Cyclization. Chemistryselect 2017, 2, 2670–2676. [Google Scholar] [CrossRef]
- Kawada, M.; Tsuyusaki, R.; Nakashima, K.; Akutsu, H.; Hirashima, S.; Matsumoto, T.; Yanai, H.; Miura, T. Diaminomethylenemalononitrile as a Chiral Single Hydrogen Bond Catalyst: Application to Enantioselective Conjugate Addition of α-Branched Aldehydes. Chem. Asian J. 2021, 16, 2272–2275. [Google Scholar] [CrossRef]
- Du, Z.-H.; Tao, B.-X.; Yuan, M.; Qin, W.-J.; Xu, Y.-L.; Wang, P.; Da, C.-S. Peptide-Catalyzed Highly Asymmetric Cross-Aldol Reaction of Aldehydes to Biomimetically Synthesize 1,4-Dicarbonyls. Org. Lett. 2020, 22, 4444–4450. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.-K.; Chen, Y.; Hang, J.; Tang, L.; McDaid, P.; Deng, L. Asymmetric Organic Catalysis with Modified Cinchona Alkaloids. Acc. Chem. Res. 2004, 37, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Palomo, C.; Oiarbide, M.; López, R. Asymmetric Organocatalysis by Chiral Brønsted Bases: Implications and Applications. Chem. Soc. Rev. 2009, 38, 632–653. [Google Scholar] [CrossRef] [PubMed]
- Ting, A.; Goss, J.M.; McDougal, N.T.; Schaus, S.E. Brønsted Base Catalysts. Top. Curr. Chem. 2010, 291, 145–200. [Google Scholar] [CrossRef]
- Singh, R.P.; Deng, L. Cinchona Alkaloid Organocatalysts. In Asymmetric Organocatalysis 2: Brønsted Base and Acid Catalysts, and Additional Topics; Maruoka, K., Ed.; Thieme: Stuttgart, Germany, 2012; pp. 41–118. [Google Scholar]
- Jang, H.B.; Oh, J.S.; Song, C.E. Bifunctional Cinchona Alkaloid Organocatalysts. In Asymmetric Organocatalysis 2: Brønsted Base and Acid Catalysts, and Additional Topics; Maruoka, K., Ed.; Thieme: Stuttgart, Germany, 2012; pp. 119–168. [Google Scholar]
- García-Urricelqui, A.; De Cózar, A.; Mielgo, A.; Palomo, C. Probing α-Amino Aldehydes as Weakly Acidic Pronucleophiles: Direct Access to Quaternary α-Amino Aldehydes by an Enantioselective Michael Addition Catalyzed by Brønsted Bases. Chem. Eur. J. 2021, 27, 2483–2492. [Google Scholar] [CrossRef]
- García-Urricelqui, A.; de Cózar, A.; Campano, T.E.; Mielgo, A.; Palomo, C. syn -Selective Michael Reaction of α-Branched Aryl Acetaldehydes with Nitroolefins Promoted by Squaric Amino Acid Derived Bifunctional Brønsted Bases. Eur. J. Org. Chem. 2021, 2021, 3604–3612. [Google Scholar] [CrossRef]
- Bernhard, Y.; Thomson, B.; Ferey, V.; Sauthier, M. Nickel-Catalyzed α-Allylation of Aldehydes and Tandem Aldol Condensation/Allylation Reaction with Allylic Alcohols. Angew. Chem. Int. Ed. 2017, 56, 7460–7464. [Google Scholar] [CrossRef]
- Franzoni, I.; Guénée, L.; Mazet, C. A General Pd-Catalyzed α- and γ-Benzylation of Aldehydes for the Formation of Quaternary Centers. Org. Biomol. Chem. 2015, 13, 6338–6343. [Google Scholar] [CrossRef] [PubMed]
- Wright, T.B.; Evans, P.A. Enantioselective Rhodium-Catalyzed Allylic Alkylation of Prochiral α,α-Disubstituted Aldehyde Enolates for the Construction of Acyclic Quaternary Stereogenic Centers. J. Am. Chem. Soc. 2016, 138, 15303–15306. [Google Scholar] [CrossRef] [PubMed]
- Trost, B.M.; Hung, C.-I. (J.); Saget, T.; Gnanamani, E. Branched Aldehydes as Linchpins for the Enantioselective and Stereodivergent Synthesis of 1,3-Aminoalcohols Featuring a Quaternary Stereocentre. Nat. Catal. 2018, 1, 523–530. [Google Scholar] [CrossRef]
- Trost, B.M.; Bartlett, M.J. ProPhenol-Catalyzed Asymmetric Additions by Spontaneously Assembled Dinuclear Main Group Metal Complexes. Acc. Chem. Res. 2015, 48, 688–701. [Google Scholar] [CrossRef]
- Trost, B.M.; Zuo, Z.; Wang, Y.; Schultz, J.E. Pd(0)-Catalyzed Chemo-, Diastereo-, and Enantioselective α-Quaternary Alkylation of Branched Aldehydes. ACS Catal. 2020, 10, 9496–9503. [Google Scholar] [CrossRef]
- Mukherjee, S.; List, B. Chiral Counteranions in Asymmetric Transition-Metal Catalysis: Highly Enantioselective Pd/Brønsted Acid-Catalyzed Direct α-Allylation of Aldehydes. J. Am. Chem. Soc. 2007, 129, 11336–11337. [Google Scholar] [CrossRef]
- Jiang, G.; List, B. Direct Asymmetric α-Allylation of Aldehydes with Simple Allylic Alcohols Enabled by the Concerted Action of Three Different Catalysts. Angew. Chem. Int. Ed. 2011, 50, 9471–9474. [Google Scholar] [CrossRef]
- Krautwald, S.; Sarlah, D.; Schafroth, M.A.; Carreira, E.M. Enantio- and Diastereodivergent Dual Catalysis: α-Allylation of Branched Aldehydes. Science 2013, 340, 1065–1068. [Google Scholar] [CrossRef]
- Krautwald, S.; Schafroth, M.A.; Sarlah, D.; Carreira, E.M. Stereodivergent α-Allylation of Linear Aldehydes with Dual Iridium and Amine Catalysis. J. Am. Chem. Soc. 2014, 136, 3020–3023. [Google Scholar] [CrossRef]
- Sandmeier, T.; Krautwald, S.; Zipfel, H.F.; Carreira, E.M. Stereodivergent Dual Catalytic α-Allylation of Protected α-Amino- and α-Hydroxyacetaldehydes. Angew. Chem. Int. Ed. 2015, 54, 14363–14367. [Google Scholar] [CrossRef]
- Wang, P.-S.; Lin, H.-C.; Zhai, Y.-J.; Han, Z.-Y.; Gong, L.-Z. Chiral Counteranion Strategy for Asymmetric Oxidative C(sp3)-H/C(sp3)-H Coupling: Enantioselective α-Allylation of Aldehydes with Terminal Alkenes. Angew. Chem. Int. Ed. 2014, 53, 12218–12221. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Brenner-Moyer, S.E. Combining Palladium and Chiral Organocatalysis for the Enantioselective Deconjugative Allylation of Enals via Dienamine Intermediates. J. Org. Chem. 2022, 87, 866–873. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.-Q.; Shen, H.-C. Nickel/Enamine Cooperative Catalysis Enables Highly Enantioselective Allylic Alkylation of α-Branched Aldehydes. ACS Catal. 2021, 11, 11849–11854. [Google Scholar] [CrossRef]
- Li, M.; Datta, S.; Barber, D.M.; Dixon, D.J. Dual Amine and Palladium Catalysis in Diastereo- and Enantioselective Allene Carbocyclization Reactions. Org. Lett. 2012, 14, 6350–6353. [Google Scholar] [CrossRef] [PubMed]
- Ballesteros, A.; Morán-Poladura, P.; González, J.M. Gold(I) Operational in Synergistic Catalysis for the Intermolecular α-Addition Reaction of Aldehydes Across Allenamides. Chem. Commun. 2016, 52, 2905–2908. [Google Scholar] [CrossRef]
- Fernández-Casado, J.; Nelson, R.; Mascareñas, J.L.; López, F. Synergistic Gold and Enamine Catalysis: Intermolecular α-Alkylation of Aldehydes with Allenamides. Chem. Commun. 2016, 52, 2909–2912. [Google Scholar] [CrossRef]
- Blieck, R.; Lemouzy, S.; van der Lee, A.; Taillefer, M.; Monnier, F. Synergistic Copper/Enamine Catalysis for the Regio-, Stereo-, and Enantioselective Intermolecular α-Addition of Aldehydes to Allenamides. Org. Lett. 2021, 23, 9199–9203. [Google Scholar] [CrossRef]
- Nicholls, L.D.M.; Wennemers, H. Synergistic Peptide and Gold Catalysis: Enantioselective Addition of Branched Aldehydes to Allenamides. Chem. Eur. J. 2021, 27, 17559–17564. [Google Scholar] [CrossRef]
- Moore, J.L.; Taylor, S.M.; Soloshonok, V.A. An Efficient and Operationally Convenient General Synthesis of Tertiary Amines by Direct Alkylation of Secondary Amines with Alkyl Halides in the Presence of Hünig’s Base. Arkivoc 2005, 2005, 287–292. [Google Scholar] [CrossRef]
- Zhou, H.; Wang, Y.; Zhang, L.; Cai, M.; Luo, S. Enantioselective Terminal Addition to Allenes by Dual Chiral Primary Amine/Palladium Catalysis. J. Am. Chem. Soc. 2017, 139, 3631–3634. [Google Scholar] [CrossRef]
- Cruz, F.A.; Dong, V.M. Stereodivergent Coupling of Aldehydes and Alkynes via Synergistic Catalysis Using Rh and Jacobsen’s Amine. J. Am. Chem. Soc. 2017, 139, 1029–1032. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Gong, L.; Meggers, E. Asymmetric Dual Catalysis via Fragmentation of a Single Rhodium Precursor Complex. Chem. Commun. 2016, 52, 7699–7702. [Google Scholar] [CrossRef] [PubMed]
- Arceo, E.; Jurberg, I.; Álvarez-Fernández, A.; Melchiorre, P. Photochemical Activity of a Key Donor–Acceptor Complex Can Drive Stereoselective Catalytic α-Alkylation of Aldehydes. Nat. Chem. 2013, 5, 750–756. [Google Scholar] [CrossRef] [PubMed]
- Matsui, H.; Murase, M.; Yajima, T. Metal-free Visible-Light Synthesis of Quaternary α-Perfluoroalkyl Aldehydes via an Enamine Intermediate. Org. Biomol. Chem. 2018, 16, 7120–7123. [Google Scholar] [CrossRef]
- Akiyama, T. Stronger Brønsted Acids. Chem. Rev. 2007, 107, 5744–5758. [Google Scholar] [CrossRef]
- Terada, M. Binaphthol-Derived Phosphoric Acid as a Versatile Catalyst for Enantioselective Carbon–Carbon Bond Forming Reactions. Chem. Commun. 2008, 4097–4112. [Google Scholar] [CrossRef]
- Kampen, D.; Reisinger, C.M.; List, B. Chiral Brønsted Acids for Asymmetric Organocatalysis. Top Curr. Chem. 2010, 291, 395–456. [Google Scholar] [CrossRef]
- Terada, M. Chiral Phosphoric Acids as Versatile Catalysts for Enantioselective Transformations. Synthesis 2010, 2010, 1929–1982. [Google Scholar] [CrossRef]
- Akiyama, T.; Mori, K. Stronger Brønsted Acids: Recent Progress. Chem. Rev. 2015, 115, 9277–9306. [Google Scholar] [CrossRef]
- James, T.; van Gemmeren, M.; List, B. Development and Applications of Disulfonimides in Enantioselective Organocatalysis. Chem. Rev. 2015, 115, 9388–9409. [Google Scholar] [CrossRef]
- Schreyer, L.; Properzi, R.; List, B. IDPi Catalysis. Angew. Chem. Int. Ed. 2019, 58, 12761–12777. [Google Scholar] [CrossRef] [PubMed]
- Arimitsu, S.; Yonamine, T.; Higashi, M. Cinchona-Based Primary Amine Catalyzed a Proximal Functionalization of Dienamines: Asymmetric α-Fluorination of α-Branched Enals. ACS Catal. 2017, 7, 4736–4740. [Google Scholar] [CrossRef]
- Kuraoku, D.; Yonamine, T.; Koja, G.; Yoshida, N.; Arimitsu, S.; Higashi, M. Effects of Water Addition on a Catalytic Fluorination of Dienamine. Molecules 2019, 24, 3428. [Google Scholar] [CrossRef] [PubMed]
- Demoulin, N.; Lifchits, O.; List, B. Organocatalytic Asymmetric α-Benzoyloxylation of α-Branched Aldehydes and Enals: A Useful Approach to Oxygenated Quaternary Stereocenters. Tetrahedron 2012, 68, 7568–7574. [Google Scholar] [CrossRef]
- Arimitsu, S.; Gima, E. Improvement of Primary-Amine-Catalyzed Asymmetric α-Benzoyloxylation of α-Branched Enals by a Synergistic Effect of Water and Sulfonic Acids. Tetrahedron Lett. 2020, 61, 152032. [Google Scholar] [CrossRef]
- Mo, X.; Hall, D.G. Dual Catalysis Using Boronic Acid and Chiral Amine: Acyclic Quaternary Carbons via Enantioselective Alkylation of Branched Aldehydes with Allylic Alcohols. J. Am. Chem. Soc. 2016, 138, 10762–10765. [Google Scholar] [CrossRef]
- Nugent, T.C.; Sadiq, A.; Bibi, A.; Heine, T.; Zeonjuk, L.L.; Vankova, N.; Bassil, B.S. Noncovalent Bifunctional Organocatalysts: Powerful Tools for Contiguous Quaternary-Tertiary Stereogenic Carbon Formation, Scope, and Origin of Enantioselectivity. Chem. Eur. J. 2012, 18, 4088–4098. [Google Scholar] [CrossRef]
- Sadiq, A.; Nugent, T.C. Catalytic Access to Succinimide Products Containing Stereogenic Quaternary Carbons. Chemistryselect 2020, 5, 11934–11938. [Google Scholar] [CrossRef]
- Ahmad, A.; Ullah, F.; Sadiq, A.; Ayaz, M.; Jan, M.S.; Shahid, M.; Wadood, A.; Mahmood, F.; Rashid, U.; Ullah, R.; et al. Comparative Cholinesterase, α-Glucosidase Inhibitory, Antioxidant, Molecular Docking, and Kinetic Studies on Potent Succinimide Derivatives. Drug Des. Dev. Ther. 2020, 14, 2165–2178. [Google Scholar] [CrossRef]
- Ahmad, S.; Mahnashi, M.H.; Alyami, B.A.; Alqahtani, Y.S.; Ullah, F.; Ayaz, M.; Tariq, M.; Sadiq, A.; Rashid, U. Synthesis of Michael Adducts as Key Building Blocks for Potential Analgesic Drugs: In vitro, in vivo and in silico Explorations. Drug Des. Dev. Ther. 2021, 15, 1299–1313. [Google Scholar] [CrossRef]
- Yoshida, M.; Sato, A.; Hara, S. Asymmetric Michael Addition of Aldehydes to Nitroalkenes Using a Primary Amino Acid Lithium Salt. Org. Biomol. Chem. 2010, 8, 3031–3036. [Google Scholar] [CrossRef]
- Zhu, S.; Yu, S.; Ma, D. Highly Efficient Catalytic System for Enantioselective Michael Addition of Aldehydes to Nitroalkenes in Water. Angew. Chem. Int. Ed. 2008, 47, 545–548. [Google Scholar] [CrossRef]
- Yoshida, M.; Masaki, E.; Ikehara, H.; Hara, S. Enantioselective Synthesis of Gabapentin Analogues via Organocatalytic Asymmetric Michael Addition of α-Branched Aldehydes to β-Nitroacrylates. Org. Biomol. Chem. 2012, 10, 5289–5297. [Google Scholar] [CrossRef]
- Yoshida, M. Asymmetric Synthesis of a Quaternary Carbon Stereogenic Center by Organocatalysis Using a Primary Amino Acid and Its Salt. Chem. Rec. 2023, e202200276. [Google Scholar] [CrossRef]
- Han, J.; Escorihuela, J.; Fustero, S.; Landa, A.; Soloshonok, V.A.; Sorochinsky, A. Asymmetric Michael Addition in Synthesis of β-Substituted GABA Derivatives. Molecules 2022, 27, 3797. [Google Scholar] [CrossRef]
- Zhu, L.; Wang, D.; Jia, Z.; Lin, Q.; Huang, M.; Luo, S. Catalytic Asymmetric Oxidative Enamine Transformations. ACS Catal. 2018, 8, 5466–5484. [Google Scholar] [CrossRef]
- Naesborg, L.; Leth, L.A.; Reyes-Rodríguez, G.J.; Palazzo, T.A.; Corti, V.; Jørgensen, K.A. Direct Enantio- and Diastereoselective Oxidative Homocoupling of Aldehydes. Chem. Eur. J. 2018, 24, 14844–14848. [Google Scholar] [CrossRef]
- Leth, L.A.; Næsborg, L.; Reyes-Rodríguez, G.J.; Tobiesen, H.N.; Iversen, M.V.; Jørgensen, K.A. Enantioselective Oxidative Coupling of Carboxylic Acids to α-Branched Aldehydes. J. Am. Chem. Soc. 2018, 140, 12687–12690. [Google Scholar] [CrossRef]
- Blom, J.; Reyes-Rodríguez, G.J.; Tobiesen, H.N.; Lamhauge, J.N.; Iversen, M.V.; Barløse, C.L.; Hammer, N.; Rusbjerg, M.; Jørgensen, K.A. Umpolung Strategy for α-Functionalization of Aldehydes for the Addition of Thiols and other Nucleophiles. Angew. Chem. Int. Ed. 2019, 58, 17856–17862. [Google Scholar] [CrossRef]
- Rezayee, N.M.; Lauridsen, V.H.; Næsborg, L.; Nguyen, T.V.Q.; Tobiesen, H.N.; Jørgensen, K.A. Oxidative Organocatalysed Enantioselective Coupling of Indoles with Aldehydes that Forms Quaternary Carbon Stereocentres. Chem. Sci. 2019, 10, 3586–3591. [Google Scholar] [CrossRef]
- Rezayee, N.M.; Enemærke, V.J.; Linde, S.T.; Lamhauge, J.N.; Reyes-Rodríguez, G.J.; Jørgensen, K.A.; Lu, C.; Houk, K.N. An Asymmetric SN2 Dynamic Kinetic Resolution. J. Am. Chem. Soc. 2021, 143, 7509–7520. [Google Scholar] [CrossRef]
- Tobiesen, H.N.; Leth, L.A.; Iversen, M.V.; Naesborg, L.; Bertelsen, S.; Jørgensen, K.A. Stereoselective Oxidative Bioconjugation of Amino Acids and Oligopeptides to Aldehydes. Angew. Chem. Int. Ed. 2020, 59, 18490–18494. [Google Scholar] [CrossRef]
- Lamhauge, J.N.; Corti, V.; Liu, Y.; Jørgensen, K.A. Enantioselective α-Etherification of Branched Aldehydes via an Oxidative Umpolung Strategy. Angew. Chem. Int. Ed. 2021, 60, 18728–18733. [Google Scholar] [CrossRef]
- Rezayee, N.M.; Rusbjerg, M.; Marx, M.; Linde, S.T.; Jørgensen, K.A. Metal-free, Oxidative α-Coupling of Aldehydes with Amine Nucleophiles for the Preparation of Congested C(sp3)–N Bonds. J. Org. Chem. 2022, 87, 1756–1766. [Google Scholar] [CrossRef]
- Zhang, M.-M.; Wang, Y.-N.; Wang, B.-C.; Chen, X.-W.; Lu, L.-Q.; Xiao, W.-J. Synergetic Iridium and Amine Catalysis Enables Asymmetric [4+2] Cycloadditions of Vinyl Aminoalcohols with Carbonyls. Nat. Commun. 2019, 10, 2716. [Google Scholar] [CrossRef]
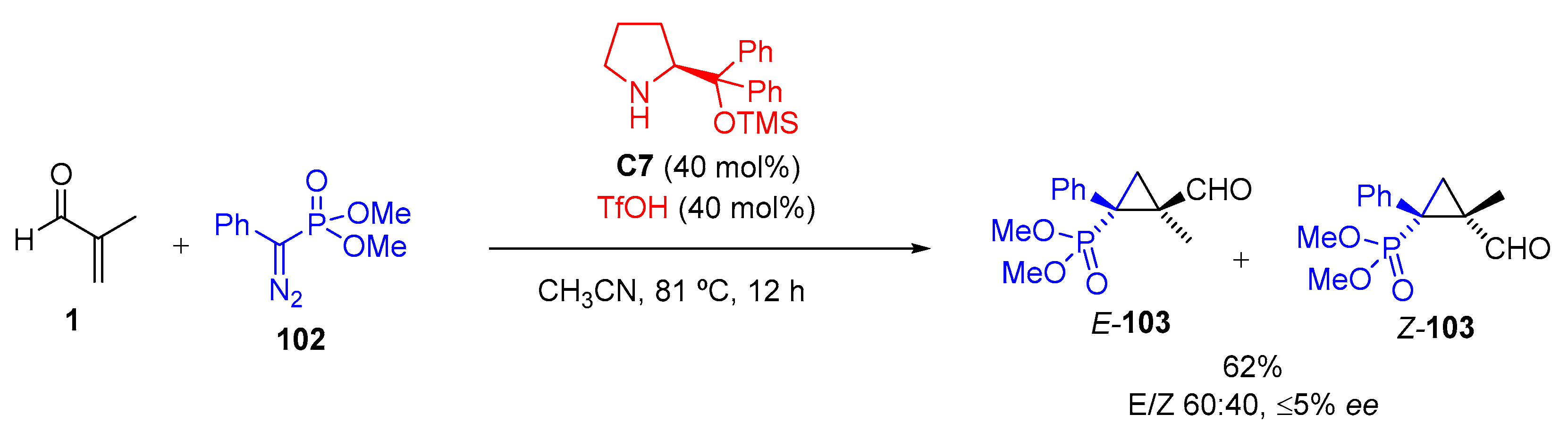
- El-Gokha, A.; Maas, G. (2-Formyl-1-phenylcyclopropyl)phosphonates as Building Blocks for (2-Aminomethyl-cyclopropyl)phosphonates. Tetrahedron 2011, 67, 2849–2857. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Klika, K.D. Terminology Related to the Phenomenon ‘Self-Disproportionation of Enantiomers’ (SDE). Helv. Chim. Acta 2014, 97, 1583–1589. [Google Scholar] [CrossRef]
- Nakamura, T.; Tateishi, K.; Tsukagoshi, S.; Hashimoto, S.; Watanabe, S.; Soloshonok, V.A.; Aceña, J.L.; Kitagawa, O. Self-disproportionation of Enantiomers of Non-racemic Chiral Amine Derivatives through Achiral Chromatography. Tetrahedron 2012, 68, 4013–4017. [Google Scholar] [CrossRef]
- Suzuki, Y.; Han, J.; Kitagawa, O.; Aceña, J.L.; Klika, K.D.; Soloshonok, V.A. A Comprehensive Examination of the Self-disproportionation of Enantiomers (SDE) of Chiral Amides via Achiral, Laboratory-Routine, Gravity-Driven Column Chromatography. RSC Adv. 2015, 5, 2988–2993. [Google Scholar] [CrossRef]
- Sorochinsky, A.E.; Katagiri, T.; Ono, T.; Wzorek, A.; Aceña, J.L.; Soloshonok, V.A. Optical Purifications via Self-Disproportionation of Enantiomers by Achiral Chromatography: Case Study of a Series of α-CF3-containing Secondary Alcohols. Chirality 2013, 25, 365–368. [Google Scholar] [CrossRef]
- Han, J.; Wzorek, A.; Kwiatkowska, M.; Soloshonok, V.A.; Klika, K.D. The Self-Disproportionation of Enantiomers (SDE) of Amino Acids and their Derivatives. Amino Acids 2019, 51, 865–889. [Google Scholar] [CrossRef]
- Wzorek, A.; Sato, A.; Drabowicz, J.; Soloshonok, V.A.; Klika, K.D. Remarkable Magnitude of the Self-Disproportionation of Enantiomers (SDE) via Achiral Chromatography: Application to the Practical-Scale Enantiopurification of β-Amino Acid Esters. Amino Acids 2016, 48, 605–613. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Sorochinsky, A.E.; Aceña, J.L. Self-Disproportionation of Enantiomers of Chiral, Non-Racemic Fluoroorganic Compounds: Role of Fluorine as Enabling Element. Synthesis 2013, 45, 141–152. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Roussel, C.; Kitagawa, O.; Sorochinsky, A.E. Self-Disproportionation of Enantiomers via Achiral Chromatography: A Warning and an Extra Dimension in Optical Purifications. Chem. Soc. Rev. 2012, 41, 4180–4188. [Google Scholar] [CrossRef] [PubMed]
- Ueki, H.; Yasumoto, M.; Soloshonok, V.A. Rational Application of Self-Disproportionation of Enantiomers via Sublimation—A Novel Methodological Dimension for Enantiomeric Purifications. Tetrahedron Asymmetry 2010, 21, 1396–1400. [Google Scholar] [CrossRef]
- Han, J.; Nelson, D.J.; Sorochinsky, A.E.; Soloshonok, V.A. Self-Disproportionation of Enantiomers via Sublimation; New and Truly Green Dimension in Optical Purification. Curr. Org. Synth. 2011, 8, 310–317. [Google Scholar] [CrossRef]
- Han, J.; Kitagawa, O.; Wzorek, A.; Klika, K.D.; Soloshonok, V.A. The Self-Disproportionation of Enantiomers (SDE): A Menace or an Opportunity? Chem. Sci. 2018, 9, 1718–1739. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Soloshonok, V.A.; Klika, K.D.; Drabowicz, J.; Wzorek, A. Chiral sulfoxides: Advances in Asymmetric Synthesis and Problems with the Accurate Determination of the Stereochemical Outcome. Chem. Soc. Rev. 2018, 47, 1307–1350. [Google Scholar] [CrossRef]
- Han, J.; Dembinski, R.; Soloshonok, V.; Klika, K. A Call for a Change in Policy Regarding the Necessity for SDE Tests to Validate the Veracity of the Outcome of Enantioselective Syntheses, the Inherent Chiral State of Natural Products, and Other Cases Involving Enantioenriched Samples. Molecules 2021, 26, 3994. [Google Scholar] [CrossRef]
- Han, J.; Wzorek, A.; Klika, K.; Soloshonok, V. Recommended Tests for the Self-Disproportionation of Enantiomers (SDE) to Ensure Accurate Reporting of the Stereochemical Outcome of Enantioselective Reactions. Molecules 2021, 26, 2757. [Google Scholar] [CrossRef]
- Soloshonok, V.A.; Wzorek, A.; Klika, K.D. A Question of Policy: Should Tests for the Self-Disproportionation of Enantiomers (SDE) be Mandatory for Reports Involving Scalemates? Tetrahedron Asymmetry 2017, 28, 1430–1434. [Google Scholar] [CrossRef]




















































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vera, S.; Landa, A.; Mielgo, A.; Ganboa, I.; Oiarbide, M.; Soloshonok, V. Catalytic Asymmetric α-Functionalization of α-Branched Aldehydes. Molecules 2023, 28, 2694. https://doi.org/10.3390/molecules28062694
Vera S, Landa A, Mielgo A, Ganboa I, Oiarbide M, Soloshonok V. Catalytic Asymmetric α-Functionalization of α-Branched Aldehydes. Molecules. 2023; 28(6):2694. https://doi.org/10.3390/molecules28062694
Chicago/Turabian StyleVera, Silvia, Aitor Landa, Antonia Mielgo, Iñaki Ganboa, Mikel Oiarbide, and Vadim Soloshonok. 2023. "Catalytic Asymmetric α-Functionalization of α-Branched Aldehydes" Molecules 28, no. 6: 2694. https://doi.org/10.3390/molecules28062694
APA StyleVera, S., Landa, A., Mielgo, A., Ganboa, I., Oiarbide, M., & Soloshonok, V. (2023). Catalytic Asymmetric α-Functionalization of α-Branched Aldehydes. Molecules, 28(6), 2694. https://doi.org/10.3390/molecules28062694