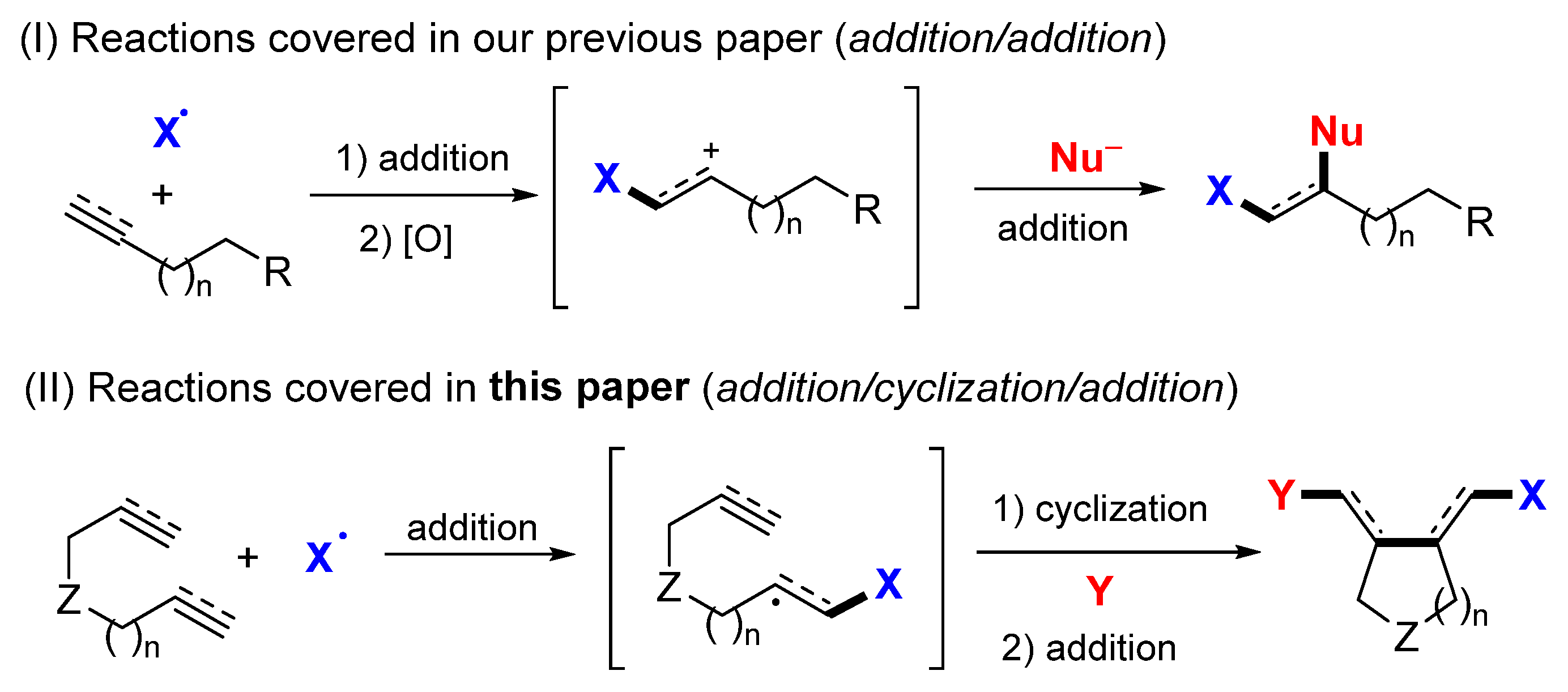
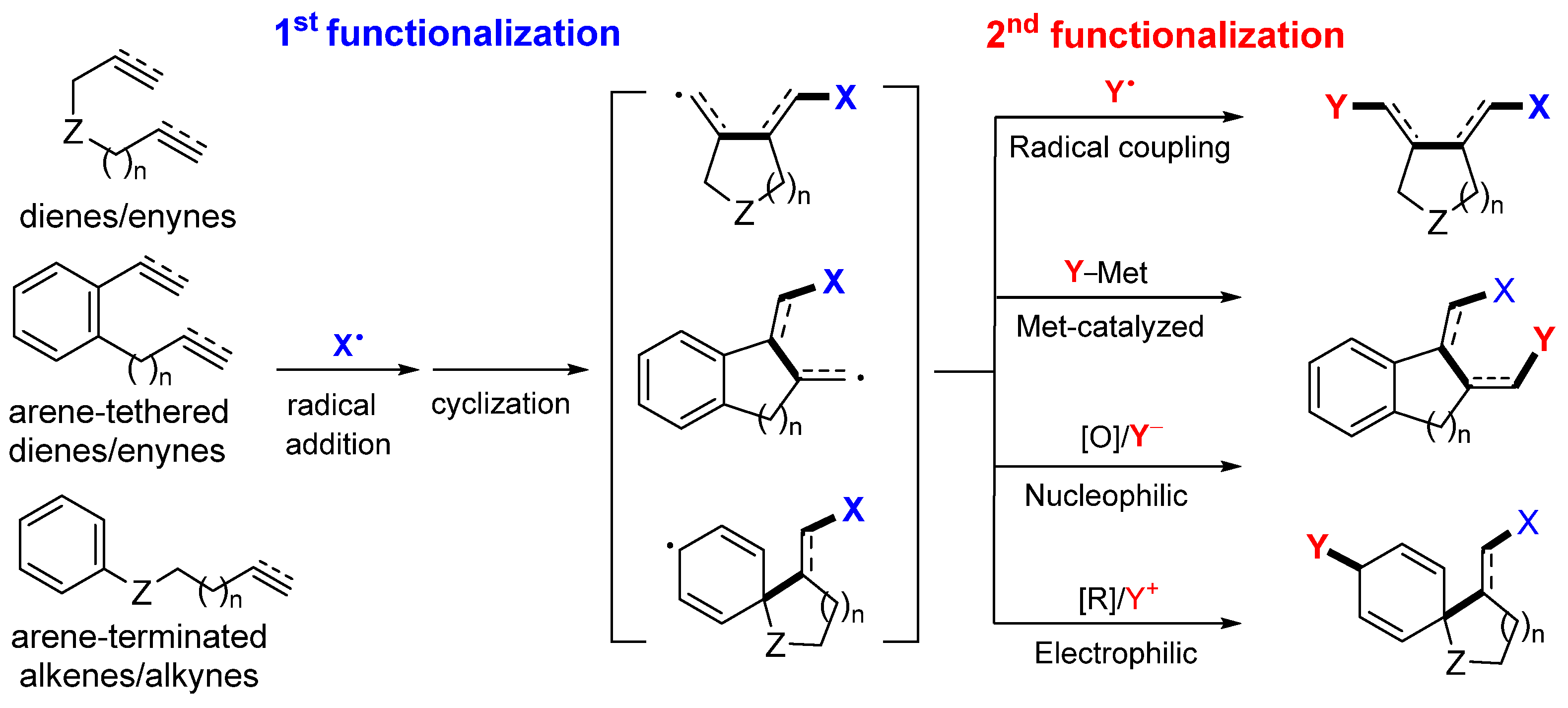
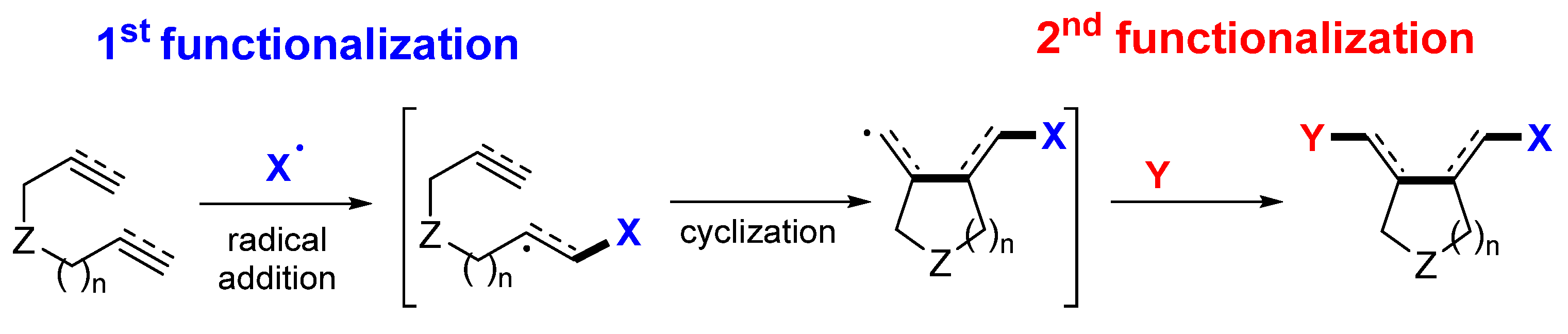
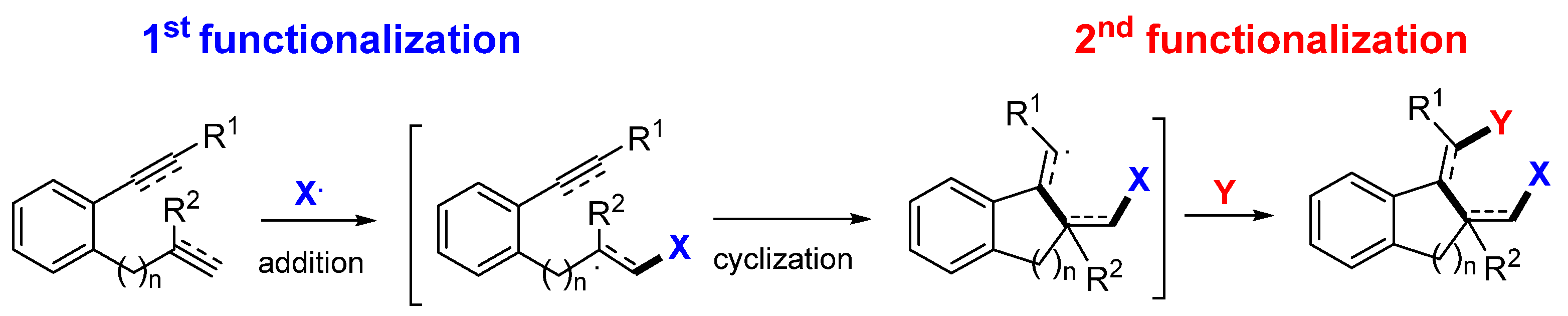
2. Reaction of Dienes and Enynes
Presented in this section are the radical addition and cyclization-initiated difunctionalization reactions of 1,n-dienes and -enynes with a reaction sequence shown in
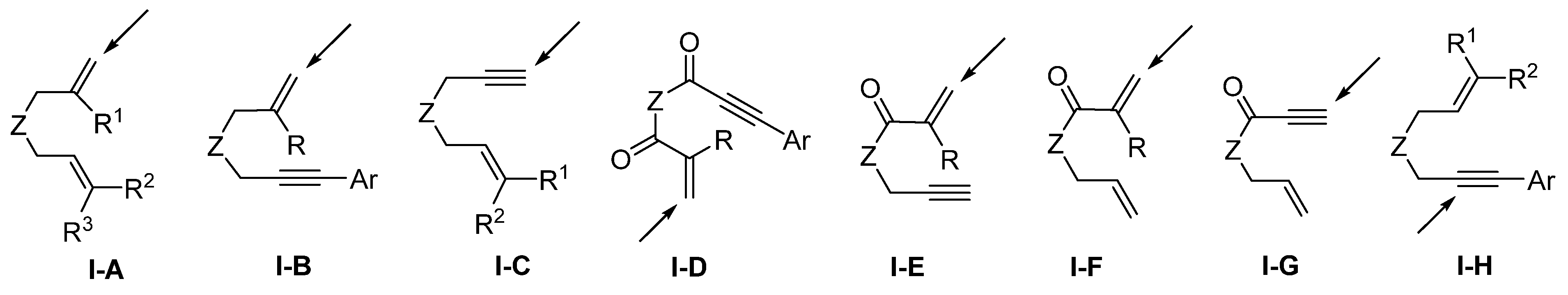
Scheme 3. The common substrates include dienes (
I-A), enynes (
I-B,
I-C,
I-H), dienyl amides (
I-F), enynyl amides (
I-D,
I-E,
I-G) with the Z as a carbon or heteroatom (
Scheme 4). Since there are two unsaturated carbon–carbon bonds in the substrates which are available for the radical addition, the regioselectivity for the initial radical addition is critical. As indicated in
Scheme 4, the steric hindrance (
I-A to
I-D) and conjugation effect of the groups, such as C=O and Ar (
I-E to
I-H), are the major factors to direct the position for the initial radical addition.
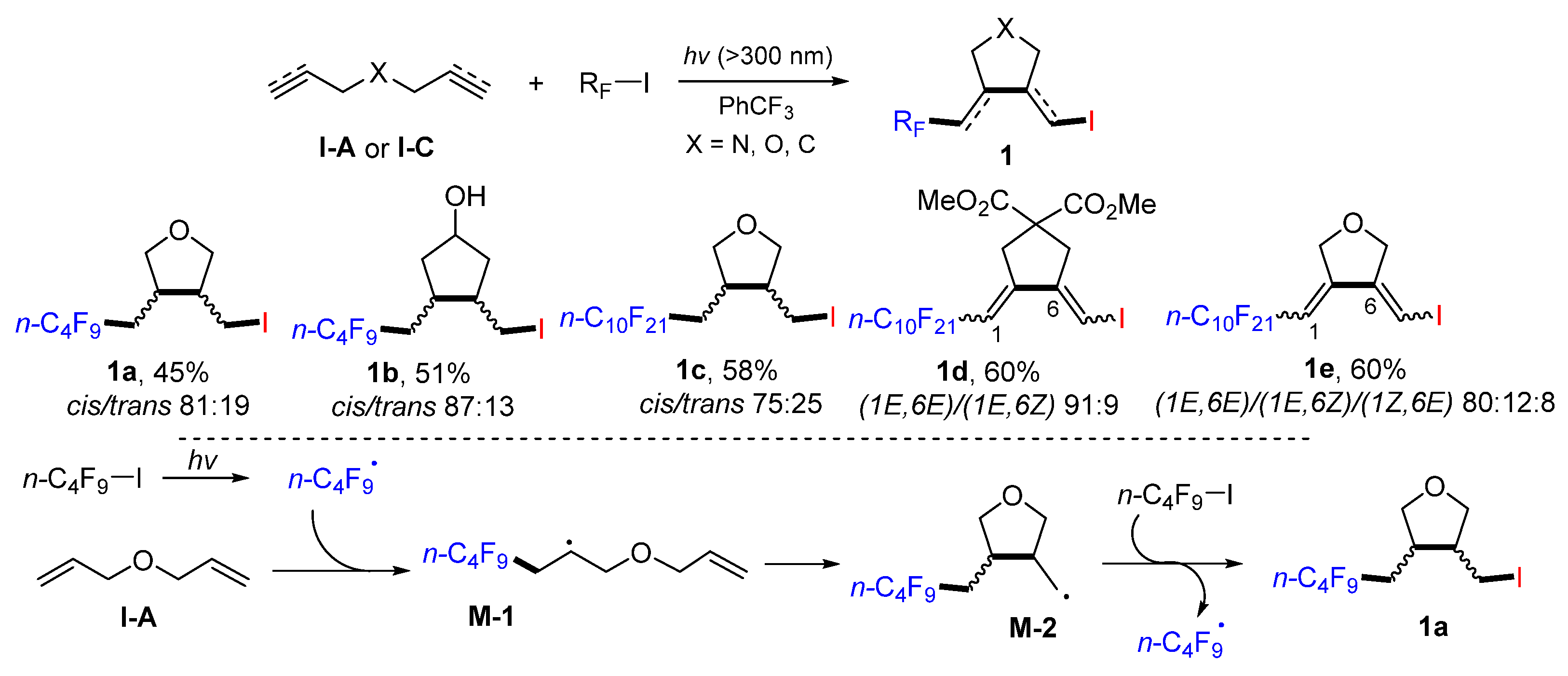
In 2005, Ogawa and coworker reported a near-UV light-mediated radical reaction of dienes, diynes, and enynes for the synthesis of iodoperfluoroalkylated cyclic products. The reactions of dienes, diynes, or enynes and perfluoroalkyl iodides in PhCF
3 under the irradiation of xenon lamp afforded products
1 as a mixture of
cis/trans isomers in moderate-to-good yields (
Scheme 5) [
18]. A proposed mechanism indicated that the
n-C
4F
9 radical generated from
n-C
4F
9I under the light adds to diene. The intermediate
M-1 undergoes 5-
exo cyclization to give alkenyl
M-2, which then reacts with
n-C
4F
9I through the iodine atom transfer to give product
1a.
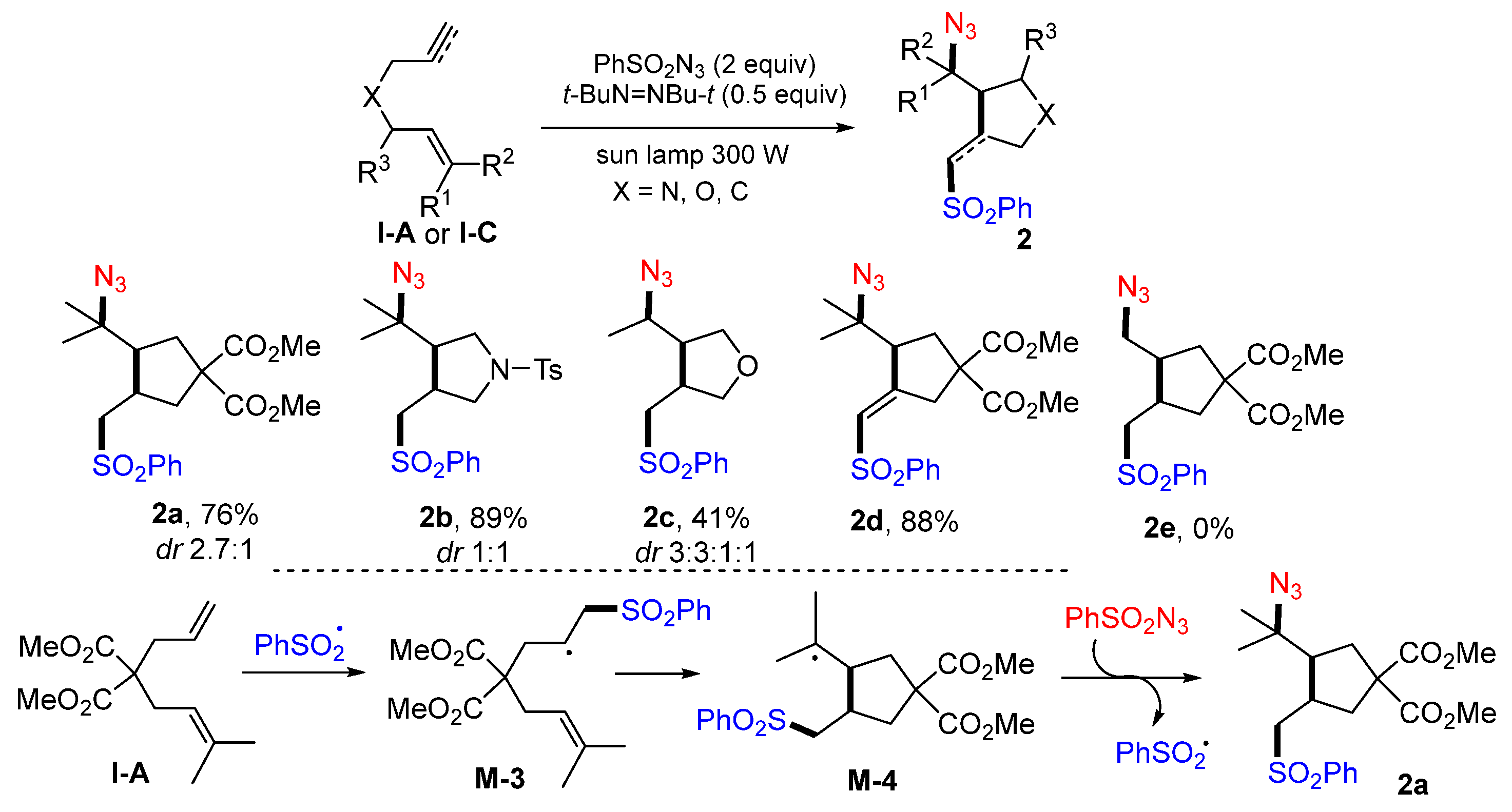
A sun lamp-mediated radical reaction for making azidosulfonylated cyclic products was reported by the Renaud group in 2008. Dienes, diynes, or enynes in dry benzene reacted with benzenesulfonyl azide with radical initiator di-
t-butyldiazene to give azidosulfone products
2 in moderate-to-excellent yields (
Scheme 6) [
19]. This method is good for the formation of tertiary and secondary azides
2a–d, but not for primary azide
2e. The reaction process involves the addition of PhSO
2 radical to the less hindered alkene to form intermediate radical
M-3, 5-
exo cyclization for radical
M-4, and N
3 radical transfer from PhSO
2N
3 to give product
2a.
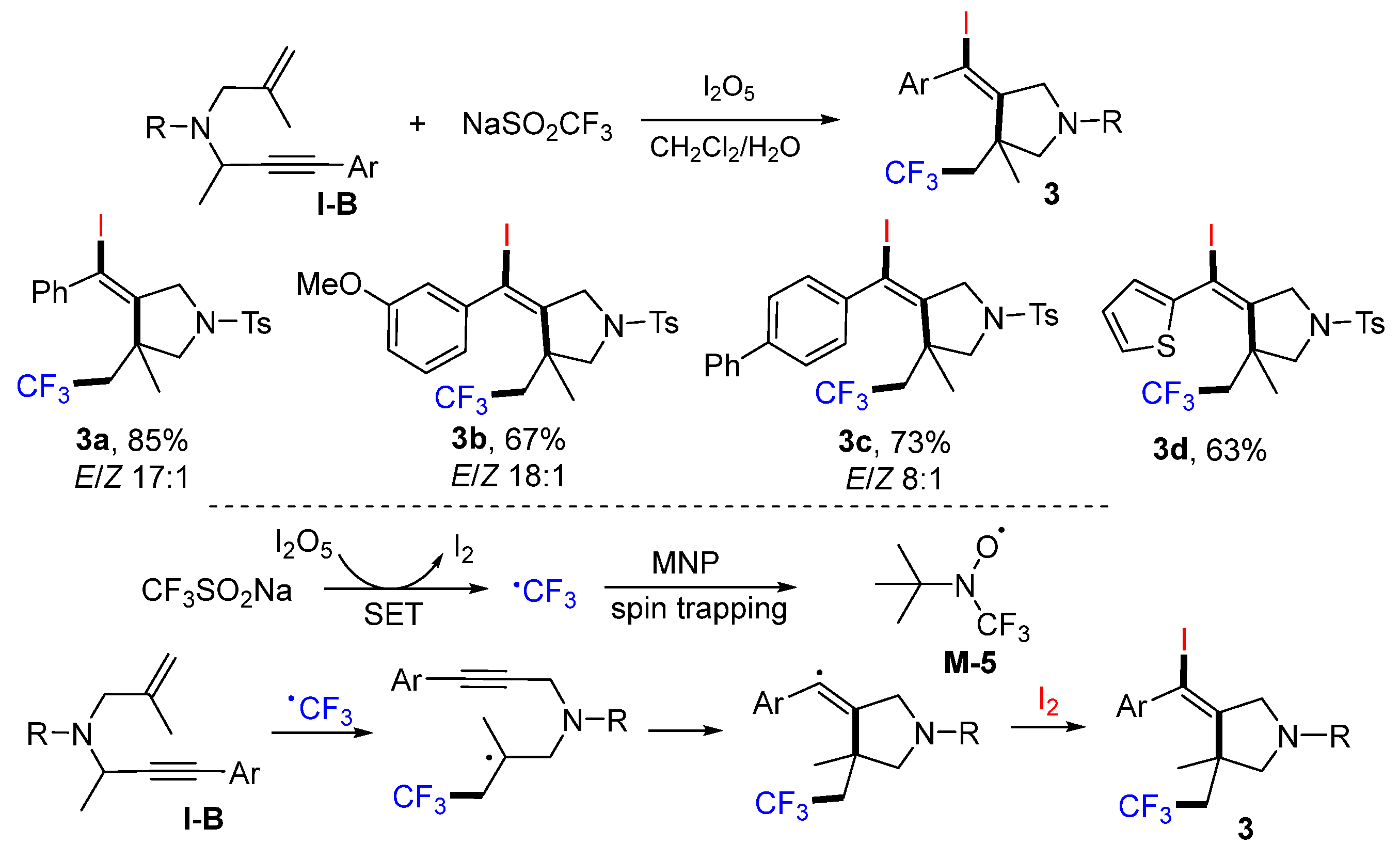
1,6-Enynes are the most popular substrates for radical reactions to make difunctionalized five-membered rings. A method for making iodotriflouromethylated
N-heterocycles was reported by the Liu group in 2014. The reaction of 1,6-enynes, NaSO
2CF
3 and I
2O
5 in CH
2Cl
2/H
2O afforded pyrrolidines products
3 in moderate-to-high yields (
Scheme 7) [
20]. The CF
3 radical generated from NaSO
2CF
3 through SET of I
2O
5 adds to the alkenyl group of 1,6-enynes followed by cyclization and the capture of iodine to give products
3. The CF
3 radical could be trapped by 2-methyl-2-nitrosopropane (MNP) to form
M-5 for ESR detection.
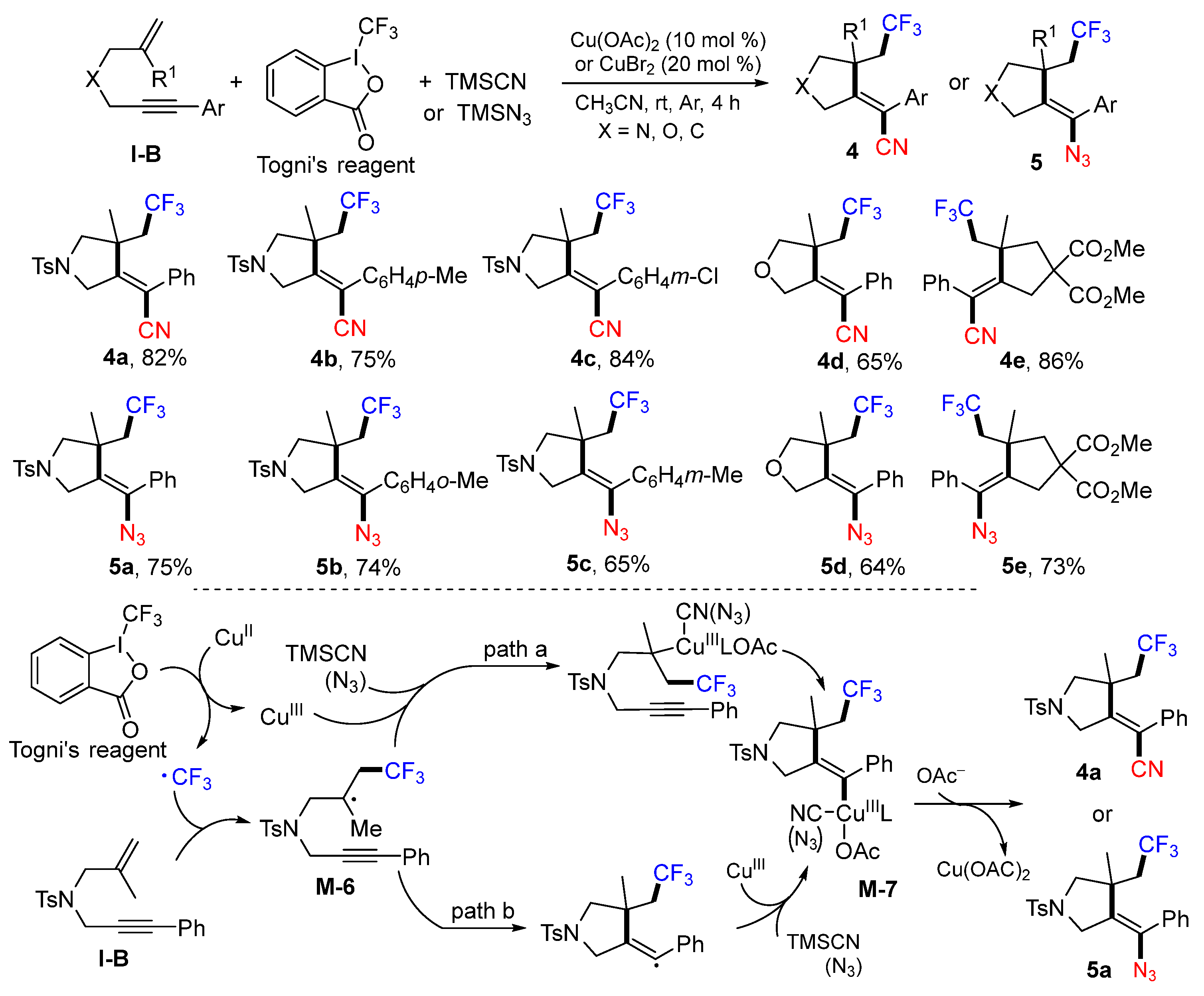
A method for cyclative trifluoromethylation of 1,6-enynes was reported by the Liang group in 2014. The reaction of 1,6-enynes, Togni’s reagent, and TMSCN (or TMSN
3) in CH
3CN under the catalysis of Cu
II gave CF
3-containing heterocycles
4 and
5 (
Scheme 8) [
21]. The CF
3 radical produced from the Togni’s reagent under the catalysis of Cu
II adds to the C=C double bond of 1,6-enyne to form the radical intermediate
M-6, which is converted to cyclized metal complex
M-7 through path a or path b. At the last step, the reaction of
M-7 with TMSCN or TMSN
3 gives corresponding cyanotrifluoromethylated or azidotrifluoromethylated five-membered ring products
4a or
5a.
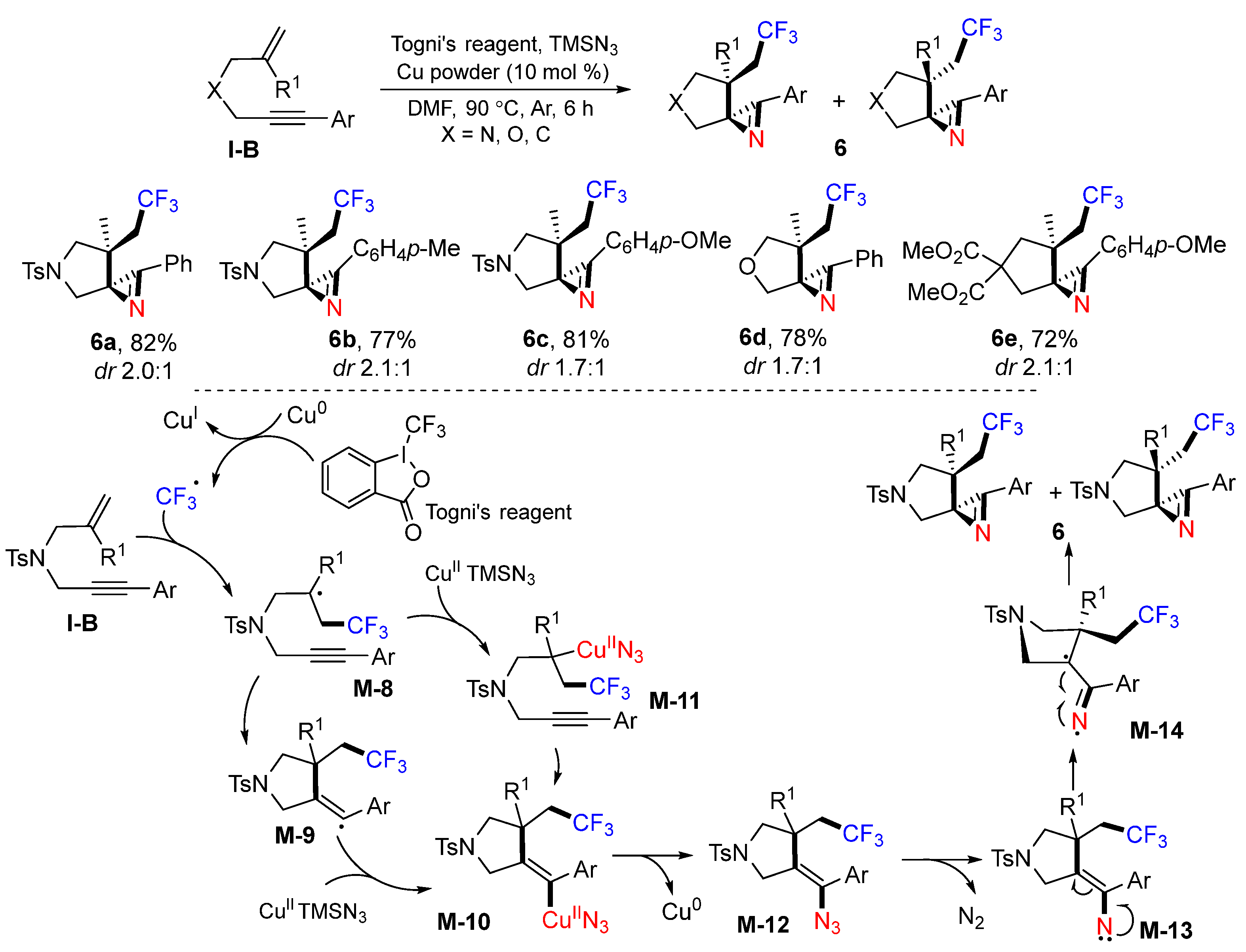
A Togni’s reagent-based synthesis of CF
3-substituted spiro 2
H-azirines was reported by the Liang group in 2015. The reaction of 1,6-enynes with Togni’s reagent and TMSN
3 in the presence of Cu
0 powder as a catalyst afforded diastereomeric products
6 in good-to-excellent yields (
Scheme 9) [
22]. A proposed mechanism suggests that the CF
3 radical generated from Togni’s reagent through SET of Cu
0 is added to the C=C bond of 1,6-enyne to produce the radical intermediate
M-8. Sequential 5-
exo cyclization and trapping of the radical
M-9 with Cu
II and TMSN
3 give Cu
II azide complex
M-10. Complex
M-10 may also be obtained from the formation of complex
M-11 and subsequent cyclization. Reductive elimination of
M-10 followed by the elimination of N
2 from azide
M-12 gives alkenyl nitrene
M-13. The cyclization of
M-14, a resonance structure of alkenyl nitrene
M-13, gives the spiroketal products
6 as a pair of diastereomers.
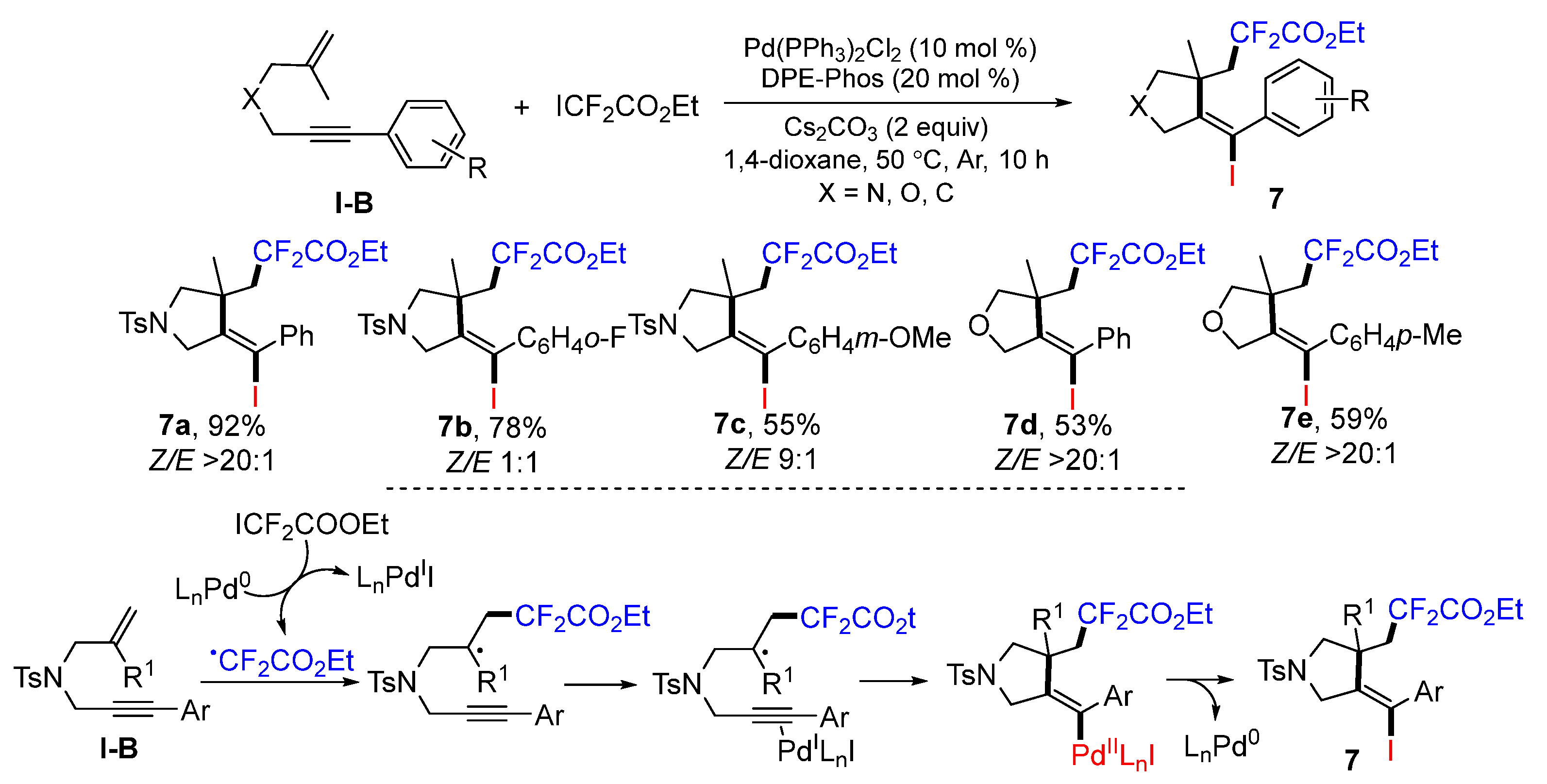
Liang’s lab introduced a method for Pd-catalyzed radical cyclative iododifluoromethylation of 1,6-enynes in 2015. The reaction of 1,6-enynes and ethyl difluoroiodoacetate in dioxane under the catalysis of Pd(PPh
3)
2Cl
2 and bis-[2-(diphenyl-phosphino)phenyl]ether (DPE-Phos) gave iododifluoromethylated heterocycles
7 in good-to-excellent yields (
Scheme 10) [
23]. The CF
2CO
2Et radical is generated from ICF
2CO
2Et through the reduction of Pd
0L
n. Radical addition to the C=C double bond of 1,6-enynes followed by the cyclization to Pd
IL
nI-activated alkyne group and reductive elimination of the Pd
0L
n gives iododifluoromethylated products
7.
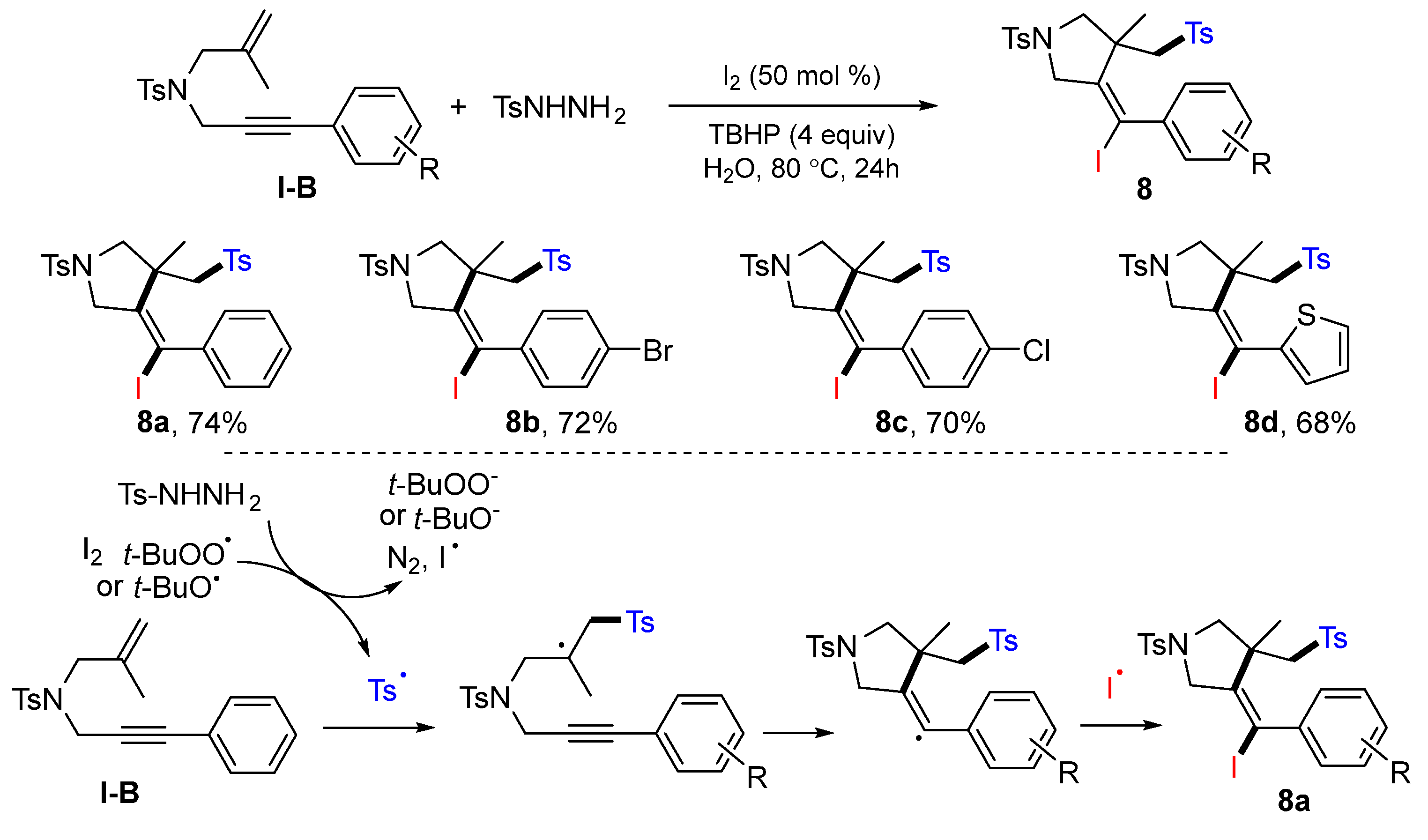
A sulfonyl radical-initiated iodosulfonylation reaction of 1,6-enynes was reported by the Liang group in 2016. The reaction of 1,6-enynes and sulfonyl hydrazide in the presence of I
2/TBHP gave five-membered heterocycles
8 in good-to-excellent yields (
Scheme 11) [
24].
A proposed mechanism indicated that the sulfonyl radical generated from the reaction of sulfonyl hydrazide and TBHP adds to the C=C double bond of 1,6-enyne, followed by the radical cyclization and coupling with iodine radical, to give product 8a.
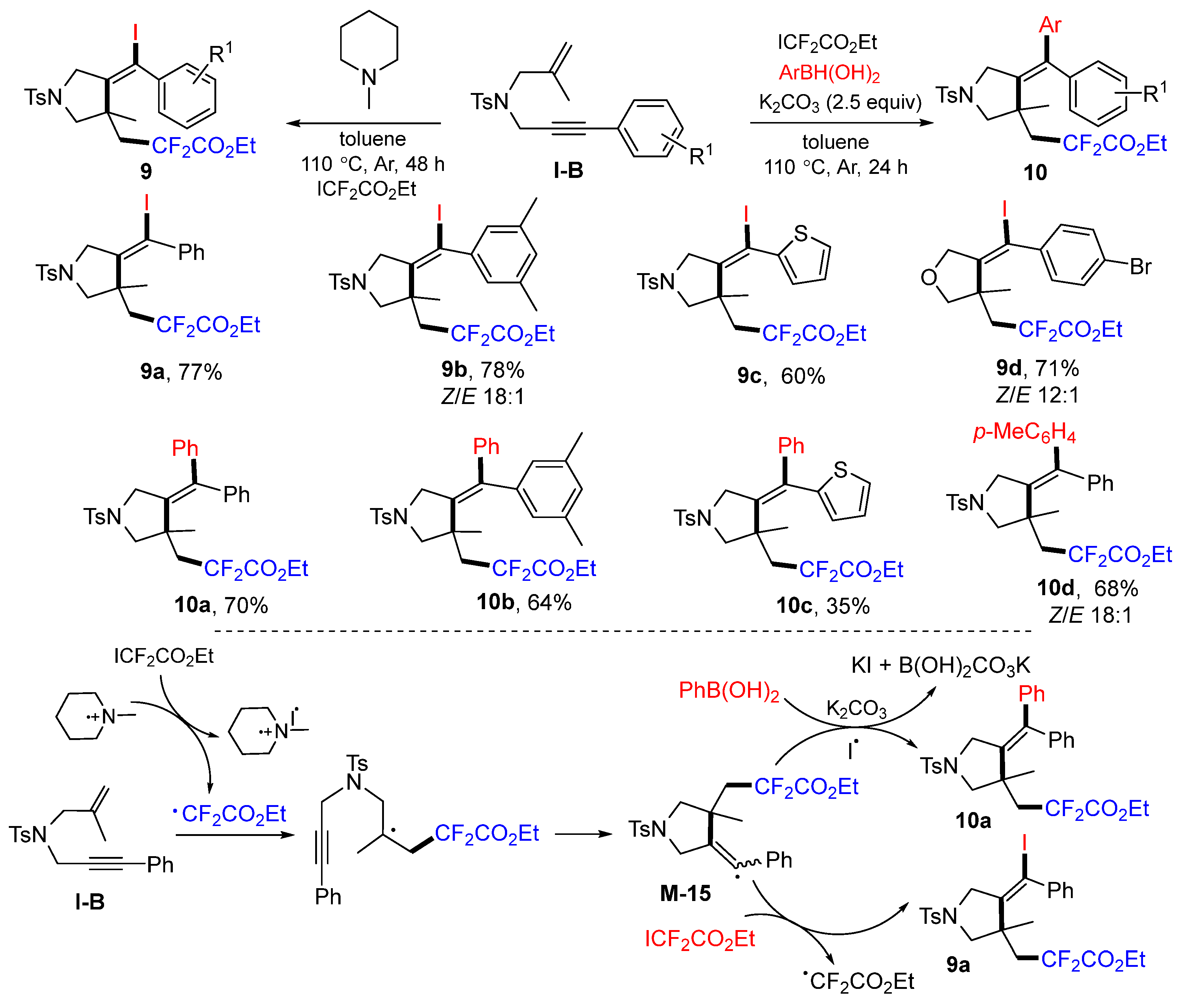
In 2018, the Liang group introduced radical cyclization of 1,6-enynes for the synthesis of substituted pyrrolidine derivatives. The reaction of 1,6-enynes, ICF
2CO
2Et in the presence of
N-methylpiperidine or borophenylic acids/K
2CO
3 afforded substituted pyrroles
9 or
10 in moderate-to-good yields (
Scheme 12) [
25]. The initial CF
2CO
2Et radical generated from the reaction of ICF
2CO
2Et adds to the C=C double bond of the 1,6-enyne followed by 5-
exo cyclization to give radical intermediate
M-15. Radical
M-15 abstracts iodo atom from iododifluoromethylation to give product
9a; otherwise, coupling of
M-15 with borophenylic acid gives product
10a.
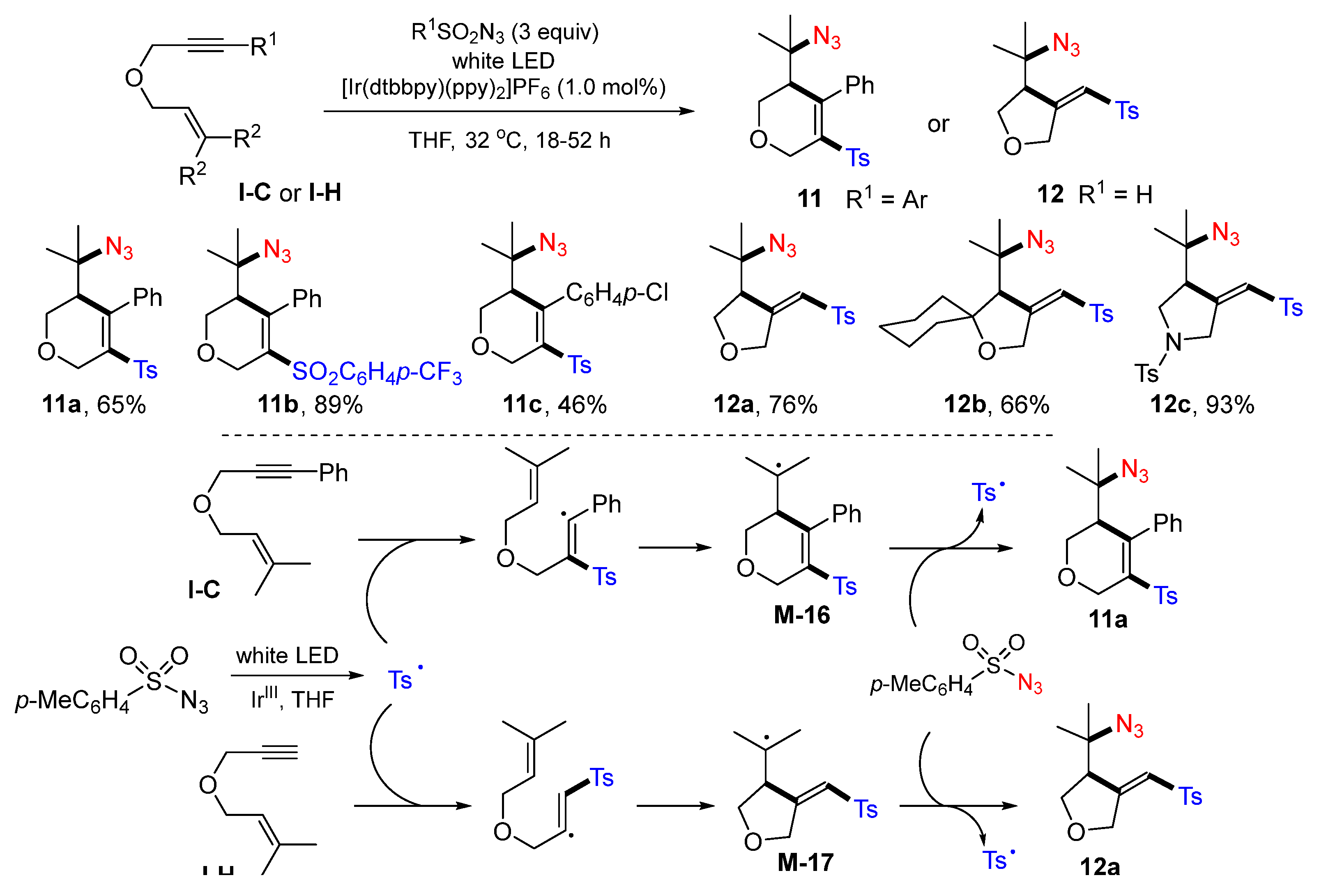
A visible light-mediated radical sulfonylative and azidosulfonylative cyclization of 1,6-enynes for the synthesis of highly functionalized heterocycles was introduced by the Lam group in 2017. The reaction of 1,6-enynes and sulfonyl azides in THF in the presence of a photoactive iridium complex afforded difunctionalized heterocycles
11 or
12 in moderate-to-excellent yields (
Scheme 13) [
26]. The use of THF as the solvent was critical for the success of the reactions. The reaction mechanism suggests that the sulfonyl radical generated from TsN
3 under the visible light catalysis of [Ir(dtbbpy)(ppy)
2]PF
6 adds to the triple bond of 1,6-enyne, followed by cyclization of the vinyl radical, giving six-membered tertiary radical
M-16. Product
11a is then obtained via azidation of
M-16 with the arylsulfonyl azide and the sulfonyl radical is regenerated. When R
1 is H, addition of the sulfonyl radical happens at the terminal carbon of the triple, followed by cyclization of the vinyl radical to give five-membered ring product
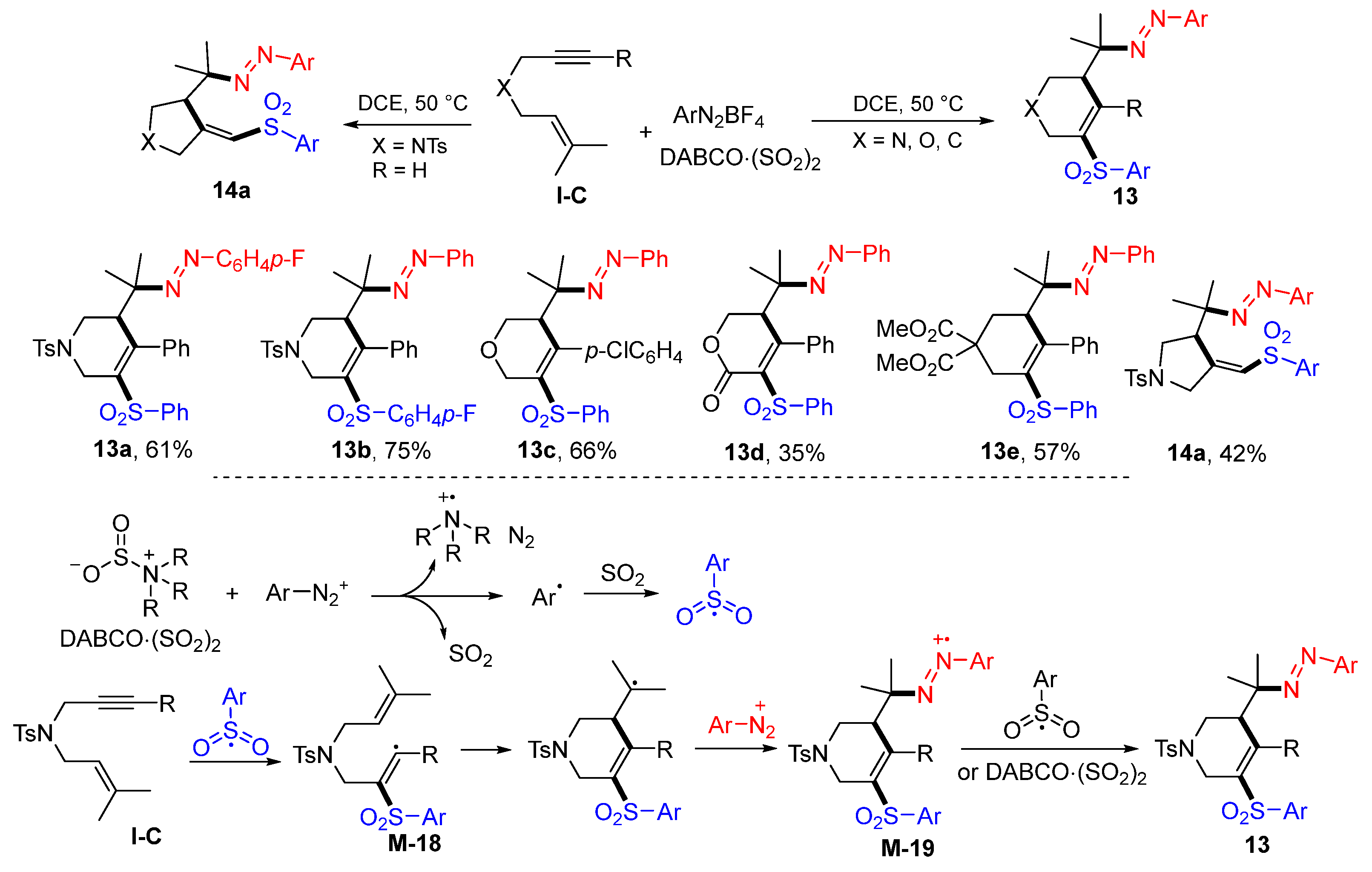
12a.The Wu group, in 2017, introduced a reaction of 1,6-enynes with DABCO·(SO
2)
2 and two equivalents of ArN
2BF
4 in DCE to give diazosulfonated six-membered heterocycles
13 in moderate-to-good yields (
Scheme 14) [
27]. Five-membered heterocycles
14a could be obtained using unsubstituted terminal alkynes as the substrates. The reaction mechanism suggests that the initially sulfonyl radicals, generated from the reaction of ArN
2BF
4 with DABCO·(SO
2)
2, adds to the C≡C bond of 1,6-enynes to form vinyl radical
M-18, followed by 6-
exo cyclization and trapping with aryldiazonium cation to give intermediates
M-19. The last step SET of arylsulfonyl radical or DABCO·(SO
2)
2 to radical
M-19 gave products
13.
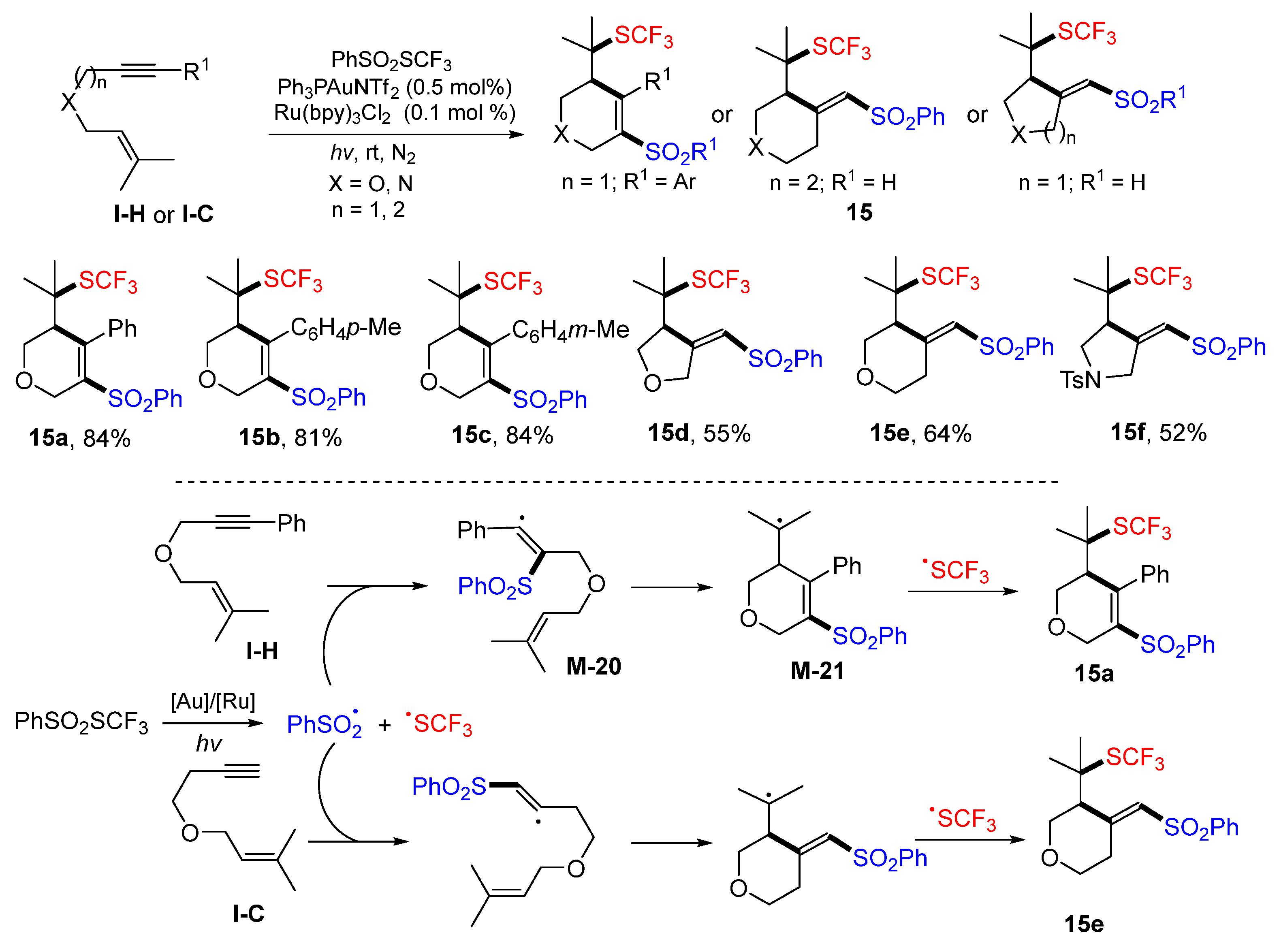
The Xu group, in 2018, introduced a visible light-mediated radical atom transfer radical cyclization (ATRC) of 1,6- and 1,7-enynes for the synthesis of sulfonyl and trifluoromethylthio functionalized vinylsulfones. In the ATRC reactions, two functional groups are from the same reagent. The reaction of enynes and PhSO
2SCF
3 in the presence of PPh
3AuNTf
2 and Ru(bpy)
3Cl
2 under the irradiation of blue LED afforded five- or six-membered vinylsulfones
15 in good yields (
Scheme 15) [
28]. A proposed mechanism for the reaction of 1,6-enyne indicated that the sulfonyl radical generated from PhSO
2SCF
3 under photocatalysis of PPh
3AuNTf
2 and Ru(bpy)
3Cl
2 adds to the triple bond to form benzyl radical
M-20, followed by 6-
exo cyclization to give tertiary radical
M-21. It then couples CF
3S radical to give product
15a. For the reaction of a 1,6-enyne without substitution on the terminal carbon (R
1 = H), sulfonyl radical adds to the terminal carbon of alkyne followed by 5-
exo cyclization, leading to product
15d. A similar process for the reaction of 1,7-enyne, which has no terminal carbon substitution on alkyne, affords product
15e.
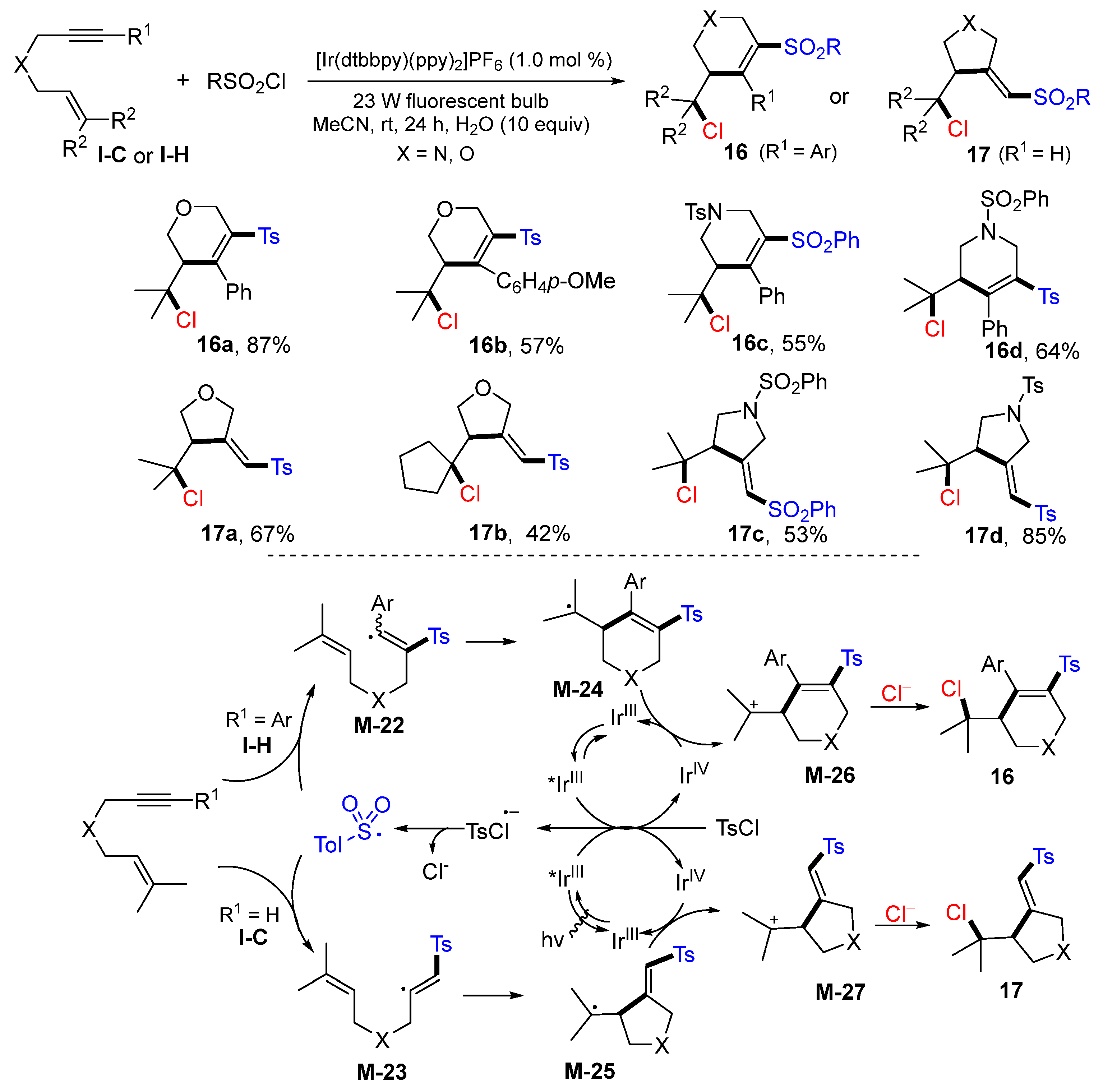
A visible light-mediated ATRC of 1,6-enyne for the preparation of chloroalkyl-substituted cyclic alkenyl sulfones using sulfonyl chlorides as the key reactants was reported by the Zhu group in 2018. The reactions of 1,6-enynes and sulfonyl chlorides in the presence of [Ir(dtbbpy)(ppy)
2]PF
6 under the irradiation of blue LED gave five- or six-membered chloroalkyl-substituted cyclic alkenyl sulfones
16 or
17 (
Scheme 16) [
29]. As the reaction mechanism indicated, the sulfonyl radical generated from TsCl under the photoredox of [Ir(dtbbpy)(ppy)
2]PF
6 adds to the C≡C bond of the 1,6-enyne followed by 5-
exo or 6-
exo cyclization to form the carbon radicals
M-24 or
M-25. They are oxidized to carbocations
M-26 and
M-27 and then react with chlorine anion to form products
16 and
17, respectively.
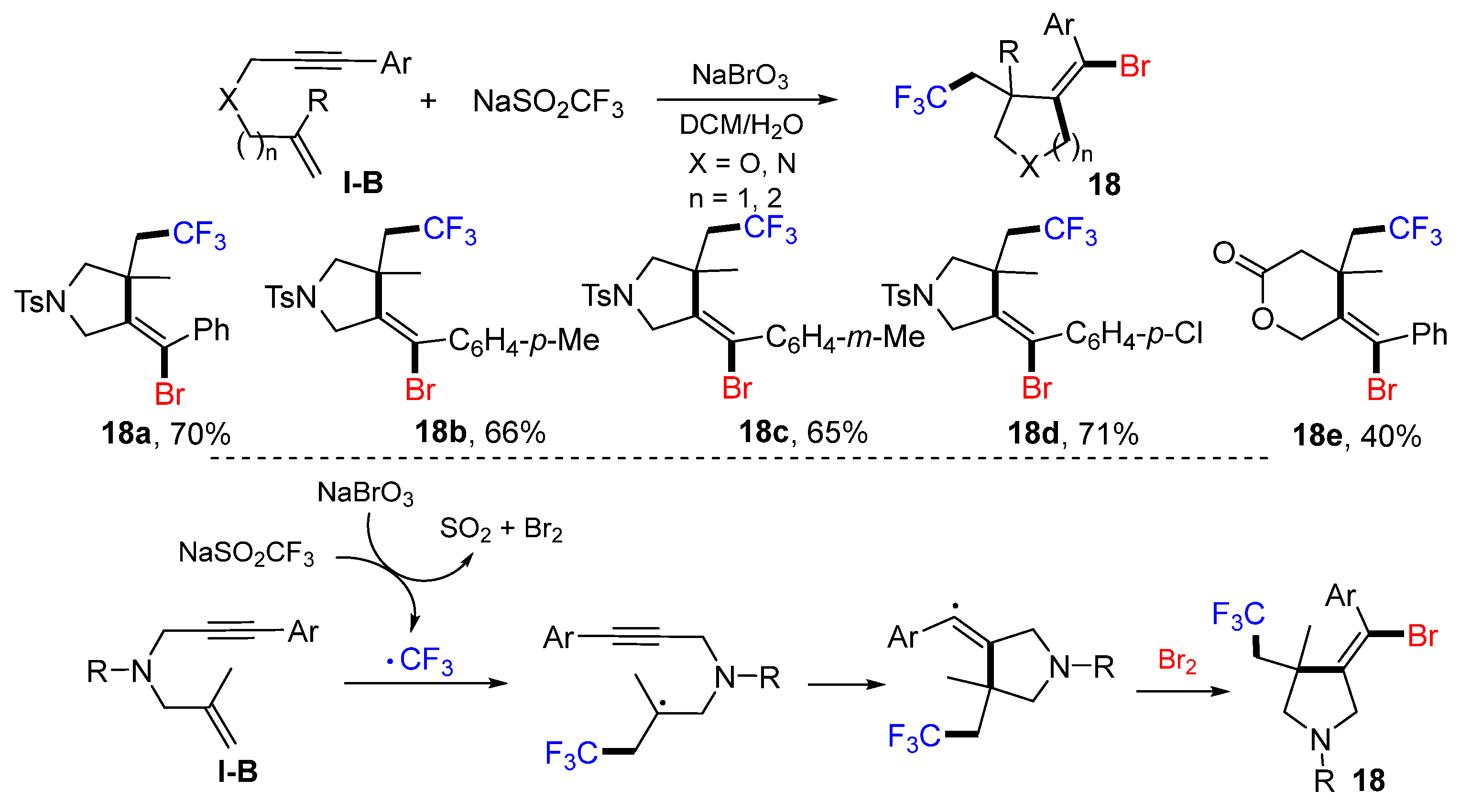
In 2018, the Liu group reported the synthesis of bromotrifluoromethylated five- and six-membered heterocycles. The reaction of 1,6- or 1,7-enynes, NaSO
2CF
3 and NaBrO
3 in DCM/H
2O produced products
18 in good yields (
Scheme 17) [
30]. The CF
3 radical, generated from the reaction of NaSO
2CF
3 and NaBrO
3, adds to the terminal carbon of alkene followed by 5-
exo or 6-
exo cyclization (n = 2) and then Br-atom abstraction to give product
18.
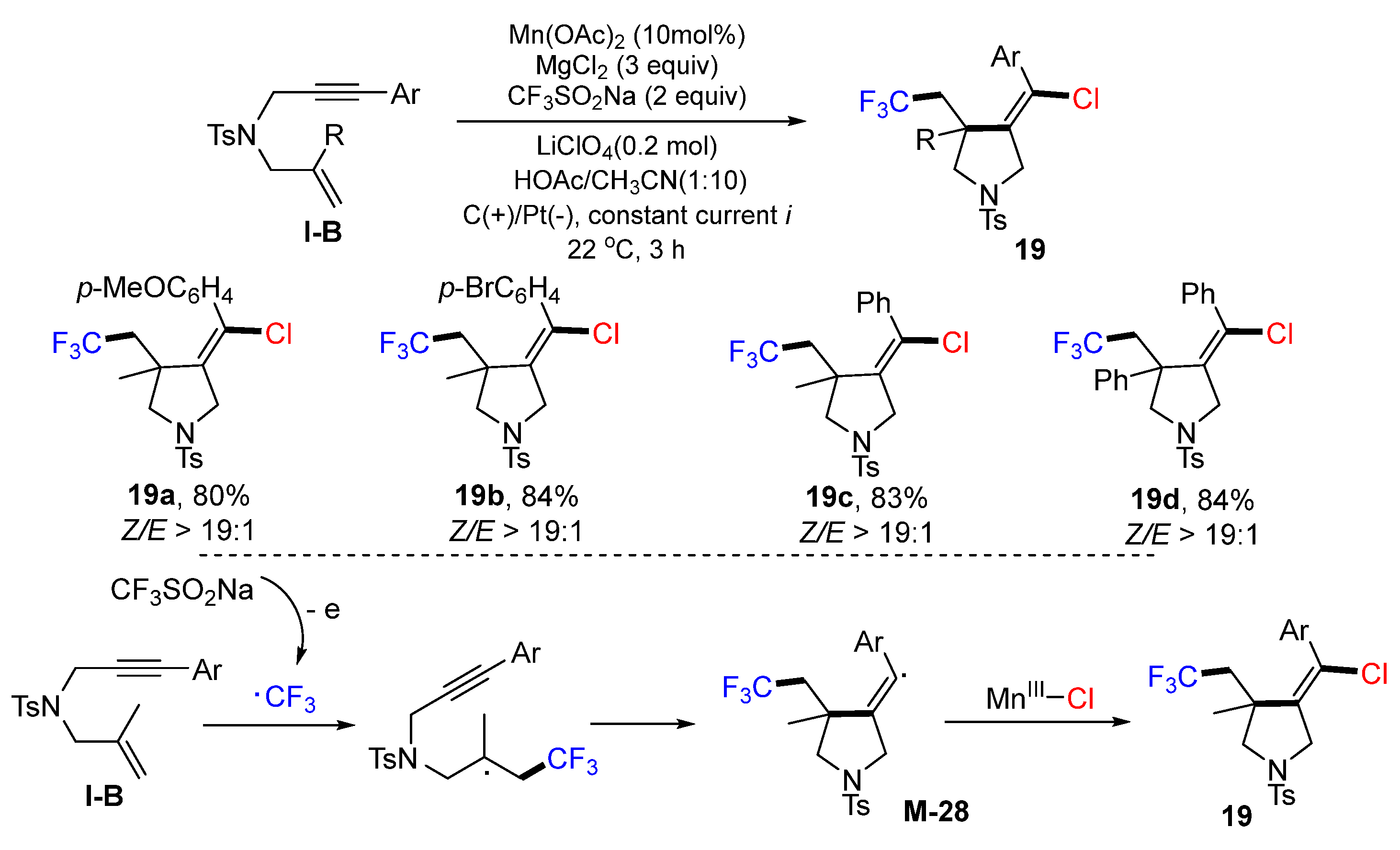
Lin and coworkers reported an electrochemical reaction for the preparation of chlorotrifluoromethylated pyrrolidines in 2018. The reaction was carried out using HOAc-MeCN as solvent at room temperature under electrochemical conditions. The reaction of 1,6-enynes, CF
3SO
2Na and MgCl
2 in the presence of LiClO
4 and Mn(OAc)
2 gave chlorotrifluoromethylated pyrrolidines
19 in excellent yields (
Scheme 18) [
31]. The initial CF
3 radical generated from the anodically coupled electrolysis adds to the C=C double bond of 1,6-enynes followed by 5-
exo cyclization to afford the vinyl radical
M-28, which couples with the Cl radical to give product
19.
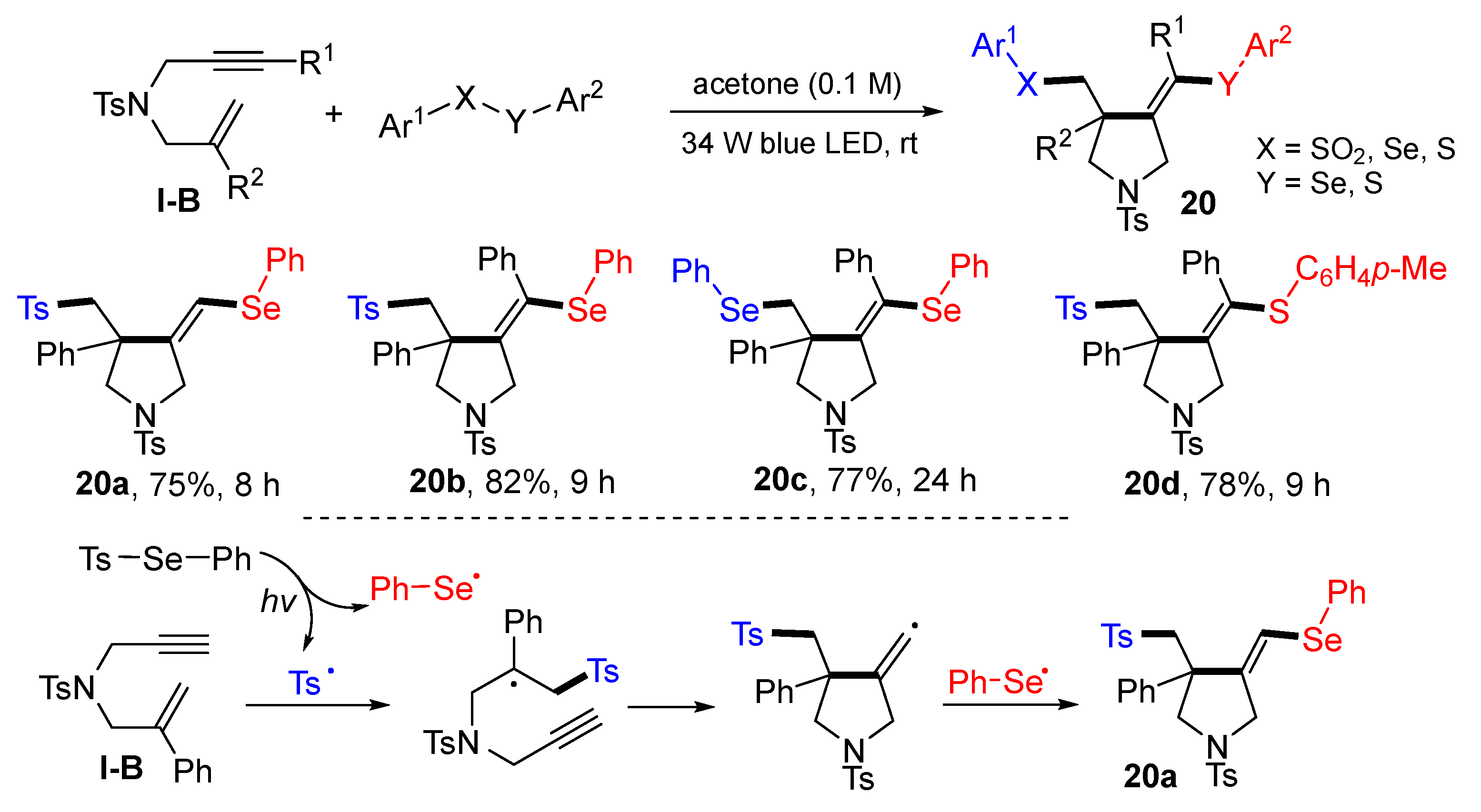
A visible light-promoted reaction of 1,6-enynes for the synthesis of difunctionalized pyrrolidines was introduced by the Wang group in 2020. The reaction of 1,6-enynes, and chalcogens (such as benzenesulfono–selenoate) in acetone at room temperature under the radiation of blue LED afforded products
20 in moderate-to-good yields (
Scheme 19) [
32]. The reaction mechanism suggests that tosyl and phenylselenyl radicals are generated from Se-phenyl 4-methylbenzenesulfonoselenoate under photo irradiation. The tosyl radical adds to the C=C bond of 1,6-enyne followed by 5-
exo cyclization and capture of phenylselenyl radical to give product
20a.
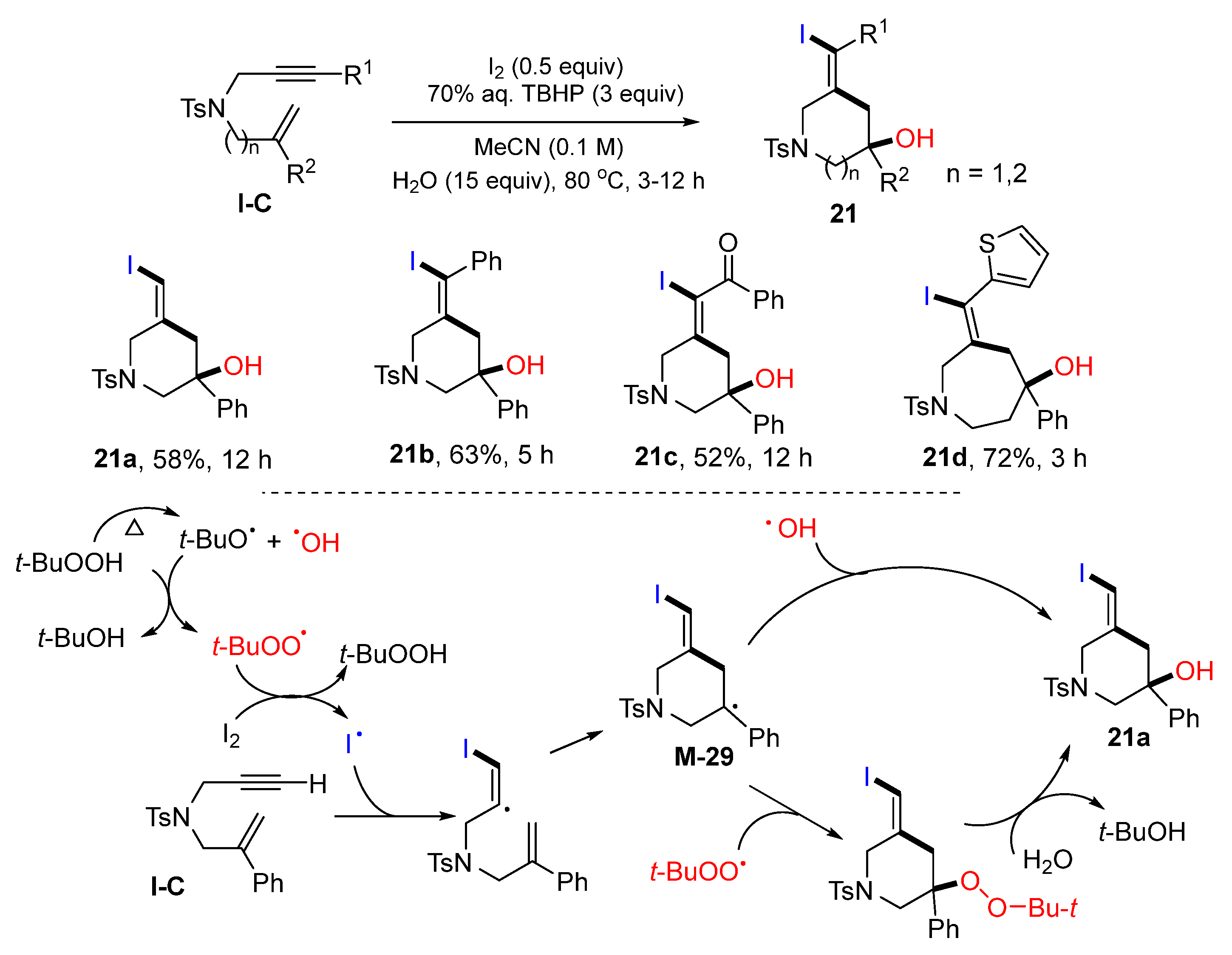
An iodine radical-initiated reaction for the synthesis of difunctionalized
N-heterocyclic compounds was reported by the Wang group in 2020. The reactions of 1,6- or 1,7-enynes, TBHP and I
2 in CH
3CN gave compound
21 in moderate-to-good yields (
Scheme 20) [
33]. The reaction mechanism suggests that iodide radical, generated from the reaction of I
2 with TBHP, adds to the C≡C triple bond of enyne followed by 6-
exo cyclization to yield tertiary radical
M-29. Addition of hydroxyl radical or
t-butylperox radical to
M-29 could lead to the formation of product
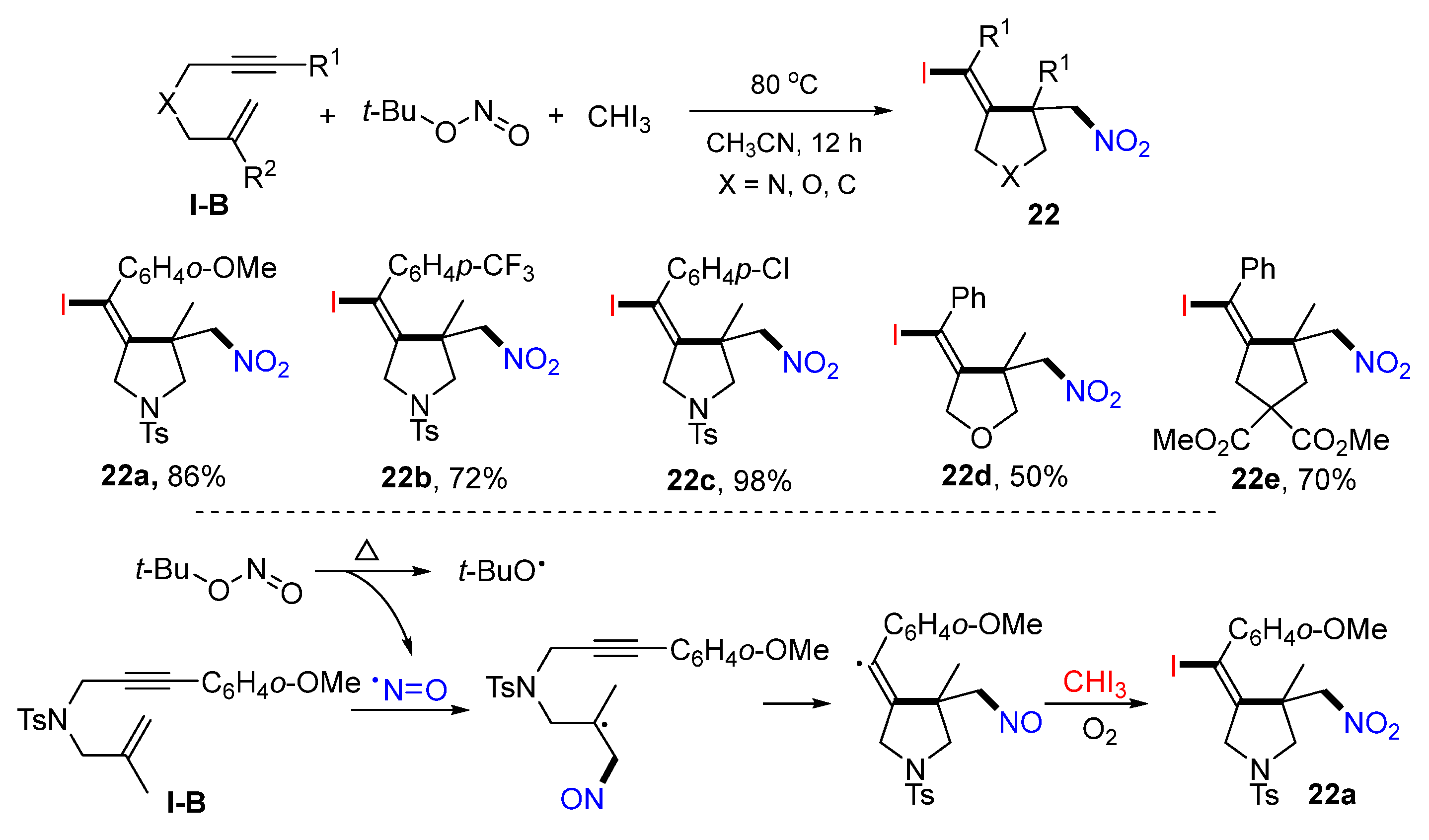
21a.In 2021, Zhu and co-workers reported the synthesis of iodo- and nitro-functionalized cyclic compounds such aspyrrolidines, tetrahydrofurans, and cyclopentanes. The reaction of 1,6-enynes,
t-BuONO, and iodoform in CH
3CN under heating gave five-membered heterocycles
22 in moderate-to-excellent yields (
Scheme 21) [
34]. The reaction mechanism suggests that nitroso radical formed from the homolysis of
t-BuONO adds to the C=C bond of the 1,6-enyne followed by 5-
exo cyclization, oxidation to cation, and then iodination with CHI
3 to give product
22a.
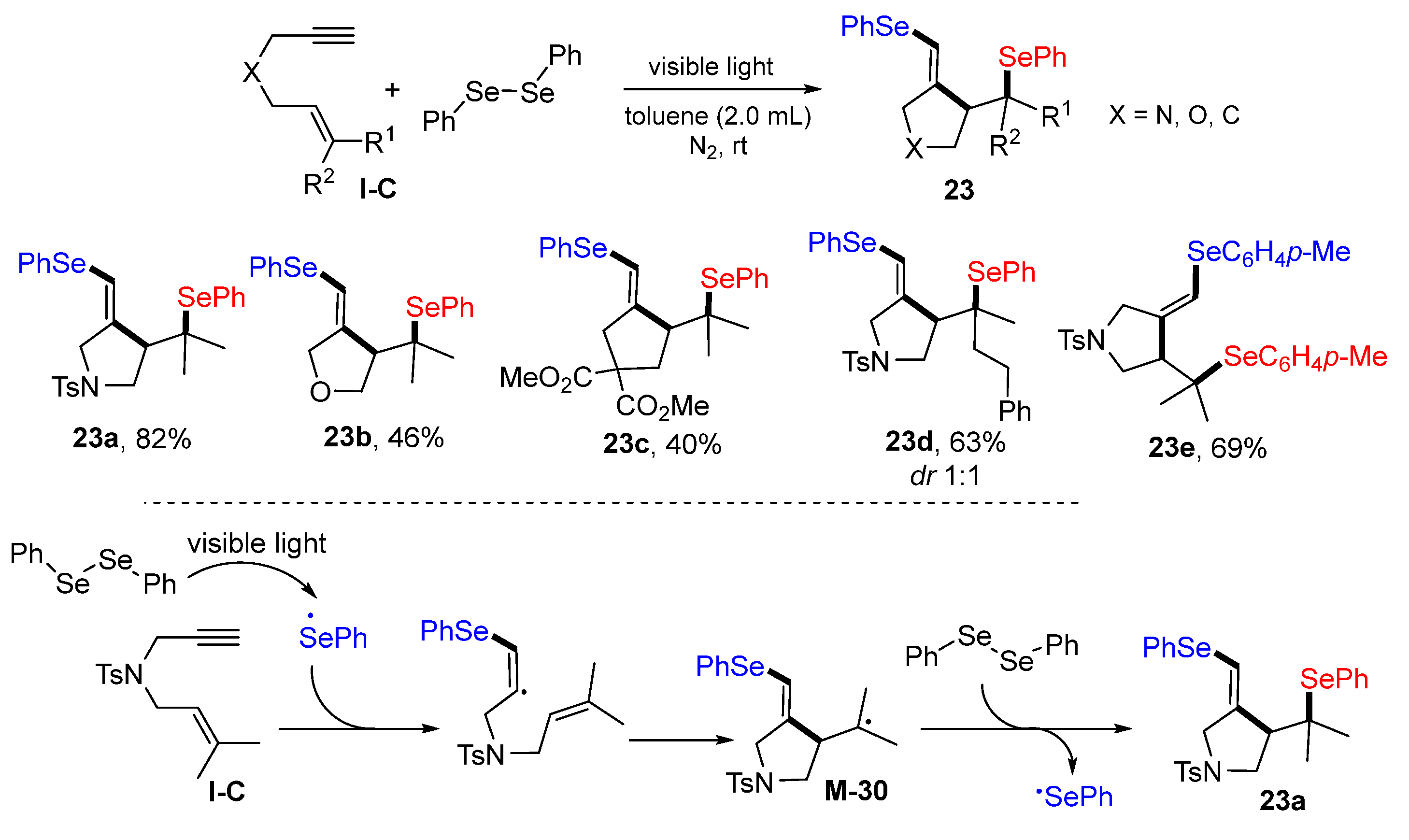
In 2021, Zhu and co-workers reported diarylselenylative cyclization reaction of 1,6-enynes for the synthesis of five-membered heterocycles. The reaction of 1,6-enyne and diaryldiselane in toluene under the radiation of light at room temperature afforded products
23 in moderate to good yields (
Scheme 22) [
35]. The reaction mechanism shows that the PhSe radical generated via photo homolytic cleavage of PhSeSePh adds to the triple bond of 1,6-enyne followed by 5-
exo cyclization to form tertiary carbon radical
M-30, which then couples with PhSe radical to give product
23a.
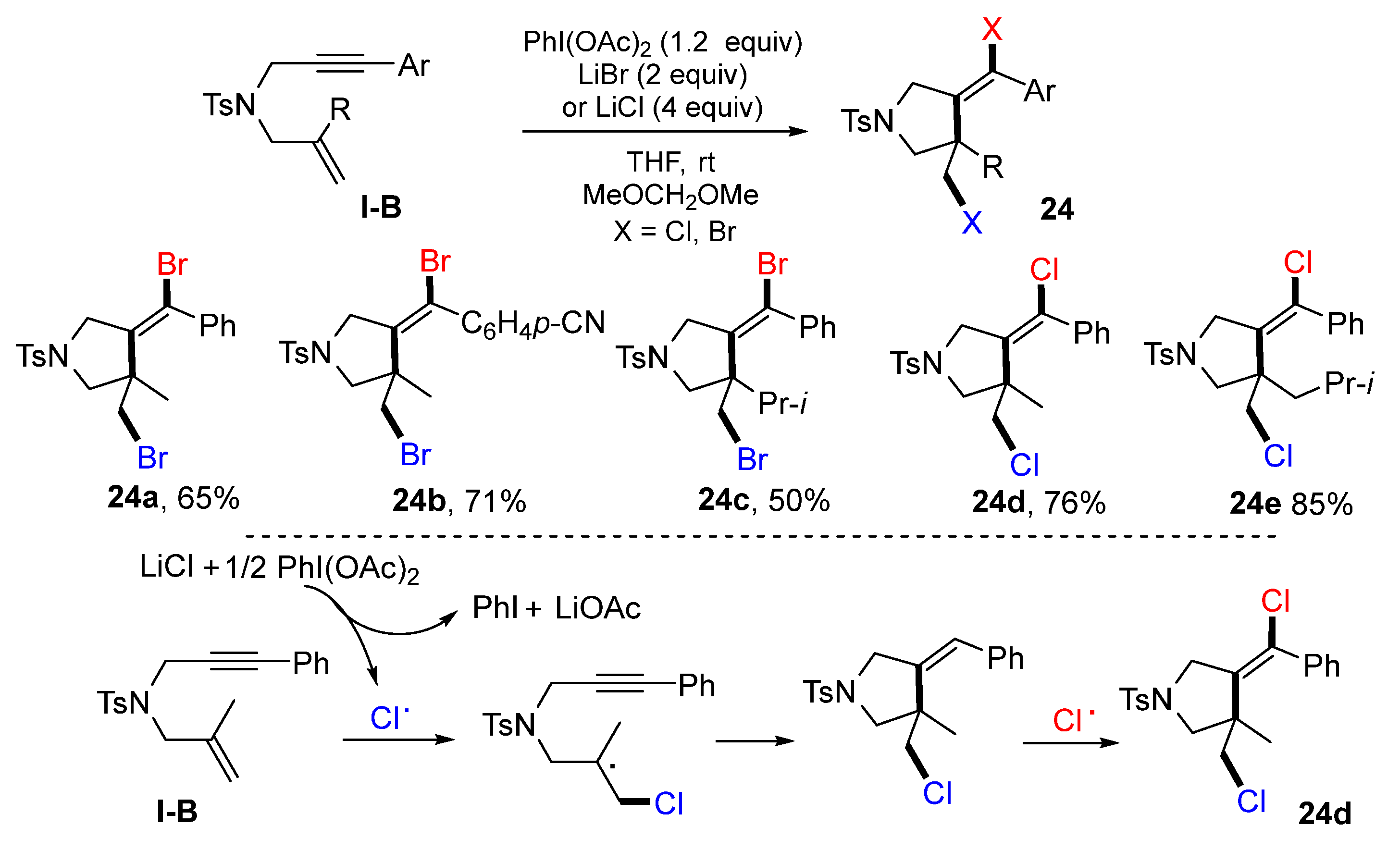
The reaction of 1,6-enynes for the synthesis of dihalogenated pyrrolidines was reported by the Tong group in 2021. The reaction of 1,6-enynes, PhI(OAc)
2 and lithium halide at room temperature gave product
24 in moderate-to-good yields (
Scheme 23) [
36]. A suggested mechanism for the reaction with LiCl indicated that the Cl radical generated via a single electron oxidation of LiCl with PhI(OAc)
2 adds to the C=C double bond of 1,6-enyne followed by 5-
exo cyclization and Cl atom abstraction to give dichloro pyrrolidine
24d.
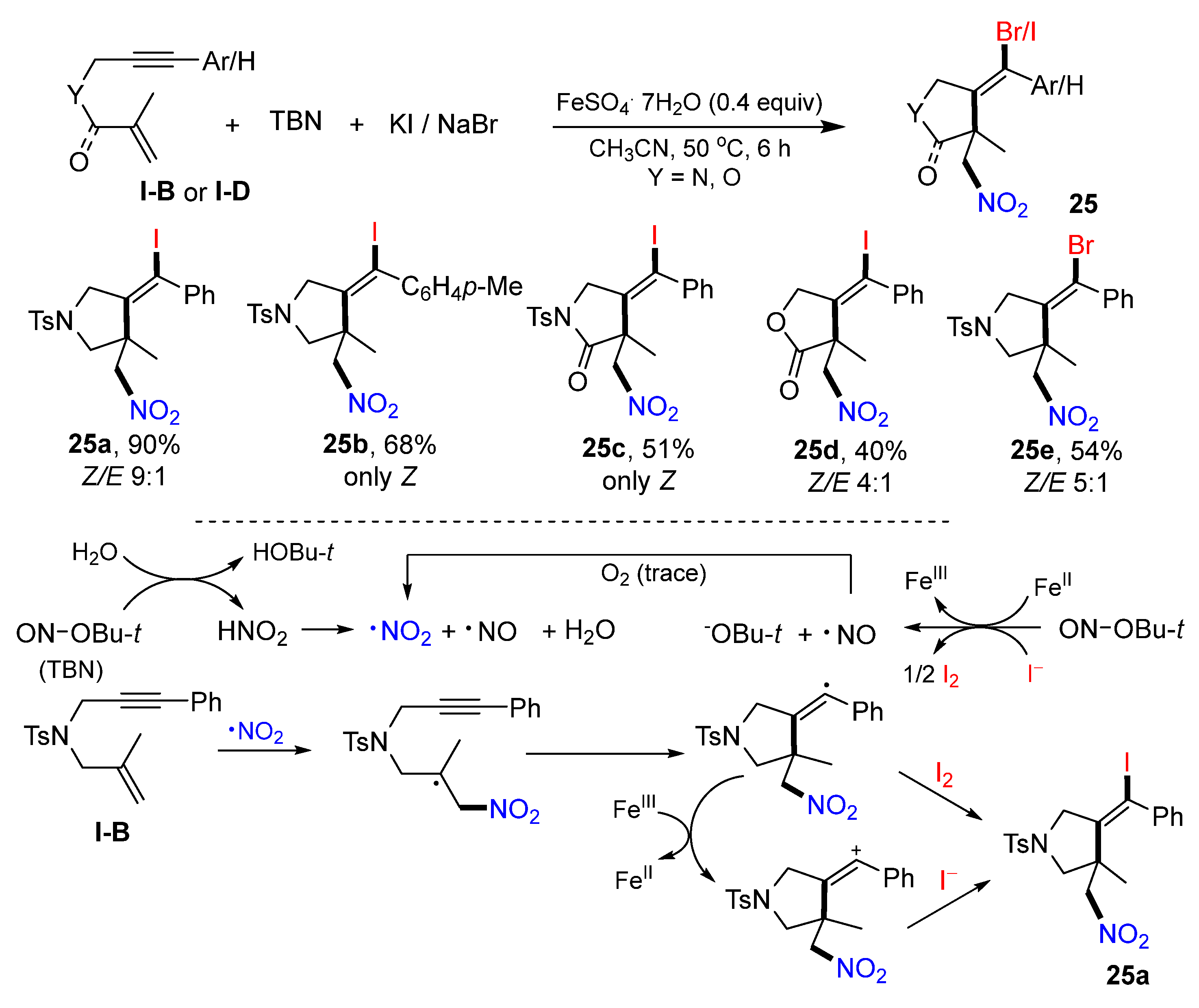
In 2021, Li and Tian’s lab reported Fe-catalyzed radical reaction of 1,6-enynes for the synthesis of difunctionalized heterocycles. The reaction of 1,6-enynes,
t-butyl nitrite (TBN) and KI or NaBr as materials in CH
3CN under the catalysis of FeSO
4·7H
2O gave products
25 in good-to-excellent yields (
Scheme 24) [
37]. As shown in the proposed mechanism, NO
2 radical produced from TBN adds to the C=C bond of 1,6-enyne followed by 5-
exo cyclization to give vinyl radical. This radical intermediate is iodinated through two possible pathways to give target product
25a.
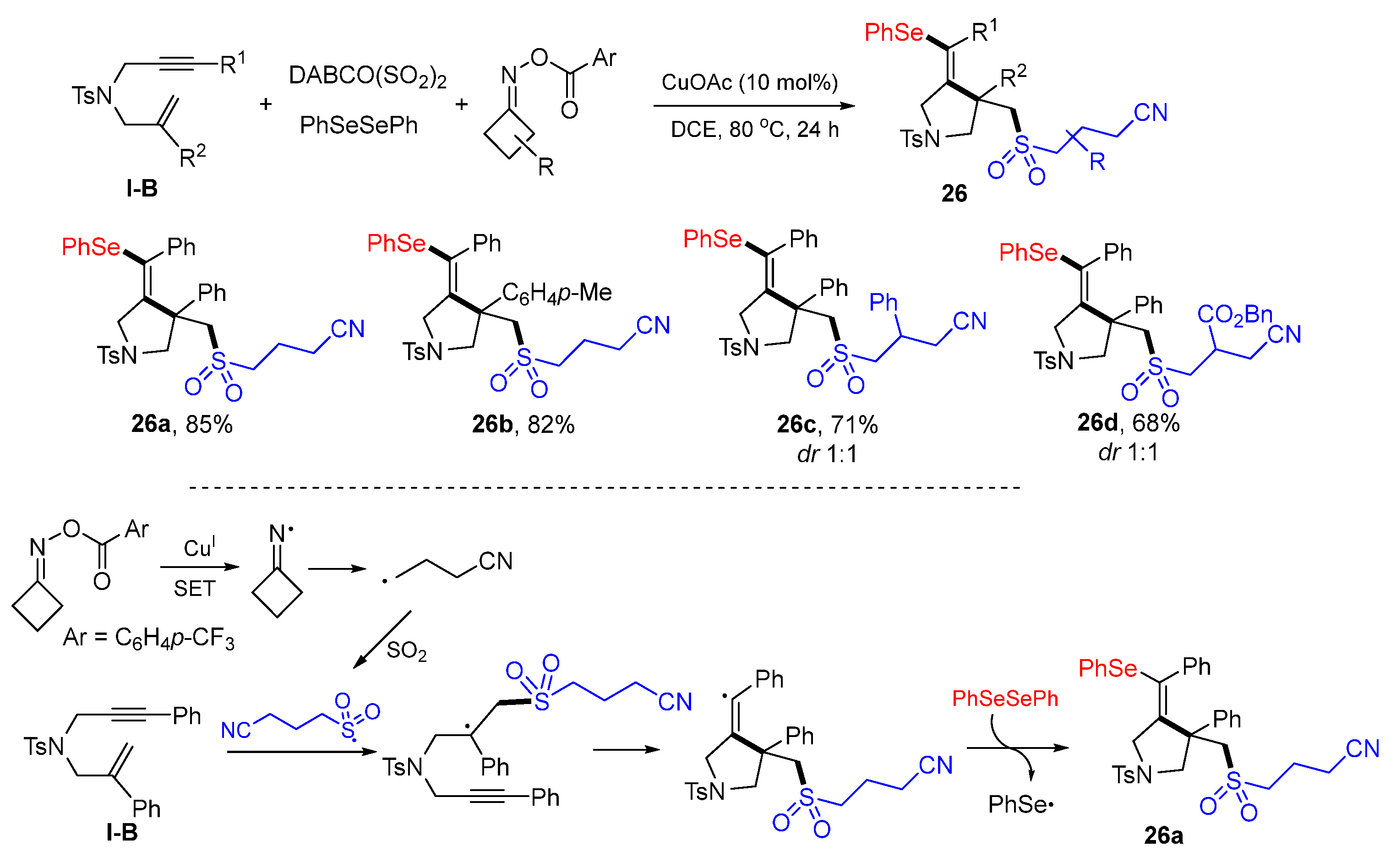
A Cu-catalyzed radical reaction of 1,6-enynes for the synthesis of cyanoalkylsulfonyl-ated pyrrolidines was introduced by He and coworkers in 2021. The reaction of 1,6-enynes, diselenides, DABCO(SO
2)
2 and cyclic ketone oxime esters in DCE with CuOAc as a catalyst afforded functionalized pyrrolidines
26 in moderate-to-good yields (
Scheme 25) [
38]. As indicated in the proposed mechanism, cyanoalkylsulfonyl radical generated from the reaction of cyclic ketone oxime esters and DABCO(SO
2)
2 adds to the C=C double bond of 1,6-enyne followed by 5-
exo cyclization and then couples with PhSe radical to give product
26a.
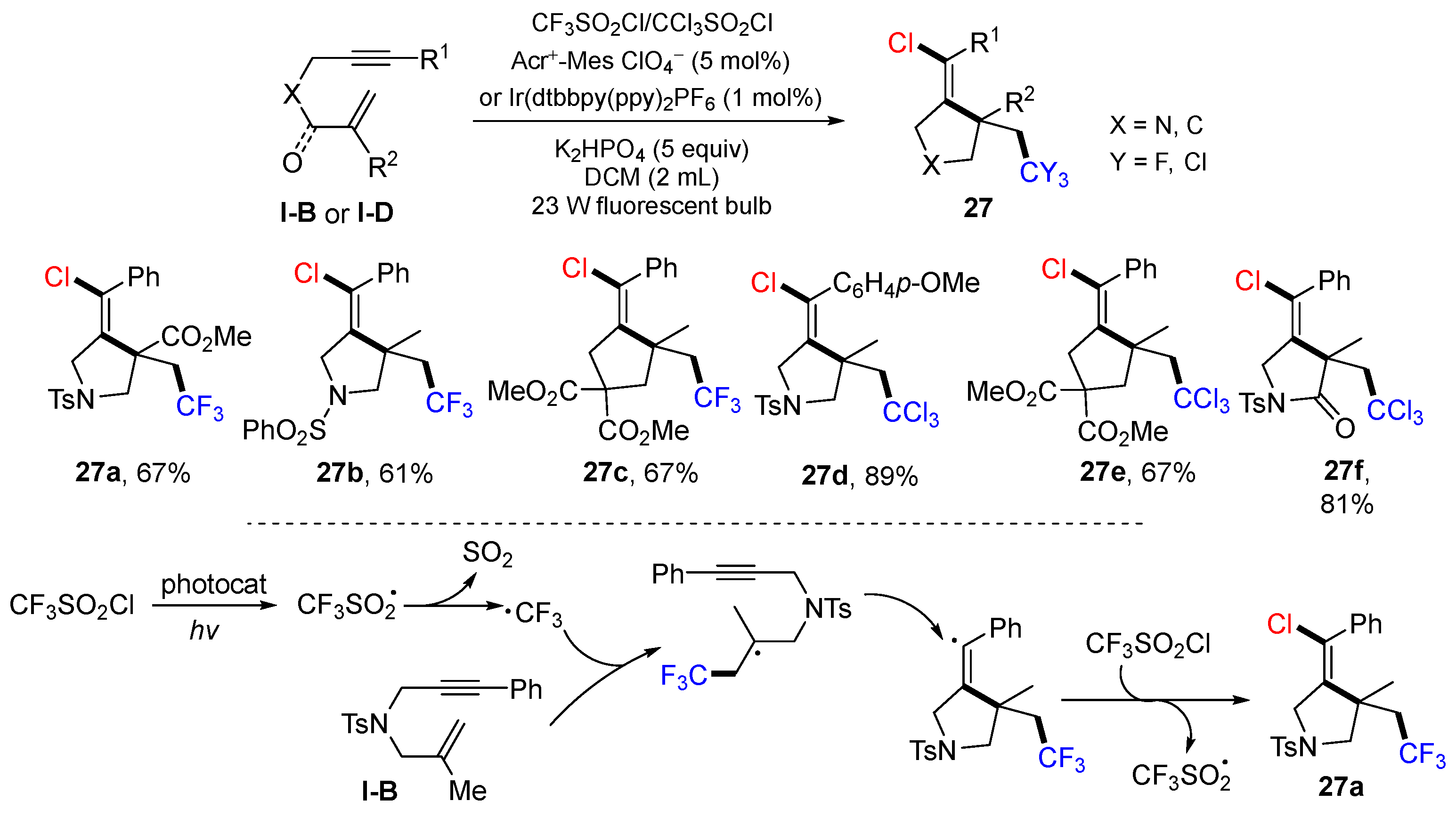
In 2019, Zhu and Hou’s group reported a visible light-mediated radical reaction for the synthesis of chlorotrifluoromethylated and chlorotrichloromethylated pyrrolidines, cyclopentanes and related compounds. The reaction of 1,6-enynes and CF
3SO
2Cl (or CCl
3SO
2Cl) in CH
2Cl
2 using Acr
+-Mes or Ir(dtbbpy(ppy)
2PF
6 as a photocatalyst gave products
27 in good-to-excellent yields (
Scheme 26) [
39]. A proposed mechanism indicated that CF
3 radical generated from CF
3SO
2Cl via SET adds to the C=C bond of 1,6-enynes, followed by 5-
exo cyclization and coupling with Cl radical, to give product
27a.
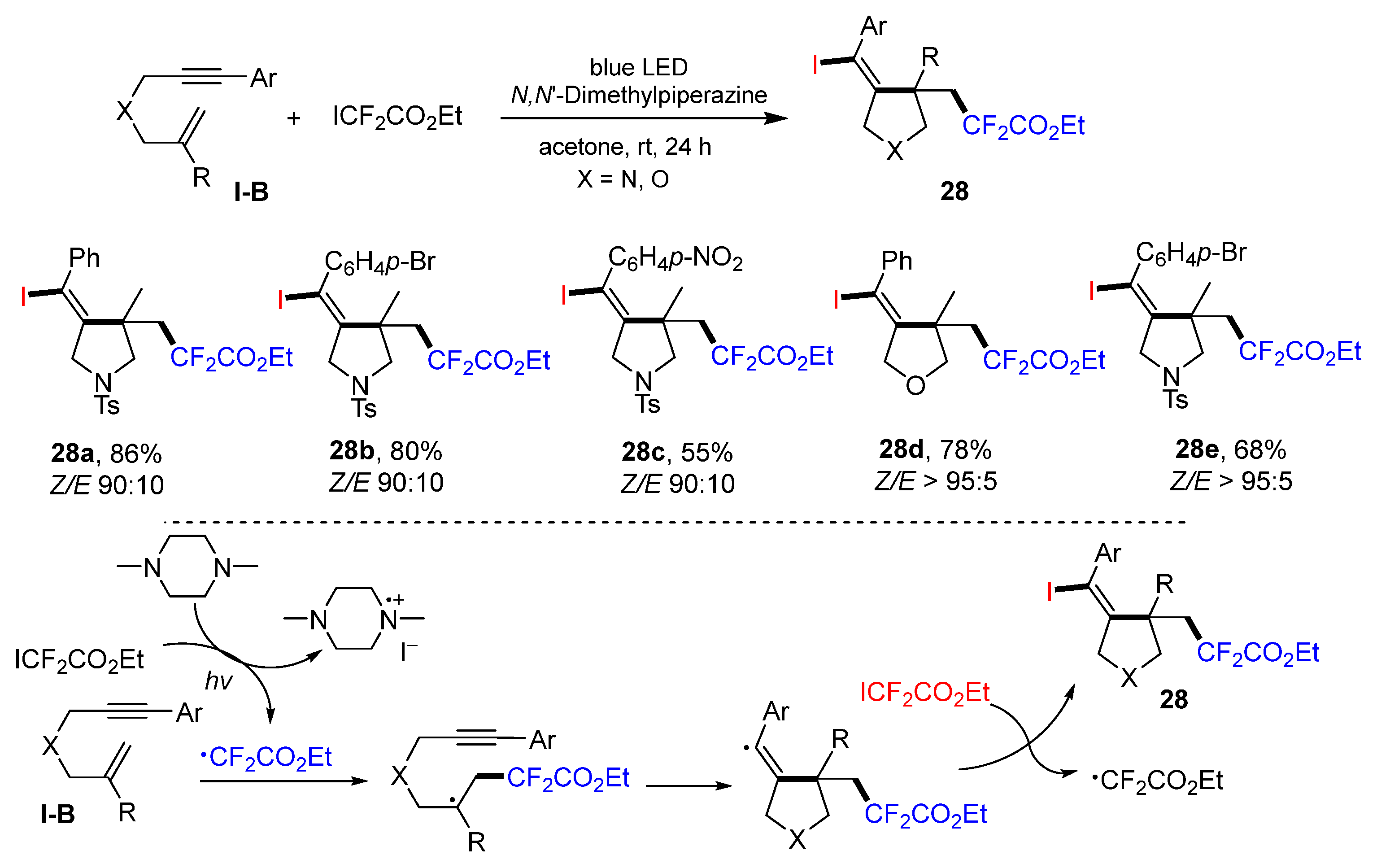
In 2022, Li and Yang reported a visible light-promoted reaction of 1,6-enynes for the synthesis of the iodovinyl- and CF
2-functionalized heterocycles. The reaction of 1,6-enynes, ICF
2CO
2Et under the radiation of blue LED afforded products
28 in good-to-excellent yields (
Scheme 27) [
40]. The reaction mechanism suggests that CF
2CO
2Et radical derived from ICF
2CO
2Et adds to the C=C double bond of 1,6-enyne, followed by 5-
exo cyclization and capture of iodine atom from ICF
2CO
2Et, to give product
28.
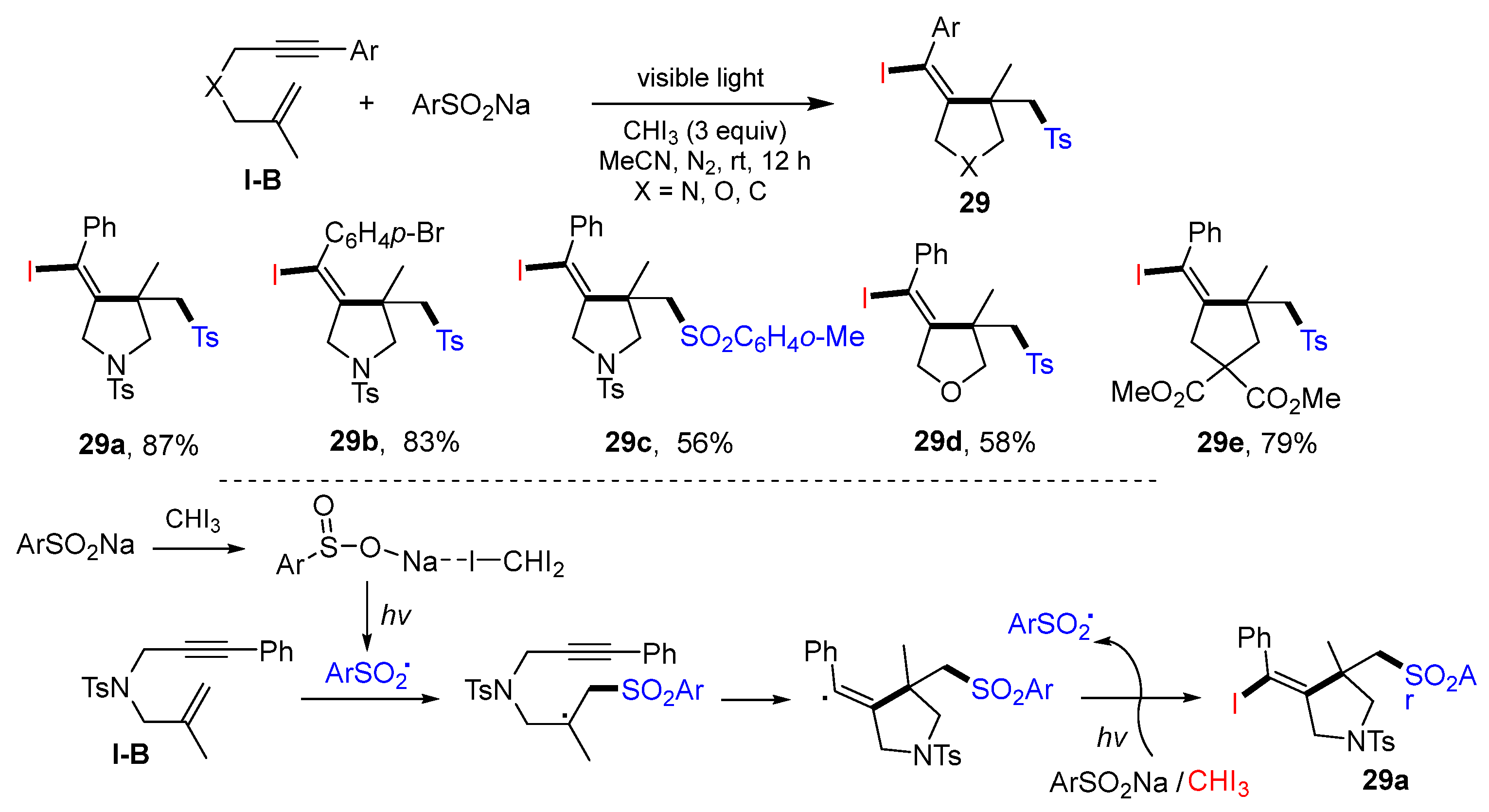
Zhu and co-workers, in 2022, reported a photo synthetic method for making iodo- and sulfonyl-containing cyclic compounds. The reaction of 1,6-enynes, ArSO
2Na, and iodoform in CH
3CN under visible light irradiation gave products
29 in good-to-excellent yields (
Scheme 28) [
41]. The reaction mechanism suggests that ArSO
2 radical derived from ArSO
2Na adds to the C=C double bond of 1,6-enyne, followed by 5-
exo cyclization and iodine atom transfer from the complex of ArSO
2Na and CHI
3, to give product
29a.
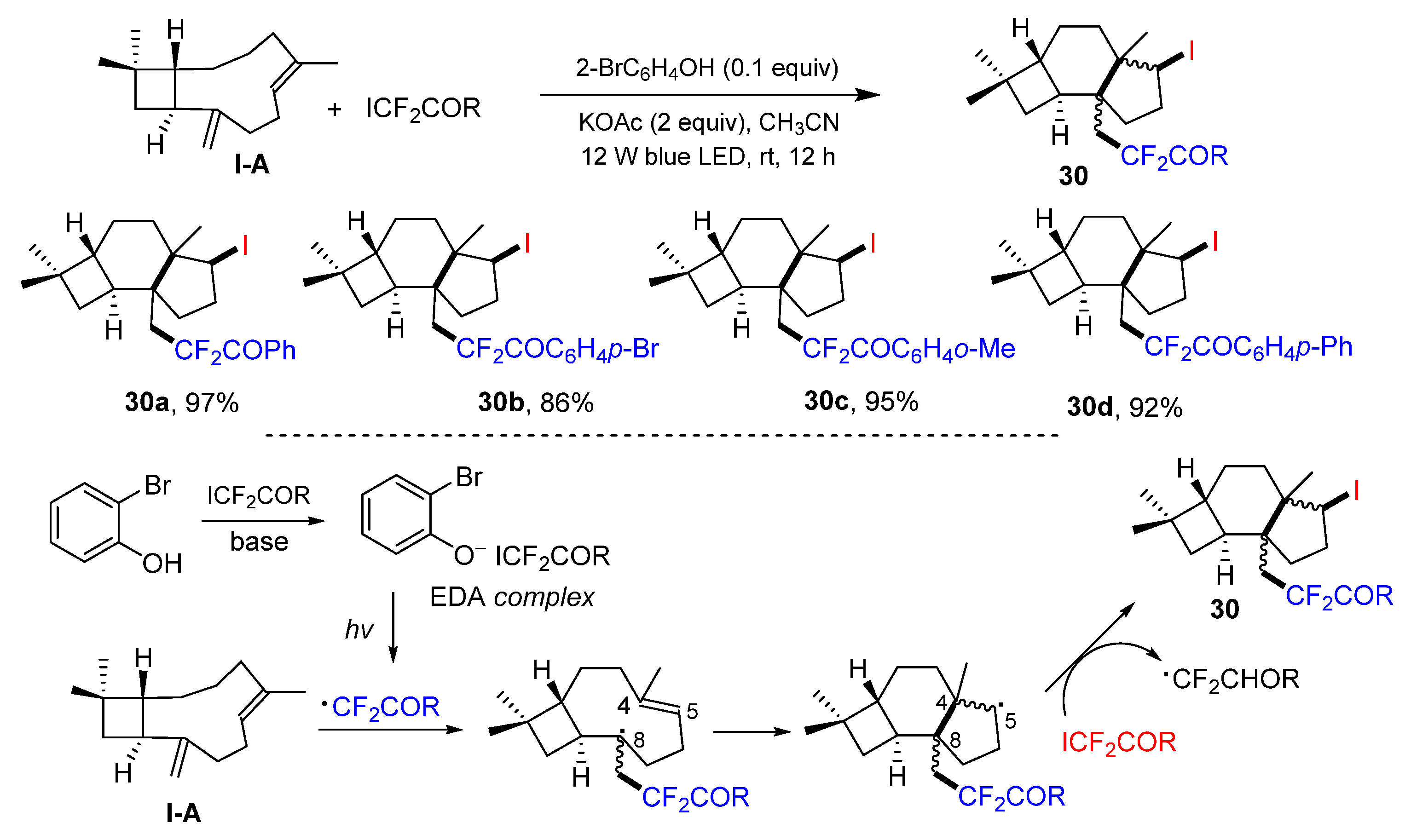
In 2022, a photo reaction of
β-caryophyllene, a 1,5-diene with one alkene in the ring and another one out of the ring, for the synthesis of iodo- and CF
2-containing protoilludanes was reported by the Huang group. The reaction of
β-caryophyllene and ICF
2COR in the presence of 2-bromophenol and base under the irradiation of blue LED afforded functionalized protoilludanes
30 in excellent yields (
Scheme 29) [
42]. A reaction mechanism suggests that the EDA complex generated from 2-bromophenol and ICF
2COR leads to the formation of CF
2COR radical. It then selectively adds to C8 of
β-caryophyllen, followed by the cyclization and abstraction of iodine atom from ICF
2COR to give the product
30.
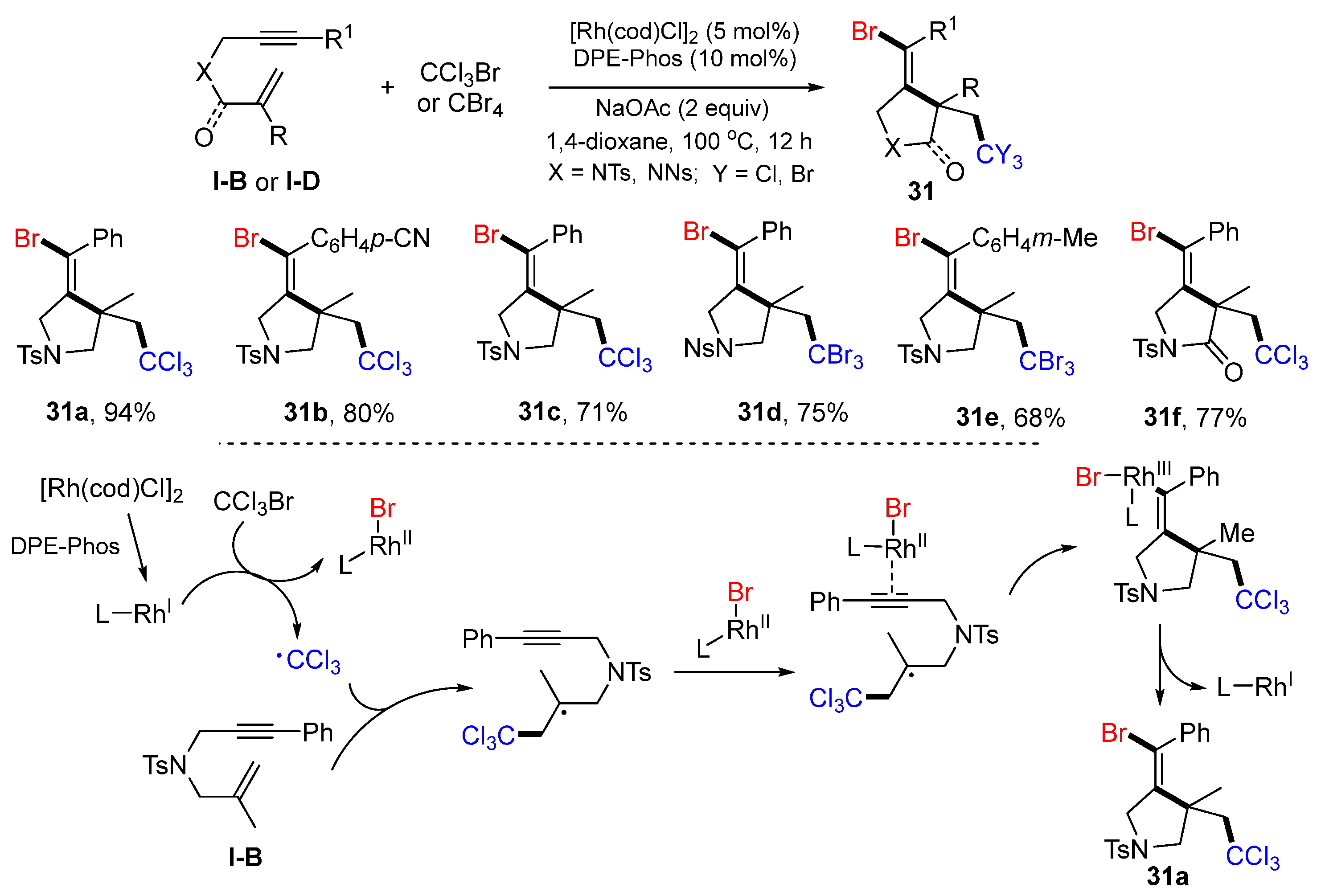
In 2019, the Liu group reported a met-catalyzed reaction of 1,6-enynes or 1,6-enynyl amides for the synthesis of bromotrihalomethylated pyrrolidines. The reaction of 1,6-enynes, and CCl
3Br or CBr
4 in 1,4-dioxane under the catalysis of [Rh(cod)Cl]
2 and DPE-Phos at 100 °C for 12 h gave products
31 in moderate-to-good yields (
Scheme 30) [
43]. The reaction mechanism suggests that CCl
3 radical, generated from CCl
3Br under the catalysis of [Rh(cod)Cl]
2 and DPE-Phos, adds to the C=C double bond of 1,6-enyne followed by 5-
exo cyclization to Rh
II-LBr activated alkyne and then L-Rh
I elimination to give product
31a.
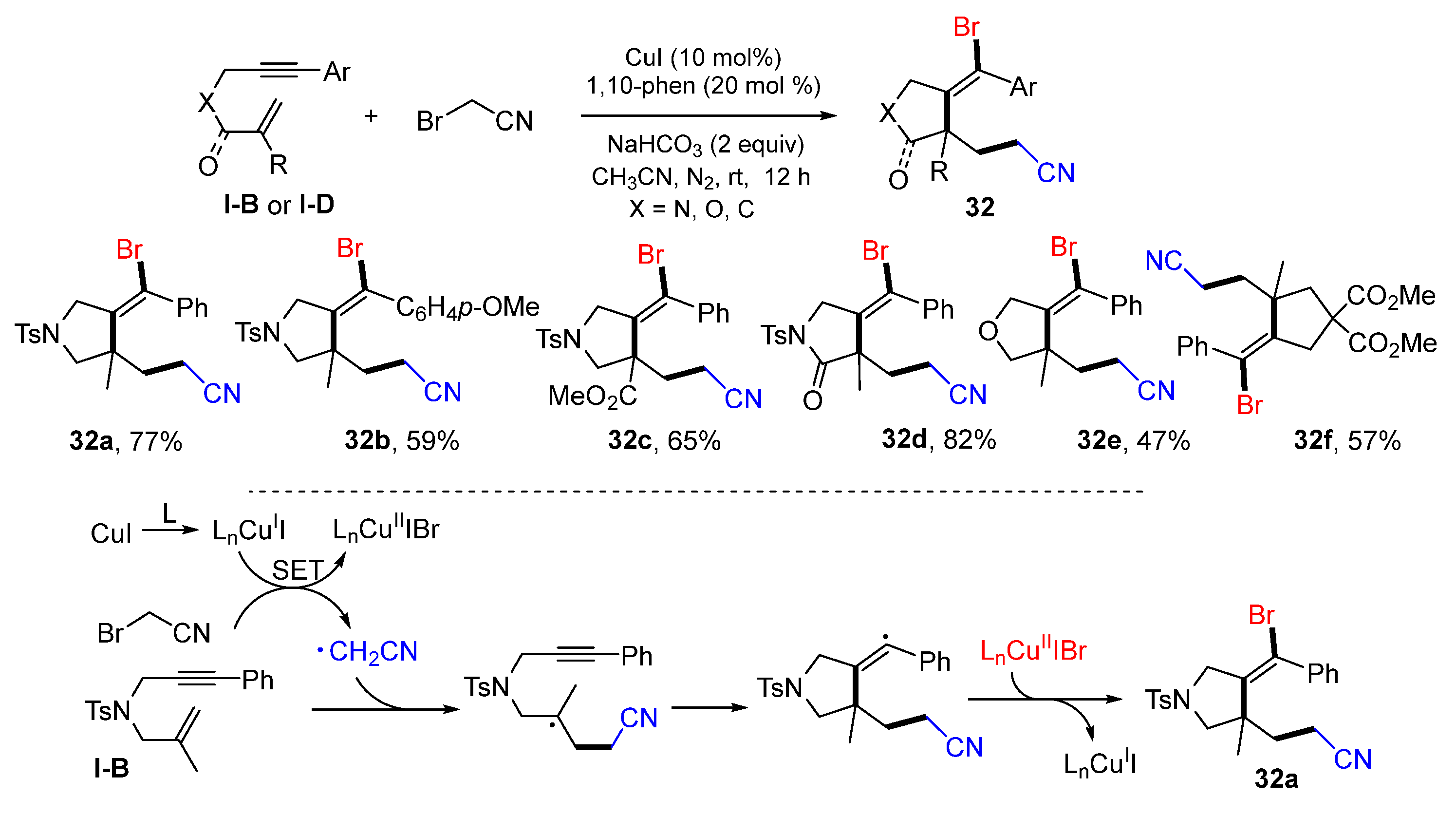
Hou and coworkers, in 2022, reported a Cu-induced radical reaction of 1,6-enynes for the synthesis of functionalized five-membered rings. The reaction of 1,6-enynes, BrCH
2CN in the presence of CuI, 1,10-phenanthroline and NaHCO
3 in CH
3CN afforded products
32 in good yields (
Scheme 31) [
44]. The reaction mechanism suggests that the CH
2CN radical derived from BrCH
2CN adds to C=C double bond of 1,6-enyne followed by 5
-exo cyclization and bromine atom-transfer to give product
32a.
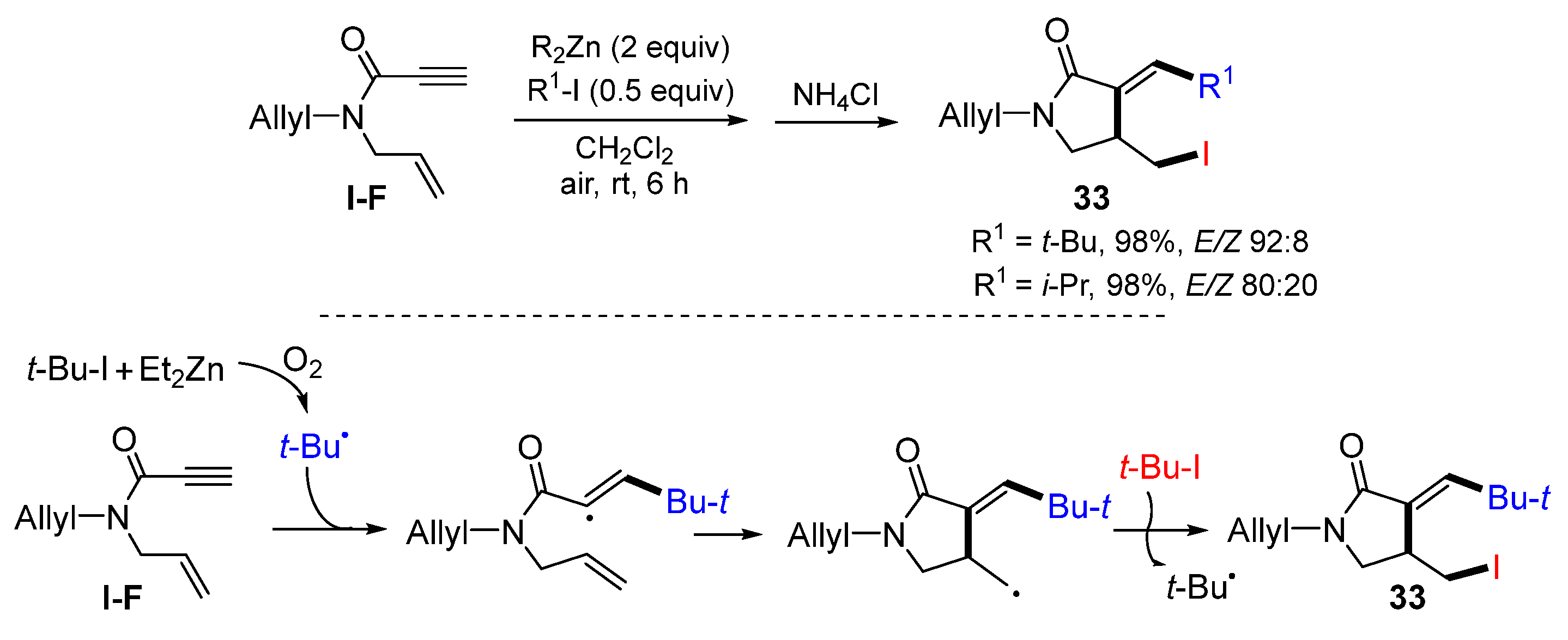
1,6-Eneynyl amides are another kind of popular substrates for radical reactions in the synthesis of functionalized 2-pyrrolidones [
45]. In 2008, Feray and Bertrand reported an R
2Zn-mediated radical reaction of 1,6-eneynyl amides for the synthesis of functionalized pyrrolidin-2-ones. The reaction of 1,6-eneynyl amides and alkyliodides in the presence dialkylzinc at room temperature gave product
33 in high yields as a mixture of
E/Z isomers (
Scheme 32) [
46]. The reaction mechanism suggests that the
t-butyl radical, generated from the reaction of
t-BuI and R
2Zn in the presence of oxygen, selectively adds to the triple bond of amide to form a stabilized vinyl radical, which then undergoes 5-
exo cyclization followed by iodine atom transfer from
t-BuI to give product
33.
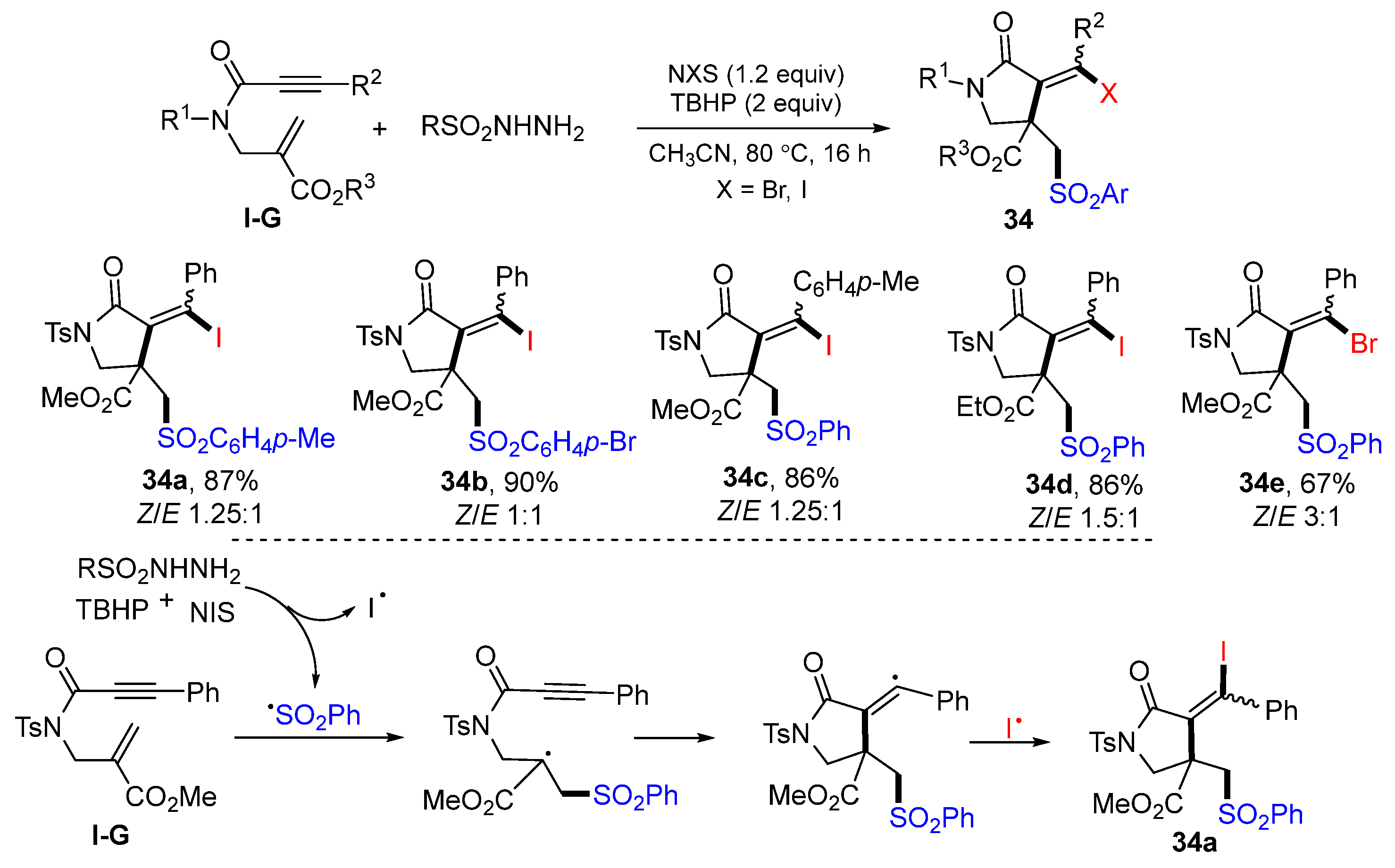
Xuan and co-workers introduced a reaction of 6-enynyl amides for the synthesis of substituted 2-pyrrolidinones in 2018. The reaction of 6-enynyl amides, NIS (or NBS), and sulfonyl hydrazide in CH
3CN and in the presence TBHP afforded
γ-lactams
34 in good to excellent yields (
Scheme 33) [
47]. The reaction mechanism suggests that sulfonyl radical generated from arylsulfonyl hydrazide adds to the C=C double bond of amide followed by 5-
exo cyclization and then coupling with iodine radical to give product
34a.
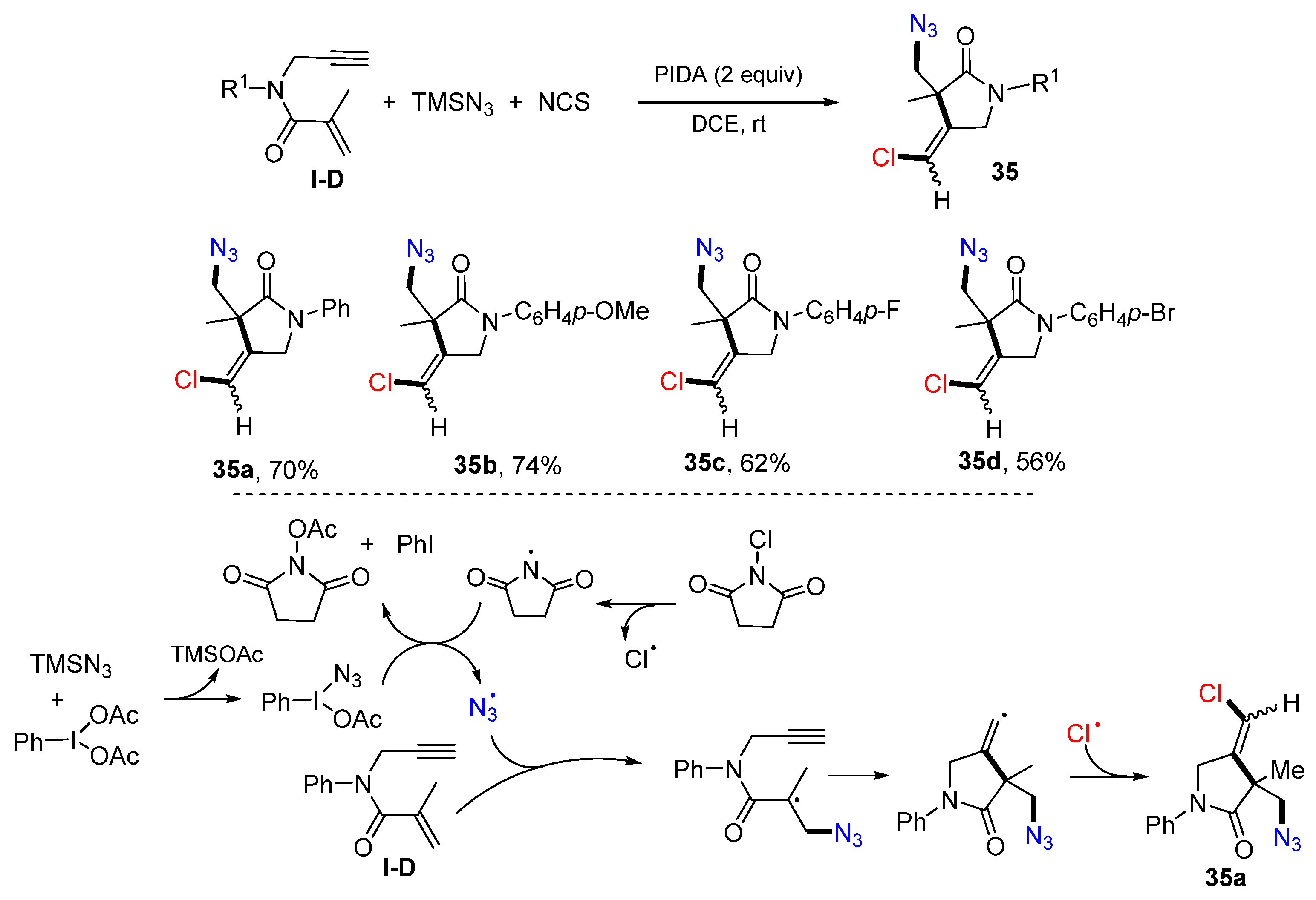
Wei and co-workers reported a protocol of cyclative chloroazidation of 1,6-enynyl amides for the synthesis of substituted 2-pyrrolidinones in 2018. The reaction of 1,6-enynyl amides, TMSN
3 and NCS in DCE in the presence of PIDA gave product
35 in moderate yields (
Scheme 34) [
48]. The reaction mechanism suggests that N
3 and Cl radicals were generated from TMSN
3 and NCS. The addition of N
3 radical to the C=C double bond of amide followed by 5-
exo cyclization and coupling with the Cl radical affords product
35a.
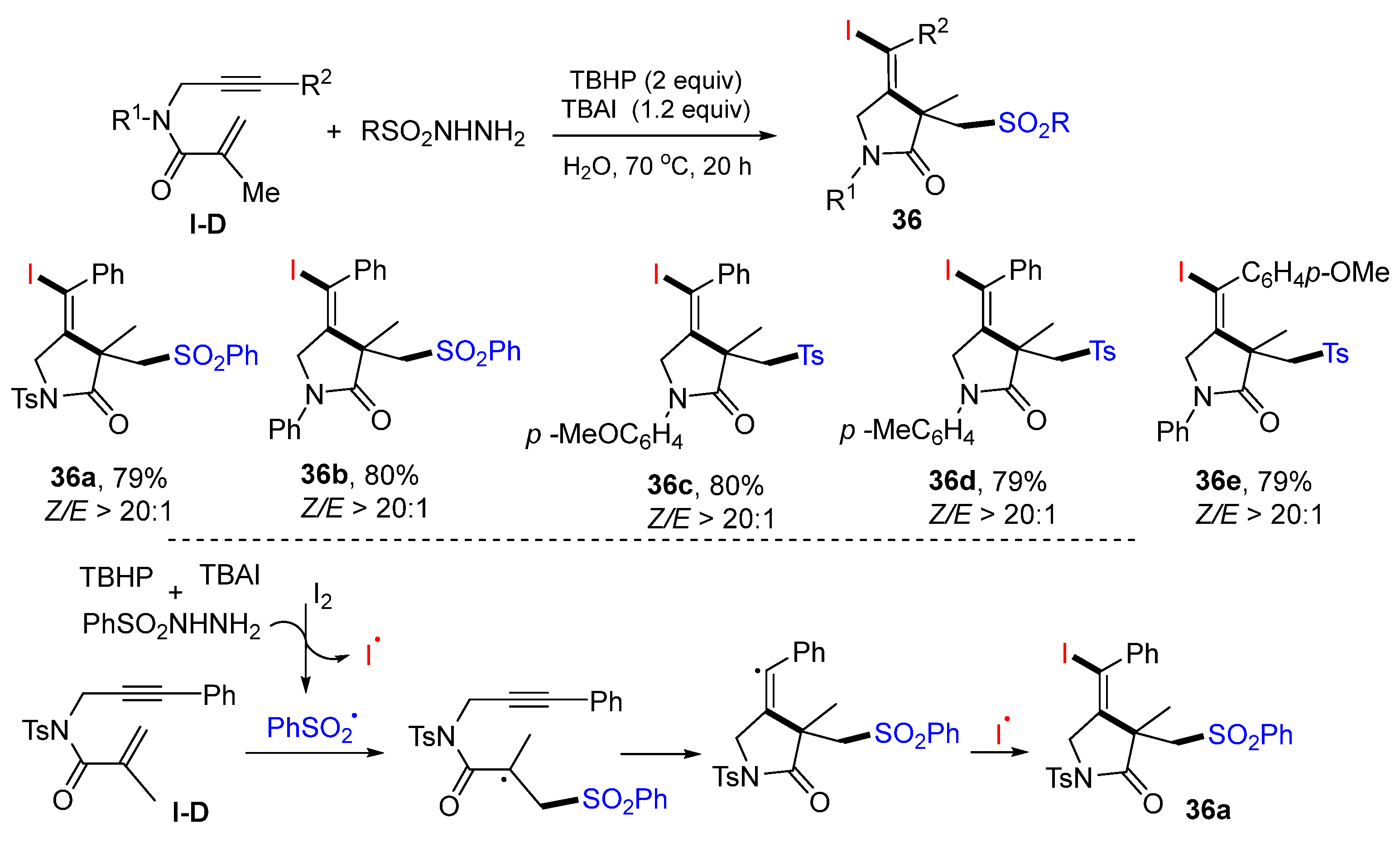
In 2022, Li and coworkers reported a reaction of 1,6-enynyl amides for the synthesis of
γ-lactams. The reaction of 1,6-enynyl amides and sulfonyl hydrazides in H
2O at 70 °C for 20 h in the presence of TBHP gave product
36 in moderate-to-good yields (
Scheme 35) [
49]. The reaction mechanism suggests that PhSO
2 radical, generated from the reaction of PhSO
2NHNH
2 with TBHP and TBAI, adds to the C=C double bond of amide followed by 5-
exo cyclization and coupling with iodine radical to give product
36a.
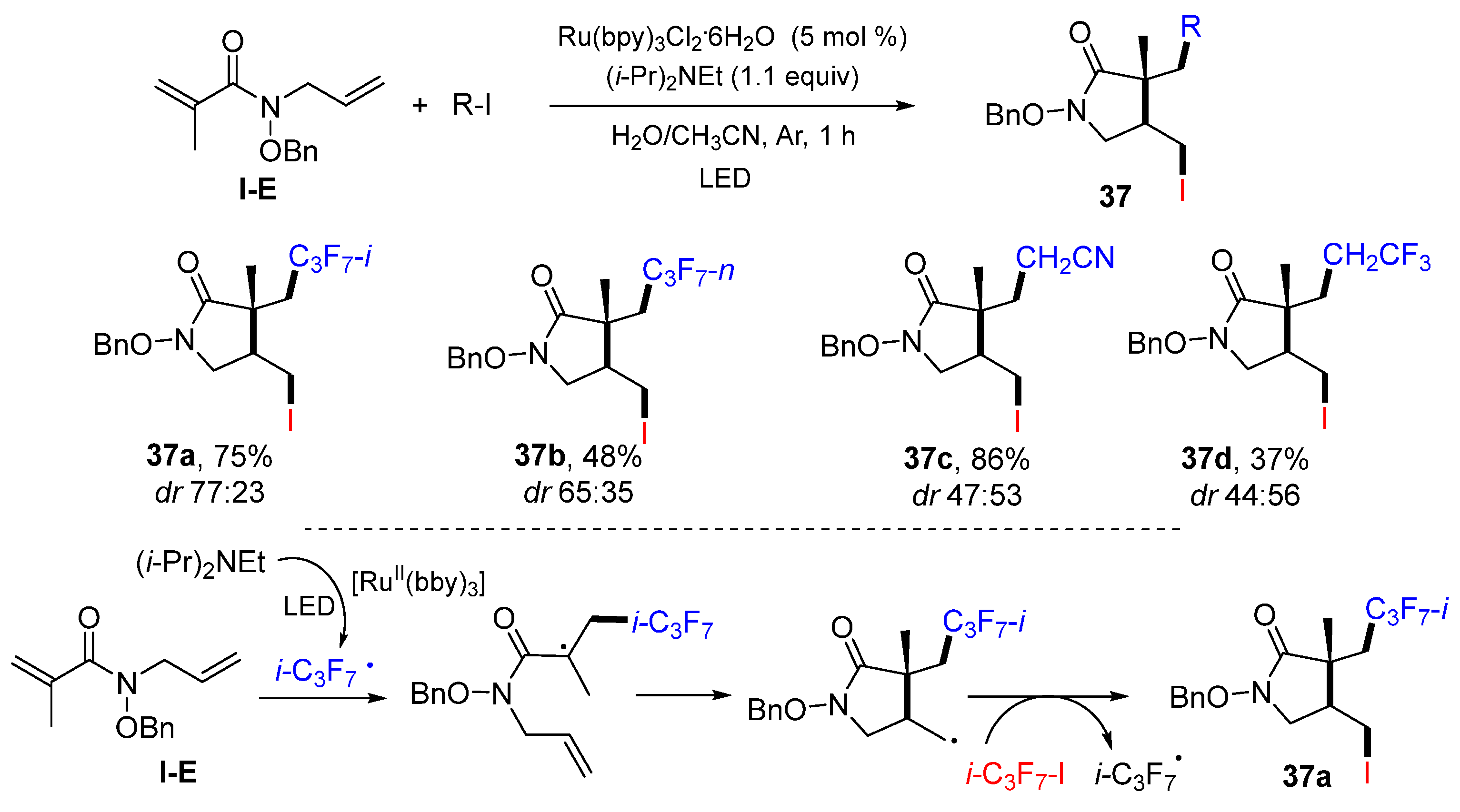
A photoredox ATRC reaction of 1,6-dienyl amides for the synthesis of functionalized pyrrolidin-2-ones was developed by the Miyabe group in 2015. The reaction of 1,6-dienyl amides and iodoalkanes in aqueous media and catalyzed by Ru(bpy)
3Cl
2·6H
2O and (
i-Pr)
2NEt gave product
37 in fair-to-good yields (
Scheme 36) [
50]. Other than
i-C
3F
7I, other iodo compounds such ICH
2CN and ICH
2CF
3 are also good radical precursors. The reaction mechanism suggests that the
i-C
3F
7 radical generated from
i-PrI via the photoredox process adds to the C=C double bond of amide, followed by 5-
exo cyclization and then iodine atom transfer from
i-PrI to give product
37a.
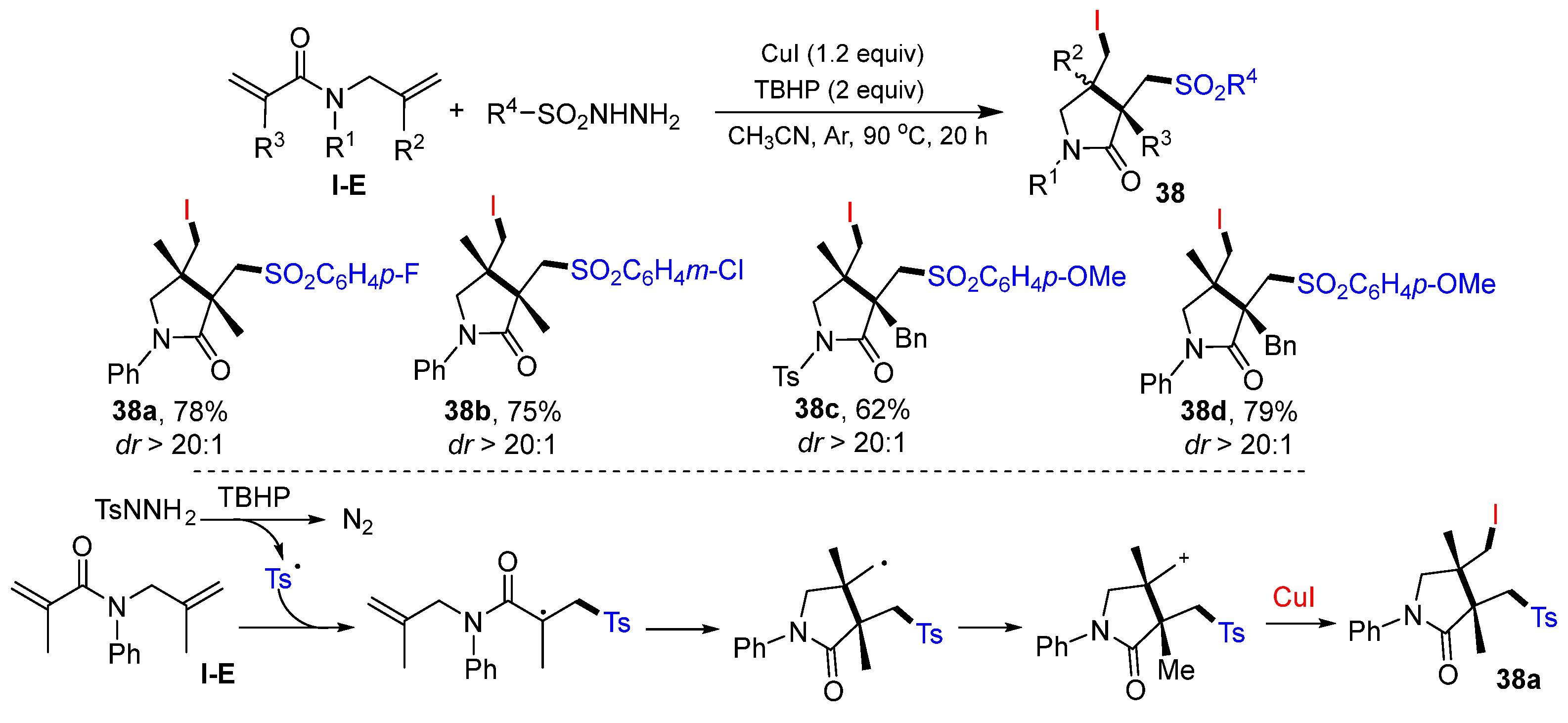
Li and Wei, in 2021, reported a Cu-catalyzed radical reaction of 1,6-dienyl amides for the synthesis of substituted
γ-lactams. The reaction of 1,6-dienyl amides and RSO
2NHNH
2 in CH
3CN in the presence of CuI and TBHP gave product
38 in moderate-to-good yields (
Scheme 37) [
51]. The reaction mechanism suggests that the sulfonyl radical, generated from the reaction of RSO
2NHNH
2 with TBHP, adds to the C=C double bond of amide followed by 5-
exo cyclization, oxidation to carbocation, and trapping I
− anion of CuI to provide iodosulfonylation of product
38a.
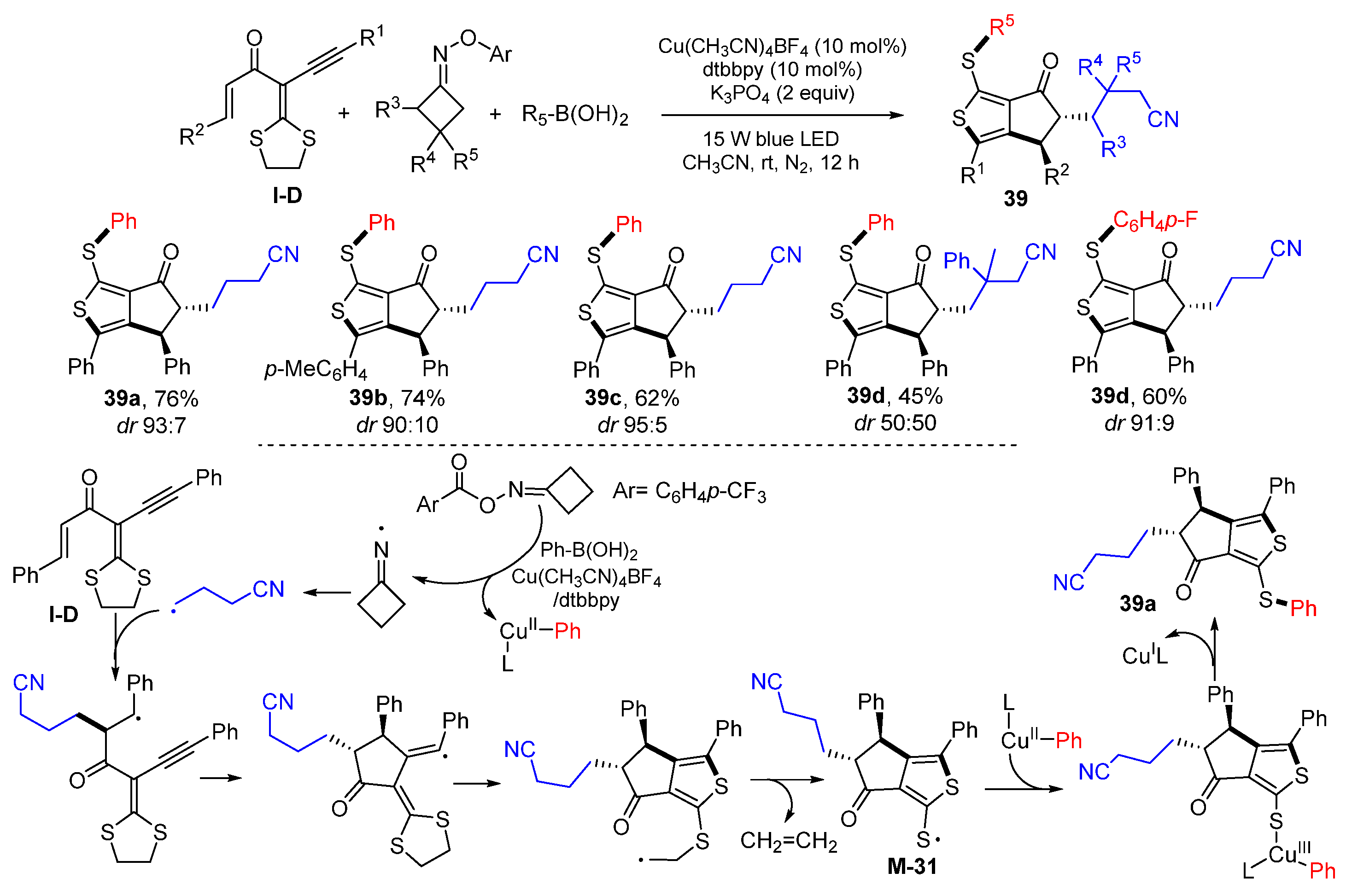
A photoredox reaction of carbonyl-containing 1,6-enynes for the synthesis of cyclopentanone derivatives was reported by Zhou, Yu and their coworkers in 2020. The reaction of
gem-dialkylthio enynes, cyclobutanone oxime esters, and boronic acids in the presence of Cu(CH
3CN)
4BF
4, dtbbpy and K
3PO
4 in CH
3CN under irradiation of blue LED gave functionalized aryl thienyl sulfide
39 in moderate-to-good yields and with good chemo- and diastereoselectivities (
Scheme 38) [
52]. The reaction mechanism suggests that
γ-cyanoalkyl radical, generated from homolytic
α,
β-C−C cleavage of
N-centered iminyl, which is derived from cyclobutanone oxime esters, adds to the C=C bond of
gem-dialkylthio 1,3-enyne followed by 5-
exo cyclization, radical rearrangement and fragment of ethylene to give sulfur-centered radical
M-31. Radical
M-31 reacts with the LCu
IIPh complex followed by reductive elimination to give product
39a.
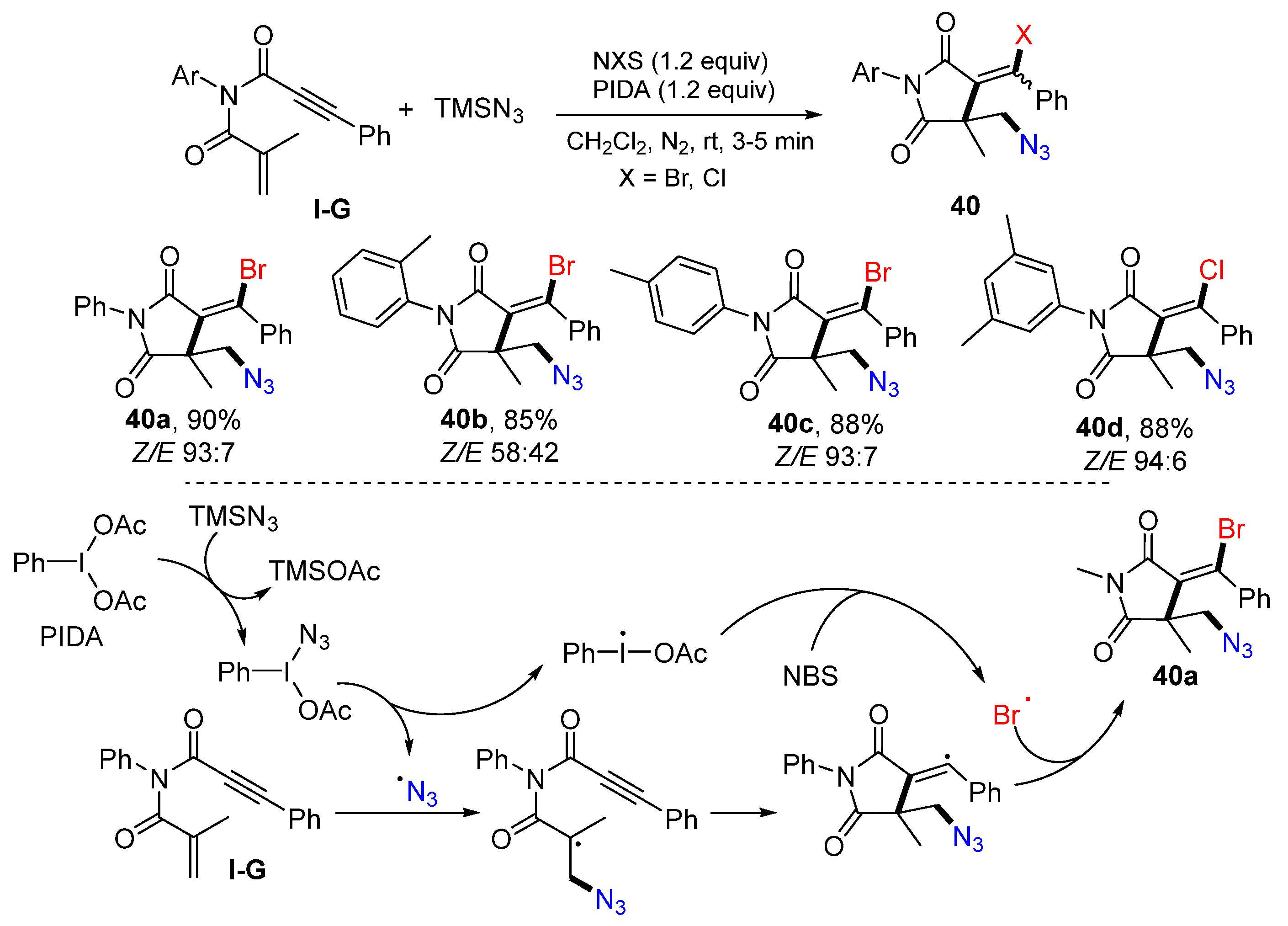
A reaction of 1,6-enynyl with two carbonyl groups for the synthesis of functionalized succinimides was introduced by the Rong group in 2020. The reaction of 1,6-enynyl amides, NBS or NCS, TMSN
3, and PIDA in DCM at room temperature for 3–5 min afforded products
40 as
E/Z isomers in excellent yields (
Scheme 39) [
53]. The reaction mechanism suggests that the azide radical, resulting from the reaction of PIDA and TMSN
3, adds to alkene moiety of 1,6-enyne, followed by 5-
exo cyclization and coupling with the bromine radical from NBS, to give product
40a.
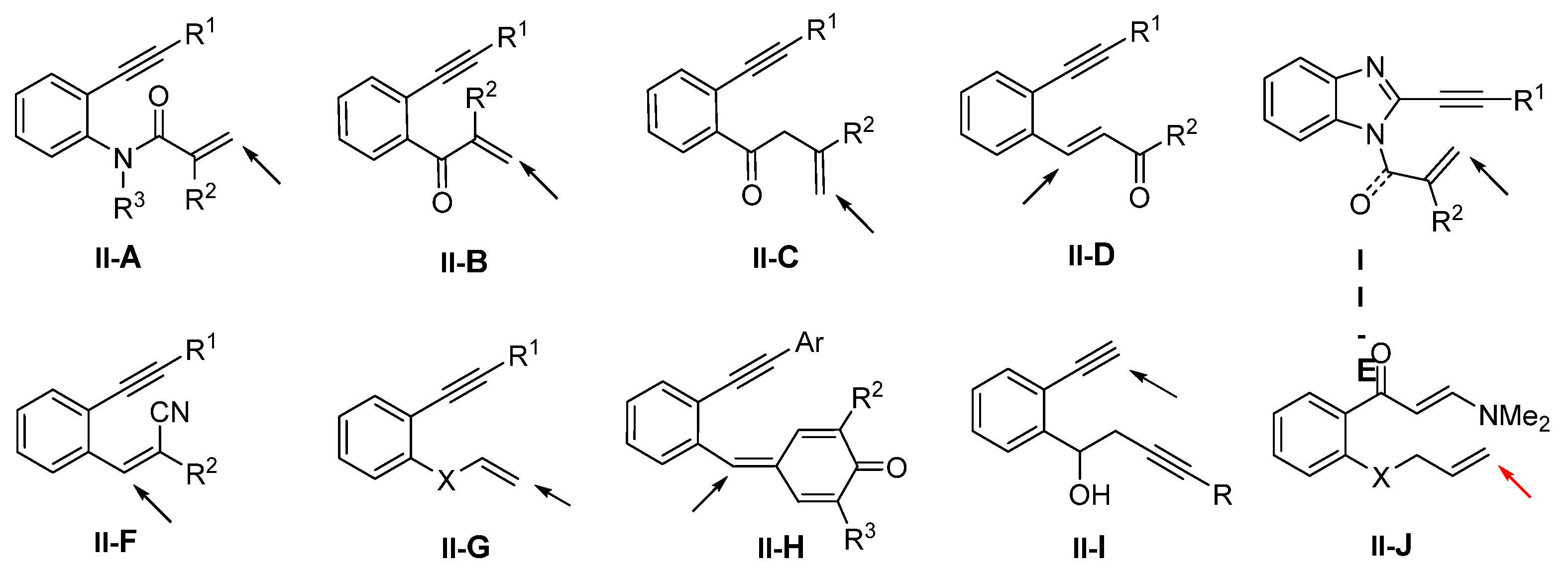
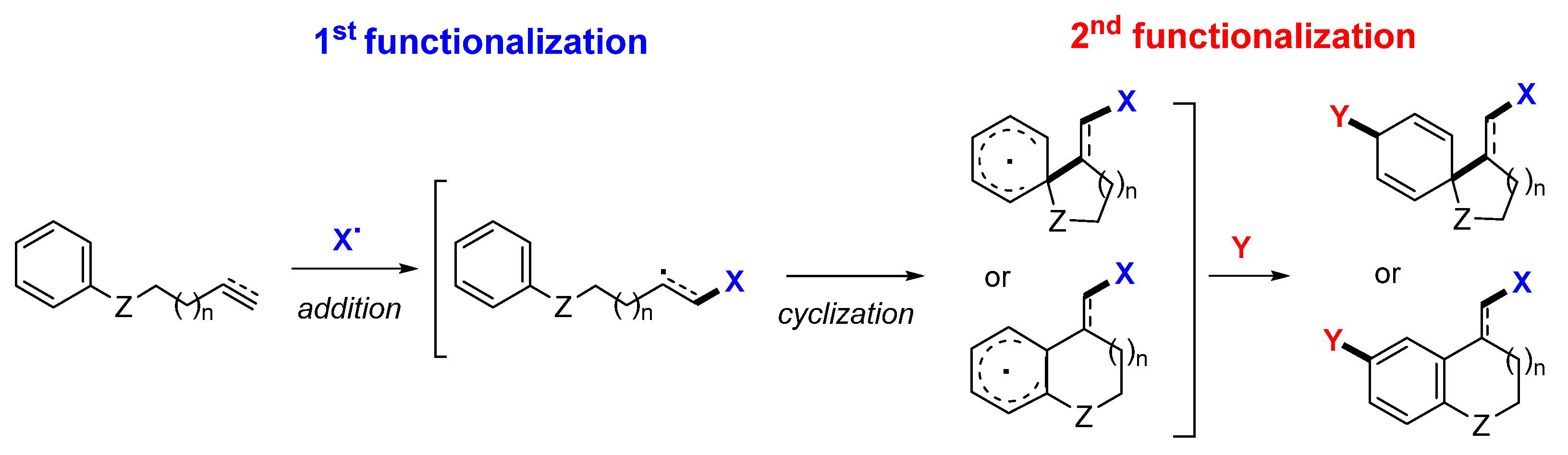
3. Reaction of Arene-Tethered Dienes and Enynes
Presented in this section are the radical addition and cyclization-initiated difunctionalization reactions of arene-bridged 1,n-dienes -diynes, and -enynes with a reaction sequence shown in
Scheme 40. It is noteworthy that most substrates found in the literature are enynes but not dienes (like
II-J) or diynes (like
II-I) (
Scheme 41). The enynyl substrates include the most popular 1,7-enynyl amides
II-A and other ones containing the carbonyl group (
II-B to
II-E). Other substrates may contain heteroatom or conjugate groups (such as CN and Ar) at the terminal carbon of the unsaturated bonds (
II-F to
II-H). Between the two unsaturated carbon–carbon bonds in the substrates, the regioselectivity for the initial radical addition is directed by the steric and the conjugation effects of the substituents. The R
1 group on the terminal carbon of alkyne is commonly employed to block the initial radical addition to the alkyne. Substrate
II-J is an exception in which the initial radical addition does not go to the conjugated alkene.
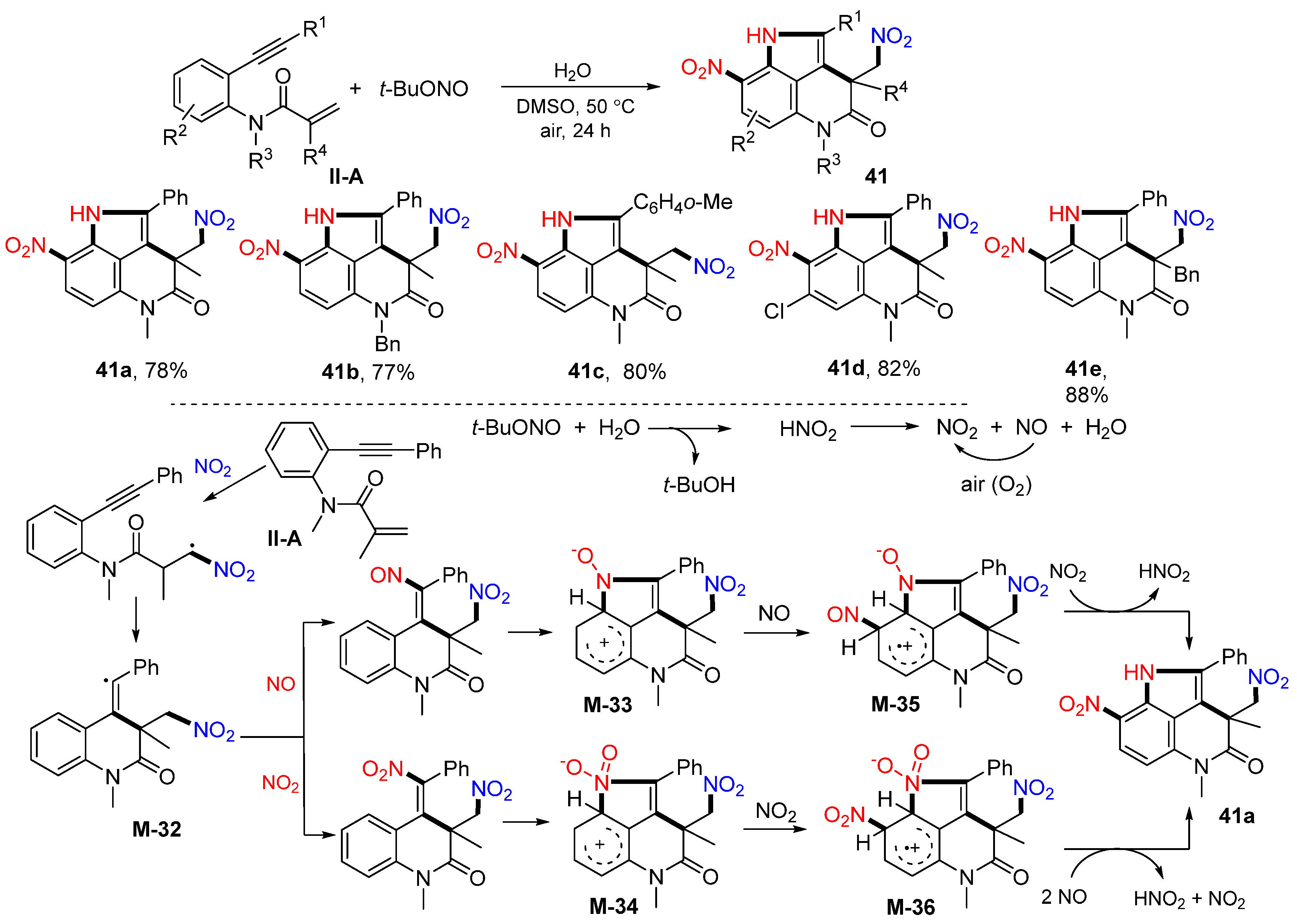
Benzene-tethered 1,7-enynyl amides are popular substrates for radical difunctionalization reactions. In 2014, the Li group introduced a reaction of such substrates for the synthesis of dinitropyrrolo[4,3,2-
de]-quinolinones. The reaction of 1,7-enynyl amides and
t-BuONO in DMSO afforded product
41 in good-to-excellent yields (
Scheme 42) [
54]. It was found that the amount of H
2O had a significant influence on the reaction. The reaction mechanism suggests that NO
2 radical generated in situ from
t-BuONO adds to the C=C double bond of amide followed by 6-
exo cyclization to form intermediate
M-32. The reaction of
M-32 with NO or NO
2 radical followed by electrophilic addition of NO or NO
2 radical to the phenyl ring gave cationic intermediates
M-33 and
M-34. Cationic radical intermediates
M-35 and
M-36 were produced through the treatment of the cationic intermediates
M-33 and
M-34 with NO or NO
2 radical and then lead to the formation of product
41a after the redox reaction.
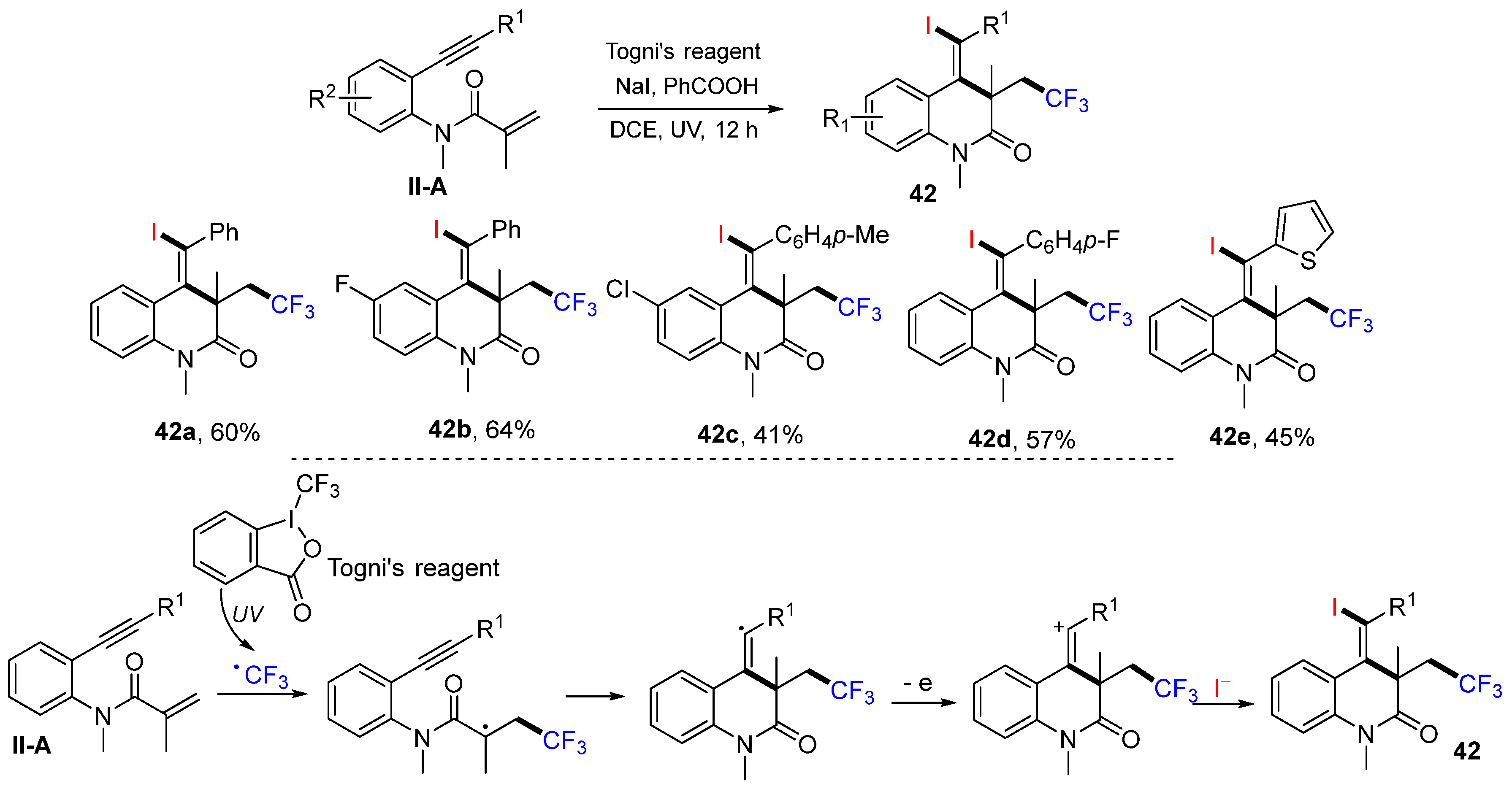
The Wu group, in 2016, introduced a photoredox reaction of benzene-tethered 1,7-enynyl amides for the synthesis of trifluoroethyl-substituted 3,4-dihydroquinolin-2(
1H)-ones. The reaction of 1,7-enynyl amides and Togni’s reagent in the presence of NaI and PhCO
2H under UV irradiation gave
42 in moderate-to-good yields (
Scheme 43) [
55]. The proposed mechanism indicated that trifluoromethyl radical derived from the Togni’s reagent adds to the C=C double bond of amide, followed by 6-
exo cyclization and oxidation to cation for the reaction with iodide anion, to give product
42.
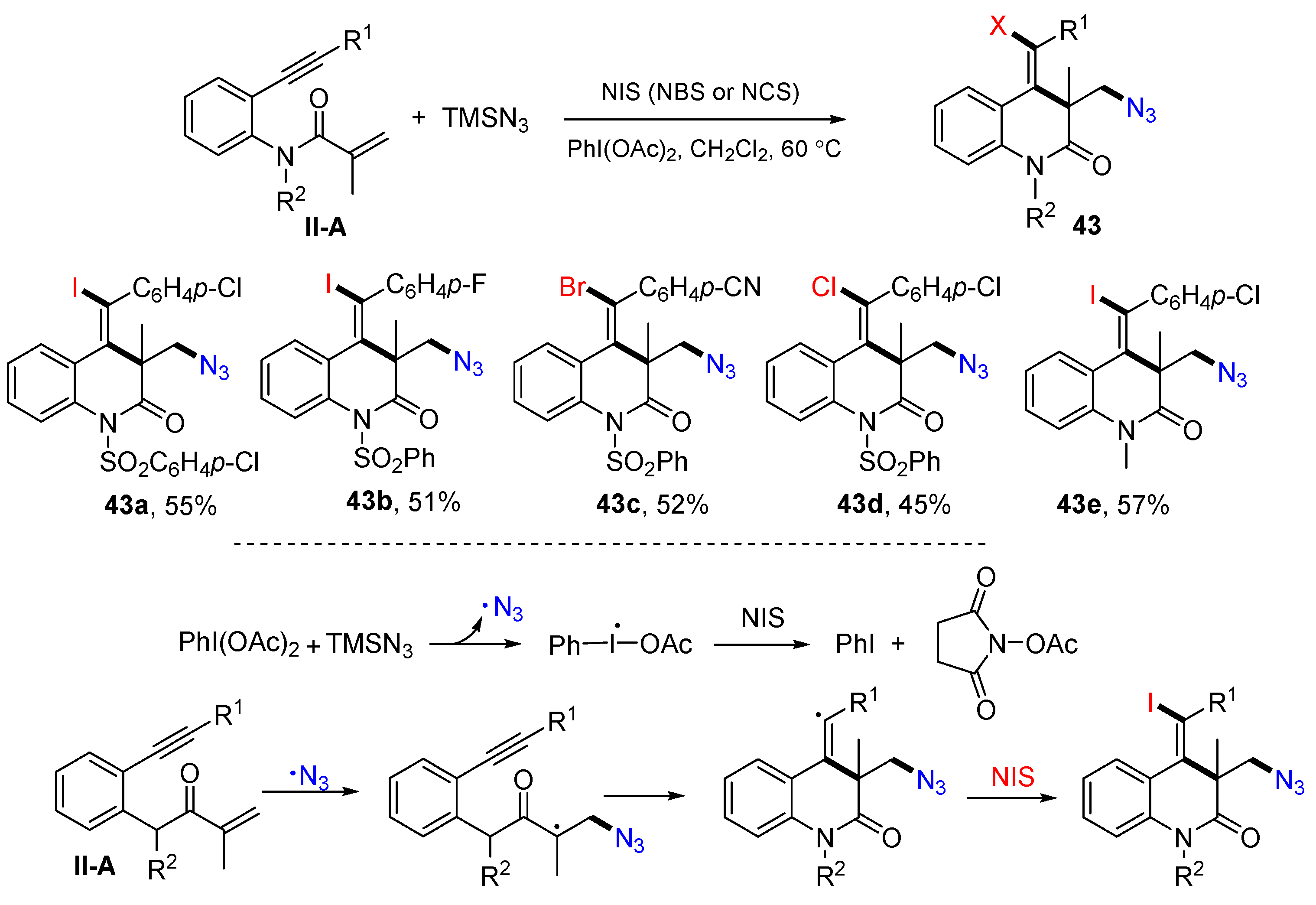
In 2016, the Jiang group reported a reaction of benzene-bridged 1,7-enynyl amides for the synthesis of substituted 3,4-dihydroquinolin-2(1
H)-ones. The reaction of 1,7-enynyl amides, TMSN
3 and NIS (or NBS and NCS) in the presence of PhI(OAc)
2 in CH
2Cl
2 gave products
43 in good-to-excellent yields (
Scheme 44) [
56]. A reaction mechanism suggests that N
3 radical generated from the reaction of PhI(OAc)
2 and TMSN
3 adds to the C=C double bond of amide followed by 6-
exo cyclization and coupling with iodine radical from NIS to give product
43.
A transition metal-mediated radical reaction of benzene-bridged 1,7-enynyl amides for the synthesis of substituted pyrrolo[3,4-
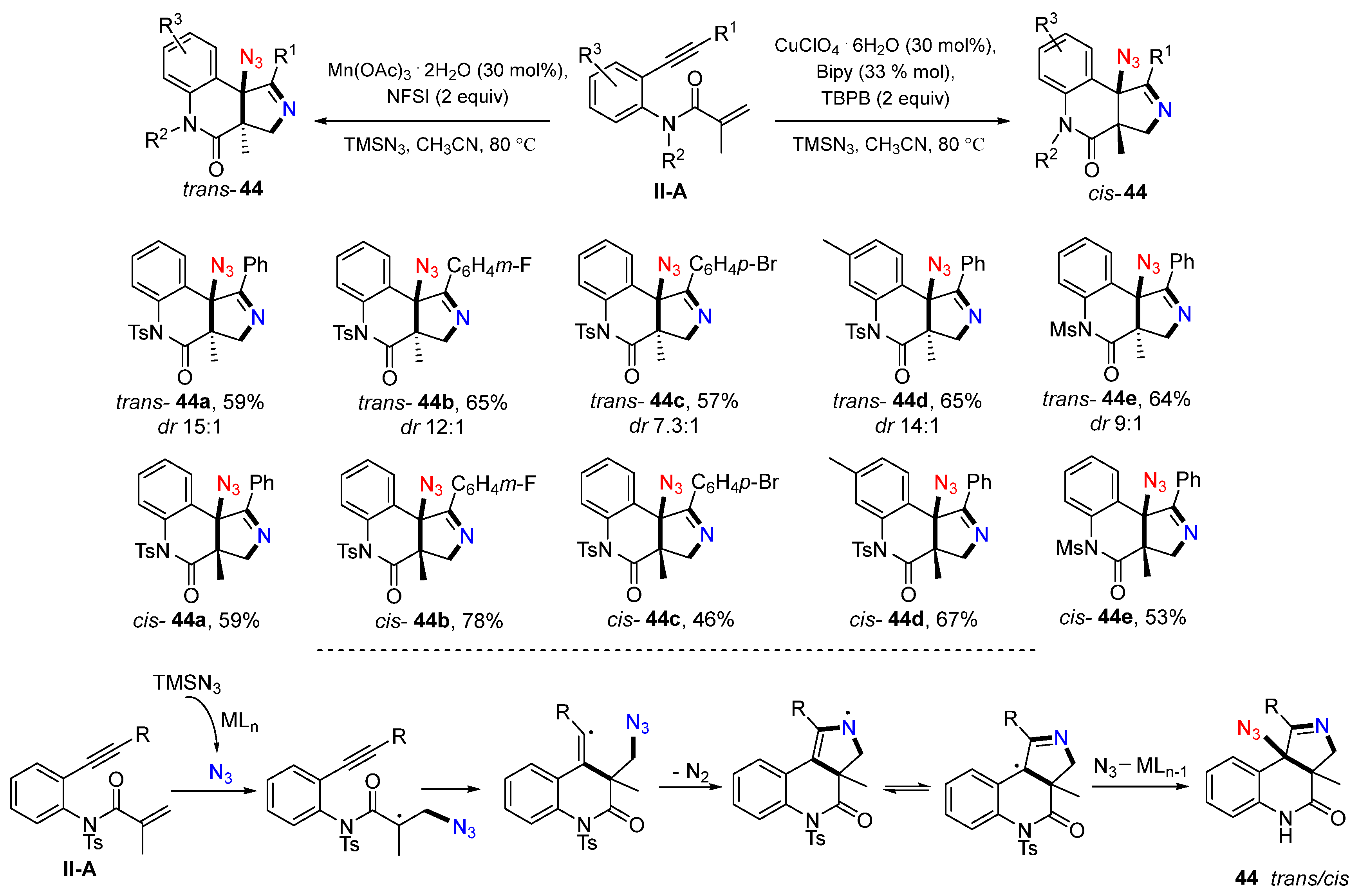
c]quinolinones was reported by the Wan group in 2016. The
trans-fused products were obtained when using Mn
III as a catalyst, whereas
cis-products were obtained using Cu
II as a catalyst. The reactions of amides and TMSN
3 in the presence of Mn(OAc)
3/NFSI or Cu(ClO
4)
2/TBPB in CH
3CN afforded
trans- or
cis-fused products
44, respectively, in good-to-excellent yields (
Scheme 45) [
57]. A reaction mechanism suggests that N
3 radical generated from TMSN
3 adds to the C=C double bond of amides followed by 6-
exo cyclization, releasing of N
2, then azido group transfer to afford the desired
trans- or
cis-fused product
44.
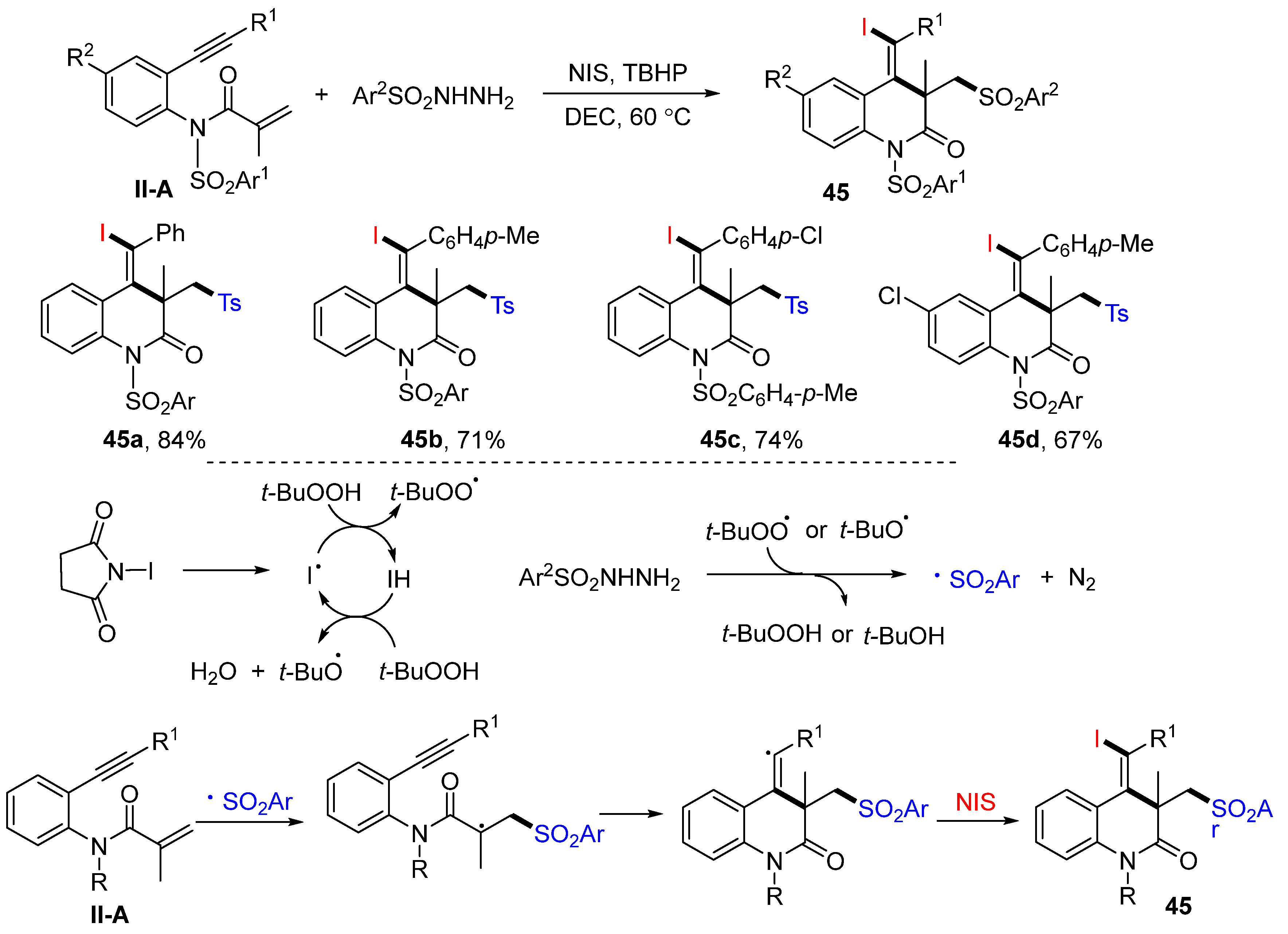
The Tu group reported a method for the synthesis of densely functionalized 3,4-dihydro-quinolin-2(1
H)-ones in 2016. The reaction of benzene-tethered 1,7-enynyl amides, arylsulfonyl hydrazides and NIS (or NBS) in DEC in the presence of TBHP afforded product
45 in good-to-excellent yields (
Scheme 46) [
58]. The reaction mechanism suggests that the sulfonyl radical derived from sulfonyl hydrazides adds to the C=C double bond of amides, followed by 6-
exo cyclization and coupling with iodine radical from NIS, to give product
45.
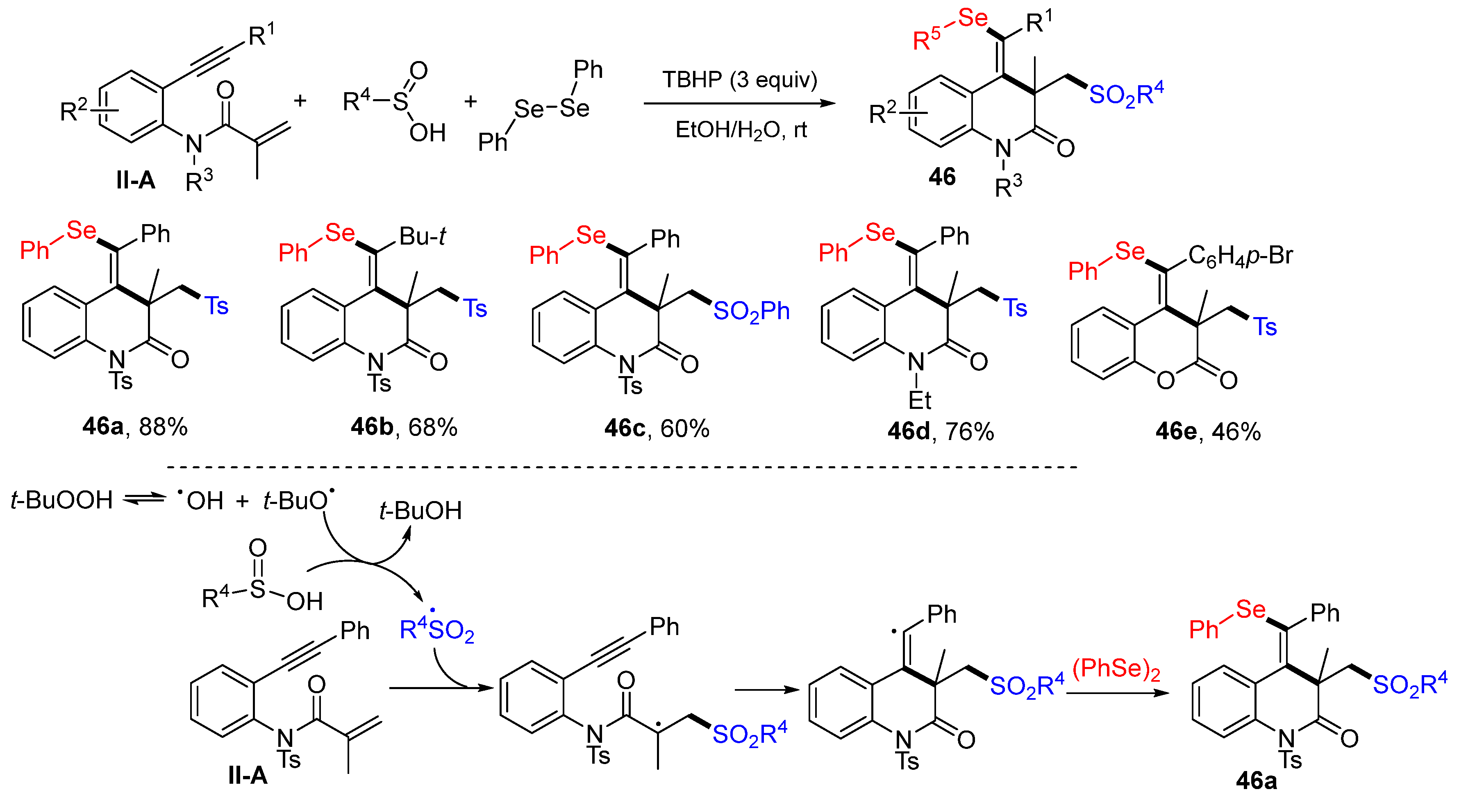
A new method for the synthesis of 3,4-dihydroquinolin-2(1
H)-ones was reported by the Guo group in 2017. The reaction of benzene-tethered 1,7-enynyl amides, sulfinic acids and diphenyl diselenides in EtOH-H
2O and in the presence of TBHB to give product
46 in moderate-to-excellent yields (
Scheme 47) [
59]. Carrying out the reaction under micro flow conditions could reduce the reaction time to less than 1 min. The reaction mechanism suggests that the sulfonyl radical, produced from the arylsulfinic acid with the oxidation of TBHP, adds to the C=C double bond of amide followed by 6-
exo cyclization and coupling with phenylselenyl radical to give product
46a.
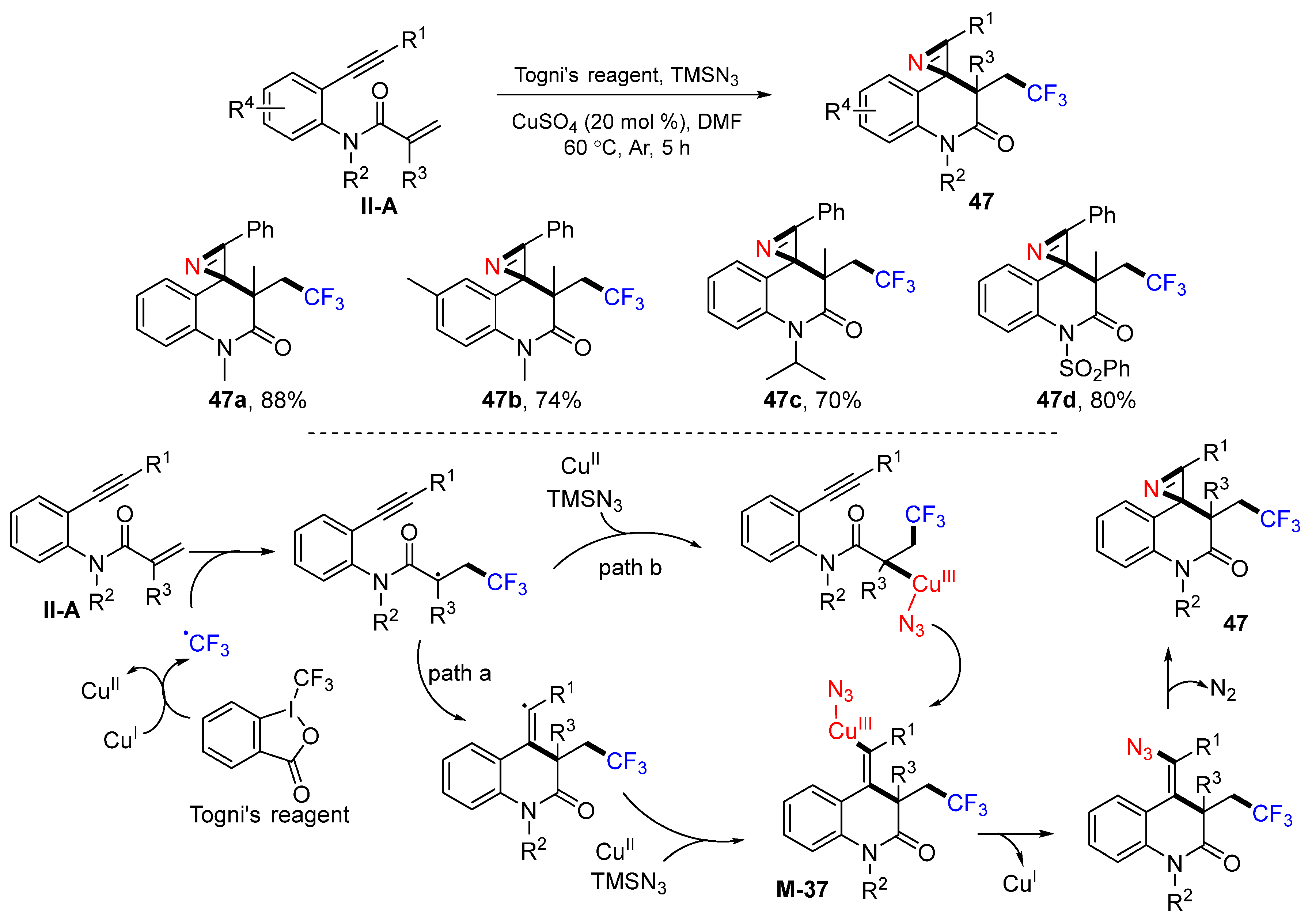
A Cu-catalyzed radical trifluoromethylative spirocyclization reaction of benzene-tethered 1,7-enynyl amides for the synthesis of trifluoromethyl-substituted 1′
H-spiro-[azirine-2,4′-quinolin]-2′(3′
H)-ones was introduced by the Han group in 2017. The reaction of amides, Togni’s reagent and TMSN
3 in DMF and in the presence of Cu
II catalyst gave product
47 in good-to-excellent yields (
Scheme 48) [
60]. The reaction mechanism suggests that the CF
3 radical from Togni’s reagent adds to the C=C double bond of amides; then, it goes through path a or b to give cyclized Cu
III-azido complex
M-37, followed by reductive catalyst elimination and denitrogenative cyclization to give product
47.
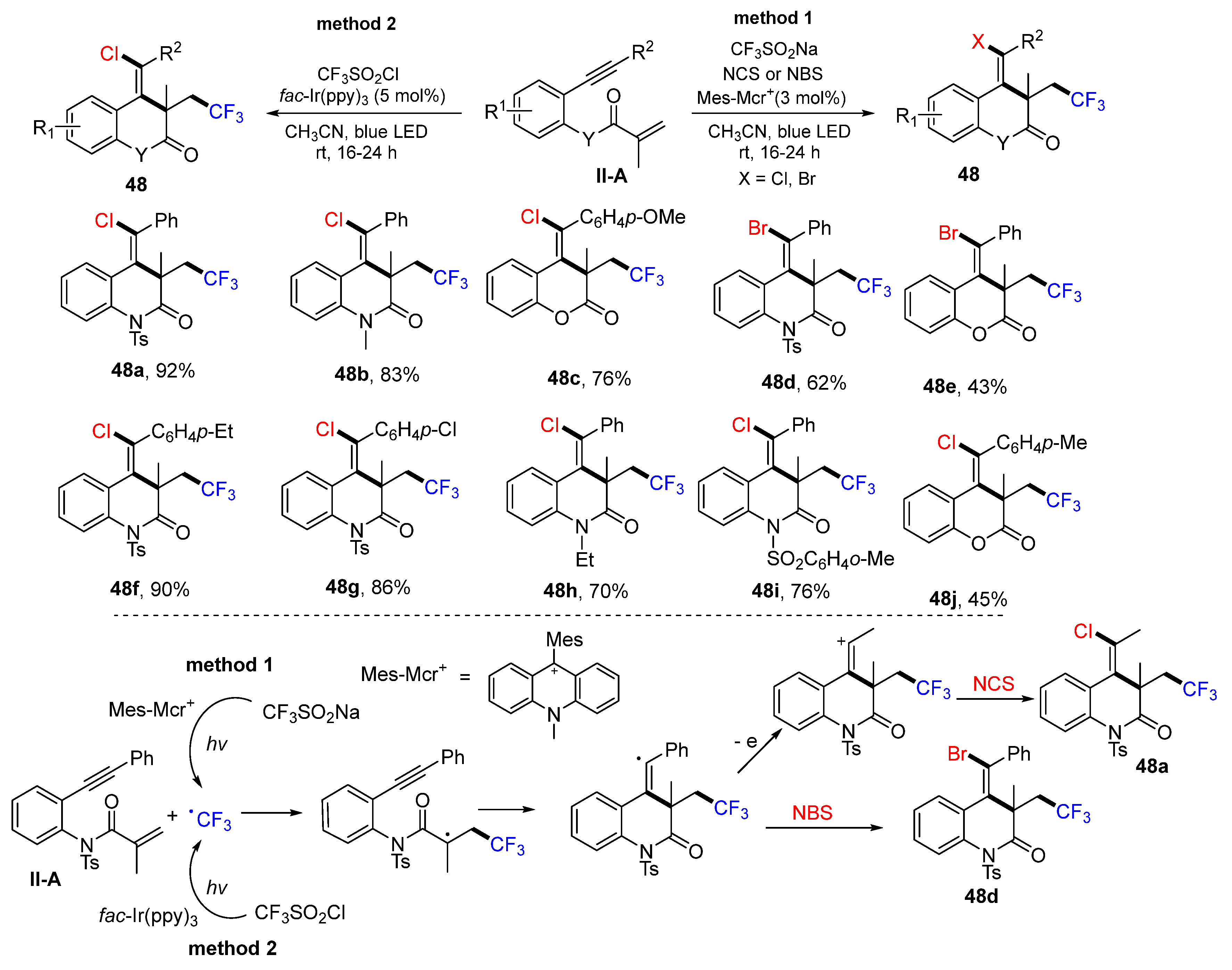
The Guo group, in 2019, reported two photoredox methods for the synthesis of trifluoroethyl-substituted 3,4-dihydroquinolin-2(
1H)-ones. Method 1 is the reaction of 1,7-enynyl amides, CF
3SO
2Na, NCS (or NBS) using photocatalyst
N-methyl-9-mesityl acridinium (Mes-Acr
+). Method 2 is the reaction of 1,7-enynyl amides and CF
3SO
2Cl using photocatalyst
fac-Ir(ppy)
3. These two methods gave product
48 in moderate-to-excellent yields (
Scheme 49) [
61]. The proposed reaction mechanism indicated that for method 1, the CF
3 radical generated from the CF
3SO
2Na under the photocatalysis of Mes-Acr
+ adds to the C=C bond of amide followed by 6-
exo cyclization and coupling with bromo radical from NBS to give product
48d. In method 2, the CF
3 radical generated from the CF
3SO
2Cl under the photocatalysis of
fac-Ir(ppy)
3 goes through similar addition, cyclization and halogen atom abstraction processes to afford product
48a.
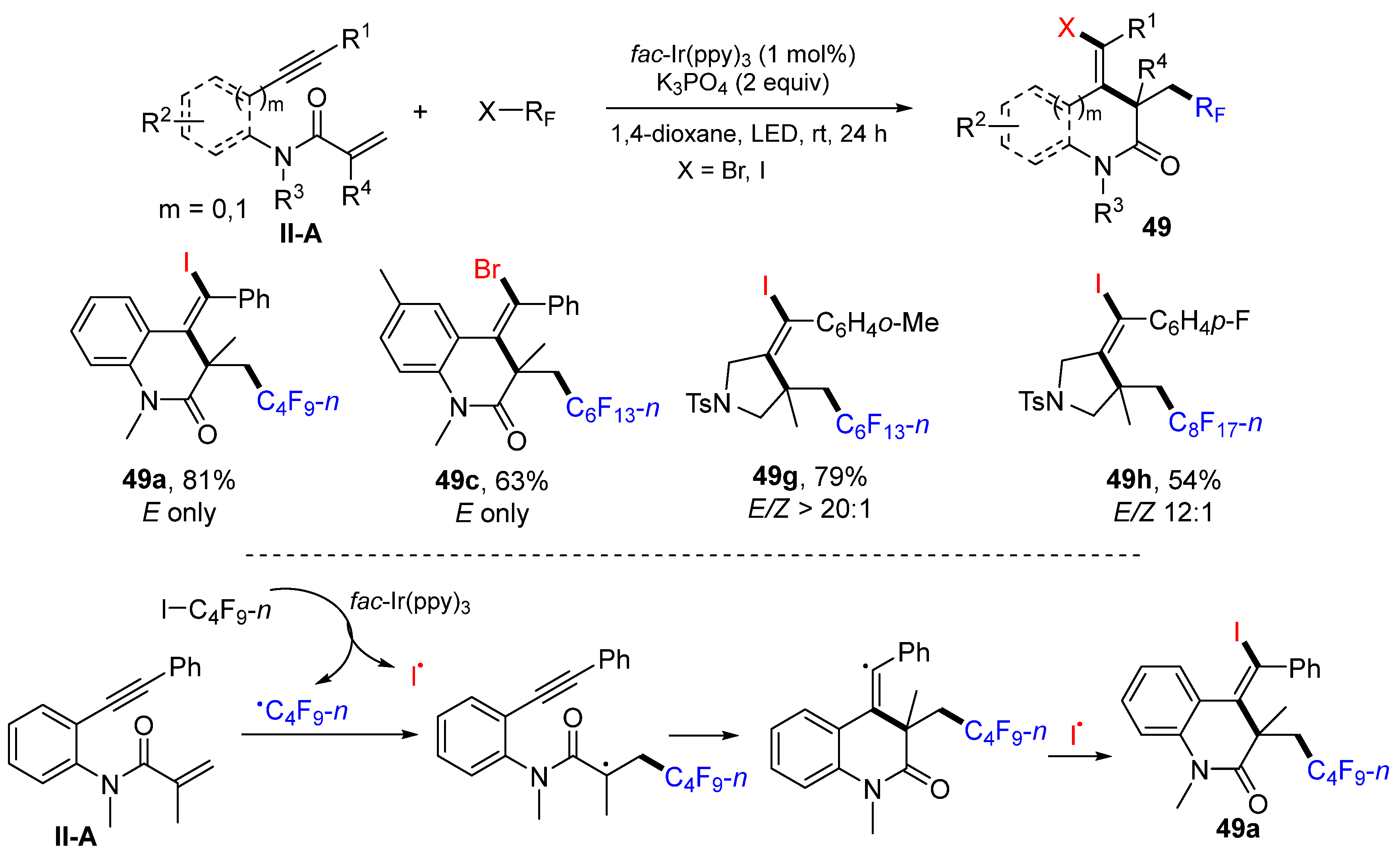
A visible light-induced radical reaction for the synthesis of haloperfluorinated
N-heterocycles was reported by the Tang group in 2019. The reaction of 1,6- or 1,7-enynyl amides, perfluoroalkyl iodides/bromides in 1,4-dioxane and in the presence of
fac-Ir(ppy)
3 and K
3PO
4 under blue LED irradiation afforded product
49 in good yields and stereoselectivity (
Scheme 50) [
62]. The reaction mechanism suggests that
n-C
4F
9 radical generated under the photocatalysis with of
fac-Ir(ppy)
3 adds to the C=C bond of amide, followed by 6-
exo cyclization and coupling with iodine radical, to selectively give product
49a as the
Z-isomer.
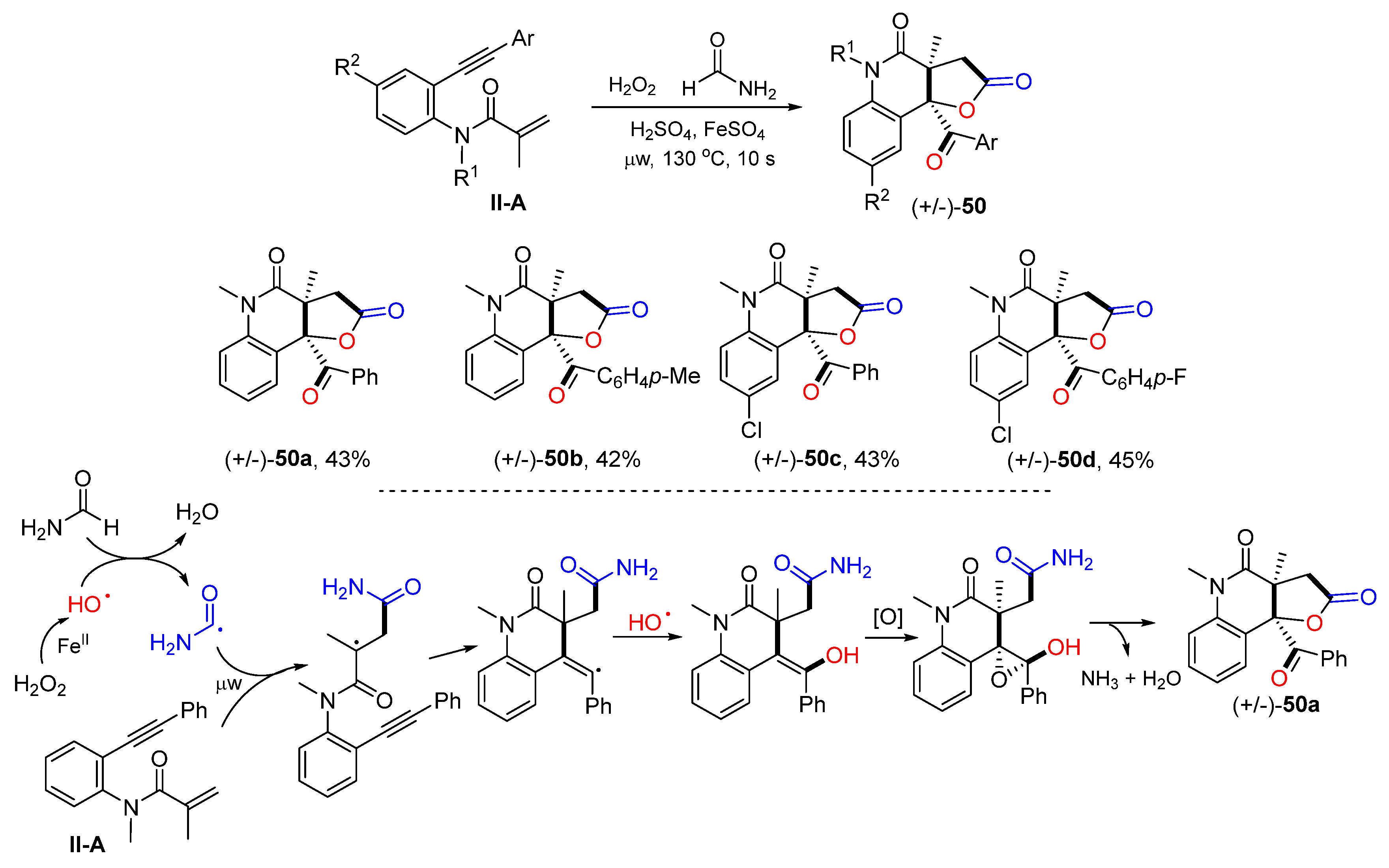
The Andrade group reported an ultrafast Fe-promoted reaction for the synthesis of 2-quinolinone-fused
γ-lactones in 2021. The reaction of benzene-tethered 1,7-enynyl amides and formamide and Fenton’s reagent under microwave irradiation for 10 s gave product
50 in a good overall yield (
Scheme 51) [
63]. The reaction mechanism suggests that the hydroxyl radical generated from Fenton’s reaction adds to the C=C double bond of amide followed by 6-
exo cyclization, coupling with hydroxyl radical, epoxidation, and lactonization to give product
50a.
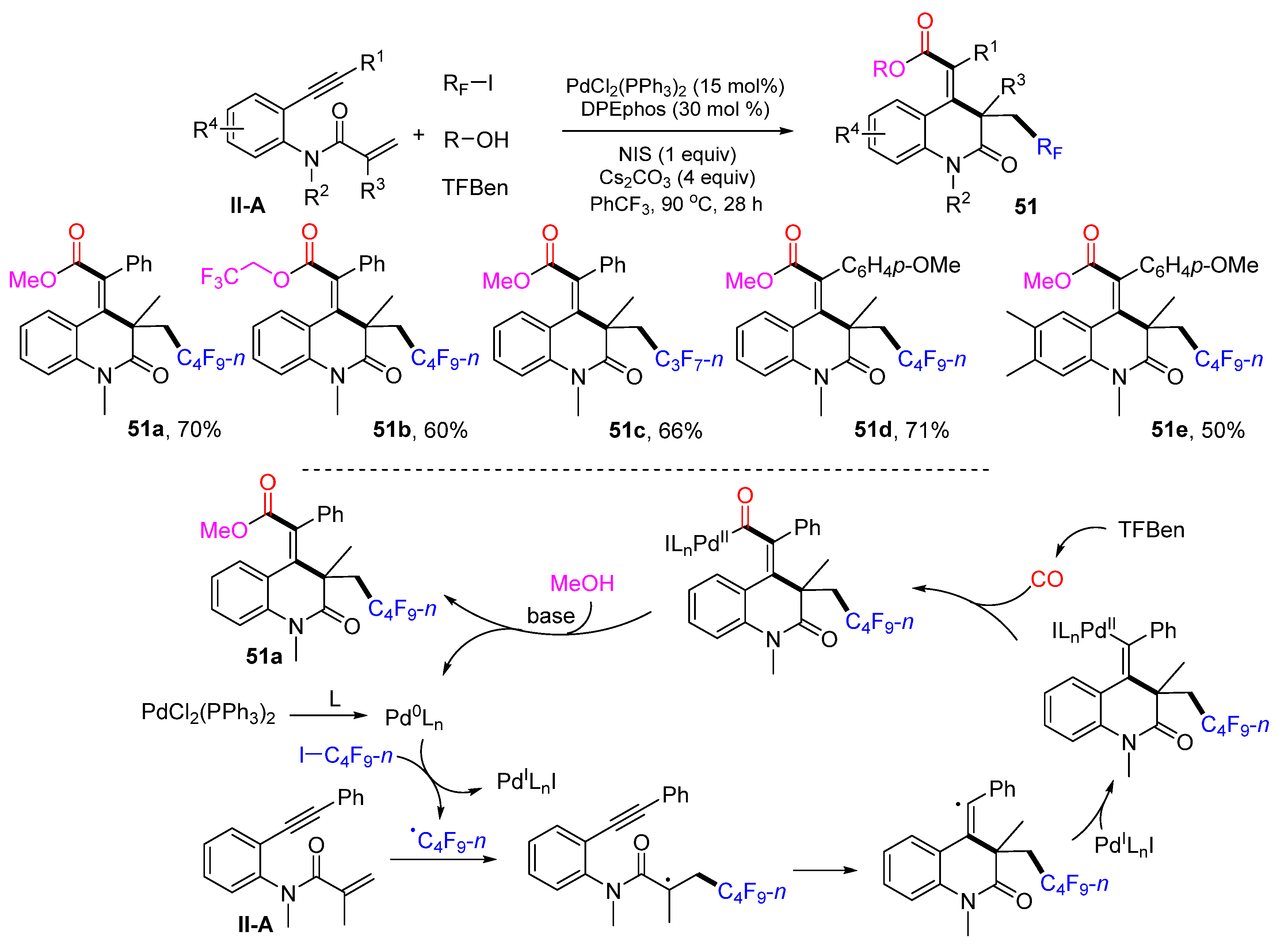
In 2022, Wu, Ying and their coworkers introduced a Pd-catalyzed reaction for the synthesis of perfluoroalkyl and carbonylated 3,4-dihydroquinolin-2(1
H)-ones. The reaction of 1,7-enynyl amides, perfluoroalkyl iodides, alcohols and benzene-1,3,5-triyl triformate (TFBen) in PhCF
3 and in the presence of PdCl
2(Ph
3P)
2, DPEphos, NIS, and Cs
2CO
3 gave product
51 in high yields with excellent
E/Z selectivity (
Scheme 52) [
64]. In this reaction, TFBen was used as the CO source and alcohols when making the ester products. A reaction mechanism suggests that the
n-C
4F
9 radical derived from
n-C
4F
9I adds to the C=C double bond of amide followed by 6-
exo cyclization, incorporation with the Pd-catalyst, CO insertion, and esterification with MeOH to afford product
51a.
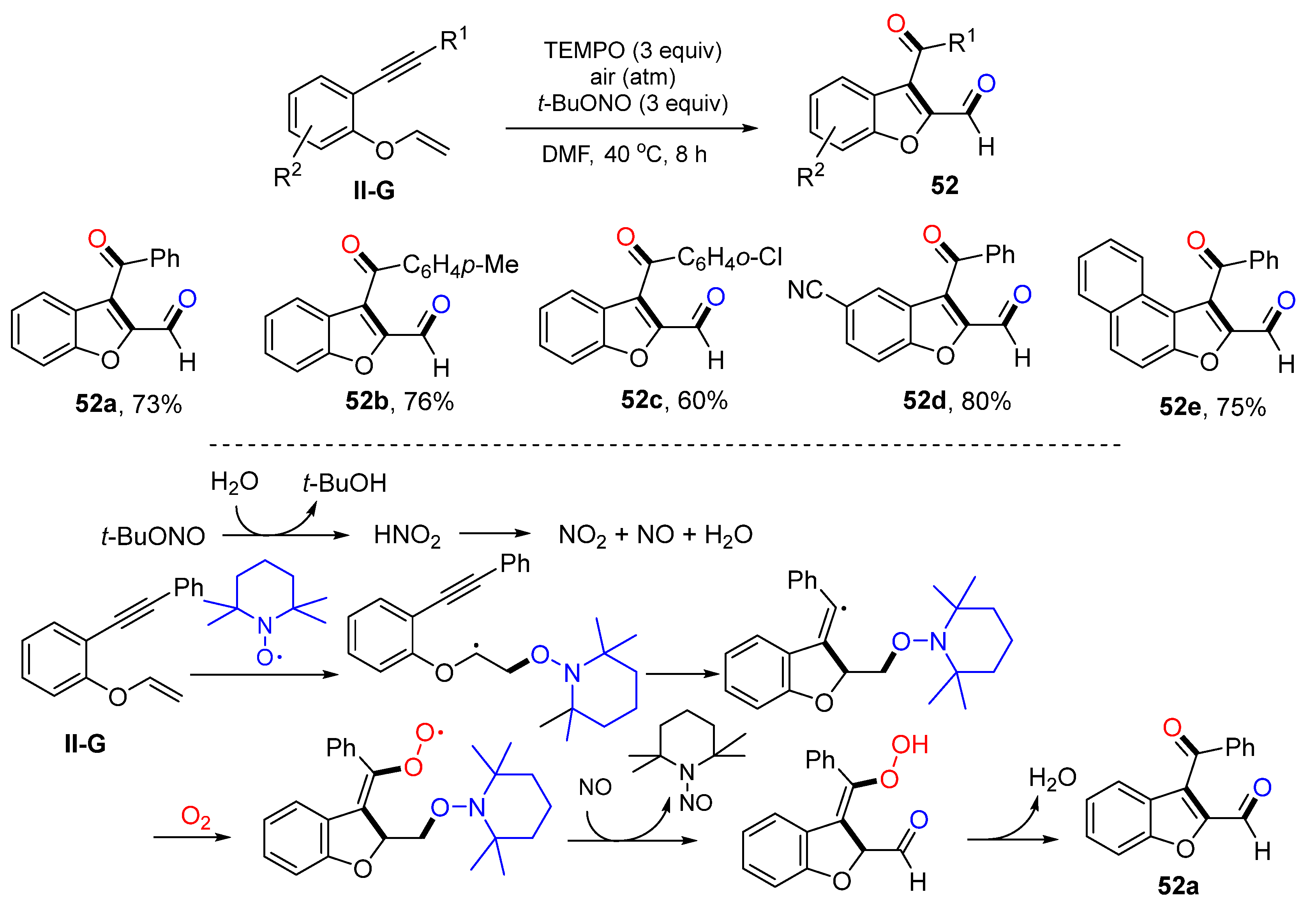
Benzene-linked 1,6-eneynyl ethers are a class of good substrates for radical difunctionalization. Li and coworkers reported a reaction of such substrates for the synthesis of dicarbonylated benzofurans in 2015. The reaction of benzene-linked 1,6-eneynyl ethers, 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO),
t-BuONO and O
2 in DMF at 40 °C for 8 h gave product
52 in moderate-to-good yields (
Scheme 53) [
65]. Two oxygen atoms were introduced to the product from O
2 and TEMPO, respectively.
t-BuONO is a key reagent which provides NO
2 and NO after decomposition of HNO
2. The reaction mechanism suggests that the addition of TEMPO to the C=C double bond of ethers followed by 5-
exo cyclization, trapping of O
2, oxidative cleavage of the N-O bond to release 2,6,6-tetramethyl-1-nitroso-piperidine, and O-O bond cleavage/isomerization to afford product
52a.
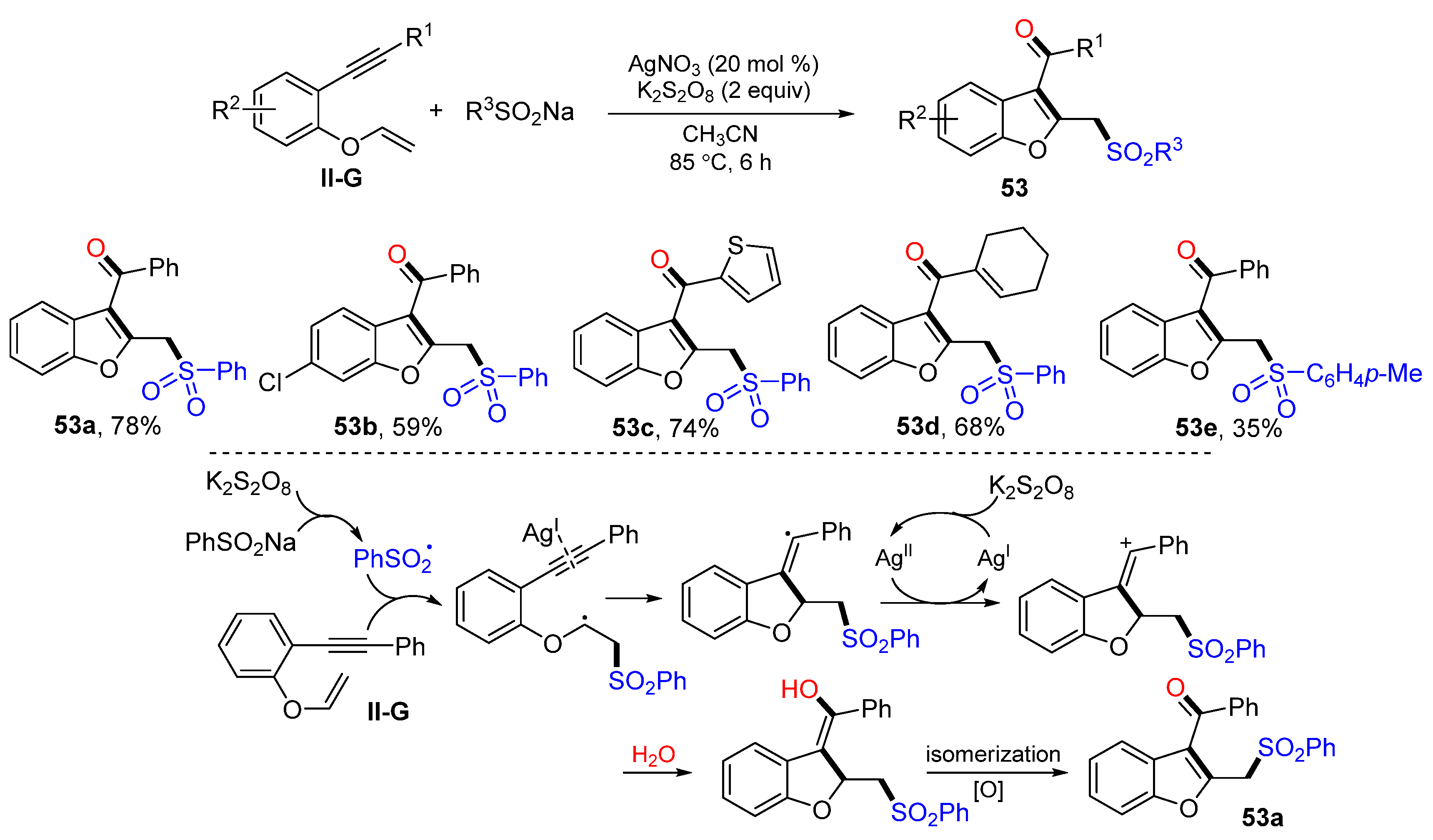
An Ag-catalyzed reaction of 1,6-eneynyl ethers for the synthesis of sulfonyl-methylated benzofurans was reported by Wu, Jiang and their coworkers in 2017. The reaction of benzene-linked 1,6-eneynyl ethers and sodium sulfinates in CH
3CN and in the presence of K
2S
2O
8 and AgNO
3 afforded product
53 in moderate-to-good yields (
Scheme 54) [
66]. The reaction mechanism suggests that the sulfonyl radical generated from the oxidation of PhSO
2Na adds to the C=C double bond of ethers followed by 5-
exo cyclization, oxidation to cation, nucleophilic addition of H
2O, and enol/ketone isomerization to give product
53a.
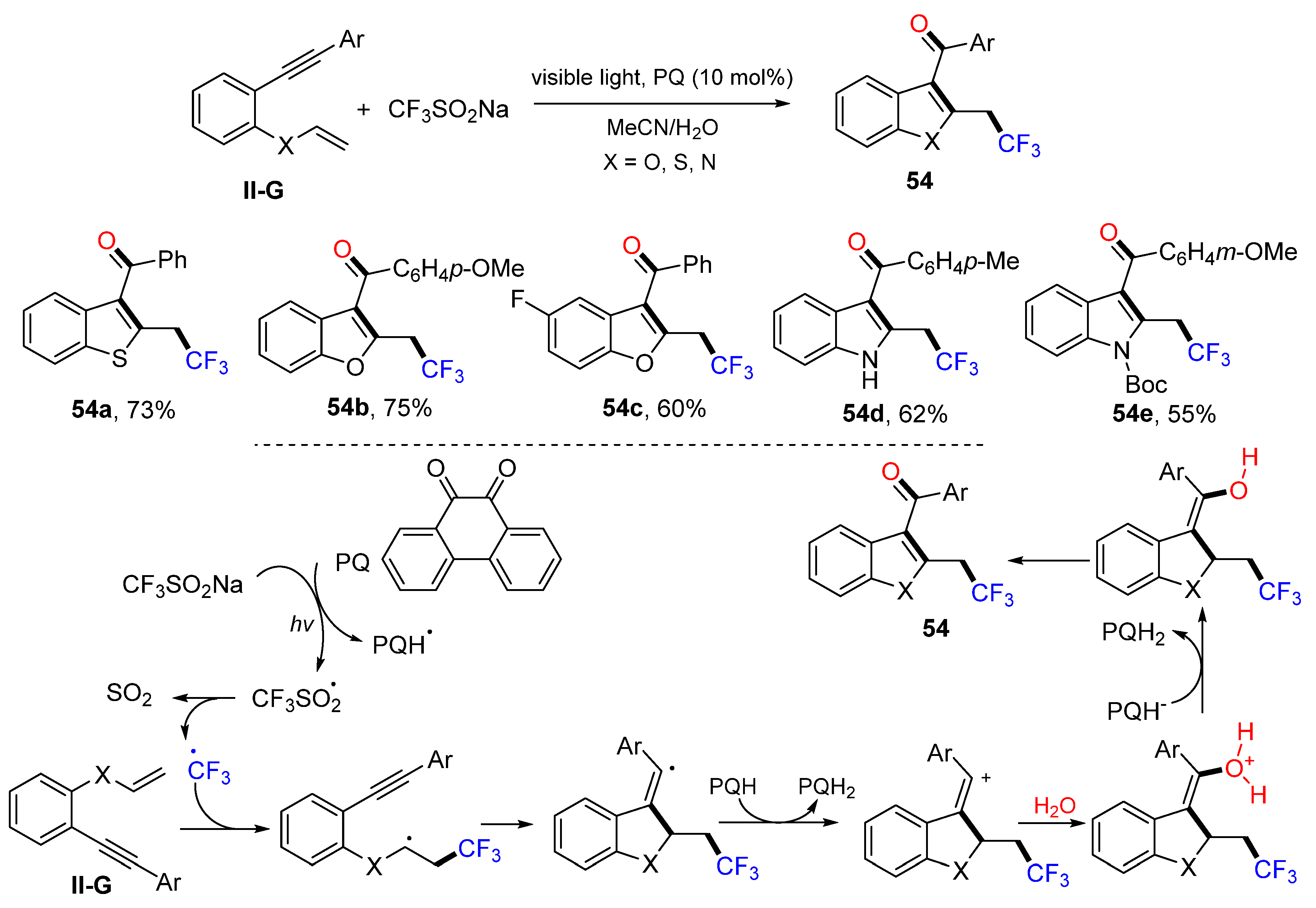
In 2017, Kumar and coworkers reported a visible light-induced reaction for the synthesis of trifluoromethylacylated benzofurans, benzothiophenes, and indoles. The reaction of 1-ethynyl-2-(vinyloxy)-benzenes and CF
3SO
2Na in CH
3CN/H
2O using phenanthrene-9,10-dione (PQ) as a photoredox catalyst gave heterocycles
54 in good yields (
Scheme 55) [
67]. The proposed reaction mechanism suggests that the CF
3 radical, generated from CF
3SO
2Na with photo-activated PQ, adds to the C=C double bond of 1-ethynyl-2-(vinyloxy)-benzenes followed by 5-
exo cyclization, electron transfer from PQH radical, H
2O addition and deprotonation, resulting in product
54.
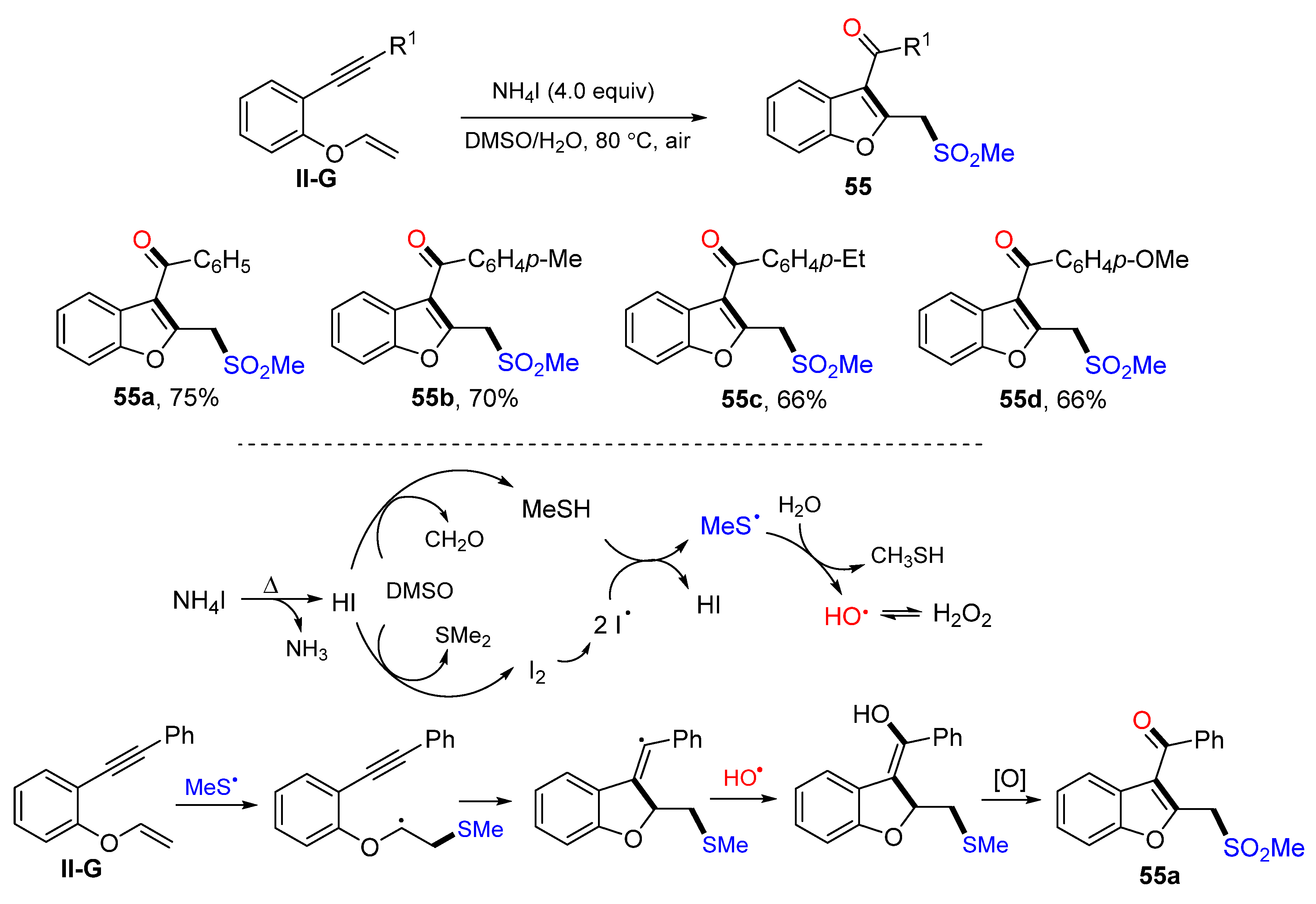
A reaction of 1,6-eneynyl ethers for the synthesis of sulfonylacylated benzofurans was introduced by the Sun group in 2018. The reaction of oxygen-linked 1,6-enynes, DMSO and H
2O in the presence of NH
4I afforded product
55 in moderate-to-high yields (
Scheme 56) [
68]. A reaction mechanism suggests that the reaction between DMSO and NH
4I produced MeS and OH radicals. Addition of MeS radical to the C=C double bond of ethers followed by 5-
exo cyclization, OH radical coupling, axidation of sulfide, and keto-enol tautomerism resulted in product
55a.
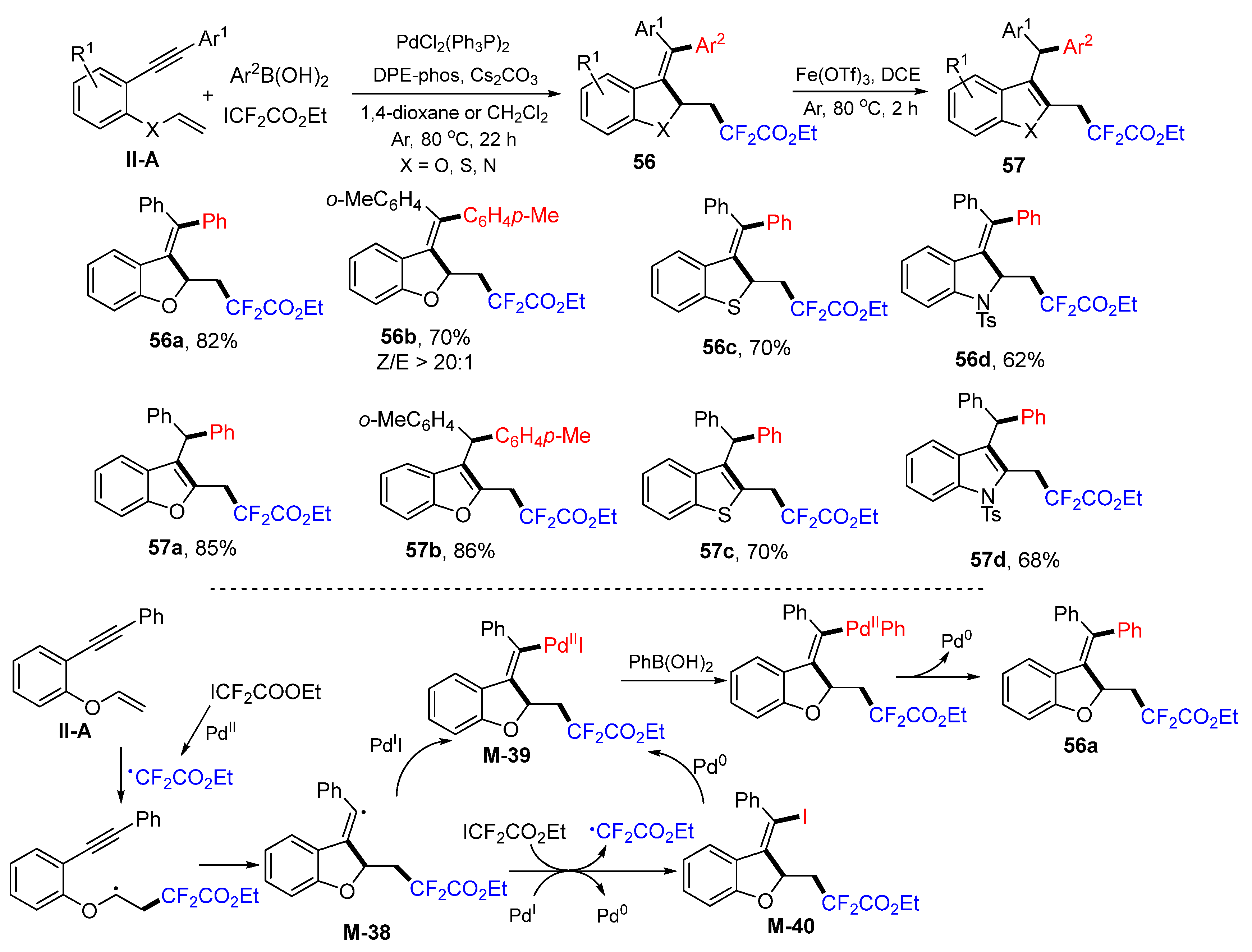
In 2020, the Zhang group introduced a Pd-catalyzed radical oxidative aryldifluoroalkylation of benzene-tethered 1,6-enynes for the synthesis of difluoroalkylated benzofuran, benzothiophene, and indole derivatives. The reaction of 1,6-enynes, ethyl difluoroiodoacetate and arylboronic acids 1,4-dioxane or DCE under the catalysis of PdCl
2(PhP
3)
2 and DPE-phos gave product
56 in moderate-to-good yields (
Scheme 57) [
69]. The resultant products can be converted into aromatic five-membered rings
57 via Fe(OTf)
3-catalyzed isomerization. A reaction mechanism suggests that the CF
2CO
2Et radical generated from ICF
2CO
2Et adds to the C=C double bond of 1,6-enyne followed by 5-
exo cyclization to form
M-38 and then reacts with Pd
II to form intermediate
M-39. Intermediate
M-39 could also be generated from
M-38 through iodine transfer with ICF
2CO
2Et and then with Pd
0. Coupling
M-39 with phenylboronic acid finishes the reaction and gives product
56a.
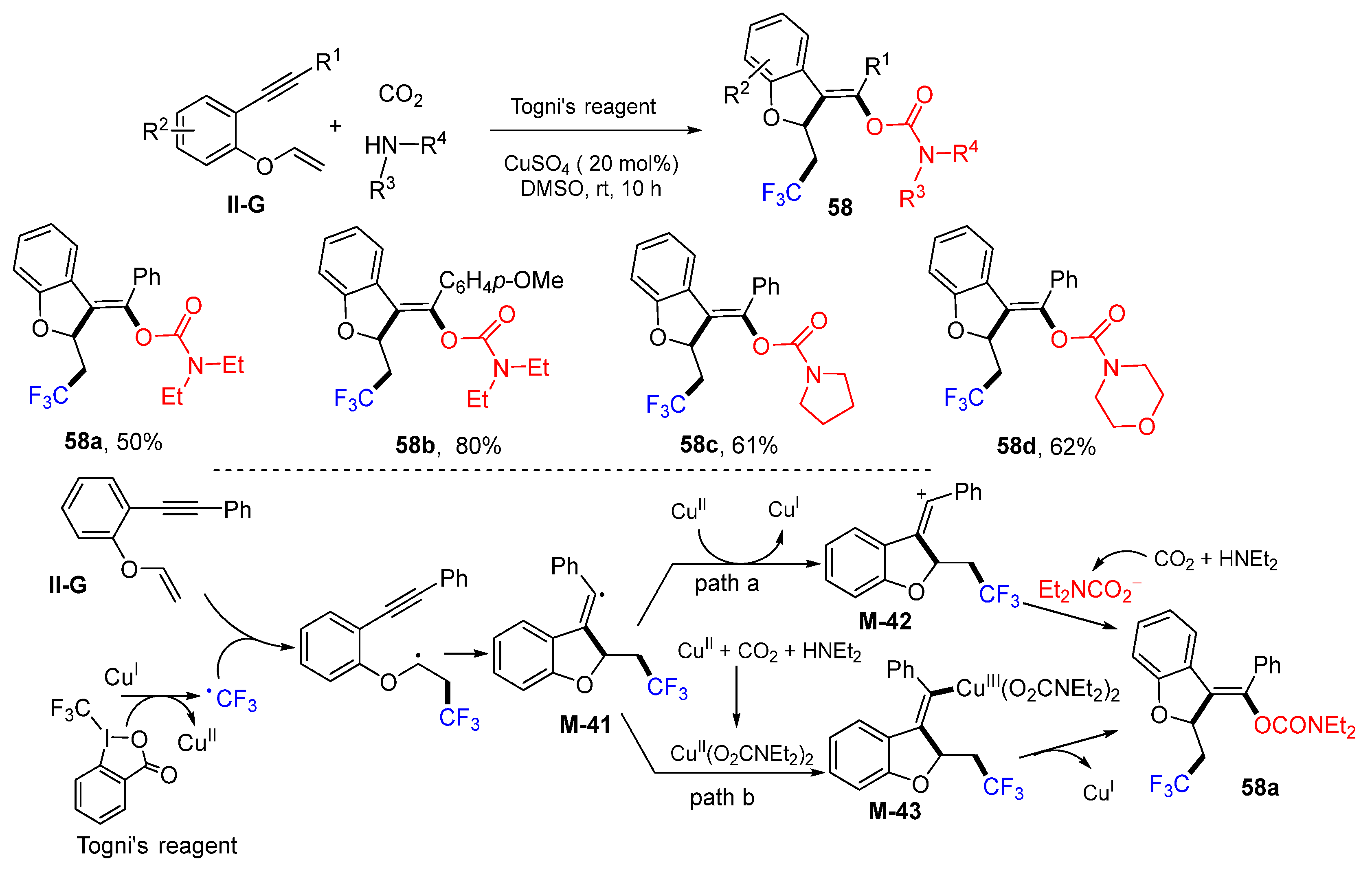
A Cu-catalyzed radical reaction of benzene-tethered 1,6-enynes for the synthesis of trifluoroethylated dihydrobenzofurans was reported by the Jiang group in 2019. The reaction of 1,6-enynes, Togni’s reagent, CO
2 and amines in DMSO under the catalysis of CuSO
4 gave products
58 in good yields (
Scheme 58) [
70]. The proposed reaction mechanism suggests that the CF
3 radical derived from the Togni’s reagent adds to 1,6-enynes followed by
5-exo cyclization to form radical
M-41. Then, it might have two pathways to form product
58a. In path a, vinyl radical
M-41 is oxidized by Cu
II to a cation
M-42, followed by trapping with carbamate anion to form
58a. Alternatively, in path b, vinyl radical
M-41 reacts with CuSO
4, CO
2, and amine to form carbamato complex
M-43, which leads to the formation of product
58a after reductive elimination of the catalyst.
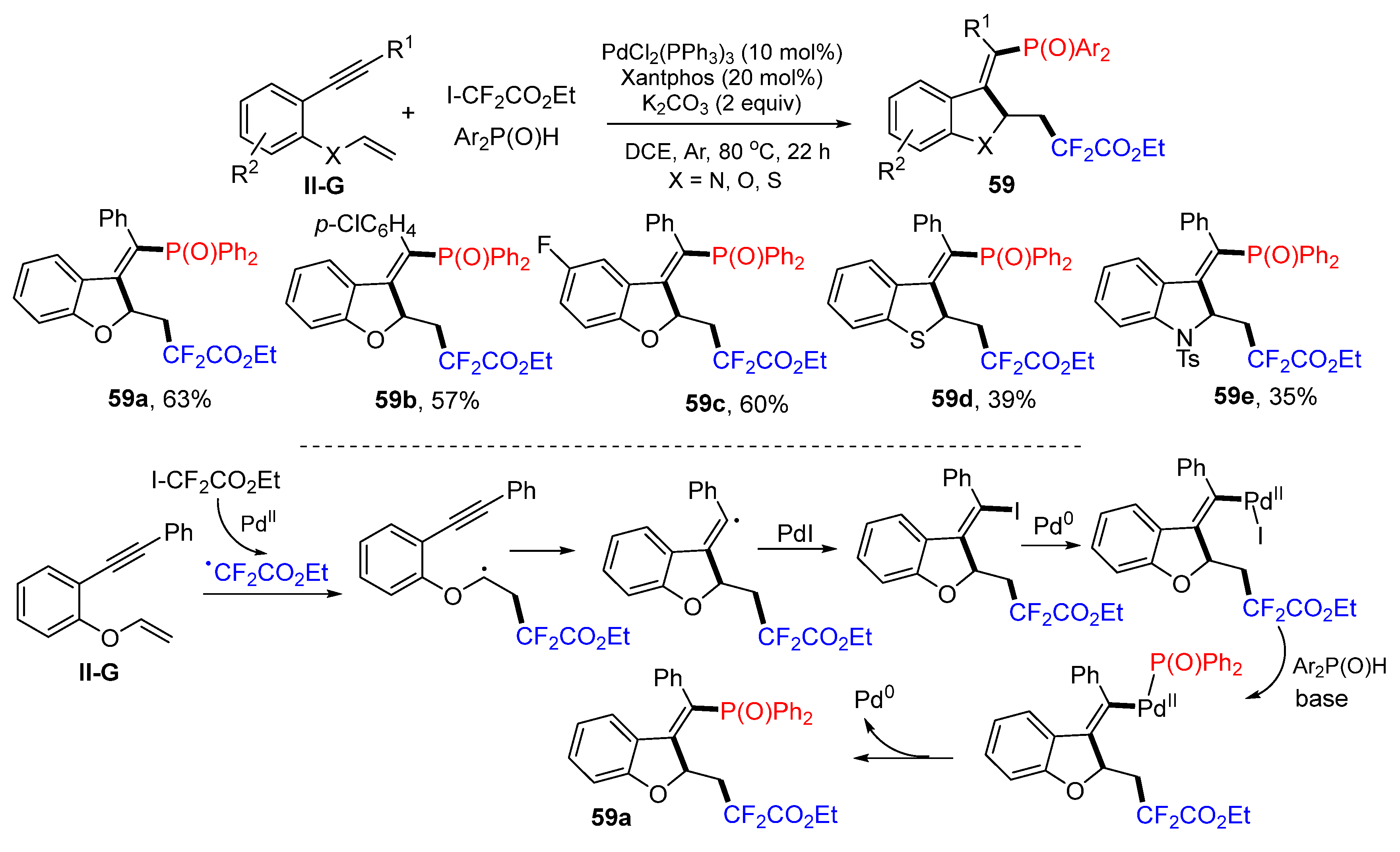
Gao, Ying and their coworkers reported a Pd-induced radical reaction for the synthesis of difluoroalkyl- and alkenylphosphinyl-functionalized heterocycles in 2021. The reaction of 2-vinyloxy arylalkynes, ICF
2CO
2Et and diphenylphosphine oxides in DCE under the catalysis of PdCl
2(PPh
3)
2 and Xantphos gave product
59 in good yields and stereoselectivity (
Scheme 59) [
71]. A reaction mechanism suggests that the CF
2CO
2Et radical derived from ICF
2CO
2Et under the catalysis of Pd
II adds to the C=C double bond of 2-vinyloxy arylalkynes followed by 5-
exo cyclization and iodine atom transfer from PdI, through the oxidative addition of Pd
0 to vinyl iodide, formation of diphenylphosphine oxide complex, reductive elimination of Pd catalyst to give product
59a.
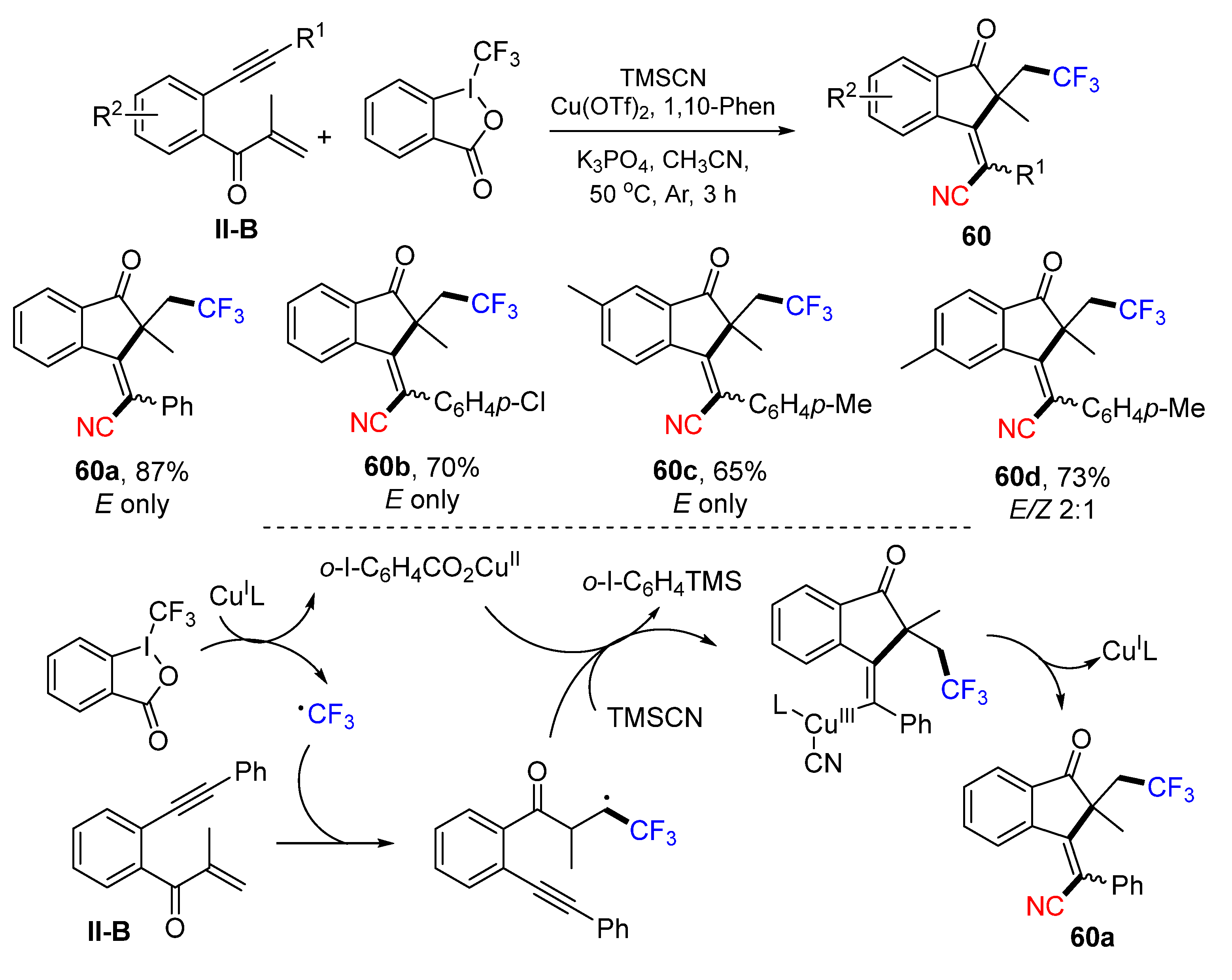
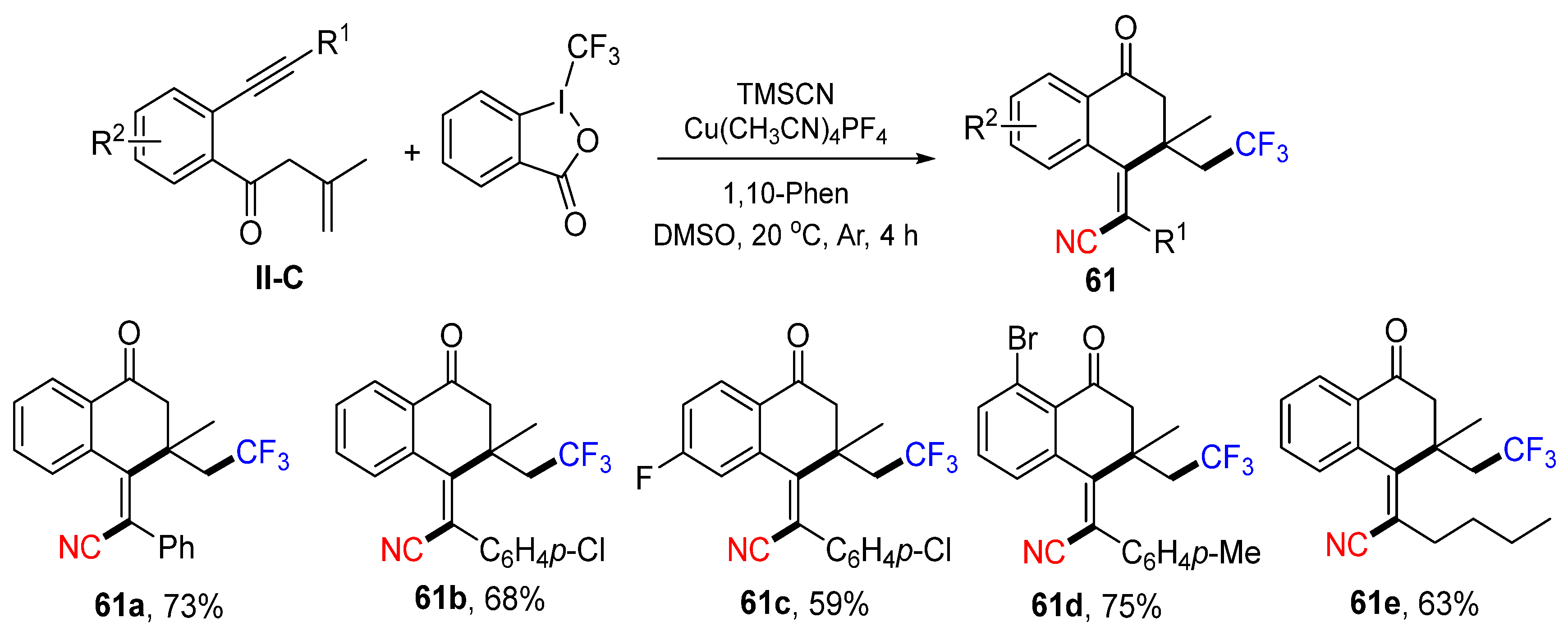
Using benzene-tethered and carbonyl-containing 1,6-enynes as a substrate for Cu-catalyzed radical reaction for the construction of cyanotrifluoromethylated 1-indanones was introduced by the Jiang group in 2020. The reaction of benzene-tethered 1,6-enynes, Togni’s reagent and trimethylsilyl cyanide (TMSCN) under the catalysis of Cu(OTf)
2 gave product
60 in good yields (
Scheme 60) [
72]. A reaction mechanism suggests that the trifluoromethyl radical generated from Togni’s reagent under the catalysis of Cu
II adds to the C=C double bond of 1,6-enyne followed by 5-
exo cyclization, formation of Cu
III-complex containing CN, and reductive elimination of the Cu-catalyst to give product
60a. By using benzene-tethered 1,7-enynes, the Jiang group extended the reaction scope for the synthesis of cyanotrifluoromethylated (
Z)-3,4-dihydronaphthalen-1(2
H)-ones
61 (
Scheme 61) [
73].
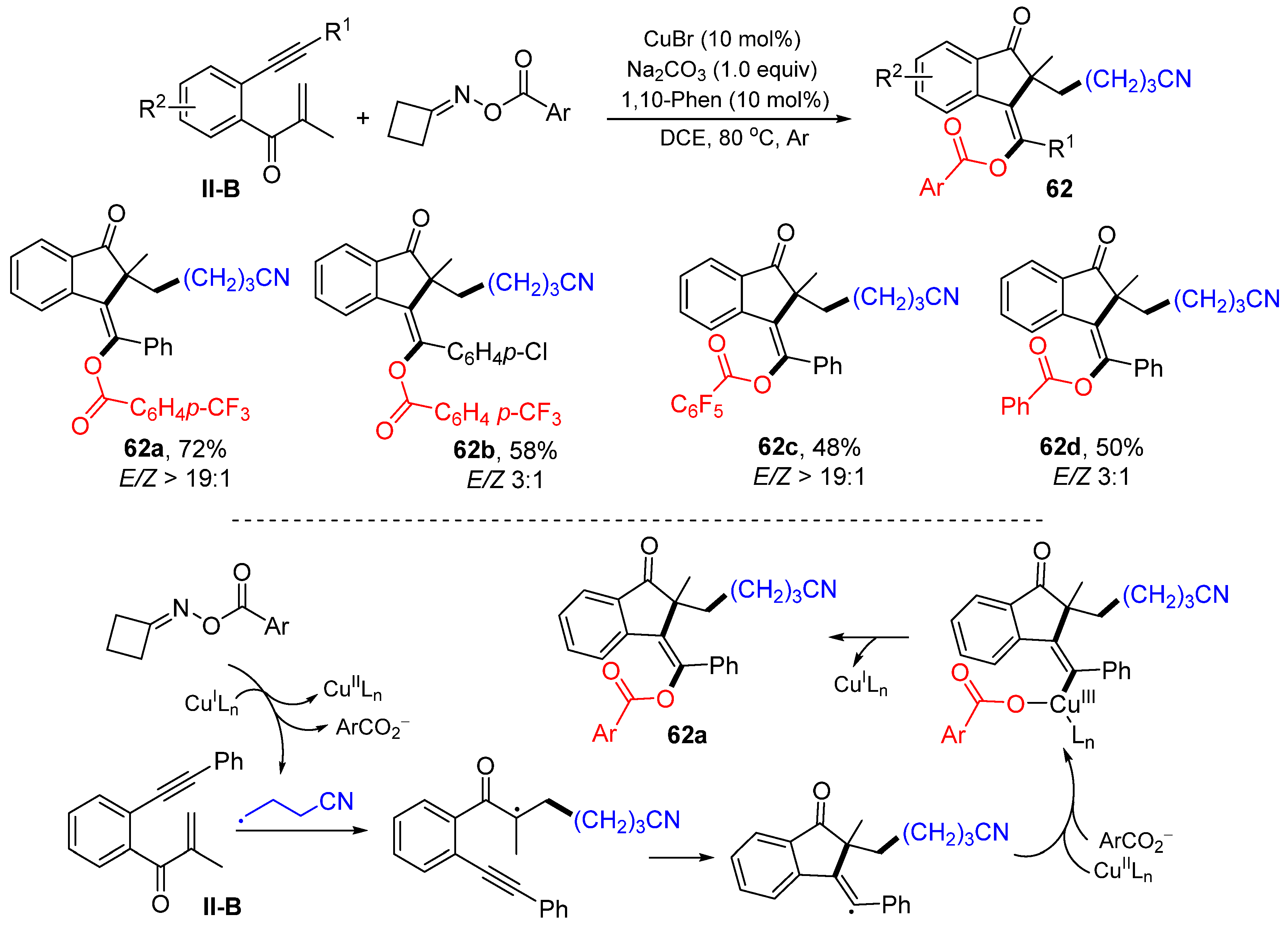
A Cu-catalyzed radical for the synthesis of cyanoalkyl and ester-functionalized 1-indanones was introduced by the Jiang group in 2021. The reaction of 1,6-enynes, cyclobutanone oxime esters in DCE at 80 °C under the catalysis of CuBr and1,10-Phen gave product
62 in good yields (
Scheme 62) [
74]. Both functional groups come from cyclic oxime esters. A reaction mechanism suggests that the γ-cyanoalkyl radical, generated from cyclic oxime ester via a SET process with Cu
IL
n, adds to the C=C double bond of 1,6-enyne followed by 5-
exo cyclization, formation of a Cu
III complex containing the ester group, and reductive elimination Cu
IL
n to give product
62a.
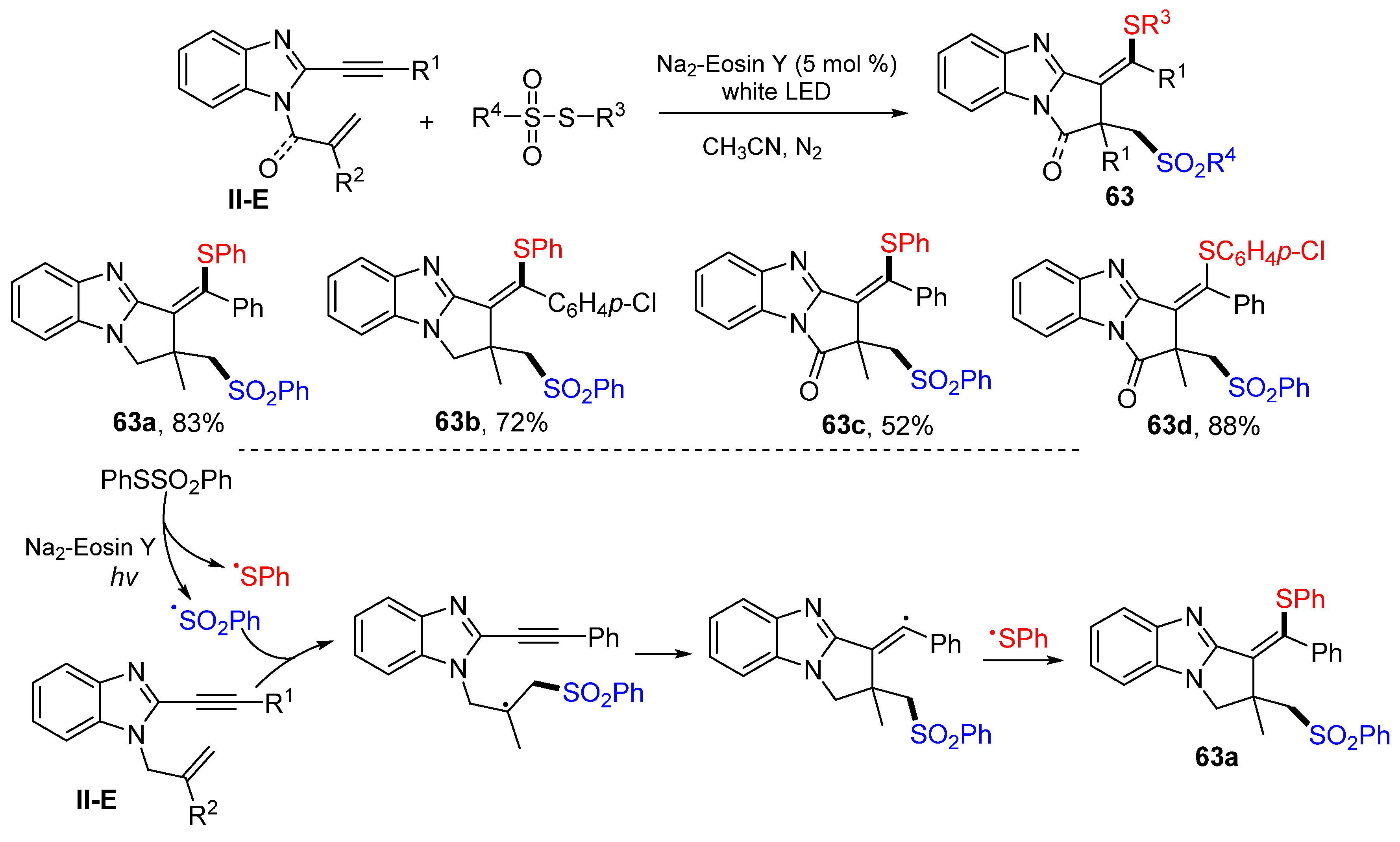
A visible light-induced radical reaction of benzene-tethered 1,6-enynes for the synthesis of the thiosulfonylated pyrrolo[1,2-
a]benzimidazoles was reported by the Chen group in 2021. The reaction of 1,6-enynes and PhSO
2SPh in CH
3CN under the photo catalysis of Na
2-Eosin Y gave
63 in moderate-to-good yields (
Scheme 63) [
75]. The reaction mechanism suggests that the sulfonyl radical derived from PhSO
2SPh adds to the C=C double bond of 1,6-enynes followed by 5-
exo cyclization and coupling with the SPh radical to afford product
63a.
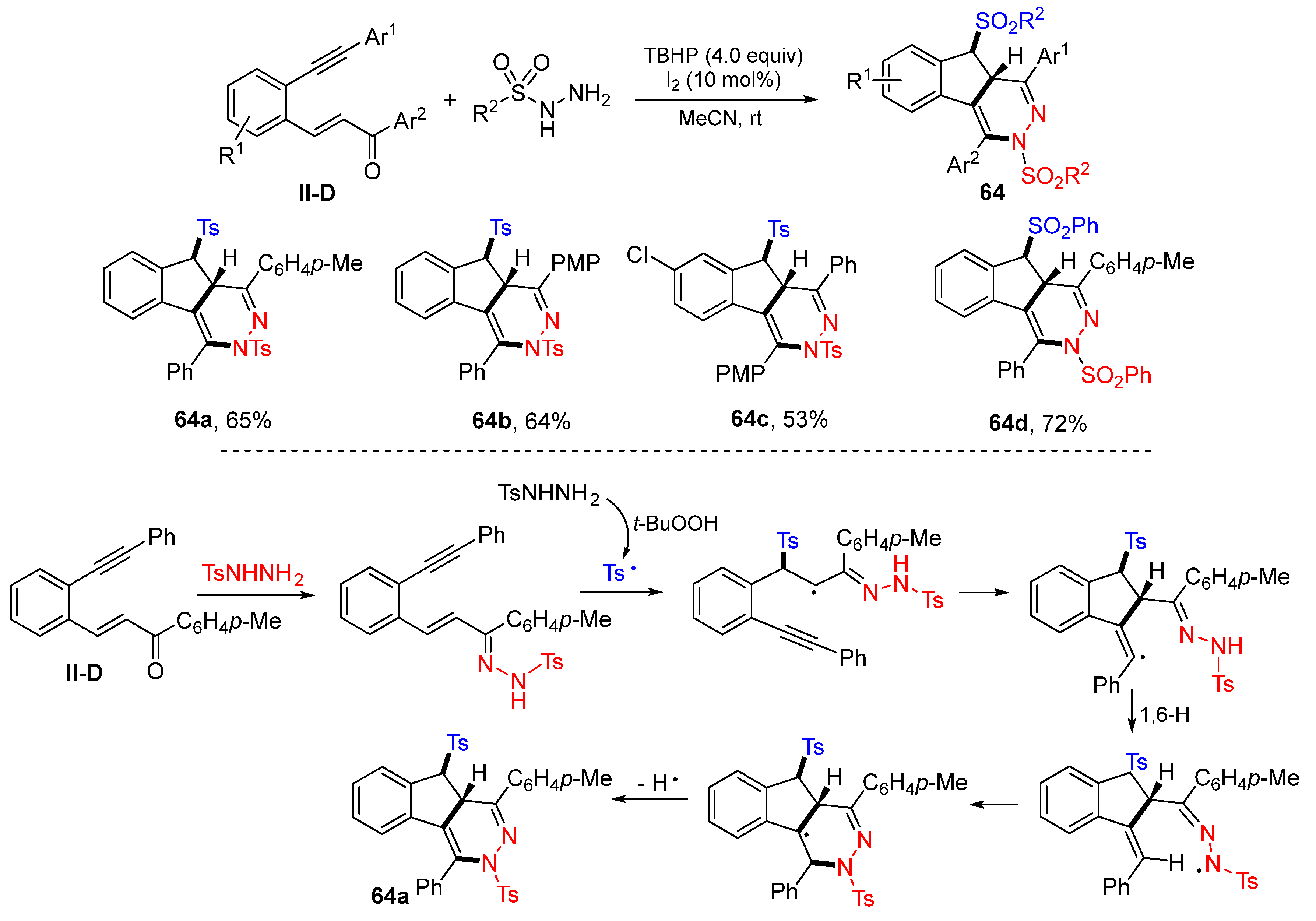
The Tu and Jiang groups, in 2016, reported a radical reaction of 1,5-enynes for the synthesis of sulfonylated indeno[1,2-
d]pyridazines. The reaction of 1,5-enynes, arylsulfonyl hydrazides in CH
3CN and in the presence of I
2 and TBHP gave products
64 in good yields (
Scheme 64) [
76]. A reaction mechanism suggests that sulfonylhydrazone, generated from the condensation of 1,5-enynes with the arylsulfonyl hydrazide, reacts with the tosyl radical, which is also derived from arylsulfonyl hydrazide followed by 5-
exo cyclization, 1,6-H atom transfer, 6-
endo cyclization of the N-radical, and aromatization to give product
64a.
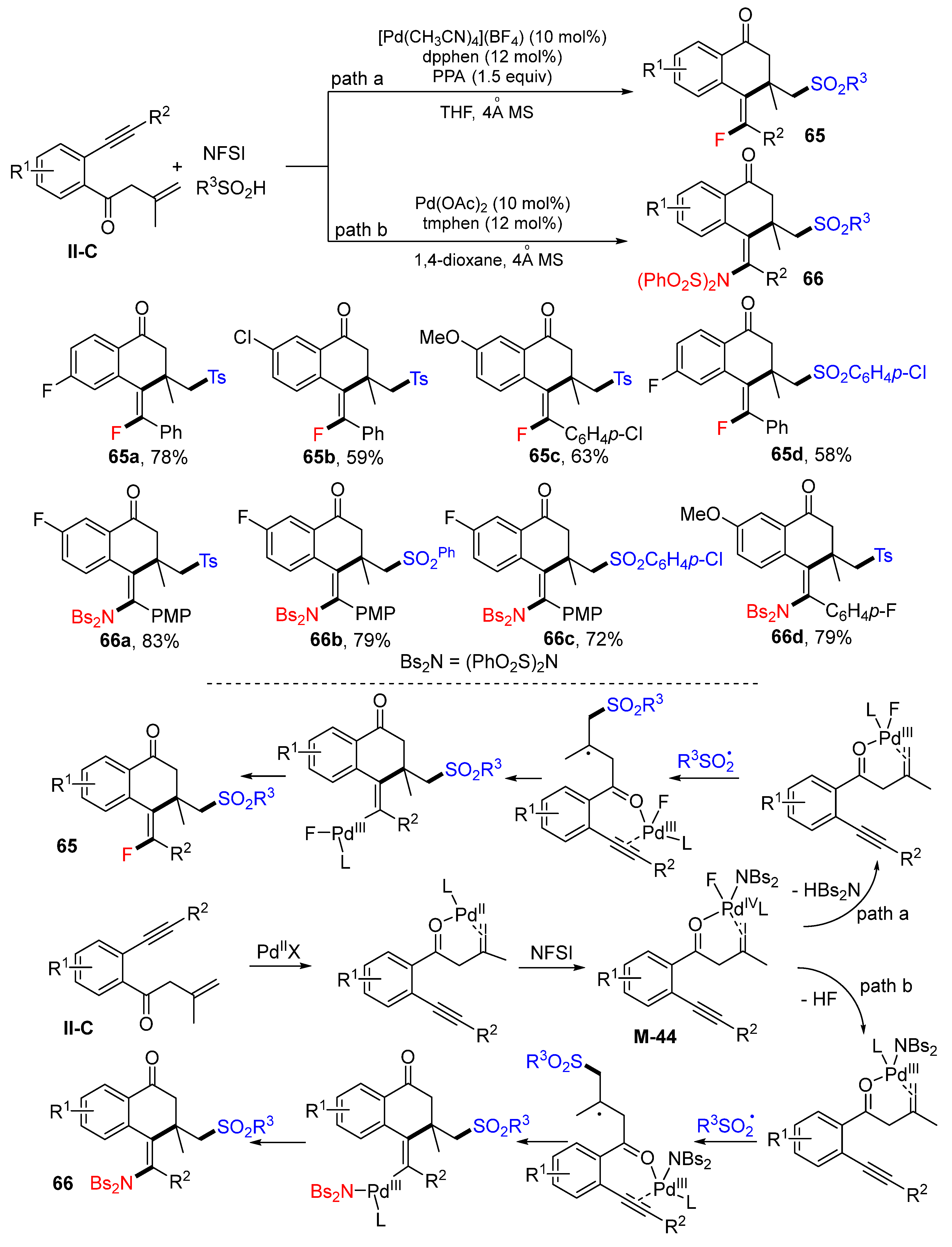
A Pd-catalyzed radical cyclization of 1,7-enynes for the synthesis of functionalized (
E)-3,4-dihydro-naphthalen-1(2
H)-ones was reported by Jiang, Tu and their coworkers in 2018. The reaction of 1,7-enynes, sulfinic acids and
N-fluorobenzenesulfonimide (NFSI) in THF under the catalysis of [Pd(CH
3CN)
4](BF
4)
2 gave
65 in good yields and high stereoselectivity (
Scheme 65) [
77]. A possible reaction mechanism suggests that 1,7-enynes generate a Pd
II complex which then reacts with NFSI to form Pd
IV complex
M-44 for following two pathways. Under the reaction conditions for path a, complex
M-44 eliminates HBs
2N, followed by the addition of R
3SO
2 radical, 6-
exo cyclization, and reductive elimination of Pd catalyst to give fluorosulfonated product
65. Under the reaction conditions for path b, HF is released from complex
M-44 followed by the similar reaction process of R
3SO
2 radical addition, 6-
exo cyclization, and reductive elimination of Pd catalyst to give benzenesulfonylated products
66.
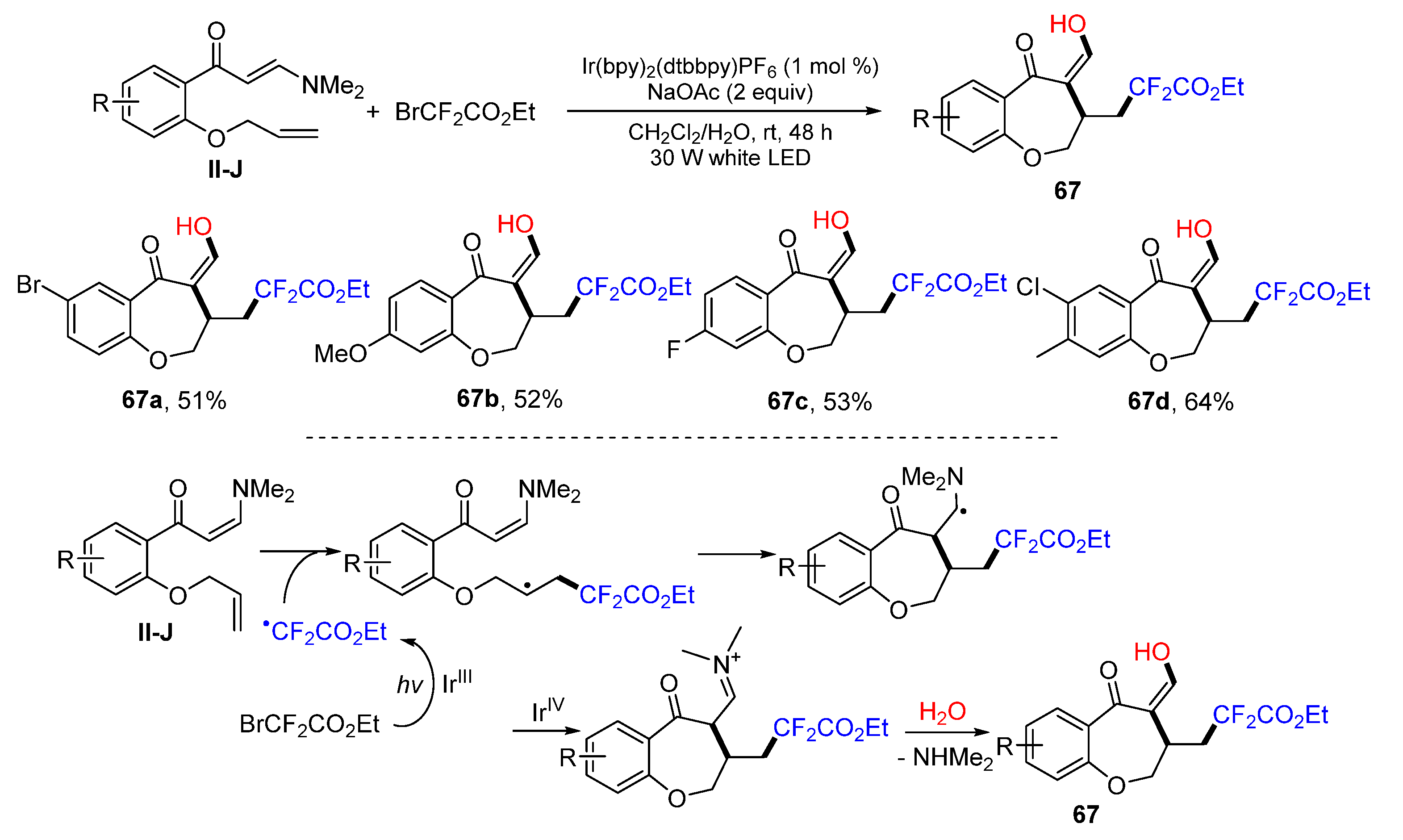
Using of benzene-tethered 1,8-dienes for Ir-catalyzed oxidative difluorinative radical cyclization for the preparation of enol and CF
2-containing benzoxepines was reported by the Yang group in 2018. The reaction of 1,8-dienes and BrCF
2CO
2Et in CH
2Cl
2/H
2O under the photoredox catalysis with Ir(dtbbpy)(bpy)
2PF
6 afforded benzoxepine product
67 in good yields (
Scheme 66) [
78]. A reaction mechanism suggests that the CF
2CO
2Et radical, generated from BrCF
2CO
2Et under the photocatalysis of Ir(dtbbpy)(bpy)
2PF
6, adds to the C=C double bond of 1,8-dienes followed by 7-
exo cyclization, the formation of an iminium ion through the oxidization of [Ir
IV(dtbbpy)(bpy)
2PF
6]
+, and iminium hydrolysis to give product
67.
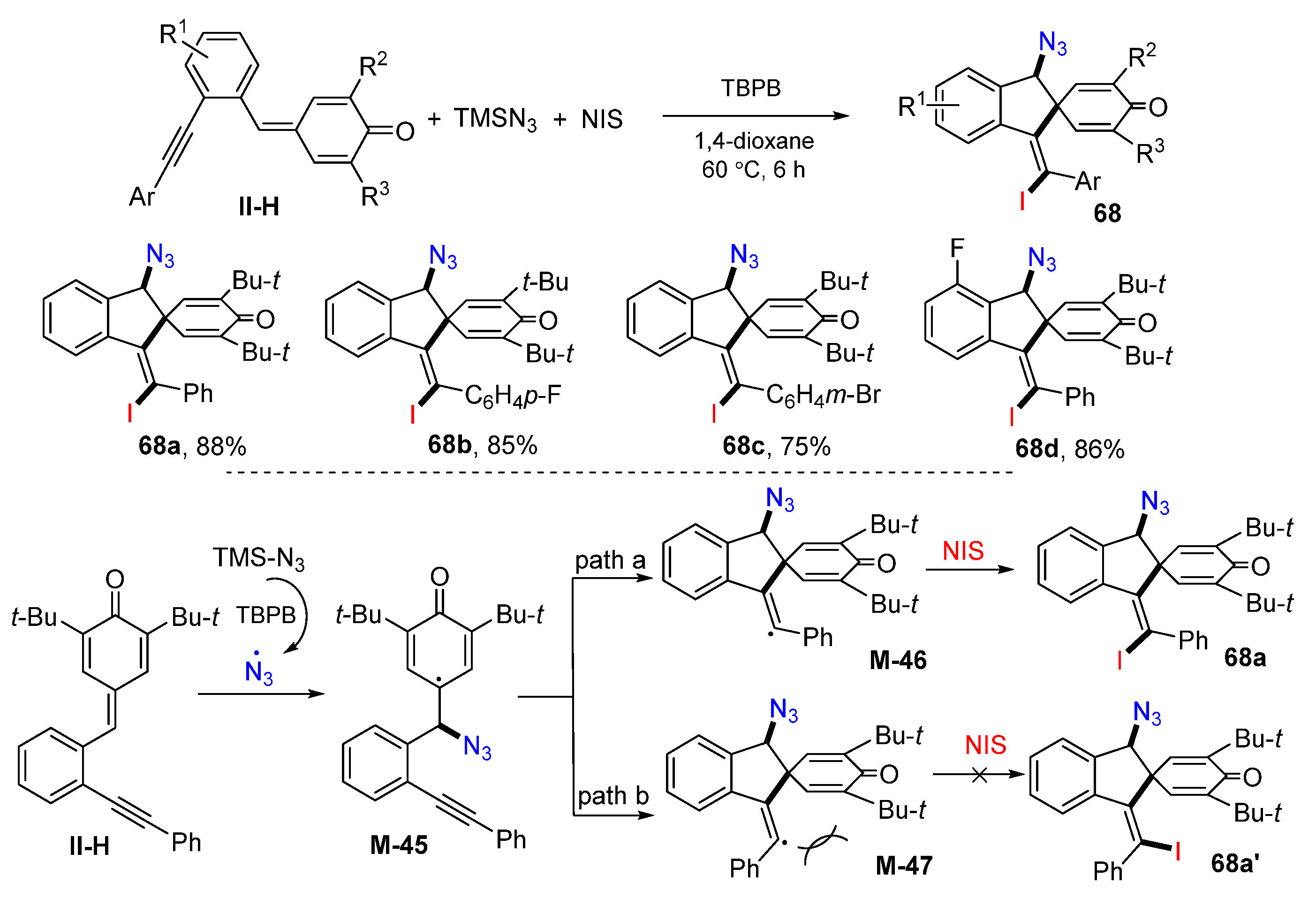
Using unique benzene-tethered 1,5-enynes, the use of 4-(2-ethynylbenzylidene)cyclohexa-2,5-dien-1-ones for the synthesis of substituted spiroindene compounds was introduced by Yao in 2018. The reaction of 1,5-enynes, TMSN
3 and NIS in dioxane in the presence of TBPB gave product
68 in good-to-excellent yields (
Scheme 67) [
79]. The suggested reaction mechanism indicated that N
3 radical derived from TMSN
3 adds to the double bond of 1,5-enynes to give cyclohexadienone radical
M-45 (path a), which then undergoes 5-
exo cyclization to form spirocyclic vinyl intermediates
M-46, followed by iodine atom transfer from NIS to selectively give iodo- and azido-functionalized spiroindene products
68a as an
E-isomer. Due to the steric hindrance of
M-47, cyclization through path b leading to the formation of
Z-product
68a’ is unfavorable.
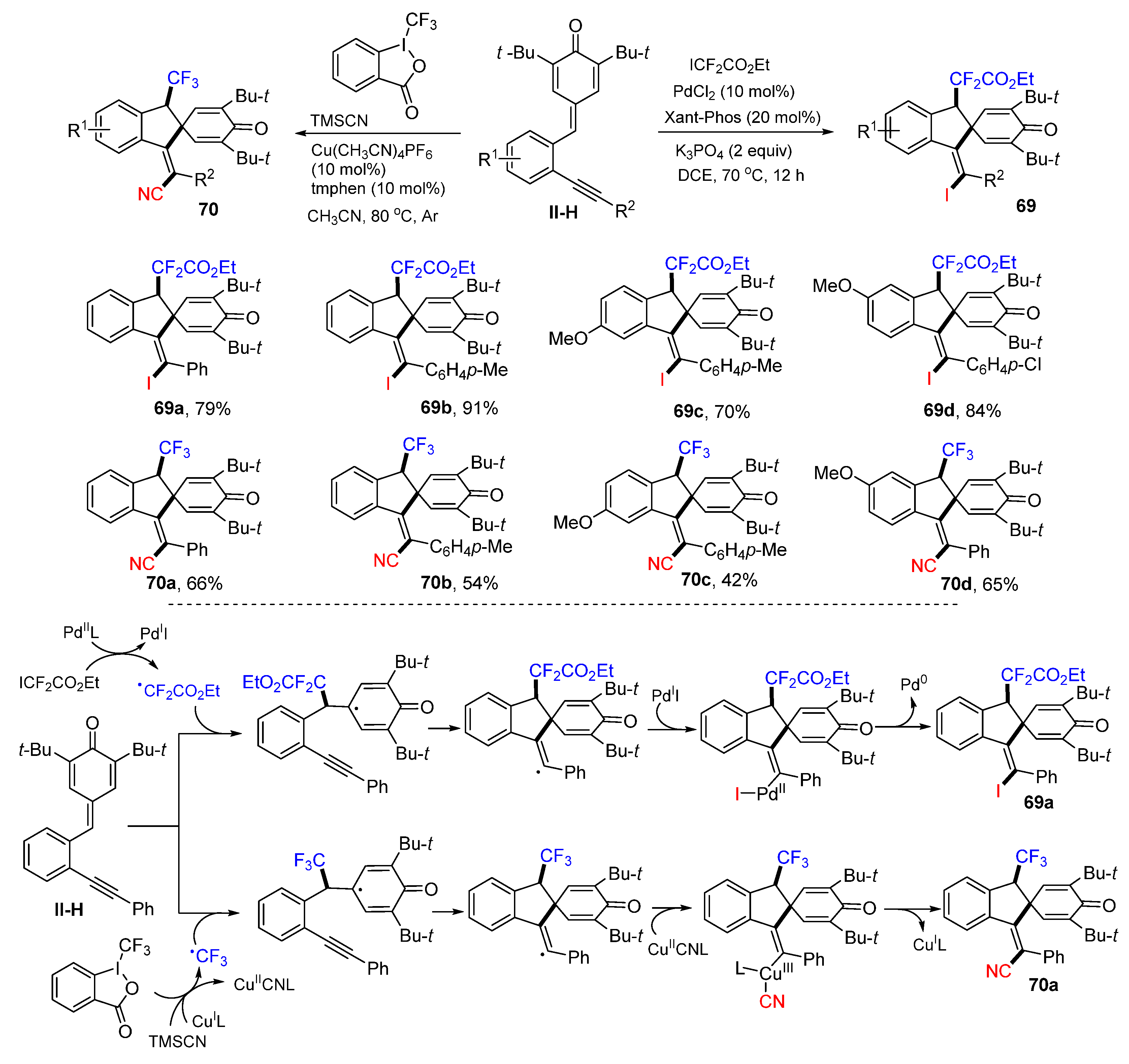
A metal-catalyzed radical spiroannulation of 1,5-enynes for the synthesis of fluorine-containing (
Z)-spiroindenes was reported by Jiang’s group in 2020. The reaction of 1,5-enynes and ICF
2CO
2Et in DCE at 70 °C under the catalysis of PdCl
2 and 9,9-dimethyl-4,5-bis(diphenylphosphino)xanthenes (Xant-Phos) gave iododifluoro-acetylated product
69 in good yields (
Scheme 68) [
80]. However, the use of BrCF
2CO
2Et or C
4F
9I as the fluoroalkylation reagents failed to give the corresponding (
Z)-spiroindenes. Another reaction of 1,5-enynes, Togni’s reagent and TMSCN in CH
3CN at 50 °C under the catalysis of Cu(OAc)
2 and 3,4,7,8-tetramethyl-1,10-phenanthroline (tmphen) gave trifluoromethylated products
70. For the synthesis of
69a, the reaction mechanism suggests that the CF
2CO
2Et radical derived from ICF
2CO
2Et adds to the C=C double bond of 1,5-enynes followed by 5-
exo spirocyclization, formation of the Pd
II-I complex, and reductive elimination of Pd catalyst to afford iododifluoroacetylated product
69a. In the synthesis of CF
3-functionalized products
70, the CF
3 radical derived from Togni’s reagent has a similar spirocyclization mechanism to form cyanotrifluoromethylated spiroindene product
70a. The Tu and Jiang groups extended this reaction in the synthesis of iodosulfonylated spiroindenes, which involves an ionic instead of a radical cyclization [
81].
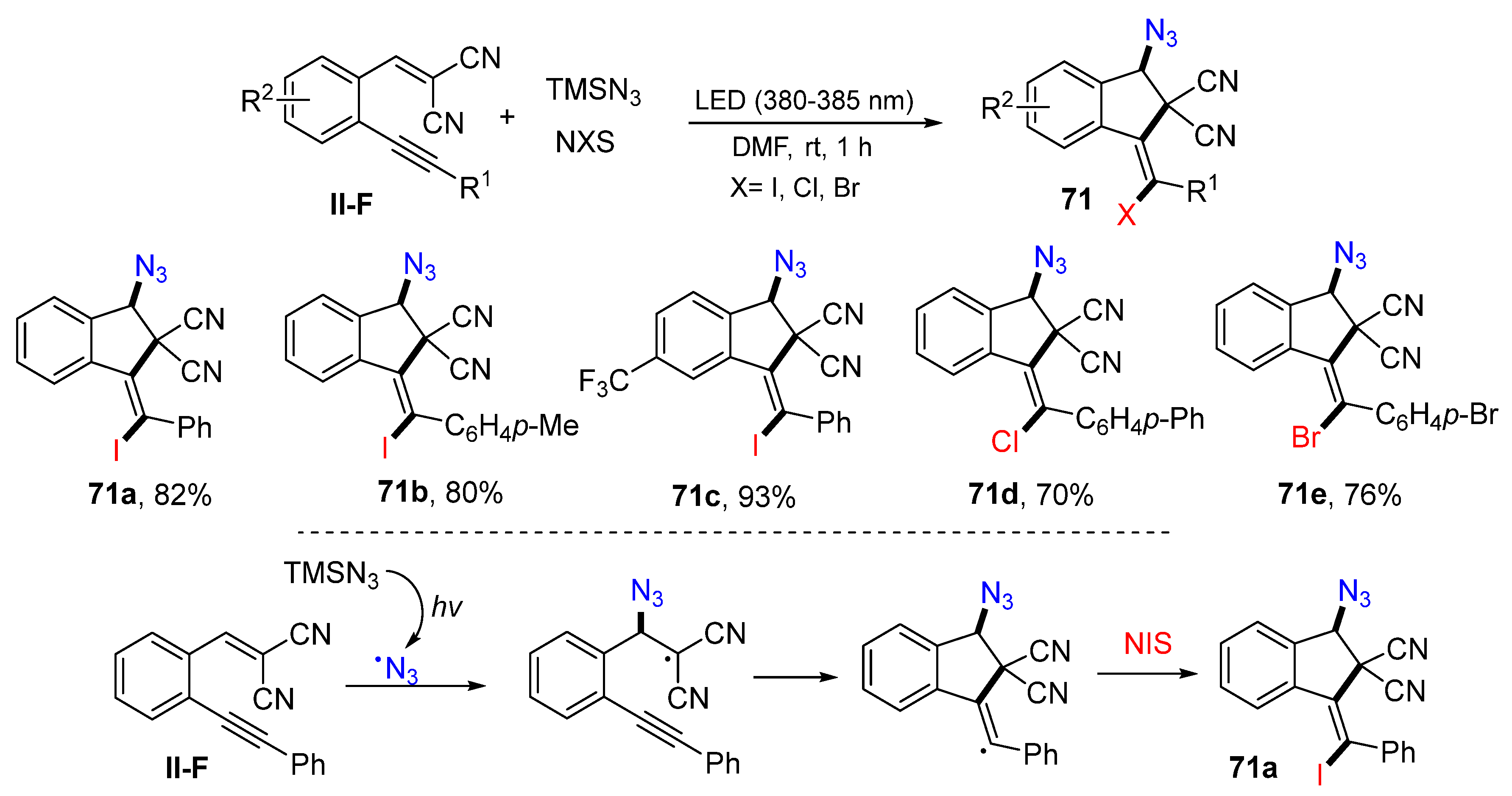
Using dicyano-substituted benzene-tethered 1,5-enynes for a visible light-driven radical haloazidative cyclization for the synthesis of holoazido-functionalized indenes was accomplished by the Li group in 2020. The reaction of 1,5-enynes, TMSN
3, and
N-iodo (bromo or chloro) succinimide in DMF under the radiation of LED (380–385 nm) afforded product
71 in moderate-to-good yields (
Scheme 69) [
82]. The suggested reaction mechanism indicated that the azide radical generated from TMSN
3 under the photo conditions adds to the double bond of 1,5-enyne followed by 5-
exo cyclization and I-atom transfer from NIS to give product
71a.
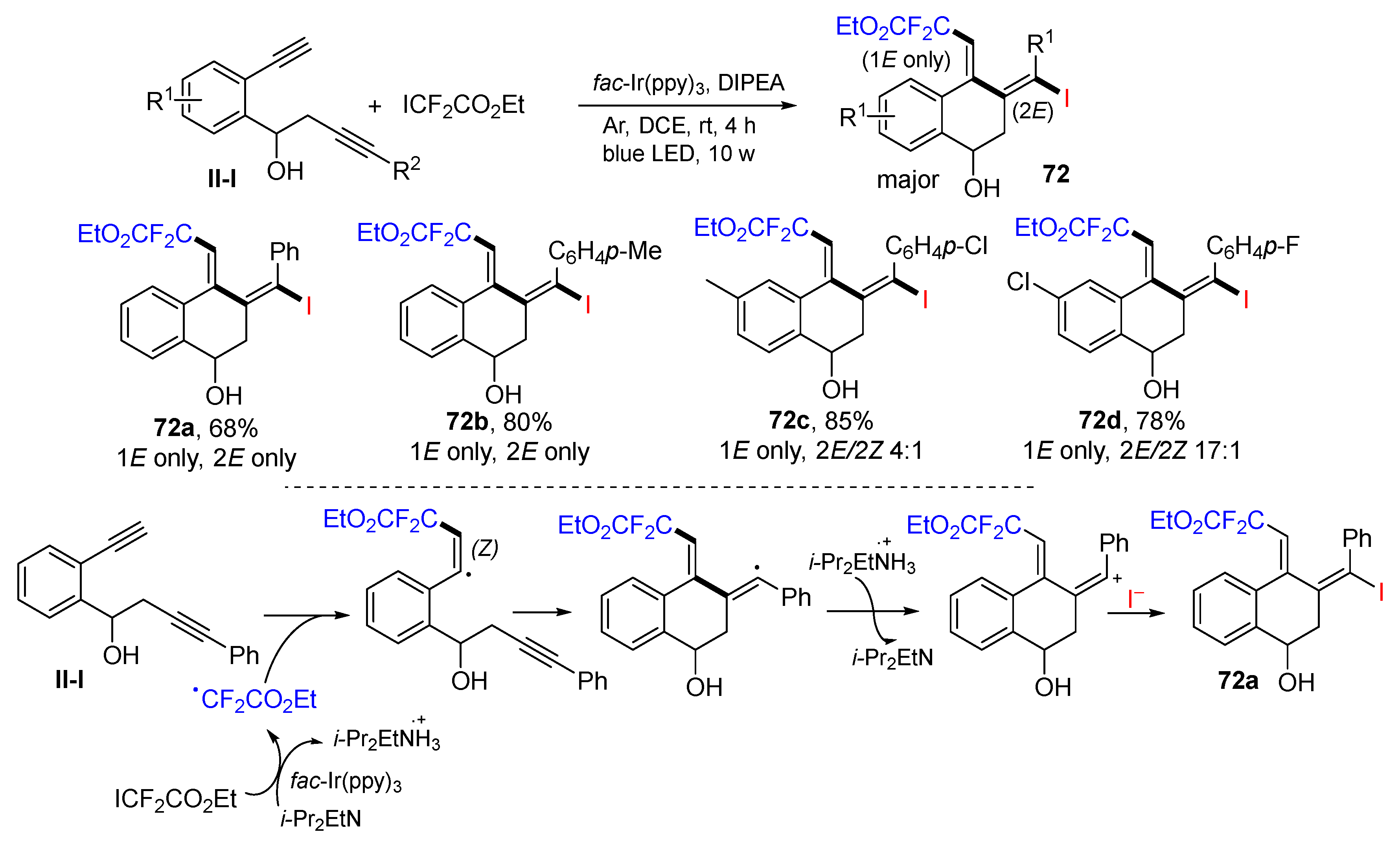
Using benzene-tethered 1,7-diynes for the synthesis of iododifluoroacetal tetrahydronaphthalen-1-ols was introduced by the Jiang group in 2021. The reaction of 1,7-diynes and ICF
2CO
2Et under photoredox catalysis with
fac-Ir(ppy)
3 gave difluoromethyl-containing (1
E,2
E)-tetrahydronaphthalen-1-ols
72 bearing two exocyclic C=C double bonds as major stereoisomers in good yields (
Scheme 70) [
83]. A reaction mechanism suggests that the CF
2CO
2E radical derived from ICF
2CO
2Et under the photocatalysis adds to the terminal alkyne of 1,7-diyne followed by 6-
exo cyclization, SET of DIPEA to form cation, and nucleophilic addition with iodide anion to give (1
E,2
E)-product
72a as a major isomer.
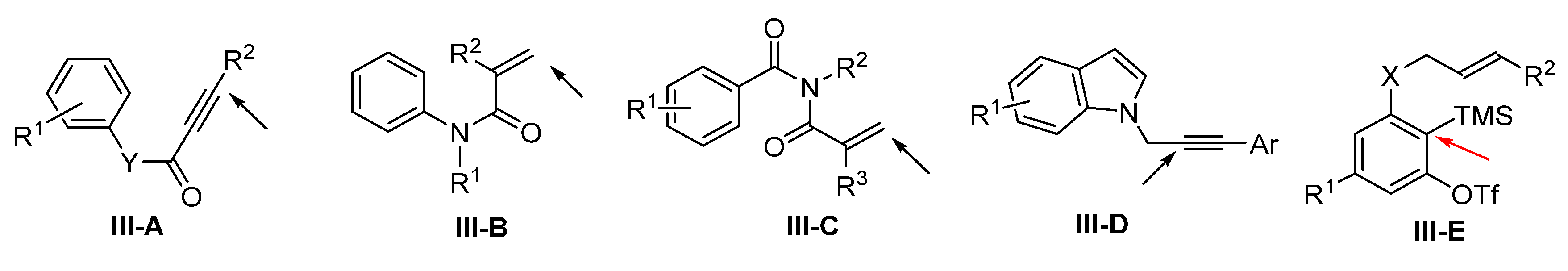
4. Reaction of Arene-Terminated Alkenes and Alkynes
Presented in this section are the radical addition and cyclization-initiated difunctionalization reactions of arene-terminated alkenes and alkynes with a reaction sequence shown in
Scheme 71. For the class of substrates shown in
Scheme 72, the initial radical addition happens at the alkene or alkyne groups instead of the arene. Sequential radical cyclization leads to the formation of spiro- or fused-ring compounds. The only exception is the reaction of substrate
III-E. The radical is added to the benzyne ring (via the benzyne intermediate). Among the general substrates, the reactions of alkynes
III-A (arylpropiolamides if Y is NR) for making spiro compounds are much more popular than those of substrates
III-B to
III-E for making fused cyclic products.
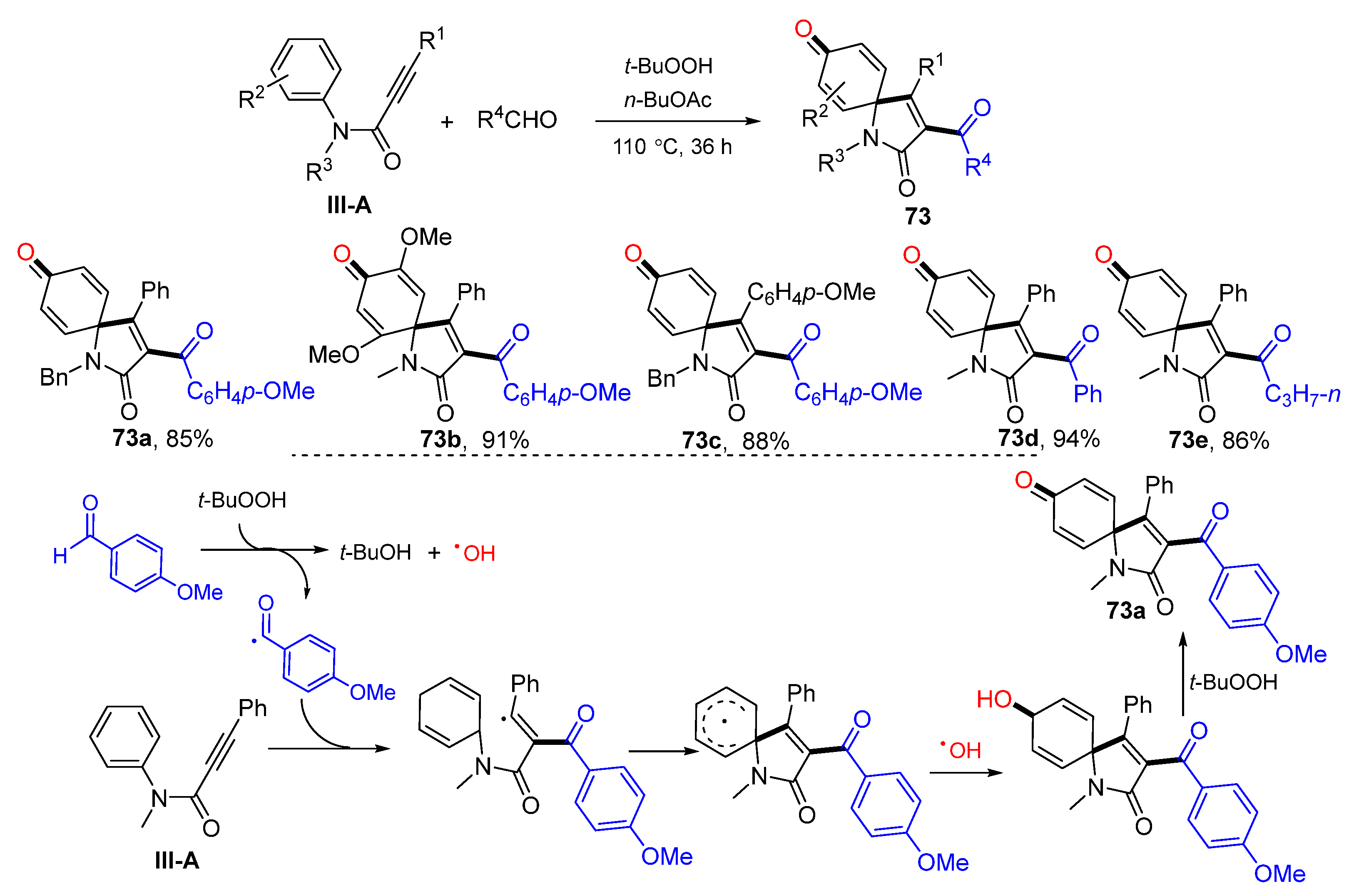
There are several reports on the reaction of arylpropiolamides for the synthesis of 3-functionalized azaspiro[4,5]trienones. In 2014, Li and co-workers reported a radical spirocyclization reaction of arylpropiolamides for the synthesis of 3-acylated azaspiro[4,5]trienones. The reaction of alkynyl amides and aldehydes in the presence of TBHP gave product
73 in good-to-excellent yields (
Scheme 73) [
84]. The reaction mechanism suggests that the carbonyl radical generated from aldehyde adds to alkyne followed by
ipso-carbocyclization, coupling with OH radical and oxidation of OH group to give 3-acylspiro[4,5]trienone
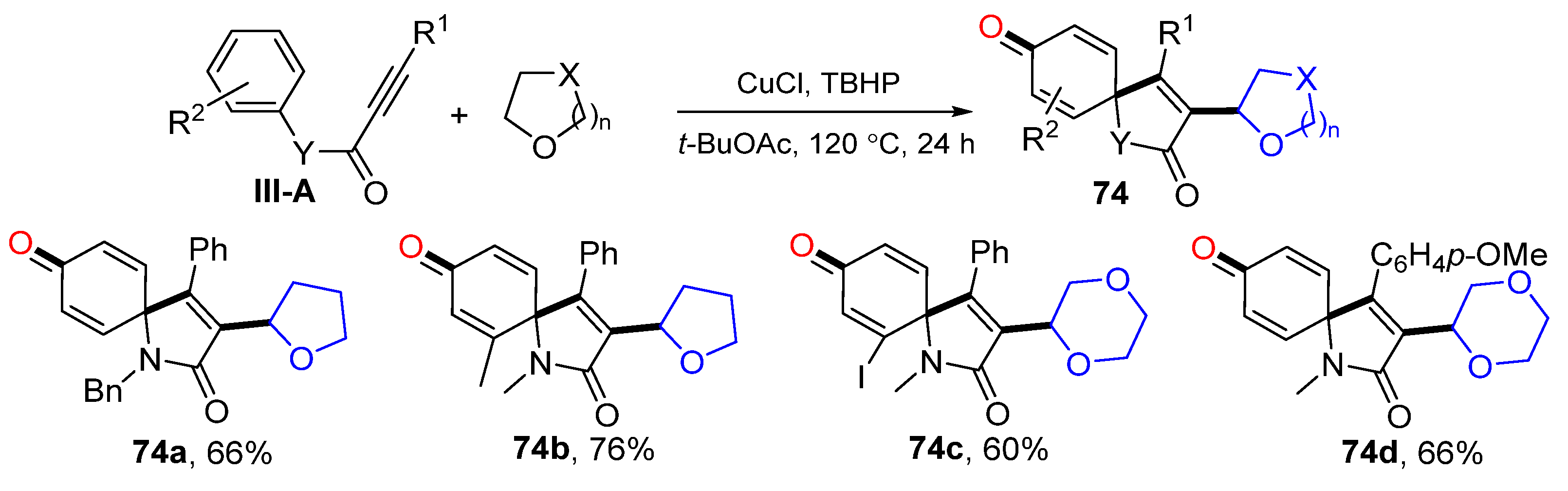
73a. In 2014, Li’s group also reported a Cu-catalyzed radical spirocyclization of aryl alkynyl amides for the synthesis of azaspiro[4,5]trienones. The reaction of arylpropiolamides and cyclic ethers in
t-BuOAc under the catalysis of Cu
II and TBHP gave product
74 in good yields (
Scheme 74) [
85].
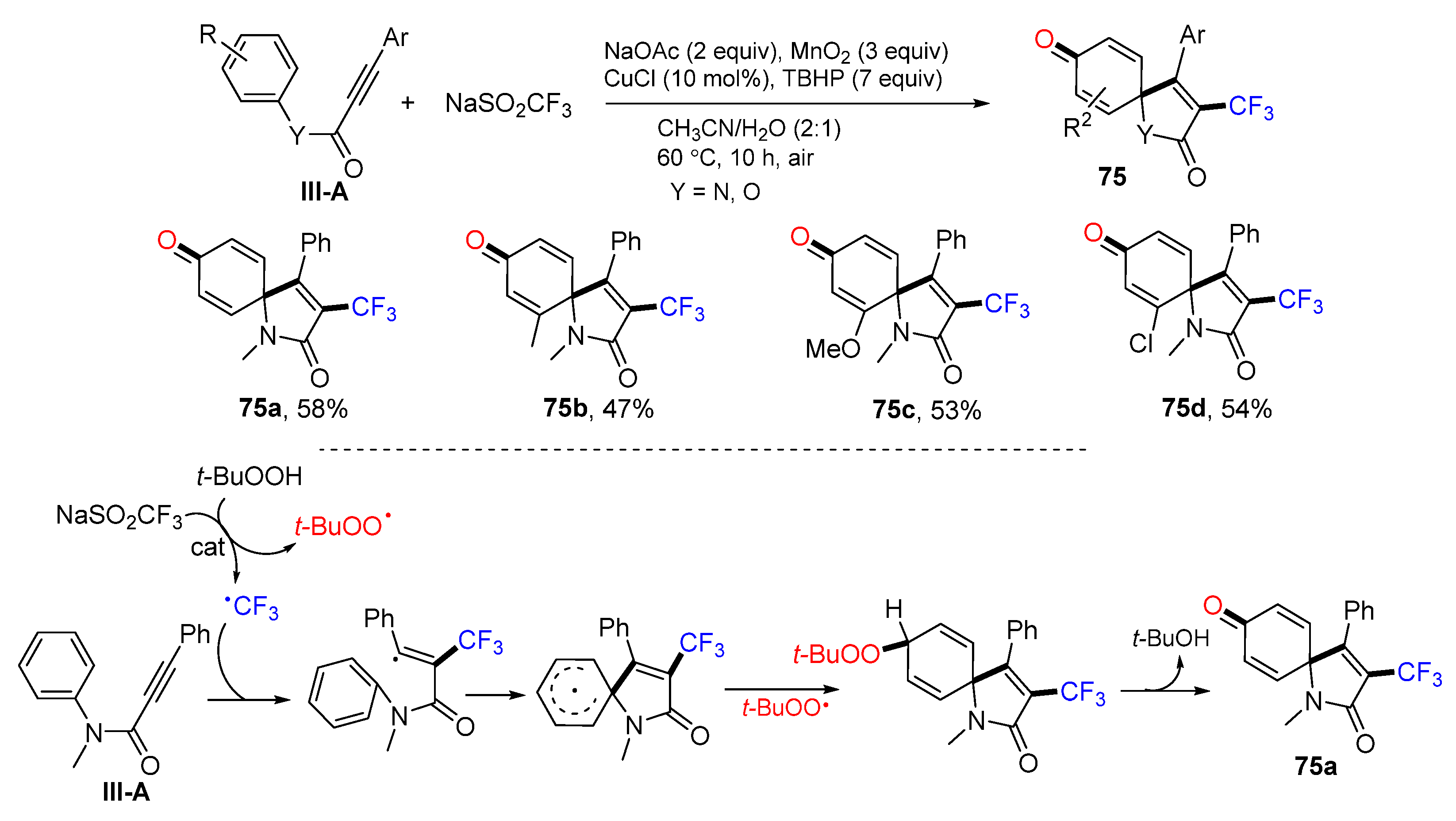
A Cu-catalyzed radical spirocyclization of arylpropiolamides for the synthesis of 3-triflouromrthylated azaspiro[4,5]trienones was reported by the Liang group in 2015. The reaction of alkynyl amides and NaSO
2CF
3 (Langlois’ reagent) in CH
3CN in the presence of TBHP, MnO
2 and CuCl gave product
75 in good-to-excellent yields (
Scheme 75) [
86]. The reaction mechanism suggests that the CF
3 radical derived from the Langlois’ reagent adds to the C≡C triple bond followed by
ipso-carbocyclization, coupling with the
t-BuOO radical, and elimination of
t-BuOH to give product
75a.
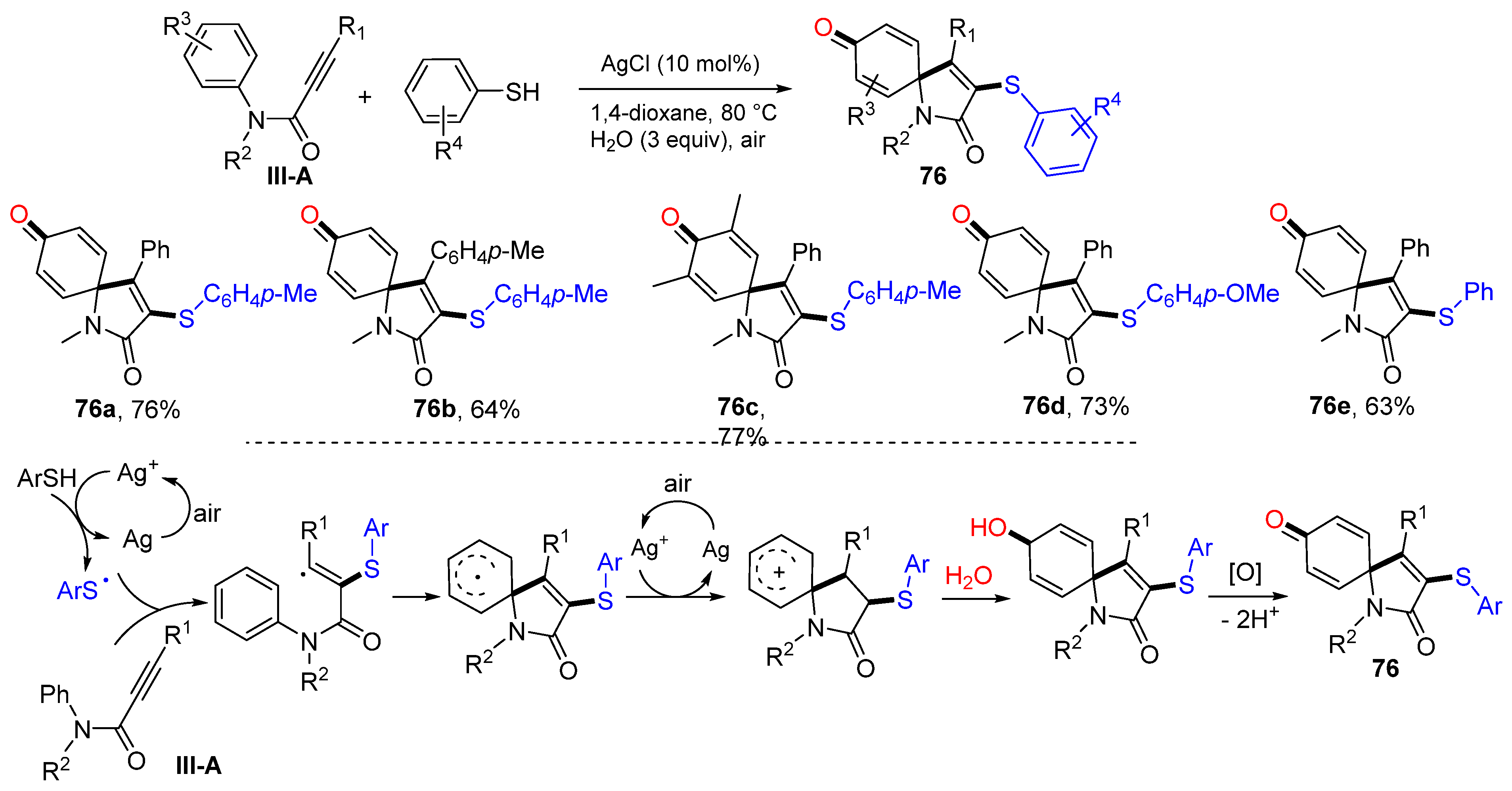
In 2015, the Wang group introduced an Ag-catalyzed radical spirocyclization of arylpropiolamides for the construction of 3-arylthiolated azaspiro[4,5]trienones. The reaction of alkynyl amides, thiophenols and H
2O in 1,4-dioxane under the catalysis Ag
I gave product
76 in moderate-to-good yields (
Scheme 76) [
87]. A proposed reaction mechanism suggests that the thiyl radical produced from thiophenol adds to the carbon triple bond of arylpropiolamides followed by the
ipso-carboncyclization, SET to form carbocation, nucleophilic addition of H
2O, and oxidization of OH to give product
76.
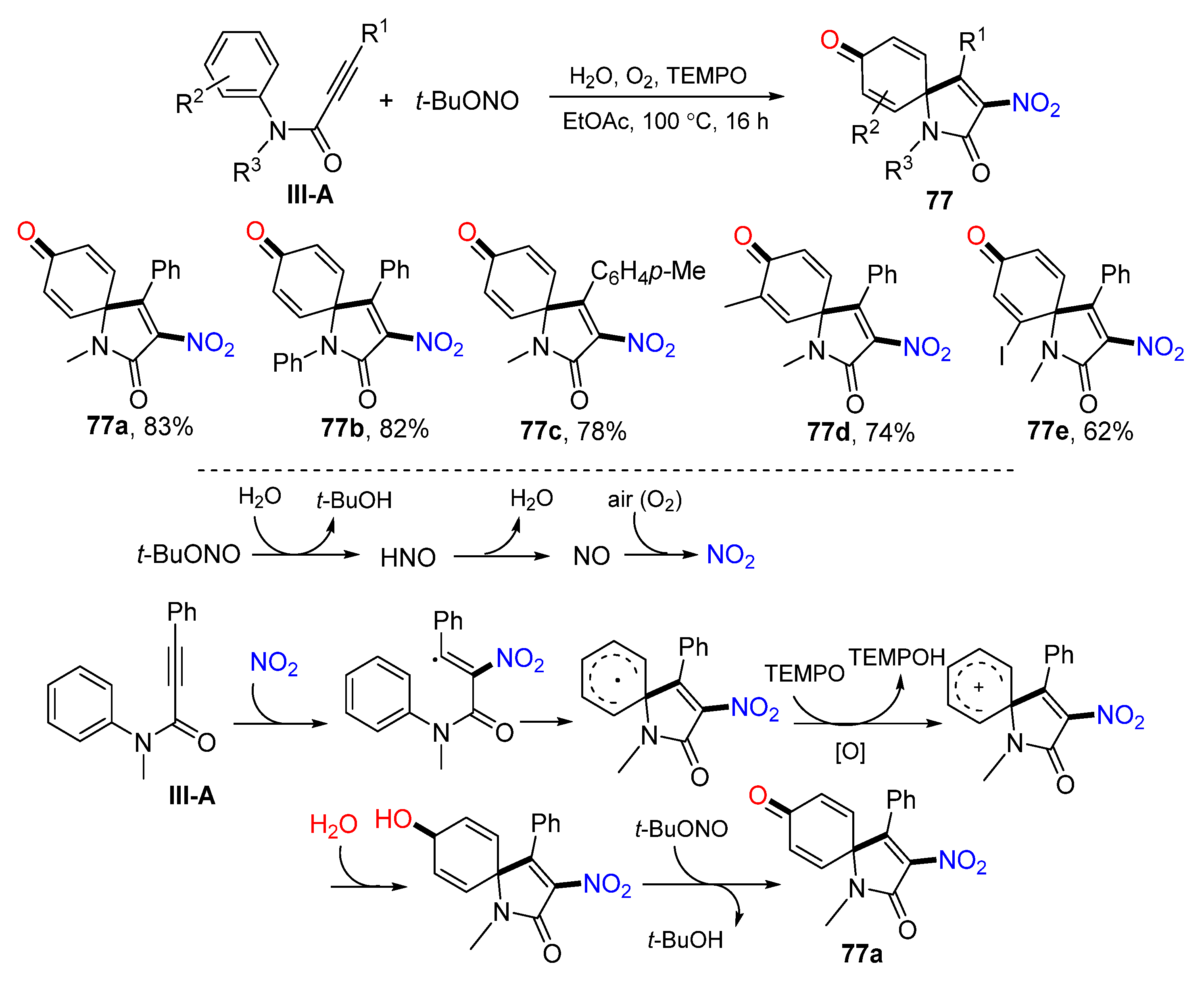
A TEMPO-mediated radical nitrative spirocyclization of arylpropiolamides for the preparation of 2-nitrated azaspiro[4,5]trienones was introduced by Li’s group in 2015. The reaction was carried out using arene-terminaled 1,5-enynes and
t-BuONO in EtOAc in the presence of O
2 and TEMPO to give nitrated spiro compound
77 in moderate-to-good yields (
Scheme 77) [
88]. A reaction mechanism suggests that NO
2 generated from the oxidization of NO adds to the carbon triple bond of arylpropiolamide followed by
ipso-carbocyclization, TEMPO oxidation to form cation, nucleophilic addition of H
2O, and oxidization to give product
77a.
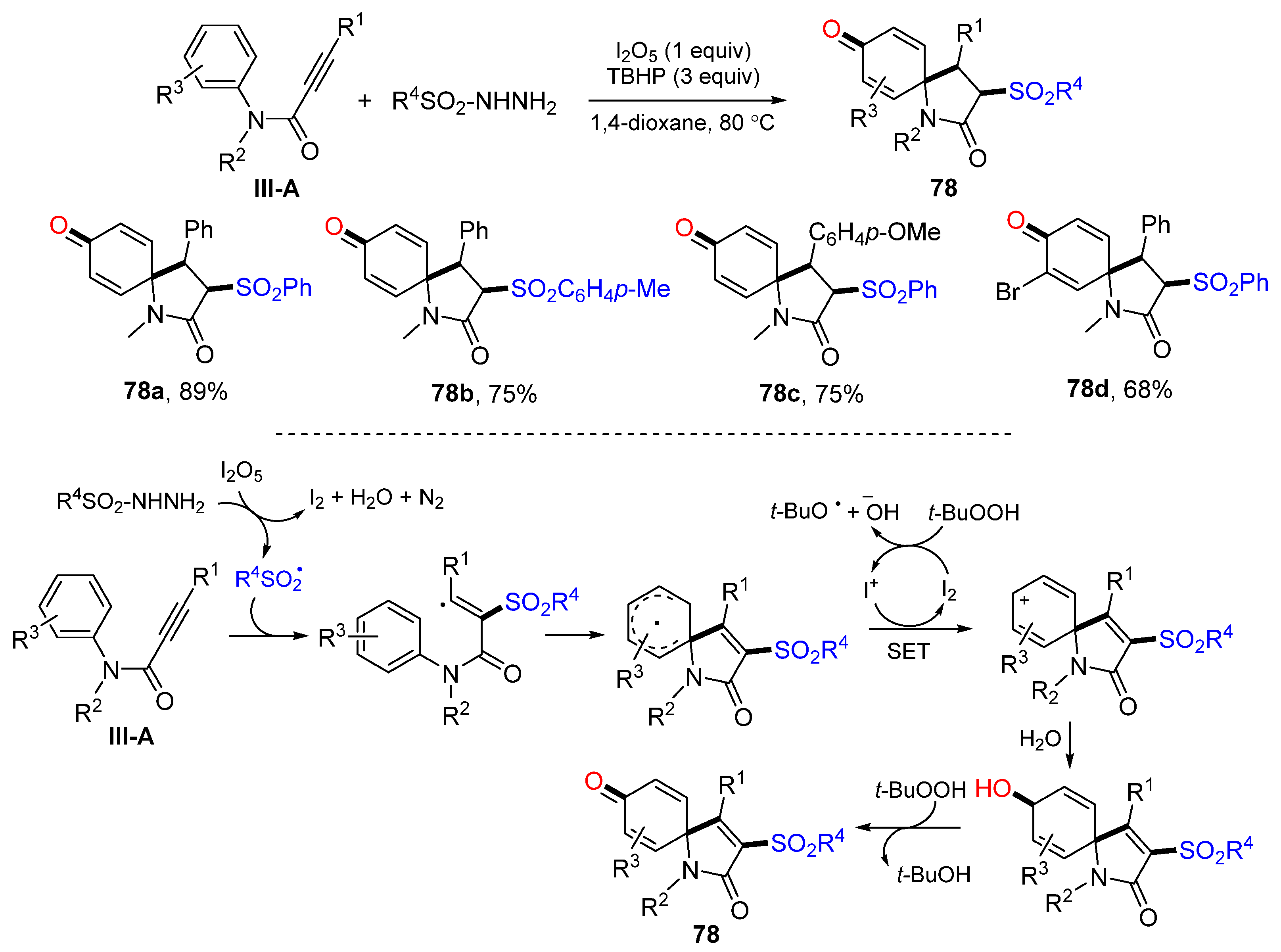
In 2015, Wang and co-workers developed an oxidative radical spirocyclization reaction of arylpropiolamides for the preparation of 3-sulfonated azaspiro[4,5]trienones. The reaction of arylpropiolamides and sulfonylhydrazide in the presence of TBHP and I
2O
5 afforded product
78 in moderate-to-good yields (
Scheme 78) [
89]. The reaction mechanism suggests that the sulfonyl radical derived from sulfonylhydrazide adds to the carbon triple bond of amides followed by
ipso-cyclization, SET to form cyclohexadienyl cation, nucleophilic addition of H
2O, and finally oxidation with TBHP to give product
78.
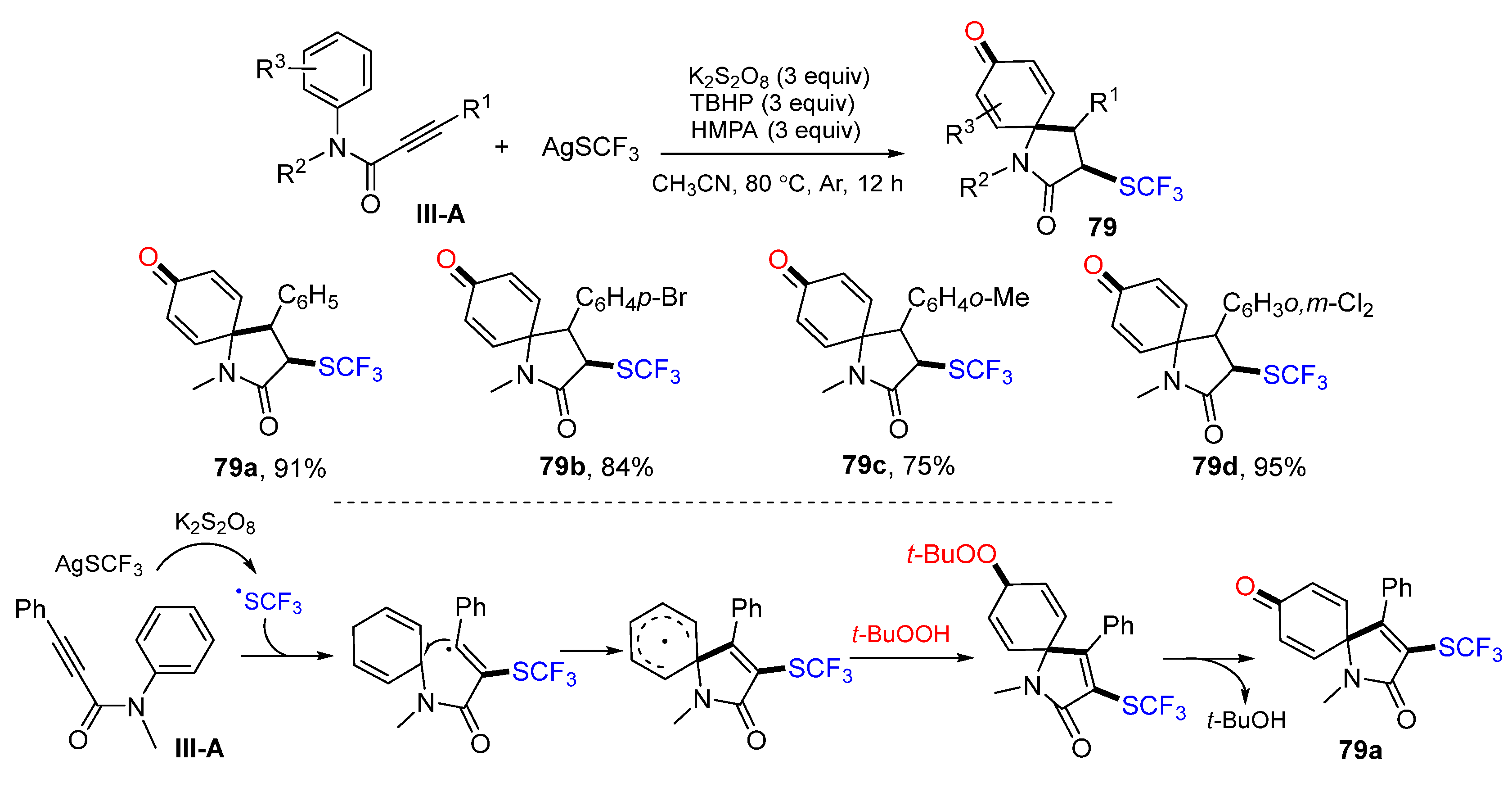
A new method for radical spirocyclization of arylpropiolamides to synthesize 3-sulfonated azaspiro[4,5]trienones was reported by Liu’s group in 2016. The reaction of amides and AgSCF
3 in CH
3CN in the presence of K
2S
2O
8 and TBHP gave product
79 in excellent yields (
Scheme 79) [
90]. A proposed reaction mechanism suggests that the CF
3S radical derived from AgSCF
3 adds to the carbon double bond of amides, followed by
ipso-carbocyclization, coupling with
t-butylperoxy radical, and elimination of
t-BuOH to give product
79a.
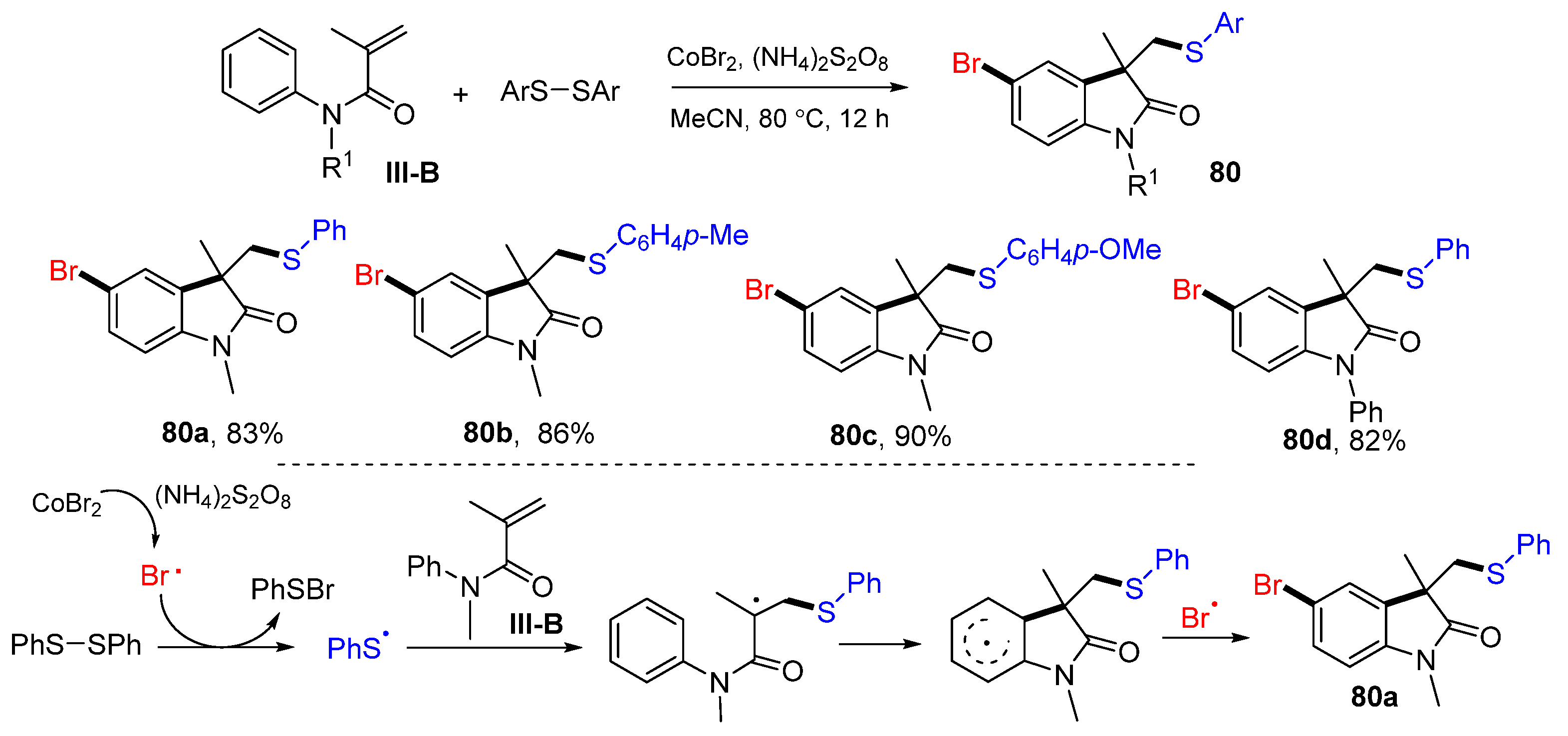
Other than the reactions of arylpropiolamides for making the spiro compounds described above, the reactions of
N-phenylacrylamides have also been developed for making fused-cyclic products. In 2022, Zhang and co-workers reported a Co-promoted reaction for the synthesis of bromoarylthiolated heterocyclic compounds. The reaction of
N-arylacrylamides and disulfides in CH
3CN in the presence of CoBr
2 and (NH
4)
2S
2O
8 gave functionalized product
80 in good-to-excellent yields (
Scheme 80) [
91]. The reaction mechanism suggests that bromine and PhS radicals for the difunctionalization are generated from the reaction of CoBr
2 and PhSSPh. The PhS radical adds to the terminal carbon of the double bond of amides, followed by cyclization and bromo radical coupling to give product
80a.
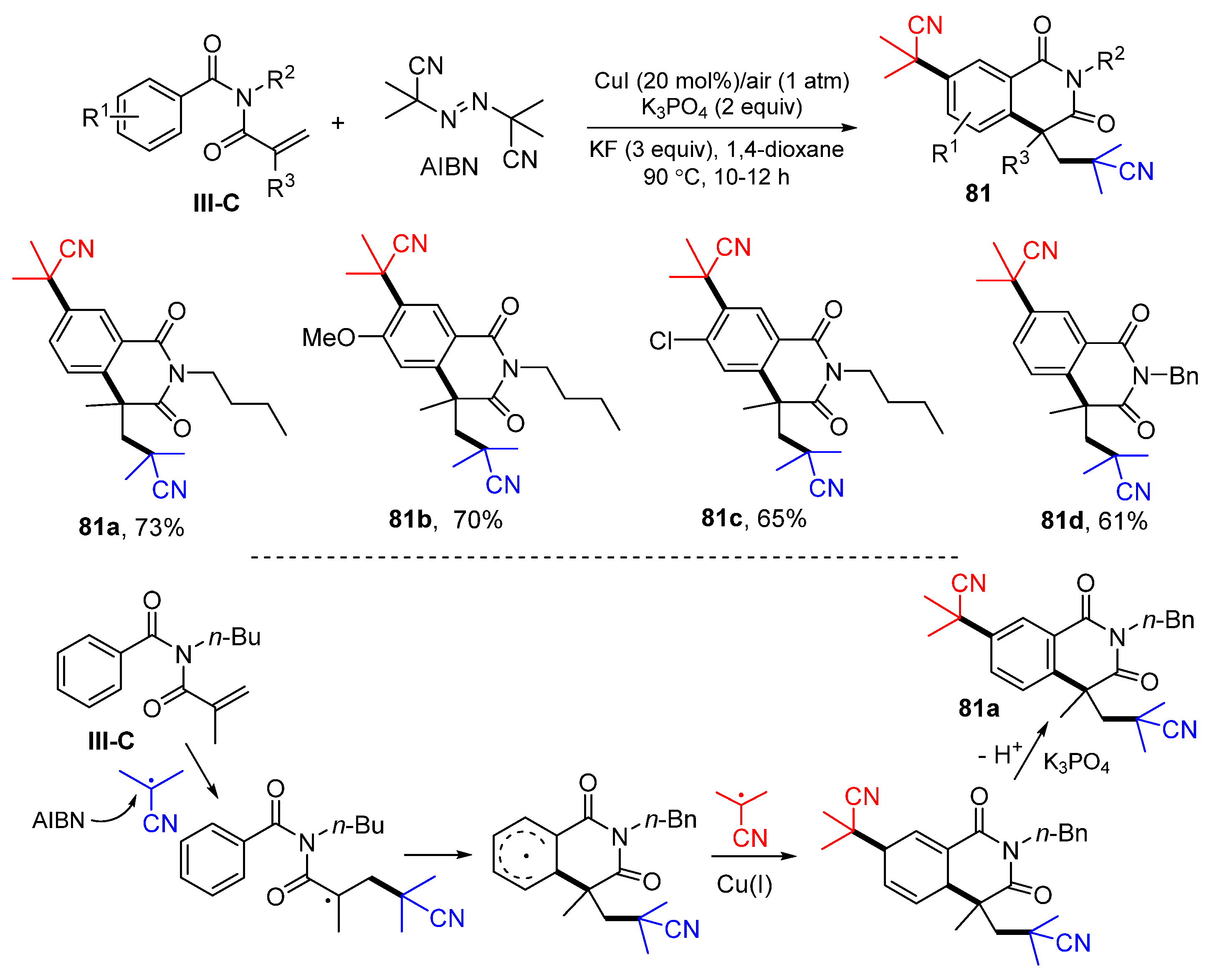
The reaction of methacryloyl benzamides could result in six-membered ring-fused products. This work was reported by Tang, Chen and their co-workers in 2016 in the development of a Cu-catalyzed radical reaction for the synthesis of dicyanoisoproylated isoquinolinediones. The reaction of methacryloyl benzamides and AIBN in dioxane in the presence of CuI, KF, and K
3PO
4 gave product
81 in good-to-excellent yields (
Scheme 81) [
92]. The reaction mechanism suggests that homolytic cleavage of AIBN gives two CNMe
2C radicals. One of them adds to the carbon double bond of amides, followed by 6
-exo cyclization to the benzene ring, selectively trapping the second CNMe
2C radical under the assistance of CuI, and final step aromatization to give isoquinoline-1,3(2
H,4
H)-dione
81a.
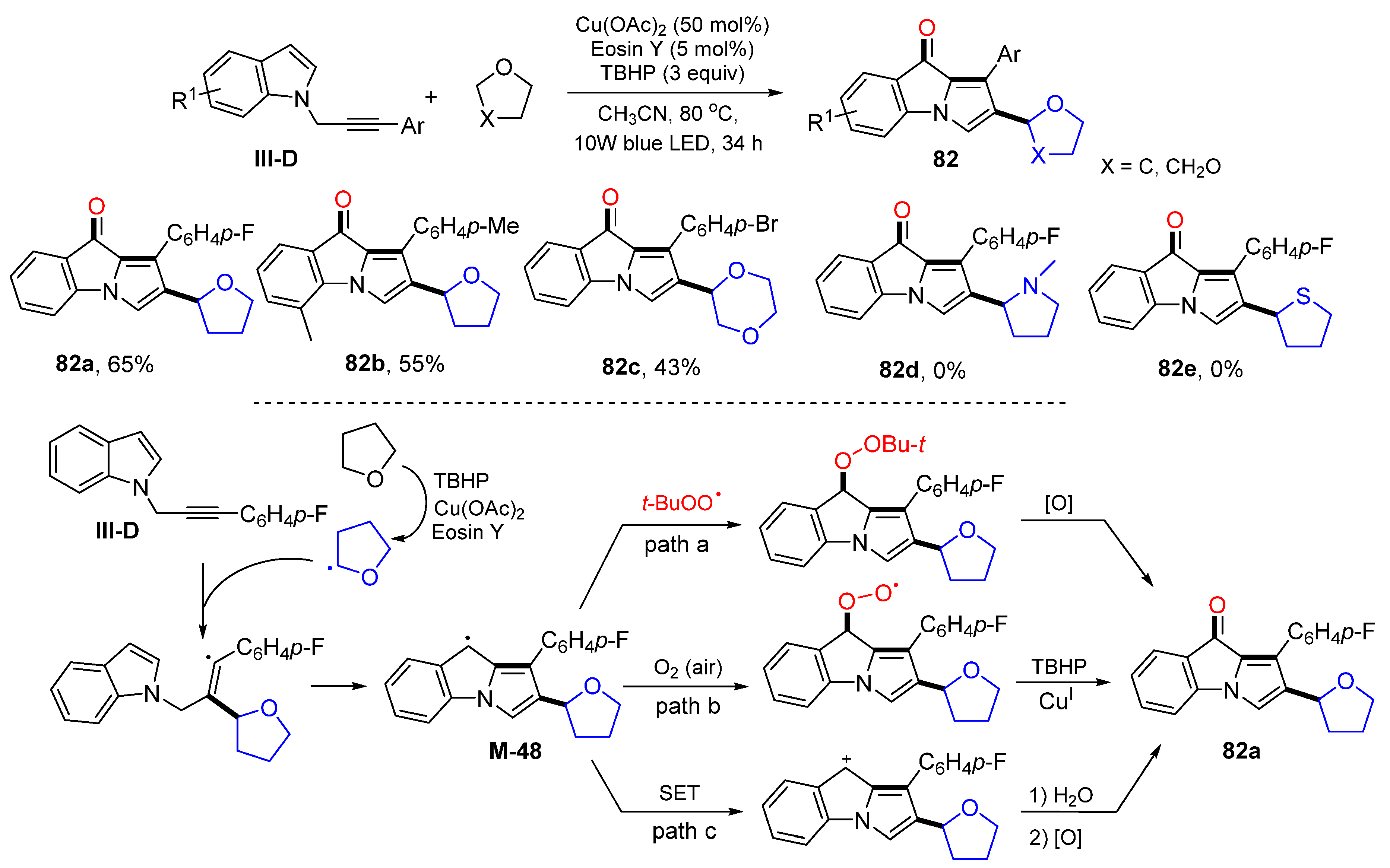
The reaction of
N-propargylindoles could result in the formation of products with a core of 9
H-pyrrolo[1,2-
a]indol-9-one. In 2022, Du and coworkers developed photoredox radical cyclization of
N-propargylindoles for the synthesis of 2-substituted 9
H-pyrrolo-[1,2-
a]indol-9-ones. The photo reaction of
N-propargylindoles and cyclic ethers in MeCN at 80 °C in the presence TBHP and dual catalysts Cu(OAc)
2 and Eosin Y give product
82 in moderate yields (
Scheme 82) [
93]. The proposed mechanism suggests that a THF radical, generated from the reaction of THF with TBHP and the catalysts, adds to the carbon triple bonds of
N-propargylindoles followed by 5-
exo cyclization to give intermediate
M-48. Intermediate
M-48 could have three paths to give product
82a, (1)
M-48 couples with
t-BuOO radical and then oxidation; (2)
M-48 traps O
2 then reacts with TBHP and CuI catalyst; (3)
M-48 oxidized to cation through SET process and then oxidized OH to C=O.
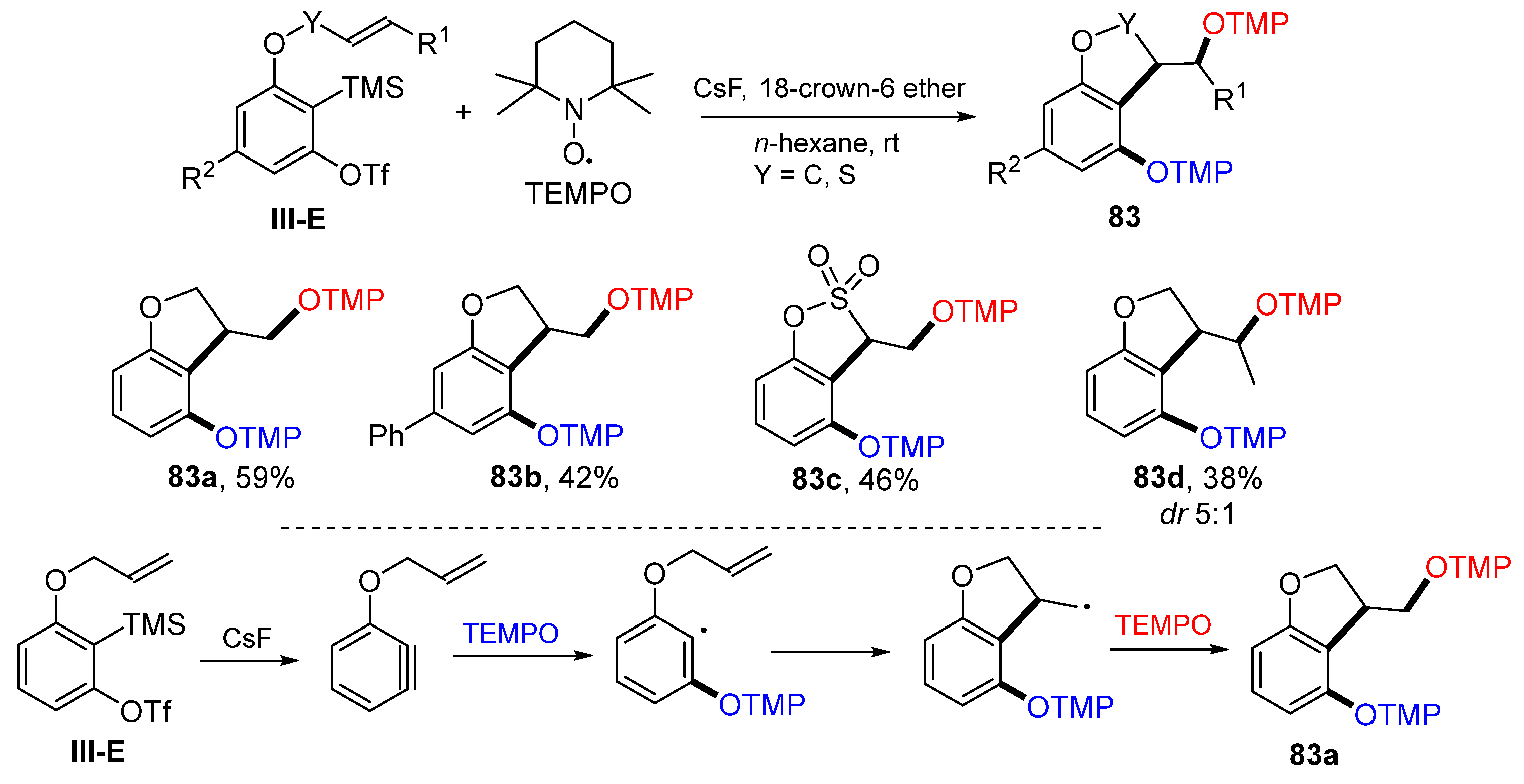
Other than the addition of an initial radical to the alkene or alkyne group on the side chain presented in previous cases, a radical could add to benzene if the ring is converted to a benzyne. In 2021, the Studer group reported such a reaction in the synthesis of substituted five-membered heterocycles. The reaction of arenes bearing 1,2-TMS and OTs groups with TEMPO in the presence of CsF and 18-crown-6 ether gave product
83 in moderate yields (
Scheme 83) [
94]. A proposed reaction mechanism suggests that arene is first converted to benzyne with the treatment of CsF and then reacts with TMPO radical followed by
5-exo cyclization and coupling with the second TEMPO to give product
83a.
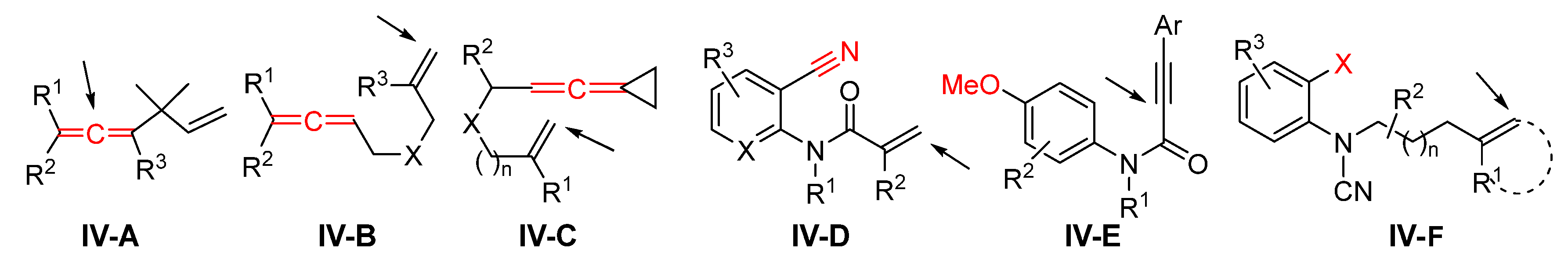
5. Reaction of Other Alkene and Alkyne Compounds
Presented in this section are the radical addition-initiated difunctionalizations of alkene- and alkyne-related compounds that cannot be fit in the previous sessions in terms of substrates or reaction mechanism. As shown in
Scheme 84, substrates
IV-A to
IV-C are 1,n-eneallenes; the cyano group in enenitrile
IV-D is responsible for the second functionalization; arene-terminated enyne
IV-E has a preexisting MeO group on the benzene ring which will be converted to a new functional group during the reaction; arene-terminated
IV-F has a leaving group X which will be displaced by a new group at the step of second functionalization. Since the reactions of these substrates are not the major focus of this paper, only selected examples are highlighted.
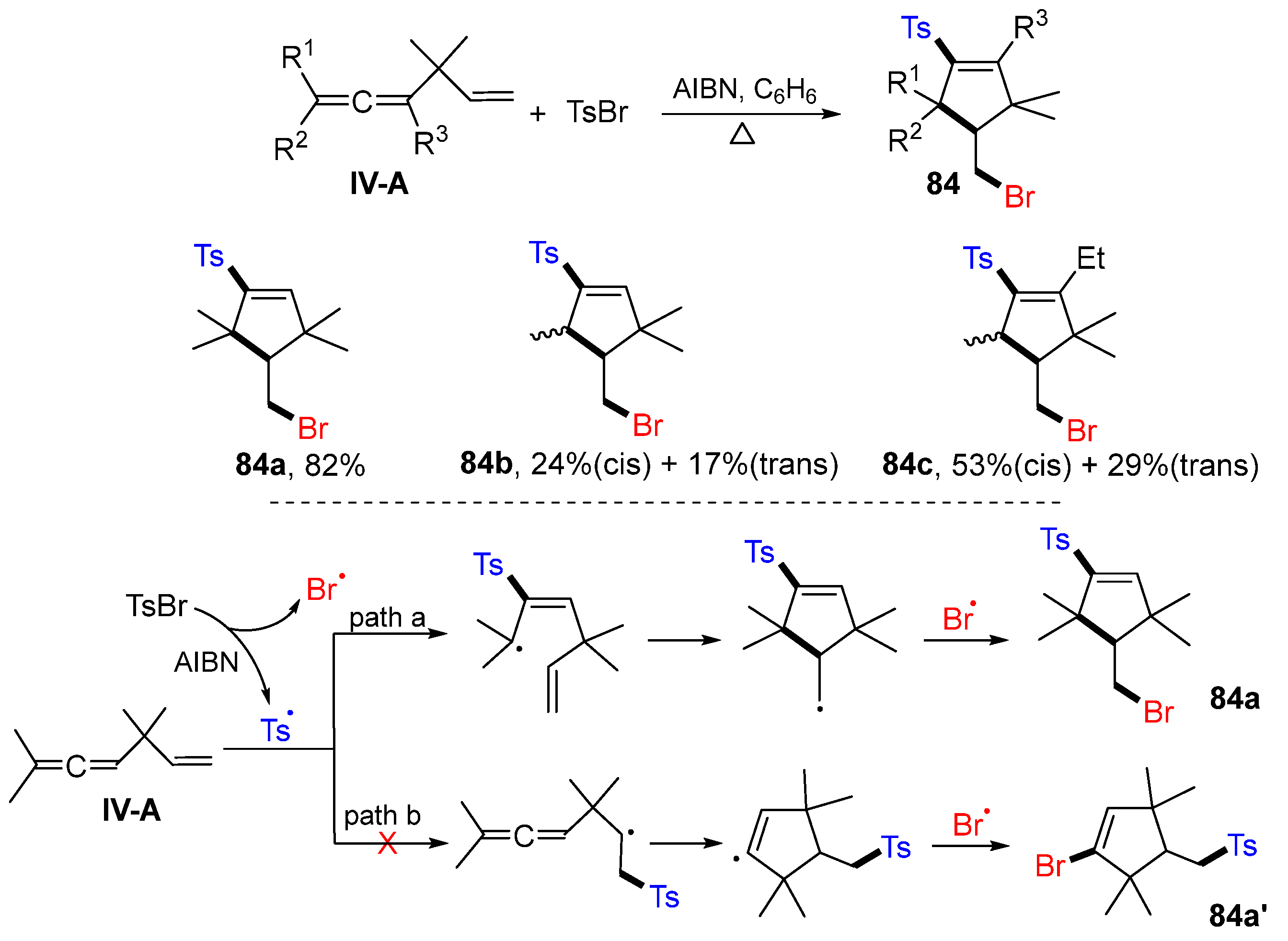
An early example of radical difunctionalization of eneallenes was reported by the Hatem group in 1995 for the synthesis of bromo- and tosyl-functionalized cyclopantenes. The reaction of eneallenes and tosyl bromide in benzene using AIBN as a radical initiator gave product
84 (
Scheme 85) [
95]. A proposed reaction mechanism suggests that the tosyl radical generated from TsBr adds the central carbon of allene, followed by 5-
exo cyclization and coupling with bromine radical, to give product
84a. Addition of tosyl radical to alkene instead of allene could be possible. However, since no expected product
84a’ was isolated, path b is less favorable than path a.
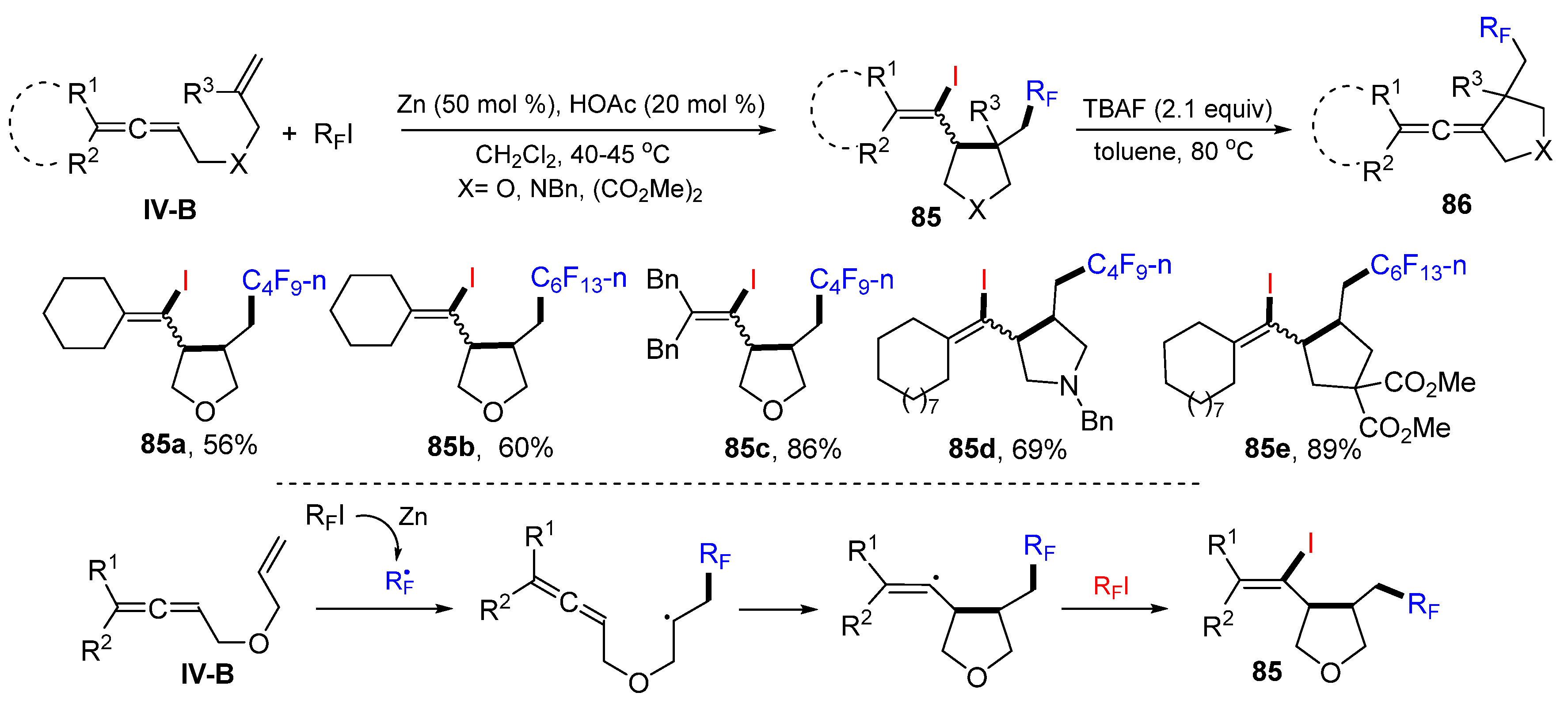
A later example for the reaction of eneallenes was reported by the Ma group in 2012. It is a Zn-catalyzed radical cyclization for the synthesis of iodoperfluoroalkylated five-membered rings. The reaction of eneallenes and R
FI in CH
2Cl
2 in the presence of Zn powder and HOAc gave product
85 in moderate-to-good yields (
Scheme 86) [
96]. It is worth mentioning that the two diastereomers of the product
85 could be converted into 3-(1-enylidene)heterocyclopentanes
86 through the TBAF-promoted dehydroiodination reaction. A mechanism for the racial reaction suggests that the perfluoroalkyl radical generated from R
FI adds to the alkene carbon of eneallenes followed by 5-
exo cyclization and coupling with the iodine radical from R
FI to give product
85.
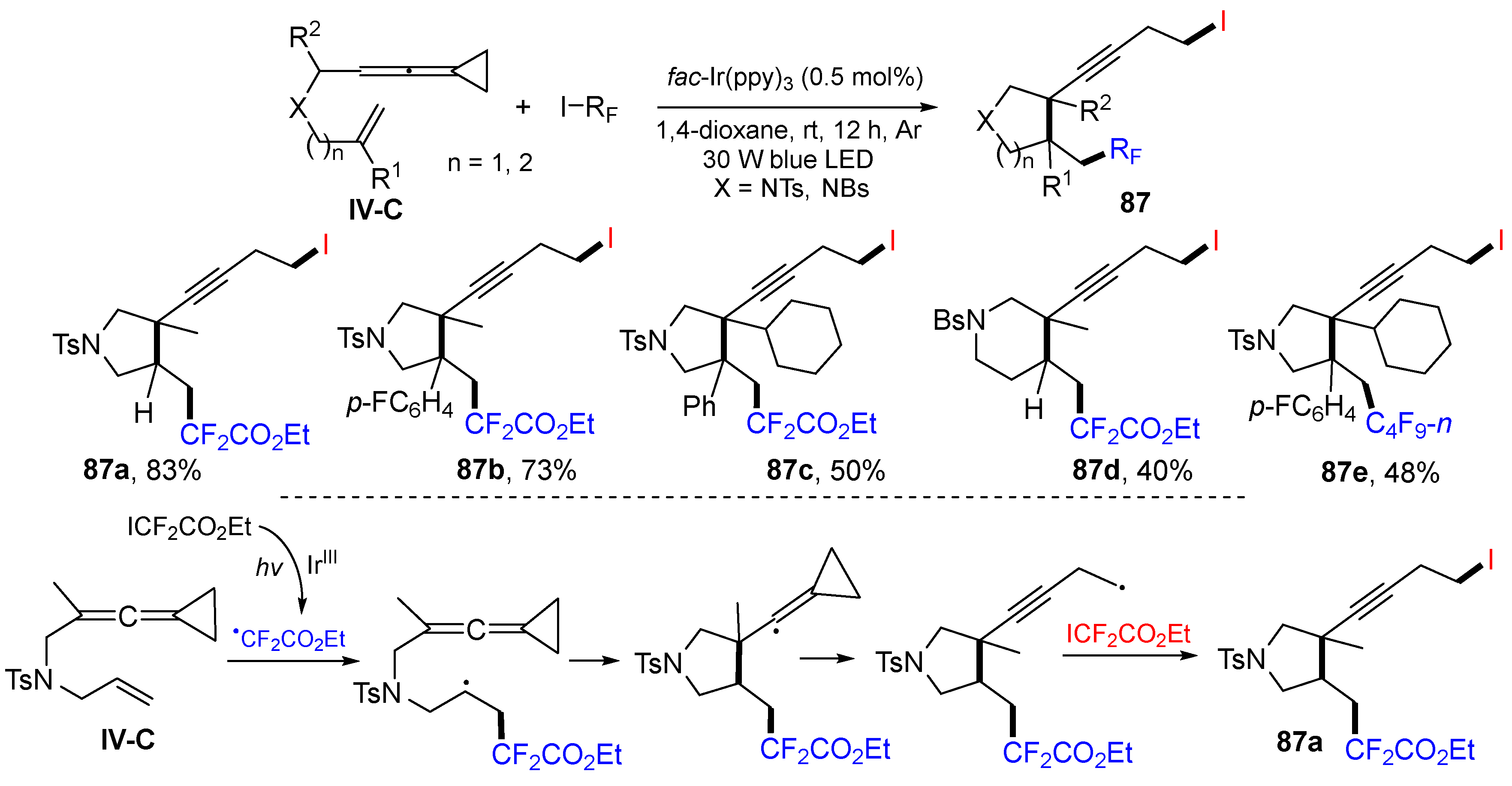
A more recent example of eneallene reaction was reported by the Shi group in 2021. It is a visible light-induced radical reaction of ene-vinylidenecyclopropanes (ene-VDCP) for the synthesis of iodoperfluoro-alkylated
N-heterocycles. The reaction of ene-VDCP, ICF
2CO
2Et or ICF
2CF
2CF
2CF
3 in 1,4-dioxane under the blue LED photocatalysis with
fac-Ir(ppy)
3 gave
87 in good yields and stereoselectivity (
Scheme 87) [
97]. The reaction mechanism suggests that the CF
2CO
2Et radical, generated from ICF
2CO
2Et under the photolysis, adds to the terminal carbon of alkene followed by 5-
exo cyclization, cyclopropane ring-opening, and extraction of iodine atom from ICF
2CO
2Et to give the final product
87a.
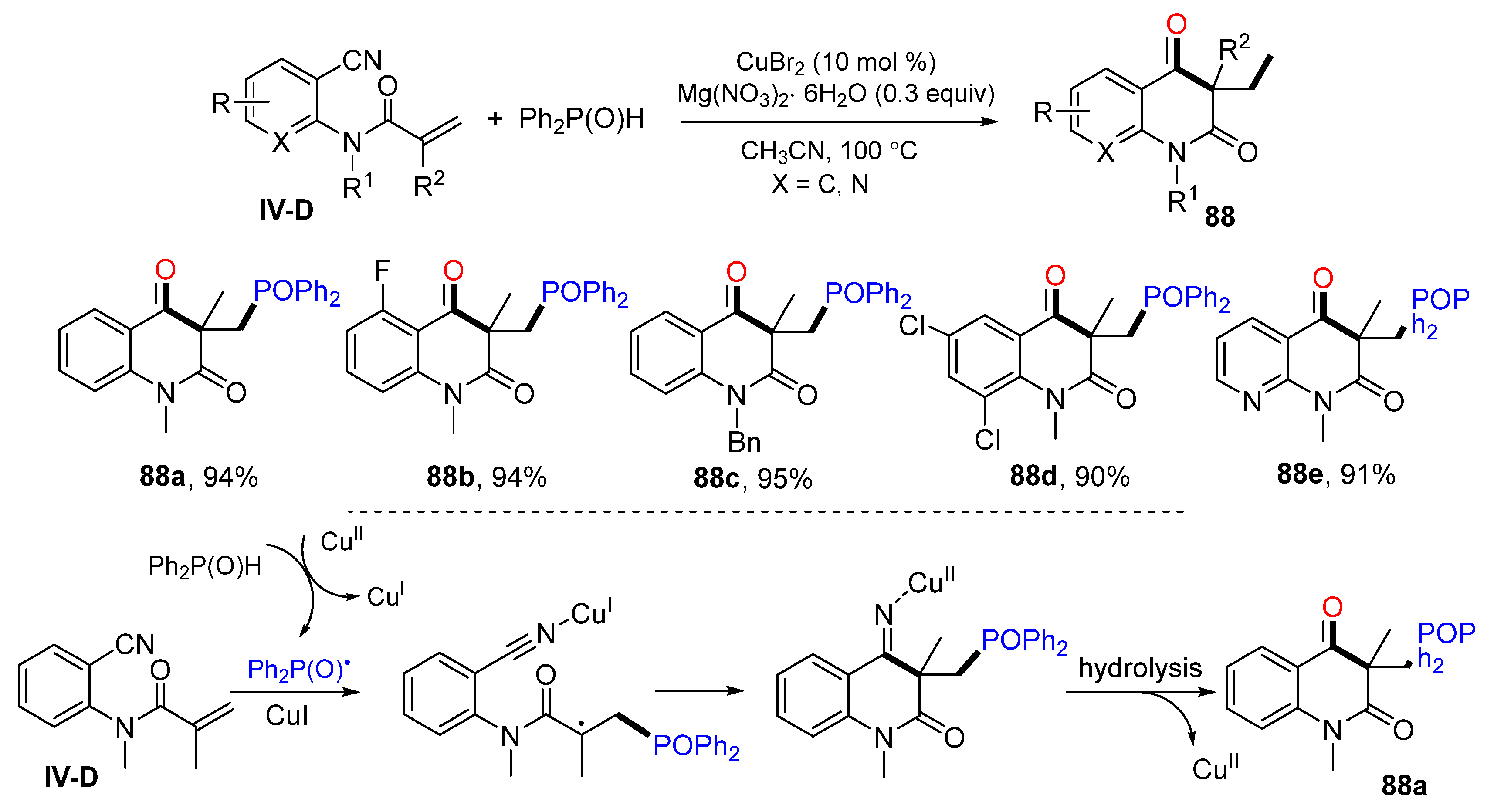
An interesting example of using the cyano group as a radical acceptor for the difunctionalization reaction was reported by the Li group in 2015. It is a Cu-catalyzed radical cyclization of arene-tethered enenitrile for the synthesis of substituted quinoline-2,4(1
H,3
H)-diones. The reaction of
o-cyanoarylacrylamide and diphenyl-phosphine oxide in CH
3CN in the presence of CuBr
2 and Mg(NO
3)
2·6H
2O gave phosphinylated quinoline-2,4(1
H,3
H)-diones
88 in good-to-excellent yields (
Scheme 88) [
98]. The reaction mechanism suggests that the Ph
2P(O) radical derived from Ph
2P(O)H under Cu
II catalysis adds to the C=C double bond of amide followed by 6-
exo cyclization to the CN group and hydrolysis with H
2O to give final product
88a.
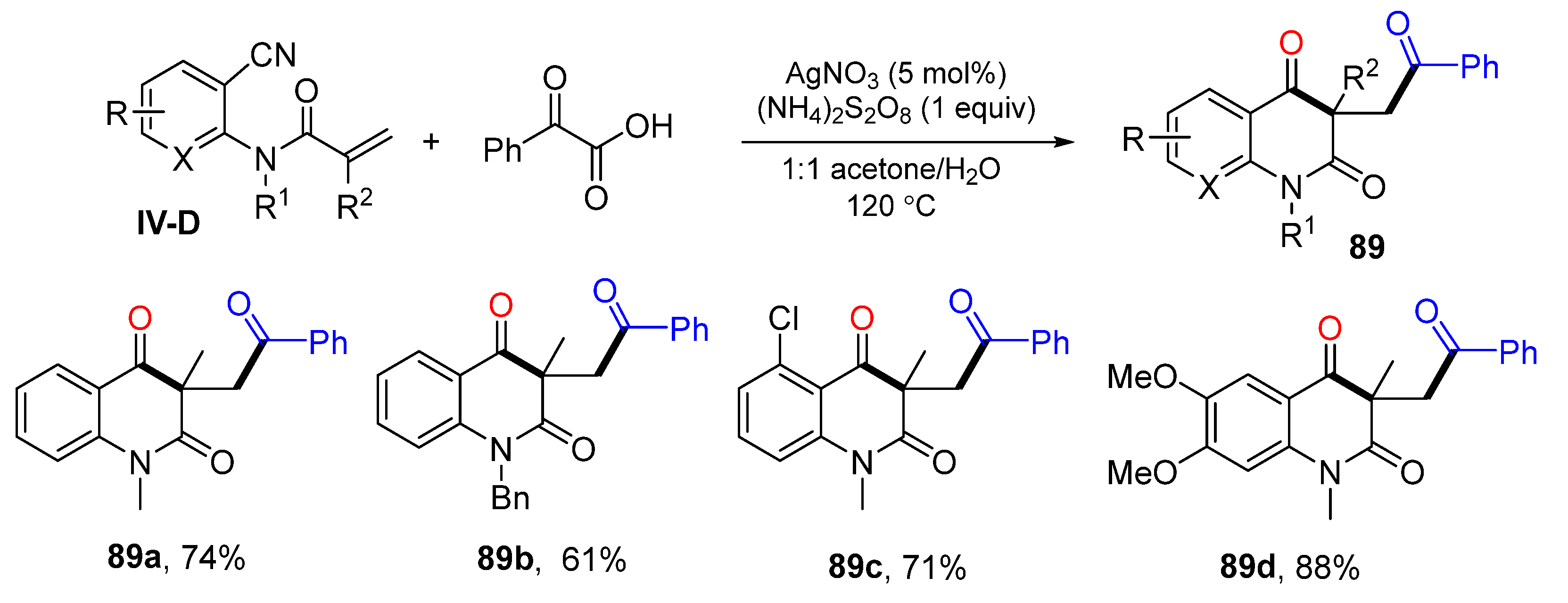
In 2016, the Li group also reported a decarboxylative radical reaction of
o-cyanoarylacrylamides for the preparation of carbonylated quinoline-2,4(1
H,3
H)-diones. The reaction of
o-cyanoarylacrylamide and
α-keto acids in acetone-H
2O at 120 °C under the catalysis of AgNO
3 and (NH
4)
2S
2O
8 gave product
89 in good yields (
Scheme 89) [
99].
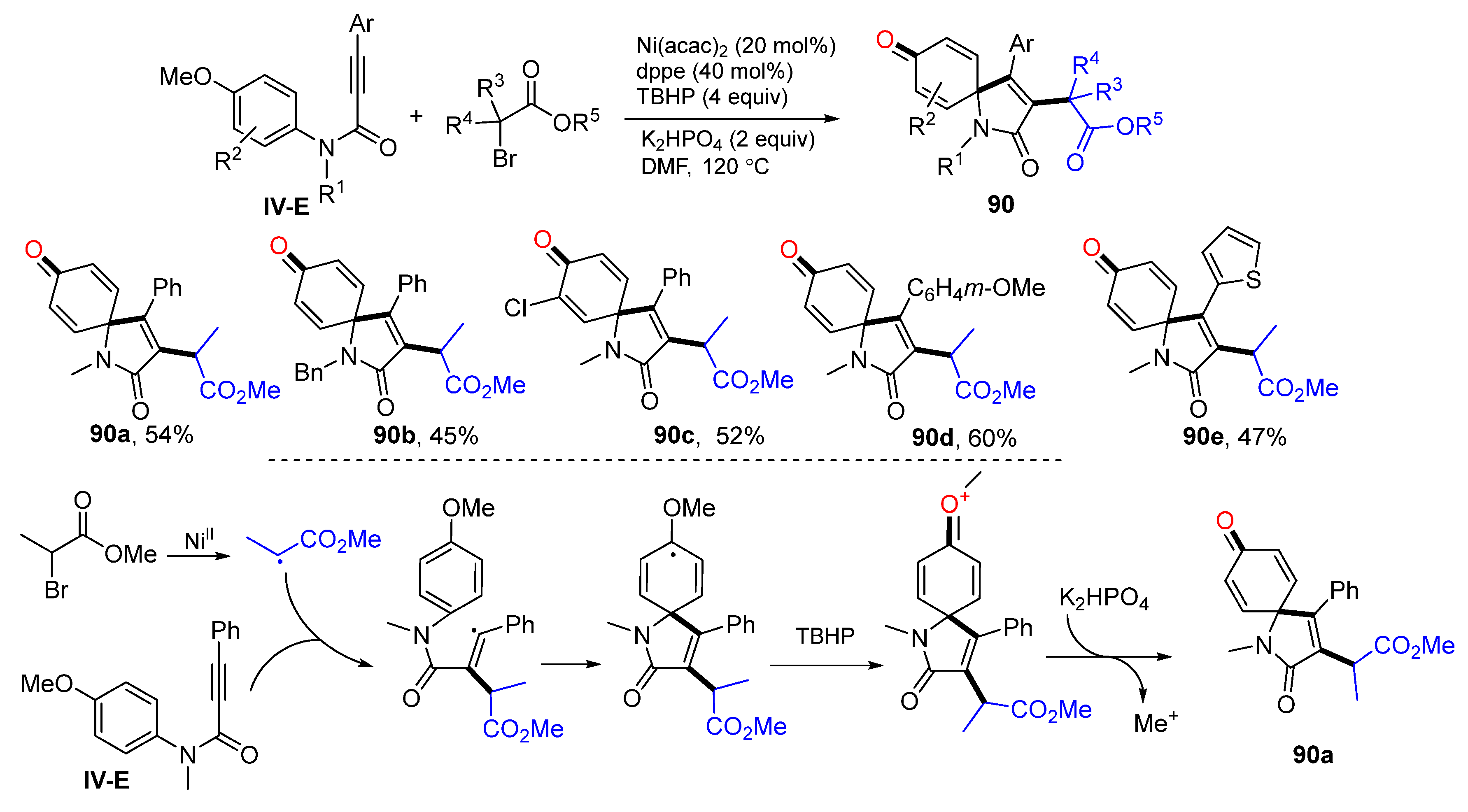
Having a MeO group on the benzene ring is a useful synthetic approach to assist radical cyclization and for dearomatization. In 2017, Li and co-workers developed a Ni-promoted radical spirocyclization of
N-(
p-methoxyaryl)propiolamides for the synthesis of 3-substituted azaspiro[4,5]trienones. The reaction of amides and
α-bromo esters in DMF in the presence of Ni(acac)
2, 1,2-bis(diphenylphosphino)ethane (dppe), TBHP and K
2HPO
4 gave product
90 in moderate yields (
Scheme 90) [
100]. A proposed mechanism suggests that alkyl radical derived from
α-bromo esters adds to the triple bond of amide followed by
ipso-carbocyclization, oxidation with TBHP to form oxonium cation, and a final step of demethylation to give product
90a. The MeO group on the aromatic ring is critical for the radical cyclization and formation of the carbonyl group through diaromatization. The product generated from this method is similar to that presented in
Scheme 73, in which there is no preexisting MeO group on the benzene ring.
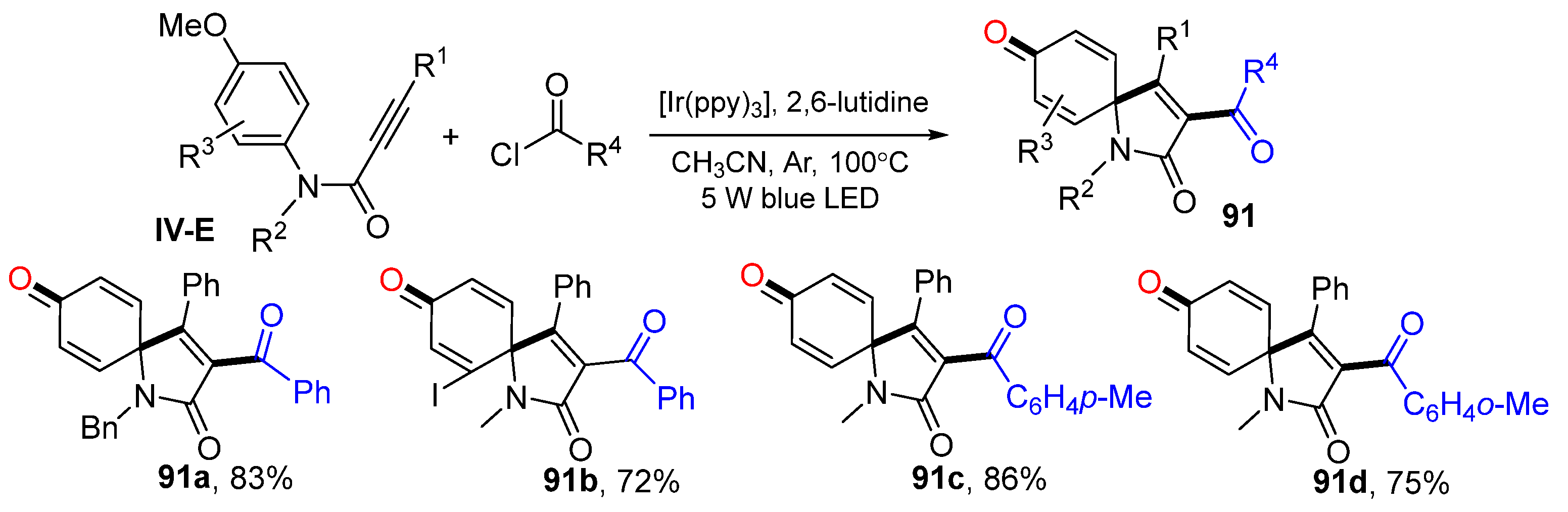
Using a similar synthetic strategy and the alkyne substrate, in 2018, Liu and co-workers reported a visible light-mediated radical spirocyclization of
N-(
p-methoxyaryl)-propiolamides for the synthesis of 3-acylspiroc (
Scheme 91) [
101]. The photo reaction of alkynes and benzoyl chloride in CH
3CN in the presence of Ir
III(ppy)
3 and 2,6-lutidine gave product
91 in good-to-excellent yields.
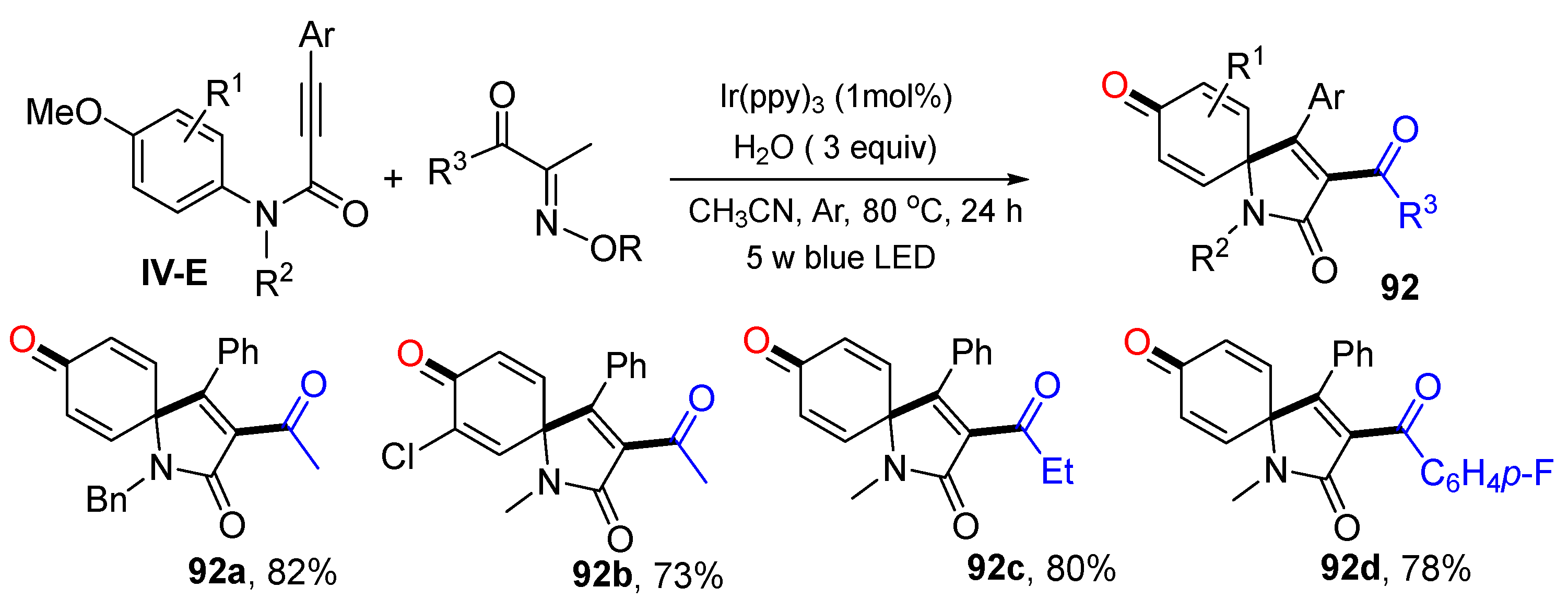
Scheme 92 shows another example of the reaction of
N-(
p-methoxyaryl)-propiolamides developed by Liu’s group also for the synthesis of 3-acylspiro[4,5]trienones [
102].The photoredox reaction of alkynes, acyl oxime esters, H
2O under the catalysis of Ir(ppy)
3 gave product
92 in good yields.
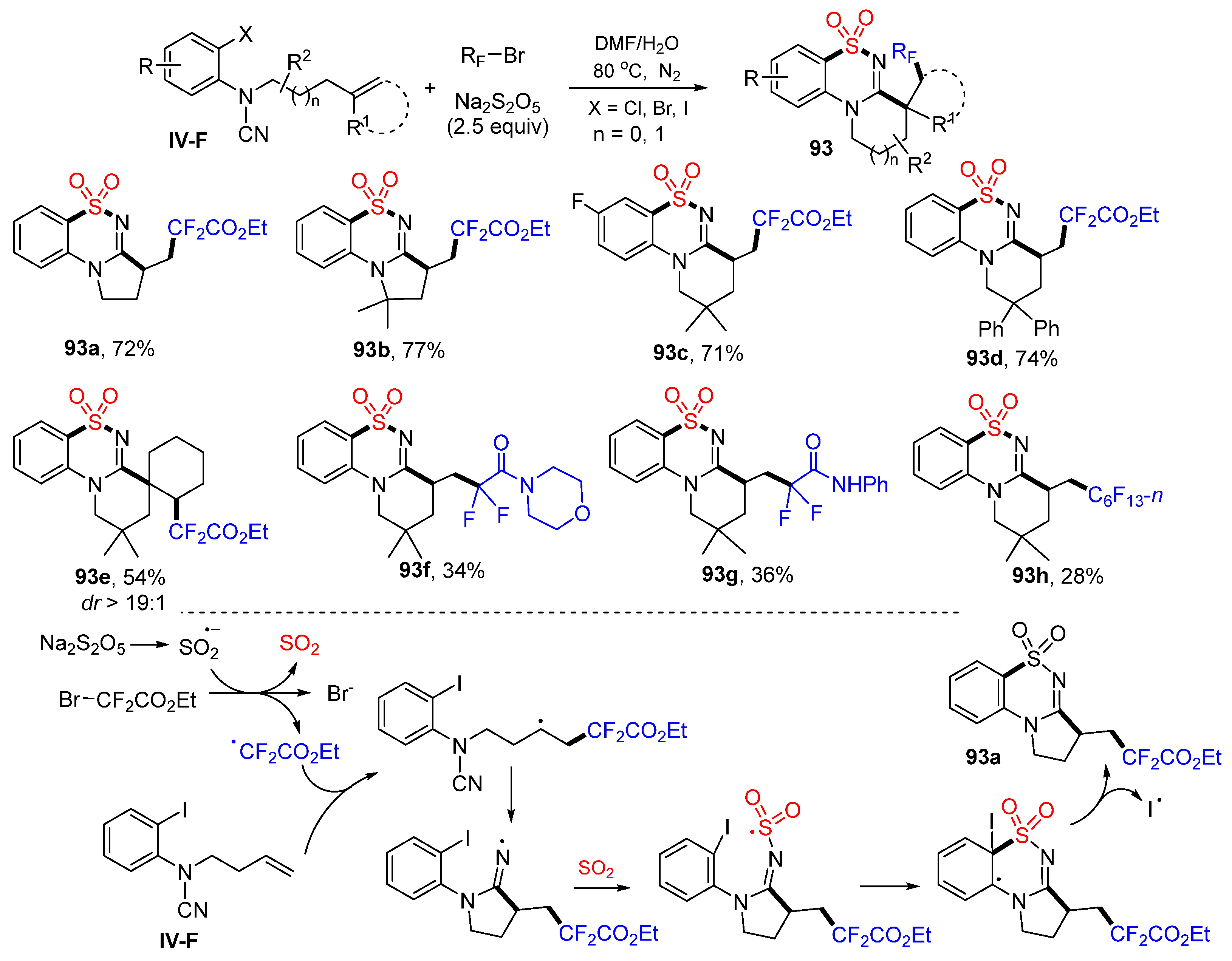
The last example in this section is the reaction of arene-terminated alkene, which has a leaving group X on the aromatic ring. Liao and coworkers employed this substrate in the synthesis of functionalized benzosultams. The reaction of
N-(2-haloaryl)cyanamide, bromodifluoroalkyl reagents and Na
2S
2O
5 in DMF and H
2O at 80 °C afforded product
93 in good yields (
Scheme 93) [
103]. A proposed reaction mechanism suggests that the CF
2CO
2Et radical derived from BrCF
2CO
2Et SO
2 adds to the carbon double bond of amide followed through 5-
exo cyclization to the CN group, capture of SO
2 (generated from Na
2S
2O
5) to form sulfonyl radicals, cyclization to the benzene ring at the carbon with iodine, and a last step of deiodo aromatization to give product
93a.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




























































































