Abstract
Our in-house ethnopharmacological knowledge directed our anti-inflammatory investigation into the leaves of Backhousia mytifolia. Bioassay guided isolation of the Australian indigenous plant Backhousia myrtifolia led to the isolation of six new rare peltogynoid derivatives named myrtinols A–F (1–6) along with three known compounds 4-O-methylcedrusin (7), 7-O-methylcedrusin (8) and 8-demethylsideroxylin (9). The chemical structures of all the compounds were elucidated by detailed spectroscopic data analysis, and absolute configuration was established using X-ray crystallography analysis. All compounds were evaluated for their anti-inflammatory activity by assessing the inhibition of nitric oxide (NO) production and tumor necrosis factor- α (TNF-α) in lipopolysaccharide (LPS) and interferon (IFN)-γ activated RAW 264.7 macrophages. A structure activity relationship was also established between compounds (1–6), noting promising anti-inflammatory potential by compounds 5 and 9 with an IC50 value of 8.51 ± 0.47 and 8.30 ± 0.96 µg/mL for NO inhibition and 17.21 ± 0.22 and 46.79 ± 5.87 µg/mL for TNF-α inhibition, respectively.
1. Introduction
Inflammation is the body’s natural defense response to various harmful stimuli including pathogens, heat, toxic chemicals, and injuries [1]. During the initial stage of trauma or infection, body initiates various cellular and molecular events which include the secretion of many proinflammatory cytokines and chemokines such as interleukin-6 (IL-6), interleukin-1β (IL-1β), tumor necrosis factor-α (TNF-α) as well as reactive oxygen species (ROS) and nitric oxide (NO) to restore tissue homeostasis and resolve acute inflammation [2,3]. The pro-inflammatory cytokines and/or bacterial lipopolysaccharides (LPS) can activate inducible nitric oxide synthase (iNOS) to produce continuously high concentrations of NO, which can then further induce injury at inflammatory sites [4]. Therefore, suppressing the overproduction of NO appears to be a promising strategy for the control of inflammatory diseases.
Backhousia myrtifolia (Myrtaceae) which is commonly referred to as carrol, neverbreak, iron wood, grey myrtle or cinnamon myrtle is a small rainforest tree species that grows in subtropical rainforests regions of Eastern Australia [5,6,7]. It was first discovered and subsequently used by the Indigenous communities of Australia, where the leaves were used to treat colic babies. B. myrtifolia is also known to harbor oils that have cinnamon-like aroma displaying both anti-bacterial and anti-fungal properties [8].
Our ongoing search to discover new anti-inflammatory molecules led us to explore the leaves of B. myrtifolia resulting in the isolation and identification of six new peltogynoid type flavonoids, myrtinols A–F (1–6), along with three known compounds, 4-O-methylcedrusin (7), 7-O-methylcedrusin (8), and 8-demethylsideroxylin (9) (Figure 1).
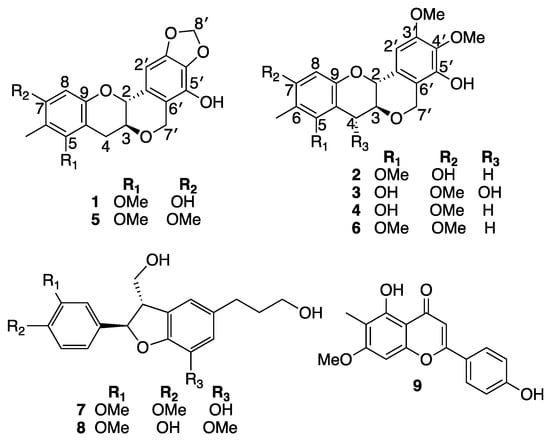
Figure 1.
Structures of compounds 1–9.
2. Structural Elucidation
Myrtinol A (1) was obtained as a green sticky mass. HRESI (−) MS analysis displayed a [M − H]− deprotonated molecular ion peak at m/z 357.0959, corresponding to a molecular formula of C19H17O7 requiring 11 double bond equivalents (DBEs). NMR data (600 MHz, CD3OD) (Table 1, Figure 2) revealed the presence of one olefinic methyl (δH 2.05), one methoxy (δH 3.71), three distinct methylenes, including a methylene dioxy, H2-8’ (δH 5.91, δC 101.9) and an oxy-methylene, H2-7’ (δH 4.89, 4.73, δC 65.8) and a more traditional downfield methylene H2-4 (δH 3.11, 2.63, δC 28.1). The presence of a downfield oxy-methine H-2 (δH 4.53, δC 74.6) and a traditional oxy methine H-3 (δH 3.64, δC 73.1) was also evident. The presence of two aromatic resonances (δH 6.21) and (δH 6.66) both as singlets showed several HMBC correlations to different quaternary carbons and oxy-carbons suggesting the presence of two separate penta-substituted benzene systems. The aromatic proton H-8 (δH 6.21) from the first of the benzene system (ring A) showed HMBC correlations to C-6 (δC 110.5), C-7 (δC 155.2), C-9 (δC 152.9) and C-10 (δC 105.1). The olefinic methyl (δH 2.05, δC 8.6) was assigned to be attached at C-6 in ring A based on having HMBC correlations to C-5 (157.6), C-6 and C-7 (Figure 1). The only methoxy (δH 3.71) in the structure was also assigned to ring A based on a HMBC correlation to C-5 (Figure 1). The aromatic proton H-2’ (δH 6.66) from the second benzene system (ring B) showed HMBC correlations to C-1’ (δC 117.7), C-3’ (δC 133.4), C-4’ (δC 147.6) and C-2 (δC 74.6). These correlations indicated the existence of another heavily substituted benzene ring. HMBC correlations from the methylene dioxy protons H2-8’ (δH 5.91, δC 101.9) to C-3’ and C-4’ confirmed the attachment of this ring system (ring E) onto the benzene ring generating a 1,3-benzodioxole moiety. COSY correlations revealed a single isolated spin system consisting of two oxymethines (H-2 (δH 4.53, δC 74.6), H-3 (δH 3.64, δC 73.1) and a downfield methylene H2-4 (δH 3.11, 2.63, δC 28.1). With the current structural fragments accounting for 9 out of the 11 DBEs, the situation required making room for two additional ring systems to be incorporated in the structure (Figure 1). Key diagnostic HMBC correlations from H2-4 to C-9 and C-10 and H2-7’ to C-1’, C-5’, C-6’ and C-3 confirmed the existence of two heterocyclic ring systems (rings C and D) where ring D was connected to ring B via an oxymethylene bridge representative of the peltogynoid type flavonoid system.

Table 1.
1H NMR data for compounds 1–6.
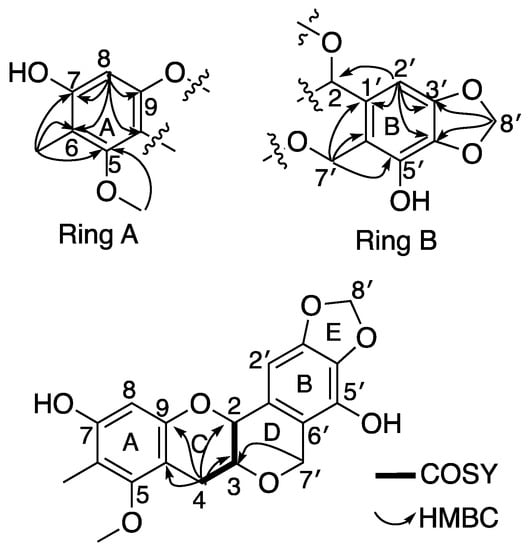
Figure 2.
Diagnostic (600 MHz, CD3OD) HMBC and COSY correlations of 1.
Myrtinol B (2) was obtained as a green solid. HRESI (+) MS analysis revealed a pseudomolecular ion [M + Na]+ ion peak at m/z 397.1247 ([M + Na]+, calcd 397.1263) in accordance with the molecular formula of C20H22O7Na requiring ten double bond equivalents. It was evident early on from the NMR spectrum and data that (2) was a close structural analogue of myrtinol A (1), and comparison of the spectroscopic data of 1 with 2 showed that 2 retained the ring system A–D. The only difference was the presence of two additional methoxy resonances at (δH 3.88 and δH 3.79) exhibiting HMBC correlations to C-3′ (δC 136.2) and C-4′ (δC 153.2), respectively, with the absence of the methylene dioxy resonance which supported the opening up of ring E (Table 1, Table 2 and Table S3 and Figure 3).
Table 2.
13C NMR data for compounds 1–6.
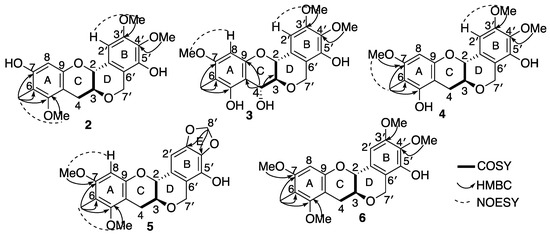
Figure 3.
Key diagnostic 2D NMR correlations of compounds 2–6.
In addition to this, we were successful in generating a crystal for compound 2, which was then subjected to X-ray crystallography and the data obtained assisted in confirmation of the structure (Figure 4, Table S1) and helped resolve the absolute stereochemistry for the chiral centers C-2 and C-3 as R and S. From the NMR data alone, a large coupling of JH-2/H-3 (9.5 Hz) also suggested a trans confirmation.
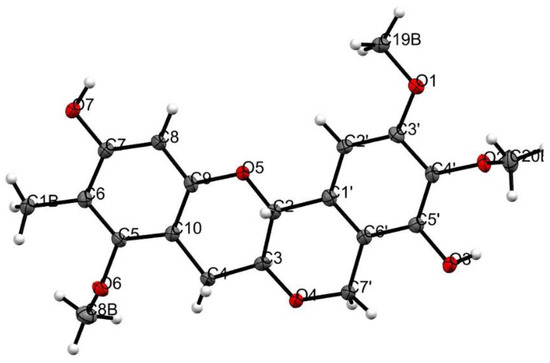
Figure 4.
ORTEP diagram of 2. The compounds in the asymmetric unit are an inversion of each other.
Myrtinol C (3) was obtained as a white solid. Its molecular formula was determined as C20 H21 O8 from HRESI (−) MS ion analysis at m/z (389.1246 ([M − H]−, calcd 389.1236). A close comparison of the spectroscopic data of 3 with 2 confirmed that myrtinol C possessed the same ring system as myrtinol B, with the only exceptions being the positional change of the hydroxy and methoxy groups present in the ring A and the introduction of a hydroxy functionality C-4 (δH 5.01, δC 70.6). This above structural changes were supported by HMBC correlation from H-4 (δH 5.01) to C-3 (δC 78.3), C- 9 (δC 154.0) and C-10 (δC 103.8), from H-8 (δH 6.15) to C- 6 (δC 106.8), C- 7 (δC 160.0), C-9 and C-10, from H-6 (δH 1.98) to C-5, C-6, C-7, and from H-7 (δH 3.77) to C-7 (Table 1, Table 2 and Table S4 and Figure 3). A large coupling of JH-3/H-4 (8.3 Hz) was suggestive of a trans confirmation, placing the absolute stereochemistry of C-4 to be R.
Myrtinol D (4) was obtained as a white solid. The molecular formula was revealed as C20H21O7 based on HRESI (−) MS ion analysis at m/z 373.1284 ([M − H]−, calcd 373.1287), which was identical to myrtinol B (2). Myrtinol D (4) was identified as a structural isomer of (2) with the only difference being the positional change between the meta coupled hydroxy and methoxy functionalities in ring A of the benzene unit. This was supported by HMBC correlations from 7-OMe (δH 3.76) to C-7 (δC 158.5) and H-8 (6.19) to C-7.
Myrtinol E (5) was obtained as a yellow solid. The molecular formula was revealed as C20H19 O7 from HRESI (−) MS ion analysis at m/z 371.1122 ([M − H]−, calcd 371.1131. Myrtinol E (5) showed the closest similarity to its analogue myrtinol A (1). The only difference in the NMR data and spectra was the detection of an additional methoxy resonance, which was confirmed to be attached at C-7 based on HMBC correlations of 7-OMe (δH 3.80) to C-7 (δC 158.8) and H-8 (δH 6.36) to C-7.
Myrtinol F (6) was obtained as a green sticky mass. The molecular formula was revealed as C21H23O7 from HRESI (−) MS ion analysis at m/z 387.1454 ([M − H]−, calcd 387.1444. Comparison of the spectroscopic data of 2 and 6 showed that myrtinol F possessed the same ring system as myrtinol B, with the only change being the replacement of hydroxy substituent at C-7 by a methoxy group, which was supported by HMBC correlation from H-8 (δH 6.36) C- 7(δC 157.8), and 7-OMe (δH 3.80) to C-7.
We also take into account that the level of purity of myrtinol F was not 100%, with the possibility of a terpene like impurity present in this fraction (Figure S35). Low yields and the level of difficulty experienced separating this two-compound mixture prevented us from obtaining an absolutely pure sample of myrtinol F. However, the HRMS data and the clear key NMR resonances attributed to myrtinol F allowed for its complete structural assignment.
The absolute stereochemistry for the chiral centers C-2 and C-3 for the remaining myrtinol analogues was assigned as R and S, respectively, based on the large coupling constant between JH-2/H-3 which was suggestive of a trans configuration along with the consideration of a likely biosynthetic relationship to myrtinol B (2) (Table 1 and Table 2) [9,10].
In addition to the discovery of the new myrtinols, three known compounds 4-O-methylcedrusin (7), 7-O-methylcedrusin (8) and 8-demethylsideroxylin (9) were identified by interpretation of their spectroscopic data (Figures S37, S39 and S41) and a close comparison with published data [11,12,13,14].
3. Anti-Inflammatory Activity
All compounds were assessed for their anti-inflammatory activity by evaluating the inhibition of NO production and TNF-α in LPS plus interferon (IFN)-γ activated RAW 264.7 macrophages. All compounds were also evaluated for their cytotoxicity using the Alamar blue assay (Table 3).
Table 3.
In vitro anti-inflammatory activity of compounds 1–9 in LPS + IFN-γ activated RAW 264.7 macrophages.
4. Discussion
As shown in Table 3, the varying anti-inflammatory activity depended mostly on the functional groups attached to rings A, B and C. Based on the slight structural variations around the tetracyclic backbone of myrtinols A–F (1–6), we have attempted to evaluate the observed structural-activity relationship (SAR) among them. A SAR was observed between compounds 1 and 5 (both having a 1,3-benzodioxole moiety attached to ring B) which showed promising inhibition of NO production and TNF-α production, with a IC50 values of 11.47 ± 0.14, 24.54 ± 0.28 and 8.51 ± 0.47, 17.21 ± 0.22 µg/mL, respectively. In comparison, compounds 2 and 6 showed NO production inhibition with an IC50 value of 16.25 ± 0.77 and 12.62 ± 0.26 µg/mL, and TNF α production inhibition with an IC50 value 52.35 ± 7.47, and 30.55 ± 5.01 µg/mL, respectively, suggesting that the presence of 1,3-benzodioxole moiety (ring E) and methoxy group at C-5 and C-7 might play an important role in their anti-inflammatory activity. Interestingly, when comparing the SAR between compounds 3 and 4, the only change between them being the introduction of a hydroxyl group at C-4 in 3, this rendered the molecule inactive compared to the baseline activity observed for compound 4 where the NO inhibition was determined to be 29.31 ± 10.95 µg/mL.
We are mindful of the fact that the experimental NO inhibition values obtained for myrtinols may not be a true reflection of their anti-inflammatory activity profile mainly due to the fact that they mostly have a low therapeutic index associated with them. This suggests that future investigations on assessing their cytotoxic activity need to be performed in order to re-evaluate their potential as cytotoxic agents.
Among the known compounds, Compound 9 displayed interesting activity with an IC50 value 8.30 ± 0.96 µg/mL and IC50 value of 46.79 ± 5.87 µg/mL, whereas 7 and 8 did not show good anti-inflammatory activity (Table 3).
Peltogynoids are a rare type of cyclized flavonoid, where a carbon atom is attached between 3-OH and C-2′ of ring B [9,15]. Robinson and Robinson isolated the first peltogynoid type flavonoid, peltogynol in 1935 [16]. Phytochemical studies of Australian and South African Acacia species including A. carnei by Roux and co-workers identified additional peltogynoid derivatives, (+)-2,3-trans-3,4- trans- peltogynols, (+)-2,3-trans-3,4-cis-peltogynol, (+)-2,3-trans-peltogynone, carnein, peltogynin, β-photomethylquercitin [17]. Peltogynoid derivates have been reported to be isolated from different genera and species including Goniorrhachis marginatu Taub [18], Pltogyne caringae, P. confertiflora; P. paniculate [19], Caesalpinia pulcherrima [20], Acacia nilotica (L.) Delile [21], Phytolacca icosandra [22], Bougainvillea spectabilis [23]. The new peltogynoid derivatives reported in this paper bear the closest resemblance to the reported pubeschin analogues [24].
5. Experimental Section
5.1. General Experimental Procedures
UV spectra were recorded on an Agilent Carry UV-Vis Multicell Peltier spectrometer. NMR spectroscopic data were recorded on a Bruker Avance 600 MHz spectrometer (Bruker Biospin GmbH, Germany). HRMS (High Resolution Mass Spectrometry) was carried out using a Waters SYNAPT G2-Si mass spectrometer operating in the positive and negative ESI mode.
5.2. Plant Material
The leaves of B. myrtifolia were collected from the Australian Botanic Garden at Mount Annan (NSW, Australia). A voucher specimen (2005-0104) has been deposited at the Australian Botanic Gardens, at Mount Annan, NSW, Australia.
5.3. Extraction and Bioactivity-Directed Isolation
The fresh leaves of B. myrtifolia (300 g) were crushed using a hand blender and extracted sequentially using organic solvents based on their polarity (n-hexane, dichloromethane (DCM), ethyl acetate (EtOAc), ethanol (EtOH), methanol (MeOH), and finally, water) using a Buchi-811 Soxhlet Extraction system. Immediately after the initial stages of sequential fractionation, each corresponding fraction was subjected to anti-inflammatory screening using the inhibition of NO in LPS plus IFN-γ treated RAW 264.7 macrophages following the Griess test. The most active extract (DCM) (Table S8) was resuspended in EtOH and was then later subjected to semi-preparative HPLC (Agilent 1260 Infinity II series) using an Agilent C18 column (5 µm, 250 × 9.4 mm) eluting at 1.8 mL/min from 10% MeCN/H2O to 100% MeCN (with a constant 0.01% FA (formic acid) modifier) over 60 mins and held for a further 6 mins and then equilibrated back to 10% MeCN/H2O in 1 min and maintained at 10% MeCN/H2O for an additional 3 mins, to give 17 fractions (Fr. 1–17) which included four pure fractions (Fr. 4 (7), Fr.8 (1), Fr.9 (2), and Fr.15 (8), corresponding to the following compounds: 7 (6.6 mg, tR 24.7 min), 1 (5.4 mg, tR 39.1 min), 2 (4.2 mg, tR 41.7 min), and 8 (2.5 mg, tR 50.3 min).
Fr.11 was re-purified by SB-C3 semipreparative column, (250 × 9.4 mm) using a gradient system of 50–60% MeCN/H2O with a flow rate of 2mL/min (with a constant 0.01% FA modifier) over 30 min to afford compounds 3 (1.4 mg, tR 22.4 min), and 4 (2.0 mg, tR 23.1 min). Fr.16 was purified using SB-Phenyl semipreparative column, (250 × 9.4 mm) with a gradient system of 10–80% MeCN/H2O over 15 mins followed by a change from 80–100% MeCN/H2O over 9 mins with a constant flow rate of 2 mL, and then held at 100% MeCN for 2 mins and equilibrated back to 10% MeCN/ H2O in 1 min and maintained at this gradient for an additional 2 min, to afford compound 5 (1.9 mg, tR 21.8 min). Fr.17 was repurified using a semi-preparative Agilent C18 column (5 µm, 9.4 × 250 mm) by employing a gradient system of 10–90% MeCN/H2O over 15 mins followed by a gradient change of 90–100% MeCN/ H2O for an additional 5 min, and then held at 100% MeCN for an additional 2 min followed by an equilibration to 10% MeCN/H2O in 3 mins, affording compound 6 (6.4 mg, tR 18.3 min). Compound 9 (4 mg, tR 13.4 min) was acquired from Fr.5 using SB-Phenyl semipreparative column, (250 × 9.4 mm) eluting with 10–55% MeCN/H2O (with a constant 0.01% FA modifier) over 8 min at 2 mL/min, followed by a gradient change from 55–70% MeCN/H2O for an additional 7 min, and finally with a 70–100% MeCN/H2O (with a constant 0.01% FA modifier) over 2 min and then equilibrated back to 10% MeCN within 1 min and maintained 10% MeCN/H2O for an additional 2 min.
Myrtinol A (1): Greenish sticky mass [α]25D +172.9 (c 0.001, MeOH); UV-Vis λmax (MeOH) nm (log ε) 208 (5.31), 232 (5.37) and 280 (5.44); 1D and 2D NMR (600 MHz, CD3OD) data (see Table 1, Table 2 and Table S2); HRESI (−) MS m/z 357.0959 [M − H]− (calcd for C19H17O7−; 357.0974).
Myrtinol B (2): Green crystal (DCM:MeOH) [α]25D +178.3 (c 0.001, MeOH); UV-Vis λmax (MeOH) nm (log ε) 208 (5.31), 233 (5.37) and 281 (5.45); 1D and 2D NMR (600 MHz, CD3OD) data (see Table 1, Table 2 and Table S3); HRESI (+) MS m/z 397.1247 [M + Na]+ (calcd for C20H22O7Na; 397.1263).
Myrtinol C (3): White solid [α]25D +176.1 (c 0.0003, MeOH); UV-Vis λmax (MeOH) nm (log ε) 208 (5.31), 233 (5.37) and 280 (5.44); 1D and 2D NMR (600 MHz, CD3OD) data (see Table 1, Table 2 and Table S4); HRESI (−) MS m/z 389.1246 [M − H]−, (calcd for C20 H21 O8 389.1236).
Myrtinol D (4): White solid [α]25D +174.6 (c 0.0005, MeOH); UV-Vis λmax (MeOH) nm (log ε) 208 (5.31), 232 (5.37) and 280 (5.44); 1D and 2D NMR (600 MHz, CD3OD) data (see Table 1, Table 2 and Table S5); HRESI (−) MS m/z 373.1284 [M − H]−, (calcd for C20H21O7−; 373.1287).
5.4. X-ray Crystallographic Analysis of 2
Crystals were obtained by slow cooled evaporation from MeOH: DCM (1:1) solution; suitable crystals were selected for X-ray crystallographic analysis using an MX1 beamline at the Australian Synchrotron, using silicon double crystal monochromated radiation (λ = 0.71073 Å) at 100 K [25]. The XDS software package [26] was used on site for data integration, processing, and scaling. SADABS [27] was used to apply an empirical absorption correction. Shelxt [28] was applied to solve the structure by the intrinsic phasing method, and a suite of SHELX programs [28,29] were used for refinement, via the Olex2 graphical interface [30]. Crystallographic data of 2 (CCDC number: 2236594) was deposited at the Cambridge Crystallographic Data Centre. Additional crystallographic information is available in the Supporting Information (Table S1).
5.5. Crystal Data for 2
C20H21O7 (M = 373.37 g/mol); triclinic, 0.2 × 0.1 × 0.1 mm3, space group P1, V = 872.6(3) Å3, Z = 2, Dc = 1.421 g/cm3, F(000) = 394.0, Mo Kα radiation, λ = 0.71073 Å, T = 100 K, μ = 0.108 mm−1; 2θrange = 3.552 to 57.202°, 20,884 reflections collected, 6838 unique (Rint = 0.0191); final GooF = 1.048, R1 = 0.0383 [I > 2σ(I)], wR2 = 0.1031; absolute structure parameter = 0.06(13).
5.6. Maintenance of RAW 264.7 Macrophages
Cells were grown in 75 cm2 flasks on Dulbecco’s Modified Eagle Medium (DMEM) containing 10% fetal bovine serum (FBS) that was supplemented with penicillin (100 U/mL), streptomycin (100 μg/mL) and L-glutamine (2 mM). The cell line was maintained in 5% CO2 at 37 °C, with media being replaced every 3–4 days. Once cells had grown to confluence in the culture flask, they were harvested using a rubber policeman, as opposed to using trypsin, which can remove membrane-bound receptors.
5.7. Pro-Inflammatory Activation of Cells
RAW 264.7 cells (1 × 106 cells/mL) were seeded in 96 well plates (Corning® Costar®, Sigma, Sydney, Australia) overnight until confluency. When the cells were confluent, each compound was serially diluted from a starting concentration of 100 μg mL−1 to construct a dose response curve (i.e., 100, 50, 25, 12.5, 6.25, and 3.13 μg mL−1) and co-incubated with cells for 1 hr prior to the addition of 1 μg mL−1 LPS and 10 U mL−1 (1 unit = 0.1 ng/mL) IFN-γ. After activation, the cells were incubated for another 24 hrs at 37 °C. The supernatant was then collected for NO, and TNF-α assays. The cells were subjected to cell viability measurement using the Alamar Blue assay. Non-activated cells (exposed to media only) were used as negative control and activated cells were positive control.
5.8. Determination of Nitrite by the Griess Assay
Nitric oxide was determined by the Griess reagent as described in previous studies [31]. Griess reagent was freshly made up of equal volumes of 1% sulfanilamide in 5% phosphoric acid and 0.1% N-1-naphthylethylenediamine dihydrochloride in Milli-Q water. From each well, 50 µL of supernatant was transferred to a fresh 96-well plate and mixed with 50 µL of Griess reagent. The production of nitrite as an indicator of NO was measured at 540 nm in a POLARstar Omega microplate reader (BMG Labtech, Mornington, Australia).
5.9. Determination of TNF-α by ELISA
The stored supernatants were analyzed for TNF-α synthesis using a commercial ELISA kit (Peprotech, Brisbane, Australia) according to the manufacturer’s instructions. The absorbance was measured at 410 nm [32]. The concentrations of TNF-α in the experimental samples were extrapolated from a standard curve.
5.10. Determination of Cell Viability by the Alamar Blue Assay
After various treatments and the stimulation by LPS and IFN-γ overnight, 100 µL of Alamar Blue solution [10% Alamar Blue (resazurin) in DMEM media] was added to cells and incubated at 37 °C for 2 hrs. The fluorescence intensity was measured with excitation at 530 nm and emission at 590 nm using a microplate reader. The results were expressed as a percentage of the intensity to that of control cells (non-activated cells).
5.11. Statistical Analysis
Data analysis was carried using GraphPad Prism 9.3.1. Calculations were performed using MS-Excel version 16.61.1. IC50 values were obtained by using the sigmoidal dose–response function in GraphPad Prism. The results were expressed as mean ± standard deviation (SD).
6. Conclusions
In conclusion, we have isolated and characterized six new rare peltogynoid flavonoids from the leaves of Backhousia myrtifolia. Myrtinols exhibited promising anti-inflammatory activity; however, their low therapeutic index warrants further cytotoxic evaluation as part of a future investigation.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28052160/s1, Figures S1-S46: NMR and HRMS spectra for compounds 1-9 Tables S1-S8: X-ray crystallography and NMR data for compounds 1-6.
Author Contributions
Conceptualization, R.R.; methodology, S.M; investigation, S.M; data curation, S.M and K.Z; resources, F.B.; writing- original draft preparation, R.R and S.M; writing-review and editing, R.R., S.M., G.M., X.Z., F.L., and K.Z. project administration, R.R.; supervision, R.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research has received no external funding.
Informed Consent Statement
Not applicable.
Data Availability Statement
NMR data can be obtained by contacting the corresponding author.
Acknowledgments
The authors would like to acknowledge Ignacio Czajkowski, Royal Botanic Garden (Mt. Annan) for the identification of the plant material. We thank Scott Willis (Biomedical Magnetic Resonance Facility Manager, Western Sydney University) for providing his technical expertise in NMR. We also would like to acknowledge the Mass Spectrometry Facility (MFS) of Western Sydney University. The crystallographic data were collected at the MX1 beamline of the Australian Synchrotron, Clayton, Victoria, Australia. We also thank the Australian Synchrotron at Australia’s Nuclear Science and Technology Organisation for travel support and their staff for beamline assistance. We are also grateful for the assistance of Meena Mikhael, Elise Wright, and Shawan Karan.
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Samples of the compounds are not available from the authors.
References
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.M.; An, J. Cytokines, inflammation, and pain. Int. Anesthesiol. Clin. 2007, 45, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Kany, S.; Vollrath, J.T.; Relja, B. Cytokines in Inflammatory Disease. Int. J. Mol. Sci. 2019, 20, 6008. [Google Scholar] [CrossRef] [PubMed]
- Rajapakse, N.; Kim, M.M.; Mendis, E.; Kim, S.K. Inhibition of inducible nitric oxide synthase and cyclooxygenase-2 in lipopolysaccharide-stimulated RAW264.7 cells by carboxybutyrylated glucosamine takes place via down-regulation of mitogen-activated protein kinase-mediated nuclear factor-kappaB signaling. Immunology 2008, 123, 348–357. [Google Scholar]
- Barrett, D.J.; Ash, J.E. Growth and carbon partitioning in rainforest and eucalypt forest species of south coastal New South Wales, Australia. Aust. J. Bot. 1992, 40, 13. [Google Scholar] [CrossRef]
- Sarana, S. Physiological and biochemical changes during heat stress induced browning of detached Backhousia myrtifolia (cinnamon myrtle) tissues. Trop. Plant Biol. 2015, 33, 31–39. [Google Scholar]
- Sinclair, R.J.; Hughes, L. Leaf mining in the Myrtaceae. Ecol. Entomol. 2008, 33, 623–630. [Google Scholar] [CrossRef]
- Hornsby Shire Council. Backhousia Myrtifolia; Hornsby Shire Council: Hornsby, NSW, Australia, 2005.
- Malan, E.; Roux, G.D. (+)-2,3-Trans-pubeschin, the first catechin analogue of peltogynoids from Peltogyne pubescens and P. venosa. Phytochemsitry 1973, 13, 1575–1579. [Google Scholar] [CrossRef]
- Drewes, S.E.; Mashimbye, M.J.; Field, J.S.; Ramesar, N. 11, 11-dimethyl-1, 3, 8, 10-tetrahydroxy-9-methoxypeltogynan and three pentacyclic triterpenes from Cassine transvaalensis. Phytochemistry 1991, 30, 3490–3493. [Google Scholar] [CrossRef]
- Pieters, L.; De Bruyne, T.; Claeys, M.; Vlietinck, A.; Calomme, M.; vanden Berghe, D. Isolation of a dihydrobenzofuran lignan from South American Dragon’s Blood (Croton spp.) as an inhibitor of cell proliferation. J. Nat. Prod. 1993, 56, 899–906. [Google Scholar] [CrossRef]
- Pieters, L.; De Bruyne, T.; De Groot, A.; Mei, G.; Dommisse, R.; Lemière, G.; Vlietinck, A. NMR study of some dihydrobenzofuran lignans. Magn. Reson. Chem. 1993, 31, 692–693. [Google Scholar] [CrossRef]
- In, S.J.; Seo, K.H.; Song, N.Y.; Lee, D.S.; Kim, Y.C.; Baek, N.I. Lignans and neolignans from the stems of Vibrunum erosum and their neuroprotective and anti-inflammatory activity. Arch. Pharm. Res. 2015, 38, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Cardona, M.L.; Seoan, E. Flavonoids and xanthonolignoids of Hypericum ericoides. Phytochemistry 1982, 21, 2759–2760. [Google Scholar] [CrossRef]
- Roux, D.G.; Ferreira, D. α-Hydroxychalcones as intermediates in flavonoid biogenesis: The significance of recent chemical analogies. Phytochemsitry 1974, 13, 2039–2048. [Google Scholar] [CrossRef]
- Robinson, G.M.; Robinson, R. Leuco-anthocyanins and leuco-anthocyanidins. Part I. The isolation of peltogynol and its molecular structure. J. Chem. Soc. 1935, 744–752. [Google Scholar] [CrossRef]
- Brandt, E.V.; Ferreira, D.; Roux, D.G. Metabolites from the Purple Heartwood of Mimosoideae. Part 2. Acacia carnei Maiden: Isolation, Synthesis, and Reactions of Crombeone. J. Chem. Soc. Perkin Trans. 1981, 1, 514–521. [Google Scholar] [CrossRef]
- Gottlieb, O.R.; De Sousa, J.R.D. Peltogynoids of Goniorrhachis marginata. Phytochemistry 1972, 11, 2841–2846. [Google Scholar] [CrossRef]
- De Almeida, M.E.; Gottlieb, O.R.; Teixeira, M.A. New peltogynoids from three Peltogyne species. Phytochemistry 1974, 13, 1225–1228. [Google Scholar] [CrossRef]
- McPherson, D.D.; Cordell, G.A.; Soejarto, D.D.; Pezzuto, J.M.; Fong, H.H. Peltogynoids and homoisoflavonoids from Caesalpinia pulcherrima. Phytochemistry 1983, 22, 2835–2838. [Google Scholar] [CrossRef]
- Ahmadu, A.; Abdulkarim, A.; Grougnet, R.; Myrianthopoulos, V.; Tillequin, F.; Magiatis, P.; Skaltsounis, A.L. Two new peltogynoids from Acacia nilotica Delile with kinase inhibitory activity. Planta Med. 2010, 76, 458–460. [Google Scholar] [CrossRef]
- Galarraga Montes, E.; Amaro-Luis, J.M. Icosandrin, a novel peltogynoid from the fruits of Phytolacca icosandra (Phytolaccaceae). Nat. Prod. Res. 2016, 30, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Do, L.T.; Aree, T.; Siripong, P.; Pham, T.N.; Nguyen, P.K.; Tip-Pyang, S.; Bougainvinones, A.-H. Peltogynoids from the Stem Bark of Purple Bougainvillea spectabilis and Their Cytotoxic Activity. J. Nat. Prod. 2016, 79, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Ngnokam, D.; Massiot, G.; Nuzillard, J.M.; Tsamo, E. (+)-7′, 7′-Dimethyl-5-hydroxy-2R, 3S-trans-pubeschin from Entandrophragma cylindricum. Phytochemistry 1994, 37, 529–531. [Google Scholar] [CrossRef]
- Cowieson, N.P.; Aragao, D.; Clift, M.; Ericsson, D.J.; Gee, C.; Harrop, S.J.; Mudie, N.; Panjikar, S.; Price, J.R.; Riboldi-Tunnicliffe, A.; et al. MX1: A bending-magnet crystallography beamline serving both chemical and macromolecular crystallography communities at the Australian Synchrotron. J. Synchrotron Rad. 2015, 22, 187–190. [Google Scholar] [CrossRef]
- Kabsch, W.X. Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J. Appl. Crystallogr. 1993, 26, 795–800. [Google Scholar] [CrossRef]
- SADABS, Version 2014/5; Bruker AXS Inc.: Madison, WI, USA, 2001.
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta. Cryst. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G. Programs for Crystal Structure Analysis; University of Göttingen: Göttingen, Germany, 2014. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Raju, R.; Mathew, S.; Reddell, P.; Munch, G. Ternstroenol F: A new pentacyclic triterpenoid saponin isolated from the Australian rainforest plant Ternstroemia cherryi. Nat. Prod. Res. 2022; Online ahead of print. [Google Scholar]
- Zhou, X.; Razmovski-Naumovski, V.; Chang, D.; Li, C.; Kam, A.; Low, M.; Bensoussan, A.; Chan, K. Synergistic Effects of Danshen (Salvia Miltiorrhiza Radix et Rhizoma) and Sanqi (Notoginseng Radix et Rhizoma) Combination in Inhibiting Inflammation Mediators in RAW264.7 Cells. Medicines 2017, 4, 85. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).