
Antimicrobial Activity of the Secondary Metabolites Isolated from a South African Red Seaweed, Laurencia corymbosa

 , and
, and
Abstract
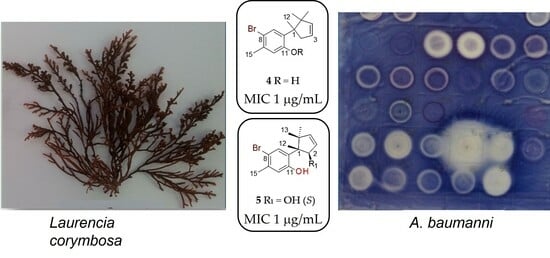
1. Introduction
2. Results and Discussion
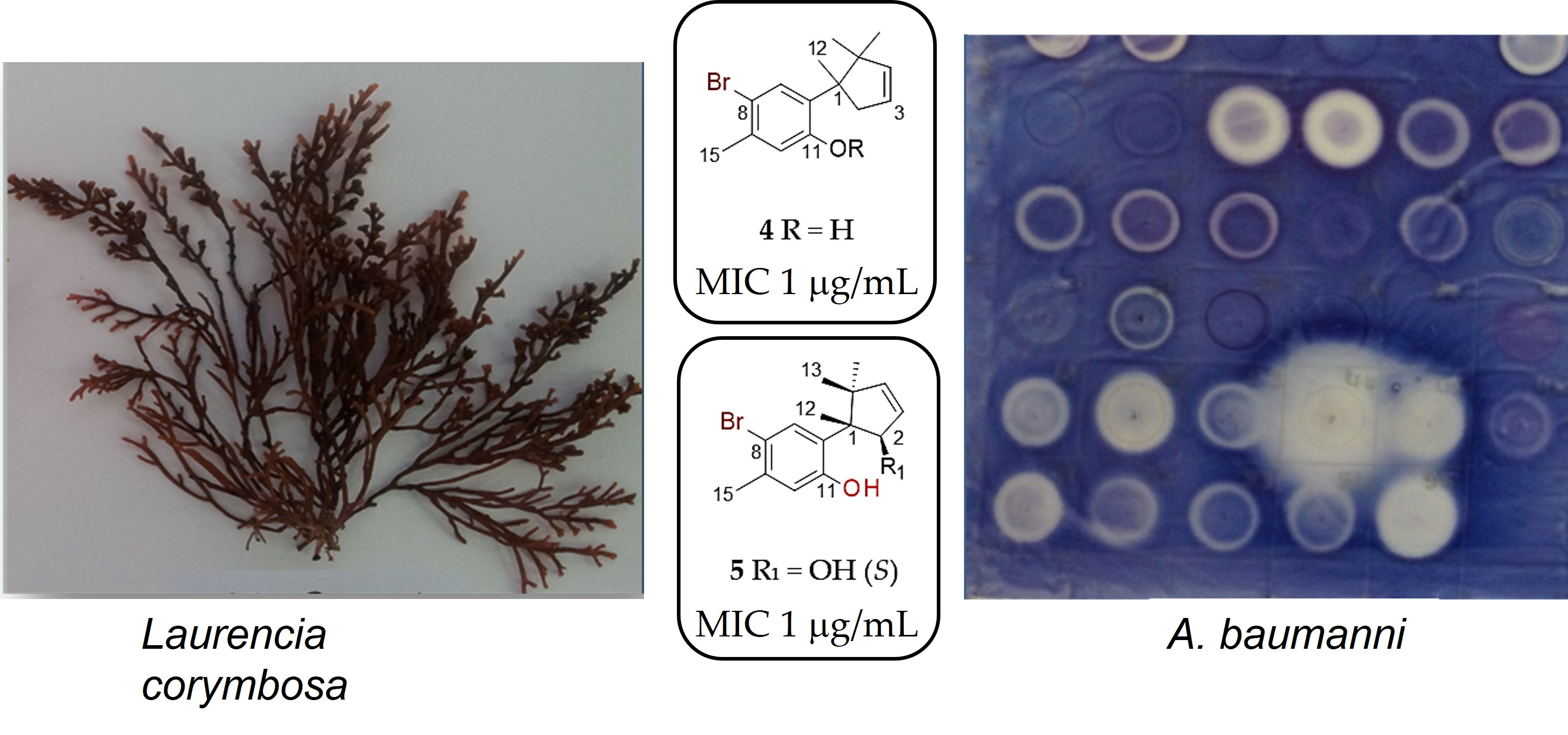
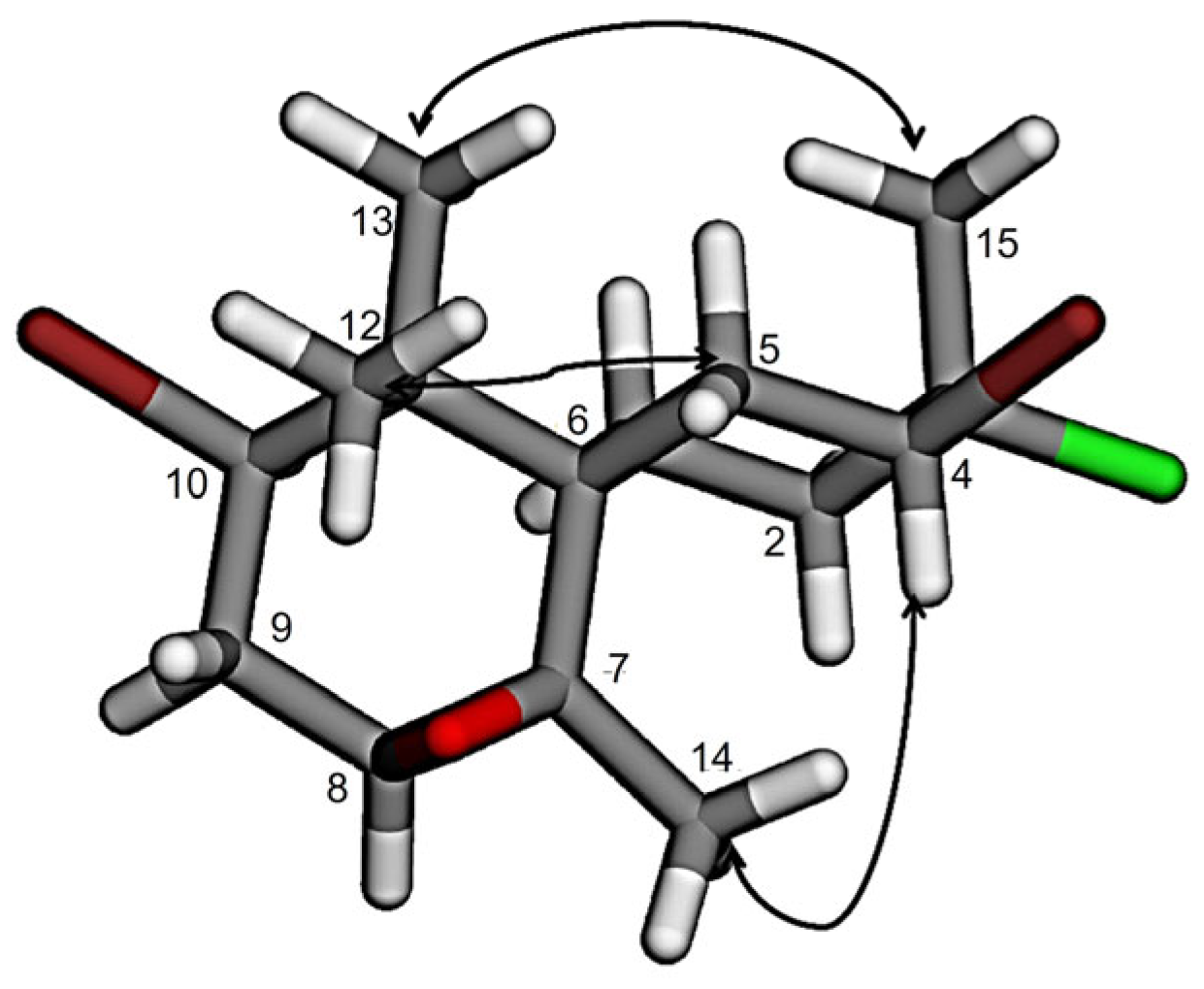
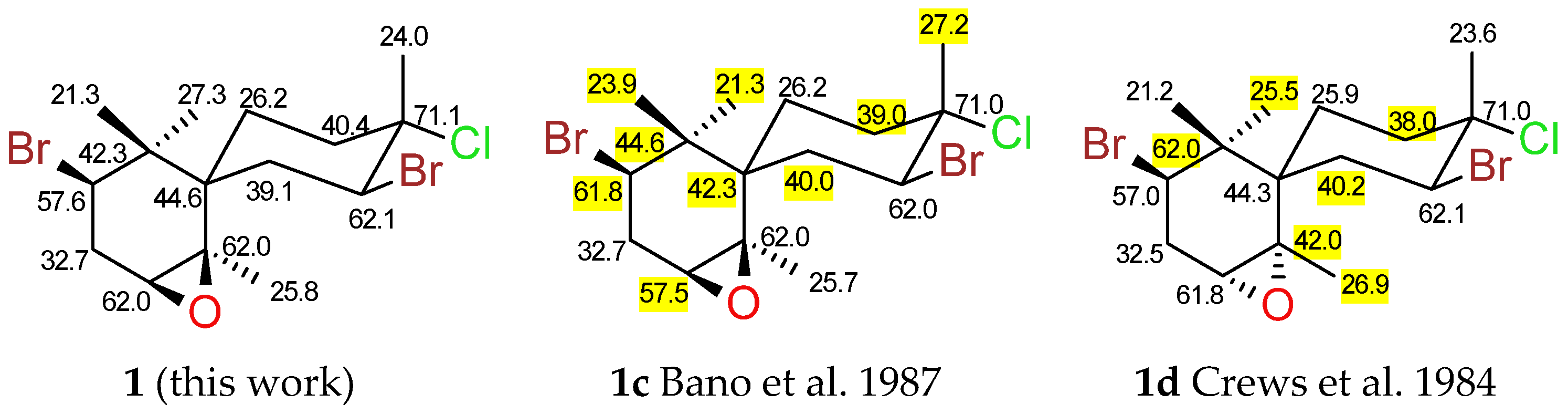
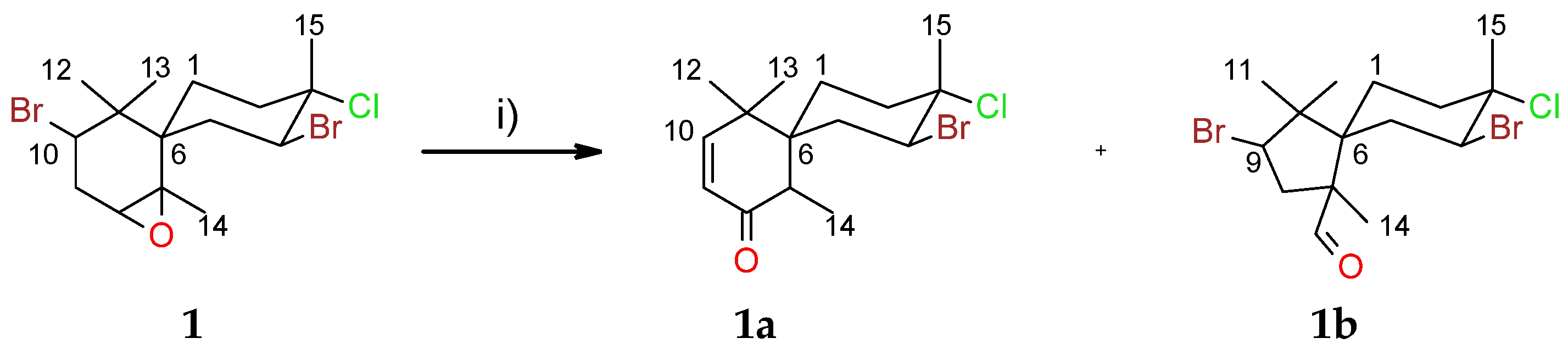
2.1. Chamigranes
2.2. Linear Acetogenin
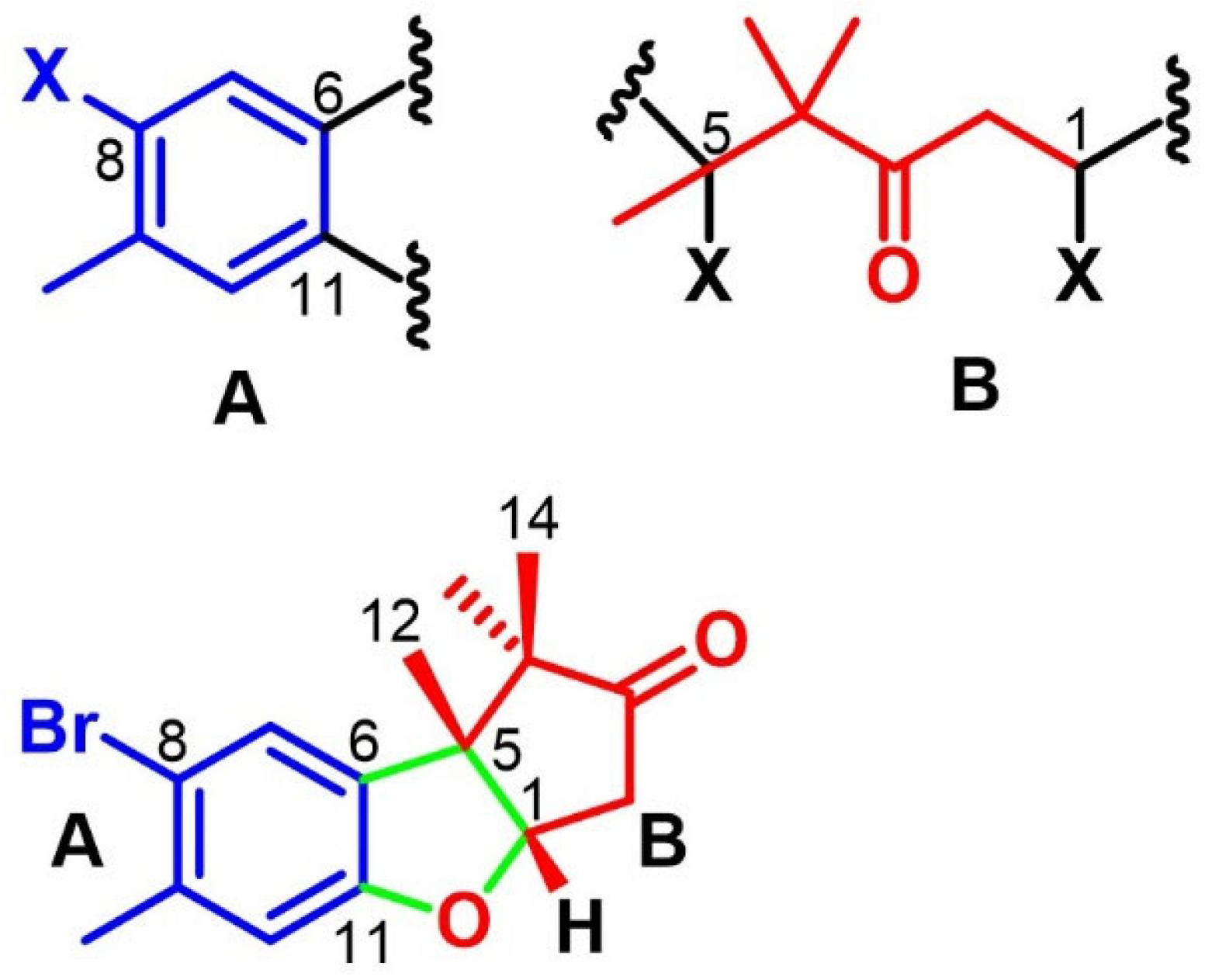
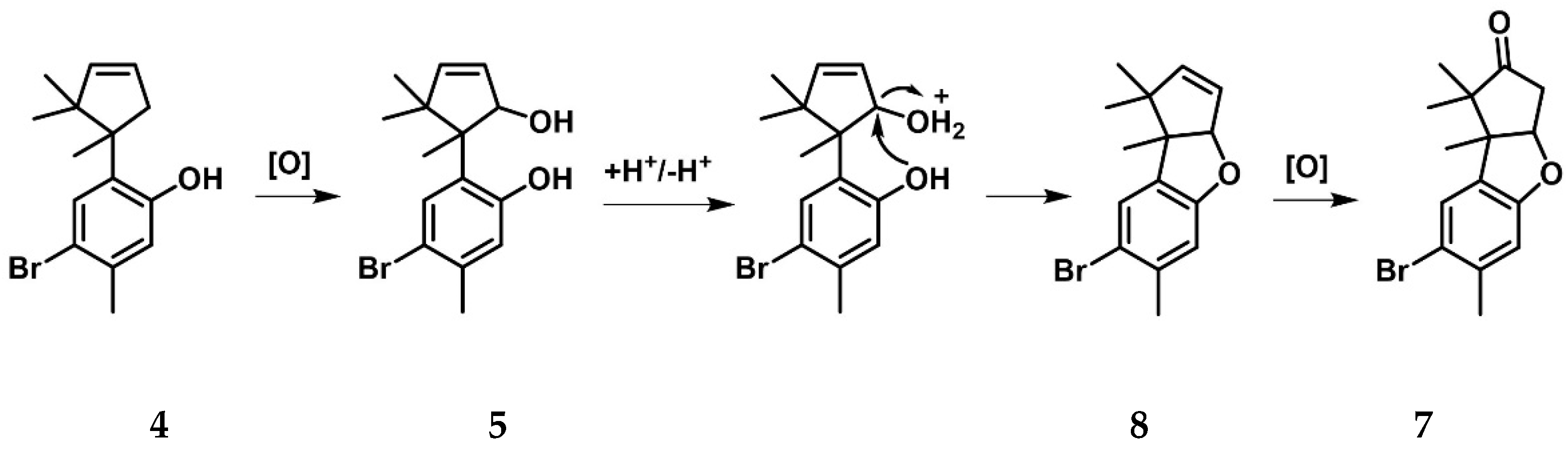
2.3. Cuparanes
2.4. Antimicrobial Study
3. Conclusions
4. Experimental Section
4.1. General Experimental Procedures
4.2. Plant Material and Taxonomy
4.3. Extraction and Isolation
Compounds Isolated from L. corymbosa
4.4. Antimicrobial Activity Study
4.4.1. Preparation of the Extracts
4.4.2. Initial Antimicrobial Screening—Disk Diffusion Assays
4.4.3. Preparation of the Test Strains
4.4.4. Bioautography
4.4.5. Liquid Assays
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Correction Statement
References
- Papon, N.; Copp, B.R.; Courdavault, V. Marine drugs: Biology, pipelines, current and future prospects for production. Biotechnol. Adv. 2022, 54, 107871. [Google Scholar] [CrossRef]
- NIH US National Library of Medicine. ClinicalTrials.gov. 2000–2022. Available online: https://clinicaltrials.gov/ (accessed on 4 February 2023).
- NCI. NCI Thesaurus. 2022. Available online: https://ncit.nci.nih.gov/ (accessed on 4 February 2023).
- ACDREVIEW. Journal of Antibody-drug Conjugates. 2022. Available online: https://www.adcreview.com/ (accessed on 4 February 2023).
- Mayer, A. Marine Pharmacology. 2022. Available online: https://www.marinepharmacology.org (accessed on 4 February 2023).
- Newman, D.J. The “utility” of highly toxic marine-sourced compounds. Mar. Drugs 2019, 17, 324. [Google Scholar] [CrossRef]
- Gerwick, W.; Moore, B. Lessons from the past and charting the future of marine natural products drug discovery and chemical biology. Chem. Biol. 2012, 19, 85–98. [Google Scholar] [CrossRef]
- Zeng, Y.; Yang, D.; Qiu, P.; Han, Z.; Zeng, P.; He, Y.; Guo, Z.; Xu, L.; Cui, Y.; Zhou, Z.; et al. Efficacy of Heparinoid PSS in treating cardiovascular diseases and beyond—A review of 27 years clinical experiences in China. Clin. Appl. Thromb. Hemost. 2016, 22, 222–229. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, L.; Jiao, W. Marine glycan-derived therapeutics in China. Prog. Mol. Biol. Transl. Sci. 2019, 163, 113–134. [Google Scholar]
- Ventola, C.I. The antibiotic resistance crisis: Part 1: Causes and threats. Pharm. Ther. 2015, 40, 277–283. [Google Scholar]
- Leal, M.C.; Munro, M.H.G.; Blunt, J.W.; Puga, J.; Jesus, B.; Calado, R.; Rosa, R.; Madeira, C. Biogeography and biodiscovery hotspots of macroalgal marine natural products. Nat. Prod. Rep. 2013, 30, 1380–1390. [Google Scholar] [CrossRef]
- Harizani, M.; Ioannou, E.; Roussis, V. The Laurencia Paradox: An Endless Source of Chemodiversity. In Progress in the Chemistry of Organic Natural Products; Kinghorn, A.D., Falk, H., Gibbons, S., Kobayashi, J., Eds.; Springer International Publishing: Cham, Switzerland, 2016; Volume 102, pp. 91–252. [Google Scholar]
- Harizani, M.; Diakaki, D.I.; Perdikaris, S.; Roussis, V.; Ioannou, E. New C15 Acetogenins from Two Species of Laurencia from the Aegean Sea. Molecules 2022, 27, 1866. [Google Scholar] [CrossRef]
- Wang, B.-G.; Gloer, J.B.; Ji, N.-Y.; Zhao, J.-C. Halogenated organic molecules of Rhodomelaceae origin: Chemistry and biology. Chem. Rev. 2013, 113, 3632–3685. [Google Scholar] [CrossRef]
- Kubanek, J.; Jensen, J.P.R.; Keifer, P.A.; Sullards, M.; Collins, D.O.; Fenical, W. Seaweed resistance to microbial attack: A targeted chemical defense against marine fungi. Proc. Natl. Acad. Sci. USA 2003, 100, 6916–6921. [Google Scholar] [CrossRef]
- Moloney, M.G. Natural products as a source for novel antibiotics. Trends Pharmacol. Sci. 2016, 37, 689–701. [Google Scholar] [CrossRef]
- Wright, G.D. Opportunities for natural products in 21st century antibiotic discovery. Nat. Prod. Rep. 2017, 34, 694–701. [Google Scholar] [CrossRef]
- Fakee, J. The Isolation, Characterisation and Chemotaxonomic Significance of Secondary Metabolites from Selected South African Laurencia spp. Rhodophyta. Ph.D. Thesis, Rhodes University, Grahamstown (Makhanda), South Africa, 2015. [Google Scholar]
- Howard, B.M.; Fenical, W. Structures and chemistry of two new halogen-containing chamigrene derivatives from Laurencia. Tetrahedron Lett. 1975, 21, 1687–1690. [Google Scholar] [CrossRef]
- Cowe, H.J.; Cox, P.J.; Howie, R.A. Structure of 2,10-dibromo-3-chloro-7R,8S-epoxychamigrene. Z. Krist. Cryst. 1989, 188, 1–4. [Google Scholar]
- Ojika, M.; Shizuri, Y.; Yamada, K. A halogenated chamigrane epoxide and six related halogen-containing sesquiterpenes from the red alga Laurencia okamurai. Phytochemistry 1982, 21, 2410–2411. [Google Scholar] [CrossRef]
- Bano, S.; Ali, M.S.; Ahmad, V.U. Marine Natural Products; VI. A Halogenated Chamigrene Epoxide from the Red Alga Laurencia pinnatifida. Planta Med. 1987, 53, 508. [Google Scholar] [CrossRef]
- Crews, P.; Naylor, S.; Hanke, J.F.; Hogue, R.E.; Kho, E.; Braslau, R. Halogen regiochemistry and substituent stereochemistry determination in marine monoterpenes by carbon-13 NMR. J. Org. Chem. 1984, 49, 1371–1377. [Google Scholar] [CrossRef]
- Elsworth, J.F.; Thomson, R.H. A new chamigrane from Laurencia glomerata. J. Nat. Prod. 1989, 52, 893–895. [Google Scholar] [CrossRef]
- Kigoshi, H.; Shizuri, Y.; Niwa, H.; Yamada, K. Laurencenyne, a plausible precursor of various nonterpenoid C15-compounds, and neolaurencenyne from the red alga Laurencia okamurai. Tetrahedron Lett. 1981, 22, 4729–4732. [Google Scholar] [CrossRef]
- Norte, M.; Gonzalez, A.G.; Cataldo, F.; Rodriguez, M.L.; Brito, I. New examples of acyclic and cyclic C-15 acetogenins from laurencia pinnatifida. Reassignment of the absolute configuration for E and Z pinnatifidienyne. Tetrahedron 1991, 47, 9411–9418. [Google Scholar] [CrossRef]
- Dias, D.A.; White, J.M.; Urban, S. Laurencia filiformis: Phytochemical profiling by conventional and HPLC-NMR approaches. Nat. Prod. Commun. 2009, 4, 157–172. [Google Scholar] [CrossRef]
- Kladi, M.; Vagias, C.; Furnari, G.; Moreau, D.; Roussakis, C.; Roussis, V. Cytotoxic cuparene sesquiterpenes from Laurencia microcladia. Tetrahedron Lett. 2005, 46, 5723–5726. [Google Scholar] [CrossRef]
- Yu, X.Q.; He, W.F.; Liu, D.Q.; Feng, M.T.; Fang, Y.; Wang, B.; Feng, L.H.; Guo, Y.W.; Mao, S.C. A seco-laurane sesquiterpene and related laurane derivatives from the red alga Laurencia okamurai Yamada. Phytochemistry 2014, 103, 162–170. [Google Scholar] [CrossRef]
- Yamada, K.; Yazawa, H.; Uemura, D.; Toda, M.; Hirata, Y. Total synthesis of (±) aplysin and (±) debromoaplysin. Tetrahedron 1969, 25, 3509–3520. [Google Scholar] [CrossRef]
- Mann, M.G.A. An Investigation of the Antimicrobial and Antifouling Properties of Marine Algal Metabolites. Master’s Thesis, Rhodes University, Grahamstown, South Africa, 2008. [Google Scholar]
- Lategan, C.; Kellerman, T.; Afolayan, A.F.; Mann, M.G.; Antunes, E.M.; Smith, P.J.; Bolton, J.J.; Beukes, D.R. Antiplasmodial and antimicrobial activities of South African marine algal extracts. Pharm. Biol. 2009, 47, 408–413. [Google Scholar] [CrossRef]
- Vlachos, V.; Critchley, A.T.; von Holy, A. Antimicrobial activity of extracts from selected southern African marine macroalgae. S. Afr. J. Sci. 1997, 93, 328–332. [Google Scholar]
- Johansson, M.; Sterner, O.; Labischinski, H.; Anke, T. Coprinol, a new antibiotic cuparane from a Coprinus species. Z. Nat. C 2001, 56c, 31–34. [Google Scholar] [CrossRef]
- Li, H.-Y.; Yang, W.-Q.; Zhou, X.-Z.; Shao, F.; Shen, T.; Guan, H.-Y.; Zheng, J.; Zhang, L.-M. Antibacterial and Antifungal Sesquiterpenoids: Chemistry, Resource, and Activity. Biomolecules 2022, 12, 1271. [Google Scholar] [CrossRef]
- Francis, C.M. Systematics of the Laurencia complex (Rhodomelaceae, Rhodophyta) in South Africa. Ph.D. Thesis, University of Cape Town, Cape Town, South Africa, 2015. [Google Scholar]
- Francis, C.M.; Bolton, J.J.; Mattio, L.; Mandiwana-Neudani, T.G.; Anderson, R.J. Molecular systematics reveals increased diversity within the South African Laurencia complex (Rhodomelaceae, Rhodophyta). J. Phycol. 2017, 53, 804–819. [Google Scholar] [CrossRef]
- Bolton, J.J. The problem of naming commercial seaweeds. J. Appl. Phycol. 2020, 32, 751–758. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Atom No. | δC (Mult.) | δH, Mult, J (Hz) | COSY | HMBC |
|---|---|---|---|---|
| 1 | 56.3 (C) | - | - | - |
| 2a | 47.6 (CH2) | 3.01, d, 16.1 | H2b | - |
| 2b | 2.50, d, 16.1 | H2a | - | |
| 3 | 126.8 (CH) | 5.83, br s | H2a, H2b | C1 1 |
| 4 | 144.3 (CH) | 5.67, br s | H2a, H2b 1 | - |
| 5 | 49.9 (C) | - | - | - |
| 6 | 133.4 (C) | - | - | - |
| 7 | 132.0 (CH) | 7.41, s | H12, H15 | C5, C8, C9, C11, C15 |
| 8 | 114.7 (C) | - | - | - |
| 9 | 136.5 (C) | - | - | - |
| 10 | 119.0 (CH) | 6.65, s | H15 | C6, C8, C11, C15 |
| 11 | 154.1 (C) | - | - | - |
| 12 | 25.3 (CH3) | 1.47, s | - | C1, C2, C5, C6 |
| 13 | 25.9 (CH3) | 1.19, s | - | C41, C5, C14 |
| 14 | 24.9 (CH3) | 0.75, s | - | C4, C5, C13 |
| 15 | 22.1 (CH3) | 2.30, s | H7, H10 | C8, C9, C10 |
| 5 | 6 | |||||
|---|---|---|---|---|---|---|
| Atom No. | δC | δH, Mult, J (Hz) | NOESY | δC | δH, Mult, J (Hz) | NOESY |
| 1 | 55.2 | - | - | 56.0 | - | - |
| 2 | 85.3 | 4.50, d, 7.4 | H12, H13 | 85.6 | 4.28, br s | H14 |
| 3 | 131.8 | 5.92, d, 5.5 | H4 | 131.0 | 6.02, d, 5.5 | H4 |
| 4 | 141.4 | 6.13, d, 5.5 | H3 | 144.6 | 6.33, d, 5.5 | H3 |
| 5 | 48.9 | - | - | 47.1 | - | - |
| 6 | 128.7 | - | - | 129.7 | - | - |
| 7 | 132.2 | 7.27, s | H12 | 132.1 | 7.26, s | H12 |
| 8 | 115.5 | - | - | 115.1 | - | - |
| 9 | 137.3 | - | - | 137.0 | - | - |
| 10 | 119.7 | 6.64, s | H15 | 119.0 | 6.62, s | - |
| 11 | 154.0 | - | - | 153.8 | - | - |
| 12 | 23.4 | 1.38, s | H2, H7, H13 | 26.4 | 1.49, s | H7, H13 |
| 13 | 23.4 | 1.19, br s | H2, H12, H14 | 18.8 | 1.22, s | H12, H14 |
| 14 | 20.3 | 0.66, s | H13 | 27.1 | 0.66, s | H2, H13 |
| 15 | 22.2 | 2.30, s | H10 | 22.3 | 2.29, s | H10 |
| Atom No. | δC (Mult.) | δH, Mult, J (Hz) | COSY | HMBC | NOESY |
|---|---|---|---|---|---|
| 1 | 89.1 (CH) | 4.86, dd, 6.9, 3.5 | H2 | - | H2, H12, H14 |
| 2 | 40.7 (CH2) | 2.82, m | H1 | C3 | H12, H13 |
| 3 | 218.5 (C) | - | - | - | - |
| 4 | 51.9 (C) | - | - | - | - |
| 5 | 55.2 (C) | - | - | - | - |
| 6 | 132.1 (C) | - | - | - | - |
| 7 | 127.6 (CH) | 7.23, s | - | C9, C11 | H12, H13, H14 |
| 8 | 115.4 (C) | - | - | - | - |
| 9 | 138.3 (C) | - | - | - | - |
| 10 | 112.4 (CH) | 6.69, s | H15 | C15 | H15 |
| 11 | 159.2 (C) | - | - | - | - |
| 12 | 21.8 (CH3) | 1.32, s | - | C1, C4, C5, C6 | H1, H2, H7 |
| 13 | 23.1 (CH3) | 0.86, s | H1, H2 | C3, C4, C5, C14 | H2, H7 |
| 14 | 20.2 (CH3) | 1.08, s | - | C3, C4, C5, C13 | H1 |
| 15 | 23.3 (CH3) | 2.34, s | - | C8, C9, C10 | - |
| Ab | Ef | Ec | Sa | Ca | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sample | MIC (μg/mL) | dd (Ba) | MIC (μg/mL) | dd (Ba) | MIC (μg/mL) | dd (Ba) | MIC (μg/mL) | dd (Ba) | MIC (μg/mL) | dd (Ba) |
| 1 | - | - | 1 | - | 100 | - | - | 18.85 | - | - |
| 2 | - | - | 100 | - | - | - | - | 30.63 | - | - |
| 3 | - | - | 1 | - | - | - | - | - | - | - |
| 4 | 1 | (rg) | 100 | (rg) | - | - | 50 | 76.18 | - | 24.74 (rg) |
| 4a | - | - | 100 | - | 100 | - | - | - | - | - |
| 5 | 1 | - | - | (rg) | - | - | - | (rg) | - | (rg) |
| 6 | - | - | 100 | (rg) | - | - | - | 24.74 (rg) | - | (rg) |
| 7 | - | - | - | - | 100 | - | - | - | - | - |
| crude | - | - | - | (rg) | 100 | - | - | 31.42 | - | - |
| Vancomycin | 100 | - | 100 | - | 100 | - | 20 | - | 100 | - |
| Ampicillin | 100 | - | 1 | - | 1 | - | >100 | - | 100 | - |
| Actidione | 100 | - | 50 | - | 100 | - | 100 | - | >100 | - |
| Atom No. | δC (Mult.) | δH, Mult, J (Hz) | COSY | HMBC | NOESY |
|---|---|---|---|---|---|
| 1a | 26.2 (CH2) | 1.67, dq, 15.2, 5.7 | H1b, H2a | - | H1b, H13, H14 |
| 1b | 1.98, dt, 15.2, 5.7 | H1a, H2a, H2b | - | H1a, H13 | |
| 2a | 40.4 (CH2) | 2.47, dt, 14.6, 5.7 | H1a, H1b | C1, C3, C15 | H2b, H14 |
| 2b | 2.38, dd, 14.6, 2.9 | H1b | C1, C3, C4, C6, C15 | H2a, H15 | |
| 3 | 71.1 (C) | - | - | - | - |
| 4 | 62.1 (CH) | 4.77, dd, 13.3, 4.7 | H5a, H5b | C3, C5, C15 | H5a, H14 |
| 5a | 39.1 (CH2) | 2.84, br d, 13.3 | H4, H5b | - | H4 |
| 5b | 2.10, t, 13.3 | H4, H5a | C4, C7, C6, C11 | H1a, H12, H13, H15 | |
| 6 | 44.6 (C) | - | - | - | - |
| 7 | 62.0 (C) | - | - | - | - |
| 8 | 62.0 (CH) | 2.92, t, 3.0 | H9 | C9, C10, C14 | H9, H14 |
| 9 | 32.7 (CH2) | 2.61, dd, 7.7, 3.0 | H8, H10 | C7/8, C10, C11 | H8, H10, H12, H13 |
| 10 | 57.6 (CH) | 4.06, t, 7.7 | H9, H12 | C6, C8, C9, C12, C13 | H1a, H9, H13 |
| 11 | 42.3 (C) | - | - | - | - |
| 12 | 21.3 (CH3) | 1.18, s | H10 | C6, C11, C13 | H5a, H5b, H9 |
| 13 | 27.3 (CH3) | 1.08, s | - | C6, C11, C2 | H1a, H1b, H5b, H15 |
| 14 | 25.8 (CH3) | 1.50, s | - | C6, C7/8 | H2a, H4, H8 |
| 15 | 24.0 (CH3) | 1.74, s | - | C2, C3, C4 | H1a, H2b, H5b, H13 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fakee, J.; Bolton, J.J.; Le Roes-Hill, M.; Durrell, K.A.; Antunes, E.; Beukes, D.R. Antimicrobial Activity of the Secondary Metabolites Isolated from a South African Red Seaweed, Laurencia corymbosa. Molecules 2023, 28, 2063. https://doi.org/10.3390/molecules28052063
Fakee J, Bolton JJ, Le Roes-Hill M, Durrell KA, Antunes E, Beukes DR. Antimicrobial Activity of the Secondary Metabolites Isolated from a South African Red Seaweed, Laurencia corymbosa. Molecules. 2023; 28(5):2063. https://doi.org/10.3390/molecules28052063
Chicago/Turabian StyleFakee, Jameel, John J. Bolton, Marilize Le Roes-Hill, Kim A. Durrell, Edith Antunes, and Denzil R. Beukes. 2023. "Antimicrobial Activity of the Secondary Metabolites Isolated from a South African Red Seaweed, Laurencia corymbosa" Molecules 28, no. 5: 2063. https://doi.org/10.3390/molecules28052063
APA StyleFakee, J., Bolton, J. J., Le Roes-Hill, M., Durrell, K. A., Antunes, E., & Beukes, D. R. (2023). Antimicrobial Activity of the Secondary Metabolites Isolated from a South African Red Seaweed, Laurencia corymbosa. Molecules, 28(5), 2063. https://doi.org/10.3390/molecules28052063