
Synthesis and Biological Evaluation of Sclareolide-Indole Conjugates and Their Derivatives



Abstract
:1. Introduction
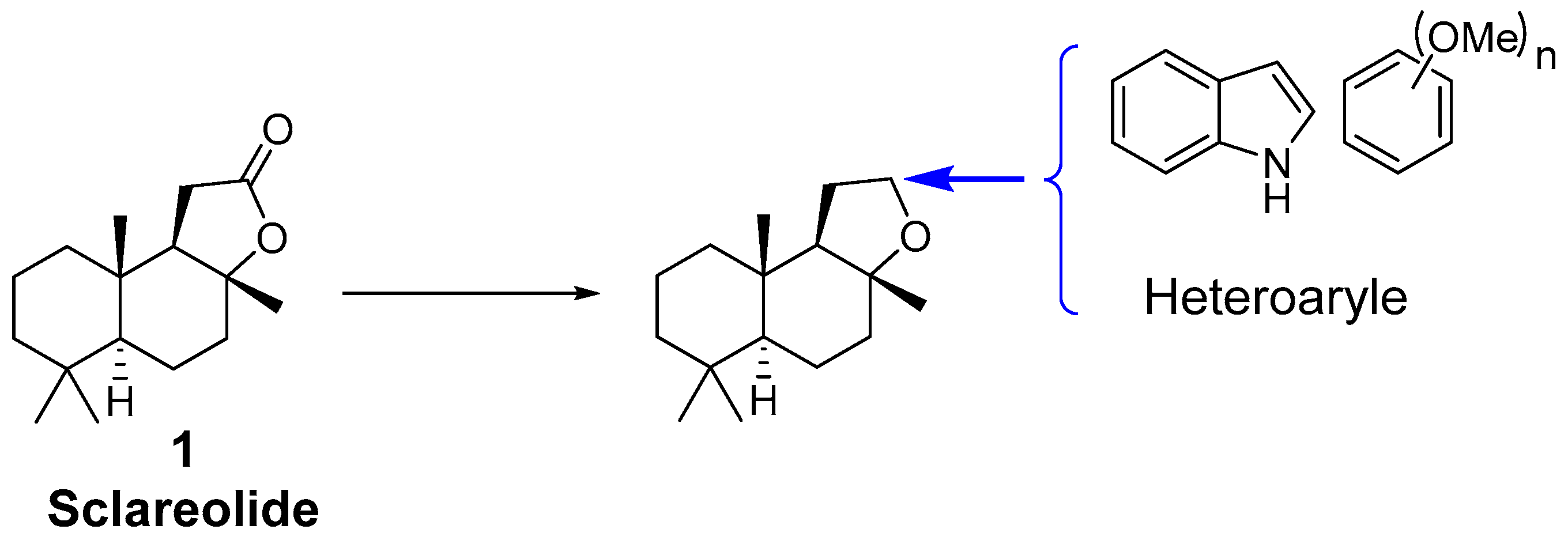
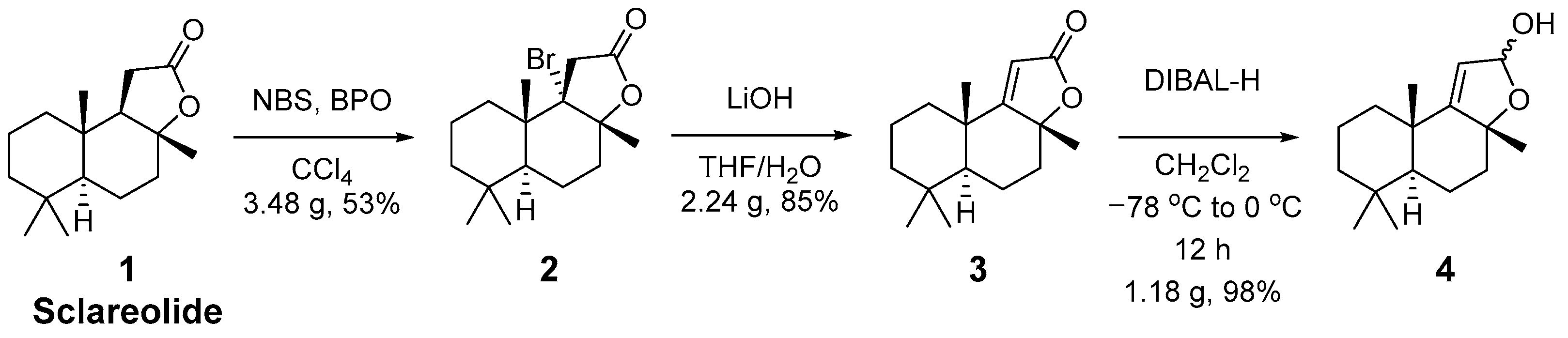
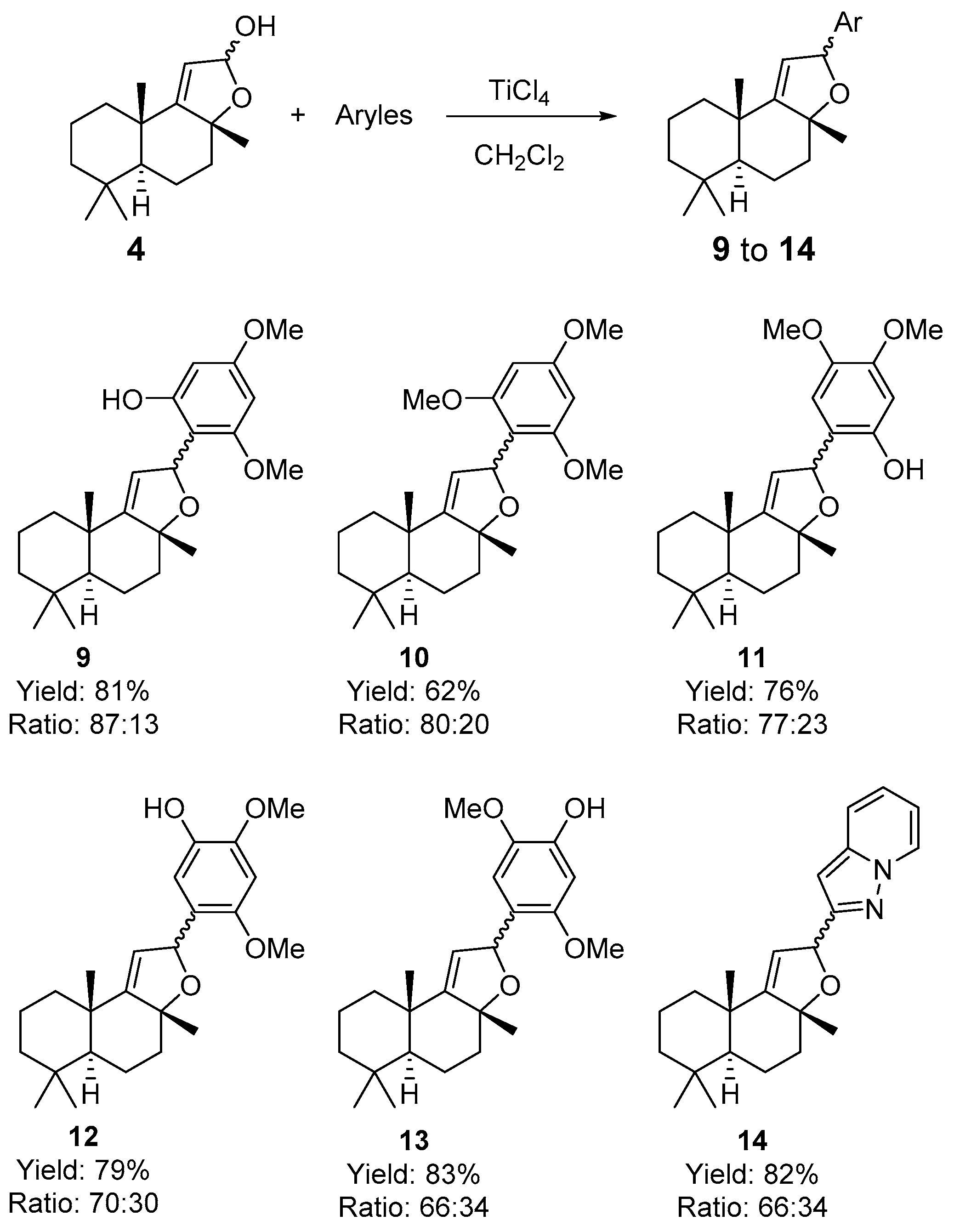
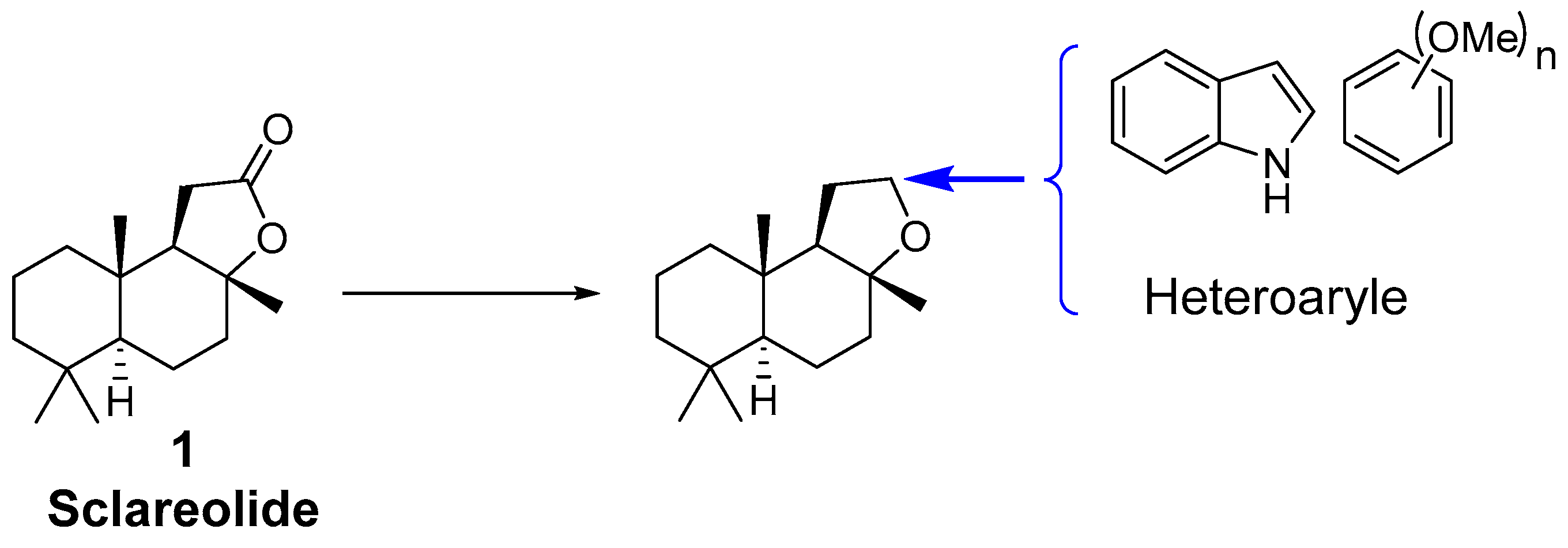
2. Results and Discussion
3. Materials and Methods
3.1. General Chemistry Methods
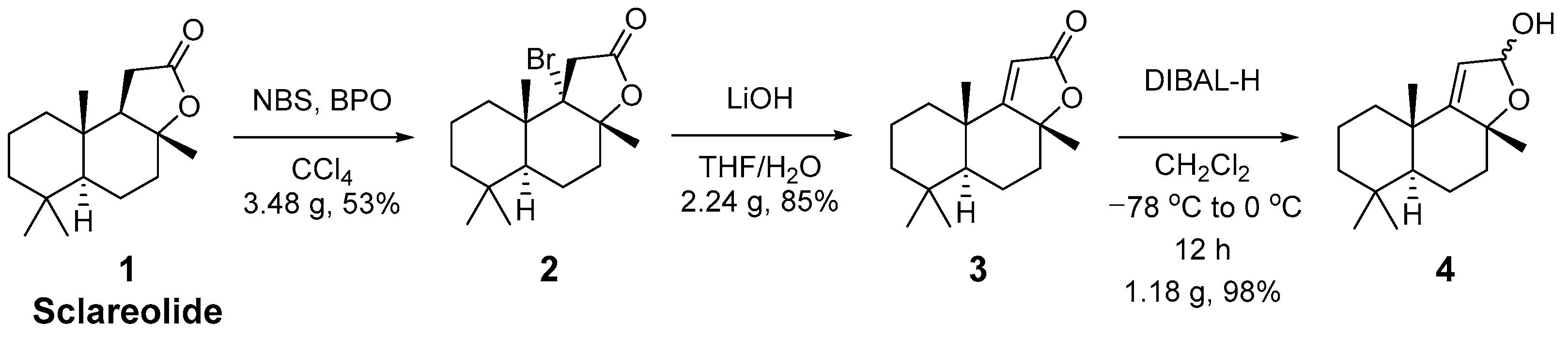
3.2. Procedures for Synthesis of Sclareolide Derivatives
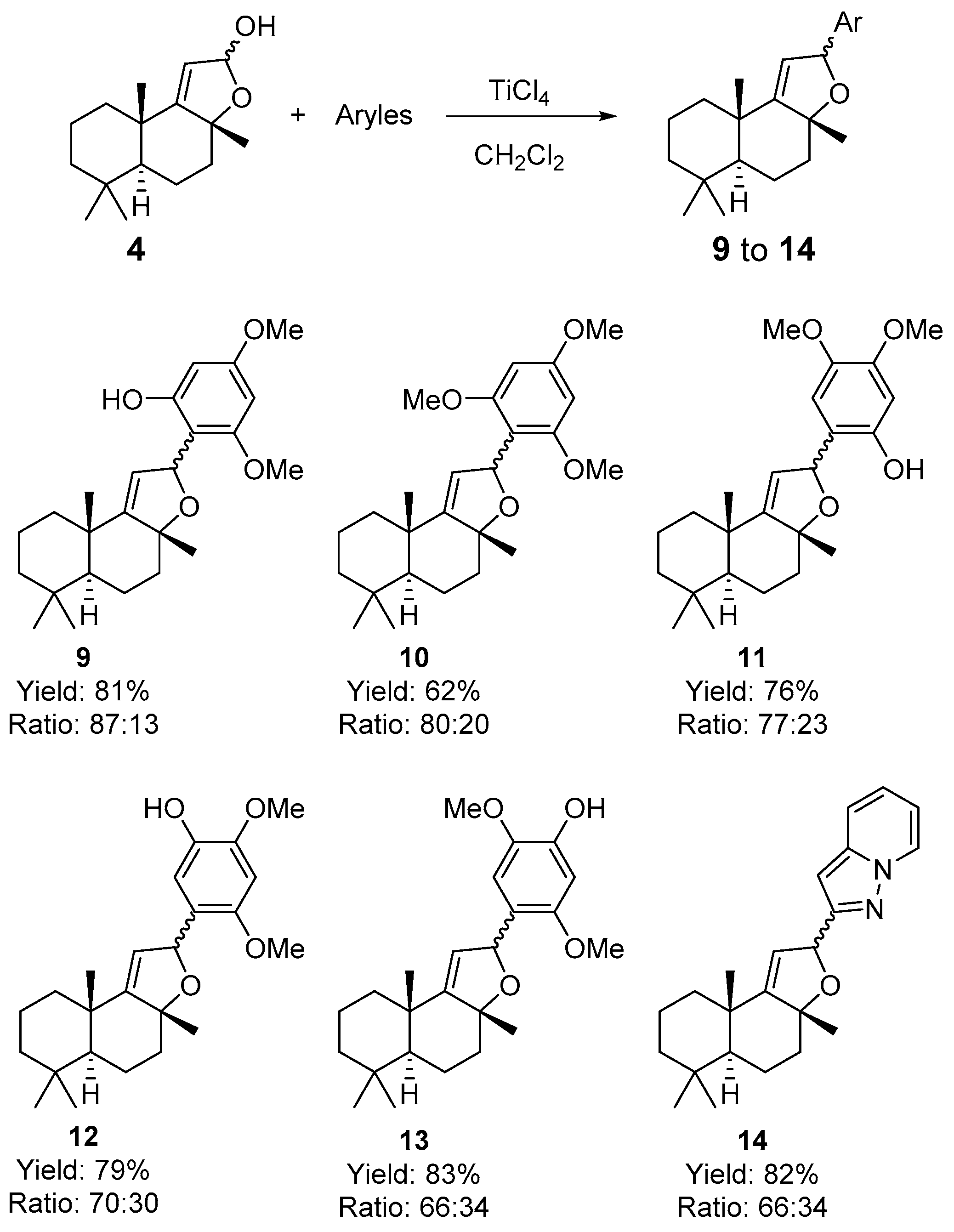
3.3. Procedure for Coupling Reactions of Sclareolide-Indole and Sclareolide-Aromatic Compounds 6, 8a-8y, 8ab-8ae, and 9–14
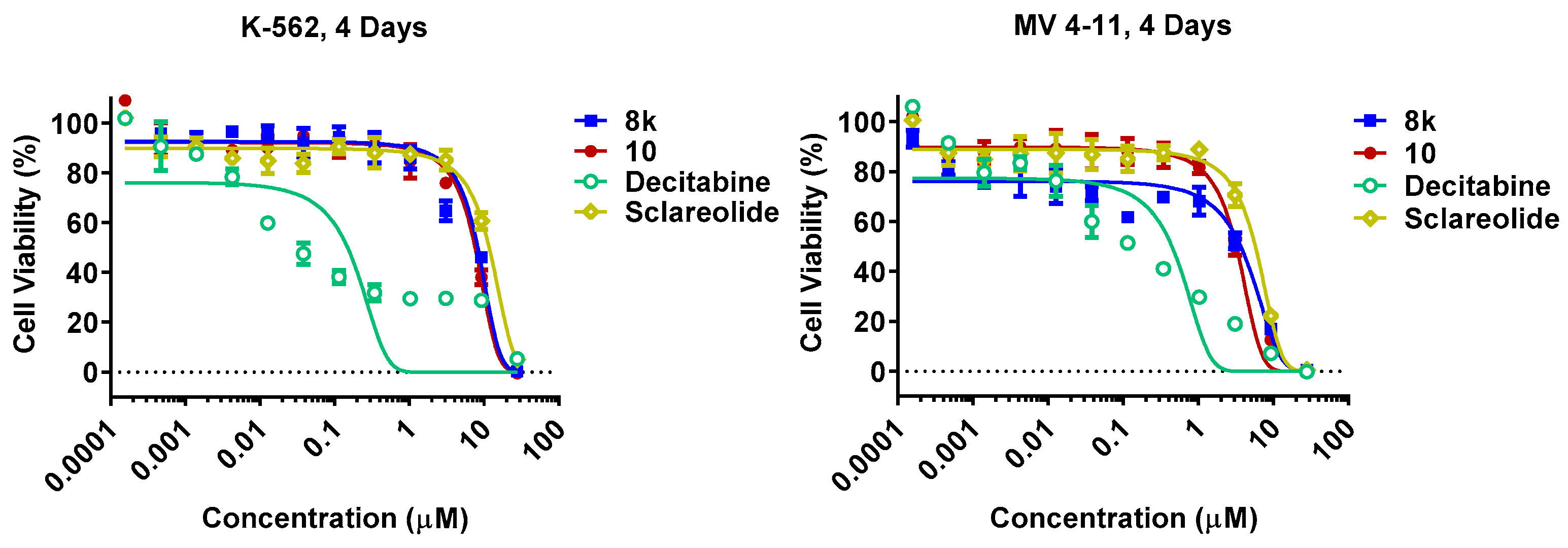
3.4. Cell Growth Inhibition Assay
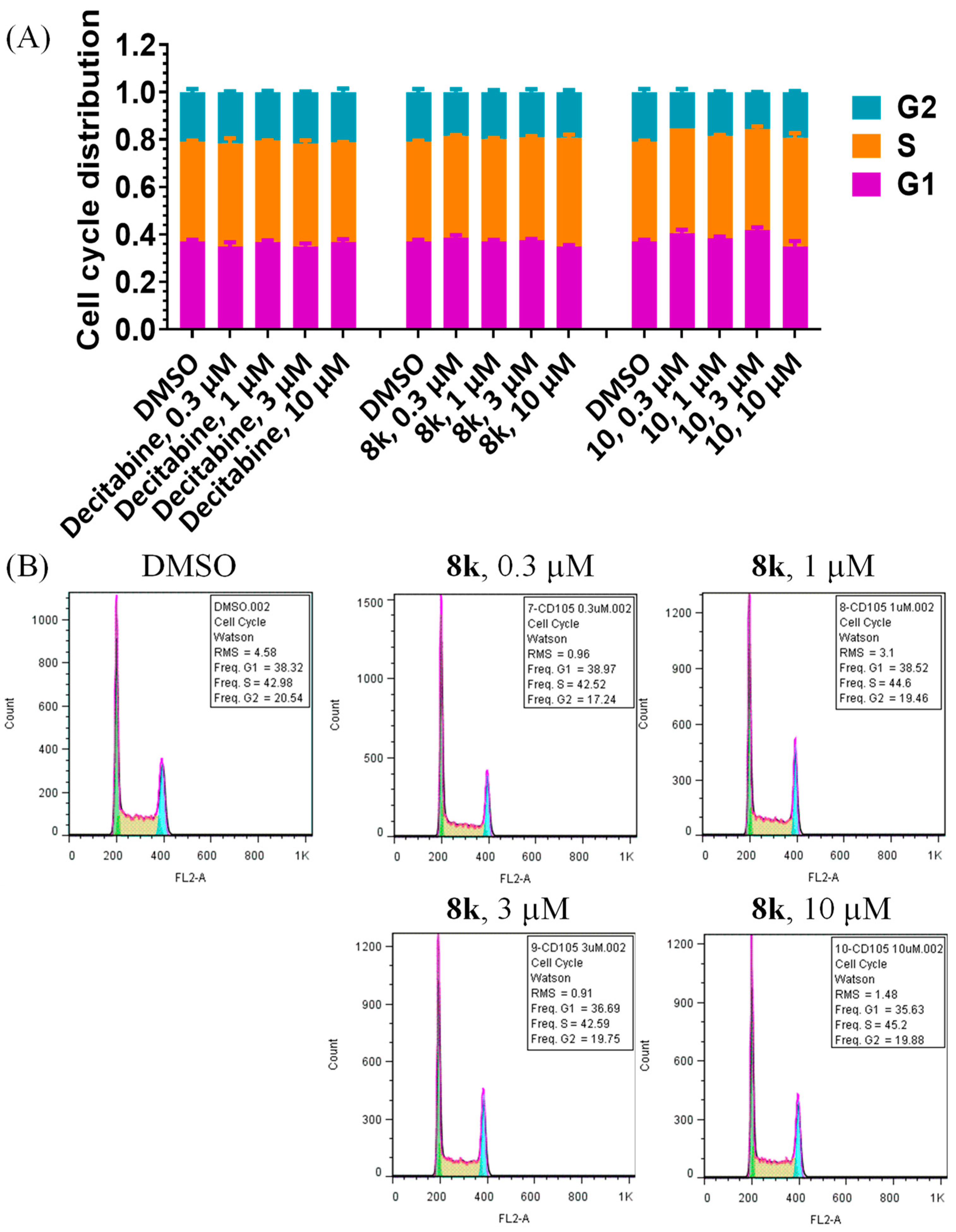
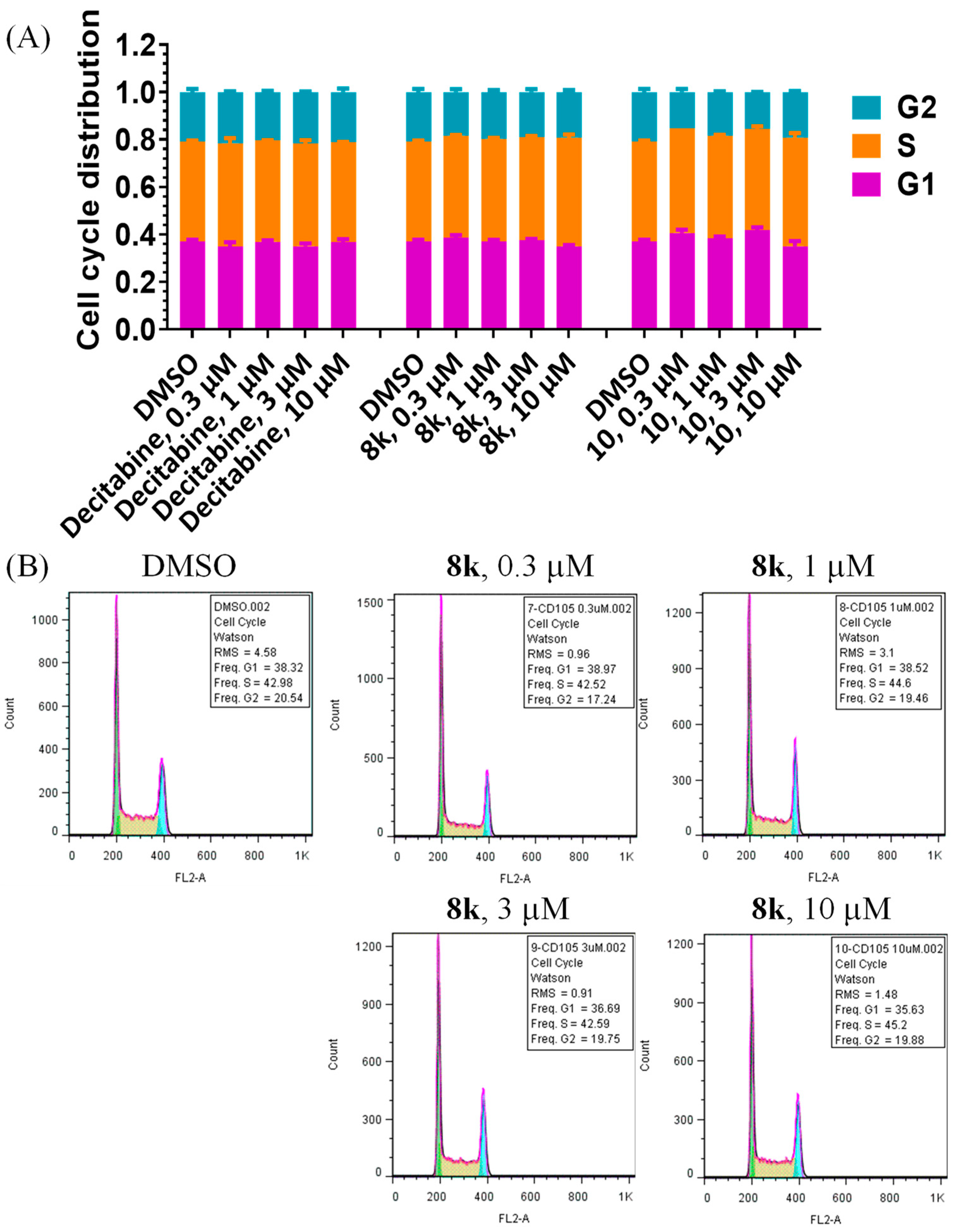
3.5. Flow Cytometry Analysis of Cell Cycle Arrest
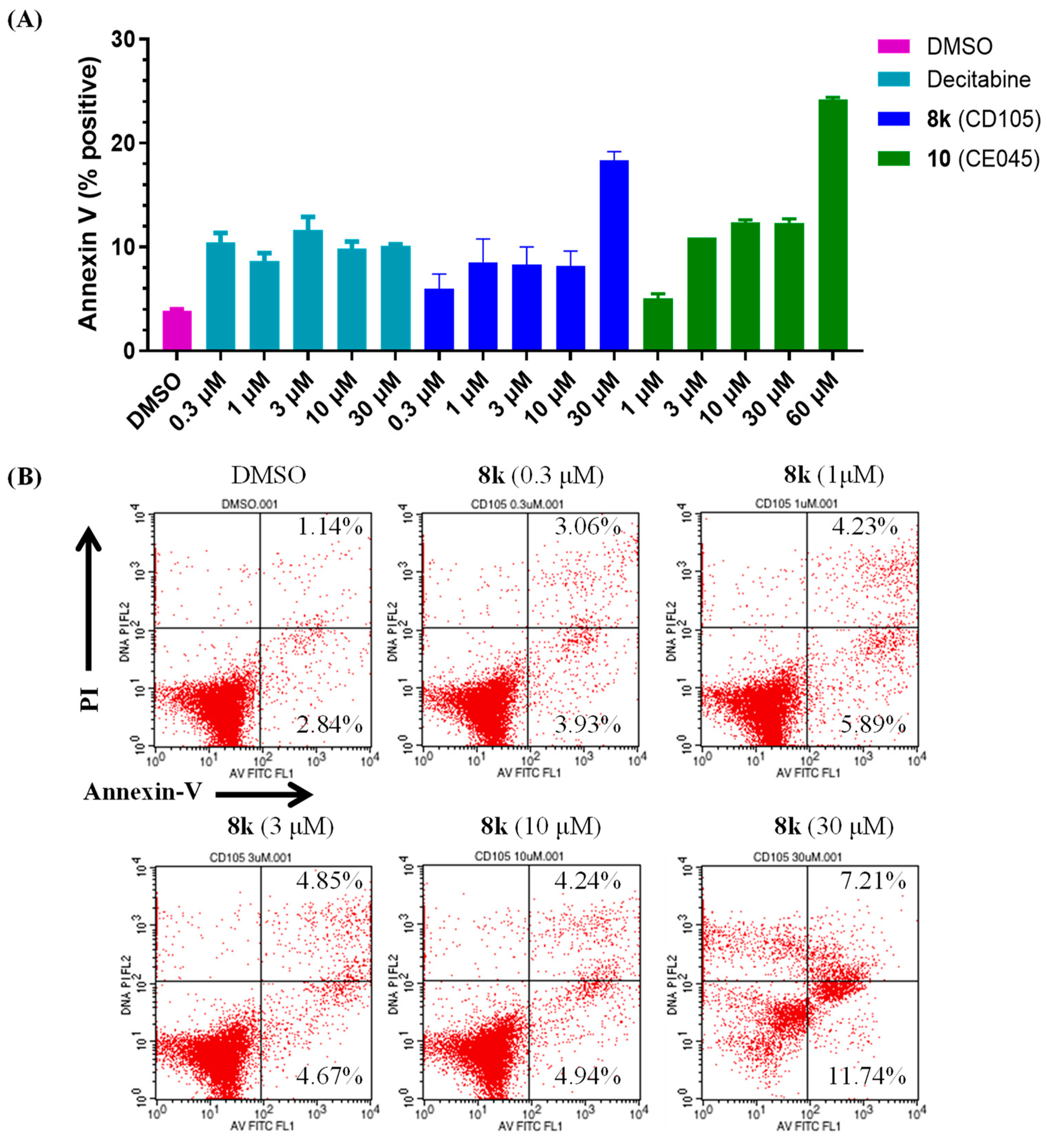
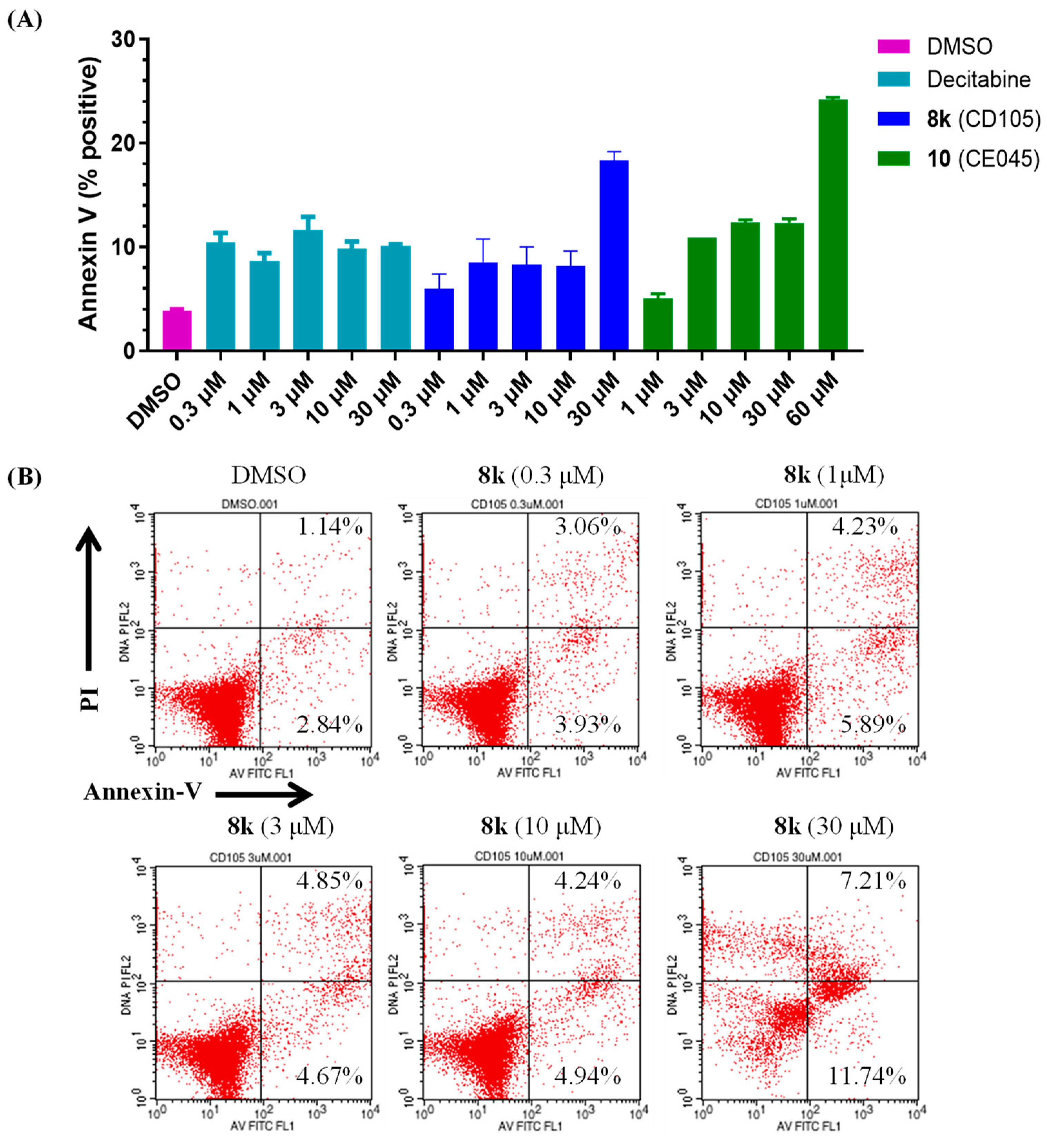
3.6. Flow Cytometry Analysis of Apoptosis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Kaneko, H. The Aroma of cigar tobacco. Agric. Biol. Chem. 1971, 35, 1461–1462. [Google Scholar]
- Frija, L.M.T.; Frade, R.; Afonso, C. Isolation, chemical, and biotransformation routes of labdane-type diterpenes. Chem. Rev. 2011, 111, 4418–4452. [Google Scholar] [CrossRef] [PubMed]
- Peres, M.; Huet, F.; Le Bonniec, M.; Guéhenneux, S.; Vié, K.; Misery, L. Evaluation of the effects on cutaneous sensitivity of a face emulsion containing sclareolide in women presenting with sensitive skin through quantitative sensory testing. J. Eur. Acad. Dermatol. Venereol. 2022, 36, e74–e75. [Google Scholar] [CrossRef]
- Api, A.M.; Belsito, D.; Botelho, D.; Bruze, M.; Burton, G.A., Jr.; Cancellieri, M.A.; Chon, H.; Dagli, M.L.; Date, M.; Tokura, Y.; et al. RIFM fragrance ingredient safety assessment, sclareolide, CAS Registry Number 564-20-5. Food Chem. Toxicol. 2022, 167, 113379. [Google Scholar] [CrossRef]
- Rangan, C.; Barceloux, D.G. Food Additives and Sensitivities. Disease-A-Month 2009, 55, 292–311. [Google Scholar] [CrossRef]
- Smith, R.L.; Ford, R.A. 16. GRAS substances. Food Technol. 1993, 47, 104–117. [Google Scholar]
- Sclareolide Market Size, Growth, Share: Global Sales Revenue, Emerging Technologies, Key Players Analysis, Development Status, Opportunity Assessment and Industry Expansion Strategies 2028. 2022. Available online: https://www.marketwatch.com/press-release/sclareolide-market-size-growth-share-global-sales-revenue-emerging-technologies-key-players-analysis-development-status-opportunity-assessment-and-industry-expansion-strategies-2028-2022-12-14 (accessed on 11 January 2023).
- Chen, S.; Wang, Y.; Zhang, W.-L.; Dong, M.-S.; Zhang, J.-H. Sclareolide enhances gemcitabine-induced cell death through mediating the NICD and Gli1 pathways in gemcitabine-resistant human pancreatic cancer. Mol. Med. Rep. 2017, 15, 1461–1470. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Tang, K.; Guo, Y. Discovery of sclareol and sclareolide as filovirus entry inhibitors. J. Asian Nat. Prod. Res. 2019, 22, 464–473. [Google Scholar] [CrossRef]
- Czaplyski, W.L.; Na, C.G.; Alexanian, E.J. C–H Xanthylation: A Synthetic Platform for Alkane Functionalization. J. Am. Chem. Soc. 2016, 138, 13854–13857. [Google Scholar] [CrossRef]
- Dixon, D.D.; Lockner, J.W.; Zhou, Q.; Baran, P.S. Scalable, divergent synthesis of meroterpenoids via “borono-sclareolide”. J. Am. Chem. Soc. 2012, 134, 8432–8435. [Google Scholar] [CrossRef] [PubMed]
- Kumar, C.N.S.S.P.; Chein, R.-J. Synthesis of Labdane Diterpenes Galanal A and B from (+)-Sclareolide. Org. Lett. 2014, 16, 2990–2992. [Google Scholar] [CrossRef]
- Kuan, K.K.W.; Pepper, H.P.; Bloch, W.M.; George, J.H. Total Synthesis of (+)-Aureol. Org. Lett. 2012, 14, 4710–4713. [Google Scholar] [CrossRef]
- Quinn, R.K.; Könst, Z.A.; Michalak, S.E.; Schmidt, Y.; Szklarski, A.R.; Flores, A.R.; Nam, S.; Horne, D.A.; Vanderwal, C.D.; Alexanian, E.J. Site-Selective Aliphatic C–H Chlorination Using N-Chloroamides Enables a Synthesis of Chlorolissoclimide. J. Am. Chem. Soc. 2016, 138, 696–702. [Google Scholar] [CrossRef]
- Duca, G.; Aricu, A.; Kuchkova, K.; Secara, E.; Barba, A.; Dragalin, I.; Ungur, N.; Spengler, G. Synthesis, structural elucidation and biological evaluations of new guanidine-containing terpenoids as anticancer agents. Nat. Prod. Res. 2018, 33, 3052–3056. [Google Scholar] [CrossRef] [PubMed]
- Carullo, G.; Saponara, S.; Ahmed, A.; Gorelli, B.; Mazzotta, S.; Trezza, A.; Gianibbi, B.; Campiani, G.; Fusi, F.; Aiello, F.; et al. Novel labdane diterpenes-based synthetic derivatives: Identification of a bifunctional vasodilator that inhibits Ca(V)1.2 and stimulates K(Ca)1.1 channels. Mar. Drugs 2022, 20, 515. [Google Scholar] [CrossRef] [PubMed]
- Tran, Q.T.N.; Lee, R.C.H.; Liu, H.J.; Ran, D.; Low, V.Z.L.; To, D.Q.; Chu, J.J.H.; Chai, C.L.L. Discovery and development of labdane-oxindole hybrids as small-molecule inhibitors against chikungunya virus infection. Eur. J. Med. Chem. 2022, 230, 114110. [Google Scholar] [CrossRef] [PubMed]
- González, M.A.; Mancebo-Aracil, J.; Tangarife-Castaño, V.; Gómez, L.S.A.; Zapata, B.; Mesa-Arango, A.; Galvis, L.A.B. Synthesis and biological evaluation of (+)-labdadienedial, derivatives and precursors from (+)-sclareolide. Eur. J. Med. Chem. 2010, 45, 4403–4408. [Google Scholar] [CrossRef] [PubMed]
- Golonko, A.; Pienkowski, T.; Swislocka, R.; Lazny, R.; Roszko, M.; Lewandowski, W. Another look at phenolic compounds in cancer therapy the effect of polyphenols on ubiquitin-proteasome system. Eur. J. Med. Chem. 2019, 167, 291–311. [Google Scholar] [CrossRef]
- Li, D.; Zhang, S.; Song, Z.; Wang, G.; Li, S. Bioactivity-guided mixed synthesis accelerate the serendipity in lead optimization: Discovery of fungicidal homodrimanyl amides. Eur. J. Med. Chem. 2017, 136, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhang, S.; Song, Z.; Li, W.; Zhu, F.; Zhang, J.; Li, S. Synthesis and bio-inspired optimization of drimenal: Discovery of chiral drimane fused oxazinones as promising antifungal and antibacterial candidates. Eur. J. Med. Chem. 2018, 143, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Umer, S.M.; Solangi, M.; Khan, K.M.; Saleem RS, Z. Indole-containing natural products 2019-2022: Isolations, reappraisals, syntheses, and biological activities. Molecules 2022, 27, 7586. [Google Scholar] [CrossRef]
- Pacheco, P.A.F.; Santos, M.M.M. Recent Progress in the Development of Indole-Based Compounds Active against Malaria, Trypanosomiasis and Leishmaniasis. Molecules 2022, 27, 319. [Google Scholar] [CrossRef] [PubMed]
- Scott, K.A.; Cox, P.B.; Njardarson, J.T. Phenols in Pharmaceuticals: Analysis of a Recurring Motif. J. Med. Chem. 2022, 65, 7044–7072. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhao, J.; Luo, L.; Gao, Y.; Bao, H.; Li, P.; Zhang, H. Research progress of indole compounds with potential antidiabetic activity. Eur. J. Med. Chem. 2021, 223, 113665. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, M.; Saxena, A.; Saha, B. An insight in anti-malarial potential of indole scaffold: A review. Eur. J. Med. Chem. 2021, 218, 113400. [Google Scholar] [CrossRef] [PubMed]
- Dvořák, Z.; Poulíková, K.; Mani, S. Indole scaffolds as a promising class of the aryl hydrocarbon receptor ligands. Eur. J. Med. Chem. 2021, 215, 113231. [Google Scholar] [CrossRef]
- Han, Y.; Dong, W.; Guo, Q.; Li, X.; Huang, L. The importance of indole and azaindole scaffold in the development of antitumor agents. Eur. J. Med. Chem. 2020, 203, 112506. [Google Scholar] [CrossRef]
- Zhou, X.; Feng, X.; Wang, D.; Chen, D.; Wu, G.; Yan, Z.; Lyu, X.; Wang, H.; Yang, J.-M.; Zhao, Y. Synthesis and bioactivity studies of covalent inhibitors derived from (-)-Chaetominine. J. Mol. Struct. 2021, 1241, 130694. [Google Scholar] [CrossRef]
- Huang, D.; Szewczyk, S.M.; Zhang, P.; Newhouse, T.R. Allyl-Nickel Catalysis Enables Carbonyl Dehydrogenation and Oxidative Cycloalkenylation of Ketones. J. Am. Chem. Soc. 2019, 141, 5669–5674. [Google Scholar] [CrossRef]
- Chen, Y.; Romaire, J.P.; Newhouse, T.R. Palladium-catalyzed α,β-dehydrogenation of esters and nitriles. J. Am. Chem. Soc. 2015, 137, 5875–5878. [Google Scholar] [CrossRef]
- Fehr, C.; Magpantay, I.; Saudan, L.; Sommer, H. trans-Tetrahydrofurans by OH-Assisted Ru-Catalyzed Isomerization of 2-Butene-1,4-diols. Eur. J. Org. Chem. 2010, 2010, 6153–6156. [Google Scholar] [CrossRef]
- Cambie, R.; Palmer, B. Chemistry of the Podocarpaceae. LVII. The preparation of some 1,3-Dioxans with ambergris-type odours. Aust. J. Chem. 1981, 34, 1265–1284. [Google Scholar] [CrossRef]
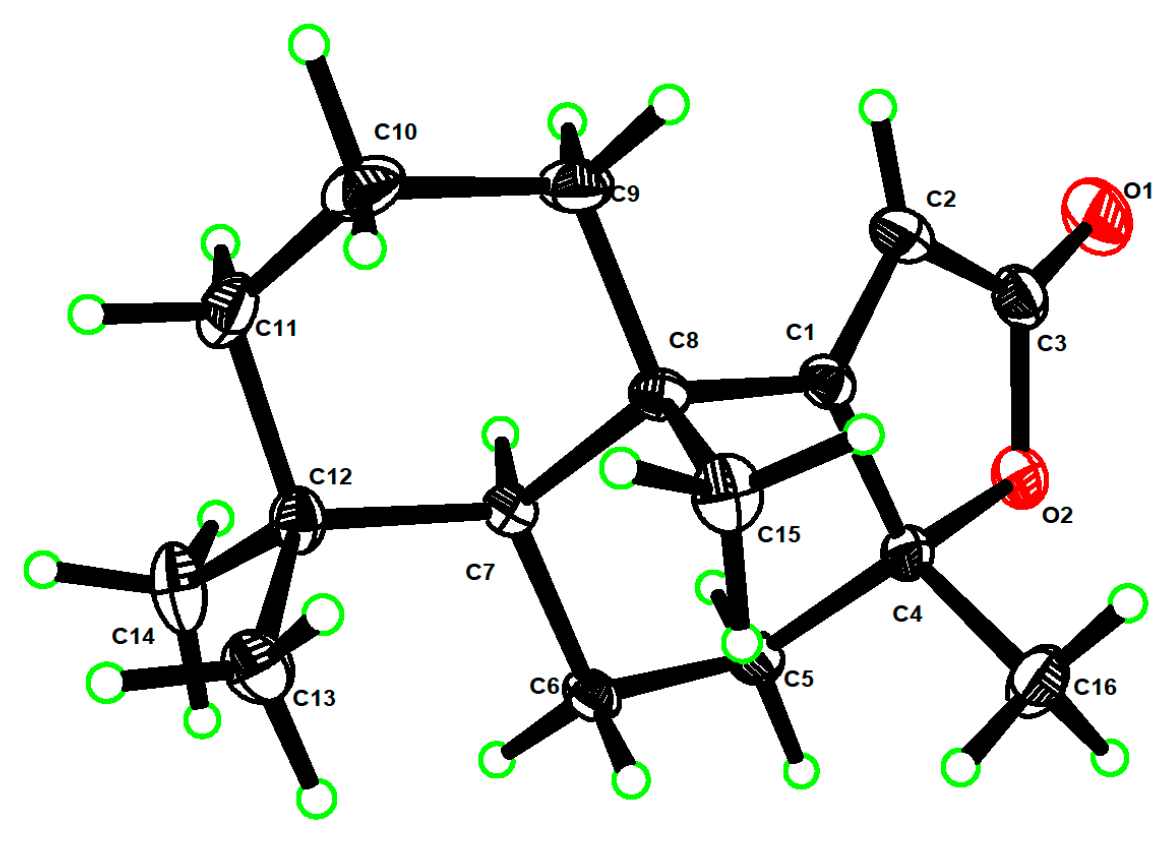
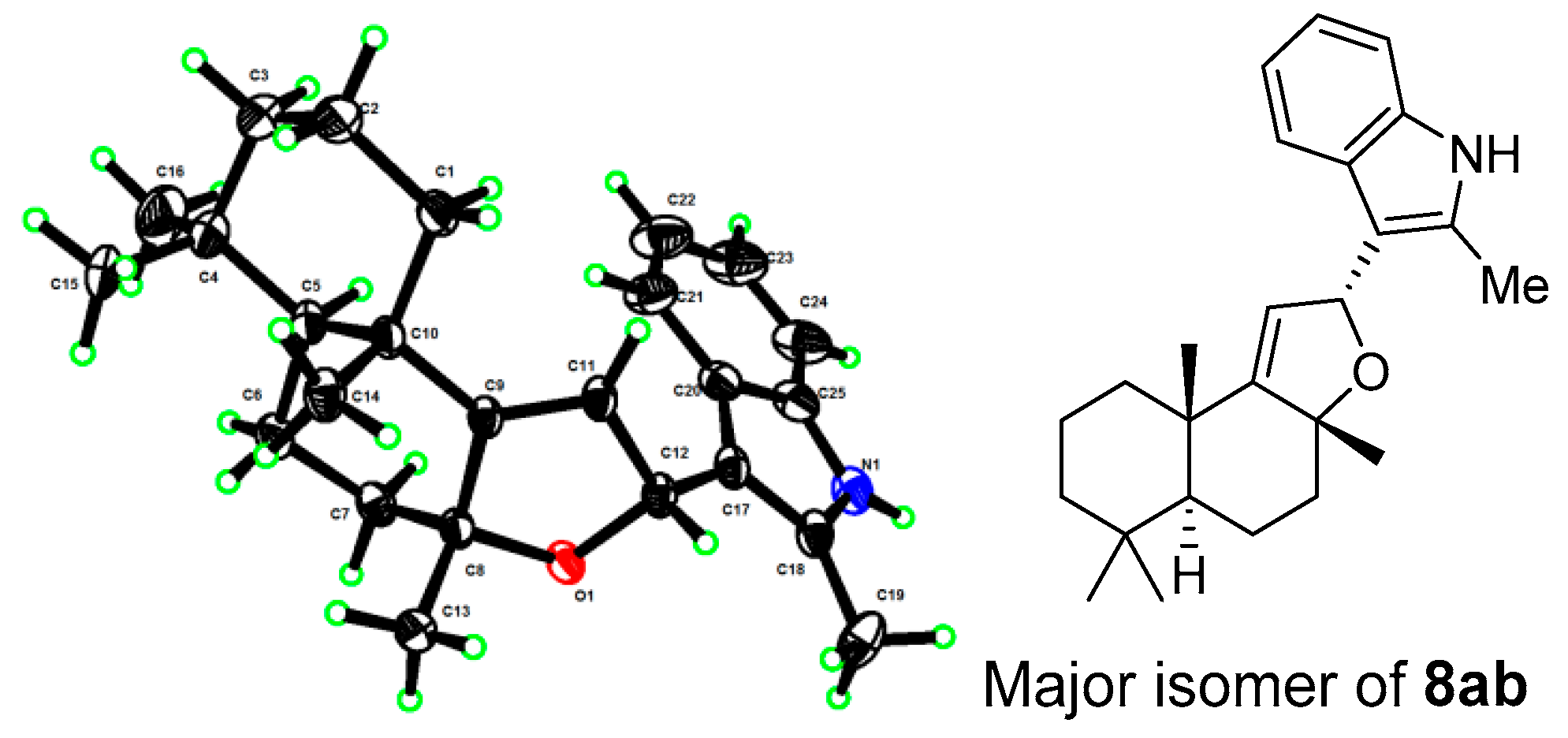
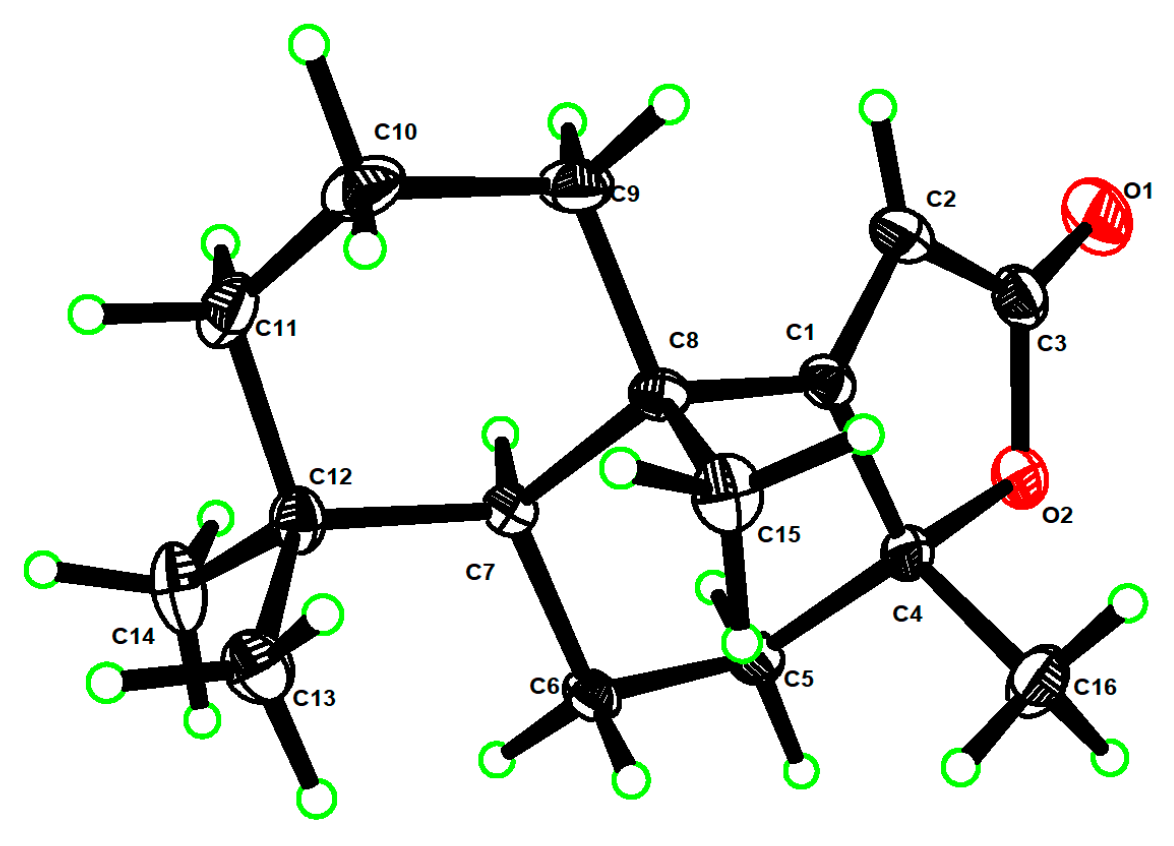
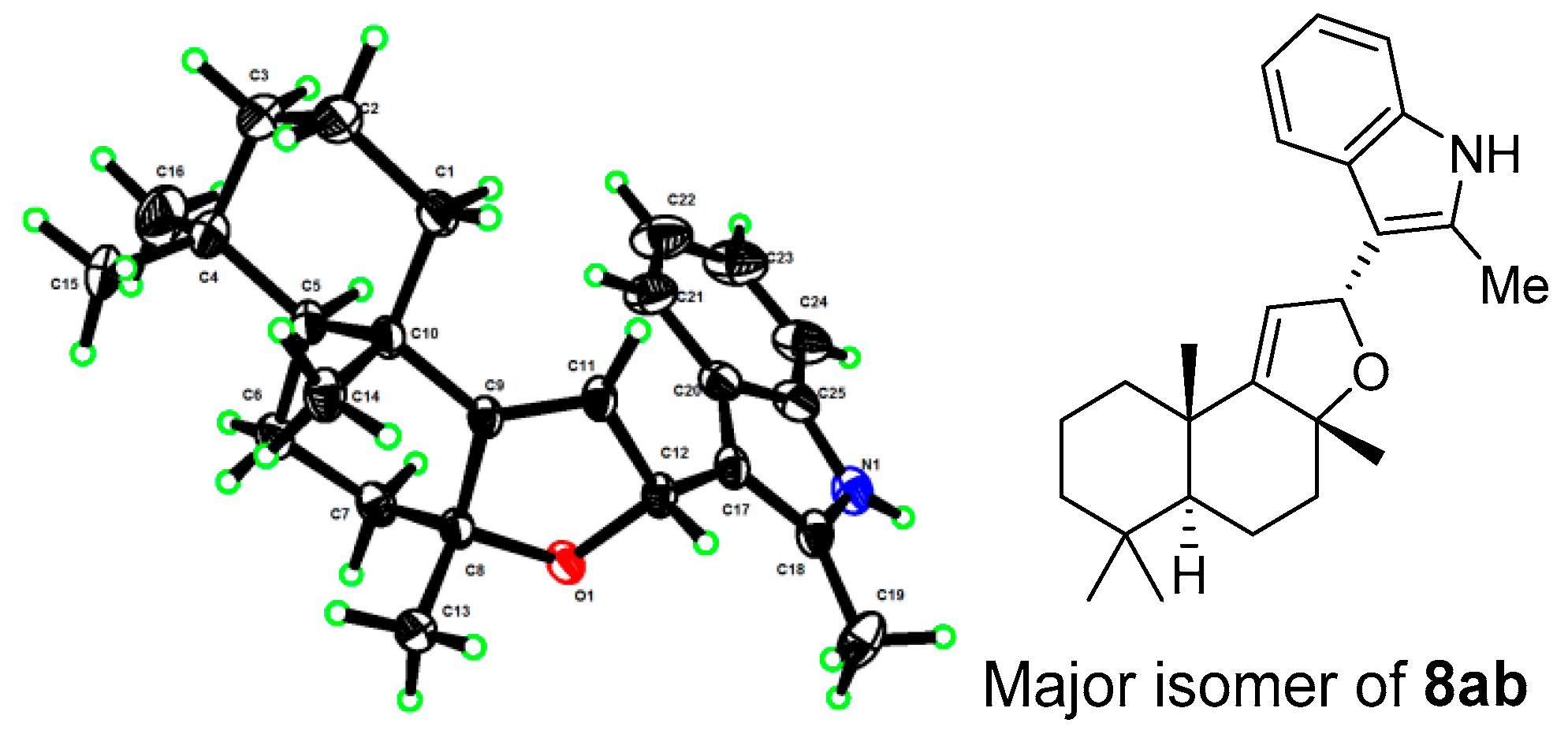
- The X-ray structures of 3 and 8ab have been deposited to CCDC (https://www.ccdc.cam.ac.uk/) with access numbers 2238307 and 2236373, respectively.
- Zhao, Y.-J.; Chng, S.-S.; Loh, T.-P. Lewis Acid-Promoted Intermolecular Acetal-Initiated Cationic Polyene Cyclizations. J. Am. Chem. Soc. 2006, 129, 492–493. [Google Scholar] [CrossRef]
- Zhao, Y.J.; Loh, T.P. Bio-inspired polyene cyclization: Synthesis of tetracyclic terpenoids promoted by steroidal acetal-SnCl4. Chem. Commun. 2008, 12, 1434–1436. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.-J.; Loh, T.-P. Asymmetric Total Synthesis of Antiochic Acid. Org. Lett. 2008, 10, 2143–2145. [Google Scholar] [CrossRef]
- Zhao, Y.J.; Loh, T.P. Bioinspired polyene cyclization promoted by intermolecular chiral acetal-SnCl4 or chiral N-acetal-TiCl4: Investigation of the mechanism and identification of the key intermediates. J. Am. Chem. Soc. 2008, 130, 10024–10029. [Google Scholar] [CrossRef]
- Chen, D.; Lu, T.; Yan, Z.; Lu, W.; Zhou, F.; Lyu, X.; Xu, B.; Jiang, H.; Chen, K.; Luo, C.; et al. Discovery, structural insight, and bioactivities of BY27 as a selective inhibitor of the second bromodomains of BET proteins. Eur. J. Med. Chem. 2019, 182, 111633. [Google Scholar] [CrossRef]
- Feng, X.; Yan, Z.; Zhou, F.; Lou, J.; Lyu, X.; Ren, X.; Zeng, Z.; Liu, C.; Zhang, S.; Zhu, D.; et al. Discovery of a selective and covalent small-molecule inhibitor of BFL-1 protein that induces robust apoptosis in cancer cells. Eur. J. Med. Chem. 2022, 236, 114327. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
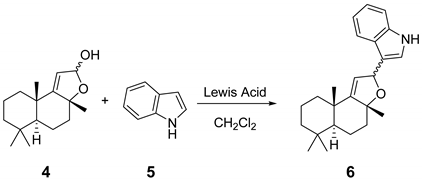
| Entry | Lewis Acid | Equiv. | 4 (Equiv.) | 5 (Equiv.) | T (oC) | Reaction Time | Yield a | Isomer Ratio b |
|---|---|---|---|---|---|---|---|---|
| 1 | BF3 OEt2 | 1.0 | 1.0 | 1.0 | −78 °C | 12 | null | null |
| 2 | InBr3 | 1.0 | 1.0 | 1.0 | rt | 12 | null | null |
| 3 | In(OTf)3 | 1.0 | 1.0 | 1.0 | rt | 12 | null | null |
| 4 | SnCl4 | 1.0 | 1.0 | 1.0 | −78 °C | 2.5 | 33% | 69:31 |
| 5 | SnCl4 | 1.0 | 1.0 | 3.0 | −78 °C | 2.5 | 27% | 69:31 |
| 6 | TiCl4 | 1.0 | 1.0 | 1.0 | −78 °C | 2.5 | 57% | 69:31 |
| 7 | TiCl4 | 1.0 | 1.0 | 1.5 | −78 °C | 2.5 | 53% | 69:31 |
| 8 | TiCl4 | 0.9 | 1.0 | 1.0 | −78 °C | 2.5 | 51% | 69:31 |
| 9 | TiCl4 | 0.8 | 1.0 | 1.0 | −78 °C | 2.5 | 59% | 69:31 |
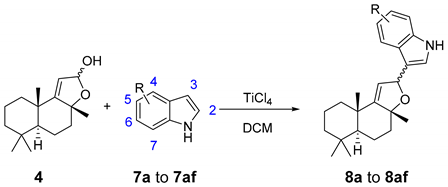
| SM | R | Product | Yield a | Isomer Ratio b | SM | R | Product | Yield a | Isomer Ratio b |
|---|---|---|---|---|---|---|---|---|---|
| 7a | 4-CO2Me | 8a | 52% | 51:49 | 7q | 6-OMe | 8q | 53% | 59:41 |
| 7b | 5-CO2Me | 8b | 92% | 76:24 | 7r | 6-Me | 8r | 67% | 69:31 |
| 7c | 6-CO2Me | 8c | 73% | 69:31 | 7s | 6-CHO | 8s | 77% | 70:30 |
| 7d | 7-CO2Me | 8d | 60% | 82:18 | 7t | 6-CF3 | 8t | 62% | 73:27 |
| 7e | 4-NO2 | 8e | 56% | 68:32 | 7u | 6-NHCbz | 8u | 78% | 69:31 |
| 7f | 5-NO2 | 8f | 67% | 85:15 | 7v | 6-BPin | 8v | 82% | 70:30 |
| 7g | 6-NO2 | 8g | 55% | 69:31 | 7w | 6-OH | 8w | 48% | 73:27 |
| 7h | 7-NO2 | 8h | 87% | 68:32 | 7x | 4-CO2Me, 6-Br | 8x | 51% | 52:48 |
| 7i | 4-Br | 8i | 43% | 62:38 | 7y | 5-BPin | 8y | 84% | 71:29 |
| 7j | 5-Br | 8j | 73% | 76:24 | 7z | 4-BPin | 8z | <1% | |
| 7k | 6-Br | 8k | 61% | 71:29 | 7ab | 2-Me | 8ab | 86% | 78:22 |
| 7l | 7-Br | 8l | 68% | 70:30 | 7ac | 2-Et | 8ac | 72% | 85:15 |
| 7m | 6-CN | 8m | 84% | 67:33 | 7ad | 2-Ph | 8ad | 82% | 82:18 |
| 7n | 6-F | 8n | 77% | 70:30 | 7ae | 2-CO2Me | 8ae | 63% | 67:33 |
| 7p | 6-Cl | 8p | 76% | 70:30 | 7af | 2-tBu | 8af | 35% | 87:13 |
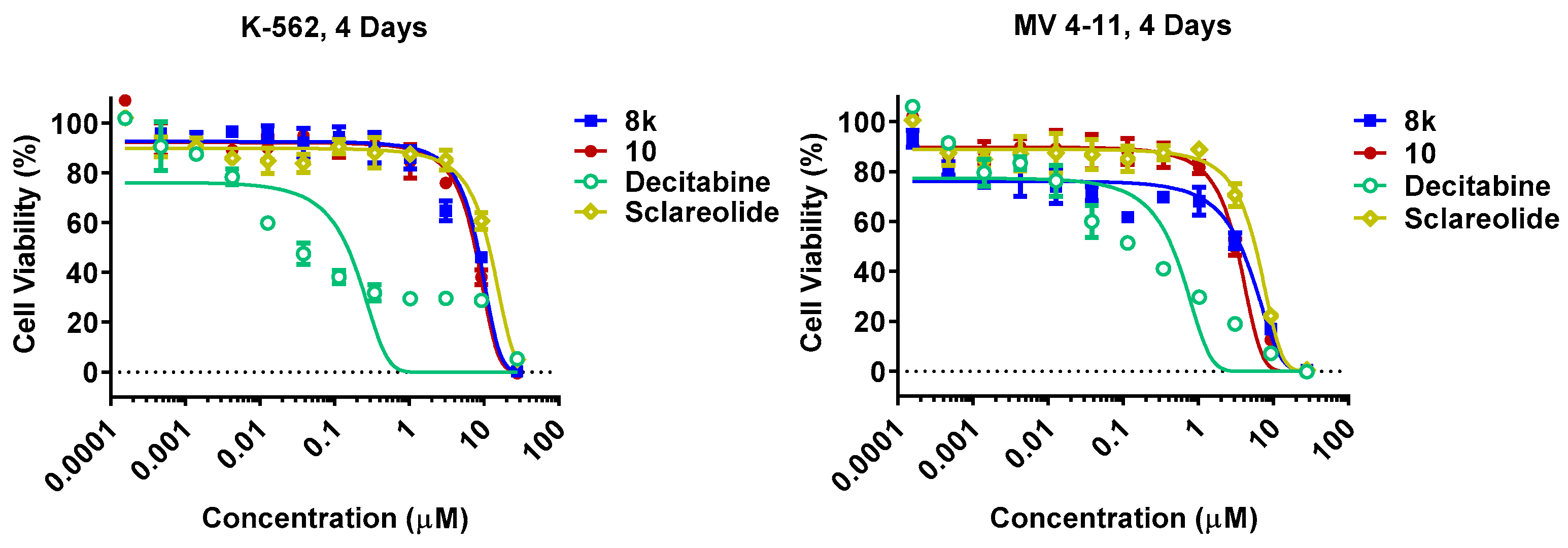
| Cmpd | K562 (IC50, μM) a | MV4-11 (IC50, μM) a | Cmpd | K562 (IC50, μM) a | MV4-11 (IC50, μM) a |
|---|---|---|---|---|---|
| Decitabine | 0.112 ± 0.035 | 0.129 ± 0.035 | 8t | 4.4 ± 0.4 | 4.2 ± 0.7 |
| Sclareolide | 10.8 ± 0.6 | 4.5 ± 0.3 | 8w | 10.4 ± 1.4 | 5.8 ± 0.7 |
| 3 | 11.6 ± 2.4 | 2.0 ± 1.7 | 8ab | 12.0 ± 1.0 | 7.9 ± 1.8 |
| 8c | 9.4 ± 0.2 | 5.2 ± 0.6 | 8ad | 9.9 ± 0.1 | 6.8 ± 0.4 |
| 8g | 4.5 ± 0.7 | 4.5 ± 2.8 | 8ae | 7.0 ± 0.8 | 4.6 ± 2.8 |
| 8k | 5.2 ± 0.6 | 0.8 ± 0.6 | 9 | 9.8 ± 0.1 | 4.7 ± 2.4 |
| 8m | 5.3 ± 0.2 | 4.4 ± 0.4 | 10 | 6.8 ± 1.1 | 2.3 ± 0.7 |
| 8n | 9.6 ± 0.1 | 7.6 ± 0.2 | 11 | 10.0 ± 0.1 | 3.9 ± 0.1 |
| 8p | 6.1 ± 0.8 | 1.7 ± 1.0 | 12 | 10.9 ± 1.8 | 6.1 ± 1.1 |
| 8q | 6.1 ± 0.9 | 1.7 ± 0.7 | 13 | 10.7 ± 1.2 | 4.6 ± 2.3 |
| 8s | 6.9 ± 0.4 | 4.1 ± 2.2 | 14 | 10.1 ± 0.8 | 3.3 ± 0.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, Y.; Lyu, X.; Liu, C.; Wang, X.; Cheng, J.; Zhang, D.; Meng, X.; Zhao, Y. Synthesis and Biological Evaluation of Sclareolide-Indole Conjugates and Their Derivatives. Molecules 2023, 28, 1737. https://doi.org/10.3390/molecules28041737
Cheng Y, Lyu X, Liu C, Wang X, Cheng J, Zhang D, Meng X, Zhao Y. Synthesis and Biological Evaluation of Sclareolide-Indole Conjugates and Their Derivatives. Molecules. 2023; 28(4):1737. https://doi.org/10.3390/molecules28041737
Chicago/Turabian StyleCheng, Ying, Xilin Lyu, Chen Liu, Xiancheng Wang, Jing Cheng, Daizhou Zhang, Xiangjing Meng, and Yujun Zhao. 2023. "Synthesis and Biological Evaluation of Sclareolide-Indole Conjugates and Their Derivatives" Molecules 28, no. 4: 1737. https://doi.org/10.3390/molecules28041737
APA StyleCheng, Y., Lyu, X., Liu, C., Wang, X., Cheng, J., Zhang, D., Meng, X., & Zhao, Y. (2023). Synthesis and Biological Evaluation of Sclareolide-Indole Conjugates and Their Derivatives. Molecules, 28(4), 1737. https://doi.org/10.3390/molecules28041737