Abstract
Mitochondria is an important drug target for ailments ranging from neoplastic to neurodegenerative diseases and metabolic diseases. Here, we describe the synthesis of chloroquine analogs and show the results of mitochondrial ATP inhibition testing. The 2,4-dinitrobenzene-based analogs showed concentration-dependent mitochondrial (mito.) ATP inhibition. The most potent mito. ATP inhibitor was found to be N-(4-((2,4-Dinitrophenyl)amino)pentyl)-N-ethylacetamide (17).
1. Introduction
Chloroquine and hydroxychloroquine (H)CQ) (Figure 1) have been used in treatment of malaria, rheumatoid arthritis, and lupus erythematosus [1]. According to the most widely accepted mechanism (H)CQ enters the red blood cells through diffusion and then moves into the digestive vacuole (DV) of the Plasmodium parasite. The Plasmodium forms these DVs for metabolism and survival inside erythrocytes, and these lysosome-like DVs are acidic. The parasite degrades host hemoglobin into amino acids by the action of proteases that operate under an acidic environment. This process generates free heme, which polymerizes into hemozoin under the acidic environment. Free heme is toxic to the parasite, while hemozoin is not. As (H)CQ is a weak base, it increases the pH inside DVs, disrupting proteolysis and formation of hemozoin. Aggregation of cytotoxic heme leads to parasite death [2,3,4,5].
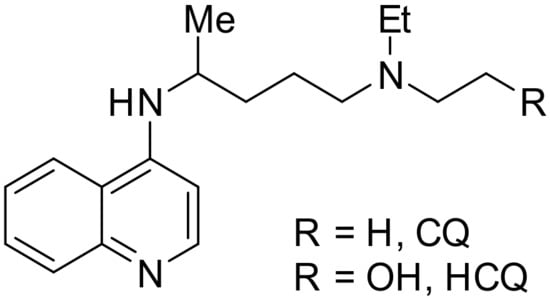
Figure 1.
Structure of CQ and HCQ.
This (H)CQ-mediated pH rise in membrane-bound organelles has been shown to inhibit viruses that require pH-dependent entry [6]. A similar pH-dependent process involving mitochondria (mito.) was later speculated to account for the possible antiviral activities of (H)CQ. In this model, (H)CQ was found to target mito. ATP production and cause a concentration-dependent decrease in ATP levels [7]. Mito. intermembrane space is more than one pH unit lower than the matrix. This difference in pH generates a proton gradient across the membrane, which is partly responsible for mito. ATP production [8]. (H)CQ was proposed to diffuse into mito. intermembrane space, where it acts as a weak base and increases the pH in the intermembrane space, thus impacting ATP production [7]. These findings suggest that (H)CQ and its analogs have the potential to be mito. inhibitors.
To understand what part of (H)CQ is responsible for inhibition of mito. ATP, we studied (H)CQ analogs. Three factors were evaluated. Which part of the (H)CQ causes inhibition of mito. ATP? The alkyl amine were aromatic amine were considered possibilities. Furthermore, is the basic nature of (H)CQ accountable for the inhibition of mito. ATP? We present here our findings.
2. Results and Discussion
2.1. Synthesis
Three types of compounds were prepared in this study:
- CQ analogs
- HCQ analogs
- A CQ analog with an amide group
We prepared the CQ analog 1 by the SNAr reaction [9] of commercially available amine 2 with 3 (Scheme 1). CQ derivatives 4 and 5 were prepared by Buchwald–Hartwig amination [10,11] of 2 with 6 and 7, respectively (Scheme 2). The HCQ analog 8 was synthesized using Scheme 3. Synthetic intermediates 9 and 10 were prepared by modifying a continuous-flow synthesis method [12]. Reductive amination of 10 gave 11, which was converted into 8 by the SNAr reaction with good yield. The amide analog of CQ was prepared as shown in Scheme 4. Amide 12 was synthesized by a modified procedure [13]. Ketalization [14] of 12 gave 13, which upon alkylation formed 14. Deprotection of the ketal 14 gave ketone 15, which was converted into amine 16 via reductive amination. Finally, the SNAR reaction between 16 and 3 gave the CQ analog 17 containing an amide group.
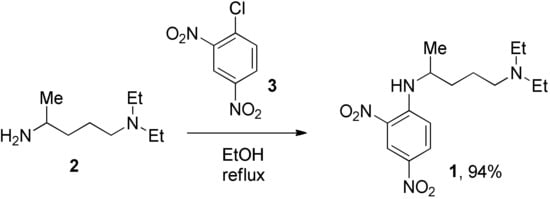
Scheme 1.
Synthesis of 2,4-dinitro CQ analog.
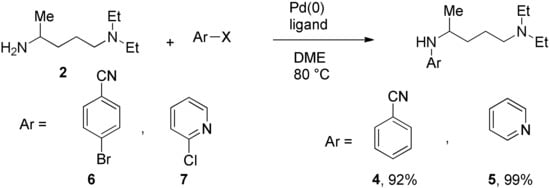
Scheme 2.
Synthesis of CQ analogs by Buchwald–Hartwig amination.
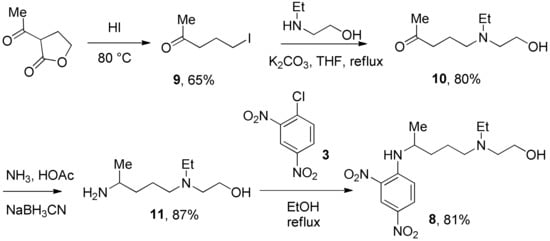
Scheme 3.
Synthesis of 2,4-dinitro HCQ analog.
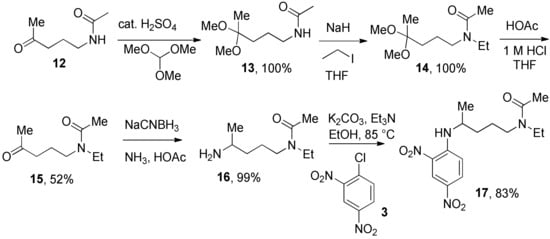
Scheme 4.
Synthesis of 2,4-dinitro CQ amide analog.
2.2. Rationale for the Selection of Analogs
(H)CQ has three nitrogen atoms, two of them are basic. The conjugate acid of the tertiary amine has a pka of 10.2, while the pka of the conjugate acid of pyridine nitrogen is 8.2 [4]. All except 17 have the tertiary amine. Due to their structural similarity with (H)CQ, these tertiary amine groups are expected to have pka values similar to (H)CQ. Compounds 1, 4, 5, 8, and 17 also contain the non-basic arylamine nitrogen function. Analog 5 has all three types of nitrogen atoms found in (H)CQ, with a calculated pka value of 6.6 for the conjugate acid of the pyridine nitrogen [15].
We selected the dinitrobenzene analogs of (H)CQ (1, 8, and 17) because, like the chloroquinoline unit of (H)CQ, dinitrobenzene is aromatic. Furthermore, 2,4-dinitrophenol (DNP) is known to block mito. ATP synthesis but permits continued electron transport along the respiratory chain to O2. In other words, it is an uncoupler [16]. DNP exists as an anion at intracellular pH. Because of the lower pH near mito., protonation of DNP anion occurs, making it a neutral specie. The neutral specie is more prone to diffuse into and through the membrane into mito. [16]. We therefore expected that the aryl part of 1, 8, and 17 should also enter mito. as it has an even less acidic arylamine function than the phenol in DNP. To test if an aryl portion is necessary for mito. inhibition, fragments 2 and 11 were selected. To deetermine whether the amine function was required for mito. ATP modulation, compound 17 was chosen as it is the same as 1, but with the alkylamine changed to an amide, which is less basic than the amine. Analogs 4 and 5 were selected because they have different aryl groups than 1 and 8, and less acidic arylamine functions (calculated pka values of the arylamine function of 4 and 5 are 20.2 and 21.7, while the pka of the arylamine function for 1 is 12.3 and CQ is 27.3) [15]. Furthermore, 5 contains a pyridine unit also present in (H)CQ. Both 4 and 5 can be prepared from commercially available starting materials in a single step.
2.3. Biochemical Data
Two major pathways for ATP production in mito. are cytosolic glycolysis and oxidative phosphorylation [17]. Cells switch to the other pathway for ATP production when one pathway is blocked [7]. Therefore, to test if a compound targets mito., it is necessary to block glycolysis, or the net ATP production from glycolysis must be zero. For this study, we chose the latter option. The net ATP production via glycolysis is zero when cells are grown in galactose instead of glucose. This happens because glycolysis generates two net ATP per molecule of glucose. However, galactose cannot be directly used in cellular metabolism and is first converted into glucose-6-phosphate. This conversion requires two ATP molecules. Therefore, the net generation of ATP by galactose molecules in glycolysis is zero [18].
Two sets of screenings were carried out for each (H)CQ analog (Table 1). In both groups, ATP production was measured in cells treated with different concentrations of an (H)CQ analog. In the first set, the cell culture media was L-15, containing galactose instead of glucose. In the second set, glucose was added to the L-15 media. Mito. ATP inhibition observed in L-15 media should be reversed in the L-15 + glucose media as the glycolysis pathways become available, resulting in net positive ATP production.

Table 1.
Mito. ATP inhibitory effects of (H)CQ analogs.
Entries 1 and 3 (Table 1) contained aryl groups, while entries 2 and 4 were non-aromatic fragments of (H)CQ. The data suggests that having an alkyl amine group is not sufficient to attain mito. ATP inhibition. However, whereas entry 1 was a potent inhibitor of mito. ATP production, entry 3 modulated ATP production only mildly. A comparison of entries 1 and 3 with 5 and 6 suggests that the nature of the arylamine portion of (H)CQ analogs is also relevant. While entries 1 and 3 seemed to inhibit mito. ATP, 5 and 6 (like entries 2 and 4) were neither potent nor specific in inhibiting mito. ATP production. Finally, the data for entry 7 suggest that the basicity of (H)CQ may also not be the cause of mito. ATP inhibition. Here, the basic function of amine has been converted into an amide, but the analog is even more potent than entry 1 in inhibiting mito. Therefore, we conclude that mito. ATP inhibition by (H)CQ and its analogs occurs via another process. More work is needed to find the mechanism of this mito. ATP inhibition process. Nonetheless, our study identified (H)CQ analogs that are mito. ATP modulators, which as such may find uses in researching pathologies linked with mito. dysfunction [19].
3. Conclusions
In this paper, we have described our efforts to target mito. through (H)CQ analogs. We attempted to understand what feature of (H)CQ analogs is responsible for ATP modulation. Neither the alkyl amine nor the basic nature of the (H)CQ analogs alone can account for the mito. modulation. The nature of the arylamine portion of the (H)CQ analogs did have an impact on mito. ATP modulation. Arylamine functions that can deprotonate more easily (such as in 2,4-dinitrobenzene-based analogs) show greater ATP inhibition, and compound 17 was the most potent mito. ATP inhibitor in the series.
4. Materials and Methods
4.1. Synthesis
4.1.1. General Experimental
Amine (2) was purchased from a commercial source. Methanol was dried by standing over on molecular sieves for 3 days. Tetrahydrofuran was dried by distillation from CaH2 onto 4 Å molecular sieves. Alumina refers to aluminum oxide 90 active, neutral from EM Reagents. TLCs were visualized using UV or stained with KMnO4. The 1H and 13C NMR spectra were performed on a Varian 400/50 (400 MHz) spectrometer and measured in ppm relative to TMS (0.00 ppm) or the residual solvent CHCl3 (1H NMR δ = 7.26 ppm and 13C NMR δ = 77.16 ppm). High-resolution mass spectrometry measurements were taken using a Thermo Scientific Fusion or Exactive spectrometer operating in a positive ion electrospray mode using an Orbitrap analyzer at a nominal resolution of 120,000. Glass silica gel plates (250 μm) were used for thin-layer chromatography, and sorbent silica gel 60 Å (40–63 μm) was used for flash chromatography. Experimntal procedures for synthesizing 9 and 10, NMR spectra of isolated compounds, and biochemical data for Table 1 are available in the Supplementary Materials.
4.1.2. N-(5-(Diethylamino)pentan-2-yl)-2,4-dinitrobenzenamine
(1). A suspension of 3 (98%, 1.93 g, 9.36 mmol) and absolute ethanol (5.90 mL) was heated at 70 °C. When this was entirely dissolved, 2 (97%, 2.06 mL, 10.3 mmol) was added with a syringe pump at a rate of 0.02 mL/min. Afterward, the reaction was refluxed for 4 h. The reaction was cooled to 70 °C and 15.0 M NH4OH (15.0 mmol) was added. The reaction was allowed to come to rt and ethanol was removed on a rotary evaporator. Distilled water (15.0 mL) was added to the reaction and the product was extracted with CH2Cl2 (20.0 mL × 4). The organic phase was dried with Na2SO4 and evaporated on a rotary evaporator. Vacuum distillation of this crude extract gave pure 1 as a dark brown liquid (2.85 g, 8.79 mmol, 94%). R f = 0.20 (EtOAc, the TLC was pretreated with 1% Et3N); 1H NMR (400 MHz, CDCl3): δ 9.11 (s, 1H), 8.54–8.53 (m, 1H), 8.26–8.24(m, 1H), 7.04 (d, J = 9.8 Hz, 1H), 3.94–3.82 (m, 1H), 2.69 (q, J = 6.8 Hz, 4H), 2.64–2.54 (m, 2H), 1.94–1.62 (m, 4H), 1.39 (d, J = 6.0 Hz, 3H), 1.12 (t, J = 6.8 Hz, 6H); 13C NMR (100 MHz, CDCl3): δ 147.8, 135.7, 130.3, 130.1, 124.5, 114.4, 52.0, 49.1, 46.6, 34.2, 22.9, 20.3, 10.8. HRMS (ESI) m/z: (M + H) calcd for C15H25N4O4, 325.18761; measured 325.18761.
4.1.3. 4-(5-(Diethylamino)pentan-2-ylamino)benzonitrile
(4). Pd2(dba)3 (97%, 2.4 mg, 2.50 μmol), BINAP (97%, 7.8 mg, 7.50 μmol) and sodium tert-butoxide (96%, 70.1 mg, 0.700 mmol) were added to an oven-dried pressure vessel in a glovebox. Amines 2 (97%, 99.7 mg, 0.600 mmol) and 6 (99%, 91.9 mg, 0.500 mmol) were transferred to the reaction vessel using anhydrous 1,2-dimethoxyethane (DME) (0.500 mL) under N2. Vials containing 2 and 6 were washed with anhydrous 1,2-dimethoxyethane (DME) (2 × 0.250 mL) and the contents were transferred to the reaction vessel under N2. The reaction was stirred and heated in an 80 °C oil bath for 24 h. The reaction was allowed to come to rt, evaporated, and flash chromatographed. The flash chromatography column was packed with 1% Et3N in EtOAc, and run with EtOAc, then MeOH in EtOAc (10%, then 20%, then 30%), giving a yellow glue-like material. This material was treated with NaOH (1.00 M, 5.00 mL) and transferred to a separatory funnel. The container was washed with CH2Cl2 (1.70 mL × 3) and the contents were transferred to the separatory funnel and extracted with CH2Cl2 (5.00 mL × 3). The combined organic layer was dried with Na2SO4 and evaporated to provide pure 4 as a dark orange oil (119 mg, 0.46 mmol, 92%). Rf = 0.21 (3:7 MeOH/EtOAc, the TLC was pretreated with 1% Et3N); 1H NMR (400 MHz, CDCl3): δ 7.38 (d, J = 8.6 Hz, 2H), 6.51 (d, J = 8.6 Hz, 2H), 4.68 (br, 1H), 3.51 (br, 1H), 2.56–2.47 (m, 4H), 2.43–2.40 (m, 2H), 1.62–1.51 (m, 4H), 1.20 (d, J = 6.0 Hz, 3H), 1.01 (t, J = 7.2 Hz, 6H); 13C NMR (100 MHz, CDCl3): δ 150.9, 133.8, 120.8, 112.3, 97.6, 52.8, 48.0, 46.7, 34.8, 23.6, 20.4, 11.5. HRMS (ESI) m/z: (M + H) calcd for C16H26N3, 260.21267; measured 260.21261.
4.1.4. N-(5-(Diethylamino)pentan-2-yl)pyridin-2-amine
(5) [20]. Pd(OAc)2 (98%, 1.1 mg, 5.00 µmol), Josiphos (97%, 4.3 mg, 7.50 μmol), and sodium tert-butoxide (96%, 70.1 mg, 700 mmol) were added to an oven-dried pressure vessel in a glovebox. Amines 2 (97%, 120 μL, 0.600 mmol) and 7 (99%, 57.3 mg, 0.500 mmol) were transferred to the reaction vessel using anhydrous 1,2-dimethoxyethane (DME) (0.500 mL) under N2. Vials containing 2 and 7 were washed with anhydrous 1,2-dimethoxyethane (DME) (2 × 0.250 mL) and the contents were transferred to the reaction vessel under N2. The reaction was stirred and heated in an 80 °C oil bath for 24 h. The reaction was allowed to come to rt, evaporated, and flash chromatographed. The flash chromatography column was packed with 1% Et3N in EtOAc, and run with EtOAc, then MeOH in EtOAc (10%, then 20%, then 30%), giving a brown glue-like material. This material was treated with NaOH (1.00 M, 5.00 mL) and transferred to a separatory funnel. The container originally holding the evaporated column fractions was washed with CH2Cl2 (1.70 mL × 3) and the contents were transferred to the separatory funnel and extracted with CH2Cl2 (5.00 mL × 3). The combined organic layer was dried with Na2SO4 and evaporated to give pure 5 as a dark orange oil (116 mg, 0.49 mmol, 99%). Rf = 0.30 (2:8 MeOH/EtOAc, the TLC was pretreated with 1% Et3N); 1H NMR (400 MHz, CDCl3): δ 8.06–8.05 (m, 1H), 7.39–7.35 (m, 1H), 6.52–6.49 (m, 1H), 6.34 (d, J = 8.4 Hz, 1H), 4.51 (s, br, 1H), 3.85–3.71 (m, 1H), 2.51 (q, J = 7.0 Hz, 4H), 2.45–2.40 (m, 2H), 1.59–1.47 (m, 4H), 1.20 (d, J = 6.8 Hz, 3H), 1.00 (t, J = 7.0 Hz, 6H); 13C NMR (100 MHz, CDCl3): δ 158.4, 148.3, 137.3, 112.3, 106.8, 52.9, 47.1, 46.8, 35.2, 23.7, 21.0, 11.7. (ESI) m/z: (M + H) calcd for C14H26N3, 236.21267; measured 236.21267.
4.1.5. (4-. Aminopentyl)(ethyl)amino)methanol
(11). Acetic acid (1.23 mL, 21.3 mmol) was added to a stirred mixture of compound 10 (1.23 g, 7.08 mmol) and NH3 (7.00 M in MeOH, 8.30 mL) under N2. Nitrogen was added through a needle passing through a rubber septum. The N2 line was removed after the addition to minimize the loss of NH3, and the mixture was stirred for 30 min. Sodium cyanoborohydride (0.703 g, 10.6 mmol) was dissolved in NH3 (7.00 M in MeOH, 4.00 mL) and added to the reaction mixture under N2. The N2 line was removed again and the reaction mixture was stirred. After stirring for 48 h at rt, the solvent was removed and aqueous NaOH (3.0 M, 15 mL) was added to the residue. The crude product was extracted with CH2Cl2 (15 mL × 4), dried over Na2SO4, and evaporated to give semi-pure 11 as a yellow oil (996 mg, 81%). The semi-pure 11 was satisfactory for the next step reaction, but highly pure 11 was required for biological tests. Further purification by vacuum distillation provided highly pure 11 as a colorless oil (mass of semi-pure = 0.652 mg, mass of pure = 0.566 mg, 87%). The 1 H and 13 C spectra matched those reported [12]: 1H NMR (400 MHz, CDCl3): δ 3.54 (t, J = 5.5 Hz, 2H), 2.89 (tq, J = 6.4, 6.4 Hz, 1H), 2.58 (t, J = 5.6 Hz, 2H), 2.55 (t, J = 7.0 Hz, 2H), 2.46 (t, J = 7.4 Hz, 2H), 2.10 (s, br, 2H), 1.56–1.40 (m, 2H), 1.36–1.27 (m, 2H), 1.07 (d, J = 6.4 Hz, 3H), 1.02 (t, J = 7.4 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 58.5, 55.1, 53.4, 47.3, 46.9, 37.9, 24.2, 24.1, 11.8.
4.1.6. 2-((4-((2,4-Dinitrophenyl)amino)pentyl)(ethyl)amino)ethanol
(8). Compound 3 (98%, 43.3 mg, 0.209 mmol) was dissolved in absolute ethanol (0.210 mL) at 65 °C, then 11 (40.1 mg, 0.230 mmol) was added to this clear solution dropwise and the reaction refluxed overnight. Ammonium hydroxide (15.0 M, 35.0 μL) was added to the reaction at rt. The solvent was evaporated and distilled water (2.00 mL) was added to the reaction mixture. The product was extracted with CH2Cl2 (4.00 mL × 3), dried over Na2SO4, and evaporated to give pure 8 as a brown liquid (57.7 mg, 81%). Spectral values were 1H NMR (400 MHz, CDCl3): δ 9.14 (s, 1H), 8.53 (d, J = 7.2 Hz, 1H), 8.25 (d, J = 10.0 Hz, 1H), 6.98 (d, J = 10.0 Hz, 1H), 4.51 (s, br, 1H), 3.87–3.80 (m, 1H), 3.65 (t, J = 5.0 Hz, 2H), 2.76–2.68 (m, 6H), 1.74–1.69 (m, 4H), 1.39 (d, J = 6.4 Hz, 3H), 1.11 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ 147.8, 135.7, 130.4, 130.2, 124.6, 114.3, 58.0, 55.2, 52.9, 49.2, 47.7, 34.1, 23.1, 20.4, 11.0. (ESI) m/z: (M + H) calcd for C15H25N4O5, 341.18253; found m/z 341.18230.
4.1.7. N-(4-Oxopentyl)acetamide
(12) [13]. A solution of acetyl chloride (0.355 mL, 4.94 mmol) in CH2Cl2 (5.00 mL) was added dropwise to a well-stirred solution of pyridine (0.800 mL, 9.79 mmol) in CH2Cl2 (10. mL) under N2. The mixture was stirred for 15 min. Then the solution of 2-methyl-1-pyrroline (0.488 mL, 4.90 mmol) in CH2Cl2 (12.5 mL) was added dropwise into this mixture. The mixture was stirred until it became clear orange, then it was stirred for another 10 min. Next, HCl (5%, 10.0 mL) was added to the mixture under vigorous stirring, and the mixture was continually stirred for 15 min. Sodium hydroxide solution (10.0 M, 3.00 mL) was added to the reaction mixture, followed by 10 min. of stirring. The organic layer was separated and the aqueous layer was extracted with CH2Cl2 (15.0 mL × 3). The combined organic phase was dried over Na2SO4, evaporated, and flash-chromatographed. The flash-chromatography column was packed with 1% Et3N in EtOAc and was run with EtOAc, then 8% MeOH in EtOAc, giving 12 as a white solid (434 mg, 3.03 mmol, 62%). Rf = 0.30 (1:9 MeOH/EtOAc). The 1H and 13C spectra matched the reported values [13].
4.1.8. N-(4,4-Dimethoxypentyl)acetamide
(13). This procedure is based on a general method reported for ketalization [14]. Trimethyl orthoformate (1.68 mL, 15.0 mmol), dry methanol (40.0 mL), and concentrated sulfuric acid (14.3 μL) were added to a round bottom flask containing 12 (0.430 g, 3.00 mmol) under N2. The reaction was stirred for 72 h. Next, NaHCO3 (86.0 mg) was added and stirred for 30 min. The solvent was removed and the residue was dissolved in EtOAc and filtered through a piece of cotton. The solvent was removed to give pure 13 as a brown-orange oil (568 mg, 100%). Spectral values were 1H NMR (400 MHz, CDCl3, basified with Na2CO3): δ 5.96 (s, br, 1H), 3.25 (dt, J = 6.0, 7.2 Hz, 2H), 3.17 (s, 6H), 1.97 (s, 3H), 1.65–1.51 (m, 4H), 1.26 (s, 3H); 13C NMR (100 MHz, CDCl3): δ 170.2, 101.4, 48.1, 39.8, 33.9, 24.5, 23.3, 21.0. (ESI) m/z: (M + H) calcd for C9H19NO3, 212.12626; found m/z 212.12648.
4.1.9. N-(4,4-Dimethoxypentyl)-N-ethylacetamide
(14). A solution of 13 (0.539 g, 2.85 mmol) in dry THF (3.00 mL) was added to a flask containing NaH (60% dispersion in mineral oil, 0.342 g, 8.55 mmol) under N2. The vial containing 13 was washed (2 × 1.00 mL) of THF and the contents were transferred to the reaction flask. The mixture was stirred and heated at 30 °C for 1 h. Iodoethane (0.917 mg, 5.70 mmol) was dissolved in THF (0.500 mL) and added to the reaction mixture under N2. The vial containing iodoethane was washed (2 × 0.250 mL) and the contents were transferred to the reaction flask. The reaction was heated at 30 °C for an additional 1 h. Next, the reaction was refluxed for 24 h under N2, using a condenser chilled at 0 °C by a circulating coolant. Finally, the reaction was allowed to come to rt and the solvent was evaporated. The residue was dissolved in Et2O, filtered through a cotton plug, and passed through a short-path alumina column. After solvent removal, 14 was obtained as a reddish-orange liquid mixture of rotamers (1.4:1) (619 mg, 2.85 mmol, 100%). Spectral values were 1H NMR (400 MHz, CDCl3): δ 3.41–3.23 (m, 4H), 3.18 (s, 6H (minor)), 3.17 (s, 6H (major)), 2.08 (s, 3H), 1.97 (s, 3H), 1.60–1.58 (m, 4H), 1.28 (s, 3H (minor), 1.26 (s, 3H (major)), 1.18 (t, J = 7.2 Hz, 3H (major)), 1.12 ((t, J = 7.2 Hz, 3H (minor)); 13C NMR (100 MHz, CDCl3): δ (major) 170.0, 101.5, 48.19, 48.15, 43.4, 33.8, 24.0, 21.7, 21.2, 13.1; (minor) 169.9, 101.3, 48.5, 45.5, 40.5, 33.7, 22.8, 21.5, 21.1, 14.2. (ESI) m/z: (M + H) calcd for C11H23NO3Na, 240.15756; found m/z 240.15773.
4.1.10. N-Ethyl-N-(4-oxopentylacetamide
(15). Glacial acetic acid (9.53 mL), 1.00 M HCl (4.77 mL), and THF (14.3 mL) were added to a flask containing 14 (0.589 g, 2.71 mmol), and the reaction was stirred for 24 h. The organic solvents were evaporated and NaOH solution (16.7 M, 12.0 mL) was added dropwise into the reaction vessel at 0 °C. The product was extracted with CH2Cl2 (30 mL × 4), dried over Na2SO4, and evaporated to obtain crude 15 as a dark yellow oil. Purification by flash chromatography (100% EtOAc, then 1% MeOH in EtOAc) yielded pure 15 as a mixture of rotamers (yellow oil, 242 mg, 1.41 mmol, 52%) in a ratio of 1.7:1. R f = 0.18 (1:9 MeOH/EtOAc). Spectral values for 1H NMR (400 MHz, CDCl3): δ 3.40–3.21 (m, 4H), 2.50–2.448 (m, 2H), 2.17 (s, 3H (minor)), 2.14 (s, 3H (major)), 2.10 (s, 3H (minor)), 2.08 (s, 3H (major)), 1.87–1.77 (m, 2H), 1.18 (t, J = 7.0 Hz, 3H (major)), 1.11 (t, J = 7.2 Hz, 3H (minor)); 13C NMR (100 MHz, CDCl3): δ (major) 208.4, 170.3, 44.1, 40.7, 39.9, 30.0, 21.8, 21.4, 14.0; (minor) 207.4, 170.0, 47.4, 43.1, 40.3, 30.1, 22.6, 21.5, 13.0. (ESI) m/z: (M + H) calcd for C9H18NO2Na, 172.13376; found m/z 172.13351.
4.1.11. N-(4-Aminopentyl)-N-ethylacetamide
(16). Acetic acid (0.234 mL, 4.20 mmol) was added to a stirred mixture of 15 (0.240 g, 1.40 mmol) and NH3 (7.00 M in MeOH, 1.63 mL) under N2. Nitrogen was added via a needle passing through a rubber septum. After the addition, the N2 line was removed to minimize the loss of NH3 and the mixture was stirred for 30 min. Sodium cyanoborohydride (0.139 g, 2.10 mmol) was dissolved in NH3 (7.00 M in MeOH, 0.800 mL) and added to the reaction mixture under N2. The N2 line was removed again and the reaction mixture was stirred. After 24 h, the solvent was removed and aqueous NaOH (4.00 M, 4.00 mL) was added to the residue. The crude product was extracted with CH2Cl2 (5 mL × 4), dried over Na2SO4, and evaporated to give 16 as a yellow oil (240 mg, 99%). The product was obtained as a 5:1 mixture of rotamers. Values for the 1H NMR spectrum (400 MHz, CDCl3): δ 3.40–3.22 (m, 4H), 2.96–2.86 (m, 1H (major)), 2.74–2.68 (m, 1H (minor)), 2.09 (s, 3H (major)), 2.08 (s, 3H (minor)), 1.90–1.48 (m, 4H), 1.39–1.26 (m, 2H), 1.18 (t, J = 7.0 Hz, 3H (major)), 1.11 (t, J = 7.0 Hz, 3H (minor)), 1.08–1.05 (m, 3H); 13C NMR (100 MHz, CDCl3): δ (major) 169.9, 48.5, 45.3, 43.3, 37.1, 26.8, 24.1, 14.1; (minor) 170.0, 46.79, 46.77, 40.4, 37.3, 26.0, 24.3, 21.5, 13.0. (ESI) m/z: (M + H) calcd for C9H21N2O, 173.16539; found m/z 173.16498.
4.1.12. N-(4-((2,4-Dinitrophenyl)amino)pentyl)-N-ethylacetamide
(17). Potassium carbonate (98%, 41.9 mg, 0.300 mmol), 3 (99%, 62.0 mg, 0.300 mmol), Et3N (41.8 μL, 0.300 mmol), 16 (62.0 mg, 0.360 mmol), and absolute ethanol (0.300 mL) were heated and stirred at 85 °C for 16 h. The reaction was allowed to come to rt and the solvent was removed on a rotary evaporator. The crude extract was purified by flash chromatography. The column was packed with 1:25:74 Et3N/EtOAc/hexanes and rinsed with 1:3 EtOAc/hexanes (350 mL). The column was then loaded with the crude extract and eluted with 9:1 EtOAc/hexanes, giving 17 (84.0 mg, 0.250 mmol, 83%) as a red oil. The product was obtained as a 3:1 mixture of rotamers. R f = 0.22 (EtOAc, the TLC was pretreated with 1% Et3N in EtOAc). Spectral values were 1H NMR (400 MHz, CDCl3): δ 9.14–9.12 (m, 1H), 8.56–8.46 (m, 1H), 8.29–8.24 (m, 1H), 6.98 (d, J = 10.0 Hz, 1H (major)), 6.93 (d, J = 9.6 Hz, 1H (minor)), 1.77–1.62 (m, 4H), 1.40 (d, J = 6.4 Hz, 3H (minor)), 1.36 (d, J = 6.4 Hz, 3H (major)), 1.21–1.17 (m, 3H (major)), 1.13–1.10 (s, 3H (minor)); 13C NMR (100 MHz, CDCl3): δ (major) 170.4, 147.9, 135.8, 130.5, 124.66, 114.3, 49.0, 44.4, 43.3, 33.8, 24.4, 21.5, 20.6, 14.1 (minor) 169.8, 147.7, 130.6, 130.2, 124.69, 114.0, 49.2, 48.0, 40.5, 33.9, 25.7, 21.8, 20.6, 13.1. HRMS (ESI) m/z: (M + H) calcd for C15H23N4O5, 339.16688; measured 339.16673.
4.2. Bioassay
H293 kidney epithelial cells were obtained from the American Tissue Culture Collection. Cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) plus penn/strep. Cultures were maintained in a 37 °C water-jacketed incubator with 5% CO2. When needed, cells were removed from the plate using trypsin, gently pelleted by centrifugation (2 rpm/min. at room temperature), aspirated, and re-suspended in the desired media. Unless noted otherwise, FBS was omitted because experiments showed it was not required for these short-term metabolic assays (data not shown). The other media used was L-15, which contained amino acids but replaced glucose with 5 mM galactose. L-15 also lacked NaHCO3, which was added to the same concentration as that in DMEM (44 mM). Its presence is essential to buffer the media and prevent acidification in the CO2 incubator.
In general, the immediate short-term effects of the indicated (H)CQ analogs on ATP levels were analyzed by removing growing cells from a p100mm plate using trypsin, washing with PBS, and re-suspending in the indicated media. After counting using a hemocytometer, 25–50 k cells in 100 microliters of media were distributed in a 96-well white flat-bottom non-treated polystyrene assay plate. The plate was typically incubated for 30 min. at 37 °C in a CO2 incubator to allow metabolic pathways to adapt to the media. Different concentrations of the drug of interest were then added, followed by shaking (700 RPM/10 sec), and the cells returned to the incubator. Incubation times were generally between 1 and 2 h so that the direct and immediate effects on ATP production could be evaluated. ATP levels were determined by adding 10 microliters of CellTiterGlo (CTG) reagent directly to the wells, followed by 5 min. shaking at 700 RPM. The light emitted by the ATP-dependent luciferase was quantitated using a photo-luminometer. Samples were tested in duplicate and the standard deviation was determined.
In some cases, the longer-term effects of the drugs were determined by seeding cells in tissue-culture-treated 96-well plates so that they could attach overnight, then treating them with the drug for longer times (e.g., 18 h). Drug effects on ATP were determined using the CTG described above, while viability was measured by the addition of 10 microliters of CellTiterBlue resazurin reagent. In the latter case, cells were incubated for 2 h at 37 °C in the CO2 incubator to allow viable cells to convert resazurin to fluorescent resorufin. Fluorescence (ex560/em590) was measured on a plate reader. In all cases, drug effects on the biological assays were evaluated in the absence of cells to ensure they were not targeting assay components or reactions.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/molecules28031161/s1, Experimental procedures for synthesizing 9 and 10, NMR spectra of isolated compounds and biochemical data for Table S1.
Author Contributions
Z.W. performed all synthesis experiments. R.J.S. conceived the project and conducted the bioassays. S.R.H. designed the synthesis, advised Z.W., and wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by an Interdisciplinary Project Grant from the University of Tulsa.
Institutional Review Board Statement
Not Applicable.
Informed Consent Statement
Not Applicable.
Data Availability Statement
Data presented in this study are available in the paper and the Supplementary Materials.
Acknowledgments
TU supported this research. HRMS studies were performed in the Oklahoma State University Center for Genomics and Proteomics.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Sample Availability
Samples of the compounds 1, 2, 4, 5, 8, 11, and 17 are available from the authors.
References
- Wolf, R.; Wolf, D.; Ruocco, V. Antimalarials: Unapproved uses or indications. Clin. Dermatol. 2000, 18, 17–35. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, D.J. Theories on malarial pigment formation and quinoline action. Int. J. Parasitol. 2002, 32, 1645–1653. [Google Scholar] [CrossRef] [PubMed]
- Coban, C. The host targeting effect of chloroquine in malaria. Curr. Opin. Immunol. 2020, 66, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Al-Bari, M.A.A. Chloroquine analogues in drug discovery: New directions of uses, mechanisms of actions and toxic manifestations from malaria to multifarious diseases. J. Antimicrob. Chemother. 2015, 70, 1608–1621. [Google Scholar] [CrossRef] [PubMed]
- Wunderlich, J.; Rohrbach, P.; Dalton, J.P. The malaria digestive vacuole. Front. Biosci. 2012, 4, 1424–1448. [Google Scholar] [CrossRef]
- Savarino, A.; Boelaert, J.R.; Cassone, A.; Majori, G.; Cauda, R. Effects of chloroquine on viral infections: An old drug against today’s diseases. Lancet Infect. Dis. 2003, 3, 722–727. [Google Scholar] [CrossRef] [PubMed]
- Sheaff, R.J.; Mitchell, I. Antiviral Therapeutic Drug Combinations. U.S. Patent Application US20220031719A1, 3 February 2022. Available online: https://image-ppubs.uspto.gov/dirsearch-public/print/downloadPdf/20220031719 (accessed on 22 January 2023).
- Mathews, C.K.; Holde, K.E.; Ahern, K.G. Biochemistry, 3rd ed.; Addison Wesley Longman: San Francisco, CA, USA, 1999. [Google Scholar]
- Bunnett, J.F.; Zahler, R.E. Aromatic Nucleophilic Substitution Reactions. Chem. Rev. 1951, 49, 273–412. [Google Scholar] [CrossRef]
- Hartwig, J.F. Evolution of a Fourth Generation Catalyst for the Amination and Thioetherification of Aryl Halides. Acc. Chem. Res. 2008, 41, 1534–1544. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, J.P.; Wagaw, S.; Buchwald, S.L. An Improved Catalyst System for Aromatic Carbon−Nitrogen Bond Formation: The Possible Involvement of Bis(Phosphine) Palladium Complexes as Key Intermediates. J. Am. Chem. Soc. 1996, 118, 7215–7216. [Google Scholar] [CrossRef]
- Yu, E.; Mangunuru, H.P.R.; Telang, N.S.; Kong, C.J.; Verghese, J.; Gilliland, S.E., III.; Ahmad, S.; Dominey, R.N.; Gupton, B.F. High-yielding continuous-flow synthesis of antimalarial drug hydroxychloroquine. Beilstein J. Org. Chem. 2018, 14, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Nenajdenko, V.G.; Zakurdaev, E.P.; Prusov, E.V.; Balenkova, E.S. Convenient synthesis of melatonin analogues: 2- and 3-substituted -N-acetylindolylalkylamines. Tetrahedron 2004, 60, 11719–11724. [Google Scholar] [CrossRef]
- Dong, J.-L.; Yu, L.-S.; Xie, J.-W. A Simple and Versatile Method for the Formation of Acetals/Ketals Using Trace Conventional Acids. ACS Omega 2018, 3, 4974–4985. [Google Scholar] [CrossRef] [PubMed]
- MarvinSketch, version 17.1.2.0; ChemAxon: Budapest, Hungry, 2017. Available online: https://www.chemaxon.com (accessed on 23 January 2023).
- Loomis, W.F.; Lipmann, F. Reversible inhibition of the coupling between phosphorylation and oxidation. J. Biol. Chem. 1948, 173, 807. [Google Scholar] [CrossRef] [PubMed]
- Abebe, F.A.; Hopkins, M.D.; Vodnala, S.N.; Sheaff, R.J.; Lamar, A.A. Development of a Rapid In Vitro Screening Assay Using Metabolic Inhibitors to Detect Highly Selective Anticancer Agents. ACS Omega 2021, 6, 18333–18343. [Google Scholar] [CrossRef] [PubMed]
- Aguer, C.; Gambarotta, D.; Mailloux, R.J.; Moffat, C.; Dent, R.; McPherson, R.; Harper, M.-E. Galactose Enhances Oxidative Metabolism and Reveals Mitochondrial Dysfunction in Human Primary Muscle Cells. PLoS ONE 2011, 6, e28536. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Faccenda, D.; Campanella, M. Pharmacological advances in mitochondrial therapy. Ebiomedicine 2021, 65, 103244. [Google Scholar] [CrossRef] [PubMed]
- Ashley, J.N.; Grove, J.F. 200. Contributions to the chemistry of synthetic antimalarials. Part I. Some δ-diethylamino-α-methylbutylamino-derivatives of pyridine and thiazole. J. Chem. Soc. (Resumed) 1945, 768–769. [Google Scholar] [CrossRef] [PubMed]



















Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).