


Fractioning and Compared 1H NMR and GC-MS Analyses of Lanolin Acid Components

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Method Validation
2.1.1. Calibration Range and Linearity
2.1.2. Accuracy and Precision
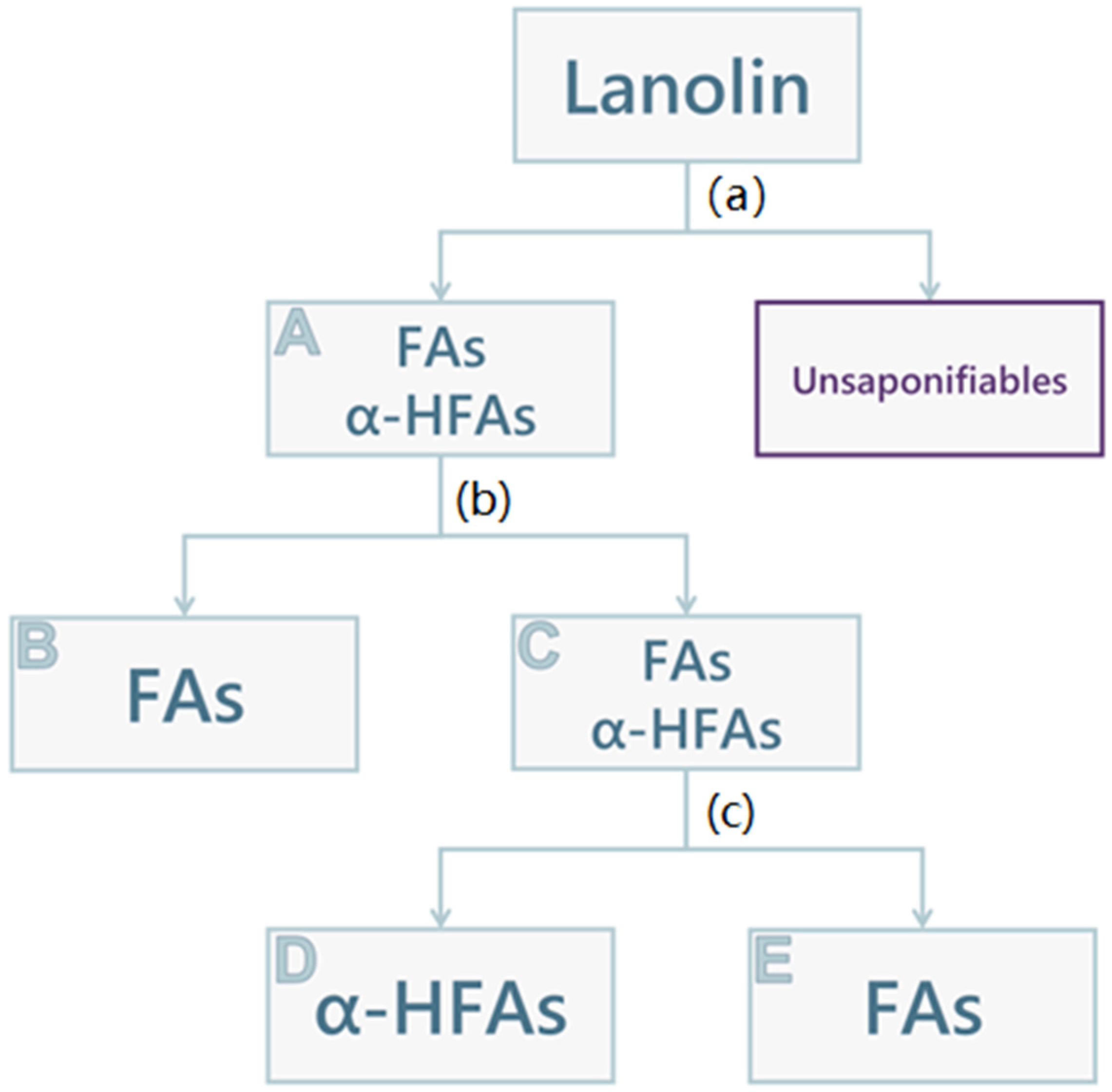
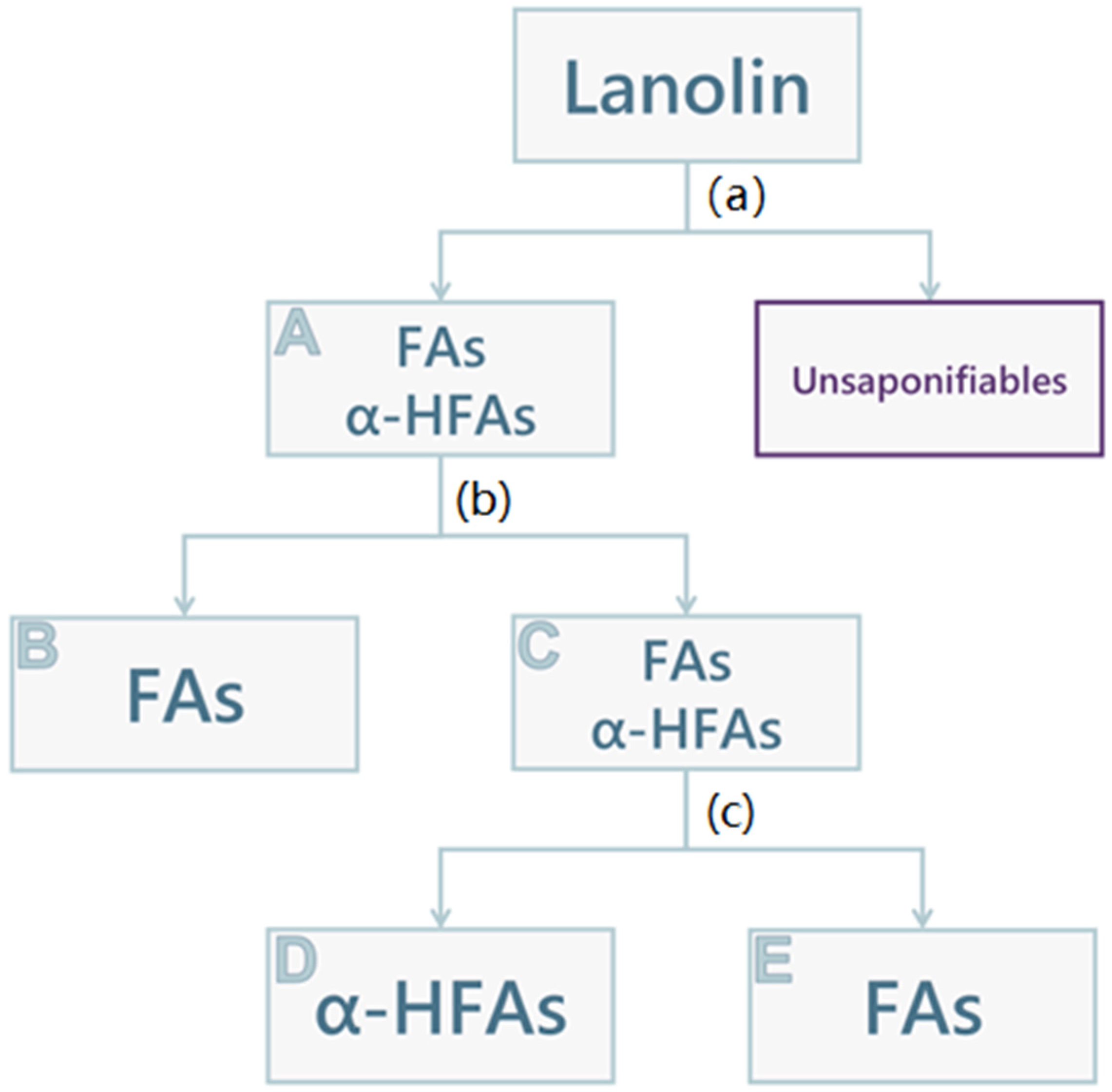
2.2. Lanolin Acid Components Fractioning
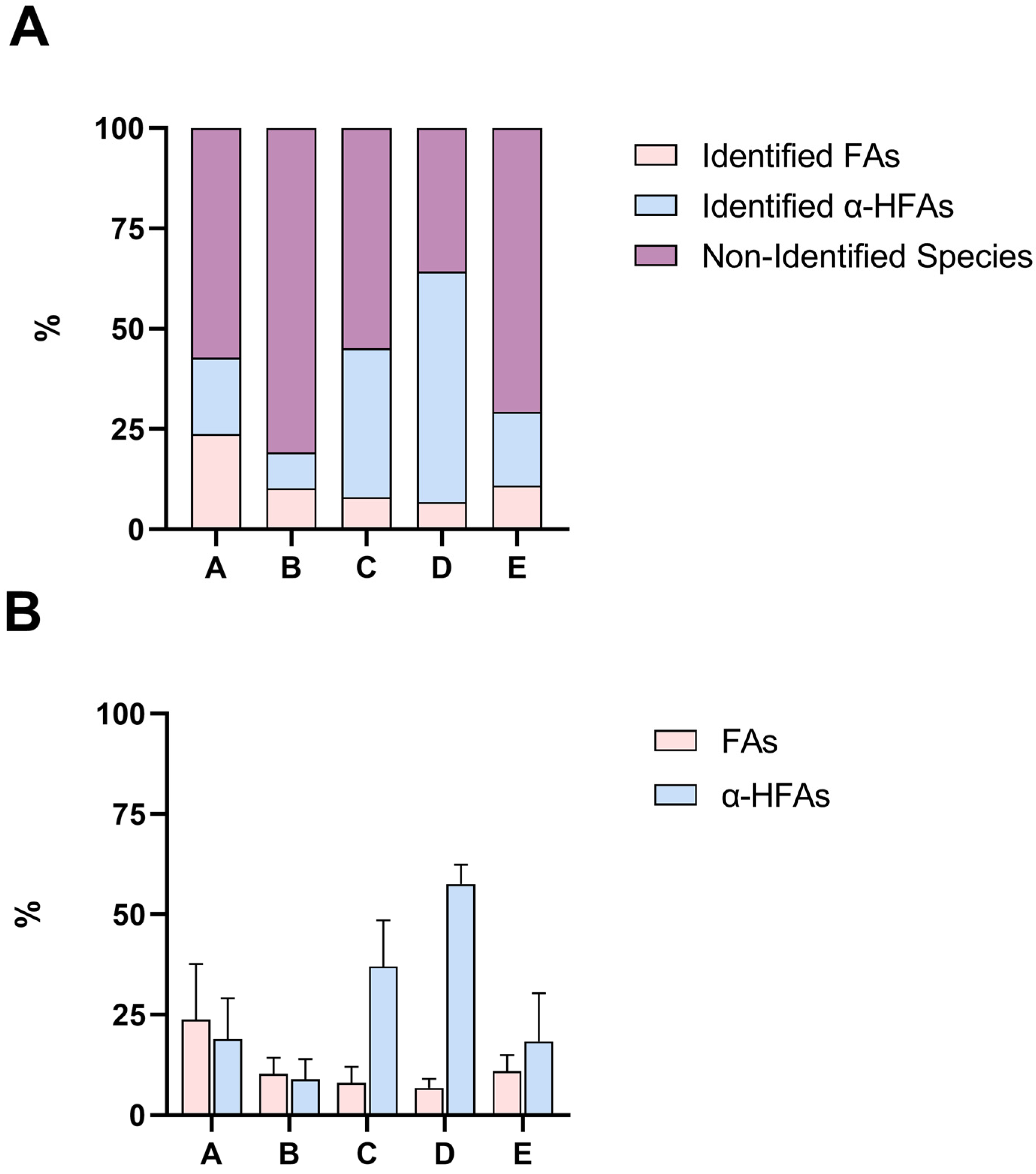
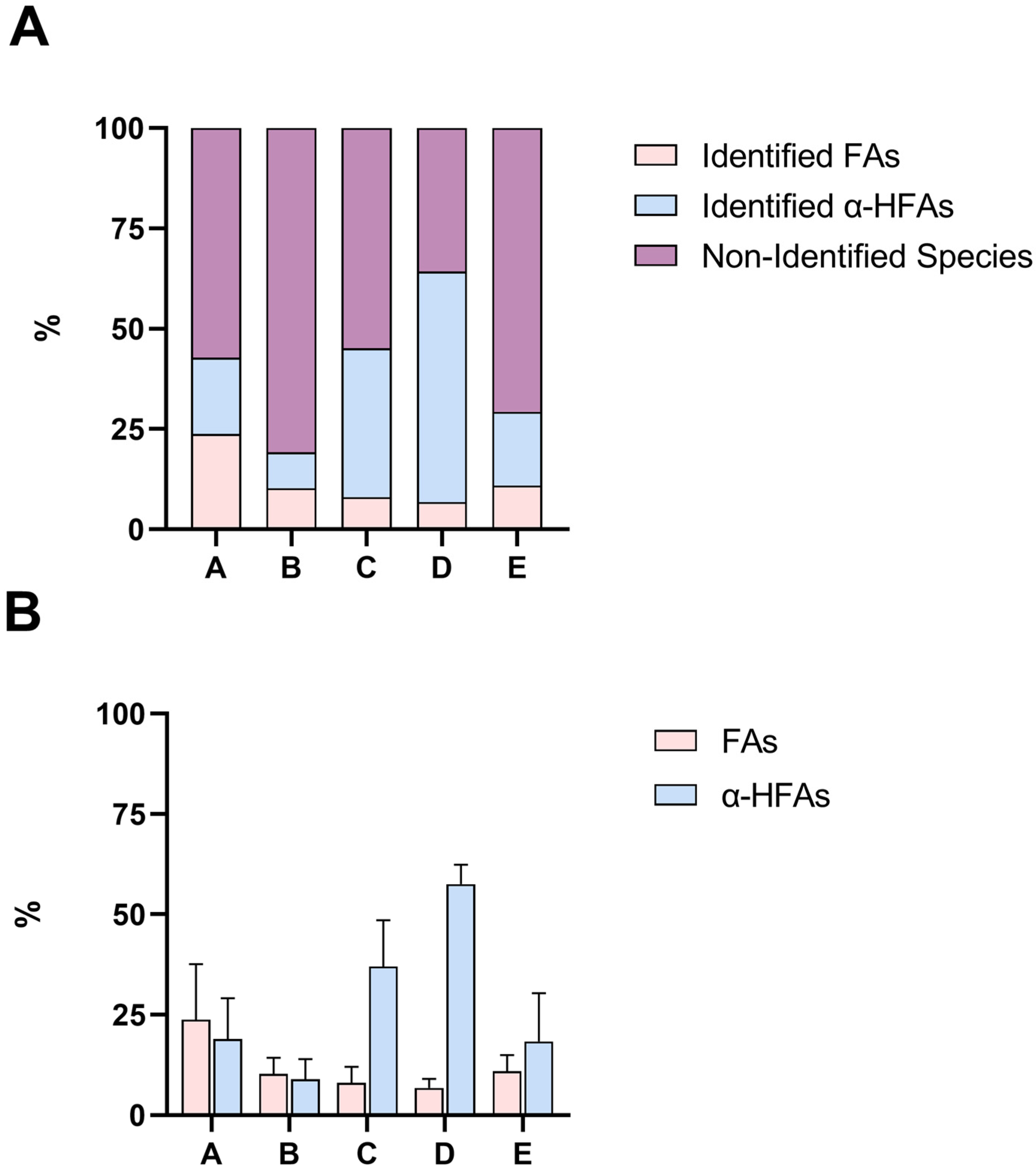
2.3. GC-MS Analysis of Lanolin Acid Components in Portions A–E
3. Materials and Methods
3.1. Chemicals
3.2. Preparation of Standard Solutions and Calibration and Quality Control Samples
3.3. Gas Chromatography Conditions
3.3.1. Fatty Acids Chromatography
3.3.2. α-Hydroxy Fatty Acids Chromatography
3.4. GC-MS Method Validation
3.4.1. Linearity
3.4.2. Sensitivity and Specificity
3.4.3. Precision and Accuracy
3.5. Lanolin Acid Components Fractioning
3.6. 1H NMR Spectroscopy Analysis
3.7. Sample Preparation for Gas Chromatography
3.7.1. Fatty Acids
3.7.2. α-Hydroxy Fatty Acids
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hamish, F.; Tom, Q.; Clementine, O. Food Waste Index Report 2021; UNEP: Nairobi, Kenya, 2021; ISBN 9789280738513. [Google Scholar]
- Ramos, J.L.; García-Lorente, F.; Valdivia, M.; Duque, E. Green Biofuels and Bioproducts: Bases for Sustainability Analysis. Microb. Biotechnol. 2017, 10, 1111–1113. [Google Scholar] [CrossRef] [PubMed]
- Sánchez Muñoz, S.; Rocha Balbino, T.; Mier Alba, E.; Gonçalves Barbosa, F.; Tonet de Pier, F.; Lazuroz Moura de Almeida, A.; Helena Balan Zilla, A.; Antonio Fernandes Antunes, F.; Terán Hilares, R.; Balagurusamy, N.; et al. Surfactants in Biorefineries: Role, Challenges & Perspectives. Bioresour. Technol. 2022, 345, 126477. [Google Scholar] [CrossRef] [PubMed]
- Campos de Bomfim, A.S.; Oliveira, D.M.d.; Voorwald, H.J.C.; Benini, K.C.C.d.C.; Dumont, M.J.; Rodrigue, D. Valorization of Spent Coffee Grounds as Precursors for Biopolymers and Composite Production. Polymers 2022, 14, 437. [Google Scholar] [CrossRef] [PubMed]
- Sawangkeaw, R.; Ngamprasertsith, S. A Review of Lipid-Based Biomasses as Feedstocks for Biofuels Production. Renew. Sustain. Energy Rev. 2013, 25, 97–108. [Google Scholar] [CrossRef]
- Petek, B.; Marinšek Logar, R. Management of Waste Sheep Wool as Valuable Organic Substrate in European Union Countries. J. Mater. Cycles Waste Manag. 2021, 23, 44–54. [Google Scholar] [CrossRef]
- Reutelingsperger, C.M.H.G.; Erutan, B.V. A Method to Wash Greasy Wool, a Method to Separate Lanolin from the Said Greasy Wool, Wool and Lanolin Obtainable by These Methods. U.S. Patent Application No. 14/350,129, 11 April 2013. [Google Scholar]
- Domínguez, C.; Jover, E.; Garde, F.; Bayona, J.M.; Erra, P. Characterization of Supercritical Fluid Extracts from Raw Wool by TLC-FID and GC-MS. J. Am. Oil Chem. Soc. 2003, 80, 717–724. [Google Scholar] [CrossRef]
- Scanes, C.G. Animal Attributes Exploited by Humans (Nonfood Uses of Animals). In Animals and Human Society; Academic Press: Cambridge, MA, USA, 2018; pp. 13–40. [Google Scholar] [CrossRef]
- Rissmann, R.; Oudshoorn, M.H.M.; Kocks, E.; Hennink, W.E.; Ponec, M.; Bouwstra, J.A. Lanolin-Derived Lipid Mixtures Mimic Closely the Lipid Composition and Organization of Vernix Caseosa Lipids. Biochim. Biophys. Acta–Biomembr. 2008, 1778, 2350–2360. [Google Scholar] [CrossRef] [PubMed]
- Jover, E.; Adahchour, M.; Bayona, J.M.; Vreuls, R.J.J.; Brinkman, U.A.T. Characterization of Lipids in Complex Samples Using Comprehensive Two-Dimensional Gas Chromatography with Time-of-Flight Mass Spectrometry. J. Chromatogr. A 2005, 1086, 2–11. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, A.; Behera, J. Comprehensive View on Chemistry, Manufacturing & Applications of Lanolin Extracted from Wool Pretreatment. Am. J. Eng. Res. 2014, 3, 33–43. [Google Scholar]
- Dei Cas, M.; Paroni, R.; Saccardo, A.; Casagni, E.; Arnoldi, S.; Gambaro, V.; Saresella, M.; Mario, C.; La Rosa, F.; Marventano, I.; et al. A Straightforward LC-MS/MS Analysis to Study Serum Profile of Short and Medium Chain Fatty Acids. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2020, 1154, 121982. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Zhang, Q.; Li, N.; Pu, Y.; Wang, B.; Zhang, T. Comparison of Critical Methods Developed for Fatty Acid Analysis: A Review. J. Sep. Sci. 2017, 40, 288–298. [Google Scholar] [CrossRef]
- Chiu, H.H.; Kuo, C.H. Gas Chromatography-Mass Spectrometry-Based Analytical Strategies for Fatty Acid Analysis in Biological Samples. J. Food Drug Anal. 2020, 28, 60–73. [Google Scholar] [CrossRef] [PubMed]
- Kramer, J.K.G.; Fellner, V.; Dugan, M.E.R.; Sauer, F.D.; Mossoba, M.M.; Yurawecz, M.P. Evaluating Acid and Base Catalysts in the Methylation of Milk and Rumen Fatty Acids with Special Emphasis on Conjugated Dienes and Total Trans Fatty Acids. Lipids 1997, 32, 1219–1228. [Google Scholar] [CrossRef] [PubMed]
- Murrieta, C.M.; Hess, B.W.; Rule, D.C. Comparison of Acidic and Alkaline Catalysts for Preparation of Fatty Acid Methyl Esters from Ovine Muscle with Emphasis on Conjugated Linoleic Acid. Meat Sci. 2003, 65, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, V.; Pallavicini, M.; Tibhe, G.; Roda, G.; Arnoldi, S.; Monguzzi, L.; Zoccola, M.; Di Nardo, G.; Gilardi, G.; Bolchi, C. Synthesis of α-Hydroxy Fatty Acids from Fatty Acids by Intermediate α-Chlorination with TCCA under Solvent-Free Conditions: A Way to Valorization of Waste Fat Biomasses. ACS Omega 2021, 6, 31901–31906. [Google Scholar] [CrossRef] [PubMed]
- Bolchi, C.; Valoti, E.; Straniero, V.; Ruggeri, P.; Pallavicini, M. From 2-Aminomethyl-1,4-Benzodioxane Enantiomers to Unichiral 2-Cyano- and 2-Carbonyl-Substituted Benzodioxanes via Dichloroamine. J. Org. Chem. 2014, 79, 6732–6737. [Google Scholar] [CrossRef] [PubMed]
- Pallavicini, M.; Bolchi, C.; Fumagalli, L.; Piccolo, O.; Valoti, E. Highly Efficient Racemisation of a Key Intermediate of the Antibiotic Moxifloxacin. Tetrahedron Asymmetry 2011, 22, 379–380. [Google Scholar] [CrossRef]
- Giuriato, D.; Correddu, D.; Catucci, G.; Di Nardo, G.; Bolchi, C.; Pallavicini, M.; Gilardi, G. Design of a H2O2-Generating P450SPα Fusion Protein for High Yield Fatty Acid Conversion. Protein Sci. 2022, 31, e4501. [Google Scholar] [CrossRef] [PubMed]
- European Medicines Agency. ICH Q2(R2) Validation of Analytical Procedures—Scientific Guideline. Available online: https://www.ema.europa.eu/en/ich-q2r2-validation-analytical-procedures-scientific-guideline (accessed on 17 December 2022).
- Biermann, U.; Bornscheuer, U.T.; Feussner, I.; Meier, M.A.R.; Metzger, J.O. Fatty Acids and Their Derivatives as Renewable Platform Molecules for the Chemical Industry. Angew. Chem. Int. Ed. 2021, 60, 20144–20165. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| Compound | ng Injected (ng inj) |
|---|---|
| FA 12:0 | 0.125–5 |
| FA 14:0 | 0.125–5 |
| FA 16:0 | 0.5–10 |
| FA 18:0 | 0.5–10 |
| FA 20:0 | 0.125–5 |
| FA 22:0 | 0.125–5 |
| FA 24:0 | 0.5–10 |
| FA 14:1 | 0.5–10 |
| FA 16:1 | 0.5–10 |
| FA 18:2 | 0.5–10 |
| FA 18:1 | 0.5–10 |
| α-HFA 14:0 | 0.5–20 |
| α-HFA 16:0 | 0.5–20 |
| α-HFA 18:0 | 0.5–20 |
| α-HFA 20:0 | 0.5–20 |
| Compound | r2 | CV (%) | LOD (ng inj) | LOQ (ng inj) |
|---|---|---|---|---|
| FA 12:0 | 0.999 | 10.2 | 0.02 | 0.08 |
| FA 14:0 | 0.998 | 11.1 | 0.02 | 0.05 |
| FA 16:0 | 0.997 | 14.0 | 0.01 | 0.04 |
| FA 18:0 | 0.999 | 13.5 | 0.09 | 0.28 |
| FA 20:0 | 0.999 | 8.5 | 0.05 | 0.18 |
| FA 22:0 | 0.999 | 14.4 | 0.03 | 0.09 |
| FA 24:0 | 0.999 | 13.8 | 0.08 | 0.26 |
| FA 14:1 | 0.994 | 14.6 | 0.10 | 0.33 |
| FA 16:1 | 0.999 | 5.1 | 0.11 | 0.36 |
| FA 18:2 | 0.998 | 9.9 | 0.08 | 0.28 |
| FA 18:1 | 0.998 | 14.2 | 0.10 | 0.32 |
| α-HFA 14:0 | 0.996 | 12.0 | 0.04 | 0.13 |
| α-HFA 16:0 | 0.999 | 10.4 | 0.05 | 0.16 |
| α-HFA 18:0 | 0.997 | 12.1 | 0.10 | 0.34 |
| α-HFA 20:0 | 0.997 | 11.2 | 0.09 | 0.30 |
| Compound | Amount (ng inj) | Precision (%) | Accuracy (%) |
|---|---|---|---|
| FA 12:0 | 0.125 | 5.0 | 6.8 |
| 0.5 | 2.5 | 6.8 | |
| 5 | 3.7 | 2.3 | |
| FA 14:0 | 0.125 | 3.1 | 11.9 |
| 0.5 | 4.1 | 3.5 | |
| 5 | 2.9 | 0.7 | |
| FA 16:0 | 0.5 | 4.9 | 14.7 |
| 2.5 | 11.8 | 12.1 | |
| 10 | 4.1 | 2.8 | |
| FA 18:0 | 0.5 | 3.9 | 4.3 |
| 2.5 | 4.1 | 5.7 | |
| 10 | 7.1 | 1.3 | |
| FA 20:0 | 0.125 | 8.6 | 8.2 |
| 0.5 | 1.8 | 11.8 | |
| 5 | 4.7 | 2.5 | |
| FA 22:0 | 0.125 | 5.5 | 3.7 |
| 0.5 | 10.3 | 9.7 | |
| 5 | 13.9 | 2.9 | |
| FA 24:0 | 0.5 | 8.3 | 10.3 |
| 2.5 | 7.2 | 4.7 | |
| 10 | 1.4 | 1.0 | |
| FA 14:1 | 0.5 | 10.9 | 14.0 |
| 2.5 | 10.5 | 10.1 | |
| 10 | 3.5 | 2.4 | |
| FA 16:1 | 0.5 | 7.1 | 8.1 |
| 2.5 | 14.1 | 12.4 | |
| 10 | 4.8 | 8.1 | |
| FA 18:2 | 0.5 | 2.2 | 10.6 |
| 2.5 | 7.0 | 4.9 | |
| 10 | 3.9 | 12.4 | |
| FA 18:1 | 0.5 | 4.1 | 5.4 |
| 2.5 | 7.1 | 6.4 | |
| 10 | 2.8 | 5.3 | |
| α-HFA 14:0 | 0.5 | 2.4 | 11.3 |
| 5 | 4.3 | 9.4 | |
| 20 | 14.8 | 0.5 | |
| α-HFA 16:0 | 0.5 | 3.7 | 14.7 |
| 5 | 14.4 | 12.3 | |
| 20 | 11.2 | 1.6 | |
| α-HFA 18:0 | 0.5 | 8.5 | 3.8 |
| 5 | 1.6 | 11.3 | |
| 20 | 3.2 | 3.1 | |
| α-HFA 20:0 | 0.5 | 14.8 | 5.3 |
| 5 | 4.6 | 7.3 | |
| 20 | 3.7 | 1.8 |
| Molar Fraction | |||
|---|---|---|---|
| Portion | Weight (g) | α-HFAs | FAs |
| A | 3.5 | 0.43 | 0.57 |
| B | 1.2 | 0 | 1 |
| C | 2 | 0.52 | 0.48 |
| D | 0.75 | 1 | 0 |
| E | 1 | 0.28 | 0.72 |
| Stock Solution | Concentration (µg/mL) |
|---|---|
| FAs | 10 |
| α-HFAs | 10 |
| IS | 1 |
| Injected Amount (ng) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compound | 0.125 | 0.25 | 0.5 | 1.0 | 1.25 | 2 | 2.5 | 5 | 10 | 20 |
| FA 12:0 | × | × | × | × | × | × | ||||
| FA 14:0 | × | × | × | × | × | × | ||||
| FA 16:0 | × | × | × | × | × | |||||
| FA 17:0 (IS) | × | |||||||||
| FA 18:0 | × | × | × | × | × | |||||
| FA 20:0 | × | × | × | × | × | × | ||||
| FA 22:0 | × | × | × | × | × | × | ||||
| FA 24:0 | × | × | × | × | × | |||||
| FA 14:1 | × | × | × | × | × | |||||
| FA 16:1 | × | × | × | × | × | |||||
| FA 18:2 | × | × | × | × | × | |||||
| FA 18:1 | × | × | × | × | × | |||||
| α-HFA 14:0 | × | × | × | × | × | × | ||||
| α-HFA 16:0 | × | × | × | × | × | × | ||||
| α-HFA 18:0 | × | × | × | × | × | × | ||||
| α-HFA 20:0 | × | × | × | × | × | × | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morano, C.; Dei Cas, M.; Bergamaschi, R.F.; Palmisano, E.; Pallavicini, M.; Bolchi, C.; Roda, G.; Casati, S. Fractioning and Compared 1H NMR and GC-MS Analyses of Lanolin Acid Components. Molecules 2023, 28, 1635. https://doi.org/10.3390/molecules28041635
Morano C, Dei Cas M, Bergamaschi RF, Palmisano E, Pallavicini M, Bolchi C, Roda G, Casati S. Fractioning and Compared 1H NMR and GC-MS Analyses of Lanolin Acid Components. Molecules. 2023; 28(4):1635. https://doi.org/10.3390/molecules28041635
Chicago/Turabian StyleMorano, Camillo, Michele Dei Cas, Roberta F. Bergamaschi, Erika Palmisano, Marco Pallavicini, Cristiano Bolchi, Gabriella Roda, and Sara Casati. 2023. "Fractioning and Compared 1H NMR and GC-MS Analyses of Lanolin Acid Components" Molecules 28, no. 4: 1635. https://doi.org/10.3390/molecules28041635
APA StyleMorano, C., Dei Cas, M., Bergamaschi, R. F., Palmisano, E., Pallavicini, M., Bolchi, C., Roda, G., & Casati, S. (2023). Fractioning and Compared 1H NMR and GC-MS Analyses of Lanolin Acid Components. Molecules, 28(4), 1635. https://doi.org/10.3390/molecules28041635